

ABSTRACT
Ferroptosis is a newly characterized form of programmed cell death, which is driven by the lethal accumulation of lipid peroxides catalyzed by the intracellular bioactive iron. Targeted induction of ferroptotic cell death holds great promise for therapeutic design against other therapy-resistant cancers. To date, multiple post-translational modifications have been elucidated to impinge on the ferroptotic sensitivity. Here we report that the Ser/Thr protein kinase ATM, the major sensor of DNA double-strand break damage, is indispensable for ferroptosis execution. Pharmacological inhibition or genetic ablation of ATM significantly antagonizes ferroptosis. Besides, ATM ablation-induced ferroptotic resistance is largely independent of its downstream target TRP53, as cells defective in both Trp53 and Atm are still more insensitive to ferroptotic inducers than the trp53 single knockout cells. Mechanistically, ATM dominates the intracellular labile free iron by phosphorylating NCOA4, facilitating NCOA4-ferritin interaction and therefore sustaining ferritinophagy, a selective type of macroautophagy/autophagy specifically degrading ferritin for iron recycling. Our results thus uncover a novel regulatory circuit of ferroptosis comprising ATM-NCOA4 in orchestrating ferritinophagy and iron bioavailability.
Abbreviations: AMPK: AMP-activated protein kinase; ATM: ataxia telangiectasia mutated; BSO: buthionine sulphoximine; CDKN1A: cyclin-dependent kinase inhibitor 1A (P21); CQ: chloroquine; DFO: deferoxamine; DFP: deferiprone; Fer: ferrostatin-1; FTH1: ferritin heavy polypeptide 1; GPX4: glutathione peroxidase 4; GSH: glutathione; MEF: mouse embryonic fibroblast; NCOA4: nuclear receptor coactivator 4; PFTα: pifithrin-α; PTGS2: prostaglandin-endoperoxide synthase 2; Slc7a11: solute carrier family 7 member 11; Sul: sulfasalazine; TFRC: transferrin receptor; TRP53: transformation related protein 53.
KEYWORDS: ATM, ferritinophagy, ferroptosis, iron, NCOA4
Introduction
During the past decade, ferroptosis has been characterized as a novel form of non-apoptotic programmed cell death [1]. This iron-driven cell death sharply differs from other well characterized forms of regulated cell death at morphological, genetic and biochemical levels [2]. A line of evidence has emerged that unrestrained ferroptosis is implicated in the pathogenesis of a group of human diseases including tissue ischemia-reperfusion injury and neurodegeneration [2], while targeted induction of ferroptosis holds great promise for therapeutic design against other therapy-resistant cancers [3]. The chemical nature of ferroptosis is fundamentally defined by the excessive production of polyunsaturated phospholipid hydroperoxides and the reduced capacity of lipid peroxidation repairing machineries [4]. Aberrant accumulation of lipid peroxides substantially disrupts the ion gradients across the bio-membrane, decreases membrane fluidity, and increases membrane permeability, leading to the bio-membrane disruption and initiating downstream death program [5]. Lipid peroxidation could be enzymatically catalyzed by lipoxygenases [6] and POR (cytochrome p450 oxidoreductase) [7,8], as well as non-enzymatically catalyzed through Fenton chemistry by using iron as the catalyst [9]. In a reverse reaction, GPX4 (glutathione peroxidase 4) [10,11], AIFM2/FSP1 (apoptosis-inducing factor, mitochondrion-associated 2) [12,13] and DHODH (dihydroorotate dehydrogenase) [14] could catalyze the reduction of peroxidized phospholipids, which constitute the three major lipid repairing systems to defense against ferroptosis.
Multiple metabolic and signaling pathways have been elucidated to manipulate ferroptotic sensitivity [15]. By building up the enzymatic cofactor heme and iron-sulfur cluster, iron constitutes the active center of numerous enzymes critical for cellular growth and metabolism. However, excessive iron bioavailability could augment oxidative damage through supporting Fenton reaction and facilitating the generation of lipid peroxidation, although how iron catalyzes lipid peroxidation and drives ferroptosis is only partially understood. Iron overload is one of the most canonical characteristics of ferroptosis. Cellular iron uptake, iron storage, iron metabolism, iron efflux and iron turnover coordinate the cellular labile iron pool and dominate lipid peroxidation [15]. TFRC (transferrin receptor) is the major protein mediating the cellular iron uptake through clathrin-dependent endocytosis of iron-loaded transferrin [16]. It has been reported that knockdown of TFRC sharply antagonizes ferroptosis by lowering down the cellular iron uptake [17]. After uptake, cytosolic iron is utilized for the biosynthesis of heme and iron-sulfur cluster [16]. Deletion of NFS1 (NFS1 cysteine desulfurase), the cysteine desulfurase abstracting the sulfur atom from cysteine for iron-sulfur cluster assembly, predisposes cancer cells to ferroptosis and slows tumor growth [18], while enhanced iron-sulfur cluster assembly by overexpression of ISCU (iron-sulfur cluster assembly enzyme) attenuates dihydroartemisinin-induced ferroptosis in leukemia cells [19]. In addition, cellular iron is largely sequestered by ferritin nanocages in cytosol or by FTMT (ferritin mitochondrial) specific inside the mitochondria. The ferritin-bound iron could be mobilized through ferritin degradation. During iron starvation, the receptor protein NCOA4 (nuclear receptor coactivator 4) selectively associates with ferritin and the autophagosomal protein MAP1LC3/LC3 (microtubule associated protein 1 light chain 3), therefore delivers ferritin to be encompassed by the phagophore, facilitates ferritin degradation in autolysosome and recycles the ferritin-bound iron to support the cellular labile iron pool. In iron replete condition, the ferritinophagy receptor NCOA4 is ubiquitinated for proteasomal degradation and thus the ferritinophagy is maintained at basal level [20,21]. Two independent studies elucidated that NCOA4-mediated ferritinophagy is activated during ferroptosis induction, while disruption of ferritinophagy by depletion of general autophagy components or specific ferritinophagy receptor NCOA4 sharply reduces the cellular labile iron and antagonizes ferroptosis [22,23]. Furthermore, genetic knockout of Fth1 (ferritin heavy polypeptide 1) specifically in cardiomyocyte aggregates high-iron diet-induced cardiac injury and hypertrophic cardiomyopathy. Ferroptosis is pathologically implicated in this heart injury as evidenced by the reduced glutathione (GSH) and the elevated lipid peroxidation in cardiomyocyte-specific fth1 null mice. Furthermore, the ferroptosis specific inhibitor, ferrostatin-1 (Fer), could alleviate these pathological phenotypes [24]. Similarly, knockout of Fth1 specifically in neurons leads to a higher vulnerability to ferroptosis-associated traumatic brain injury [25]. Collectively, these studies demonstrated a critical importance of ferritin in restraining labile iron pool, mitigating lipid peroxidation and antagonizing ferroptosis by sequestering intracellular iron. Besides, the lipid metabolism, including lipid synthesis, storage, transport and oxidation, substantially dominates the cellular lipidomic signature and finely tunes the levels of peroxidable polyunsaturated fatty acids, thus determines ferroptotic sensitivity [15].
Post-translational modifications influence the conformation, activity, stability and sub-cellular localization of the targeted proteins. Dynamic phosphorylation mediated by kinase and phosphatase is one of the most important post-translational modifications, which almost regulates all cellular behaviors. To date, several kinases have been documented to engage in the ferroptotic regulation. AMP-activated protein kinase (AMPK), the cellular ATP sensor, is one of the most important guardians for cell metabolism [26]. It has been reported that energy stress could antagonize ferroptotic cell death through activating AMPK, leading to slow down the biosynthesis of intracellular peroxidable polyunsaturated fatty acids by phosphorylating and inhibiting ACACA/ACC1 (acetyl-Coenzyme A carboxylase alpha)-ACACB/ACC2 [27,28]. However, another study reported that AMPK activation could facilitate ferroptosis by phosphorylating the autophagy core component BECN1/Beclin 1, and this phosphorylation strengthens BECN1 interacting with and inhibiting the cystine/glutamate antiporter system Xc-, leading to the reduced cystine uptake and restrained GSH biosynthesis [29]. MTOR (mechanistic target of rapamycin kinase) also manipulates the ferroptotic sensitivity in tumor cells. Similarly, the effect is context-dependent [30,31]. Furthermore, other studies revealed that the MAP3K5/ASK1-MAPK14/p38 kinase cascade [32] and PRKC/PKC phosphorylation HSPB1 [33] could also manipulate ferroptosis in corresponding circumstance. Recently, Chen and colleagues performed a genetic kinase screen using a siRNA kinome library to systemically identify critical kinases essential for ferroptosis. The authors found some alternative kinases which could potentially determine ferroptotic sensitivity. Among them, the Ser/Thr protein kinase ATM (ataxia telangiectasia mutated), an apical activator and transducer of the DNA damage response in the face of DNA double-strand breaks [34], is indispensable for ferroptosis execution in MDA-MB-231 breast cancer cells triggered by cystine deprivation. ATM dominates the intracellular labile iron pool by regulating the transcription of iron storage- and export-associated genes through manipulating the nuclear translocation of MTF1 (metal regulatory transcription factor 1) [35]. To seek the potential protein kinases and the corresponding post-translational phosphorylated modifications in the ferroptosis regulation, we performed a kinase inhibitor screen, and similarly we found that the ATM kinase is indispensable for ferroptosis execution. Genetic ablation or pharmacological inhibition of ATM antagonizes ferroptosis in multiple cell lines in response to diverse ferroptosis inducers, which is greatly independent of its downstream target TRP53 (transformation related protein 53; the mouse homolog of human TP53/p53). Mechanistically, we found that ATM kinase dominates the intracellular labile iron pool, alternatively by phosphorylating ferritinophagy receptor NCOA4 and thus manipulating ferritinophagy for iron turnover. This study further suggests the possibility of manipulating ATM activity to maintain tissue fitness by finely tuning human diseases-associated ferroptosis.
