

ABSTRACT
Ferroptosis is a type of iron-dependent regulated cell death characterized by unrestricted lipid peroxidation and membrane damage. Although GPX4 (glutathione peroxidase 4) plays a master role in blocking ferroptosis by eliminating phospholipid hydroperoxides, the regulation of GPX4 remains poorly understood. Here, we report an unexpected role for copper in promoting ferroptotic cell death, but not cuproptosis, by inducing macroautophagic/autophagic degradation of GPX4. Copper chelators reduce ferroptosis sensitivity but do not inhibit other types of cell death, such as apoptosis, necroptosis, and alkaliptosis. Conversely, exogenous copper increases GPX4 ubiquitination and the formation of GPX4 aggregates by directly binding to GPX4 protein cysteines C107 and C148. TAX1BP1 (Tax1 binding protein 1) then acts as an autophagic receptor for GPX4 degradation and subsequent ferroptosis in response to copper stress. Consequently, copper enhances ferroptosis-mediated tumor suppression in a mouse model of pancreatic cancer tumor, whereas copper chelators attenuate experimental acute pancreatitis associated with ferroptosis. Taken together, these findings provide new insights into the link between metal stress and autophagy-dependent cell death.
Abbreviations: CALCOCO2, calcium binding and coiled-coil domain 2; GPX4, glutathione peroxidase 4; MAP1LC3A/B, microtubule associated protein 1 light chain 3 alpha/beta; MPO, myeloperoxidase; NCOA4, nuclear receptor coactivator 4; OPTN, optineurin; PDAC, pancreatic ductal adenocarcinoma; RIPK1, receptor interacting serine/threonine kinase 1; ROS, reactive oxygen species; SLC40A1, solute carrier family 40 member 1; SQSTM1, sequestosome 1; TAX1BP1, Tax1 binding protein 1; TEPA, tetraethylenepentamine; TM, tetrathiomolybdate.
KEYWORDS: Autophagy, copper, cuproptosis, ferroptosis, GPX4, TAX1BP1
Introduction
To maintain tissue and organ structure, multicellular organisms carefully coordinate macroautophagy (to which we refer as “autophagy”) and cell death for development, differentiation, and homeostatic maintenance of many cell types [1]. Based on morphological, biochemical, and mechanistic criteria, regulated cell death is divided into apoptotic and non-apoptotic modalities [2]. Ferroptosis constitutes a non-apoptotic cell death routine that mainly relies on the iron-dependent Fenton reaction and subsequent unrestricted intracellular lipid peroxidation [3]. Impaired ferroptosis is involved in several pathological conditions, especially cancer and tissue damage [4]. GPX4 (glutathione peroxidase 4), a downstream mediator of amino acid antiporter system xc−, is an antioxidant enzyme involved in reducing phospholipid hydroperoxides in membranes, thus conferring cytoprotection against ferroptosis [5]. The inhibition of GPX4 activity or the downregulation of GPX4 expression can induce ferroptosis [5–8]. Although the anti-ferroptotic function of GPX4 has been extensively studied, the regulation of GPX4 remains poorly understood [9].
Copper serves as an essential trace element for human physiology and its altered metabolism contributes to the pathogenesis of various diseases. Elevated serum and tumor copper levels have been observed in patients with a large number of different types of cancer, including lymphoma, breast and lung cancer [10]. Thus, copper chelation has potential inhibitory effects on tumor development and angiogenesis [11]. However, excessive copper uptake can also lead to cell damage and death. In particular, supraphysiological levels of intracellular copper reportedly trigger apoptosis by inducing reactive oxygen species (ROS) or by inhibiting proteasome function [12–14]. Moreover, intracellular copper accumulation may cause a specific cell death modality dubbed “cuproptosis” that differs from apoptosis, necroptosis, and ferroptosis [15–17]. At the actual state-of-the-art, it appears that copper can stress and kill cells through several distinct, likely context-dependent pathways.
Although iron plays an important role in inducing ferroptosis [18], little is known about whether other metals modulate the ferroptosis process. Here, we report a novel role for copper in facilitating ferroptosis sensitivity in vitro and in vivo. We demonstrate that copper directly binds to GPX4 protein, leading to the formation of GPX4 aggregates and subsequent autophagic degradation of GPX4. We identified TAX1BP1 (Tax1 binding protein 1) as an autophagy receptor for GPX4 protein degradation. These findings may provide new insights into the mechanism of autophagy-dependent ferroptosis.
Results
Copper chelators inhibit ferroptosis
Pancreatic ductal adenocarcinoma (PDAC) cells are highly dependent on system xc−-mediated antioxidant defenses for growth [19]. Inhibition of system xc− by erastin is a classical approach to induce ferroptotic PDAC cell death [20–23]. To determine whether copper regulates ferroptosis, we treated four human PDAC cell lines (AsPC-1, PANC-1, MIA PaCa-2, and SW 1990) with erastin in the presence or absence of copper chelators tetrathiomolybdate (TM) or tetraethylenepentamine (TEPA). Cell viability assays revealed that TM and TEPA attenuated erastin-induced cytotoxicity in these PDAC cell lines (Figure 1A). The broad-spectrum protective activity of copper chelators against ferroptosis inducers (including the system xc− inhibitor sulfasalazine and GPX4 inhibitors RSL3, ML162, and ML210) was further confirmed (Figure 1B). As a control, TM failed to inhibit bortezomib-induced apoptosis [24], CCT137690-triggered necroptosis [25], and JTC801-initiated alkaliptosis [26], arguing against a nonspecific cytoprotective effect of copper chelators (Figure 1C-E).
Figure 1.
