

ABSTRACT
Microglial phagocytosis of apoptotic debris prevents buildup damage of neighbor neurons and inflammatory responses. Whereas microglia are very competent phagocytes under physiological conditions, we report their dysfunction in mouse and preclinical monkey models of stroke (macaques and marmosets) by transient occlusion of the medial cerebral artery (tMCAo). By analyzing recently published bulk and single cell RNA sequencing databases, we show that the phagocytosis dysfunction was not explained by transcriptional changes. In contrast, we demonstrate that the impairment of both engulfment and degradation was related to energy depletion triggered by oxygen and nutrient deprivation (OND), which led to reduced process motility, lysosomal exhaustion, and the induction of a protective macroautophagy/autophagy response in microglia. Basal autophagy, in charge of removing and recycling intracellular elements, was critical to maintain microglial physiology, including survival and phagocytosis, as we determined both in vivo and in vitro using pharmacological and transgenic approaches. Notably, the autophagy inducer rapamycin partially prevented the phagocytosis impairment induced by tMCAo in vivo but not by OND in vitro, where it even had a detrimental effect on microglia, suggesting that modulating microglial autophagy to optimal levels may be a hard to achieve goal. Nonetheless, our results show that pharmacological interventions, acting directly on microglia or indirectly on the brain environment, have the potential to recover phagocytosis efficiency in the diseased brain. We propose that phagocytosis is a therapeutic target yet to be explored in stroke and other brain disorders and provide evidence that it can be modulated in vivo using rapamycin.
Abbreviations: AIF1/IBA1: allograft inflammatory factor 1; AMBRA1: autophagy/beclin 1 regulator 1; ATG4B: autophagy related 4B, cysteine peptidase; ATP: adenosine triphosphate; BECN1: beclin 1, autophagy related; CASP3: caspase 3; CBF: cerebral blood flow; CCA: common carotid artery; CCR2: chemokine (C-C motif) receptor 2; CIR: cranial irradiation; Csf1r/v-fms: colony stimulating factor 1 receptor; CX3CR1: chemokine (C-X3-C motif) receptor 1; DAPI: 4’,6-diamidino-2-phenylindole; DG: dentate gyrus; GO: Gene Ontology; HBSS: Hanks’ balanced salt solution; HI: hypoxia-ischemia; LAMP1: lysosomal-associated membrane protein 1; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MCA: medial cerebral artery; MTOR: mechanistic target of rapamycin kinase; OND: oxygen and nutrient deprivation; Ph/A coupling: phagocytosis-apoptosis coupling; Ph capacity: phagocytic capacity; Ph index: phagocytic index; SQSTM1: sequestosome 1; RNA-Seq: RNA sequencing; TEM: transmission electron microscopy; tMCAo: transient medial cerebral artery occlusion; ULK1: unc-51 like kinase 1.
KEYWORDS: Autophagy, ischemia, lysosomes, microglia, phagocytosis, rapamycin, stroke, tMCAo
Introduction
Stroke is one of the most pervasive neurological diseases, with a global estimated yearly incidence of 13.7 million people that suffer disabling motor and neurophysiological deficits. It is also one of the most lethal diseases, claiming 5.5 million lives every year [1]. The pathophysiology of stroke is particularly complex, with a cascade of events initiated by the lack of blood supply resulting from a broken or blocked blood vessel. A key element throughout this process is the immune system and particularly the macrophages residing in the brain parenchyma, microglia [2].
The dual role of microglia as a double-edged sword orchestrator of the brain immune response is well-recognized [2,3]. This dichotomy is evident in the contradictory results obtained by different studies using microglial depletion paradigms in rodent stroke models, with either detrimental [4,5] or beneficial [6] consequences. On one hand, microglia orchestrate inflammation and infiltration of peripheral immune cells, with deleterious effects on neurons when sustained or uncontrolled. On the other hand, microglia have beneficial effects by engulfing monocytes [7], neutrophils 7 [8],, and damaged blood vessels [9], controlling astrocytic inflammatory cytokines [5], or releasing growth factors such as IGF1 [4]. In spite of this handful of examples, functional studies of microglia in stroke remain scarce. Many studies still analyze microglia in terms of M1/M2 phenotypes (presumably pro- and anti-inflammatory), which in recent years are being substituted with more sophisticated phenotyping methods, such as RNA sequencing (RNA-Seq) [10–14]. In contrast, there is a strong consensus in the microglial cell biology community that M1/M2 are outdated and should be abandoned; and that RNA-Seq signatures do not equate with cell function, which needs to be directly assessed [15].
One indispensable microglial function that has received little attention is the phagocytosis of cellular debris generated during stroke. Efficient phagocytosis is crucial for the recovery of the homeostasis of the brain parenchyma, not only because it removes apoptotic corpses before they progress into secondary necrosis, releasing toxic intracellular compounds; but also because it modulates the phagocyte´s inflammatory response [16]. We do know that phagocytosis is beneficial in stroke because its genetic [17] or pharmacological [18,19] inhibition results in larger infarct areas. While there have been some attempts to analyze it [20], we still lack a basic understanding of how successful phagocytosis during stroke is and whether it needs to be recovered.
The high efficiency of microglial phagocytosis of apoptotic cells under physiological conditions [21] is in sharp contrast to the dysfunction we recently reported in mouse and human epilepsy [22,23]. Epilepsy and stroke share some cardinal neuropathological events such as inflammation and excitotoxicity, thus raising the possibility of microglial phagocytosis impairment during stroke. To test this hypothesis, we used in vivo models of transient medial cerebral artery occlusion (tMCAo) and hypoxia-ischemia (HI) in mice and in preclinical models of stroke (Macaca fascicularis and Callithrix jacchus) [24,25], as well as in vitro models of oxygen and nutrient deprivation (OND) in organotypic cultures and primary microglia. We found a pervasive impairment of microglial phagocytosis of apoptotic cells in all models studied that was related to several cellular mechanisms, including the induction of a protective macroautophagy/autophagy response due to the energy depletion associated with stroke. We found that inhibition of basal autophagy using genetic and pharmacological approaches was essential to maintain microglial survival and function. Moreover, the autophagy inducer rapamycin prevented to some extent the impairment of phagocytosis induced by tMCAo, supporting the possibility of pharmacological modulation of microglial phagocytosis in vivo.
Results
Phagocytosis impairment in mouse and monkey models of stroke
We studied the impact of stroke on microglial phagocytosis in mouse and preclinical monkey models of transient occlusion of the MCA and branches (tMCAo). In mice we focused on the hippocampus, where we can establish the baseline of microglial phagocytosis efficiency in control conditions because of the ongoing apoptosis of neural progenitors in the hippocampal neurogenic niche [21], which allows us to quantitatively compare it with disease models [22]. To ensure that the hippocampus was systematically affected by the arterial occlusion we used an extended tip filament that blocked the collateral branches of the MCA, including the anterior choroidal artery, which irrigates the hippocampus (Figure 1A). We used laser Doppler flowmetry to confirm occlusion (60 min) and subsequent reperfusion (Figure 1B). In agreement with other studies, ischemic lesions were found in the cortex, striatum, thalamus and hippocampus after tMCAo [26]. It is important to note that while the occlusion was transient, these brain areas were maintained in hypoxia over the time course of the experiment (6 h and 1 d) (Figure 1C and D).
Figure 1.
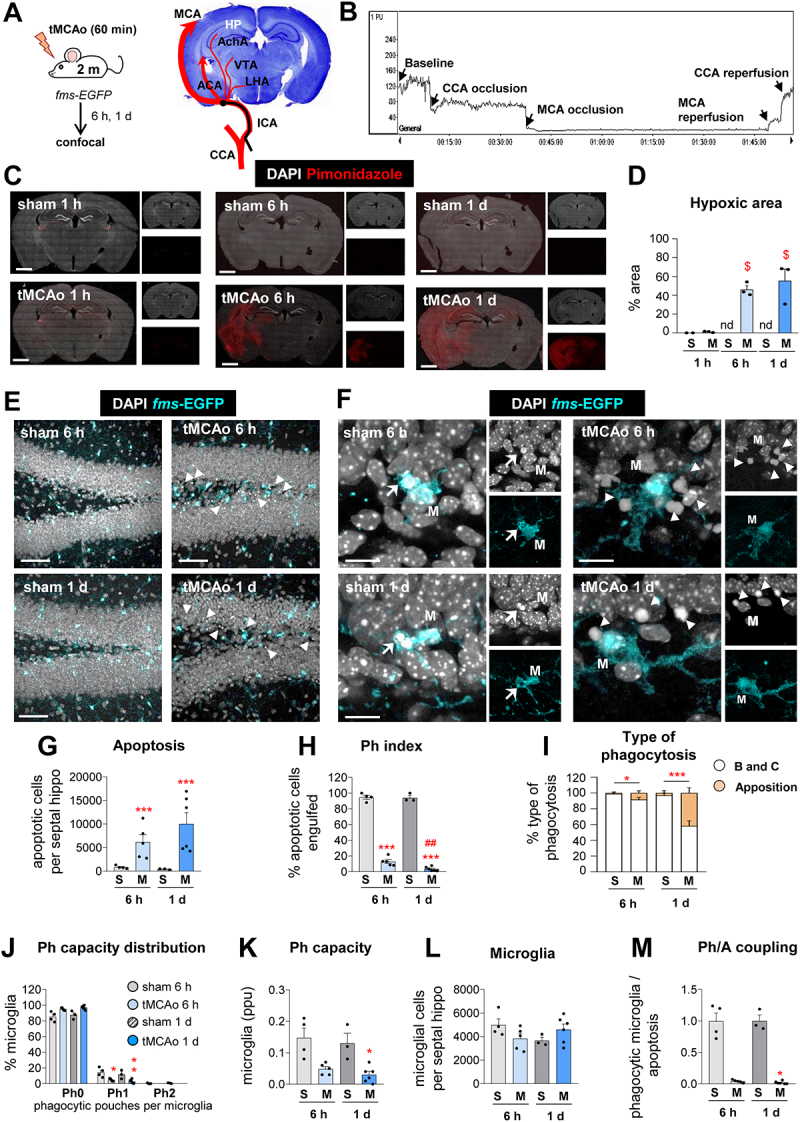
Microglial phagocytosis is impaired in mice exposed to tMCAo. (A) Experimental design of tMCAo in 2 mo fms-EGFP mice and coronal slice with cresyl violet showing the areas irrigated by the MCA. (B) Laser Doppler signal graph showing CBF in the territory supplied by MCA during baseline, CCA and MCA occlusion, and reperfusion. Successful MCA occlusion, determined by CBF > 70% drop from the baseline, recovers after reperfusion. The values are expressed in arbitrary Perfusion Units (PU). (C) Representative tiled confocal image of coronal hippocampi showing cell nuclei (with DAPI, in white) and hypoxic areas labeled with the hypoxic probe pimonidazole (in red) in 2-mo fms-EGFP mice after sham (S) and tMCAo (M) treatment at 1 h, 6 h and 1 d. (D) Percentage of hypoxic brain area determined by pimonidazole (in red) after tMCAo at 6 h and 1 d. The pimonidazole signal was not detected (nd) in sham animals. (E) Representative confocal z-stacks of the DG of fms-EGFP mice at 6 h and 1 d after tMCAo. Cell nuclei were visualized with DAPI (in white) and microglia by EGFP (in cyan). Apoptotic cells are marked with arrowheads. (F) Representative confocal z-stacks from the septal DG of a sham and tMCAo-treated mice at 6 h and 1 d, showing apoptotic cells non-phagocytosed (arrowheads) or phagocytosed (arrows) by microglia (fms-EGFP+, in cyan; M). (G) Number of apoptotic cells per septal hippocampus in sham and tMCAo. (H) Ph index in the septal hippocampus (% of apoptotic cells engulfed by microglia). (I) Type of microglial phagocytosis (% of “ball and chain” or “apposition” mechanism). (J) Histogram showing the Ph capacity of microglia (% of microglia with phagocytic pouches). (K) Weighted Ph capacity (% of microglia with phagocytic pouches). (L) Number of fms-EGFP+ microglia per septal hippocampus. (M) Ph/A coupling (in fold change) in the septal hippocampus. Bars show mean ± SEM. In (D), n = 2 (sham at 1 h), n = 3 (sham 6 h and 1 d), and n = 3 (tMCAo at 1 h, 6 h, and 1 d); (G-M), n = 3 mice (sham at 6 h), n = 4 mice (sham at 1 d), n = 5 mice (tMCAo at 6 h) and n = 6 mice (tMCAo at 1 d).Data were analyzed using one-way ANOVA using Holm-Sidak post hoc test (D). The effect of sham/tMCAo at 6 h and 1 d on apoptosis (G), Ph index (H), Type of phagocytosis (I), Ph capacity distribution (J), Ph capacity (K), Microglia (L) and Ph/A coupling (M) was analyzed using two-way ANOVA. Significant interactions were found between the two factors (tMCAo treatment x time); therefore, data were split into two one-way ANOVAs to analyze statistical differences due to the time after sham/ tMCAo at each time. Holm-Sidak was used as a post hoc test. To comply with homoscedasticity, some data were Log10 (G, K) or (Log10 + 1) transformed (I, J, M). In case that homoscedasticity was not achieved with the transformation, data were analyzed using a Kruskal-Wallis ranks test, followed by Dunn method as a post hoc test (I, J, M). (* and #) represent significance compared to sham and/or tMCAo at 6 h respectively. One symbol represents p < 0.05, two p < 0.01, and three p < 0.001. Only significant effects are shown. Scale bars: 500 μm (C); 50 μm, z = 18.9 μm (E); 14 μm, z = 16 μm (F).
We then performed immunofluorescence and confocal imaging to observe microglia, expressing the green reporter EGFP under the Csf1r (colony stimulating factor 1 receptor) promoter (fms-EGFP mice) [27]; and apoptotic cells, with abnormal nuclear morphology (pyknosis, karyorrhexis) using the DNA stain, 4’-6-diamidino-2-phenylindole (DAPI) (Figure 1E and F). Abnormal nuclear morphology is the gold standard for assessing apoptosis and, in our hands, more reliable than markers such as activated CASP3 (caspase 3) [21,22], which has alternative functions to apoptosis. Compared to control (sham-operated mice), tMCAo mice showed more apoptotic cells (Figure 1E) but few cases of phagocytosis (Figure 1F), as determined by a microglial process forming a three-dimensional pouch that surrounded the apoptotic cell and connected to the microglial cell body [21]. The contralateral hippocampus was not used as control because we found fluctuating levels of apoptosis. In the ipsilateral hippocampus, apoptosis increased significantly at 6 h and 1d after tMCAo compared to control (Figure 1G), whereas the phagocytic index (Ph index, % of apoptotic cells engulfed) dropped from 94.1 ± 3.0% and 94.8 ± 2.5% in control to 13.0 ± 2.5% and 3.5 ± 1.1% in tMCAo (at 6 h and 1d, respectively; Figure 1H). Another indication of phagocytosis impairment was that whereas in control mice phagocytosis was executed by terminal branches of microglia (“ball-and-chain” mechanism), one day after tMCAo up to 42% of phagocytosis was performed by direct apposition to the microglial soma (Figure 1I). We had already observed phagocytosis by apposition in models of epilepsy, where it was related to dysfunctional phagocytosis due to reduced microglial process motility and impaired apoptotic cell recognition [22].
To counteract the increased number of apoptotic cells, microglia would be expected to increase their phagocytic capacity (Ph capacity, % of microglia with one or more pouches) [22]. Instead, we found that microglia reduced their Ph capacity without altering their cell numbers (Figure 1J-L). Altogether, these changes led to a reduced coupling between phagocytosis and apoptosis (Ph/A coupling, ratio between net phagocytosis (Ph capacity x microglia) and apoptosis) in the hippocampus after tMCAo (Figure 1M). Similarly, microglial phagocytosis efficiency was consistently reduced in other brain regions analyzed including the striatum and cortex (Figure 2A-E), wherein apoptotic cells tended to be more abundant after tMCAo but almost non-detectable in basal conditions (Figure 2D). In tMCAo mice, but not in control mice, we also observed occasional cases of presumptive phagoptosis, or engulfment of seemingly normal nuclei, albeit at much lower levels than apoptosis (Fig. S1A, B). However, we did not observe compensatory phagocytosis by other presumed phagocytic cells of the hippocampus, such as astrocytes or the radial neural stem cells (rNSCs) (Fig. S1C, D); nor by invading monocytes, identified by their lack of expression of the pan-microglial marker P2RY12/P2Y12 [28], which were not present in the hippocampus at the time points studied (Fig. S1E-G).
Figure 2.
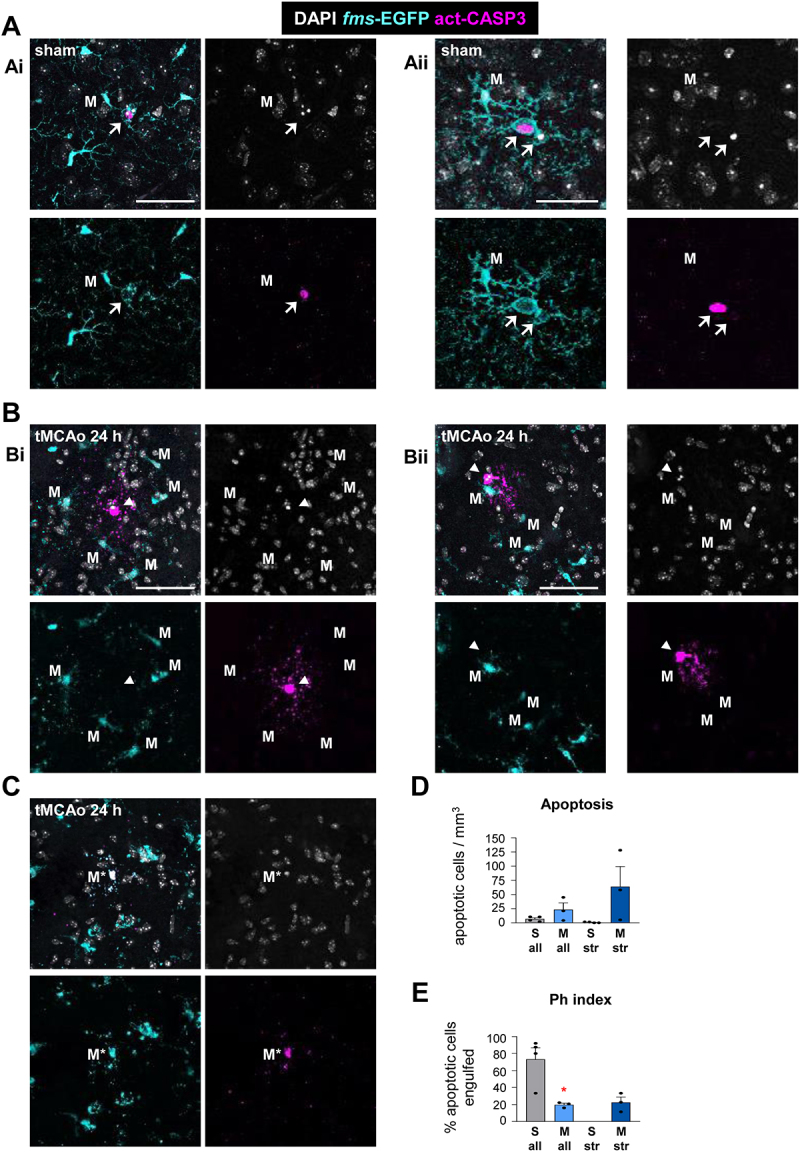
Microglial phagocytosis after tMCAO in the striatum, cortex, and subventricular zone. (A) Representative confocal z-stacks of the cortex of sham-operated 2 mo fms-EGFP mice. Normal or apoptotic (pyknotic-karyorrhectic) nuclear morphology was visualized with DAPI (white), microglia b EGFP (cyan), and apoptosis was confirmed by activated CASP3 staining (magenta). Images show apoptotic cells (pyknotic-karyorrhectic and act-CASP3+) engulfed (arrows) by microglia (M) (EGFP+) (Ai, Aii). (B, C) Representative confocal z-stacks of the striatum of tMCAo-exposed 2 mo fms-EGFP mice after 24 h. Normal or apoptotic (pyknotic-karyorrhectic) nuclear morphology was visualized with DAPI (white), microglia by EGFP (cyan), and apoptosis was confirmed by act-CASP3 staining (magenta). Images show apoptotic cells (pyknotic-karyorrhectic and act-CASP3+) not-engulfed (arrowheads) by microglia (M) (EGFP+) (Bi, Bii) and apoptotic microglia (M*) (pyknotic-karyorrhectic, act-CASP3+, EGFP+) (C). (D, E) Quantification of microglial phagocytosis in the striatum and cortex. Apoptotic cells were scarce in the striatum and cortex in sham-operated control mice. To establish a baseline level of microglial phagocytosis in these regions, apoptotic cells from the striatum and cortex were combined with apoptotic cells from the subventricular zone, which were more abundant. (D) Number of apoptotic cells per mm3 analyzed in the striatum (str) and the combination of striatum, cortex and subventricular zone (all). (E) Ph index (% of apoptotic cells engulfed by microglia) in the striatum (str) or the combination of striatum, cortex and subventricular zone (all). Bars show mean ± SEM (D, E). n = 3–4 mice per group (D, E). Data were analyzed by Student’s t-test (D, E). * represents significance between tMCAo and sham (p < 0.05). Scale bars: 50 µm, z = 9 µm (Ai), 10 µm, z = 7 µm (Aii), 50 µm, z = 25 µm (Bi), 50 µm, z = 10 µm (Bii), 50 µm, z = 9 µm (C).