Results
ATM kinase is indispensable for ferroptotic cell death
To seek critical protein kinases and the corresponding phosphorylated modifications in the ferroptosis regulation, we performed a kinase inhibitor screen in mouse embryonic fibroblast (MEF) cells. KU55933, the specific inhibitor of ATM kinase, was listed as a top ferroptosis inhibitor in this screen. As expected, exposure of erastin and RSL3, the two typical ferroptosis inducers, triggered notable cell death in a dose-dependent manner, and this cell death could be almost fully antagonized by the specific ferroptosis inhibitor Fer. Similarly, supplementation of KU55933 strikingly decreased erastin- and RSL3-induced cell death, as shown by the cell viability assay and PI staining (Figure S1A-S1D). Furthermore, KU55933 administration substantially decreased lipid peroxidation induced by erastin and RSL3 (Figure S1E and S1F). ATM inhibition suppressing ferroptosis was not restricted in MEF cells. Supplementation of KU55933 also significantly decreased erastin- and RSL3-induced ferroptosis in HT-1080 human fibrosarcoma cells and other cancer cells (Figure S1G, S1H, and data not shown). Notably, ATM inhibition restored the colony formation capacity of HT-1080 under erastin or RSL3 challenge (Figure S1I and S1J). Erastin targets and inhibits the cystine/glutamate antiporter system Xc-, leading to the restrained cystine uptake and decelerated GSH biosynthesis. GSH exhaustion is one of the typical hallmarks of ferroptosis. Interestingly, ATM inhibition by KU55933 failed to impinge on GSH depletion induced by erastin (Figure S1K). Collectively, these data suggest that inhibition of ATM activity by KU55933 antagonizes ferroptosis in a GSH-independent manner.
To confirm the ferroptosis-regulatory capacity of ATM kinase, the atm knockout MEF cells were utilized [36]. We found that atm knockout protected MEF from cell death triggered by erastin and RSL3, as evidenced by the cell viability assay (Figure 1A and 1B). Supplementation of erastin or RSL3 resulted in obvious PI-positive staining in WT cells, which could be almost completely blocked by Fer. atm knockout significantly antagonized this PI-positive staining as shown by the fluorescence microscopy scanning and flow cytometry analysis (Figure 1C-1F). Furthermore, exposure of erastin and RSL3 induced excessive production of lipid peroxides, which was dramatically suppressed in atm knockout MEF cells (Figure 1G and 1H). Similar to the ATM inhibition, ATM deletion had no influence on cellular GSH level, both under basal and erastin-challenged conditions (Figure S1L). Collectively, these data confirm the indispensable role of ATM kinase in ferroptotic program. In addition, we also confirmed the relevance of ATM kinase in other inducers-induced ferroptosis. Buthionine sulphoximine (BSO) and sulfasalazine (Sul) could initiate ferroptosis by depleting cellular GSH through irreversibly inhibiting γ-glutamylcysteine synthase and potently targeting system Xc-, respectively. atm deletion also counteracted BSO- and Sul-induced ferroptosis, as shown by the cell viability assay (Figure 1I and 1J), PI staining (Figure 1K-1N), and lipid peroxidation analysis (Figure 1O and 1P).
Figure 1.
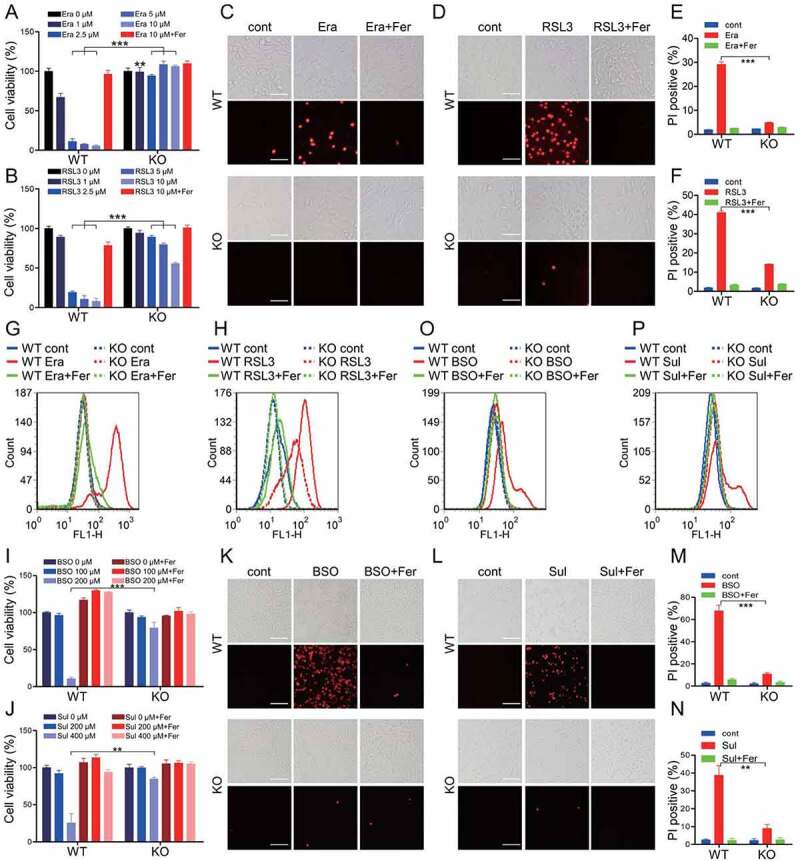
ATM is indispensable for ferroptotic cell death. (A, B) WT and atm KO MEF cells were treated with indicated dose of erastin (Era) (A) or RSL3 (B) with or without 10 μM ferrostatin-1 (Fer). The relative cell viability was measured with CCK8 assay. (C-F) WT and atm KO MEF cells were treated with 2.5 μM erastin (C, E) or 1 μM RSL3 (D, F) with or without 10 μM Fer. The cell death was analyzed with PI staining followed by fluorescence imaging (C, D) and flow cytometry (E, F). Representative images are presented. Scale bar: 50 μm. (G, H) WT and atm KO MEF cells were treated with 2.5 μM erastin (G) or 1 μM RSL3 (H) with or without 10 μM Fer. The lipid peroxidation was measured by C11 BODIPY 581/591 staining and flow cytometry analysis. (I, J) WT and atm KO MEF cells were treated with indicated dose of BSO (I) or Sul (J) with or without 10 μM Fer. The relative cell viability was analyzed with CCK8 assay. (K-N) WT and atm KO MEF cells were treated with 200 μM BSO (K, M) or 400 μM Sul (L, N) with or without 10 μM Fer. The cell death was analyzed with PI staining followed by fluorescence imaging (K, L) and flow cytometry (M, N). Representative images are presented. Scale bar: 100 μm. (O, P) WT and atm KO MEF cells were treated with 200 μM BSO (O) or 400 μM Sul (P) with or without 10 μM Fer. The lipid peroxidation was measured. Data represent the mean ± SEM from three independent experiments. ** P < 0.01, *** P < 0.001 was determined by student’s t-test.
To dissect the ferroptotic regulation by ATM in cancer cells, the human fibrosarcoma HT-1080 cells stably expressing shRNA targeting to ATM were constructed (Figure S2A). Similarly, we found that ATM silence in HT-1080 protected cells from both erastin- and RSL3-induced cell death as evidenced by the PI staining and cell viability assay (Figure S2B-S2G). The elevated transcription of PTGS2/COX2 (prostaglandin-endoperoxide synthase 2) is regarded as a typical hallmark of ferroptosis [10]. ATM knockdown in HT-1080 resulted in a decreased expression of PTGS2, both under basal and erastin- or RSL3-challenged conditions (Figure S2H and S2I). Notably, ATM silence also protected the colony formation capacity of HT-1080 under the supplementation of erastin or RSL3 (Figure S2J and S2K). Collectively, these findings suggest that ATM kinase is indispensable for ferroptosis execution.
ATM determines ferroptotic cell death independent of TRP53
Tumor suppressor TRP53 is one of the most important downstream targets of ATM kinase. TRP53 phosphorylation by ATM has been widely documented to dominate cell cycle and DNA damage-repair [37]. More importantly, TRP53 has been reported to manipulate ferroptosis through multiple mechanisms beyond its well-characterized regulatory effects on apoptosis and autophagy, although discrepancy still exists [38]. To dissect the potential role of TRP53 in ATM-mediated ferroptosis, we analyzed the ferroptotic sensitivity of atm knockout MEFs and the control MEFs in the genetic background of Trp53 ablation. Herein, the AP29 cells are trp53 single knockout, while AP26 cells are atm trp53 double knockout (Figure 2A) [36]. Compared to the WT MEFs, the trp53 single knockout AP29 cells were more resistant to erastin- and RSL3-induced cell death to a certain extent (Figure 2B and 2C), suggesting a ferroptosis-promoting activity of TRP53, which is consistent with some previous studies [17,39–41]. When compared to the trp53 single knockout AP29 cells, the atm trp53 double-knockout AP26 cells appeared much more insensitive to erastin- and RSL3-induced ferroptotic cell death, as shown by the cell viability assay (Figure 2B and 2C), PI staining (Figure 2D) and lipid peroxidation analysis (Figure 2E and 2F). Notably, a decreased expression of Ptgs2 was observed in AP26 cells with exposure of erastin and RSL3 (Figure 2G and 2H).
Figure 2.