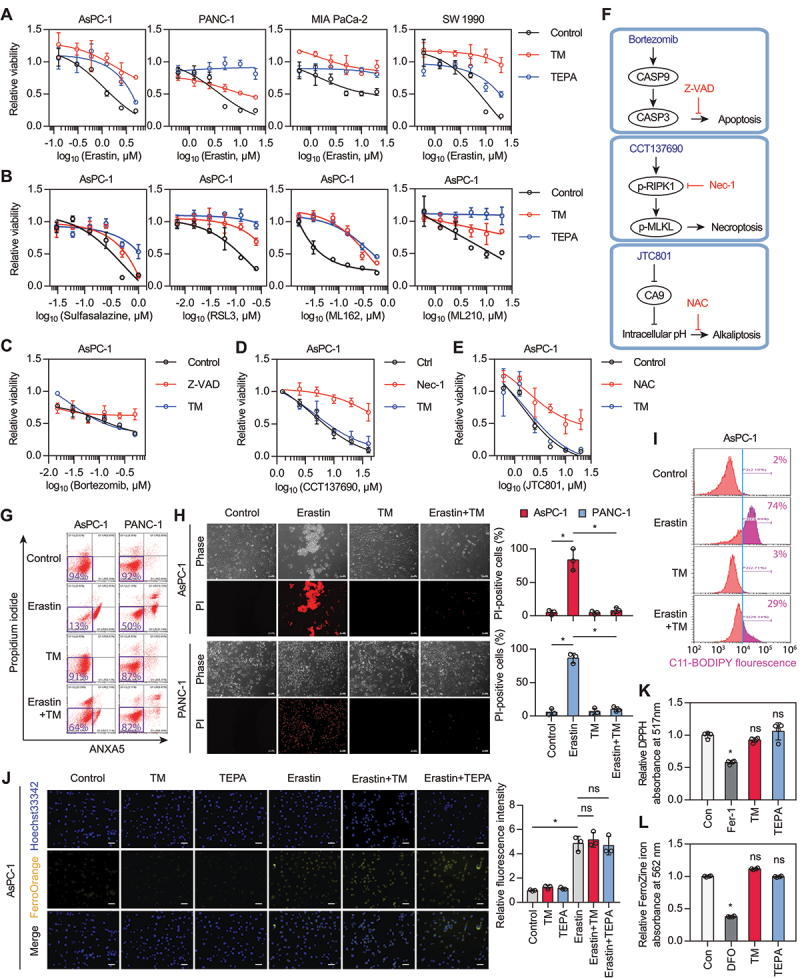
Copper chelators inhibit ferroptosis. (A) Cell viability of PDAC cells treated with erastin in the presence or absence of copper chelators (tetrathiomolybdate [TM], 50 μM; tetrethylenepentamine [TEPA], 1 mM) for 24 h. (B) Cell viability of AsPC-1 cells treated with different doses of sulfasalazine, RSL3, ML162, or ML210 in the presence or absence of copper chelators (TM, 50 μM; TEPA, 1 mM) for 24 h. (C-E) Cell viability of AsPC-1 cells treated with apoptosis inducer (b, bortezomib), necroptosis inducer (c, CCT137690), or alkaliptosis inducer (d, JTC801) in the presence or absence of the corresponding inhibitors (Z-VAD-FMK/Z-VAD, 50 μM; necrostatin-1/Nec-1, 10 μM; N-acetyl-L-cysteine/NAC, 1 mM) or TM (50 μM) for 24 h. (F) Schematic diagram of the inducers and inhibitors of various types of cell death. (G) Cell death of PDAC cells treated with erastin (5 μM for AsPC-1 cells; 10 μM for PANC-1 cells) in the presence or absence of TM (50 μM) for 24 h. (H) Representative phase-contrast and propidium iodide (PI) staining images of PDAC cells treated with erastin (5 μM for AsPC-1 cells; 10 μM for PANC-1 cells) in the presence or absence of TM (50 μM) for 24 h. Scale bar: 50 µm. Quantification of PI-positive cells was shown. *P < 0.05; ns, no significance. (I) Lipid peroxidation of AsPC-1 cells treated with erastin (5 μM) in the presence or absence of TM (50 μM) for 6 h. (J) Confocal imaging of FerroOrange (Fe2+-selective fluorescent probe) in AsPC-1 cells treated with erastin (5 μM) in the presence or absence of copper chelators (TM, 50 μM; TEPA, 1 mM) for 6 h. Hoechst 33,342 (blue) was used as a nuclear counterstain. Scale bar: 50 µm. Data are presented as mean ± SD, n = 3 biologically independent samples; *P < 0.05. (K) The antioxidant activity of copper chelators (TM, 50 μM; TEPA, 1 mM) or ferrostatin-1 (Fer-1, 10 μM) was analyzed by using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay. *P < 0.05 versus control group; ns, no significance. (L) The iron chelator activity of copper chelators (TM, 50 μM; TEPA, 1 mM) or deferoxamine (DFO, 50 μM) was analyzed by using a ferrozine Fe2+ binding assay. *P < 0.05 versus control group; ns, no significance.
As expected, the caspase inhibitor Z-VAD-FMK, the RIPK1 (receptor interacting serine/threonine kinase 1) inhibitor necrostatin-1, and the anti-oxidant acidic compound N-acetyl-L-cysteine alleviated apoptosis, necroptosis, and alkaliptosis, respectively (Figure 1C-1F). As an alternative to cell viability assays, we determined cell death induction by cytofluorometry, confirming that the percentage of erastin-induced cell death was downregulated after TM treatment (Figure 1G and H). Functionally, TM diminished lipid peroxidation (Figure 1I), but failed to inhibit intracellular Fe2+ accumulation (Figure 1J) during ferroptosis. Unlike the effects of the radical-trapping antioxidant ferrostatin-1 or the iron chelator deferoxamine, the copper chelators did not exhibit any antioxidative (Figure 1K) and iron-chelating abilities (Figure 1L). Altogether, these results demonstrate an unexpected role for copper chelators in blocking ferroptosis, but not other types of cell death.
Copper amplifies ferroptosis by promoting autophagic degradation of GPX4
Next, we sought to determine whether copper (II) ions (Cu2+) can enhance the susceptibility of cells to the induction of ferroptosis. The addition of Cu2+ in cell culture medium amplified cell death (Figure 2A) and lipid peroxidation (Figure 2B) in response to low-dose erastin treatment, and this process was blocked by ferrostatin-1 (Figure 2C). To understand the mechanism of Cu2+-mediated sensitization to ferroptosis, we first determined whether Cu2+ directly facilitates ferroptosis through the Fenton reaction in a Fe2+-like manner (Figure 2D). However, when free Fe2+ was removed by the specific Fe2+ chelator deferoxamine, the addition of Cu2+ to erastin failed to induce cell death (Figure 2E), suggesting that Cu2+ cannot replace Fe2+ to mediate Fenton reactions.
Figure 2.
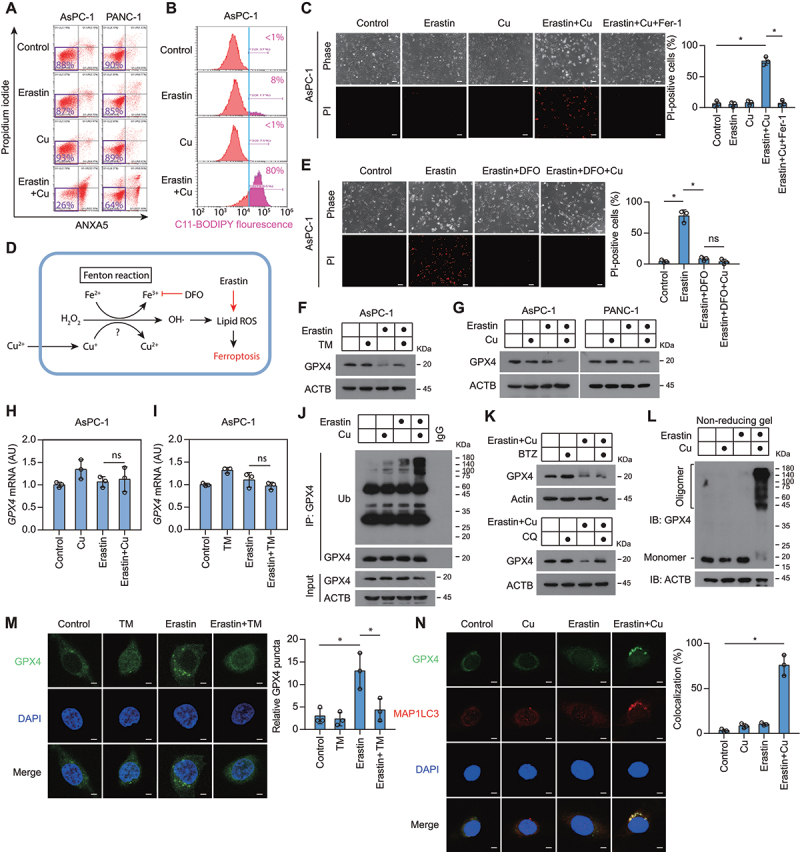
Copper amplifies ferroptosis by promoting autophagic degradation of GPX4. (A) Cell death of PDAC cells treated with low dose of erastin (1.25 μM for AsPC-1 cells; 2.5 μM for PANC-1 cells) in the presence or absence of copper sulfate (50 μM) for 24 h. (B) Lipid peroxidation of AsPC-1 cells treated with erastin (1.25 μM) in the presence or absence of copper sulfate (Cu, 50 μM) for 6 h. (C) Representative phase-contrast and propidium iodide (PI) staining images of AsPC-1 cells treated with erastin (1.25 μM), copper sulfate (Cu, 50 μM), or ferrostatin-1 (Fer-1, 10 μM) for 24 h. Scale bar: 50 µm. Quantification of PI-positive cells was shown. *P < 0.05. (D) Schematic diagram of the putative mechanism of copper-mediated Fenton reaction in ferroptosis. (E) Representative phase-contrast and propidium iodide (PI) staining images of AsPC-1 cells treated with erastin (1.25 μM), deferoxamine (DFO, 50 μM), or copper sulfate (Cu, 100 μM) for 24 h. Scale bar: 50 µm. Quantification of PI-positive cells was shown. *P < 0.05; ns, no significance. (F) Western blot of lysates from AsPC-1 cells treated with erastin (1.25 μM) in the presence or absence of TM (50 μM) for 12 h. (G) Western blot of lysates from PDAC cells treated with erastin (1.25 μM for AsPC-1 cells; 2.5 μM