We confirmed the microglial phagocytosis impairment and the lack of involvement of monocytes using a related model of HI in mice by ligation of the right common carotid artery (CCA) and exposure to 10% oxygen for 75 min [29]. We used Cx3cr1GFP/+/Ccr2RFP/* mice to discriminate microglia (RFP−) from monocytes (RFP+). As in tMCAo, microglia did not respond to the increased number of apoptotic cells with an increased Ph capacity, resulting in many apoptotic cells not phagocytosed (reduced Ph index) and disrupting the phagocytosis-apoptosis crosstalk (reduced Ph/A coupling) (Figure 3). In this model we observed that monocytes, which expressed CCR2 and not P2RY12 (Fig. S2A-C), invaded the hippocampus 3 d after HI but did not engage in phagocytosis (Fig. S2D-G). As in tMCAo, we also observed a few cases of microglial and monocyte phagoptosis (Fig. S2H, I). The phagocytosis impairment that microglia suffered in tMCAo and HI was more evident when we analyzed the microglial response in a model of cranial irradiation [30,31](Figure 4). Here, mice received a single dose of 8 Gy that led to a huge increase of apoptotic cells in the subgranular zone of the dentate gyrus (DG) (presumably, proliferating newborn cells, more susceptible to irradiation). In contrast to tMCAo and HI, microglia now responded by increasing phagocytosis and clearing out the dead cells within 1 d. Therefore, the phagocytosis impairment observed in tMCAo and HI was not merely a saturation of the phagocytic response but rather an active phenomenon related to the pathophysiological mechanisms operating in tMCAo and HI.
Figure 3.
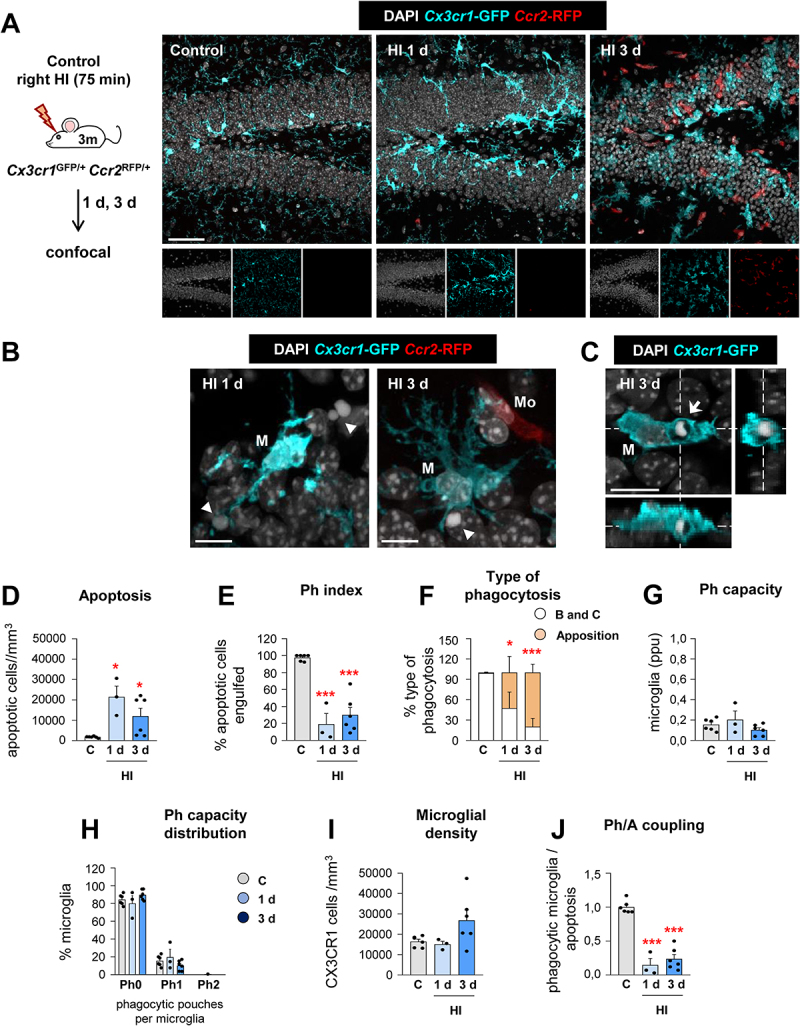
Microglial phagocytosis is impaired during HI. (A) Experimental design and representative confocal z-stack of the DG of 3 mo Cx3cr1-GFP Ccr2-RFP mice at 1 and 3 d under HI. Cell nuclei were visualized with DAPI (in white), microglia (Cx3cr1-GFP+, in cyan) and monocytes (Ccr2-RFP+, in red). (B) Representative confocal z-stack from the DG of a HI-treated mouse at 1 and 3 d showing apoptotic cells (arrowheads) non-phagocytosed by microglia (Cx3cr1-GFP+, in cyan; M), close to monocytes (Ccr2-RFP+, in red; Mo). (C) Orthogonal projection of a confocal z-stack from the septal DG of a HI-treated mouse at 3 d showing an apoptotic cell (arrow) phagocytosed by microglia (Cx3cr1-GFP+, in cyan; M). (D) Density of apoptotic cells (cells/mm3) in the septal DG under control and HI treatment. (E) Ph index in the septal DG (in % of apoptotic cells engulfed by microglia). (F) Type of microglial phagocytosis (% of “ball and chain” or “apposition” mechanism). (G) Weighted Ph capacity under control and HI conditions. (H) Histogram showing the Ph capacity of microglia (% of microglia with pouches). (I) Density of Cx3cr1-GFP+ microglia (cells/mm3) in the septal DG. (J) Ph/A coupling (in fold change) in the septal DG. Bars represent mean ± SEM. n = 5 (control), n = 3 (at 1 d) and n = 6 (at 3 d). Data were analyzed by one-way ANOVA, using Holm-Sidak as post hoc test. To comply with homoscedasticity, some data were Log10 (D, H) and/or Log10 + 1 (G) transformed. In the case that homoscedasticity was not achieved with a logarithmic transformation data were analyzed using a Kruskal-Wallis rank test, followed by Dunn method as a post hoc test (D, G). One (*) symbol indicates p < 0.05, and three p < 0.001 (vs control). Scale bars: 50 μm, z = 16 μm (A); 20 μm, z = 15 μm (B); 10 μm, z = 14 μm (C).
Figure 4.
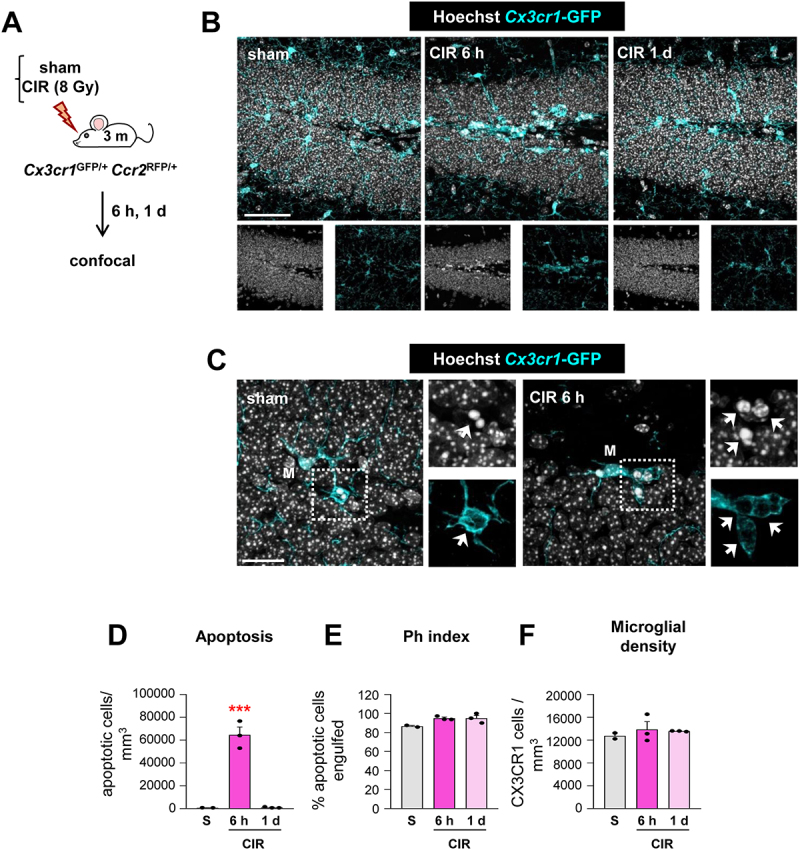
Microglial phagocytosis increases after CIR exposure. (A, B) Experimental design and representative confocal z-stacks of the DG of 3 mo Cx3cr1-GFP/Ccr2-RFP mice at 6 h and 1 d after CIR (8 Gy). Apoptotic nuclei were detected by pyknosis/karyorrhexis (in white, Hoechst), and microglia and blood-derived macrophages by the transgenic expression of Cx3cr1-GFP (in cyan) and Ccr2-RFP (in red), respectively. (C) Representative confocal z-stack of apoptotic cells (pyknotic-karyorrhectic, Hoechst, in white, arrow) phagocytosed by microglia (Cx3cr1-GFP+, in cyan; M) in the septal DG of sham and CIR-treated mice. (D) Density of apoptotic cells (cells/mm3) in the septal DG. (E) Ph index (% of apoptotic cells engulfed by microglia) in the septal DG. (F) Density of CX3CR1+ microglia (cells/mm3) in the septal DG. Bars represent mean ± SEM. In (C, D, E), n = 2 (sham) and n = 3 (at 6 h and 1 d). Data were analyzed by one-way ANOVA, using Holm-Sidak as a post hoc test. To comply with homoscedasticity some data were Log10 transformed (D). *** indicates p < 0.001 (vs CIR at 1 d). Scale bars: 50 µm, z = 21 µm (sham, 6 h), z = 17.5 µm (1 d) (B); 20 µm, z = 13.3 µm (sham), z = 18.9 µm (6 h) (C).
We next addressed the relevance of the phagocytosis impairment for human stroke using a model of tMCAo in macaques (Macaca fascicularis) [32], a validated preclinical model of human stroke [24,25] (Figure 5A-G). Analysis of autopsy human tissue is constrained by the postmortem delay, which affects both phagocytosis and apoptosis [22], microglial process motility [33], and microglial gene expression [34]. We therefore analyzed phagocytosis in the cortical temporal region of three macaques with varying levels of apoptosis, 30 days after the stroke (Figure 5C and D). Apoptosis was lower in the contralateral hemisphere compared to the ipsilateral hemisphere in the most damaged animal (#892) (Figure 5D), with largest infarct size and sustained behavioral deficits (Figure 5E). We analyzed phagocytosis using P2RY12 to label microglial processes and found that it was lower in the ipsilateral than in the contralateral hemisphere in animals #638 and #892, as determined by the Ph index (Figure 5F). Overall, apoptosis inversely correlated with the Ph index (Figure 5G), indicating a higher phagocytosis impairment in the most damaged tissue. We also analyzed cortical and hippocampal tissue from three marmoset monkeys (Callithrix jacchus), 45 days after tMCAo [35](Figure 5H-J). Although uninjured controls were not available, we found low absolute levels of phagocytosis, with a Ph index of 41.1% (Figure 5J). Altogether, data obtained in mice and monkey models strengthens our hypothesis that stroke impairs microglial phagocytosis.
Figure 5.
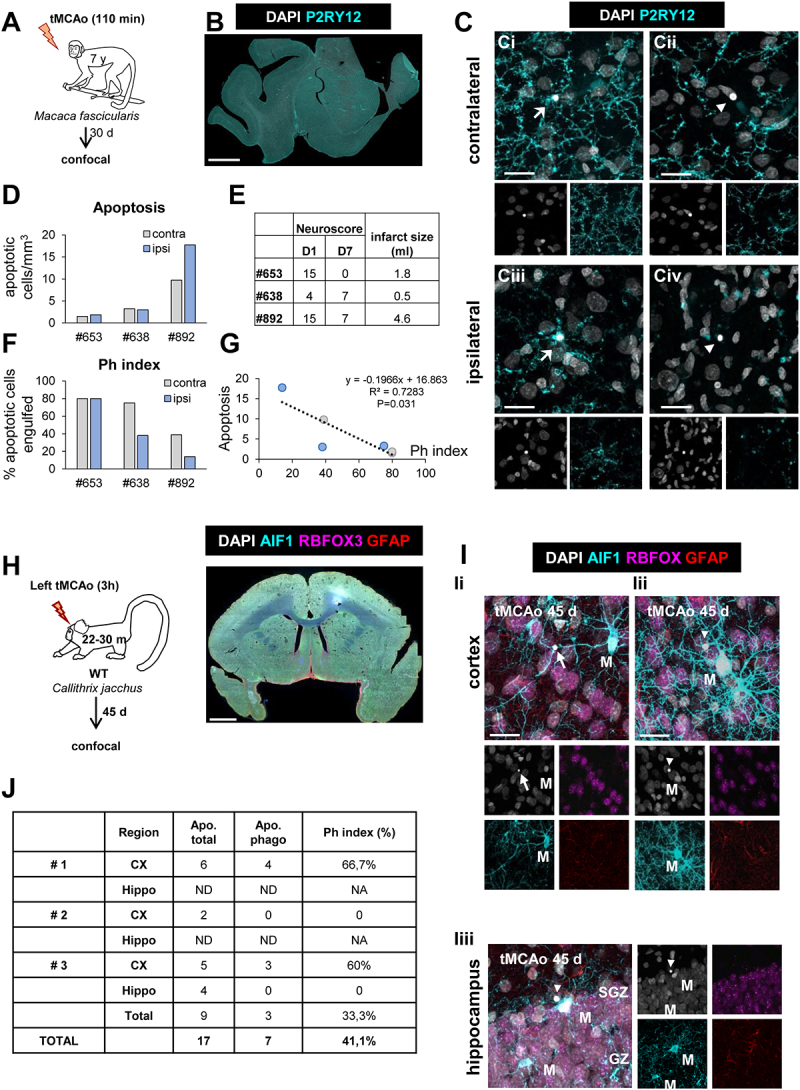
Microglial phagocytosis after tMCAo in Macaca fascicularis and Callithrix jacchus. (A) Experimental design of tMCAo in Macaca fascicularis. Data from these animals were previously published here [80]. (B) Epifluorescent image of the cerebral cortex of Macaca fascicularis showing DAPI (nuclei, white) and P2RY12 (microglia, in cyan). (C) Representative confocal z-stacks of the cortical regions of macaques at 30 d after tMCAo from the contralateral (Ci, Cii) or ipsilateral (Ciii, Civ) hemispheres. Cell nuclei were visualized with DAPI (in white) and microglia with P2RY12 (in cyan). Arrows and arrowheads point to phagocytosed (Ci, Ciii) and non-phagocytosed apoptotic cells (Cii, Civ), respectively. (D) Number of apoptotic cells per mm3 in the ipsi- and contralateral hemispheres. (E) Table summarizing the behavioral impairment (neuroscore; higher numbers indicate more impairment) at days (D) 1 and 7 after the tMCAo, and the infarct size determined by magnetic resonance imaging [80]. (F) Ph index (% of apoptotic cells engulfed by microglia) in the ipsi- and contralateral hemispheres. (G) Correlation between apoptosis and Ph index in the ipsi- (light blue) and contralateral (grey) hemispheres of the three macaques. The regression line, the regression coefficient R2, and the adjusted p-value are shown. (H) Experimental design of tMCAo in Callithrix jacchus and low magnification epifluorescent image of common marmoset brain showing nuclei (DAPI, white), microglia (AIF1/IBA1, in cyan), neurons (RBFOX3/NeuN, in magenta), and astrocytes (GFAP, in red). (I) Representative confocal z-stacks of the cortical regions (Ii, Iii) and hippocampus (Iiii) of marmosets at 45 d after tMCAo showing phagocytosed (arrow) and non-phagocytosed (arrowheads) apoptotic cells. M, microglia. (J) Table summarizing the number of apoptotic cells (total and phagocytosed by microglia) in the three marmosets analyzed. ND, not-detected. NA, not applicable. Scale bars: 5 mm (B); 20 μm (C), z = 18.9 μm (Ci), 14.1 μm (Cii), 14.1 μm (Ciii), 15.4 μm (Civ); 1 mm (H); 20 μm, z = 19.6 μm (Ii, Iii); 20 μm, z = 23.8 μm (Iiii). RBFOX3/NeuN: RNA-binding fox homolog 3; GFAP: glial fibrillary acidic protein.
In silico analysis of microglial phagocytosis-related genes
To delve into potential mechanisms underlying the microglial phagocytosis impairment, we first analyzed recently published RNA sequencing (RNA-Seq) databases from mice or rat MCAo models [10–14]. While none of these studies originally discussed alterations in phagocytosis-related genes in their unbiased analysis, we used the functional annotation tool DAVID to determine whether the significantly regulated genes in each of those databases was related to the Gene Ontology (GO) term Phagocytosis (Fig. S3). We found that the Phagocytosis GO term was only significantly regulated in a bulk RNA-Seq study of permanent MCAo in mice at 3 d by Androvic and cols [11] and in a microglial cluster in single cell (sc) RNA-Seq study of transient (90 min) MCAo in mice at 1 d [13]. In addition, the sub-term Positive regulation of phagocytosis was regulated in the Androvic study [11] and in another bulk RNA-Seq study of transient MCAo (60 min) at 1d by Guo and cols [12]. However, in these three studies, the percentage of phagocytosis-related genes over the total significant genes was very small (0.7–3.9%), which is likely the reason why the original papers did not report it as a relevant finding. In addition, the most recent scRNA-Seq study of FACS-sorted leukocytes by Beuker and cols. did not identify changes in phagocytosis in either of the microglial clusters [14]. They also discovered a new cluster of stroke-associated myeloid cells (SAMCs), in which they reported changes in lipid metabolism genes and increased lipid uptake in vitro, suggestive of myelin engulfment, but in which we found no significant regulation of the Phagocytosis GO term (Fig. S3A).