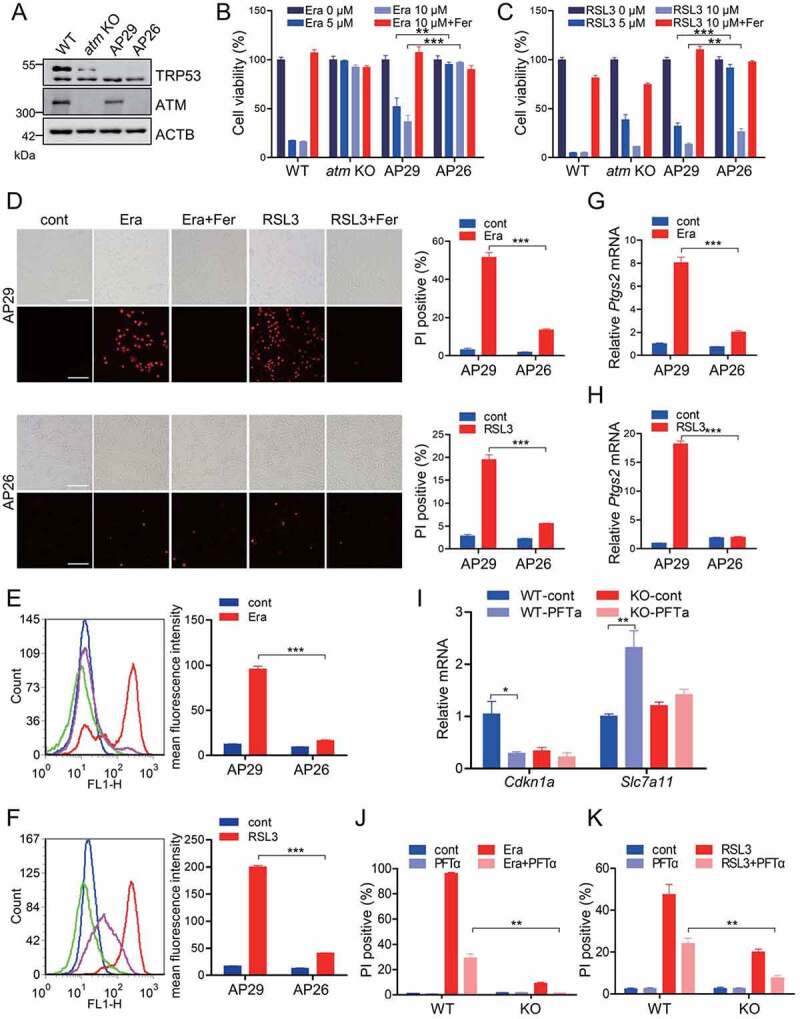
ATM determines ferroptotic cell death independent of TRP53. (A) Western blot for the WT, atm KO, AP29 (trp53 KO) and AP26 (atm trp53 double KO) MEF cells. (B, C) WT, atm KO, AP29 and AP26 MEF cells were treated with the indicated dose of erastin (B) or RSL3 (C) with or without 10 μM Fer. The relative cell viability was analyzed with CCK8 assay. (D-H) AP29 and AP26 cells were treated with 5 μM erastin or 1 μM RSL3 with or without 10 μM Fer. The cell death was analyzed with PI staining followed by fluorescence imaging and flow cytometry (D). Representative images are presented. Scale bar: 100 μm. The lipid peroxidation was measured by C11 BODIPY 581/591 staining and flow cytometry (E, F). The relative mRNA level of Ptgs2 was analyzed by quantitative RT-PCR (G, H). (I) WT and atm KO MEF cells were treated with TRP53 inhibitor PFTα (25 μM), and the relative mRNA levels of Cdkn1a/p21 and Slc7a11 were analyzed by quantitative RT-PCR. (J, K) WT and atm KO cells were treated with 5 μM erastin (J) or 1 μM RSL3 (K) with or without 25 μM PFTα. The cell death was analyzed with PI staining followed by flow cytometry analysis. Data represent the mean ± SEM from three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 was determined by student’s t-test.
TRP53 has been reported to facilitate ferroptosis by transcriptionally inhibiting the expression of Slc7a11 (solute carrier family 7 member 11; encoding the catalytic subunit of system Xc-) or by transcriptionally elevating the expressions of Sat1 (spermidine/spermine N1-acetyltransferase 1; which correlates with the expression of Alox15 for lipid peroxidation) and Gls2 (glutaminase 2; a mitochondrial glutaminase essential for glutaminolysis to fuel TCA cycle, mitochondrial respiration, and ATP generation). To further confirm the relevance of TRP53 in ATM-mediated ferroptosis, pifithrin-α (PFTα), a specific TRP53 inhibitor suppressing TRP53-dependent transactivation of TRP53-responsive genes, was utilized. PFTα supplementation led to an obvious downregulation of Cdkn1a/p21 (cyclin dependent kinase inhibitor 1A (P21); an important TRP53 targeting gene) and upregulation of Slc7a11 in MEF cells (Figure 2I), suggesting that PFTα did inhibit the transcriptional factor activity of TRP53. A marginal downregulation of Cdkn1a and upregulation of Slc7a11 was observed in atm KO cells during PFTα challenge (Figure 2I), which may be due to the lower expression of TRP53 in atm KO cells (Figure 2A). Notably, PFTα supplementation drove ferroptotic resistance both in WT and atm KO cells (Figure 2J, 2K and Figure S3A-S3C), further suggesting the ferroptosis-promoting capacity of TRP53. Additionally, a longer treatment of erastin or RSL3 resulted in more cell death both in WT and atm KO cells. Specifically, when compared to the PFTα-treated WT cells, the PFTα-treated atm KO cells performed a much higher insensitivity to both erastin and RSL3 (Figure 2J, 2K and Figure S3A-S3C). Furthermore, a similar phenotype was observed in human colon cancer cell line HCT116. Pharmacological inhibition of ATM activity by KU55933 also protects HCT116 TP53−/− cells from erastin-induced ferroptosis, as shown by the PI staining (Figure S3D). Collectively, these studies demonstrate that ATM determines ferroptotic cell death, at least partially, independent of TRP53.
It is well understood that ATM is an apical sensor and signaling transducer of the DNA double-strand breaks response [34]. We found that both erastin and RSL3 exposure led to the elevated phosphorylation of ATM at Ser1981 residue in MEF cells and HT-1080 cells in a dose-dependent manner (Figure S4A and S4B), suggesting ATM activation during ferroptosis induction. γH2AX (phosphorylated H2A.X variant histone) and TRP53BP1 (transformation related protein 53 binding protein 1) foci are regarded as two typical hallmarks of DNA damage response [34]. Neither erastin nor RSL3 could induce γH2AX or TRP53BP1 foci formation, while bleomycin, a family of natural glycopeptides clinically used as antitumor agents, triggered apparent γH2AX and TRP53BP1 foci formation (Figure S4C-S4F). Besides DNA damage, ATM kinase could also be activated by oxidative stress. More importantly, the underlying mechanisms for DNA damage- and oxidative stress-induced ATM activation are entirely different. During the presence of DNA damage, the MRE11, RAD50 and NBN/NBS1 form a stable MRN complex in the damage site, where this MRN complex recruits ATM and facilitates ATM activation [42]. Mutation or ablation of MRN component MRE11 markedly reduces DNA damage-induced ATM activation [42]. Herein, we found that the erastin- and RSL3-induced ATM activation could not be disturbed by Mirin (Figure S4G), a MRE11 chemical inhibitor which could suppress ATM activation in response to DNA damage [43]. On the contrary, Mirin potently inhibited camptothecin-induced ATM phosphorylation (Figure S4G). Meanwhile, administration of Mirin failed to change erastin- and RSL3-induced ferroptosis as shown by the PI staining (Figure S4H). Furthermore, the nuclear/cytosol fractionation assay showed that the activated ATM in response to erastin and RSL3 mainly localized in the cytosolic fraction (Figure S4I). Consistent to the previous report [44], we confirmed that 4-oxo-2-nonenal (4-ONE), the end product of lipid peroxides, could activate ATM kinase (Figure S4J). These data collectively highlight that the ferroptosis inducers activate ATM kinase independent on DNA damage response, and possibly dependent on the lipid peroxides and/or the downstream metabolic products.
ATM manipulates ferritinophagy
The intracellular labile free iron is one of the most important factors determining the redox homeostasis. Excessive iron bioavailability supports the Fenton reaction and facilitates the production of lipid peroxidation to initiate ferroptosis. The cell-permeable FerroOrange is a novel fluorescent iron sensor that enables live-cell fluorescent imaging of intracellular ferrous iron. Ferroptosis inducers led to an elevated fluorescence of FerroOrange in WT MEF cells, while this fluorescence was largely attenuated in atm KO MEF cells under the identical treatments (Figure 3A-3C). Furthermore, a similar phenotype was also observed in HT-1080 with stable ATM knockdown (Figure 3D-3F), suggesting the notion that ATM dominates intracellular labile free iron during ferroptosis execution. To characterize the underlying mechanism for which ATM dominates the intracellular iron, the iron metabolic and regulatory proteins were analyzed in atm ablated cells. The iron uptake-associated TFRC and SLC11A2/DMT1 (solute carrier family 11 member 2), as well as the iron regulatory proteins ACO1/IRP1 (aconitase 1) and IREB2/IRP2 (iron responsive element binding protein 2), had only marginal or no obvious changes when atm is ablated in MEF and HT-1080 cells, both under basal and erastin- or RSL3-challenged conditions (Figure S5). However, the iron efflux exporter SLC40A1/FPN (solute carrier family 40 member 1) and the iron storage-associated FTH1 were higher expressed in atm KO MEFs and ATM knockdown HT-1080 (Figure 3G-3L and Figure S5), which is similar to a recent study [35].
Figure 3.