for PANC-1 cells) in the presence or absence of copper sulfate (50 μM) for 12 h. (H, I) GPX4 mRNA level in AsPC-1 cells treated with erastin (1.25 μM), copper sulfate (Cu, 50 μM), or TM (50 μΜ) for 12 h. (J) Endogenous GPX4 was immunoprecipitated from AsPC-1 cells treated with erastin (1.25 μM) in the presence or absence of copper sulfate (50 μM) for 6 h, followed by immunoblotting using the indicated antibody. Ub, ubiquitin. (K) Western blot of lysates from AsPC-1 cells treated with erastin (1.25 μM) plus copper sulfate (Cu, 50 μM) in the presence or absence of bortezomib (BTZ, 50 nM) or chloroquine (CQ, 20 μM) for 12 h. (L) Western blot under non-reducing conditions of lysates from AsPC-1 cells treated with erastin (1.25 μM) in the presence or absence of copper sulfate (50 μM) for 6 h. (M) Confocal imaging of GFP-GPX4 in AsPC-1 treated with erastin (1.25 μM), copper sulfate (Cu, 50 μM), or TM (50 μΜ), for 6 h. DAPI (blue) is used as a nuclear counterstain. Scale bar: 5 μm. Quantification of GFP-GPX4 puncta per cell was shown. *P < 0.05. (N) Colocalization of endogenous MAP1LC3A/B with GFP-GPX4 in AsPC-1 cells treated with erastin (1.25 μM) in the presence or absence of copper sulfate (50 μM) for 6 h. Scale bar: 5 μm. Quantification of colocalization of GXP4 and MAP1LC3A/B was shown. *P < 0.05.
Because the intracellular level of GPX4 is closely related to ferroptosis sensitivity [4], we then further examined the effect of Cu2+ on GPX4 protein expression. Western blotting indicated that TM prevented, while Cu2+ amplified, the erastin-induced downregulation of GPX4 protein (Figure 2Fand G). In parallel, the levels of GPX4 mRNA were not affected (Figure 2Hand I). Importantly, Cu2+ plus erastin (hereinafter referred to as Cu2+-erastin) induced the ubiquitination of GPX4 protein in AsPC-1 cells (Figure 2J), suggesting that copper may promote GPX4 protein degradation. Previous studies have shown that GPX4 protein degradation in ferroptosis can be mediated by proteasomal and autophagic pathways [27,28]. However, only pharmacological inhibition of autophagy (with chloroquine), not inhibition of proteasomes (with bortezomib), impeded the GPX4 protein degradation triggered by Cu2+-erastin (Figure 2K). Thus, autophagy may be involved in GPX4 protein degradation.
In response to Cu2+-erastin stimulation, GPX4 protein was detected in a high-molecular-weight complex, suggesting that copper induces GPX4 oligomerization (Figure 2L). To further test this hypothesis, we transfected GFP-tagged GPX4 plasmid into AsPC-1 cells. Immunofluorescence studies revealed that erastin induced GPX4 protein aggregation in the cytoplasm during ferroptosis, which was blocked by TM (Figure 2M). The colocalization of autophagosome marker MAP1LC3A/B (microtubule associated protein 1 light chain 3 alpha/beta)-II with GPX4 puncta was increased in response to Cu2+-erastin treatment (Figure 2N and Fig. S1), further supporting that GPX4 becomes a substrate of autophagy.
TAX1BP1 acts as an autophagy receptor for GPX4 degradation
To further explore the relationship between autophagy and copper-mediated ferroptosis, we measured autophagic flux in the presence or absence of chloroquine [29]. Western blot analysis of MAP1LC3A/B turnover and SQSTM1/p62 (sequestosome 1) degradation showed that copper treatment alone had little effect on autophagy, but the combination of copper and erastin significantly activated autophagy (Figure 3A and B). Furthermore, chloroquine inhibited, whereas the autophagy activator rapamycin enhanced, Cu2+-erastin-induced ferroptosis (Fig. S2A and S2B). The knockdown of ATG5 (autophagy related 5) also attenuated Cu2+-erastin-induced ferroptosis (Figure 3C and Fig. S3). These findings support the conclusion that copper can promote autophagy-dependent ferroptosis.
Figure 3.
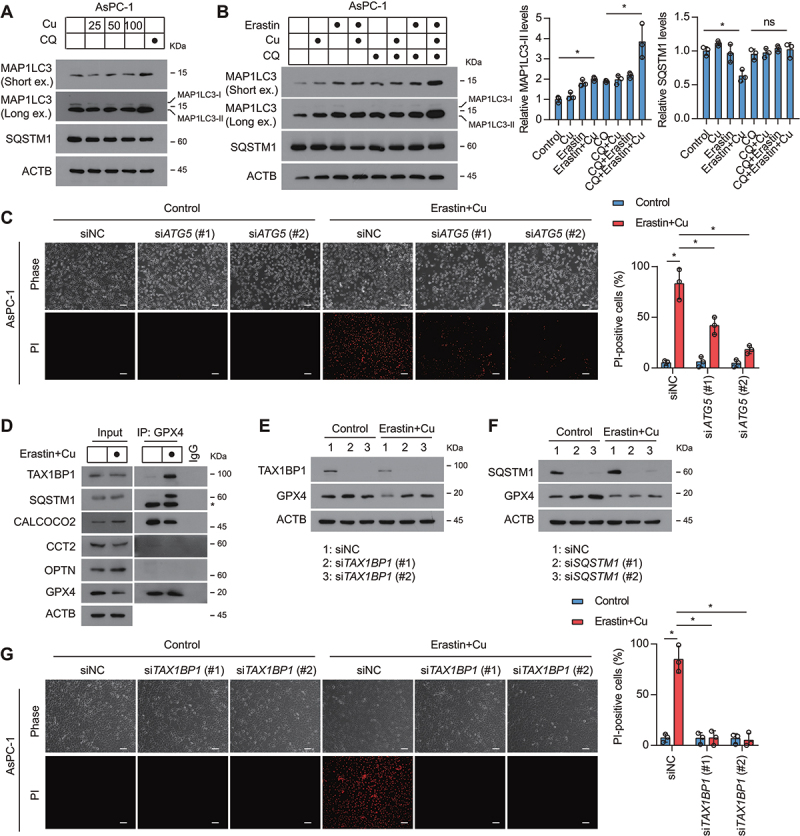
TAX1BP1 is essential for GPX4 degradation during ferroptosis. (A) Western blot analysis of the expression of indicated proteins in AsPC-1 cells after treatment with dose-dependent copper sulfate (Cu) for 12 h in the absence or presence of chloroquine (CQ, 20 μM). (B) Western blot analysis of the expression of indicated proteins in AsPC-1 cells after treatment with copper sulfate (Cu, 50 μM) and erastin (1.25 μM) for 12 h in the absence or presence of chloroquine (CQ, 20 μM). (C) Representative phase-contrast and propidium iodide (PI) staining images of indicated AsPC-1 cells treated with or without erastin (1.25 μM) plus copper sulfate (50 μM) for 24 h. (D) Endogenous GPX4 was immunoprecipitated from AsPC-1 cells treated with or without erastin (1.25 μM) plus copper sulfate (Cu, 50 μM) for 6 h, followed by immunoblotting using the indicated antibody. *, nonspecific band. (E, F) AsPC-1 cells were transfected with non-targeting scrambled siRNA (siNC), TAX1BP1 siRNA (siTAX1BP1), or SQSTM1 siRNA (siSQSTM1). Western blot of lysates from the indicated AsPC-1 cells treated with or without erastin (1.25 μM) plus copper sulfate (50 μM) for 12 h. (G) AsPC-1 cells were transfected with non-targeting scrambled siRNA (siNC) or TAX1BP1 siRNA (siTAX1BP1). Representative phase-contrast and propidium iodide (PI) staining images of the indicated AsPC-1 cells treated with or without erastin (1.25 μM) plus copper sulfate (Cu, 100 μM) for 24 h. Scale bar: 50 µm. Quantification of PI-positive cells was shown. *P < 0.05.