To examine deeper the expression of individual genes related to phagocytosis, we cross-referenced the 380 genes listed in the Phagocytosis GO term with the five databases and found that the Androvic study reported significant changes in expression in only 24.2% of the Phagocytosis GO genes. This percentage dropped in the rest of the studies, down to 0.5% in some of the microglial clusters of the Beuker study (Fig. S3B). Even more importantly, the majority of the genes with significant differential expression in either of the five studies had a very small FC (Fig. S3C). In addition, most of the significantly regulated genes related to the Phagocytosis GO term in these two databases were positive regulators of phagocytosis with increased expression. In light of the consistent dysfunction of phagocytosis we found across models (Figures 1–5), these gene changes could be interpreted as a compensatory mechanism to recover phagocytosis efficiency. Altogether, this analysis suggests that RNA-Seq studies are not necessarily the optimal strategy to identify functional changes in microglia, and that in MCAo models microglial phagocytosis is not regulated at the transcriptional level.
Energy depletion impairs engulfment and degradation via alterations in motility and lysosomal function
We then studied whether the effects of stroke on phagocytosis were related to the energy depletion induced by the lack of blood supply, using an in vitro model of oxygen and nutrient deprivation (OND: culture medium salt solution and 1% O2) (Figure 6). In organotypic cultures, 3 h and 6 h of OND resulted in a phagocytosis phenotype similar to that induced by tMCAo and HI: increased apoptosis and reduced Ph index and Ph capacity, resulting in Ph/A uncoupling (Figure 6A-H; Fig. S4A). Importantly, engulfment was recovered rapidly as early as 1 h after reperfusion (complete medium and normoxia) indicating that the effect of energy depletion on phagocytosis was reversible in vitro. In contrast, in tMCAo we found sustained phagocytosis impairment 24 h after reperfusion, possibly related to the buildup of hypoxia over time (Figure 1C and D), which has been associated with the death in rigor of pericytes and the irreversible constriction of brain capillaries [36]. The reduced engulfment induced by OND was likely related to a reduction in microglial process motility, as determined by 2-photon microscopy in organotypic slices under OND (Figure 6I-L).
Figure 6.
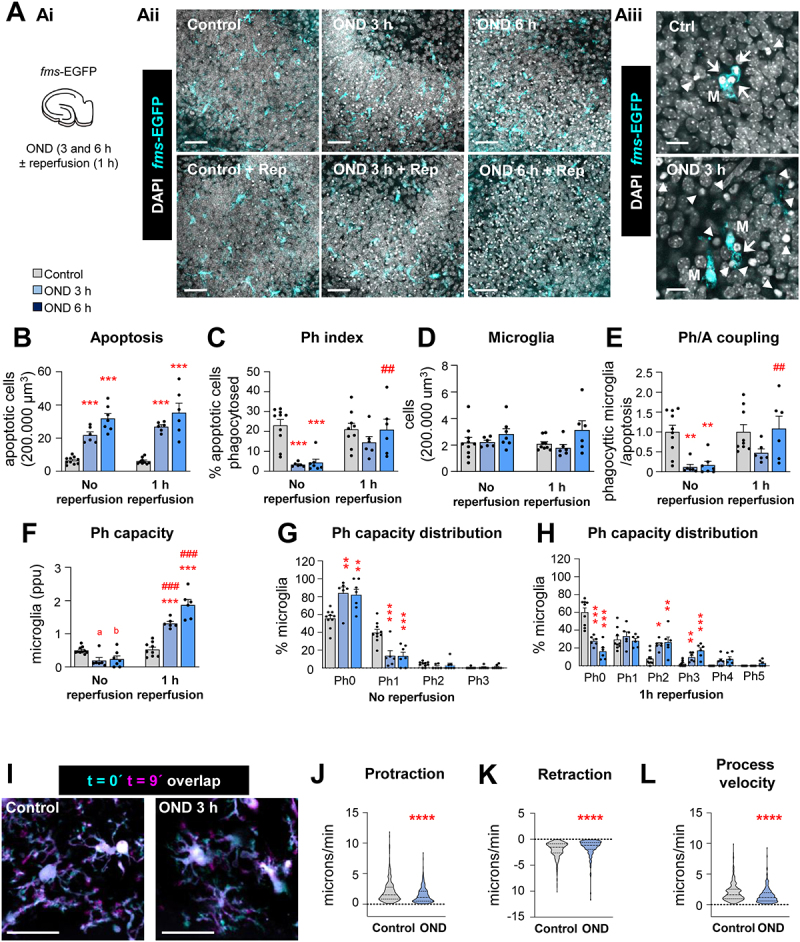
Engulfment of apoptotic cells is impaired after OND in hippocampal organotypic slices due to alterations in motility. (A) Experimental design showing the exposure of hippocampal organotypic slices (fms-EGFP) to OND (3 and 6 h) in the presence and absence of 1 h reperfusion (Ai). Representative confocal images of the DG after OND (Aii). Normal or apoptotic (pyknotic-karyorrhectic) nuclear morphology was visualized with DAPI (white) and microglia by the transgenic expression of fms-EGFP (cyan). High magnification images show apoptotic cells (pyknotic-karyorrhectic) engulfed (arrows) or not-engulfed (arrowheads) by microglia (M) (EGFP+) (A3). High magnification images for reperfusion experiments in Aii are shown in Fig. S4A. (B) Number of apoptotic cells in 200,000µm3 of the DG. (C) Ph index (% of apoptotic cells phagocytosed by microglia). (D) Number of microglia in 200,000 µm3 of the DG. (E) Ph/A coupling expressed as fold-change; ratio between net phagocytosis and total levels of apoptosis. (F) Weighted Ph capacity (number of phagocytic pouches containing an apoptotic cell per microglia, in parts per unit (ppu). (G, H) Ph capacity histogram after OND in non-reperfused (G) and reperfused (H) conditions. (I) Representative projections of 2-photon images of microglial cells at t0 (cyan) and t9 (magenta) from hippocampal organotypic slices (Cx3cr1GFP/+) under control and OND conditions. (J, K, L) Microglial process motility: protraction (J), retraction (K), and process velocity (L). Bars show mean ± SEM (B-H). Violin plots show the data distribution including extreme values; lower and upper hinges correspond to the first and third quartile respectively (J-L). n = 6–10 mice per group (B-H); n = 355 processes from 98 cells from 12 animals (control), and n = 222 processes from 57 cells from 9 animals (OND) (J-L). Data were analyzed by two-way ANOVA followed by Holm-Sidak post hoc tests (B, D). When an interaction between factors was found, one-way ANOVA (factor: treatment) was performed followed by Holm-Sidak post hoc tests (C, E, F). To comply with homoscedasticity, some data were Log10 transformed (B) or Ln transformed (F). Other data were analyzed by Kruskal-Wallis rank test (J-L). (* and #) represent significance between control and OND, or no reperfusion vs reperfusion, respectively: one symbol represents p < 0.05, two symbols represent p < 0.01, three symbols represent p < 0.001, and four symbols represent p < 0.0001; (a) represents p = 0.06 and (b) represents p = 0.07 (control vs OND). Scale bars: 50 µm, z = 10.5 µm (Aii); 15 µm, z = 11.9 µm (Aiii), 20 µm, z = 22 μm (I).
To identify the mechanisms of phagocytosis impairment during energetic depletion we used primary cultures of microglia co-incubated with a neuronal cell line in which we induced apoptosis (SH-SY5Y-Vampire pre-treated with staurosporine). We analyzed the amount of microglia with Vampire+ DAPI particles at two time points to discriminate engulfment (1 h of co-incubation) and degradation (3 h after washout) in cells treated with OND (Figure 7A-D). This model uncovered an effect of OND on degradation (Figure 7C), which could not be assessed in vivo or in organotypic cultures because it is downstream of engulfment. Nonetheless, our in vitro model did not reveal the expected effect on engulfment, not even when we co-incubated microglia in more stringent conditions (fewer apoptotic SH-SY5Y cells, or reduced co-incubation time) (Fig. S4B, C), possibly because in vitro models of phagocytosis do not fully mimic the complexity of the “find-me” and “eat-me” signals that regulate engulfment in vivo [37]. We therefore focused on understanding the basis of degradation impairment by studying lysosomes, the degradative organelles. We found that under OND microglia had a small but significant increase in lysosomal pH (Figure 7E and F) and reduced lysosomal number (Figure 7G-I). Although individual lysosomes had a similar enzymatic activity (Figure 7J), these changes resulted in a reduced degradative capacity of microglia under OND. In conclusion, the energetic depletion associated with stroke activated several cell processes that affected both engulfment and degradation of apoptotic cells that included alterations in motility and in lysosomal pH and number.
Figure 7.
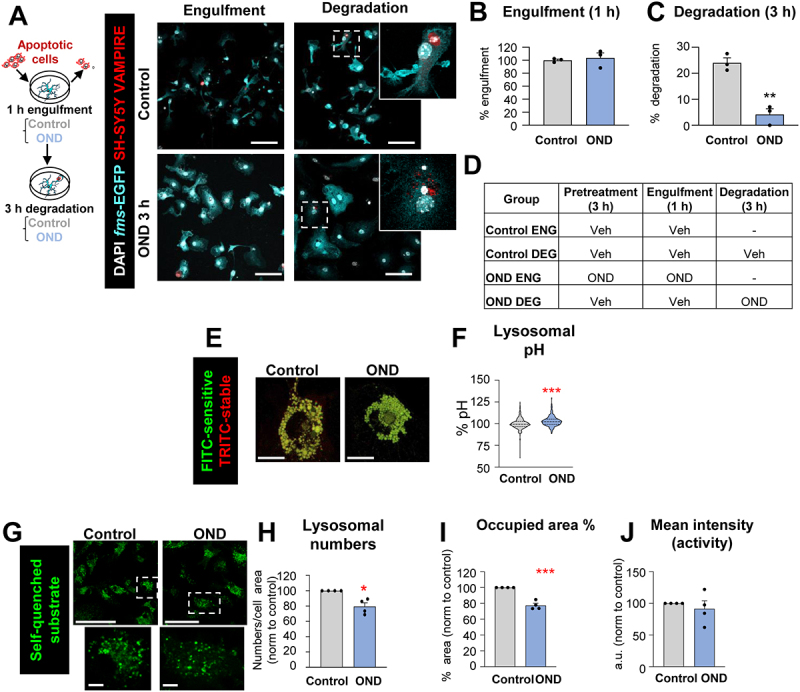
Degradation of apoptotic cells is impaired after OND in primary microglia due to alterations in lysosomal function. (A) Experimental design of the phagocytosis assay to assess engulfment and degradation of apoptotic cells under control and OND conditions. Representative images of primary microglia fed with apoptotic SH-SY5Y VAMPIRE cells during engulfment and degradation. Nuclei were visualized with DAPI (white), microglia by expression of EGFP (cyan), and SH-SY5Y neurons by expression of the red fluorescent protein VAMPIRE. (B, C) Percentage of phagocytic microglia after engulfment (1 h) and degradation (3 h after engulfment). Only particles fully enclosed by microglia were identified as being phagocytosed. Raw % of phagocytic microglia are shown in Fig. S4B. Phagocytosis under more stringent conditions (shorter incubation, fewer apoptotic cells) is shown in Fig. S4C. (D) Summary of the experimental groups. (E) Representative confocal images of microglia incubated with dextran molecules conjugated to two fluorophores: FITC (pH sensitive) and TRITC (pH stable) located in the lysosomes, whose ratio serves as an indirect measurement of the lysosomal pH. (F) Lysosomal pH expressed as % normalized to control values. Note the truncated Y axis. (G) Representative confocal images of microglia loaded with the self-quenched substrate. (H) Percentage of lysosomal numbers normalized to control values under control and OND conditions. (I) Percentage of the area occupied by lysosomes, referred to control values. (J) Mean intensity (representative of lysosomal activity) represented in arbitrary units and referred to control values. Bars show mean ± SEM (B, C, H-J). Violin plot shows the data distribution including extreme values; lower and upper hinges correspond to the first and third quartile respectively (F); n = 3 independent experiments (B, C); n = 530 cells (control) and 452 cells (OND) from 4 independent experiments (F). n = 4 independent experiments (H-J). Data were analyzed by Student´s t-test (B, C, F, H-J). (*) one symbol represents p < 0.05, two symbols represent p < 0.01, three symbols represent p < 0.001, and four symbols represent p < 0.0001. Scale bars: 5 µm, z = 8.5 μm (A), 7 µm (E); 50 µm low magnification, 5 µm high magnification, z = single plane (G).
Autophagy is induced in microglia after stroke and OND
The lysosomal alterations induced by OND motivated us to focus on autophagy, a major intracellular degradative pathway with several reported alterations in models of stroke [38]. Autophagy and phagocytosis are relatively similar processes, whose goal is to degrade intracellular (autophagy) or extracellular (phagocytosis) cargo, and they share some mechanisms and cellular machinery [39]. To study the autophagy status in microglia we used in vivo and in vitro complementary approaches (Figure 8, 9): in vivo, we used confocal imaging of the autophagosome marker MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) and colocalization with the autophagy substrate SQSTM1 (sequestosome 1) and the lysosomal marker LAMP1 (lysosomal-associated membrane protein 1). In vitro, we analyzed LC3 by confocal imaging and western blot, and direct visualization of autophagosomes by transmission electron microscopy (TEM). First, we evaluated autophagy flux in microglia 6 h and 1 d after tMCAo (Figure 8A-H). The number of LC3-positive puncta (autophagosomes) tended to increase after tMCAo (Figure 8B) and the area they occupied was significantly larger both at 6 h and 24 h (Figure 8C), indicative of an expansion of the autophagosome compartment. The composition of these LC3 puncta, determined by colocalization, changed after tMCAo, with autophagosomes containing more SQSTM1 autophagy substrate (Figure 8D) and similar levels of LAMP1 (Figure 8E), which suggested effective autophagosome-lysosome fusion and ongoing autophagic turnover. These effects were accompanied by a progressive reduction of total SQSTM1 puncta numbers (Figure 8F), confirming enhanced autophagy flux after tMCAo. Similar to OND (Figure 7H), tMCAo tended to reduce LAMP1-containing puncta (Figure 8G), suggesting the consumption of lysosomes for autophagic purposes in detriment of phagocytosis.
Figure 8.
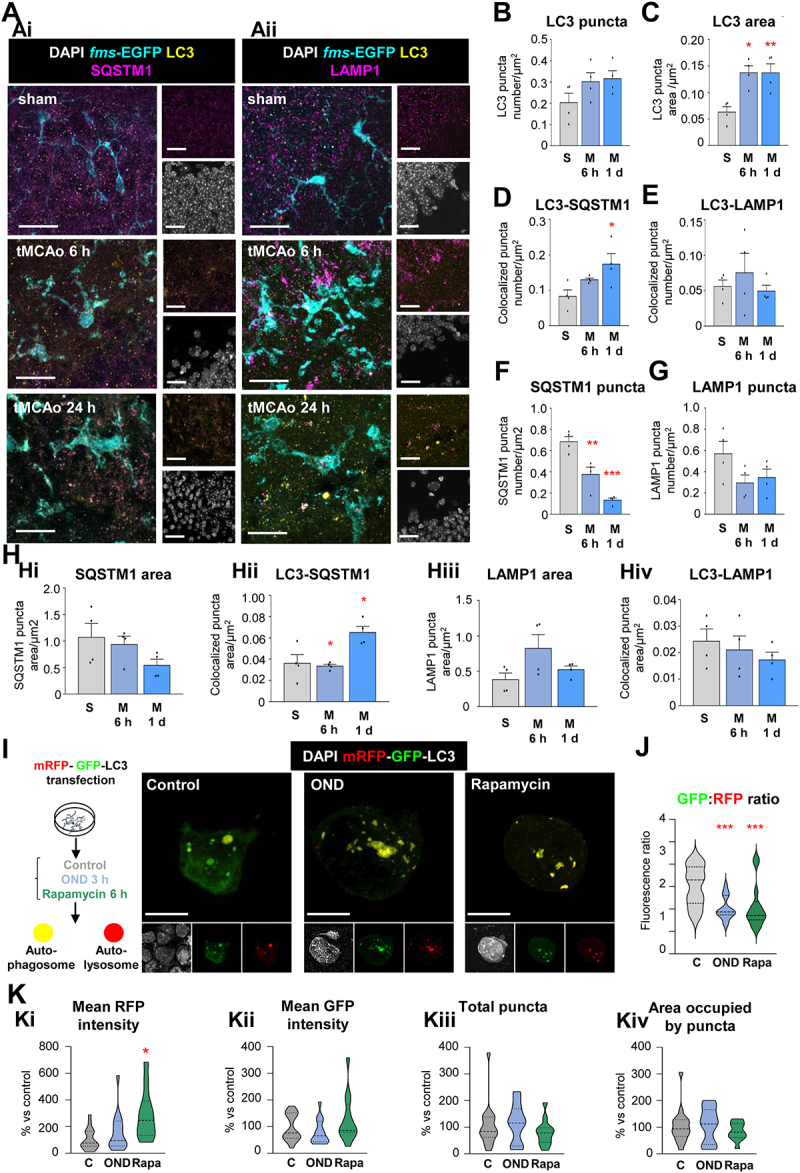
Autophagy flux is induced in microglia after tMCAo and OND. (A) Representative confocal images of the DG of 2 mo fms-EGFP mice exposed to sham and tMCAo surgery at 6 h and 1 d. LC3 present in autophagosomes was immunostained and observed as puncta (yellow). Microglia were visualized by the transgenic expression of fms-EGFP (cyan) and cellular nuclei were identified by DAPI (white). SQSTM1 (Ai) and LAMP1 (Aii) were immunostained and visualized as puncta (magenta). (B) Number of LC3 puncta normalized to microglial cytoplasmic area (LC3 puncta number/µm2) in sham and tMCAo (6 h and 1 d) mice. (C) Total area of LC3 puncta normalized to microglial cytoplasmic area (LC3 puncta area/µm2) in sham and tMCAo (6 h and 1 d) mice. (D) Number of LC3 and SQSTM1 puncta that colocalize normalized to microglial cytoplasmic area (LC3-SQSTM1 puncta number colocalization/µm2) in sham and tMCAo (6 h and 1 d) mice. (E) Number of LC3 and LAMP1 puncta that colocalize normalized to microglial cytoplasmic area (LC3-LAMP1 puncta number colocalization/µm2) in sham and tMCAo (6 h and 1 d) mice. (F) Number of SQSTM1 puncta normalized to microglial cytoplasmic area (SQSTM1 puncta number/µm2) in sham and tMCAo (6 h and 1 d) mice. (G) Number of LAMP1 puncta normalized to microglial cytoplasmic area (LAMP1 puncta number/µm2) in sham and tMCAo (6 h and 1 d) mice. (H) Quantified total SQSTM1 puncta area normalized by microglial cytoplasmic area (SQSTM1 puncta area/µm2) (Hi), the colocalized area of LC3 and SQSTM1 puncta normalized by microglial cytoplasmic area (colocalized LC3-SQSTM1 puncta area/µm2) (Hii), total LAMP1 puncta area normalized by microglial cytoplasmic area (LAMP1 puncta area/µm2) (Hiii), and the colocalized area of LC3 and LAMP1 puncta normalized by microglial cytoplasmic area (colocalized LC3-LAMP1 puncta area/µm2) (Hiv). (I) Experimental design used to transfect BV2 microglia-like cells with the fluorescent tandem mRFP-GFP-LC3 to assess autophagy flux in control conditions (C) and after OND (3 h) or rapamycin (Rapa, 100 nM, 6 h) treatments. Representative confocal images of control, OND and rapamycin treated microglia. Nuclei are stained with DAPI (white), autophagosomes and autolysosomes are differentiated according to the tandem expression (yellow and red, respectively). (J) GFP:RFP mean fluorescence intensity ratio, indicative of autophagy flux. (K) Mean RFP fluorescence intensity (Ki), mean GFP fluorescence intensity (Kii), total number of puncta (Kiii), and area occupied by puncta (Kiv) per cell and normalized to control conditions (expressed as % change versus control conditions). Bars show mean ± SEM (B-H). Violin plots show the data distribution, including extreme values; lower and upper hinges correspond to the first and third quartile, respectively (J, K). n = 4 mice per experimental condition (B-H); n = 12–17 cells from 3 independent experiments (J, K). Data were analyzed by one-way ANOVA followed by Holm-Sidak post hoc test (B-H), Bonferroni post hoc test (J); by Kruskal-Wallis one- way ANOVA on ranks followed by Dunn’s multiple comparisons (K). * represents significance between sham or control and tMCAo, OND or rapamycin: one symbol represents p < 0.05, two symbols represent p < 0.01 and three symbols represent p < 0.001. Scale bars: 20 µm, z = 20 µm (A); 10 µm, z = 1.9 µm (control), 3.3 µm (OND), and 3.9 µm (rapamycin) (I).