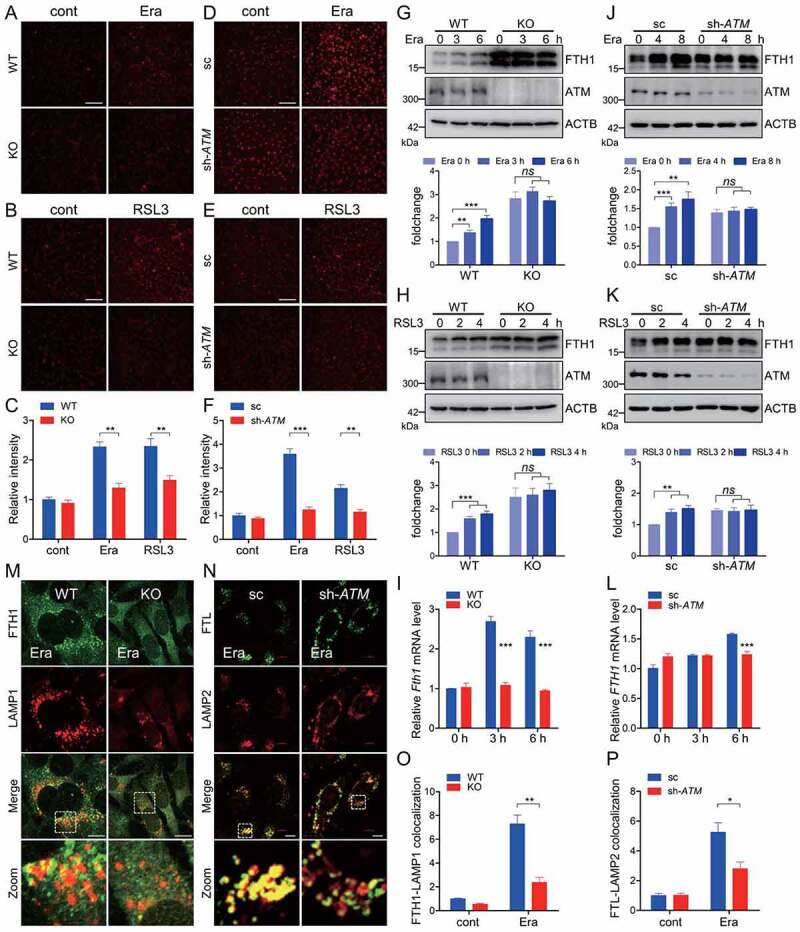
ATM determines ferroptosis-associated ferritinophagy. (A-C) WT and atm KO cells were treated with 2.5 μM erastin (A) or 1 μM RSL3 (B), and the labile free iron was analyzed with FerroOrange staining. Representative images are presented. Scale bar: 200 μm. The relative fluorescence intensity was quantified (C). (D-F) Stable ATM knockdown HT-1080 and the scramble control cells were treated with 5 μM erastin (D) or 2.5 μM RSL3 (E), and the labile free iron was analyzed with FerroOrange staining. Representative images are presented. Scale bar: 200 μm. The relative fluorescence intensity was quantified (F). (G-I) WT and atm KO cells were treated with 2.5 μM erastin (G, I) or 1 μM RSL3 (H) for the indicated time. Cells were then harvested for immunoblotting analysis (G, H) and quantitative RT-PCR (I) to check the FTH1 expression. Quantification of the relative FTH1 protein levels in (G) and (H) is shown in the corresponding below histogram. (J-L) Stable ATM knockdown HT-1080 and the scramble control cells were treated with 5 μM erastin (J, L) or 2.5 μM RSL3 (K) for the indicated time. Cells were then harvested for immunoblotting analysis (J, K) and quantitative RT-PCR (L) to check the FTH1 expression. Quantification of the relative FTH1 protein levels in (J) and (K) is shown in the corresponding below histogram. (M-P) WT and atm KO cells (M, O), as well as sc and ATM knockdown HT-1080 cells (N, P) were treated with erastin. Cells were then fixed, stained by indicated antibodies, and imaged with a confocal microscope. Representative images are presented (M, N). Scale bar: 10 μm. The relative colocalization between ferritin and lysosome was quantified (O, P). Data represent the mean ± SEM from three independent experiments, each with >200 cells counted per condition. * P < 0.05, ** P < 0.01, *** P < 0.001, ns non-significant, was determined by student’s t-test.
Cytosolic iron is largely sequestered by ferritin nanocages. It was reported that NCOA4-mediated ferritinophagy is activated during ferroptosis induction, while depletion of general autophagy components or specific ferritinophagy receptor NCOA4 sharply decreases iron mobilization, leading to decrease cellular labile iron pool and antagonize ferroptosis [22,23]. Erastin exposure resulted in an increase in FTH1 protein in WT MEF cells (Figure 3G). The elevated Fth1 expression at mRNA level was also observed during erastin exposure in WT MEF cells, as shown by the quantitative RT-PCR assay (Figure 3I). This is consistent with the previous notion that ferritinophagy-mediated ferritin degradation and subsequent iron release would transcriptionally enhance the expression of Fth1 [22,23]. FTH1 was highly expressed in atm KO MEF cells in steady state. However, no obvious increase in FTH1 protein or mRNA was observed in atm KO cells during erastin exposure, suggesting a decelerated ferritin turnover during ATM ablation (Figure 3G and 3I). Additionally, this was similar in atm KO cells with RSL3 challenge (Figure 3H). To further confirm the important role of ATM in determining ferroptosis-associated ferritin turnover, the ATM knockdown HT-1080 cells were utilized and we found that FTH1 was also highly expressed when ATM is silenced (Figure 3J and 3K). Similarly, erastin and RSL3 exposure led to an increase in FTH1 in the control cells. ATM knockdown resulted in an inert FTH1 elevation in HT-1080 cells both under erastin and RSL3 exposure (Figure 3J and 3K). Furthermore, erastin exposure led to an obvious increase in the transcription of FTH1, which was not observed in ATM knockdown HT-1080 (Figure 3L). Immunofluorescence staining showed that erastin treatment facilitated the co-localization of ferritin and lysosome, while this co-localization was sharply restrained in atm KO MEF cells and in ATM knockdown HT-1080 cells (Figure 3M-3P). Collectively, these data suggest that ATM dominates the ferroptosis-associated ferritinophagy for ferritin turnover.
Ferritinophagy was originally regarded as a critical regulatory mechanism for iron homeostasis. Iron starvation stabilizes NCOA4 to boost ferritinophagy for ferritin degradation and iron recycling. Whether ATM only manipulates the specific ferroptosis-associated ferritinophagy or ATM could generally regulate the iron starvation-induced ferritinophagy is the next open question. Exposure of iron chelators, including deferiprone (DFP) and deferoxamine (DFO), resulted in rapid degradation of FTH1 in MEF cells. atm knockout blunted both DFP- and DFO-induced FTH1 degradation (Figure 4A and 4B). Additionally, pre-treatment of ATM inhibitor, KU55933, also blocked this FTH1 degradation (Figure 4C and 4D). Moreover, the iron chelator facilitated the colocalization of FTH1 and lysosomal marker LAMP1, as well as the colocalization of ferritinophagy receptor NCOA4 and autophagosomal marker GFP-MAP1LC3. However, both these colocalizations were significantly suppressed during ATM inhibition (Figure 4E and 4F).
Figure 4.
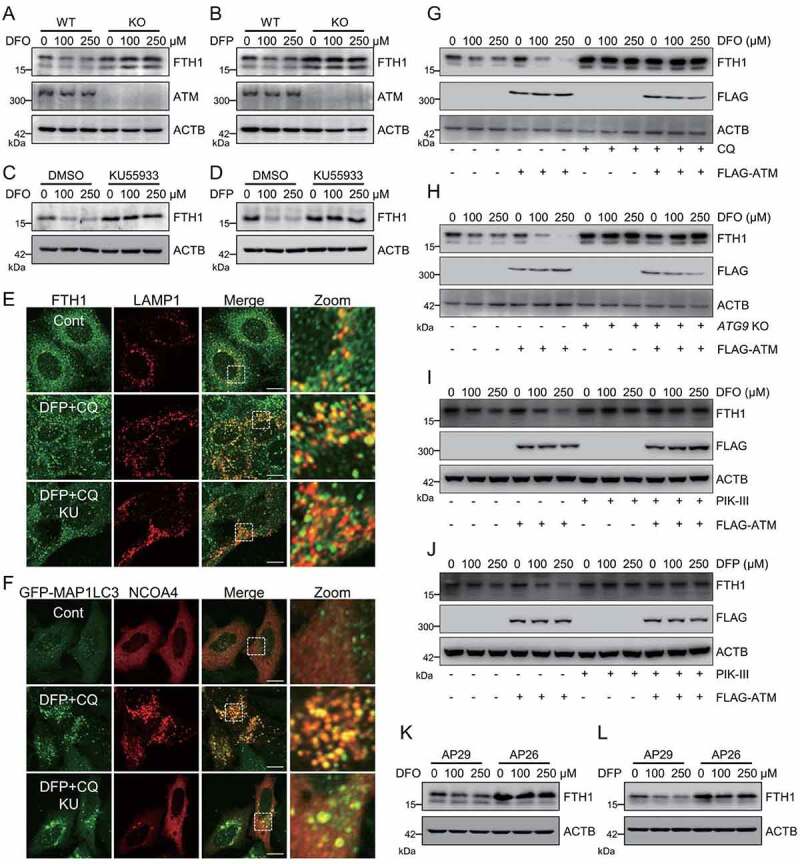
ATM dominates iron starvation-induced ferritinophagy. (A, B) WT and atm KO cells were treated with indicated dose of iron chelator DFO (A) or DFP (B) for 6 h. Cells were then harvested for immunoblotting analysis. (C, D) Cells were pretreated with 10 μM KU55933 for 2 h, and then treated with indicated dose of DFO (C) or DFP (D) for 6 h. Cells were then harvested for immunoblotting analysis. (E) MEF cells were pretreated with 10 μM KU55933, and then treated with 250 μM DFP plus autophagy inhibitor CQ (10 μM). Cells were then fixed, stained by anti-FTH1 and anti-LAMP1 antibody, and imaged with a confocal microscope. Representative images are presented. Scale bar: 10 μm. (F) MEF cells were transfected with GFP-MAP1LC3. 24 h after transfection, the cells were treated with 10 μM KU55933 for 2 h, following by 250 μM DFP and 10 μM CQ. Cells were then fixed, stained by anti-NCOA4 antibody, and imaged with a confocal microscope. Representative images are presented. Scale bar: 10 μm. (G-J) WT (G, I, J) and ATG9 KO (H) cells were transfected with FLAG-ATM, and then treated with indicated dose of iron chelators with or without CQ (G) or PIK3C3/VPS34 inhibitor PIK-III (I, J). Cells were then harvested for immunoblotting analysis. (K, L) AP29 and AP26 cells were treated with indicated dose of DFO (K) or DFP (L). Cells were then harvested for immunoblotting analysis.