The recognition of particular cargos by phagophores requires different autophagy receptors, such as CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), CCT2 (chaperonin containing TCP1 subunit 2), OPTN (optineurin), SQSTM1, and TAX1BP1 [30–32]. To determine which autophagy receptor is involved in GPX4 protein degradation during Cu2+-erastin-induced ferroptosis, we performed immunoprecipitation experiments. In this assay, Cu2+-erastin selectively increased the binding of GPX4 to TAX1BP1 and SQSTM1, but not to CALCOCO2, CCT2, or OPTN (Figure 3D). Functionally, the knockdown of TAX1BP1, but not the knockdown of SQSTM1, inhibited Cu2+-erastin-induced GPX4 protein degradation (Figure 3E and F), indicating that TAX1BP1 acts as the key autophagy receptor for GPX4 protein degradation. Consistently, the knockdown of TAX1BP1 decreased cell sensitivity to ferroptosis (Figure 3G). These findings indicate that TAX1BP1-mediated GPX4 degradation is required for Cu2+-induced sensitization to ferroptosis.
Copper-induced GPX4 aggregation depends on the cysteine residues of GPX4
Heavy metal-induced ROS production is an important mechanism of cell killing. However, the ROS scavenger N-acetyl-L-cysteine failed to inhibit Cu2+-erastin-induced GPX4 aggregation, ruling out the possibility that Cu2+-mediated ROS generation would be involved in this phenomenon (Figure 4A). We next examined whether Cu2+ directly binds to GPX4 to induce GPX4 aggregation. To test this hypothesis, we performed fluorescent spectra-based studies of conformational changes affecting recombinant GPX4 protein. A noticeable spectral shift (from 328 nm to 334 nm) occurred under Cu2+ treatment, suggesting a direct interaction between copper and GPX4 (Figure 4B). In addition, Cu2+ promoted the oligomerization of recombinant GPX4 protein in vitro (Fig. S4), indicating that the binding of copper to GPX4 might promote GPX4 oligomerization.
Figure 4.
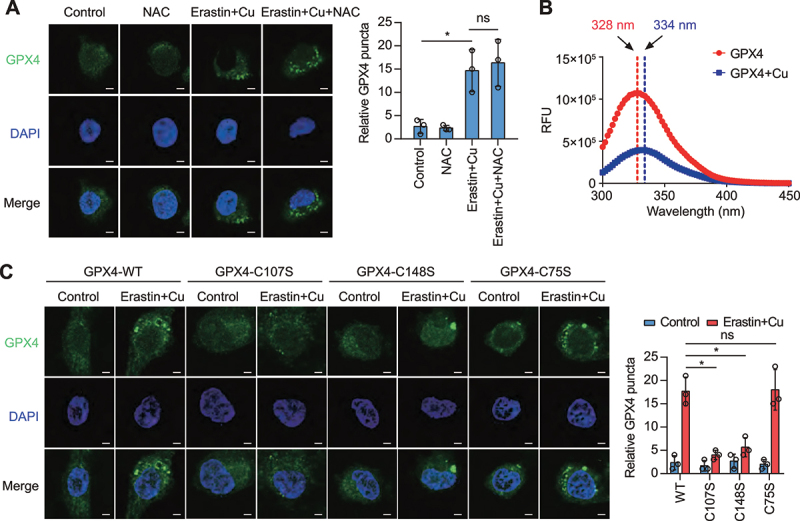
Copper promotes GPX4 aggregation during ferroptosis. (A) Confocal imaging of GFP-GPX4 in AsPC-1 cells treated with erastin (1.25 μM) plus copper sulfate (Cu, 50 μM) in the presence or absence of N-acetyl-L-cysteine (NAC, 1 mM) for 6 h. DAPI (blue) is used as a nuclear counterstain. Scale bar: 5 μm. Quantification of GFP-GPX4 puncta per cell was shown. *P < 0.05; ns, no significance. (B) Change in fluorescence spectra of recombinant protein GPX4 (1 mg/ml) with or without copper sulfate (50 μM), excited at 230 nm. Arrow indicates the maximum of fluorescence spectrum. (C) AsPC-1 cells were transfected with a plasmid expressing GFP-tagged WT or mutant GPX4. Confocal imaging of GFP puncta in the indicated AsPC-1 cells treated with erastin (1.25 μM) plus copper sulfate (Cu, 50 μM) for 6 h. Quantification of GFP-GPX4 puncta per cell was shown. *P < 0.05; ns, no significance.
Cu2+ can interact with highly reactive non-oxidized cysteine residues [33]. A prior study found two surface-accessible cysteine residues C107 and C148 in GPX4 [34]. Site-directed mutagenesis to replace these cysteine residues by serine residues revealed that both C107 and C148 were necessary for Cu2+-induced GPX4 aggregation to occur in response to Cu2+-erastin (Figure 4C). In contrast, mutations in C75 that is not surface-exposed, failed to affect the Cu2+-induced GPX4 protein aggregation (Figure 4C). These data suggest that Cu2+ selectively binds to cysteine residues C107 and C148 of GPX4.