Figure 9.
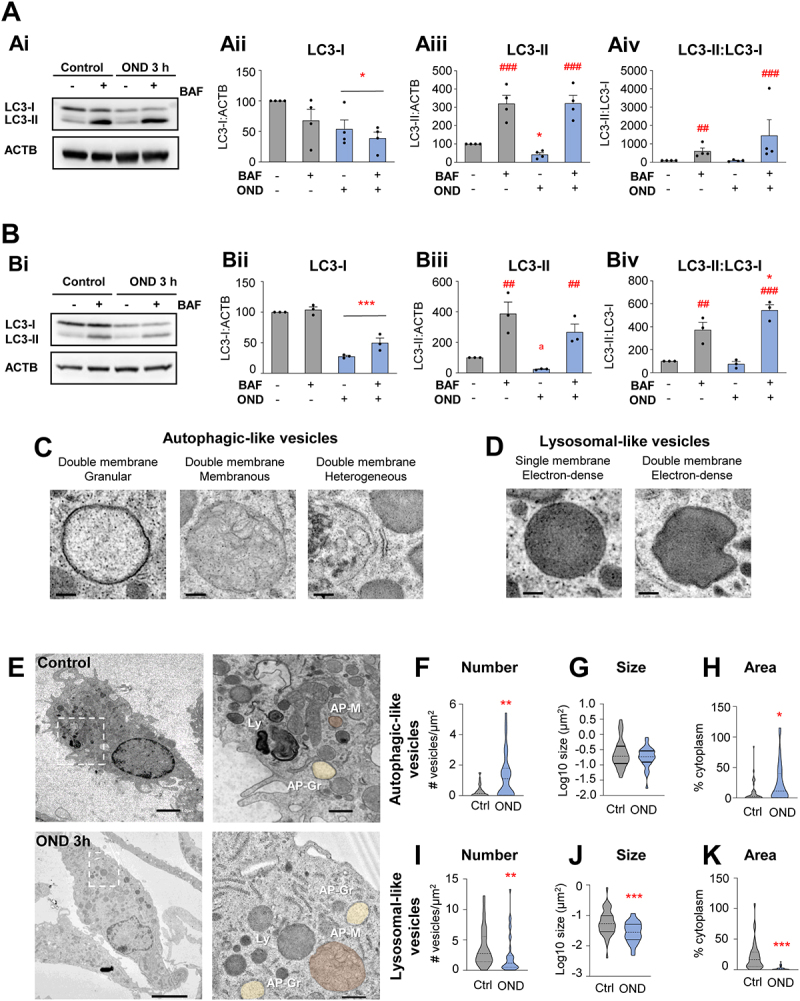
Complementary autophagy analysis in microglial cultures after OND. (A) Primary microglia were exposed to OND for 3 h in the presence and absence of bafilomycin A1 (BAF, 100 nM, 3 h) to assess autophagy flux by LC3 turnover assay. Delipidated (~1 KDa) and lipidated (~17 KDa) LC3 levels were analyzed by western blot. ACTB (~42 KDa) was used as a loading control. Representative blots showing LC3-I, LC3-II and ACTB bands (Ai), LC3-I levels normalized to ACTB (Aii), LC3-II levels normalized to ACTB (Aiii), LC3-II levels normalized to LC3-I levels (Aiv). (B) BV2 cells were exposed to OND for 3 h in the presence and absence of bafilomycin A1 (BAF, 100 nM, 3 h) to assess autophagy flux by LC3 turnover assay. Delipidated (~1 KDa) and lipidated (~17 KDa) LC3 levels were analyzed by western blot. ACTB (~42 KDa) was used as loading control. Representative blots showing LC3-I, LC3-II and ACTB bands (Bi), LC3-I levels normalized to ACTB (Bii), LC3-II levels normalized to ACTB (Biii), LC3-II levels normalized to LC3-I levels (Biv). (C) Details of autophagic-like vesicles identified as containing at least a portion of double membrane with different types of cargo (granular, membranous, heterogeneous). (D) Details of lysosomal-like vesicles identified as electron-dense vesicles with single or double membrane. (E) Representative TEM images of primary microglia in control and OND conditions. Ly: lysosomes; AP-Gr: autophagosomes with granular cargo (yellow); AP-M: autophagosomes with membranous cargo (Orange). (F-H) Quantification of autophagic-like vesicle number per µm2 (F), size in µm2 (in logarithmic scale) (G), and percentage of cytoplasm occupied (H). (I-K) Quantification of lysosomal-like vesicle number per µm2 (I), size in µm2 (in logarithmic scale) (J), and percentage of cytoplasm occupied (K). Bars show mean ± SEM (A, B). Violin plots show the data distribution, including extreme values; lower and upper hinges correspond to the first and third quartile, respectively (F-H, I-K). n = 4 independent experiments (A); n = 3 independent experiments (B); n = 36–38 cells from 3 independent experiments (F-H, I-K). Data were analyzed by two-way ANOVA followed by Holm-Sidak post hoc test (A). When a trend for a significant interaction between factors was found (Aiii, OND x BAF interaction: p = 0.08), one-way ANOVA (factor: treatment) was performed followed by Holm-Sidak post hoc test (Aiii). To comply with homoscedasticity some data were square root (Aiii) or Log10 transformed (Aiv). Data were analyzed by two-way ANOVA followed by Holm-Sidak post hoc test (B). When a trend for a significant effect of factors was found (Biii, OND factor: p = 0.067), analysis was followed by Holm-Sidak post-hoc tests (Biii). When an interaction between factors was found, one-way ANOVA (factor: treatment) was performed followed by Holm-Sidak post hoc tests (Biv). Data were analyzed by one-way ANOVA followed by non-parametric Mann-Whitney test (F-H, I-K) (* and #) represent significance: one symbol represents p < 0.05, two symbols represent p < 0.01 and three symbols represent p < 0.001. Scale bars: 500 nm (C, D); 2 µm (control), 5 µm (OND), 500 nm (high magnification) (E).
To confirm that autophagy induction was a consequence of the drop of energy supplies in microglia, we next analyzed the autophagy flux in microglial cultures after OND. First, we used a tandem plasmid mRFP-EGFP-LC3 to transfect the microglial cell line BV2 (Figure 8I-K). Cytoplasmic LC3 was observed as a diffuse signal, whereas autophagosome LC3 was located in puncta. As GFP fluorescence is pH-sensitive (whereas RFP is pH-stable), a reduced GFP:RFP fluorescence ratio indicated the fusion of the autophagosomes with lysosomes (autolysosomes) [40]. Microglial cells under OND, similar to cells treated with the autophagy inducer rapamycin, showed a trend to increased RFP intensity in puncta (Figure 8Ki), suggesting increased LC3 presence in autophagic vesicles. However, GFP intensity did not increase proportionally (Figure 8Kii) and the GFP:RFP ratio was reduced in both OND and rapamycin-treated cells compared to the control group (Figure 8J), suggesting increased LC3 degradation in autolysosomes. Similar to rapamycin treatment, OND tended to decrease both the number and area of LC3 puncta (Figure 8Kiii, Kiv), although significant changes were not evident due to the high variability of data. Overall, these results indicate that OND and rapamycin, similar to tMCAo, enhance autophagy flux in microglia.
This effect was, however, not evident by western blot of LC3, which differentiates LC3-I (unconjugated, cytosolic) from LC3-II (conjugated to phagophore or autophagosomal lipids). Primary microglia (Figure 9A) or BV2 cells (Figure 9B) were maintained in OND in the presence or absence of the lysosomal inhibitor bafilomycin A1 to assess the autophagic flux, i.e. the accumulation of autophagosomes [41]. We detected OND-induced reduction in LC3-I that was more evident in BV2 cells (Figure 9Aii, Bii) suggestive of LC3-I lipidation to phagophores. Indeed, LC3-II:LC3-I tended to increase after OND (Figure 9Aiv, Biv), reaching significance in BV2 cells. Moreover, LC3-II:ACTB levels tended to decrease after OND (Figure 9Aiii, Biii), similar to LC3 imaging experiments (Figure 8Kiii, Kiv), suggesting increased LC3 turnover after OND. However, no significant additive effects were observed after OND on LC3-II:ACTB levels after treatment with bafilomycin A1 as would be expected in a classic autophagy induction (Figure 9Aiii, Biii). We attribute these inconclusive results to a lower sensitivity of western blot compared to imaging techniques in detecting autophagy induction, at least in our hands.
We next used TEM to directly visualize autophagosomes (double membrane vesicles with granular, membranous or heterogeneous contents) and lysosomes (electrondense vesicles) in primary microglia (Figure 9C-K). Control microglia had evident autophagosomes, suggestive of ongoing autophagy in basal conditions (Figure 9E). After OND, we found more autophagosomes and they occupied a larger area of the cytoplasm (Figure 9F-H), supporting the induction of autophagy after OND similar to tMCAo (Figure 8A-H). In addition, we found a reduced number of smaller lysosomes that occupied less cytoplasm (Figure 9I-K), in agreement with tMCAo in vivo data (Figure 8G) and the increased pH and reduced lysosomal number (Figure 7E-J). We speculated that their fusion with autophagosomes would lead to their consumption in the short-term, as has been observed before [42], resulting in reduced numbers and a reduced degradative capacity available for phagocytosis. Overall, our data indicates that autophagy flux is induced in microglia after ischemic stroke, which may interfere with microglial phagocytic function. This idea prompted us to study the functional relationship between autophagy and phagocytosis in basal conditions and during energy depletion.
Basal autophagy sustains microglial physiology, including phagocytosis
We first focused on basal autophagy and used transgenic mice deficient in several autophagic genes to assess their impact on phagocytosis (Figure 10, Fig. S5A): the LC3 protease ATG4B (autophagy related 4B, cysteine peptidase); and the phagophore extending class III phosphatidylinositol 3-kinase proteins BECN1/Beclin1 (beclin 1, autophagy related) and AMBRA1 (autophagy/beclin 1 regulator 1). In the hippocampus of mice deficient in ATG4B, we found a reduction of phagocytosis (Ph index) and the number of microglia (Figure 10A-E). Similarly, in mice with inducible BECN1 deficiency in microglia (driven by Tmem119; becn1 iKO), we found a tendency to reduced phagocytosis (Fig. S5B-E). In contrast, we found no effect on phagocytosis in mice with heterozygous deficiency in AMBRA1 (Fig. S5F-I). We did not use Ambra1 full KO mice because they are embryonically lethal [43]. This data suggests that basal autophagy, which may be controlled by a specific set of genes in microglia compared to other cell types [44], is necessary for an efficient phagocytosis.
Figure 10.
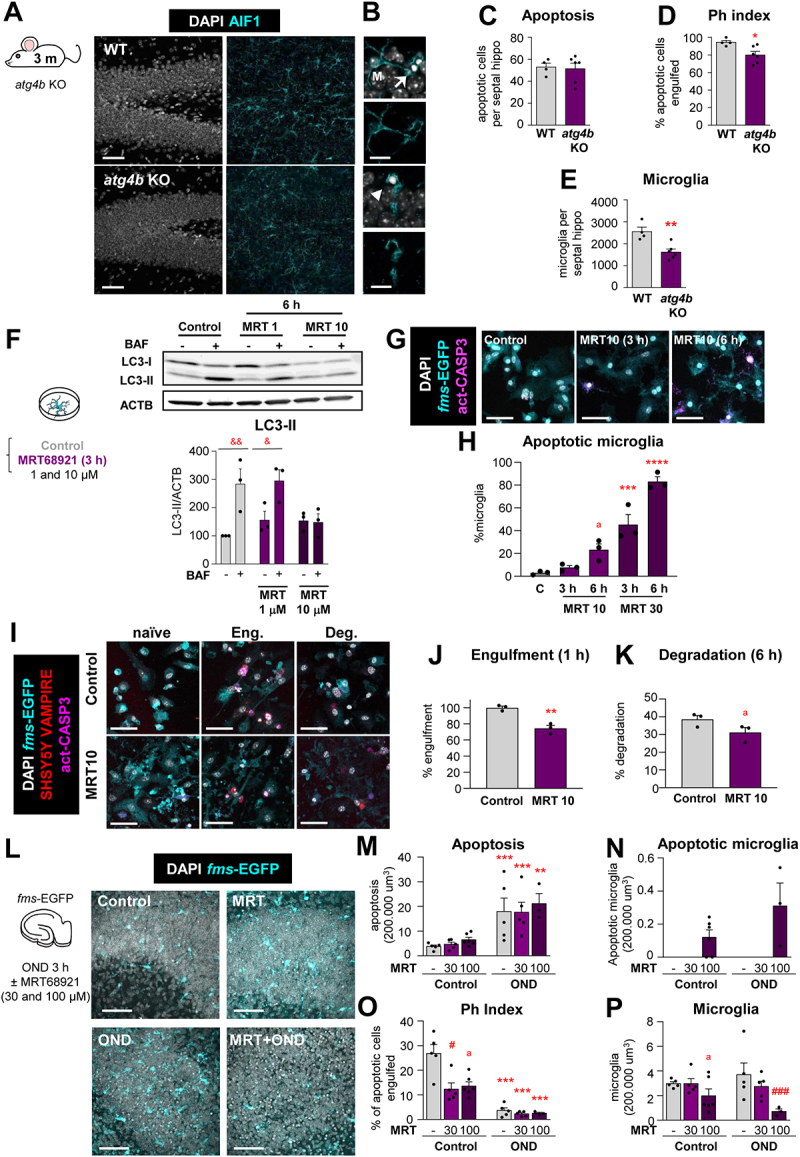
Basal autophagy is essential for microglial survival and function. (A) Representative confocal z-stacks of the DG of 3 mo WT and atg4b KO mice. Healthy or apoptotic nuclei (pyknotic-karyorrhectic) were visualized with DAPI (white) and microglia were stained for AIF1/IBA1 (cyan). A cartoon representing the proteins involved in the autophagic response is shown in Fig. S5A. Phagocytosis in Tmem119-becn1 iKO and Ambra1± mice are shown in in Fig. S5B-E and F-I, respectively. (B) High magnification examples of phagocytosed (arrows) and nonphagocytosed (arrowheads) apoptotic cells. (C) Number of apoptotic cells per septal hippocampus in WT and atg4b KO mice. (D) Ph index in the septal hippocampus (% of apoptotic cells engulfed by microglia). (E) Number of microglial cells per septal hippocampus in WT and atg4b KO mice. (F) Experimental design of the dose-response administration of ULK1-ULK2 inhibitor MRT68921 to primary microglia. Representative blot showing relative levels of LC3-I and LC3-II after 1 and 10 μM MRT68921 administration for 6 h. Quantification of the LC3-II levels (referred to ACTB) after 1 and 10 μM MRT68921 in the presence and absence of the lysosomal inhibitor, bafilomycin A1 (BAF, 100 nM). The quantification of LC3-I is shown in Fig. 6SA, B. The analysis of LC3-I and II after 3 h of MRT68921 (10 and 30 μm) is shown in Fig. S6C. This data is reprinted with permission from Frontiers in Immunology [41]. (G) Representative confocal images of primary fms-EGFP microglia treated with MRT68921 (10 and 30 μM). Nuclei were visualized with DAPI (white), microglia with their constitutive EGFP expression (cyan), and apoptotic cells with activated CASP3 (act-CASP3+, magenta). Images of all experimental groups are shown in Fig. S6D. (H) Percentage of apoptotic microglia assessed by their healthy or apoptotic nuclei (pyknotic-karyorrhectic). (I) Representative confocal images of naïve (non-phagocytic), engulfing and degrading fms-EGFP microglia (cyan), after the addition of apoptotic SH-SY5Y vampire with RFP (red); nuclear morphology (pyknotic-karyorrhectic) was assessed with DAPI (white). A table summarizing the treatments is shown in Fig. S7A. (J, K) Percentage of phagocytic microglia after engulfment (1 h) and degradation (6 h after engulfment). Only particles fully enclosed by microglia were identified as phagocytosis. Raw % of phagocytic microglia is shown in Fig. S7B and the % of apoptotic microglia is shown in Fig. S7C. (L) Experimental design and representative confocal images of the DG after treatment with MRT68921 (100 µM) for 3 h in the presence and absence of OND. Normal or apoptotic (pyknotic-karyorrhectic) nuclear morphology was visualized with DAPI (white) and microglia by the transgenic expression of fms-EGFP (cyan). (M) Number of apoptotic cells in 200,000 µm3 of the DG. (N) Number of apoptotic microglia. Apoptotic microglia were discriminated from apoptotic cells contained in microglial pouches thanks to their expression of EGFP within the nuclei and the lack of a process connecting it to a healthy microglial soma. An example is shown in Fig. S7D. (O) Ph index (% of apoptotic cells phagocytosed by microglia). (P) Number of microglia in 200,000 µm3 of the DG. The weighted Ph capacity and distribution, and the Ph/A coupling are shown in Fig. S7E-G. Bars show mean ± SEM. n = 4–6 mice per group (C-E), n = 3 independent experiments (F, H, J, K), n = 3–6 mice per group (M, N, O, P). Data were analyzed using Student´s t-test analysis (C-E, J-K), by two-way ANOVA followed by Holm-Sidak post hoc tests (F) after logarithmic transformation (M, O, P), or by one-way ANOVA followed by Tukey´s multiple comparisons (H). (&) represents significance between bafilomycin A1 treated and non-treated groups: one symbol represents p < 0.05 and two symbols represent p < 0.01. (* and #) represent significance compared to the control group and between MRT-treated vs MRT-untreated, respectively: one symbol represents p < 0.05, two symbols represent p < 0.01, three symbols represent p < 0.001 and four symbols represent p < 0.0001; (a) represents significance between MRT-treated vs MRT-untreated groups in control conditions; p = 0.051 (H), p = 0.1080 (K), p = 0.055 (O) and p = 0.127 (P). Scale bars = 50 μm, z = 36.4 μm (A); 10 μm (B); 50 μm, z = 8.5 μm (G, I); 50 µm and z = 11.2 µm (L).