Ectopic expression of ATM accelerated DFO-induced FTH1 degradation (Figure 4G). The autophagy inhibitor chloroquine (CQ) sharply prevented this DFO-induced FTH1 degradation, even in the ATM overexpressed cells (Figure 4G). It was reported that ferritinophagy is a specific type of selective autophagy, which is highly dependent on certain autophagy proteins, especially ATG9 and PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), but not on other critical autophagy components, including ATG16L1, ATG14, ATG7, and ATG3 [45]. We found that ATG9 knockout [46] and PIK3C3/VPS34 inhibition by its inhibitor PIK-III [21] could disturb iron chelators-induced FTH1 degradation, both in the control cells and in ATM overexpressed cells (Figure 4H-4J). These data further confirmed the regulatory role of ATM kinase in iron starvation-induced ferritinophagy. Previous studies reported that TRP53 is functionally important for autophagy [47,48] and even ferritinophagy [49,50]. However, ATM manipulating iron starvation-induced ferritinophagy seemed TRP53-independent, as the decelerated FTH1 degradation was still observed in atm trp53 double knockout AP26 cells when compared to the trp53 single knockout AP29 cells (Figure 4K and 4L). Taken together, these findings indicate that ATM is a master regulator of ferritinophagy.
ATM phosphorylates ferritinophagy receptor NCOA4
During iron starvation, the ferritinophagy receptor NCOA4 escapes from the E3 ubiquitin ligase HERC2-mediated ubiquitination and proteasomal degradation [51]. The stabilized NCOA4 thus binds to ferritin via a direct interaction between the conserved arginine residue (Arg23) of FTH1 and the C-terminal domain of NCOA4 [51]. During erastin supplementation, an apparent increase in NCOA4-FTH1 interaction was observed as evidenced by the co-immunoprecipitation analysis (Figure 5A). At the meantime, an obvious NCOA4 phosphorylation was detected by using the pan-phosphorylated-serine (p-Ser) antibody. Importantly, this NCOA4 phosphorylation was elevated during erastin challenge (Figure 5A). We thus assumed that ATM kinase may be responsible for this NCOA4 phosphorylation. Erastin treatment led to the elevation in NCOA4 phosphorylation, while this phosphorylation was almost completely blocked in atm knockout MEF cells, atm trp53 double knockout AP26 cells and ATM knockdown HT-1080 cells, when compared to the corresponding control cells (Figure 5B-5D), suggesting that ATM kinase is crucial for this NCOA4 phosphorylation in response to erastin.
Figure 5.
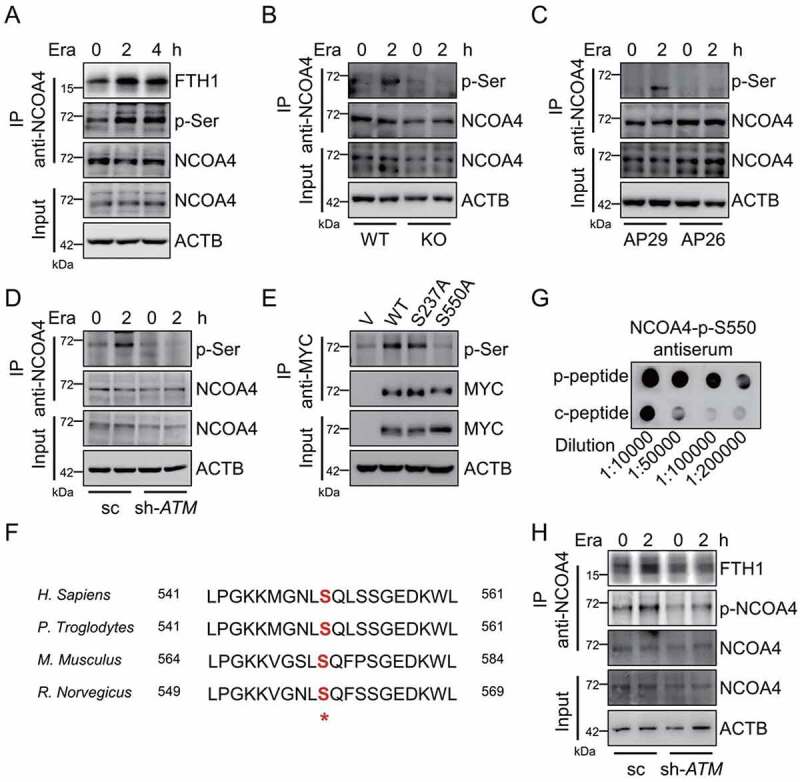
ATM phosphorylates ferritinophagy receptor NCOA4. (A) MEF cells were treated with 2.5 μM erastin. The cells were harvested for Co-IP by using anti-NCOA4 antibody. The input and IP fractions were immunoblotted to analyze NCOA4 phosphorylation and NCOA4-FTH1 interaction. (B-D) WT and atm KO MEF cells (B), AP29 and AP26 cells (C), as well as control and ATM stable knockdown HT-1080 cells (D) were treated with erastin. The cells were harvested for Co-IP by using anti-NCOA4 antibody. The input and IP fractions were immunoblotted to analyze NCOA4 phosphorylation. (E) HT-1080 cells were transfected with MYC tagged WT NCOA4 or the indicated mutants. The cells were harvested for Co-IP by using anti-MYC antibody. The input and IP fractions were immunoblotted to analyze the NCOA4 phosphorylation. (F) Alignment of NCOA4 amino acid sequences from multiple species suggests that Ser550 is evolutionally conserved and locates in a signature motif recognized by ATM kinase. (G) The NCOA4-p-S550 antiserum was generated. The dot blot was carried out to check the specificity of this antiserum to the NCOA4 phospho-peptide (p-peptide) and control unphospho-peptide (c-peptide). (H) The sc and ATM stable knockdown HT-1080 cells were treated with 5 μM erastin for 2 h. The cells were harvested for Co-IP by using anti-NCOA4 antibody. The input and IP fractions were immunoblotted to analyze NCOA4 phosphorylation and NCOA4-FTH1 interaction.
ATM kinase preferentially phosphorylates the substrates on serine or threonine residues that precede glutamine residues (SQ or TQ motifs). Four such serine or threonine residues are found in human NCOA4 protein, namely T109, T229, S237 and S550. To characterize the exact phosphorylable residues by ATM kinase, we therefore constructed the unphosphorylated T109A, T229A, S237A and S550A mutants. The co-immunoprecipitation analysis showed that mutation in S550 almost abolished the NCOA4 phosphorylation (Figure 5E). To further confirm that the evolutionarily conserved S550 residue (Figure 5F) is the critical site phosphorylated by ATM kinase, a polyclonal antibody against phosphorylated S550 was generated by immunizing mice with the synthesized NCOA4 phospho-peptide followed by affinity purification. The dot blot assay showed that this antibody was much more specific against the phospho-peptide (p-peptide) than the control unphospho-peptide (c-peptide) (Figure 5G). By using this antibody, we found that NCOA4 phosphorylation at S550 was elevated in response to erastin treatment in control HT-1080 cells. A pronounced reduction of NCOA4 phosphorylation at S550 was observed in ATM silenced HT-1080 cells, both under steady state and erastin challenged state (Figure 5H). Collectively, these data indicate that ATM phosphorylates NCOA4 at the S550 residue.
NCOA4 phosphorylation is crucial for ferritinophagy and ferroptosis
Whether ATM kinase-mediated NCOA4 phosphorylation at S550 residue impinges on the ferritinophagy or not is the next open question. ATM knockdown antagonized NCOA4 phosphorylation. At the meantime, we noticed that ATM knockdown significantly attenuated NCOA4-FTH1 interaction, especially under the erastin-challenged state (Figure 5H). The co-immunoprecipitation analysis showed that mutation in the S550 residue obviously disrupted the NCOA4-FTH1 interaction (Figure 6A), suggesting that the NCOA4 phosphorylation at S550 by ATM kinase may strengthen NCOA4 interaction with ferritin. To further elucidate the relevance of NCOA4 phosphorylation in ferritinophagy, the NCOA4 stable knockdown HT-1080 was constructed (Figure 6B). In line with the previous study [20], NCOA4 silence elevated FTH1 level due to the impaired ferritinophagy (Figure 6B). Moreover, NCOA4 knockdown profoundly abrogated erastin-induced FTH1 increase, while this FTH1 increase was restored by ectopic expression of the WT NCOA4 and the S237A mutant, but not the S550A mutant (Figure 6B). Ferritinophagy could turnover ferritin and complement the intracellular labile iron pool. The intracellular labile iron was detected by the FerroOrange staining. NCOA4 knockdown blunted the erastin-induced fluorescence enhancement of FerroOrange probe. In line, ectopic expression of the WT NCOA4 and the S237A mutant, but not the S550A mutant, could restore the FerroOrange fluorescence enhancement in NCOA4 knockdown HT-1080 cells (Figure 6C), suggesting that NCOA4 phosphorylation at S550 residue is crucial for ferroptosis-associated ferritinophagy and the elevation of intracellular bioactive iron. Furthermore, erastin-induced ferroptosis was sharply abrogated in NCOA4 knockdown cells, while ectopic expression of the WT NCOA4 and the S237A mutant, but not the S550A mutant, could restore the ferroptotic cell death in NCOA4 knockdown HT-1080 cells (Figure 6D and 6E). Similarly, ectopic expression of the WT NCOA4, but not the S550A mutant, could restore the erastin-induced elevation of lipid peroxidation, which was strikingly abolished by NCOA4 knockdown (Figure 6F). Collectively, these data suggest that ATM kinase phosphorylating NCOA4 at the S550 residue is critically important for the NCOA4-ferritin interaction, ferritinophagy-mediated elevation of cellular free iron and the subsequent ferroptosis execution.
Figure 6.