Copper enhances the anticancer activity of IKE in vivo
We next examined whether Cu2+ enhances ferroptosis sensitivity in vivo. For this purpose, immunodeficient mice bearing human PDAC tumors were treated with imidazole ketone erastin (IKE), an analog of erastin with high metabolic stability, alone or together with copper. We randomized mice injected with PANC-1 cells into five groups: (i) vehicle; (ii) IKE; (iii) copper gluconate (an orally bio-available form of copper); (iv) IKE plus copper gluconate; and (v) IKE, copper gluconate plus the ferroptosis inhibitor liproxstatin-1. Copper enhanced the sensitivity of PANC-1 xenograft to IKE treatment, and this effect was inhibited by liproxstatin-1 (Figure 5A-C). The combination of copper gluconate and IKE resulted in a stronger suppression of GPX4 levels than single-agent treatments, as determined by immunohistochemical staining (Figure 5D). Additionally, the combination treatment augmented the tumor level of 4-hydroxy-2-noneal (4-HNE, the aldehydic product of lipid peroxidation), but not cleaved-CASP3 (caspase 3; one of the main drivers of apoptosis) (Figure 5D). However, copper failed to affect erastin-induced increases in TFRC (transferrin receptor) and Fe2+ level (Figure 5D and Fig. S5), suggesting that copper does not affect ferroptosis-related iron metabolism in tumors. Consistent with in vitro experiments, combination treatment of copper and IKE increased autophagy in xenograft tumors (Fig. S6). These results confirm that copper promotes the ferroptosis of cancer cells in vivo.
Figure 5.
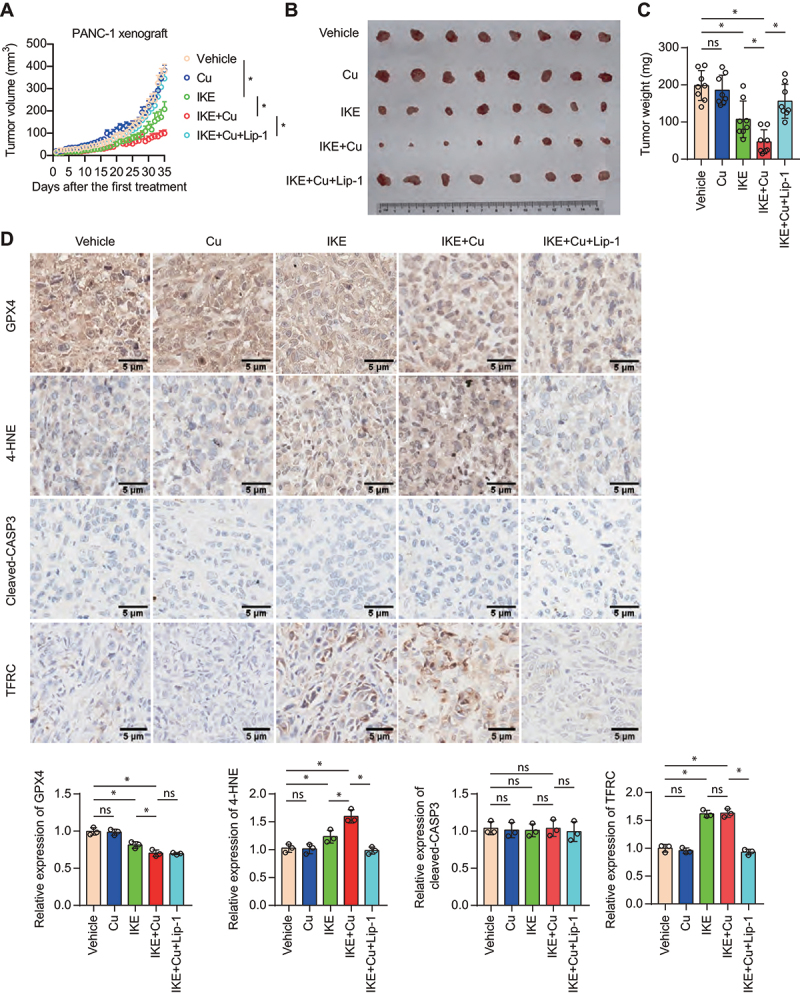
Copper promotes ferroptosis in nude mice bearing a PANC-1 xenograft. (A-C) Subcutaneous growth of PANC-1 xenografts in nude mice treated with IKE (30 mg/kg, i.p., once every other day), copper gluconate (Cu, 0.15 mg/kg, i.g., once every day), or liproxstatin-1 (Lip-1, 10 mg/kg, i.p., once every day). (A) Tumor growth curves of PANC-1 xenografts in nude mice treated with the indicated agents. *P < 0.05. Representative images of tumors (B) and tumor weight (C) were shown. Data are presented as mean ± SD, n = 8 mice per group; *P < 0.05; ns, no significance. (D) Immunohistochemical staining and relative staining intensity of the indicated proteins in the tumor sections. *P < 0.05; ns, no significance.
Copper chelator protects against ferroptosis-induced acute pancreatitis in mice
Because ferroptosis is a therapeutic target for sterile inflammation caused by tissue damage [35], we also evaluated the role of copper in acute pancreatitis, a disease that is closely related to excessive ferroptotic damage [36]. Of note, the copper chelator TM attenuated acinar cell death in L-arginine-induced acute pancreatitis in mice, as determined by histopathological examination (Figure 6A). Consistently, TM reduced various indicators of pancreatitis, including serum AMY (amylase) (Figure 6B), pancreatic MPO (myeloperoxidase) activity (Figure 6C), and tissue damage-induced release of HMGB1 (high mobility group box 1) into the circulation (Figure 6D). Pancreatic lipid peroxidation, TFRC expression, and inflammation-associated iron accumulation was decreased by TM (Figure 6E). Most importantly, the downregulation of pancreatic GPX4 expression was prevented by TM administration (Figure 6E). These data indicate that copper contributes to ferroptosis-induced acute pancreatitis.
Figure 6.
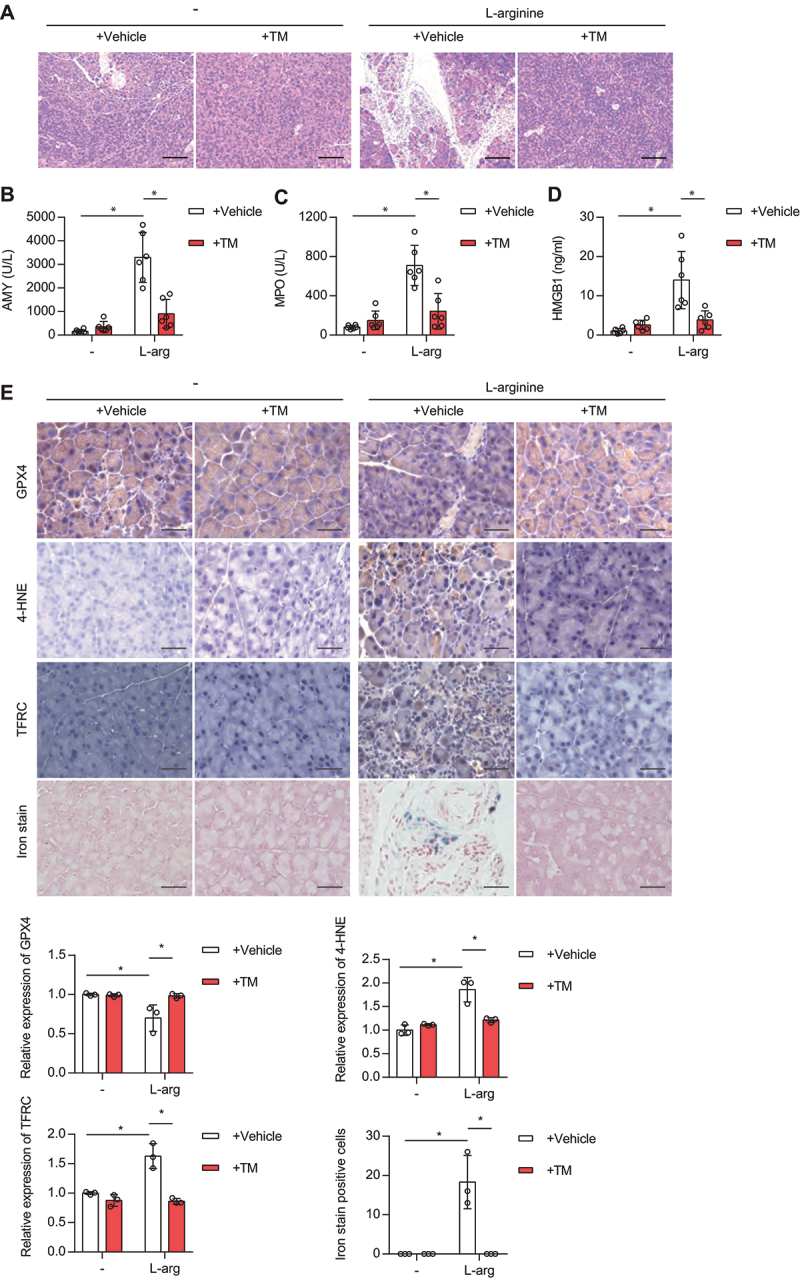
Copper chelator inhibits ferroptosis-associated acute pancreatitis in mice. (A) Representative images of pancreatic histology in L-arginine-induced pancreatitis in mice with or without TM treatment. Scale bar: 200 µm. (B-D) Serum AMY (B), pancreatic MPO activity (C), or serum HMGB1 (D) in control or L-arginine-induced-acute pancreatitis mice treated with or without TM (0.7 mg per mouse, i.g., once every day). Data are presented as mean ± SD; n = 6 mice/group; *P < 0.05 versus control group. (E) Representative immunohistochemical (IHC) staining of pancreatic tissues from control or acute pancreatitis mice treated with or without TM. In addition, iron content (blue) in tissues was detected using an iron staining kit. The relative expression of GPX4, 4-HNE, TFRC or iron was quantified from 3 mice. *P < 0.05.