We further confirmed the functional relationship between autophagy and phagocytosis with MRT68921, an inhibitor of the upstream autophagy regulator ULK1 (unc-51 like kinase 1)-ULK2 that we used at a range of concentrations based on prior reports [45] (Figure 10F-K; Fig. S6). Here, western blot of LC3 was sufficiently sensitive to detect a blockade of autophagy flux in primary cultures (Figure 10F, Fig. S6A-C). While high concentrations of MRT led to microglial apoptosis (Figure 10G and H), we identified a dose of MRT (10 μM, 6 h) that blocked autophagy without inducing microglial apoptosis (Figure 10F-H, Fig. S6A-D) and used it to test its effects on phagocytosis (Figure 10I-K; Fig. S7A-C). MRT reduced very effectively apoptotic cell engulfment and showed a trend to reduced degradation (Figure 10I-K). Therefore, autophagy inhibition with MRT impaired both survival and phagocytosis in vitro, confirming the effects of ATG4B deficiency in vivo.
Additionally, we used organotypic cultures to test the effect of autophagy inhibition with MRT on both basal and OND-treated cultures (Figure 10L-P). We used a higher concentration of MRT (30 μM and 100 μM) to account for the drug diffusivity throughout the tissue slice. At these concentrations, MRT did not induce global apoptosis (Figure 10L and M). However, in control conditions, MRT induced a small amount of microglial apoptosis (Figure 10N) and reduced phagocytosis (Figure 10O; Fig. S7D-G), further supporting the relevance of basal autophagy on microglial health and phagocytosis efficiency. Under OND, MRT led to a stronger induction of microglial apoptosis (Figure 10N; Fig. S7D) and to reduced microglial numbers (Figure 10P), indicating that the autophagy induction after OND was protective for microglia. However, MRT did not further enhance the OND-induced phagocytosis impairment (Figure 10O; Fig. S7E-G), showing that either inhibiting autophagy or OND led to a similar phagocytosis blockade.
Rapamycin prevents phagocytosis impairment
Finally, we tested the opposite strategy and induced autophagy using rapamycin to address whether it could prevent phagocytosis dysfunction during tMCAo in vivo and OND in vitro (Figure 11). We selected rapamycin because of its beneficial reported effects in several brain disease models [38], including models of stroke [46], and because while it is largely known as an inhibitor of the MTOR (mechanistic target of rapamycin kinase) pathway [47], it also promotes lysosomal biogenesis [48,49] thus enhancing autophagy at different levels of the process (Fig. S5A). Mice were pretreated for two consecutive days with rapamycin (10 mg/kg) [49] or vehicle before being exposed to tMCAo and were sacrificed 6 h later (Figure 11A). To control for surgery heterogeneity, in each surgery session a pair of one vehicle and one rapamycin mouse were operated. All mice had MCA occlusion and reperfusion confirmed with laser Doppler flowmetry (Figure 11B). At the time point tested, rapamycin had no overall effect on the tMCAo-induced damage, as it had no effect on hippocampal apoptosis or on microglial numbers (Figure 11C-E). Importantly, however, rapamycin partially recovered phagocytosis globally (Figure 11F) and in 5/6 mouse pairs (Figure 11G), although the Ph index did not reach the expected values of untreated animals (Figure 1H).
Figure 11.
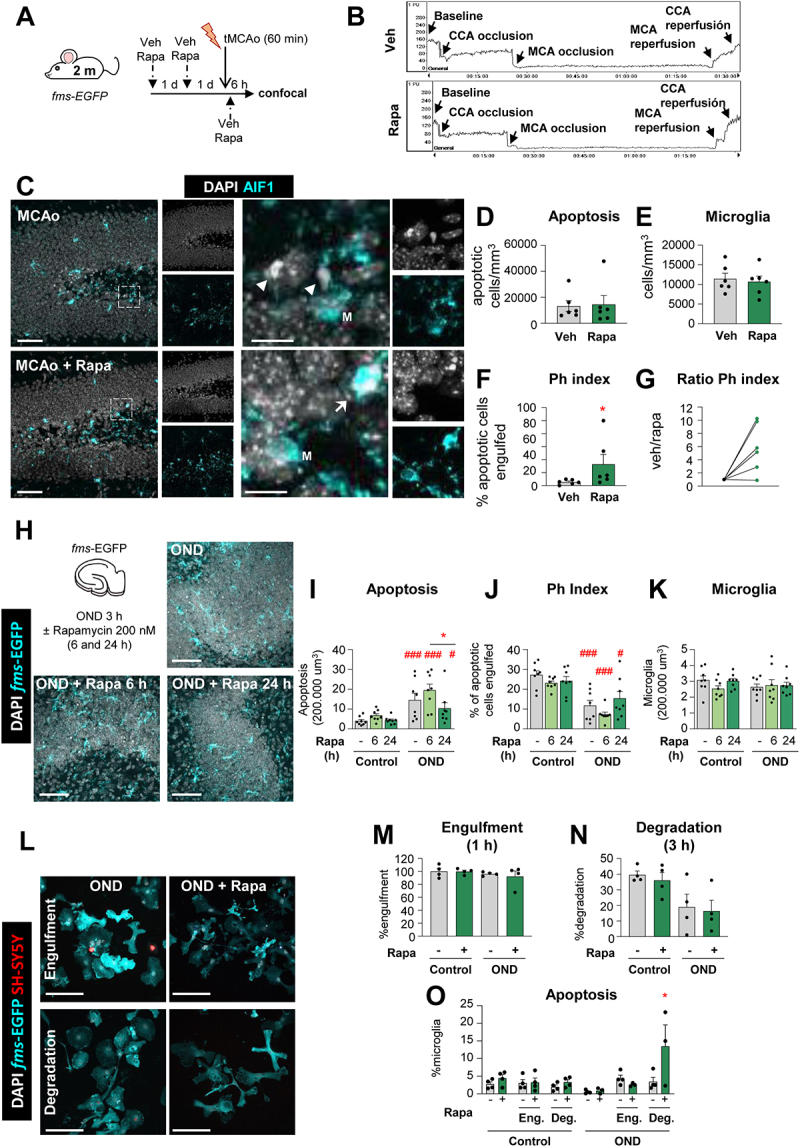
Rapamycin reverts the tMCAo-induced phagocytic dysfunction in vivo but not in vitro. (A) Experimental design showing the daily administration of rapamycin (10 mg/kg, ip) two days prior to the tMCAo in 2 mo fms-EGFP mice. Mice received a third rapamycin injection right after reperfusion, and were sacrificed 6 h later. (B) Representative laser Doppler signal graph showing the effective MCA occlusion and reperfusion in both vehicle and rapamycin-treated mice. (C) Representative confocal z-stacks of the DG of fms-EGFP mice 6 h after tMCAo, treated with vehicle or rapamycin (10 mg/kg, ip). Cell nuclei were visualized with DAPI (in white) and microglia (fms-EGFP+, in cyan). Arrowheads point to non-phagocytosed apoptotic cells and arrows to phagocytosed apoptotic cells. M labels a microglial soma. (D) Density of apoptotic cells in the septal hippocampus. (E) Density of microglial cells in the septal hippocampus. (F) Ph index in the septal hippocampus (% of apoptotic cells engulfed by microglia). (G) Normalized ratio of Ph index change in each rapamycin-treated mice over its same day vehicle-treated mice. (H) Experimental design and representative confocal images of hippocampal organotypic cultures treated with vehicle or rapamycin (200 nM; 6 and 24 h) exposed to OND (3 h). Normal or apoptotic (pyknotic-karyorrhectic) nuclear morphology was visualized with DAPI (white) and microglia by the transgenic expression of fms-EGFP (cyan). Rapamycin- and vehicle- treated control cultures are shown in Fig. S8A. (I) Number of apoptotic cells in 200,000 µm3 of the DG. (J) Ph index (% of apoptotic cells phagocytosed by microglia). The weighted Ph capacity and the Ph/A coupling are shown in Fig. S8B, C. (K) Number of microglia in 200,000 µm3 of the DG. (L) Representative confocal images of microglia (fms-EGFP+, cyan) engulfing (1 h) and degrading (3 h) apoptotic SH-SY5Y VAMPIRE neurons (red) under OND conditions in the presence of rapamycin. Nuclear morphology was assessed with DAPI (white). Only particles fully enclosed by microglia were identified as phagocytosis. A table summarizing the treatments is shown in Fig. S8D, the effect of rapamycin in LC3 levels is shown in Fig. S8E, and control cells are shown in Fig. S8F. (M, N) Percentage of phagocytic microglia after engulfment (1 h) and degradation (3 h after engulfment). (O) Percentage of apoptotic (pyknotic-karyorrhectic) microglia after the phagocytosis assay and rapamycin treatment in both control and OND. Bars show mean ± SEM. Dot and line plot represents the normalized ratio between rapamycin- and vehicle-treated animals (by pairs). n = 6 mice per group (D-G), n = 8 mice per group (I-K), and n = 4 independent experiments (M-O). Data were analyzed using a Student´s t-test (D-G); by two-way ANOVA followed by Holm-Sidak post hoc tests when appropriate (I-K, M-N); or by one-way ANOVA followed by Tukey´s post hoc test when a significant interaction in two-way ANOVA was found (O). Some data (I) was Log10 transformed to comply with homoscedasticity. Asterisks represent significance between untreated and rapamycin-treated mice or cultures: (*) represents p < 0.05. (#) represent significance between OND and control cultures: # represents p < 0.05, and ### represents p < 0.001. Scale bars = 50 µm, z = 19.6 µm; inserts bar = 10 µm z = 9.8 µm (C); 50 µm, z = 11.2 µm (H); 50 µm, z = 8.5 µm (L).
This partially protective effect on phagocytosis was however not recapitulated in organotypic cultures (Figure 11H-K; Fig. S8A-C). We selected a 200 nM concentration of rapacymin based on the cell culture experiments, a pilot experiment in organotypics with 200 nM and 600 nM (data not shown), and previous reports of rapamycin in brain slices [50]. Here, rapamycin reduced global apoptosis in the longest time point tested (24 h, 200 nM) but had variable effects on phagocytosis that were not significant. Similarly, rapamycin had no effect on either engulfment or degradation in primary cultures (Figure 11L-N; Fig. S8D) at a concentration that induced autophagy (100 nM), as observed by tandem mRFP-GFP-LC3 in BV2 cells (Figure 8I-K) but not by LC3 western blot in primary microglia (Fig. S8E). In addition, we detected a detrimental effect of rapamycin in phagocytic (degradative) microglia in OND conditions that led to their demise (Figure 11O; Fig. S8F), possibly related to an over-stimulation of microglial autophagy in vitro. While we cannot disregard the possibility that the time points and concentrations were not optimized in vitro or that different pathways regulate microglial autophagy in vitro and in vivo, another possibility to explain the discrepant effects of rapamycin after tMCAo and OND is that the partial protection afforded by rapamycin in tMCAo mice was not due to a direct effect on microglia. Thus, while blocking autophagy was clearly detrimental, enhancing autophagy in microglia proved to be more challenging. Overall, our results confirm that the phagocytosis impairment can be modulated in vivo and is a druggable target to be explored.
Discussion
In this paper, we demonstrate that microglial phagocytosis is an early therapeutic target in stroke, amenable for drug interventions. We first show evidence of phagocytosis dysfunction in mouse and monkey preclinical models of stroke, both in vivo and in vitro. Next, we reveal some of the underlying cellular mechanisms that affect both engulfment and degradation of apoptotic cells, including reduced microglial process motility and lysosomal alterations. Then we show that the energy depletion associated with stroke leads to increased autophagy, whose basal levels are essential for microglial survival and function both in vivo and in vitro. Finally, we demonstrate that the autophagy inducer rapamycin partially prevents the stroke-induced phagocytosis impairment in vivo (Figure 12). Overall, we shed light onto two unappreciated microglial activities with key roles during stroke and high therapeutic potential: autophagy, responsible for intracellular recycling, and the cell´s well-being and function; and phagocytosis, responsible for extracellular laundering and controlling inflammatory responses.
Figure 12.
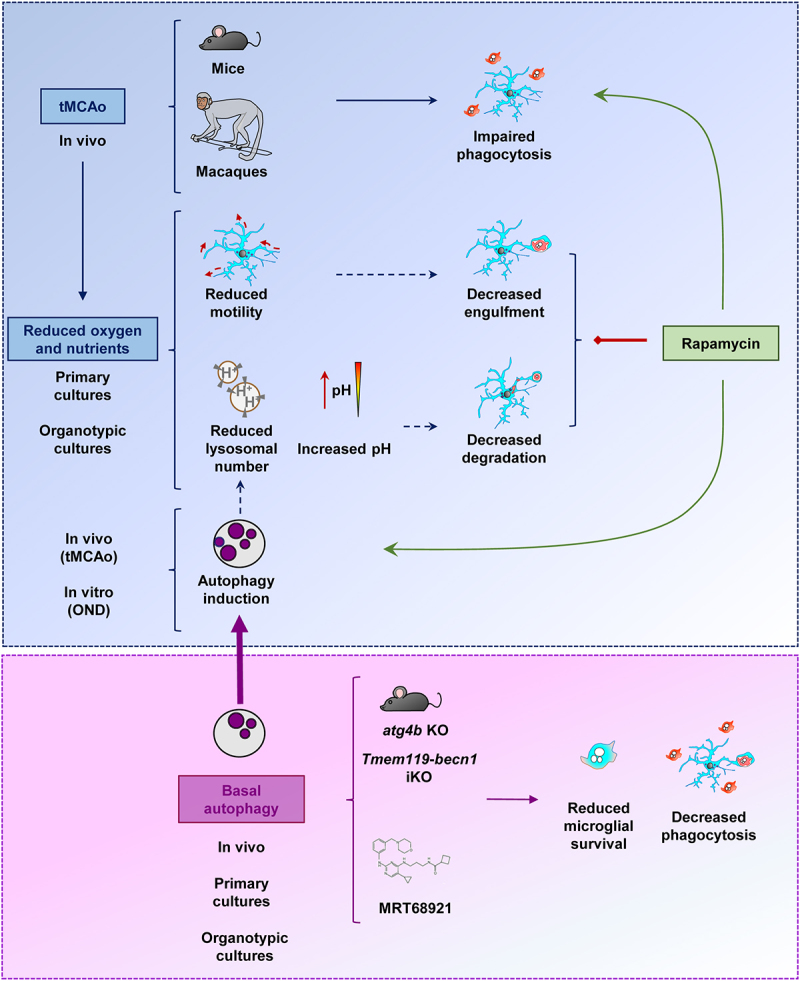
Microglial phagocytosis of apoptotic cells was impaired in mouse and macaque models of stroke induced by tMCAo. This impairment was related to the lack of oxygen and nutrients, which lead to reduced process motility, possibly related to the decreased engulfment; and reduced lysosomal numbers and increased pH, possibly related to the decreased degradation in primary and hippocampal organotypic cultures. In vivo tMCAo and in vitro energetic depletion also induced a protective autophagy response, possibly related to the lysosomal depletion. The maintenance of basal autophagy was critical for microglial survival and phagocytosis, as shown in mice that lacked expression of autophagy genes such as atg4b KO or Tmem119-becn1 iKO mice, or in primary and organotypic cultures treated with the ULK1-ULK2 inhibitor MRT68921. While the autophagy inducer rapamycin did not improve the phagocytosis blockade in vitro, it was effective in preventing the phagocytosis impairment induced by tMCAo, supporting the possibility of pharmacological modulation of microglial phagocytosis in vivo.
Exploiting microglial phagocytosis as a future therapeutic target
The profits of microglial phagocytosis for the diseased brain are evident: prevention of intracellular content spillover and immunomodulatory effects [16]. However, its therapeutic potential in brain diseases has been largely unappreciated, likely because phagocytosis was presumed to occur rather than directly assessed [37]. Here we have used a quantitative approach that has allowed us to discover microglial phagocytosis dysfunction in stroke in mice and monkeys, similar to what we had observed in mouse and human epilepsy [22,23]. In these diseases, it is necessary not only to prevent neuronal death, which in stroke is currently achieved by the intravenous administration of tissue plasminogen activator (tPA) at early time points, but also to accelerate the removal of neuronal debris by developing new strategies to harness phagocytosis.
Pioneer work in cancer has catapulted macrophage phagocytosis as a consolidated target with several ongoing clinical trials [51]. Here, deficient phagocytosis is due to tumor cells escaping recognition by macrophages. As such, most efforts have been put into developing opsonizing antibodies to coat the tumor cells and facilitate their interaction with macrophages [52]. Another rising idea is to interfere with phagocytosis checkpoints such as CD47, a “don´t-eat-me” signal that interacts with SIRPA/SIRPα receptors on the macrophage [53]. In the brain, however, CD47 expressed by healthy synapses prevents their excessive phagocytosis by microglia, at least during development [54]. Unhinged phagocytosis could lead to phagoptosis, i.e. the engulfment of stressed but viable cells [55], responsible for delayed neuronal death in stroke models [56]. These examples highlight that translating cancer-based approaches into effective brain therapies may not be straightforward and requires a deeper understanding of the mechanisms operating on microglial phagocytosis dysfunction, as we will discuss next.
Impairment of microglial phagocytosis during stroke
One key difference between cancer and the diseased brain is that in the first case, tumor cells are the ones to blame for deficient phagocytosis, as they develop mechanisms to escape engulfment. In contrast, in stroke the problem does not seem to lie on the target cells but on the phagocytes, as microglia is severely compromised due to the energy depletion. We found that the lack of oxygen and nutrients reduced the motility of microglial processes and interfered with the engulfment phase of phagocytosis. It also altered the microglial lysosomal number, pH, and the cell´s degrading capacity, which was likely related to reduced apoptotic cell degradation. Finally, it induced autophagy, a cell process that we found necessary for microglial survival and phagocytosis. In addition, environmental-related factors are also likely to play a role on microglial phagocytosis dysfunction. An example is extracellular ATP, one of the major “find-me” signals from apoptotic cells sensed by purinergic receptors on microglia [57]. However, ATP is also a neurotransmitter, widely released during pathological conditions such as epilepsy and ischemia [58]. These two sources of ATP put microglia in conflict and disrupt their targeting of apoptotic cells, resulting in impaired phagocytosis during epilepsy [22] and possibly during stroke. Inhibitors of the purinergic receptor P2RY12, such as clopidogrel, are currently used to prevent platelet aggregation in several cardiovascular diseases [59], but a side effect not considered is their inhibitory action on microglial phagocytosis [37]. Another potential mechanism underlying the impairment of microglial phagocytosis during stroke, which was not studied here, is endoplasmic reticulum (ER) stress, which leads to reduced phagocytosis of apoptotic cells in peripheral macrophages [60,61]. Autophagy induction in hippocampal neurons alleviates ER stress [62], possibly also contributing to the modulation of microglial phagocytosis. In sum, this combination of mechanisms that relate to the target, the phagocyte, and/or the environment should be considered when designing effective therapies to recover or potentiate phagocytosis.