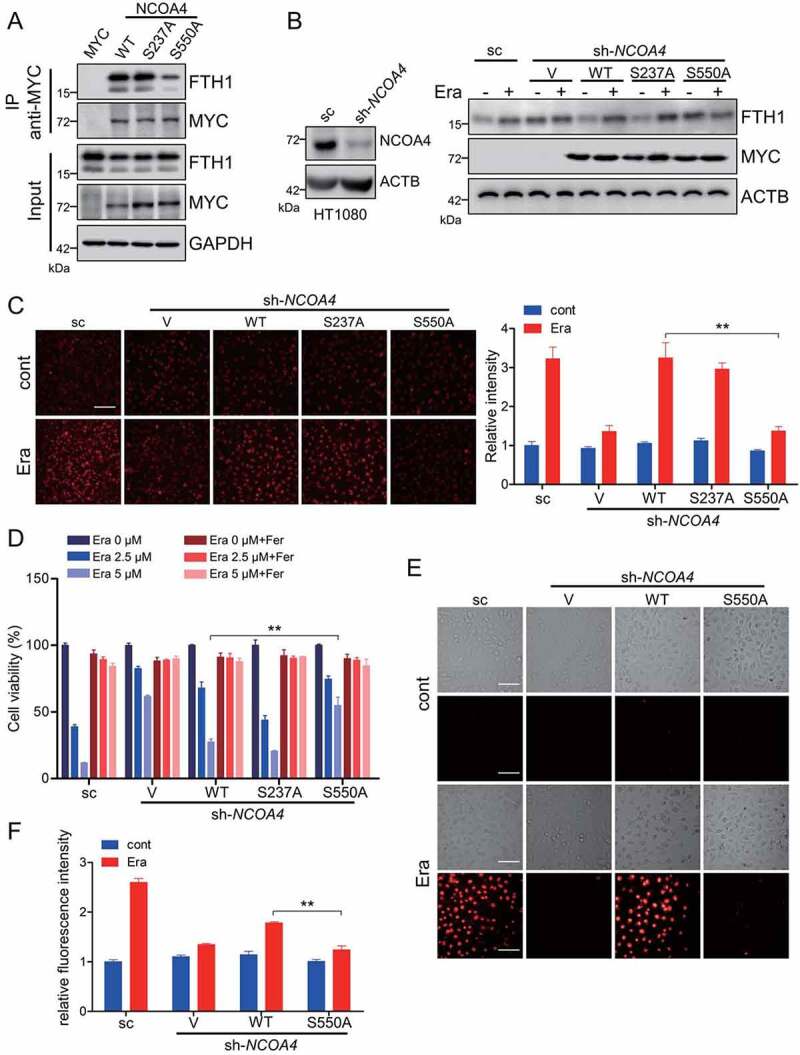
NCOA4 phosphorylation is crucial for ferritinophagy and ferroptosis. (A) HT-1080 cells were transfected with MYC tagged WT NCOA4 or the indicated mutants. The cells were harvested for Co-IP by using anti-MYC antibody. The input and IP fractions were immunoblotted to analyze NCOA4-FTH1 interaction. (B) The NCOA4 stable knockdown HT-1080 cells were transfected with empty vector, MYC tagged WT NCOA4 or the indicated mutants. Cells were treated with 5 μM erastin and then harvested for immunoblotting analysis. (C) The NCOA4 stable knockdown HT-1080 cells were transfected with empty vector, MYC tagged WT NCOA4 or the indicated mutants. Cells were treated with 5 μM erastin and the labile free iron was analyzed with FerroOrange staining. Representative images are presented. Scale bar: 200 μm. The relative fluorescence intensity was quantified and shown in the right histogram. Data from three independent experiments are shown as the mean ± SEM. (D-F) The NCOA4 stable knockdown HT-1080 cells were transfected with empty vector, MYC tagged WT NCOA4 or the indicated mutants. Cells were treated with the indicated dose of erastin with or without 10 μM Fer and then the cell viability (D), cell death (E), and lipid peroxidation (F) were analyzed. Scale bar: 50 μm. Data from three independent experiments are shown as the mean ± SEM. ** P < 0.01 was determined by student’s t-test.
Discussion
Ferroptosis is a newly characterized form of regulated cell death. Several kinases-mediated phosphorylated modifications have been documented to modulate the ferroptotic sensitivities [52]. In this study, we show that ATM kinase, the master sensor of DNA damage response [34], is a critical upstream regulator of ferroptosis. Mechanistically, ATM kinase phosphorylates ferritinophagy receptor NCOA4, facilitates the NCOA4-FTH1 interaction to drive ferritinophagy-mediated iron mobilization and exacerbate lipid peroxidation.
The serine/threonine protein kinase ATM is one of the master sensors and regulators of DNA damage response [34]. Upon DNA double-strand breaks, ATM rapidly translocates onto the DNA damage sites and phosphorylates a number of substrates, including BRCA1 (BRCA1 DNA repair associated), CHEK2 (checkpoint kinase 2) and TP53, therefore dominates cell cycle arrest, DNA damage repair and apoptosis [34]. Besides, ATM is also responsive to the intracellular oxidative stress. Oxidative insults facilitate the disulfide bond formation of ATM, which directly drives ATM activation in the absence of DNA damage signaling [53]. Herein, we found that ferroptosis inducers, including erastin and RSL3, activate ATM kinase independent on DNA damage signaling. To our knowledge, a feedback machinery may exist to explain ATM activation. The basal intracellular labile iron catalyzes Fenton chemistry to generate lipid peroxides. These lipid peroxides and the end products (especially 4-ONE [44]) would likely activate the ATM kinase. The following NCOA4 phosphorylation and ferritinophagy would amplify the lipid peroxidation to further activate ATM by releasing free iron. Under normal condition, this loop would be terminated by the cellular antioxidant systems (GSH, GPX4 …). When cells are challenged with the ferroptotic stimuli (erastin-induced GSH depletion, or RSL3-induced GPX4 inactivation), the reduced antioxidant capacity would disrupt this redox balance and then trigger a propagation reaction, leading to ATM activation and ferroptosis. ATM kinase phosphorylates and activates several downstream molecules to defense against oxidative stress [54]. Specifically, ATM dominates bulk autophagy [55,56], as well as selective autophagy including pexophagy [57], lipophagy [58] and mitophagy [59,60], which are important for the maintenance of cellular redox homeostasis. Herein, we present substantial evidence to show that ATM is a powerful regulator of both ferroptosis-associated and iron starvation-induced ferritinophagy, a selective autophagy specifically degrading ferritin for iron recycling. Mechanistically, the ferritinophagy receptor NCOA4 is phosphorylated at S550 residue by ATM kinase, and this phosphorylation facilitates the NCOA4-FTH1 interaction and thus boosts ferritinophagy for ferritin degradation. It was reported that the C-terminal domain in NCOA4 is indispensable for the NCOA4-FTH1 interaction and the subsequent ferritinophagy. Specifically, the W497, I489, S492, L494, and L498 in NCOA4 are important residues for NCOA4-ferritin binding [51]. The S550 site locates adjacently to these residues. However, how S550 phosphorylation by ATM kinase modulates the molecular affinity between NCOA4 and FTH1 needs further investigation. Therefore, our study expands the physiological function of ATM kinase in ferritinophagy beyond its well characterized functions in general autophagy and other forms of selective autophagy.
Chen and colleagues recently performed a forward genetic-based kinase screen against ferroptosis and nominated ATM as an essential kinase for ferroptosis execution [35]. Chemical inhibition or siRNA-mediated knockdown of ATM leads to predominant resistance to cystine deprivation- and erastin challenge-induced ferroptosis in MDA-MB-231 triple negative breast cancer cells. Mechanistically, ATM inhibition reduces the level of intracellular labile iron through facilitating the nuclear localization of MTF1 and transcriptionally activating the expression of iron efflux-associated SLC40A1, as well as the iron storage-associated FTH1 and FTL (ferritin light chain) [35]. Actually, we report a similar mechanism for ATM kinase in determining ferroptotic sensitivity in the current study. Pharmacological inhibition or genetic ablation of ATM strikingly abolishes ferroptosis in several types of cultured cells. In line with the previous report [35], we also found that ATM manipulates the expression of FTH1 and SLC40A1. Alternatively, our study suggests that ATM is a master regulator of ferritinophagy through phosphorylating NCOA4. Defective ferritinophagy suppresses the elevation of intracellular bioactive iron and leads to ferroptotic resistance in atm ablated and inhibited cells. Furthermore, ATM dominates ferroptotic sensitivity independent of TRP53. Knockout or chemical inhibition of TRP53 fails to alter the ferroptotic insensitivity of atm knockout cells. It is noteworthy an interaction between TRP53 and iron metabolism exists [61]. TRP53 transcriptionally modulates the expression of iron-sulfur cluster assembly-associated genes, including ISCU [62], FDXR (ferredoxin reductase) [63] and FXN (frataxin) [64], which were all identified as ferroptosis regulators [63,65,66]. An early study reported that TRP53 expression leads to an increase in both FTH1 and FTL by suppressing ACO1/IRP1 [67], possibly due to that TRP53 facilitates iron-sulfur cluster biogenesis. Furthermore, as an important regulator of general autophagy [47], TRP53 is proposed to fine-tune the specific ferritinophagy [49,50], although our data show that ATM-mediated ferritinophagy is TRP53-indenpednt (Figure 4K and 4L). In a reverse reaction, it has been well documented that the cellular iron status and iron homeostasis could modulate TRP53 stability and transcriptional activity [61]. Collectively, the mechanism for which TRP53 regulates ferroptosis is thus complicated. Whether the crosstalk between TRP53 and iron metabolism could modulate ATM-associated ferroptotic vulnerability needs further investigation.
It should be noted that ATM deficiency leads to a lower ferroptotic resistance to RSL3 than erastin (Figure 1B, 1H and 2C), although the resistance to RSL3-induced ferroptosis mediated by ATM knockout is obvious enough. The mechanism of erastin- and RSL3-induced ferroptosis differs sharply. Erastin targets the system Xc-, suppresses cystine uptake, and then reduces GSH synthesis, while RSL3 directly inhibits GPX4 [52]. To the difference between erastin- and RSL3-induced ferroptosis in response to ATM deficiency, the following possibility may exist. It was reported that ATM deficiency leads to the accumulation of dysfunctional mitochondria with decreased respiration and reduced ATP production [68], which would desensitize ferroptotic vulnerability. This mitochondria-associated ferroptotic regulation was recently reported to be restricted to GSH depletion-associated ferroptosis (erastin- and cysteine deprivation-induced ferroptosis), but not to GPX4 inhibition-induced ferroptosis [69]. Therefore, when cells are challenged with erastin, ATM depletion or inhibition would reduce the intracellular labile free iron by disrupting ferritinophagy and decrease mitochondrial activity, leading to a greater ferroptotic resistance. When GPX4 is directly inactivated by RSL3, the low amount of lipid ROS catalyzed by basal intracellular labile iron or by other machinery will rapidly amplify through the Fenton chain reaction, leading to the ferroptosis execution. In this case, ATM knockout or inhibition would perform relatively lower inhibitory capacity.