Discussion
Autophagy is an evolutionarily conserved mechanism for the intracellular degradation of organelles and macromolecules [37]. In a highly context-dependent fashion, autophagy can mediate cytoprotection against various pathogenic agent, but may also facilitate cell death induction via the release of lysosomal hydrolases or the selective destruction of anti-cell death proteins [38]. Specifically, ferroptosis is considered a model for studying autophagy-dependent cell death [20,39–43]. For example, ferritinophagy, a type of selective autophagy, promotes iron accumulation and subsequent ferroptosis through receptor NCOA4 (nuclear receptor coactivator 4)-mediated ferritin degradation [44]. SQSTM1-mediated autophagic degradation of SLC40A1 (solute carrier family 40 member 1) blocks Fe2+ export, leading to intracellular Fe2+ accumulation and ferroptosis [45]. Some other types of selective autophagy, such as lipophagy and clockophagy, have also been reported to regulate ferroptosis by affecting lipid supply and cellular metabolism [46–48]. Here, we demonstrate that Cu2+ increases ferroptosis sensitivity by inducing TAX1BP1-mediated autophagic degradation of GPX4 independent from ROS generation. The knockdown of TAX1BP1 not only largely inhibits the Cu2+-induced downregulation of GPX4, but also limits ferroptosis sensitivity of PDAC cells. A previous study showed that the knockdown of TRIM11 (tripartite motif containing 11) enhances the expression of TAX1BP1, leading to increased autophagic signaling, including ferritinophagy, in PDAC cells [49]. In our current work, TAX1BP1-mediated ferroptosis may not be related to ferritinophagy, as copper chelators fail to reduce intracellular Fe2+ accumulation during ferroptosis. Rather, it appears plausible that TAX1BP1-mediated autophagic degradation of GPX4 protein reduces the ability to detoxify oxidized lipids, hence accounting for the copper-mediated sensitization to ferroptosis [50].
GPX4 is a key anti-ferroptotic enzyme that is highly regulated at multiple levels. NFE2L2/NRF2 (NFE2 like bZIP transcription factor 2) is a well-known transcription factor that protects cells against oxidative stress by inducing the expression of antioxidant genes, including GPX4 [51,52]. Beyond its transcriptional regulation, GPX4 expression can also be regulated at the protein level [53]. Heat shock proteins have been reported as key regulators of GPX4 protein degradation. For instance, HSP90AB1/HSP90 (heat shock protein 90 alpha family class B member 1) acts as a molecular chaperone to promote the degradation of GPX4 by chaperone-mediated autophagy, while HSPA5 (heat shock protein family A (Hsp70) member 5) protects against GPX4 protein degradation [28,54]. In addition to autophagy, the ubiquitin-proteasome pathway can mediate GPX4 protein degradation. Several anti-cancer compounds induce ferroptosis through the ubiquitination of GPX4 and its subsequent proteasomal degradation [7,27]. The E3 ubiquitin ligase TRIM46 (tripartite motif containing 46) contributes to high glucose-induced ferroptosis through interacting with GPX4 and promoting GPX4 degradation [55]. Here, we found yet another mechanism of GPX4 depletion that is triggered by copper. Copper acts as an inducer of TAX1BP1-dependent autophagic degradation of GPX4, thereby promoting lipid peroxidation for the induction of ferroptosis. It is unclear whether copper is involved in other regulated cell death by interfering with GPX4 expression [56–58].
Excessive or deficient ferroptotic response is associated with a variety of human diseases [4]. The induction of ferroptosis may be a goal of antineoplastic therapies, although sterile inflammation (e.g., pancreatitis) associated with ferroptosis can promote tumor growth [36,59–61]. Notably, some anticancer drugs (e.g., sorafenib, sulfasalazine, artesunate, and cisplatin) not only induce apoptosis, but also cause ferroptosis [62,63]. This fact highlights the challenge of developing specific ferroptosis inducers for clinical use. In addition, the assessment of copper-facilitated ferroptosis in the tumor microenvironment may contribute to the development of new tumor immunotherapy strategies [64]. Indeed, a recent study found that copper sulfide nanoparticles enhanced the sensitivity of cancer cells to immune checkpoint blockers (anti-CD274/PD-L1) in a mouse 4T1 breast tumor model [65].
In a tumor xenograft nude mouse model, we found that copper did not affect erastin-induced increases in TFRC and iron levels in tumor tissue. However, the copper chelator TM could inhibit TFRC upregulation and iron accumulation in a pancreatitis model. In addition to differences in animal models, these in vivo findings suggest that the inflammatory microenvironment may interfere with the effects of copper on iron accumulation.
In conclusion, we discovered a novel pro-ferroptotic effect of Cu2+ that involves the binding of Cu2+ to specific surface-exposed cysteine residues of the GPX4 protein and its subsequent TAX1BP1-dependent autophagic degradation (Figure 7). Our findings may provide potential therapeutic strategies for the amplification of the desirable ferroptosis of cancer cells by administering extra Cu2+, as well as for the suppression of tissue-damaging ferroptosis by Cu2+ chelation.
Figure 7.