Here, we have used rapamycin to prove the principle that phagocytosis dysfunction during stroke can be modulated in vivo. The autophagy inducer rapamycin has become an increasingly popular drug since its discovery in the early nineties in soil samples from Easter Island [63]. Due to its immunosuppressant properties, rapamycin (sirolimus) is currently used to prevent kidney transplant rejection and to treat certain lung diseases, and several clinical trials are testing its efficacy in pathologies such as Alzheimer´s disease and aging (Clinical Trials NCT04629495, NCT04488601). In stroke, rapamycin prevents neuronal cell death [47,64,65]. Here we have observed a neuroprotective effect on apoptosis induced by OND at 24 h, but not at earlier time points either after OND or tMCAo. We have also shown that rapamycin pretreatment partially prevented the impairment of microglial phagocytosis in the tMCAo model at 6 h, but not in vitro in the OND model. We speculate that, in vivo, rapamycin may have indirectly improved microglial phagocytosis by reducing the tMCAo-induced ischemic damage in astrocytes, pericytes and endothelial cells, promoting blood-brain barrier integrity and neurovascular unit function, ultimately facilitating reperfusion [47]. In addition, rapamycin may have exerted autophagy-independent effects on microglia. For example, systemic administration of rapamycin may modulate peripheral immune cell inflammatory profile and activities, including their migration to the CNS, indirectly affecting microglial functions [66]. It is also important to take into account the early therapeutic time window to promote microglial phagocytosis, preferably in the first 6 h after the stroke, when pathological alterations in microglial autophagy are already evident. While rapamycin may not be the optimal drug to target microglia, these results suggest that preventing phagocytosis impairment in stroke models in vivo is possible.
Microglial autophagy is indeed a challenging target. We achieved autophagy inhibition using pharmacological blocking of ULK1-ULK2 or by genetic manipulation of ATG4B and BECN1, demonstrating that basal autophagy was essential to sustain microglial survival and phagocytosis of apoptotic cells. In agreement with our results, disruption of basal autophagy has been involved in the phagocytosis of myelin [67] and beta amyloid deposits [68], in mice deficient in ATG7 (autophagy related 7) and BECN1, respectively. However, we found no significant effects in Ambra1 heterozygous mice, suggesting that microglial autophagy may have a unique set of regulators compared to other cell types [44]. Nonetheless, it is important to note that the different transgenic models (inducible vs constitutive) used here are not directly comparable and may have different impact on microglial autophagy. For instance, the constitutive and systemic down-regulation of autophagy in atg4b KO mice could mainly influence non-microglial cell type fitness, indirectly affecting microglial phagocytosis and survival. In spite of this apparent disagreement between the different transgenic models, in vitro MRT68921 experiments clearly showed that autophagy inhibition was detrimental for microglia, which reinforced the phagocytosis impairment obtained in atg4b KO mice and becn1 iKO mice. Nevertheless, the relatively high concentrations of MRT needed to inhibit autophagy in vitro and the small effects found in autophagy deficient KO mice point to a tight regulation of autophagy in microglia by redundant mechanisms, which complicates its exogenous (genetic or pharmacologic) regulation.
Inhibition of the protective autophagy response mounted after OND was also detrimental for microglial survival. Unexpectedly, promoting this response with rapamycin not only did not recover phagocytosis but even had a deleterious effect on e survival of phagocytic microglia during OND. These results point to the complex regulation of autophagy in microglia, whose beneficial or detrimental effects may depend on the timing and the amount of autophagy. Nonetheless, microglial autophagy is a promising target to be explored. Autophagy controls the microglial inflammatory response in rodent stroke models [69,70] through ANXA1 (annexin A1), which activates the inflammatory transcription factor NFKB/NF-κB (nuclear factor of kappa light polypeptide gene enhancer in B cells) by directing its inhibitor IKBK (inhibitor of kappaB kinase) to phagophores for degradation [71]. Future research will identify the target organelles or subcellular substrates that need to be recycled in microglia to maintain its health status, and whether microglial autophagy can be therapeutically exploited to support phagocytosis.
Microglia beyond the inflammatory paradigm
For too long, the field of stroke has focused on microglial inflammatory responses with little attention to their other functions. The field is still categorizing pro- or anti-inflammatory microglia using outdated terms [72,73], such as M1 and M2 to define presumed beneficial or detrimental subtypes [3,70,71,74,75]. In contrast, both bulk and single cell RNA-Seq studies have clearly shown that microglia do not polarize to either of these categories in rodent stroke models [10–14]. Sequencing methods, particularly when applied to isolated microglia, can powerfully capture the cell´s transcriptional profile. However, their predictive value of microglial function is limited [15], and it is worth noting that they failed to identify phagocytosis dysfunction, likely because it was not regulated at the transcriptional level. The stagnation in M1/M2 and the excessive focus on phenotyping instead of function has led to a shortage of microglial targets for clinical trials, which are to this day still largely focused on inflammation. In addition, studies in stroke patients have also oversimplified the role of human microglia by studying its “activation” in imaging studies, whereas functional studies are largely missing [3]. Our results demonstrate that microglial phagocytosis is a promising new target in stroke, with a solid therapeutic potential of microglial phagocytosis to restore brain homeostasis that grants further exploration.
Materials and methods
Mice
All experiments were performed in adult 2-month-old (2 mo) fms-EGFP (MacGreen) mice, except where indicated, in which microglia constitutively express the green fluorescent reporter under the expression of the fms promoter [27,76]. Two-photon microscopy experiments were performed in Cx3cr1GFP/+ where microglia express GFP [77]. Brain tissue from Cx3cr1GFP/+ Ccr2RFP/+ mice was provided by K. Blomgren (Karolinska Institute, Sweden), in which peripheral monocytes express the red fluorescent protein RFP [78]. Brain tissue from constitutive atg4b KO was provided by G. Mariño, Universidad de Oviedo, Spain [79]; Ambra1± heterozygous mice by P. Boya, Centro de Investigaciones Biológicas CIB, Spain [43]; and tamoxifen inducible KO of Becn1 under the Tmem119-CreERT2 by D. Schafer, University of Massachussetts, USA. To induce deletion of Becn1, two doses of tamoxifen dissolved in corn oil (75 mg/kg) or corresponding volume of corn oil alone (control mice) were administered intraperitoneally at postnatal days (P)21 and P23. All animals used were on a C57BL/6 background except atg4b KO mice and Ambra1± heterozygous mice, which were on a mixed C57BI6/129 Sv and CD1 background, respectively. Mice were housed in 12:12 h light cycle with ad libitum access to food and water. Both males and females were used and pooled together, unless otherwise noted. All procedures followed the European Directive 2010/63/EU, NIH guidelines and were approved by the Ethics Committees of the University of the Basque Country EHU/UPV (Leioa, Spain; CEBA/205/2011, CEBA/206/2011, CEIAB/82/2011, CEIAB/105/2012) and Karolinska Institute (Stockholm, Sweden; protocol number N249/13).
Non-human primates
Brain samples from cynomolgus macaque (Macaca fascicularis) and common marmoset (Callithrix jacchus) under transient MCA occlusion were provided by Emmanuelle Canet-Soulas (University of Lyon, France) [32] and Omar Touzani (University of Caen, France) [35], respectively. Animals were housed in a 12-h light-dark cycle. All procedures were approved by European Directives and approved by the Regional Ethics Committee. For tMCAo in non-human primates, 7-year-old (7 yo) cynomolgus macaque and 2–2.5 yo common marmosets were anaesthetized with sevoflurane and isofluorane, respectively, as previously described [35,80], and MCA was occluded for 110 min in macaque and 3 h in marmosets. Animals were sacrificed at 30 d and 45 d after tMCAo, respectively.
Primary microglia
Primary microglia was obtained as previously [81] described. Briefly, P0-P1 fms-EGFP or WT mice were sacrificed by decapitation and brains were extracted. Meninges were removed in Hanks’ balanced salt solution (HBSS; Gibco, 14,175,095) under a binocular magnifier and the cerebellum and olfactory bulbs were removed. The remaining brain was manually chopped and enzymatically digested in the presence of papain (20 U/mL; Sigma-Aldrich, P3125), a cysteine protease enzyme extracted from the papaya latex, and desoxyribonuclease I (DNAse I, 150 U/μL; Invitrogen, 18,047,019) for 10–15 min at 37°C after which the remaining fragments were mechanically homogenized by gentle pipetting. The resulting cell suspension was filtered through a 40-μm polypropylene cell strainer (Fisher Scientific, 11,587,522) to obtain individualized cells and transferred to a 50-mL Falcon tube containing 20% fetal bovine serum (FBS; Ibian Technologies, P30-3306) in HBSS to inactivate the papain after the enzymatic digestion. Next, the cell suspension was centrifuged at 200 g for 5 min and the resulting pellet was resuspended in 1 mL of DMEM (Gibco, 11,995,065) supplemented with 10% FBS and 1% antibiotic-antimycotic (Gibco, 15,240,062). Cells were seeded in poly-L-Lysine (15 μL/mL; Sigma-Aldrich, P4832)-coated T-75 flasks with a density of two brains per flask. The medium was changed the day after the culture and every 3–4 d, enriched with CSF2/GM-CSF (colony stimulating factor 2 (granulocyte-macrophage); 5 ng/mL; Sigma-Aldrich, SRP3201) to promote microglia proliferation at 37°C, 5% CO2. When confluence was reached, after approximately 11–14 days, microglia cells were harvested by shaking at 120–140 rpm, at 37°C for 4 h. The isolated cells were counted and plated at the desired density; 100,000 cells/well in 24-well plates for immunofluorescence experiments, 500,000 cells/well in glass-bottom dishes (Ibidi) for live imaging and 2,000,000 cells/well in 6-well plates for western blot purposes; all coated with poly-L-Lysine to guarantee optimal cell adhesion. Before performing any experiment, microglia were allowed to settle for at least 24 h.
BV2 cell line
BV2 cells (Interlab Cell Line Collection San Martino-Instituto Scientifico Tumori-Instituto Nazionale per la Ricerca sul Cancro), a cell line derived from RAF1-MYC-immortalized rat neonatal microglia was used in autophagy assessing experiments wherein transfection of mRFP-EGFP-LC3 plasmid was required as well as some western blot experiments. Cells were grown as an adherent culture in non-coated 90-mm2 Petri dishes in the presence of 10 mL of DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic. When confluence was reached, cells were trypsinized (Trypsin-EDTA 0.5% no phenol red; Gibco, 15,400,054) and replated at a 1:5 density. Before performing any experiment, BV2 cells were allowed to settle for at least 24 h.
Red fluorescent SH-SY5Y cell line (VAMPIRE SH-SY5Y)
The SH-SY5Y cell line is a human neuroblastoma cell line developed as a stable transfection of the SH-SY5Y cell line with the red fluorophore tFP602 (InnoProt, P20303). It derives from neuroepithelioma cell line SK-N-SH, generated from the bone marrow of 4 yo female with metastatic neuroblastoma. Cells were grown as adherent culture in non-coated T-75 flasks in the presence of 10–15 mL of DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic and 0.25 mg/ml Geneticin (G418; Gibco, 10,131,027) to select the transfected cells. When confluence was reached, cells were trypsinized (Trypsin-EDTA 0,5% no phenol red) and replated at a 1:3 density. SH-SY5Y cells were used to perform phagocytosis assay for which they were trypsinized and replated at the same density 24 h prior to the phagocytosis assay to avoid the addition of cell clusters to the microglia culture.
Organotypic hippocampal slice cultures
Organotypic hippocampal slice cultures were prepared as described previously [81]. Briefly, P7 fms-EGFP pups were sacrificed, and brains removed and placed in cold HBSS. The hippocampi were extracted and cut into 350 µm slices with a tissue chopper (McIlwain). Slices were then placed in 0.4-µm culture plate inserts (Merck, PICM0RG50), each containing 3–5 sections. These inserts were placed in 6-well culture plates, each well containing 1 ml of fresh organotypic slice medium. The medium consisted of 50% Neurobasal medium (Gibco, 21,103,049) supplemented with 0.5% B27 (Gibco, 17,504,044), 25% horse serum (Ibian Technologies, P30-0702), 1% Glutamax (Gibco, 35,050,038), 1% antibiotic-antimycotic, and 1% glucose solution in HBSS. The medium was changed the first day after culturing the slices and every 2 d afterwards for 7 d. The experiments were performed on day in vitro 7. For OND, slices were rinsed twice with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) to remove medium remainders, nutrient deprived in salt solution, and transferred to a hypoxia chamber (1% O2) for 3 or 6 h. For reperfusion experiments, slices were placed back in fresh complete medium containing propidium iodide (PI, 5 µg/ml; Sigma-Aldrich, P4864) for an additional hour in a regular cell culture incubator (20% O2, 5% CO2). The autophagy inhibitor MRT68921 (30 and 100 µM; Sigma-Aldrich, SML1644) was added to the culture media during the 3 h challenge with OND. The autophagy inducer rapamycin (200 nM; Selleckchem, S1039) was tested at 6 and 24 h time points and was added for 3 or 21 h before the 3 h challenge with OND. Immediately after finishing the experiments, the slices were rinsed with PBS and fixed in cold 4% paraformaldehyde solution for 40 min. After a couple of rinses with PBS, slices were stored at 4°C until immunofluorescence processing.
tMCAo and rapamycin treatment
Two-mo fms-EGFP mice were anesthetized with isofluorane (2% for induction and 1% for maintenance). tMCAo was induced using the intraluminal filament method as previously described [82]. Briefly, a midline-neck incision was performed to expose the left CCA. The left external and internal carotid arteries (ECA and ICA, respectively) were carefully isolated, and a monofilament (silicon coated silk with an extended tip of width of 9–10 mm and 0.21 mm diameter; Doccol Corporation, 7021910PK5Re) was inserted into the ECA and advancing it via the ICA to the origin of the MCA. After a 60-min embolization, the monofilament was removed and the CCA reperfused to restore blood flow. The successful occlusion and reperfusion were confirmed by a Laser-Doppler flowmetry (LDF) with a cerebral blood flow (CBF) less than 70%. Mice that did not demonstrate a rapid restoration of the CBF signal during reperfusion were excluded. Body temperature was kept at 37 ± 0.5°C throughout the experiment by an electrical blanket. Mice were sacrificed 6 h and 1 d after the damage. Sham animals were anesthetized but did not suffer arteries ligation. Some mice received an intraperitoneal injection of rapamycin (10 mg/kg, in 20 μl/10 g body weight) or vehicle (5% DMSO, 5% Tween-80 (Fisher BioReagents, 10,592,955), 15% polyethylene glycol 400 (PEG400; Thermo Scientific Acros, 192,230,010) in 0.9% NaCl solution) during 3 consecutive days. Two injections were administered as a pre-treatment prior to tMCAo surgery and a third injection was given post-tMCAo. Mice were sacrificed 6 h later.
Pimonidazole treatment
Mice received one intravascular tail injection of pimonidazole (60 mg/kg, diluted in saline) 30 min before MCA reperfusion and were sacrificed 30 min later. A second set of animals were injected with pimonidazole 30 min before sacrifice at 6 h and 1 d after tMCAo. The percentage of hypoxic area was estimated in confocal tiled images of coronal slices from sham and tMCAo-treated animals. First, hypoxic areas were manually selected in each image using the “Threshold” tool (FIJI) to mask only the pixels of the image with pimonidazole staining. Then, the percentage of pixels occupied by pimonidazole was calculated using the Area Fraction parameter of the “Measure” tool (FIJI). All commands were automated in an ImageJ macro (FIJI). The average area fraction of 4–5 tiled images per animal was calculated.
Hypoxia-ischemia (HI)
Three-mo Cx3cr1GFP/+ Ccr2 RFP/+ mice were anesthetized with isoflurane (5% for induction and 2% for maintenance) and the right common carotid artery was ligated with a 6–0 silk suture according to the Rice-Vannucci model [29,83]. After ligation, the animals were returned to their home cage for 1 h, and then placed in a chamber perfused with a humidified gas mixture (10% oxygen in nitrogen) for 75 min at 36°C. Animals were sacrificed 1 d and 3 d after injury. Control mice were neither subjected to ligation nor hypoxia.
Cranial irradiation (CIR)
Three-mo Cx3cr1GFP/+ Ccr2 RFP/+ mice were irradiated in X-ray irradiator [30]. Animals were anesthetized with isofluorane (5% for induction and 2% for maintenance) and the whole brain was irradiated with a single dose of 8 Gy (0.72 Gy/min). The acute exposure of 8 Gy is equivalent to approximately 18 Gy when delivered in repeated 2 Gy fractions, which represents a clinically relevant dose used in treatment protocols such as the medulloblastoma study (PNET5 MB) designed for children (https://clinicaltrials.gov/ct2/show/NCT02066220). Animals were sacrificed 6 h and 1 d after irradiation. Sham mice were anesthetized but did not receive any CIR.
OND assay
Primary microglia cultures and hippocampal organotypic slices were treated with oxygen and nutrient deprivation to mimic a hypoxia-like state. Cultures were treated with a salt solution (130 mM NaCl, 5,4 mM KCl, 1.8 mM CaCl2, 26 mM NaHCO3, 0.8 mM MgCl2, 1.18 mM NaH2PO4, pH 7.4 in milliQ water) [84] without glucose nor amino acids. After adding the salt solution, cultures were placed in a hypoxia chamber coupled to a Cytocentric® oxygen controller (Biospherix, ProOx 110) inside a standard thermal incubator for 3 or 6 h with an oxygen concentration between 1–3% and no CO2.
In vitro rapamycin, MRT68921 and bafilomycin A1 treatment
The autophagy inducer rapamycin was dissolved in DMSO (3.65 mM) and diluted in water to 10 µM. Rapamycin was added to hippocampal organotypic cultures at a final concentration of 200 nM for 6 and 24 h. In primary microglia and BV2 cells rapamycin was used at a final concentration of 100 nM for 24 h and 6 h, respectively. The autophagy inhibitor MRT68921 was dissolved in water (1 mM). Hippocampal organotypic slices were treated with MRT68921 for 3 h at a final concentration of 30 and 100 µM. Primary microglia were treated with MRT68921 for 3 and 6 h at 1, 10, and 30 µM concentrations. The lysosomal inhibitor bafilomycin A1 (Selleckchem, S1413) was dissolved in DMSO (500 µM) and added to primary microglial and BV2 cell cultures at 100 nM concentration for 3 h.
In vitro phagocytosis assay
The in vitro phagocytosis assay was adapted from [81]. Phagocytosis was performed in high glucose DMEM, supplemented with 1% antibiotic-antimycotic and 10% FBS. Primary microglia were plated and allowed to rest 24 h prior to the experiment. Cells were fed with apoptotic VAMPIRE SH-SY5Y, previously treated with staurosporine (3 μM, 4 h; Sigma-Aldrich, S4400) to induce apoptosis. To ensure collecting only apoptotic cells, the floating fraction was added to primary microglia in a proportion of 1:1 except in the engulfment ratio assay where apoptotic cells were added in a 1:9 proportion. Microglia were left to interact with the apoptotic cells for 1 h and then fixed to assess the engulfment of the dead cells, or, instead, the non-phagocytosed apoptotic cells were removed by washing twice with PBS and primary microglia left to degrade the engulfed cells for 3 more h before fixation to assess degradation.
Two-photon imaging on hippocampal organotypic slices
Organotypic hippocampal slices were obtained from P7 Cx3cr1GFP/+ mice. Briefly, slices were placed in a fluidic chamber under a Femto-2D microscope equipped with a tunable Ti:Sapphire MaiTai DeepSee laser (Spectra Physics) and GaAsP PMT detectors. Control slices were perfused with oxygenated complete culture medium whereas OND pre-treated (3 h) slices were constantly perfused with nitrogenated OND medium throughout the imaging. The laser was tuned at 920 nm to image GFP with a 20X water immersion lens (1.00 N.A.; Olympus) and a 490/60 nm filter. Stacks of 22 µm (z-step = 1 µm) were acquired at 60s intervals for 10 min in a 512 × 512 pixel format (1 pixel = 0.434 µm).