ATM loss-of-function due to the nonsense mutation in the ATM gene leads to ataxia-telangiectasia, a rare autosomal recessive human hereditary disorder. Besides the typical feature of cerebellar degeneration, ataxia-telangiectasia is also clinically manifested by cancer predisposition [54]. Moreover, diverse ATM inhibitors are currently in preclinical or clinical development as the sensitizers of tumor radiotherapy and chemotherapy, due to their apoptosis-promoting activity [70,71]. As targeted induction of ferroptosis is widely regarded to provide the next generation of therapeutic strategy for other therapy-resistant cancers, it is noteworthy that the ferroptosis-related therapies would have a more potent efficacy to those ATM positive cancers than those ATM deficient cancers. Furthermore, ATM activation is implicated in the pathogenesis of tissue injury associated diseases, including the doxorubicin-induced cardiotoxicity [72], pressure overload-induced heart failure [73], nonalcoholic fatty liver disease [74] and Huntington disease [75]. Genetic ablation or pharmacological inhibition of ATM has been proved to ameliorate these diseased phenotypes in the corresponding models in vivo and/or in vitro [72–75]. Moreover, a line of evidence has emerged that ferroptosis is also implicated in the pathogenesis of these diseases [2]. Our study herein suggests that ATM kinase orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4, which may be pathologically important for these diseases. Therefore, further understanding of the pathological function of ATM kinase-mediated ferroptosis could help to develop novel therapeutic strategies to these diseases.
Materials and methods
Antibodies and reagents
The rabbit antibodies against ATM (2873), FTH1 (4393), TRP53/TP53 (2527), MYC (3946), ACO1/IRP1 (20272), IREB2/IRP2 (37135) and ACTB/actin (8457) were purchased from Cell Signaling Technology. The mouse antibody against LAMP1 (15665) was purchased from Cell Signaling Technology. The rabbit antibody against NCOA4 (A302-272A) was purchased from Bethyl Laboratories. The rabbit antibodies against p-ATM (ab81292), γH2AX (ab11174) and TRP53BP1 (ab21083) were purchased from Abcam. The rabbit antibody against p-Ser (sc-81514) and mouse antibody against LAMP2 (sc-18822) were purchased from Santa Cruz Biotechnology. The rabbit antibodies against LMNB1/lamin B1 (12987-1-AP), FTL (10727-1-AP), TFRC (10084-2-AP), SLC11A2/DMT1 (20507-1-AP) and SLC40A1/FPN (26601-1-AP) were purchased from ProteinTech. Polyclonal antibodies against phosphorylated NCOA4 were generated by immunizing mice with synthesized NCOA4 phospho-peptide followed by affinity purification (Abclonal Technology). The secondary antibodies used for immunoblotting in this study were purchased from Cell Signaling Technology as follows: HRP-conjugated anti-mouse IgG (7076) and HRP-conjugated anti-rabbit IgG (7074). The secondary antibodies used for immunofluorescence in this study were purchased from Thermo Fisher Scientific as follows: Goat anti-rabbit IgG-Cy3 (A10520), goat anti-mouse IgG-Cy3 (A10521) and goat anti-rabbit IgG-FITC (31635). The biochemical reagents used in this study are as follows: Erastin (S7242), RSL3 (S8155), Fer (S7243), BSO (S9728), Sul (S1576), KU55933 (S1092), PFTα (S2929), Mirin (S8096), camptothecin (S1288), PIK-III (S7683) and bleomycin (S1214) were purchased from Selleck; 4-ONE (HY-114524) was purchased from MedChemExpress; DFO (D9533) and DFP (379409) were purchased from Sigma-Aldrich.
Cell culture, transfection and construction of stable cell lines
The ATM WT, atm KO, AP29 (trp53 KO) and AP26 (trp53 and atm double KO) MEFs were kindly gifted by Dr. Bao-hua Liu from Shenzhen University, China. HT-1080 was kindly gifted by Dr. Zhen-yu Ju from Jinan University, China. HCT116 TP53+/+ and HCT116 TP53−/− cells were kindly gifted by Dr. Tie-shan Tang from Institute of Zoology, Chinese Academy of Sciences, China. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, C11995500BT) supplemented with 10% fetal bovine serum (FBS; Gibco, 10099–141C) and 1% penicillin-streptomycin at 37°C in 5% CO2.
To construct the stable ATM and NCOA4 knockdown HT-1080, plasmids expressing hairpin siRNA were constructed by inserting pairs of annealed DNA oligonucleotides into the pLKO.1 vector (Addgene, 8453; deposited by Dr. Robert Weinberg lab from Massachusetts Institute of Technology) according to the manufacturer’s instructions. The targeting sequence to ATM was 5′-AAGCACCAGTCCAGTATTGGC-3′, the targeting sequence to NCOA4 was 5′- GGCCCAGGAAGTATTACTTAA-3′, and a nontargeting sequence, 5′- ACTACCGTTGTTATAGGTG-3′, was used as a scramble control (sc). To generate the stable knockdown cells, HT-1080 cells were transfected with pLKO.1-ATM, pLKO.1-NCOA4, or pLKO.1-sc. 24 h after transfection, 0.5 μg/ml puromycin (Sigma-Aldrich, P9620) was added to the cell medium for selection. Stable clones were analyzed by western blotting to confirm the knockdown efficiency.
Cell viability measurement
Cell viability measurement was performed by CCK-8 assay. Briefly, cells (8,000/well) were seeded in a 96-well plate, and cells were treated as indicated. DMEM (100 μl) containing 10 μl CCK8 reagent (Beyotime Biotechnology, C0042) was added to each well and the 96-well plate was incubated at 37°C for 1 h. The absorbance of each well was detected at 450 nm by using a CYTATION 5 Imaging Reader (BioTek, USA). The relative cell viability to the control group was calculated.
Cell death analysis by PI staining
For image capture, cells (40,000/well) were seeded in a 24-well plate, and the cells were treated as indicated. Then the medium was discarded, and 500 μl PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) containing 1 μg/ml PI (propidium iodide; Invitrogen, P1304MP) was added to each well and the 24-well plate was incubated for 1 min. Images were captured by using a fluorescence microscope (Nikon, Japan). For quantitative flow cytometry analysis, cells (40,000/well) were seeded in a 24-well plate, and the cells were treated as indicated. Then both the floating cells in the medium and the adherent cells were collected by using trypsin digestion and centrifugation. The cell pellets were resuspended with 200 μl PBS containing 1 μg/ml PI, and then analyzed by using a flow cytometry (BD, USA).
Lipid peroxides measurement
Cells (100,000/well) were seeded in a 12-well plate, and the cells were treated as indicated. Then the medium was discarded, and cells were collected by using trypsin digestion. The cell pellets were resuspended with 200 μl PBS containing 1 μM C11 BODIPY 581/591 (Thermo Fisher Scientific, D3861), and analyzed by using the 488-nm laser of a flow cytometer (BD, USA) for excitation. Data was analyzed by using FCS Express V3 and FlowJo software.
GSH determination
Cells (1x106/dish) were seeded in 10-cm dishes, and the cells were treated as indicated. GSH content was measured by using a GSH detection kit (Beyotime Biotechnology, S0053) according to the manufacturer’s protocol. Protein quantification was performed by using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225) as described in the manufacturer’s guidelines. The GSH levels were normalized to the corresponding protein concentration.
qRT-PCR
Total RNA was extracted by using the Trizol reagent (Tiangen Biotech, DP424) according to the manufacturer’s instruction. After reverse transcription by using PrimeScript RT-PCR Kit (Takara, RR014B), qRT-PCR was performed by using Power SYBR Green PCR Mix (Thermo Fisher Scientific, 4367659) to detect the relative mRNA levels of indicated genes. The relative expression levels of target genes were normalized by subtracting the corresponding Actb/ACTB threshold cycle value. The primers used in this study are as follows:
| gene | sequence (5’-3’) |
|---|---|
| Mm-Actb | Forward: CATTGCTGACAGGATGCAGAAGG |
| Reverse: TGCTGGAAGGTGGACAGTGAGG | |
| Mm-Ptgs2 | Forward: GCGACATACTCAAGCAGGAGCA |
| Reverse: AGTGGTAACCGCTCAGGTGTTG | |
| Mm-Fth1 | Forward: GCCGAGAAACTGATGAAGCTGC |
| Reverse: GCACACTCCATTGCATTCAGCC | |
| Mm-Cdkn1a/p21 | Forward: TCGCTGTCTTGCACTCTGGTGT |
| Reverse: CCAATCTGCGCTTGGAGTGATAG | |
| Mm-Slc7a11 | Forward: CTTTGTTGCCCTCTCCTGCTTC |
| Reverse: CAGAGGAGTGTGCTTGTGGACA | |
| Hs-ACTB | Forward: CACCATTGGCAATGAGCGGTTC |
| Reverse: AGGTCTTTGCGGATGTCCACGT | |
| Hs-PTGS2 | Forward: CGGTGAAACTCTGGCTAGACAG |
| Reverse: GCAAACCGTAGATGCTCAGGGA | |
| Hs-FTH1 | Forward: TGAAGCTGCAGAACCAACGAGG |
| Reverse: GCACACTCCATTGCATTCAGCC |
Immunofluorescence analysis
Cells were plated in a 6-well plate with sterilized glass coverslips. After indicated treatments, cells were fixed with PBS containing 3.7% formaldehyde at 37°C for 15 min, then washed three times with ice-cold PBS. Subsequently, cells were permeabilized by PBS containing 0.2% Triton X-100 (Beyotime Biotechnology, ST795) at room temperature for 15 min, blocked by PBS containing 5% FBS, then incubated with indicated primary and corresponding secondary conjugated antibodies at room temperature for 120 min and 60 min, respectively. Images were captured with a LSM 510 Zeiss confocal microscope.
Intracellular iron quantification
Cells (100,000/well) were seeded in a 12-well plate and treated as indicated. Then the medium was discarded, and cells were stained with HBSS containing 1 μM FerroOrange labile ferrous iron detecting probe (Dojindo, F374) at 37°C for 30 min. Images were captured by using a fluorescence microscope (Nikon, Japan).