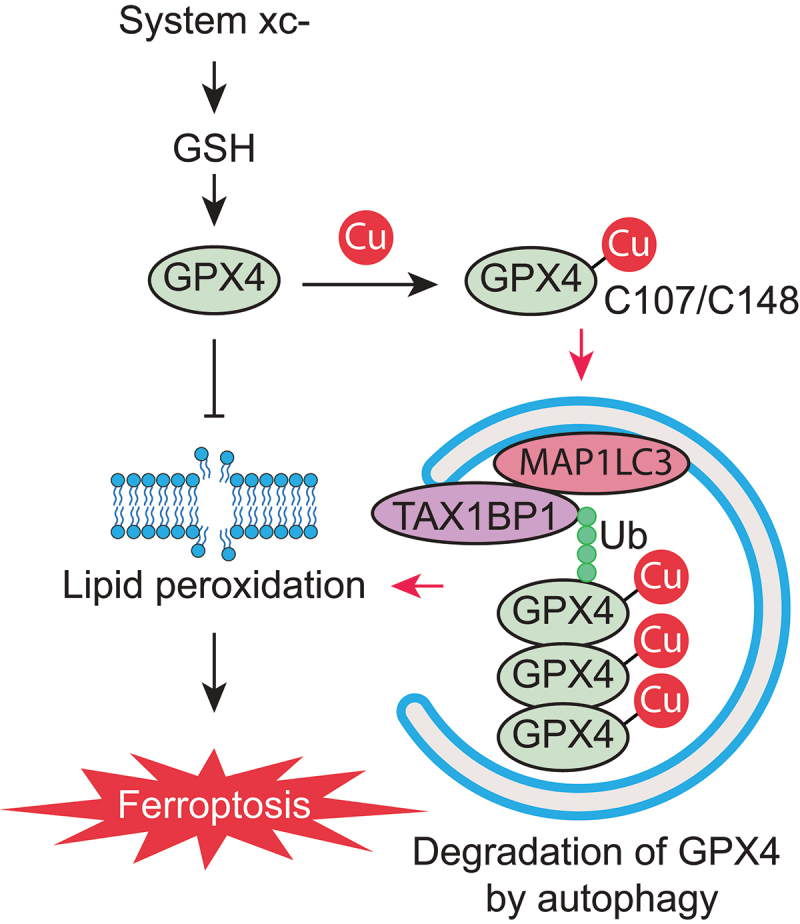
A schematic representation of copper-mediated autophagic GPX4 degradation to amplify lipid peroxidation during ferroptosis.
Materials and methods
Reagents
Cell culture and treatment
AsPC-1, PANC-1, MIA PaCa-2, and SW 1990 cell lines were obtained from the American Type Culture Collection (ATCC, CRL-1682; CRL-1469; CRM-CRL-1420; CRL-2172) and cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco, C11995500BT) or Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco, C11875500BT) with 10% fetal bovine serum (FBS) and penicillin and streptomycin (100 U/ml) at 37°C in an incubator containing 5% CO2. All cells were mycoplasma free and authenticated using short tandem repeat DNA profiling analysis. Dimethyl sulfoxide (DMSO) was used to prepare the stock solution of drugs. The final concentration of DMSO in the drug working solution in the cells was < 0.01%. DMSO of 0.01% was used as a vehicle control in all cell culture assays.
Cell viability assay
Cell viability was analyzed with a Cell Counting Kit-8 (CCK8; DOJINDO, CK04). Cells were seeded at 1 × 104 cells per well into 96-well plates and incubated with the indicated drugs. Subsequently, 100 μl of fresh medium was added to cells containing 10 μl of CCK-8 solutions and incubated for 1 h in an incubator of 5% CO2 at 37°C. Absorbance at 450 nm was measured using a microplate reader (Thermo Scientific, Varioskan Flash).
Lipid peroxidation assay
Lipid peroxidation was analyzed by BODIPY 581/591 C11 (Thermo Fisher Scientific, D3861) staining. Cells were seeded at 1 × 105 cells per well into 12-well plates and incubated with the indicated treatments in an incubator of 5% CO2 at 37°C. During the last 30 min of incubation, 1.5 μM BODIPY 581/591 C11 dyes were added. Then the cells were washed twice with Hanks’ balanced salt solution (HBSS; Macgene, cc025), resuspended in HBSS, and analyzed using a flow cytometer.
Cell death assay
Cell death was analyzed by ANXA5/annexin V-FITC-propidium iodide (PI) or PI staining. Cells were seeded at a density of 2 × 105 cells/well in 6-well plates. The next day, cells were incubated with the indicated treatments. For ANXA5-FITC-PI staining, cells were collected, washed with binding buffer, and then incubated in binding buffer with ANXA5-FITC and PI (KeyGEN BioTECH, KGA108) for 15 min in the dark. Dead cells were determined by flow cytometry. For PI staining, the cells were stained with PI for 30 min in an incubator of 5% CO2 at 37°C. The PI-positive cells were imaged by using a fluorescence microscope (ZEISS, Axio Observer 5).
RNA interference and gene transfection
The transfection of interfering RNA (siRNA) or cDNA was performed with Lipofectamine 3000 (Thermo Fisher Scientific, L3000-015) according to the manufacturer’s protocol. Commercial sources of siRNA and cDNA are listed in Table 1.
Table 1.
Reagent sources.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GPX4 | Abcam | ab125066 or ab16739 |
| ACTB/actin | Bioworld | BS6007M |
| 4-HNE | Abcam | ab48506 |
| Cleaved-CASP3 | Cell Signaling Technology | 9661 |
| Ubiquitin | Santa Cruz Biotechnology | sc-8017 |
| TAX1BP1 | Proteintech | 14,424-1-AP |
| SQSTM1/p62 | Abcam | ab56416 |
| TFRC | Abcam | ab137844 |
| CALCOCO2/NDP52 | Proteintech | 66,401-1-IG |
| CCT2 | Proteintech | 24,896-1-AP |
| OPTN | Proteintech | 60,293-1-IG |
| MAP1LC3A/B | Cell Signaling Technology | 4108 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tetrathiomolybdate (TM) | Sigma-Aldrich | 323,446 |
| Tetraethylenepentamine (TEPA) | Sigma-Aldrich | 375,683 |
| Copper sulfate | Sigma-Aldrich | 451,657 |
| Copper gluconate | MACKLIN | C832356 |
| Erastin | Selleck Chemicals | S7242 |
| RSL3 | Selleck Chemicals | S8155 |
| Sulfasalazine | Selleck Chemicals | S1576 |
| ML162 | Sigma-Aldrich | SML2561 |
| ML210 | Cayman Chemical | 23,282 |
| Imidazole ketone erastin (IKE) | Selleck Chemicals | S8877 |
| Ferrostatin-1 (Fer-1) | Selleck Chemicals | S7243 |
| Liproxstatin-1 (Lip-1) | Selleck Chemicals | S7699 |
| Necrostatin-1 (Nec-1) | Selleck Chemicals | S8251 |
| Z-VAD-FMK (Z-VAD) | Selleck Chemicals | S7023 |
| Deferoxamine (DFO) | Selleck Chemicals | S5685 |
| Chloroquine (CQ) | Selleck Chemicals | S6999 |
| Rapamycin | Selleck Chemicals | S1039 |
| Bortezomib (BTZ) | Selleck Chemicals | S1013 |
| N-Acetyl-L-cysteine (NAC) | Selleck Chemicals | S1623 |
| DPPH | Sigma-Aldrich | D9132 |
| 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonic acid monosodium salt hydrate (FerroZine Iron Reagent) | Sigma-Aldrich | 160,601 |
| Iron (II) chloride | Sigma-Aldrich | 372,870 |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 |
| Hoechst 33,342 | Thermo Fisher Scientific | 62,249 |
| DAPI | Abcam | ab104139 |
| Western blotting luminol reagent | Santa Cruz Biotechnology | sc-2048 |
| PBS | Thermo Fisher Scientific | AM9625 |
| HBSS | Macgene | cc025 |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000-015 |
| Cell lysis buffer | Beyotime | P0013 |
| Recombinant human GPX4 protein | Cayman Chemical | 26,906 |
| Critical Commercial Assays | ||
| Cell Counting Kit-8 (CCK8) solutions | DOJINDO | CK04 |
| BCA assay kit | Thermo Fisher Scientific | 23,225 |
| ANXA5/annexin V-Propidium iodide staining kit | KeyGEN BioTECH | KGA108 |
| BODIPY 581/591 C11 | Thermo Fisher Scientific | D3861 |
| Dynabeads™ antibody coupling kit | Thermo Fisher Scientific | 14311D |
| Iron Stain Kit | Thermo Fisher Scientific | 87,006 |
| Iron Assay Kit | Abcam | ab83326 |
| Amylase assay kit | Abcam | ab102523 |
| HMGB1 ELISA kit | Sino-Test Corporation | 326,070,442 |
| MPO ELISA kit | Thermo Fisher Scientific | EMMPO |