Lysosomal pH measurement assay (live imaging)
The measurement of lysosomal pH is based on the ratiometric measurement of two fluorophores by confocal microscopy [85]: fluorescein (FITC) and tetramethylrhodamine (TRITC) conjugated to a dextran molecule (70,000 MW, anionic; Invitrogen, D1951). Microglia were incubated with 2 V mg/mL of dextran for 15 h for its internalization. After that time, the excess of dextran was removed by thorough washing with PBS and the cells incubated in complete medium high glucose DMEM, supplemented with 1% antibiotic-antimycotic and 10% FBSfor 3.5 h allowing the dextran to be delivered to the lysosomes (chase pulse). For the OND group the chase was partially done under OND conditions (3 h). Next, control cells were washed with imaging medium (150 mM NaCl, 20 mM HEPES, pH 7.4, 1 mM CaCl2, 5 mM KCl, 1 mM MgCl2, 0.2% glucose) and the OND group was imaged in OND buffer (130 mM NaCl, 5,4 mM KCl, 1.8 mM CaCl2, 26 mM NaHCO3, 0.8 mM MgCl2, 1.18 mM NaH2PO4, pH 7.4 in milliQ water) to avoid the loss of the treatment effect. Single plane images were taken using a Leica TCS STED CW SP8 laser scanning microscope using the 63X oil-immersion objective and 2X zoom. Images were analyzed using the FIJI/ImageJ free software and fluorescence intensity was measured for each individual cell. In each experiment, calibration curves were generated from fixed and equilibrated dextran loaded-cells to a range of pH buffers. Calibration groups were fixed in 4% paraformaldehyde for 10 min at RT and equilibrated for 20 min in the corresponding pH buffer. Buffers contained 50 mM Tris maleate adjusted to 3.5, 4.0, 4.5 and 5.5 pH. Data are shown normalized to the control of each experiment to reduce interexperimental variability.
Lysosomal enzymatic activity assay
Lysosomal activity was measured using the commercial Lysosomal Intracellular Activity Assay Kit according to the manufacturer’s instructions (Abcam, ab234622). Cells were incubated with a self-quenched substrate, washed with 1 mL of ice-cold 1X assay buffer and immediately imaged. Single plane images were taken using a Leica TCS STED CW SP8 laser scanning microscope using the 63X oil-immersion objective and 1.5X zoom. Images were analyzed with FIJI/ImageJ and mean fluorescence intensity was measured for each individual lysosome. Lysosomal fluorescence intensity values proportionally correlate with the amount of lysosomal activity [86]. Intensity, lysosomal number and occupied area values were normalized to the cell area. Data are shown normalized to the control of each experiment to reduce interexperimental variability
mRFP-EGFP-LC3 transfection in BV2 cells
The mRFP-EGFP-LC3 plasmid (Addgene, 21,074; depositing lab: Dr. Tamotsu Yoshimori) was transfected in Escherichia coli DH5α competent cells (Invitrogen, 18,258,012) using the heat shock method and kanamycin (50 µg/ml) was used for the selection of transformed bacteria. The plasmid was purified using a maxiprep commercial kit (PureLink HiPure Plasmid Filter Maxiprep Kit; Invitrogen, K210016). BV2 cells were seeded in 24-well plates at 70–90% confluence 24 h prior to the experiment in high glucose DMEM (Gibco, 11,995,065) supplemented with 10% FBS without antibiotics. Transfection assay was performed following manufacturer´s instructions. Lipofectamine 2000 (2 µg/mL; Invitrogen, 11,668,030) and plasmid DNA (0.9 µg/µL) in a 1:5 ratio were separately pre-incubated with Opti-MEM (Gibco, 31,985,070). Lipofectamine 2000 and plasmid DNA were left to complex for 20 min at room temperature and added to the plated BV2 cells in low glucose DMEM supplemented with 5% FBS and no antibiotics to ensure full transfection efficiency. Lipofectamine 2000-plasmid complexes were added to BV2 cells for 6 h and then replaced with the regular growth medium, high glucose DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic. mRFP-EGFP-LC3 expression was allowed for 24 h after the transfection. Cells were fixed in 4% PFA and imaged in a Zeiss LSM 880 Fast Airy Scan microscope under a 40X objective and 2x electronic zoom. A single z-plane was analyzed in FIJI/ImageJ. First, individual puncta were identified using the Find Maxima plugin, after manually optimizing the following parameters: minimum Sigma = 2.5, maximum Sigma = 3, threshold method Moments, prominence = 40 and minimum size = 0.032 mm. In these puncta, we calculated the GFP:RFP fluorescence ratio as a measure of the degradation of autophagosomes due to the sensitivity of GFP to the lysosomal pH. In addition, we calculated the mean RFP and GFP fluorescence intensity, the total number of puncta, and the area occupied by the puncta per cell and normalized to control conditions (expressed as % change over control).
Western blot
Primary microglia and BV2 cells were lysed in RIPA buffer (Sigma-Aldrich, R0278) containing protease and phosphatase inhibitor cocktail (Thermo Scientific, 78,440). The cell lysate was collected and centrifuged (10,000 x g, 10 min). The solubilized protein was quantified by BCA Assay Kit (Thermo Scientific, 23,225) in triplicates at 590 nm using a microplate reader (Synergy HT, BioTek). ß-mercaptoethanol denatured protein (15 to 20 µg) were loaded in Tris-glycine polyacrylamide gels (14%) and run for 90 min at 120 V. The resolved proteins were then transferred to a nitrocellulose membrane at 220 mA for 2 h and the transfer efficiency was verified by Pounceau staining (Sigma-Aldrich, P7170). The membranes were then blocked for 1 h in 5% milk prepared in Tris-buffered saline (Tris 20 mM, NaCl 150 mM) with 0.1% Tween-20 (Sigma-Aldrich, P1379; TBS-T) buffer. Membranes were afterwards incubated with rabbit primary antibody to LC3 (1:3,000; Novus Biologicals, NB100-2220), and mouse primary antibody to ACTB/β-actin (1:5,000; Sigma-Aldrich, A2228), in TBS-T containing 4% bovine serum albumin (BSA; Sigma-Aldrich, A7906) overnight (4°C, shaker). The next day, membranes were rinsed and incubated with the fluorescent secondary antibodies StarBright Blue 700 anti-mouse (1:5,000; Bio-Rad, 12,004,159) and StarBright Blue 700 anti-rabbit (1:5,000; Bio-Rad, 12,004,162) in TBS-T containing 5% milk powder. After rinsing membranes, protein bands were imaged in a ChemiDoc imaging system (Bio-Rad). Band intensity was quantified using the Gel Analyzer method of FIJI software.
Immunofluorescence
Mice were transcardially perfused with 30 ml of PBS followed by 30 ml of 4% PFA. The brains were removed and post-fixed with the same fixative for 3 h at RT, then washed in PBS and kept in cryoprotectant (30% sucrose (Fisher Bioreagents, 177,140,010), 30% ethylene glycol (Thermo Scientific, 146,750,250) at −20°C. Six series of 50-µm-thick brain sections were cut using a Leica VT 1200S vibrating blade microtome (Leica Microsystems GmbH, Wetzlar, Germany). Brains obtained from the Karolinska Institute were processed slightly differently. HI-treated mice were anesthetized with 50 mg/kg sodium pentobarbital and transcardially perfused-fixed with 6% formaldehyde solution. The tissue was immersion-fixed in the same fixative for 24 h at 4°C after perfusion and then soaked overnight in graded concentrations of sucrose solution (10%, 20%, and 30%). The right hemisphere was cut into 40 µm-thick sagittal sections in a series of 10 using a sliding microtome (Leica SM2010R, Wetzlar, Germany). The sections were stored in a cryoprotection solution at −20°C for further use. CIR mice were anesthetized with 50 mg/kg sodium pentobarbital and transcardially perfused with 4% PFA. The brains were immersion-fixed in the same fixative for 3 d at 4°C after perfusion and then soaked overnight in graded concentrations of sucrose solution (10%, 20%, and 30% in 0.1 M phosphate buffer, pH 7.4). One of the hemispheres was cut into 12 series of 25-µm-thick sagittal sections using a sliding microtome (Leica SM2010R, Wetzlar, Germany). The sections were stored in a cryoprotection solution (25% ethylene glycol and 25% glycerin in 0.1 M phosphate buffer) at −20°C for further use. Common marmosets (Callithrix jacchus) were deeply anesthetized with isoflurane (5% during 10 min) and transcardially perfused with a heparinized solution of saline followed by a solution of 4% PFA. The brains were removed from the skull and post-fixed for 4 h in the same fixative. Then the brains were cut in the coronal plane and sections (50 μm) were used for staining. The sections were stored in a cryoprotection solution at −20°C for further use. Cynomolgus macaque were deeply anesthetized before lethal injection. The brain was perfused with a saline solution through bilateral carotid catheters, removed from the skull, cut in the coronal plane, post-fixed in a 4% PFA solution and stored at 4°C. It was then soaked in graded concentrations of sucrose solutions in 0.1 M PBS (15% for 24 h followed by 30% for 24 h) and cut 40-µm sections were stored at −20°C for further use.
Free-floating sections from mice and non-human primates were incubated in permeabilization solution (0.3% Triton X-100, 0.5% BSA in PBS) for 2 h at RT, and then incubated overnight with the primary antibodies diluted in the permeabilization solution at 4°C (a list of primary and secondary antibodies can be found in Table 1). For immunofluorescent detection of autophagy markers, the permeabilization solution consisted on 0.1% Triton X-100, 5% normal goat serum (Sigma-Aldrich, G9023) in PBS for 1 h at room temperature, and labelling of each primary with its corresponding secondary was done sequentially, first SQSTM1 or LAMP1 (overnight), followed by LC3 (overnight), and finally GFP (overnight).
Table 1.
List of reagents and/or resources used in this article.
| REAGENT and/or RESOURCE | |||
|---|---|---|---|
| Antibodies | |||
| Primary (host) | Dilution | Source | Cat. Number |
| Cleaved CASP3 (rabbit) | 1:1000 | Cell Signaling Technology | 9664 |
| DsRed (rabbit) | 1:1000 | Clontech | 632,496 |
| GFAP (glial fibrillary acidic protein; goat) | 1:1000 | Abcam | ab53554 |
| GFAP (rabbit) | 1:1000 | DakoCytomation | Z0334 |
| Green fluorescent protein (GFP; chicken) | 1:1000 | Aves Labs | GFP-1020 |
| AIF1/IBA1 (allograft inflammatory factor 1; rabbit) | 1:1000 | Wako Chemicals | 019–19,741 |
| LAMP1 (rat) | 1:200 | DSHB | AB528127 |
| LC3 (rabbit; western) | 1:3000 (western) 1:200 (immuno) |
Novus Biologicals | NB100-2220 |
| RBFOX3/NeuN (mouse), clone A60 | 1:750 | Millipore | MAB377 |
| RBFOX3/NeuN (rabbit) | 1:1000 | Abcam | ab177487 |
| P2RY12/P2Y12 (rabbit) | 1:1000 | Anasec | AS-55043A |
| SQSTM1 (rabbit) | 1:200 | Novus Biologicals | NBP1-48,320 |
| Pimonidazole (mouse) | 1:50 | Hypoxyprobe | HP1-100Kit |
| ACTB/ß-actin (mouse) | 1:5000 | Sigma | A5441 |
| Secondary | |||
| Goat anti-chicken (Alexa Fluor 488) | 1:500 | Life Technologies | A11039 |
| Goat anti-mouse (Alexa Fluor Rhodamine Red X, RRX) | 1:500 | Jackson Immunology Research | 115,295,166 |
| Goat anti-mouse (Alexa Fluor 647) | 1:500 | Jackson Immunology Research | 115,605,003 |
| Goat anti-rabbit (Alexa Fluor 488) | 1:500 | Jackson Immunology Research | 111,545,144 |
| Goat anti-rabbit (Alexa Fluor Rhodamine Red X, RRX) | 1:500 | Jackson Immunology Research | 111,295,144 |
| Goat anti-rabbit (Alexa Fluor 647) | 1:500 1:200 |
Jackson Immunology Research ThermoFisher |
111,605,003 A-21245 |
| Goat anti-rat (Alexa Fluor 680) |
1:200 | ThermoFisher | A-21096 |
| Donkey anti-goat (Alexa Fluor 568) | 1:500 | Fisher (Spain) | A11057 |
| Donkey anti-mouse (Alexa Fluor 568) | 1:500 | Fisher (Spain) | A10037 |
| Donkey anti-mouse (Alexa Fluor 647) | 1:500 | Fisher (Spain) | A31571 |
| DAPI | 1:1000 | Sigma-Aldrich (Spain) | D9542-10 mg |
| StarBright Blue 700 Goat anti-rabbit IgG | 1:5000 | Bio-Rad | 12,004,162 |
| StarBright Blue 700 Goat anti-mouse IgG | 1:5000 | Bio-Rad | 12,004,159 |
| Chemicals | |||
| Absolute ethanol | Panreac | 1,310,861,211–1 L | |
| Bovine serum albumin (BSA) | Sigma-Aldrich (Spain) | A7906-50 G | |
| Buprenorphine | Dechra | 582,039.2 | |
| Ethylene glycol | Sigma-Aldrich (Spain) | 324,558–2 L | |
| Dextran (Fluorescein and Tetramethylrhodamine, 70,000 MW, Anionic | Invitrogen | 11,580,236 | |
| Fluorescent mounting medium | Agilent | S302380-2 | |
| Isofluorane (Isovet) | S. Revet | 469,860–250 ml | |
| Lysosomal Intracellular Activity Assay Kit | Abcam | Ab234622 | |
| mRFP-GFP-LC3 tandem (ptfLC3) | Addgene | 21,074 | |
| MRT68921 dihydrochloride | Sigma | SML1644-5 MG | |
| Paraformaldehyde (PFA) | Sigma-Aldrich (Spain) | 441,244–3 Kg | |
| Pimonidazole | Hypoxyprobe (USA) | Mouse-Mab | |
| Propidium iodide | Sigma | P4864-10ML | |
| Rapamycin | Selleckchem | AY-22989 | |
| Triton™ X-100 | Sigma-Aldrich (Spain) | T8787-100 ml | |
| Experimental Models: Organisms/Strains | |||
| Ambra1± brain samples | Bred at Centro de Investigaciones Biológicas (CIB) | N/A | |
| atg4b KO brain samples | Bred at Universidad de Oviedo | N/A | |
| Common marmoset (Callithrix jacchus) brain samples | Bred at University of Caen (France) | N/A | |
| Cx3cr1GFP/+Ccr2RFP/+ brain samples | Bred at Karolinska Institute (Sweden) | N/A | |
| Cx3cr1GFP/+ mice | Bred at Achucarro Basque Center for Neuroscience | N/A | |
| fms-EGFP (MacGreen) mice | The Jackson Laboratories | 018549 | |
| Macaque (Macaca fascicularis) brain samples | Bred at University of Lyon (France) | N/A | |
| Microglia specific becn1 KO (Tmem119-CreER) brain samples | Bred at University of Massachussetts | N/A | |
| Software and Algorithms | |||
| ImageJ (FIJI) | https://imagej.nih.gov/ij/ | N/A | |
| Adobe Photoshop 7.0 | Adobe Systems Incorporated | N/A | |
| SigmaPlot | http://sigmaplot.co.uk/index.php | N/A | |
| Other | |||
| PF5010, Laser Doppler Perfusion Monitoring (LDPM) Unit | Perimed Company | N/A | |
| FACS Jazz (2B/4YG) | BD | N/A | |
| Hypoxia chamber | Biospherix | N/A | |
| TCS STED CW SP8 laser scanning microscopy | Leica | N/A | |
| LSM880 Fast Airyscan | Zeiss | ||
| VT 1200S vibrating blade microtome | Leica | N/A | |
| Axiovert epifluorescent microscopy | Zeiss | N/A | |
| Master Probe 418 | Perimed Company | 91–00053 | |
| Filament 2 (F2) | Doccol corporation | 6021910PK10Re | |
| X-ray irradiator | XRAD320, Precision X-Ray, North Branford, CT, U.S | N/A | |
Hippocampal organotypic slices were processed similarly, using 0.2% Triton X-100, 2% BSA in PBS as permeabilization solution. After thorough washing with PBS the sections were incubated with fluorochrome-conjugated secondary antibodies and DAPI (5 mg/ml) diluted in the permeabilization solution for 2 h at RT. After washing with PBS, the sections were mounted on glass slides with DakoCytomation Fluorescent Mounting Medium (Agilent, S3023).
Phagocytosis and autophagy image analysis
Fluorescence immunostaining images were collected in a Leica TCS STED CW SP8 laser scanning microscope using 40X or 63X oil-immersion objectives and a z-step of 0.7 µm (phagocytosis) or 0.34 μm (autophagy). All images were imported into Adobe Photoshop 7.0 (Adobe Systems Incorporated, San Jose, CA) in tiff format. Brightness, contrast, and background were adjusted equally for the entire image using the “brightness and contrast” and “levels” controls from the “image/adjustment” set of options without any further modification. For mouse tissue sections, 2–5 20 µm-thick z-stacks located at random positions containing the DG were collected per hippocampal section, and a minimum of 6 (sagittal) and 4–5 (coronal) sections per series were analyzed. For macaque and common marmoset tissue, one coronal section was fully scanned under the microscope to find all apoptotic cells. In hippocampal organotypic slices, 2–3 images of the DG were acquired per slice and each experimental condition consisted of 3–4 slices. In macaques, the areas covered were cortical regions of the precentral and superior temporal gyri. For primary cultures 3–5 random z-stacks were analyzed from 3 independent coverslips.
Apoptosis and phagocytosis analysis was performed using unbiased stereology methods as previously described [81]. Apoptotic dead cells were determined by their nuclear morphology visualized with the DNA dye DAPI. These cells lost their chromatin structure (euchromatin and heterochromatin) and appeared condensed and/or fragmented (pyknosis/karyorrhexis). Phagocytosis was defined as the formation of a three dimensional pouch, usually located in the terminal or passant branches of microglia, completely surrounding an apoptotic cell [21]. In tissue sections and hippocampal organotypic slices the number of apoptotic cells, phagocytosed cells, microglia, and/or monocytes were estimated in the volume of the DG contained in the z-stack (determined by multiplying the thickness of the stack by the area of the DG at the center of the stack using ImageJ [FIJI]). To obtain the absolute numbers (cells per hippocampus), this density value was then multiplied by the volume of the septal hippocampus (spanning from −1 mm to −2.5 mm in the anteroposterior axes, from Bregma; approximately 6 slices in each of the 6 series), which was calculated using FIJI from images collected at 20x with a Zeiss Axiovert epifluorescent microscope.
In the case of HI and CIR experiments, a limited tissue and/or unrepresentative number of samples was provided by Karolinska Institute (Sweden). Due to this reason, the total volume of the septal hippocampus could not be calculated, and therefore the number of apoptotic, phagocytosed, microglial cells and peripheral monocytes was estimated as cell density (cells per mm3).
The following formulae were used to estimate microglial phagocytic efficiency in tissue and organotypic slices:
Ph index =
Ph capacity =
Ph/A coupling =
where apoPh is the number of apoptotic cells phagocytosed; apotot is the total number of apoptotic cells; and mgPhn is the proportion of microglia with “n” phagocytic pouches.
In primary cultures phagocytosis assays the number of total microglia was calculated, and microglial processes were scanned for DAPI or RFP (from apoptotic SH-SY5Y) inclusions. The number of microglia with inclusions was divided by the total microglial numbers and calculated as percentage of phagocytic microglia at a given time point. In the engulfment-degradation experiments, the engulfment was calculated as above, normalized by the control group. The degradation in each experimental condition was calculated by subtracting the percentage of phagocytic microglia at 3 h from the percentage of phagocytic microglia in the control group at 1 h.