SDS-PAGE and western blotting
Cells were collected by trypsin digestion after indicated treatment and then washed with ice-cold PBS twice. Cell pellets were lysed with appropriate volume of lysis buffer (20 mM Tris, 1% NP40 [Sangon Biotech, A100109-0100], 10% glycerol, 2 mM EDTA, 137 mM NaCl, pH 7.2, 1 mM PMSF [Beyotime Biotechnology, ST505], 1 mM Na3VO4, 10 mM NaF) on ice for 40 min. The lysis was centrifuged and the supernatant as the cell lysate was collected to measure the protein concentration by using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225) according to the manufacturer’s guidelines. An equivalent quantity (20 μg) of protein was subjected to SDS-PAGE and then transferred to nitrocellulose membranes. The membrane was blocked with 5% fat-free milk, and subsequently incubated with the indicated primary antibodies and HRP-conjugated secondary antibodies overnight and 2 h, respectively. Finally, immunoreactive protein bands were visualized with a chemiluminescence kit (Tiangen Biotech, PA110). The relative protein levels normalized to ACTB were quantitatively analyzed via the band density by the ImageJ software.
Nuclear/cytosol fractionation assay
After treatment with erastin or RSL3, the cells were collected with trypsin digestion, washed with ice-cold PBS twice, and then centrifuged at 600 g for 5 min. The cell pellet was resuspended with 200 μl cytosolic extract buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA, pH 7.6, 0.075% NP40, 1 mM DTT, 1 mM PMSF), incubated on ice for 5 min, and then centrifuged at 1,000 g for 5 min. The supernatant was collected as cytosolic extract. The pellet was resuspended with 60 μl nuclear extract buffer (20 mM Tris, 400 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, pH 8.0, 1 mM PMSF and 25% glycerol), incubated on ice for 10 min, and the nuclear fraction was collected by centrifugation at 10,000 g for 10 min. The nuclear and cytosol fractions were subjected to SDS-PAGE for further immunoblotting analysis.
Immunoprecipitation
After the indicated treatments, cells plated in a 10-cm dish were lysed in 0.8 ml lysis buffer (20 mM Tris, 1% NP40, 10% glycerol, 2 mM EDTA, 137 mM NaCl, pH 7.2) plus protease and phosphatase inhibitors (1 mM PMSF, 1 mM Na3VO4, 10 mM NaF) for 40 min on ice. After centrifugation at 12,000 g for 15 min, the supernatants as the cell lysate were collected to measure the protein concentration by using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225) according to the manufacturer’s guidelines. An equivalent quantity (1000 μg) of protein was immunoprecipitated with the specific antibody or IgG control and protein A/G plus-Sepharose (Santa Cruz Biotechnology, sc-2003) overnight at 4°C. Thereafter, the precipitants were washed 4 times with ice-cold lysis buffer, and the immune complexes were eluted with 1*loading buffer for 3 min at 100°C. The input and IP fraction was analyzed via SDS-PAGE and western blotting.
Colony formation assay
For the colony formation assay, cells (250/well) were seeded into 6-well plates. After overnight adhesion, cells were treated with DMSO, erastin, or RSL3 with or without Fer as indicated. Then the medium was changed with fresh and drug-free medium. Medium was changed every 3 days. After 14 days of incubation, colonies were fixed with 4% paraformaldehyde for 15 min and then stained with 0.5% crystal violet for 10 min. The colony numbers were calculated.
Statistical analysis
For quantitative analysis presented as histograms, values were obtained from at least three independent experiments and are shown as the mean ± SEM. Statistical significances were determined by student’s t-test. P < 0.05 was considered as statistically significant. All statistical analysis were performed with GraphPad Prism software.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China (32070738), the State Key Laboratory of Medicinal Chemical Biology, Nankai University (2020015), Agricultural Microbiology of Large Research Infrastructures (Project NO. 463119009), and the Fundamental Research Funds for the Central Universities (Project NO. 2662020DKPY009).
Disclosure statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2170960
References
- [1].Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stockwell BR, Angeli JPF, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–296. [DOI] [PubMed] [Google Scholar]
- [4].Kagan VE, Mao GW, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Feng HZ, Stockwell BR.. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16:e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences of the United States of America 2016; 113:E4966–E75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355–69 e10. [DOI] [PubMed] [Google Scholar]
- [9].Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–1147. [DOI] [PubMed] [Google Scholar]
- [10].Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Angeli JPF, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bersuker K, Hendricks JM, Li ZP, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. [DOI] [PubMed] [Google Scholar]
- [14].Mao C, Liu XG, Zhang YL, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–937. [DOI] [PubMed] [Google Scholar]
- [16].Bogdan AR, Miyazawa M, Hashimoto K, et al. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Du J, Wang TT, Li YC, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–369. [DOI] [PubMed] [Google Scholar]
- [20].Mancias JD, Wang XX, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–1079. [DOI] [PubMed] [Google Scholar]
- [22].Hou W, Xie YC, Song XX, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao MH, Monian P, Pan QH, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fang XX, Cai ZX, Wang H, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127:486–501. [DOI] [PubMed] [Google Scholar]
- [25].Rui TY, Wang HC, Li QQ, et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J Pineal Res. 2021;70:e12704. [DOI] [PubMed] [Google Scholar]
- [26].Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee H, Zandkarimi F, Zhang YL, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li CZ, Dong X, Du WJ, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther. 2020;5:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Song XX, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X-c(-) activity. Curr Biol. 2018;28:2388–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yi J, Zhu J, Wu J, et al. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A. 2020;117:31189–31197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Conlon M, Poltorack CD, Forcina GC, et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol. 2021;17:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hattori K, Ishikawa H, Sakauchi C, et al. Cold stress-induced ferroptosis involves the ASK1-p38 pathway. EMBO Rep. 2017;18:2067–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34:5617–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817. [DOI] [PubMed] [Google Scholar]
- [35].Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al . Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Qian M, Liu Z, Peng L, et al. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife. 2018;7:e34836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hafner A, Bulyk ML, Jambhekar A, et al. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Bio. 2019;20:199–210. [DOI] [PubMed] [Google Scholar]
- [38].Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jiang L, Kon N, Li TY, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ou Y, Wang SJ, Li D, et al. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806–E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hu W, Zhang C, Wu R, et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Uziel T, Lerenthal Y, Moyal L, et al. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dupre A, Boyer-Chatenet L, Sattler RM, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shibata T, Iio K, Kawai Y, et al. Identification of a lipid peroxidation product as a potential trigger of the p53 pathway. J Biol Chem. 2006;281:1196–1204. [DOI] [PubMed] [Google Scholar]
- [45].Goodwin JM, Dowdle WE, DeJesus R, et al. Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017;20:2341–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhou C, Ma K, Gao R, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27:184–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Maiuri MC, Galluzzi L, Morselli E, et al. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. [DOI] [PubMed] [Google Scholar]
- [49].Xu Z, Feng J, Li Y, et al. The vicious cycle between ferritinophagy and ROS production triggered EMT inhibition of gastric cancer cells was through p53/AKT/mTor pathway. Chem Biol Interact. 2020;328:109196. [DOI] [PubMed] [Google Scholar]
- [50].Feng J, Li C, Xu R, et al. DpdtC-induced EMT inhibition in MGC-803 cells was partly through ferritinophagy-mediated ROS/p53 pathway. Oxid Med Cell Longev. 2020;2020:9762390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mancias JD, Vaites LP, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4:e10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Guo Z, Kozlov S, Lavin MF, et al. ATM activation by oxidative stress. Science. 2010;330:517–521. [DOI] [PubMed] [Google Scholar]
- [54].Lee JH, Paull TT. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol. 2021;22:796–814. [DOI] [PubMed] [Google Scholar]
- [55].Guo QQ, Wang SS, Zhang SS, et al. ATM-CHK2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020;39:e103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy. 2010;6:672–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang JW, Tripathi DN, Jing J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Le Guezennec X, Brichkina A, Huang YF, et al. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012;16:68–80. [DOI] [PubMed] [Google Scholar]
- [59].Fang EF, Kassahun H, Croteau DL, et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016;24:566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cirotti C, Rizza S, Giglio P, et al. Redox activation of ATM enhances GSNOR translation to sustain mitophagy and tolerance to oxidative stress. EMBO Rep. 2021;22:e50500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhang J, Chen X. p53 tumor suppressor and iron homeostasis. FEBS J. 2019;286:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Funauchi Y, Tanikawa C, Yi LPH, et al. Regulation of iron homeostasis by the p53-ISCU pathway. Sci Rep. 2015;5:16497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang YH, Qian YJ, Zhang J, et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017;31:1243–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shimizu R, Lan NN, Tai TT, et al. p53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene. 2014;551:79–85. [DOI] [PubMed] [Google Scholar]
- [65].Terzi EM, Sviderskiy VO, Alvarez SW, et al. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7:eabg4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Du J, Zhou Y, Li YC, et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020;32:101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhang F, Wang W, Tsuji Y, et al. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J Biol Chem. 2008;283:33911–33918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Valentin-Vega YA, Maclean KH, Tait-Mulder J, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119:1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354–63 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Riches LC, Trinidad AG, Hughes G, et al. Pharmacology of the ATM inhibitor AZD0156: potentiation of irradiation and olaparib responses preclinically. Mol Cancer Ther. 2020;19:13–25. [DOI] [PubMed] [Google Scholar]
- [71].Durant ST, Zheng L, Wang Y, et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv. 2018;4:eaat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhan H, Aizawa K, Sun J, et al. Ataxia telangiectasia mutated in cardiac fibroblasts regulates doxorubicin-induced cardiotoxicity. Cardiovasc Res. 2016;110:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nakada Y, Nhi Nguyen NU, Xiao F, et al. DNA damage response mediates pressure overload-induced cardiomyocyte hypertrophy. Circulation. 2019;139:1237–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Mahata T, Sengar AS, Basak M, et al. Hepatic regulator of G protein signaling 6 (RGS6) drives non-alcoholic fatty liver disease by promoting oxidative stress and ATM-dependent cell death. Redox Biol. 2021;46:102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lu XH, Mattis VB, Wang N, et al. Targeting ATM ameliorates mutant huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med. 2014;6:268ra178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.