For autophagy puncta numbers and area quantification, a custom FIJI/ImageJ script was developed (LC3_LAMP_quant, freely available at https://github.com/SoriaFN/Tools), based on the scripts used in [87]. Briefly, the script enables segmentation of fms-GFP+ microglia by manual thresholding, and LC3, LAMP1 and SQSTM1 puncta by automatic thresholding, in confocal z-stacks of slices processed for immunofluorescence. The script then quantifies the number, size and fluorescent intensity of LC3, LAMP1 and SQSTM1 puncta within the cellular (microglial) ROI, and calculates the colocalization percentage.
Transmission electron microscopy
Five-million primary microglial cells were cultured in 60-mm diameter plates (Nunclon; Thermo Scientific, 150,288) and exposed to OND in a hypoxia chamber (1% O2) with no nutrients (salt solution) during 3 h. Subsequently, primary microglia were rinsed in PBS and pre-fixed as an adherent cell monolayer using 0.5% glutaraldehyde solution in Sörenson buffer 0.1 M pH = 7.4 (SB; 19% 0.1 M NaH2PO4, 81% 0.185 M Na2HPO4 in milliQ water; 10 min, room temperature). After scraping the pre-fixed cells, primary microglia were centrifuged (800 g, 5 min, room temperature) to form a pellet and fixed in 2% glutaraldehyde solution in SB (overnight, 4°C). Primary microglia were then rinsed with 4% sucrose in SB and post-fixed with 1% osmium tetroxide in SB (1 h, 4°C, darkness). After rinsing, primary microglia were dehydrated in a growing concentration series of acetone. After dehydration, primary microglia samples were embedded in epoxy resin EPON Polarbed 812 (Electron Microscopy Sciences, 14,901). Semi-thin sections (1-µm thick) were stained with toluidine blue to identify the regions of interest. Ultra-thin sections were cut using a LEICA EM UC7 ultramicrotome and contrasted with uranyl-acetate and lead citrate. The ultrastructural analysis was done with a Transmission Electron Microscope Jeol JEM 1400 Plus at 100 kVs equipped with a sCMOS digital camera. Image analysis (4–6 images per cell) was performed in 36–38 cells per experimental condition using FIJI. Autophagic-like vesicles (containing at least a portion of double membrane with granular, membranous and heterogeneous cargo) and lysosomal-like vesicles (electron-dense vesicles with single or double membrane) were identified manually and their perimeter was selected to generate regions of interest (ROIs) in each image. The area of the cellular cytoplasm (µm2) was also identified and selected manually to generate a ROI in each image. Subsequently, the number (vesicles) and area (vesicles and cytoplasm, µm2) of ROIs were measured. Finally, the quantitative data coming from ROIs of the images belonging to the same cell were grouped and the number of autophagic-and lysosomal-like vesicles per µm2 was calculated dividing the number of vesicles present in the cell by its cytoplasm area (µm2). The percentage of cytoplasm area occupied by autophagic- and lysosomal-like vesicles per each cell was calculated by summing up the areas of individual vesicles and relating it to the cellular cytoplasmic area, which was considered 100%. The mean size (area, µm2) of the vesicles per cell is represented in logarithmic scale for illustrative purposes.
Analysis of RNAseq databases
Unbiased analysis of recent RNA-Seq databases [10–14] was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID; https://david.ncifcrf.gov/summary.jsp). To compare between databases, the differentially expressed genes we pre-filtered according to their significant expression by adjusted p-value < 0.05, and a fold change > 0.5 or < −0.5. Then, the annotated Gene Ontology (GO) terms were obtained from DAVID using the Fisher Exact statistics and the thresholds Max.Prob. ≤ 0.1 or p-value and Min.Count ≥ 2 or minimum number of genes per GO term group. The p-values associated to the GO terms of Phagocytosis (GO: 0006909) and Positive regulation of phagocytosis (GO: 0050766) were calculated using the linear step-up method of Benjamini and Hochberg.
Statistical analysis
SigmaPlot (San Jose, CA, USA) was used for statistical analysis. Data were tested for normality and homoscedasticity (Levene´s test). When the data did not comply with these assumptions, a logarithmic transformation (Log10, Log10 + 1, of Ln) or a square root was performed and the data were analyzed using parametric tests. Two-sample experiments were analyzed by Students’ t-test and more than two sample experiments with one-way or two-way ANOVA. In two-way ANOVAs, the interaction between factors was assessed prior to analyzing the effect of individual factors. In case that homoscedasticity or normality were not achieved with a logarithmic transformation, data were analyzed using a Kruskal-Wallis ranks test, followed by Dunn method as a post hoc test. Two sample non-parametric data were analyzed using Mann Whittney U test. Only p < 0.05 is reported to be significant.
Supplementary Material
Acknowledgments
We dedicate this paper to Takashi Umekawa, who generated the HI model at the Karolinska Institute, and unfortunately passed away in 2018. This work was supported by grants from the Spanish Ministry of Science and Innovation Competitiveness MCIN/AEI/10.13039/501100011033 (https://www.ciencia.gob.es/) and ERDF “A way to make Europe” (RTI2018-099267-B-I00 and RYC-2013-12817 to AS; RTI2018–097948-A-100 and RYC-2016–20480 to OP), a Tatiana Foundation Award (P-048-FTPGB 2018) to AS a Basque Government Department of Education project (PIBA 2020_1_0030; http://www.euskadi.eus/basque-government/department-education/) to AS, a Basque Government Department of Economic development, Sustainability and environment (ELKARTEK KK-2020/00034; https://www.spri.eus/en/) to ECZ, and . SB is recipient of predoctoral fellowship from the Spanish Ministry of Economy and Competitiveness, and VST is recipient of predoctoral fellowship from the Basque Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. UPV/EHU SGIker technical and human support is gratefully acknowledged.
Funding Statement
This work was supported by the Basque Government [ELKARTEK KK-2020/00034;]; Department of Education, Basque Government [PIBA 2020_1_0030]; Spanish Ministry of Science and Innovation [RYC-2016–20480]; Tatiana Foundation [P-048-FTPGB 2018]; Spanish Ministry of Science and Innovation [RTI2018-099267-B-I00, RYC-2013-12817]; Spanish Ministry of Science and Innovation [RTI2018–097948-A-100].
Disclosure statement
Authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2165313
References
- [1].Collaborators GBDS, Nguyen M, Roth GA.. Global, regional, and national burden of stroke, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019. May;18(5):439–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iadecola C, Buckwalter MS, Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest. 2020. Jun 1;130(6):2777–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ma Y, Wang J, Wang Y, et al. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017. Oct;157:247–272. [DOI] [PubMed] [Google Scholar]
- [4].Lalancette-Hebert M, Gowing G, Simard A, et al. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007. Mar 7;27(10):2596–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jin WN, Shi SX, Li Z, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017. Jun;37(6):2224–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li T, Zhao J, Xie W, et al. Specific depletion of resident microglia in the early stage of stroke reduces cerebral ischemic damage. J Neuroinflammation. 2021. Mar 23;18(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Neumann J, Sauerzweig S, Ronicke R, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. 2008. Jun 4;28(23):5965–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Otxoa-de-Amezaga A, Miro-Mur F, Pedragosa J, et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019. Feb;137(2):321–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jolivel V, Bicker F, Biname F, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015. Feb;129(2):279–295. [DOI] [PubMed] [Google Scholar]
- [10].Rajan WD, Wojtas B, Gielniewski B, et al. Dissecting functional phenotypes of microglia and macrophages in the rat brain after transient cerebral ischemia. Glia. 2019. Feb;67(2):232–245. [DOI] [PubMed] [Google Scholar]
- [11].Androvic P, Kirdajova D, Tureckova J, et al. Decoding the transcriptional response to ischemic stroke in young and aged mouse brain. Cell Rep. 2020. Jun 16;31(11):107777. [DOI] [PubMed] [Google Scholar]
- [12].Guo K, Luo J, Feng D, et al. Single-cell RNA sequencing with combined use of bulk RNA sequencing to reveal cell heterogeneity and molecular changes at acute stage of ischemic stroke in mouse cortex penumbra area. Front Cell Dev Biol. 2021;9:624711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zheng K, Lin L, Jiang W, et al. Single-cell RNA-seq reveals the transcriptional landscape in ischemic stroke. J Cereb Blood Flow Metab. 2022. Jan;42(1):56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Beuker C, Schafflick D, Strecker JK, et al. Stroke induces disease-specific myeloid cells in the brain parenchyma and pia. Nat Commun. 2022. Feb 17;13(1):945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022. Nov 2;110(21):3458–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morioka S, Maueroder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. 2019. May 21;50(5):1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kawabori M, Kacimi R, Kauppinen T, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. 2015. Feb 25;35(8):3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wen RX, Shen H, Huang SX, et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci Ther. 2020. Apr;26(4):416–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang S, Chen T, Suo Q, et al. BK channel-mediated microglial phagocytosis alleviates neurological deficit after ischemic stroke. Front Cell Neurosci. 2021;15:683769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rudolph M, Schmeer CW, Gunther M, et al. Microglia-mediated phagocytosis of apoptotic nuclei is impaired in the adult murine hippocampus after stroke. Glia. 2021. Aug;69(8):2006–2022. [DOI] [PubMed] [Google Scholar]
- [21].Sierra A, Encinas JM, Deudero JJ, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010. Oct 8;7(4):483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abiega O, Beccari S, Diaz-Aparicio I, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 2016. May;14(5):e1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sierra-Torre V, Plaza-Zabala A, Bonifazi P, et al. Microglial phagocytosis dysfunction in the dentate gyrus is related to local neuronal activity in a genetic model of epilepsy. Epilepsia. 2020. Nov;61(11):2593–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Higo N. Non-human primate models to explore the adaptive mechanisms after stroke. Front Syst Neurosci. 2021;15:760311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kaiser EE, West FD. Large animal ischemic stroke models: replicating human stroke pathophysiology. Neural Regen Res. 2020. Aug;15(8):1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Morris GP, Wright AL, Tan RP, et al. A comparative study of variables influencing ischemic injury in the longa and koizumi methods of intraluminal filament middle cerebral artery occlusion in mice. PLoS One. 2016;11(2):e0148503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sierra A, Gottfried-Blackmore AC, McEwen BS, et al. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007. Mar;55(4):412–424. [DOI] [PubMed] [Google Scholar]
- [28].Mildner A, Huang H, Radke J, et al. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia. 2017. Feb;65(2):375–387. [DOI] [PubMed] [Google Scholar]
- [29].Umekawa T, Osman AM, Han W, et al. Resident microglia, rather than blood-derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia-ischemia. Glia. 2015. Dec;63(12):2220–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Han W, Umekawa T, Zhou K, et al. Cranial irradiation induces transient microglia accumulation, followed by long-lasting inflammation and loss of microglia. Oncotarget. 2016. Dec 13;7(50):82305–82323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Osman AM, Sun Y, Burns TC, et al. Radiation triggers a dynamic sequence of transient microglial alterations in juvenile brain. Cell Rep. 2020. Jun 2;31(9):107699. [DOI] [PubMed] [Google Scholar]
- [32].Debatisse J, Eker OF, Wateau O, et al. PET-MRI nanoparticles imaging of blood-brain barrier damage and modulation after stroke reperfusion. Brain Commun. 2020;2(2):fcaa193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dibaj P, Steffens H, Nadrigny F, et al. Long-lasting post-mortem activity of spinal microglia in situ in mice. J Neurosci Res. 2010. Aug 15;88(11):2431–2440. [DOI] [PubMed] [Google Scholar]
- [34].Dachet F, Brown JB, Valyi-Nagy T, et al. Selective time-dependent changes in activity and cell-specific gene expression in human postmortem brain. Sci Rep. 2021. Mar 23;11(1):6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Freret T, Bouet V, Toutain J, et al. Intraluminal thread model of focal stroke in the non-human primate. J Cereb Blood Flow Metab. 2008. Apr;28(4):786–796. [DOI] [PubMed] [Google Scholar]
- [36].Hall CN, Reynell C, Gesslein B, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014. Apr 3;508(7494):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Diaz-Aparicio I, Beccari S, Abiega O, et al. Clearing the corpses: regulatory mechanisms, novel tools, and therapeutic potential of harnessing microglial phagocytosis in the diseased brain. Neural Regen Res. 2016. Oct;11(10):1533–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Galluzzi L, Bravo-San Pedro JM, Blomgren K, et al. Autophagy in acute brain injury. Nat Rev Neurosci. 2016. Aug;17(8):467–484. [DOI] [PubMed] [Google Scholar]
- [39].Plaza-Zabala A, Sierra-Torre V, Autophagy SA. Microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. 2017. Mar 9;18(3):598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lopez A, Fleming A, Rubinsztein DC. Seeing is believing: methods to monitor vertebrate autophagy in vivo. Open Biol. 2018. Oct 24;8(10). DOI: 10.1098/rsob.180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Plaza-Zabala A, Sierra-Torre V, Sierra A. Assessing autophagy in microglia: a two-step model to determine autophagosome formation, degradation, and net turnover. Front Immunol. 2020;11:620602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010. Jun 17;465(7300):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007. Jun 28;447(7148):1121–1125. [DOI] [PubMed] [Google Scholar]
- [44].Corona Velazquez AF, Jackson WT. So many roads: the multifaceted regulation of autophagy induction. Mol Cell Biol. 2018. Nov 1;38(21):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015. May 1;290(18):11376–11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Buckley KM, Hess DL, Sazonova IY, et al. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hadley G, Beard DJ, Couch Y, et al. Rapamycin in ischemic stroke: old drug, new tricks? J Cereb Blood Flow Metab. 2019. Jan;39(1):20–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Civiletto G, Dogan SA, Cerutti R, et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med. 2018. Nov;10(11). doi: 10.15252/emmm.201708799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dehay B, Bove J, Rodriguez-Muela N, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010. Sep 15;30(37):12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Palazuelos J, Ortega Z, Diaz-Alonso J, et al. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J Biol Chem. 2012. Jan 6;287(2):1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen S, Lai SWT, Brown CE, et al. Harnessing and enhancing macrophage phagocytosis for cancer therapy. Front Immunol. 2021;12:635173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012. Mar 22;12(4):278–287. [DOI] [PubMed] [Google Scholar]
- [53].Matlung HL, Szilagyi K, Barclay NA, et al. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol Rev. 2017. Mar;276(1):145–164. [DOI] [PubMed] [Google Scholar]
- [54].Lehrman EK, Wilton DK, Litvina EY, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. 2018. Oct 10;100(1):120–134 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014. Apr;15(4):209–216. [DOI] [PubMed] [Google Scholar]
- [56].Neher JJ, Emmrich JV, Fricker M, et al. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A. 2013. Oct 22;110(43):E4098–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Calovi S, Mut-Arbona P, Sperlagh B. Microglia and the purinergic signaling system. Neuroscience. 2019. May;1(405):137–147. [DOI] [PubMed] [Google Scholar]
- [58].Dale N, Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol. 2009. Sep;7(3):160–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ha ACT, Bhatt DL, Rutka JT. Ha ACT, Bhatt DL, Rutka JT, et al . Intracranial hemorrhage during dual antiplatelet therapy: JACC review topic of the week. J Am Coll Cardiol.2021. Sep 28;78(13):1372–1384. [DOI] [PubMed] [Google Scholar]
- [60].Ito H, Yamashita Y, Tanaka T, et al. Cigarette smoke induces endoplasmic reticulum stress and suppresses efferocytosis through the activation of RhoA. Sci Rep. 2020. Jul 28;10(1):12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Linton MF, Babaev VR, Huang J, et al. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J. 2016. Oct 25;80(11):2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Papadakis M, Hadley G, Xilouri M, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013. Mar;19(3):351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].A long and winding sTORy. Nat Cell Biol. 2017. Sep 28;19(10):1131. https://www.nature.com/articles/ncb3624 [DOI] [PubMed] [Google Scholar]
- [64].Wu M, Zhang H, Kai J, et al. Rapamycin prevents cerebral stroke by modulating apoptosis and autophagy in penumbra in rats. Ann Clin Transl Neurol. 2018. Feb;5(2):138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li L, Huang J. Rapamycin pretreatment alleviates cerebral ischemia/reperfusion injury in dose-response manner through inhibition of the autophagy and NFkappaB pathways in rats. Dose Response. 2020;Jul-Sep;18(3):1559325820946194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [66].Xie L, Sun F, Wang J, et al. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol. 2014. Jun 15;192(12):6009–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Berglund R, Guerreiro-Cacais AO, Adzemovic MZ, et al. Microglial autophagy-associated phagocytosis is essential for recovery from neuroinflammation. Sci Immunol. 2020. Oct 16;5(52):52. [DOI] [PubMed] [Google Scholar]
- [68].Lucin KM, O’Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013. Sep 4;79(5):873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yang Z, Zhong L, Zhong S, et al. Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp Mol Pathol. 2015. Apr;98(2):219–224. [DOI] [PubMed] [Google Scholar]
- [70].He T, Li W, Song Y, et al. Sestrin2 regulates microglia polarization through mTOR-mediated autophagic flux to attenuate inflammation during experimental brain ischemia. J Neuroinflammation. 2020. Nov 5;17(1):329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li X, Xia Q, Mao M, et al. Annexin-A1 SUMOylation regulates microglial polarization after cerebral ischemia by modulating IKKalpha stability via selective autophagy. Sci Adv. 2021. Jan;7(4). https://www.science.org/doi/10.1126/sciadv.abc5539?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016. Jul 26;19(8):987–991. [DOI] [PubMed] [Google Scholar]
- [73].Paolicelli R, Sierra V, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads Neuron (in press). Neuron. 2022. Nov 2;110(21):3458–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lauro C, Limatola C. Metabolic reprograming of microglia in the regulation of the innate inflammatory response. Front Immunol. 2020;11:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jiang CT, Wu WF, Deng YH, et al. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol Med Rep. 2020. May;21(5):2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sasmono RT, Oceandy D, Pollard JW, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003. Feb 1;101(3):1155–1163. [DOI] [PubMed] [Google Scholar]
- [77].Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000. Jun;20(11):4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Saederup N, Cardona AE, Croft K, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010. Oct 27;5(10):e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Marino G, Fernandez AF, Cabrera S, et al. Autophagy is essential for mouse sense of balance. J Clin Invest. 2010. Jul;120(7):2331–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Debatisse J, Wateau O, Cho TH, et al. A non-human primate model of stroke reproducing endovascular thrombectomy and allowing long-term imaging and neurological read-outs. J Cereb Blood Flow Metab. 2021. Apr;41(4):745–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Beccari S, Diaz-Aparicio I, Sierra A. Quantifying microglial phagocytosis of apoptotic cells in the brain in health and disease. Curr Protoc Immunol. 2018. Aug;122(1):e49. [DOI] [PubMed] [Google Scholar]
- [82].Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989. Jan;20(1):84–91. [DOI] [PubMed] [Google Scholar]
- [83].Rice JE 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981. Feb;9(2):131–141. [DOI] [PubMed] [Google Scholar]
- [84].Cuartero MI, Ballesteros I, de la Parra J, et al. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation. 2014. Dec 2;130(23):2040–2051. [DOI] [PubMed] [Google Scholar]
- [85].Majumdar A, Cruz D, Asamoah N, et al. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. 2007. Apr;18(4):1490–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].WHt H, Payne CK. Imaging lysosomal enzyme activity in live cells using self-quenched substrates. Anal Biochem. 2012. May 15;424(2):178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Arotcarena ML, Soria FN, Cunha A, et al. Acidic nanoparticles protect against alpha-synuclein-induced neurodegeneration through the restoration of lysosomal function. Aging Cell. 2022. Apr;21(4):e13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.