| FerroOrange | DOJINDO | F347 |
| Evo M-MLV RT kit with gDNA clean for qPCR kit | Accurate Biology | AG11705 |
| SYBR® Green Premix Pro Taq HS qPCR kit | Accurate Biology | AG11701 |
| Experimental Models: Cell Lines | ||
| AsPC-1 | ATCC | CRL-1682 |
| PANC-1 | ATCC | CRL-1469 |
| MIA PaCa-2 | ATCC | CRM-CRL-1420 |
| SW 1990 | ATCC | CRL-2172 |
| Oligonucleotides | ||
| Human TAX1BP1 siRNA#1: 5′-CAGTGATGCTGTCAACGTA-3′ |
RiboBio | This paper |
| Human TAX1BP1 siRNA#2: 5′-GGAACACGAACTAAGAAGA-3′ |
RiboBio | This paper |
| Human SQSTM1/p62 siRNA#1: 5′-GGAGTCGGATAACTGTTCA-3′ |
RiboBio | This paper |
| Human SQSTM1/p62 siRNA#2: 5′-TGAGGAAGATCGCCTTGGA-3′ |
RiboBio | This paper |
| Human ATG5 siRNA#1: 5′-CAACTTGTTTCACGCTATA-3′ |
RiboBio | This paper |
| Human ATG5 siRNA#2: 5′-TGACGTTGGTAACTGACAA-3′ |
RiboBio | This paper |
| Human GPX4 cDNA (with GFP) | Kidan Bio Co. Ltd | This paper |
| Human GPX4 C107S cDNA (with GFP) | Kidan Bio Co. Ltd | This paper |
| Human GPX4 C1148S cDNA (with GFP) | Kidan Bio Co. Ltd | This paper |
| Human GPX4 C75S cDNA (with GFP) | Kidan Bio Co. Ltd | This paper |
| Human RNA18S RNA primers: F: AACCCGTTGAACCCCATT R: CCATCCAATCGGTAGTAGCG |
Sigma-Aldrich | This paper |
| Human GPX4 primers: F: AGAGATCAAAGAGTTCGCCGC R: TCTTCATCCACTTCCACAGCG |
Sigma-Aldrich | This paper |
| Software | ||
| GraphPad Prism 8.4.3 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Western blot analysis
Cells or lysosomes were lysed three times with cell lysis buffer (Beyotime, P0013) containing protease inhibitor (Sigma-Aldrich, P8340) on ice for 10 min. Protein was quantified using a bicinchoninic acid (BCA; Thermo Fisher Scientific, 23,225) assay, and 20 to 40 μg of each sample was fractionated by 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in SDS running buffer and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5% nonfat dry milk in PBS (Thermo Fisher Scientific, AM9625) with Tween 20 (Sigma-Aldrich, SLCG3047; PBST) for 1 h and incubated with primary antibodies at 4°C overnight. Following three washes in PBST, membranes were incubated with goat anti-rabbit or anti-mouse immunoglobulin G (IgG) horseradish-peroxidase conjugated secondary antibody (Cell Signaling Technology, 7076S or 7074S) at room temperature for 1 h and washed with PBST. Chemiluminescence substrate was applied using western blotting luminol reagent (Santa Cruz Biotechnology, sc-2048).
Immunoprecipitation analysis
Cells were lysed at 4°C in ice-cold cell lysis buffer (Beyotime, P0013), and cell lysates were cleared by brief centrifugation (13,000 g, 15 min) [66]. Concentrations of proteins in the supernatant fraction were determined using the BCA assay. Before immunoprecipitation, samples containing equal amounts of proteins were precleared with Dynabeads (Thermo Fisher Scientific, 14311D) at 4°C for 3 h and subsequently incubated with various irrelevant IgG or specific antibodies (5 μg/ml) in the presence of Dynabeads for 2 h or overnight at 4°C with gentle shaking. Following incubation, Dynabeads were washed extensively with PBS, and proteins were eluted by boiling in loading buffer (Cell Signaling Technology, 56036S) before SDS-PAGE.
Immunofluorescence assay
The cells were fixed with 4% paraformaldehyde (Servicebio, G1101), permeabilized with 0.3% Triton X-100 (Solarbio, T8200), and incubated with primary antibodies in PBS with 1% bovine serum albumin (Sigma-Aldrich, 9048–46-8) overnight at 4°C, followed by washing with PBS three times and the application of secondary antibodies. Cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Abcam, ab104139) for 10 min. After washing with PBS three times, sections were protected with coverslips with an anti-fading mounting medium sealed with nail polish. Immunofluorescence images were acquired using a confocal laser scanning microscope (ZEISS LSM 980).
qPCR analysis
Total RNA was extracted and purified from cultured cells using the standard TRIZOL RNA extraction protocol (AG, AG21102). First-strand cDNA was synthesized from 1 µg of RNA using the Evo M-MLV RT Kit with gDNA Clean for qPCR Kit (Accurate Biology, AG11705). Briefly, 20-µl reactions were prepared by combining 10 µl of reaction mix, 1 µl of Evo M-MLV RTase Enzyme Mix, 1 µl of RT Primer Mix, 4 µl of 5 × RTase Reaction Buffer Mix I, and 4 µl of RNA-free water. The cDNA from various cell samples was then amplified by realtime qPCR with specific primers using the SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, AG11701). The gene expression was calculated via the 2−ΔΔCt method and normalized to RNA18S. The relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1. The primers, which were synthesized and desalted from Sigma-Aldrich, are shown in Table 1.
DPPH assay
2,2-Diphenyl-1-picrylhydrazyl (DPPH; Sigma-Aldrich, D9132) were dissolved in methanol to a final concentration of 100 μM. The tested compounds were added to 1 ml of DPPH solution with a final concentration of 10 μM and incubated at room temperature for 1 h [67]. The absorbance at 517 nm (indicating the concentration of nonreduced DPPH) was measured using a microplate reader (Thermo Scientific, Varioskan Flash). Results were normalized to control group (DMSO vehicle; set as 100%).
FerroZine iron chelation assay
Fe2+ chloride (Sigma-Aldrich, 372,870) was dissolved in water to a final concentration of 10 μM. The tested compounds were added to 1 ml of Fe2+ chloride solution with a final concentration of 40 μM and incubated at room temperature for 10 min. Next, FerroZine Iron Reagent (Sigma-Aldrich, 160,601) was added with a final concentration of 20 μM and incubated at room temperature for 1 h [67]. The absorbance at 562 nm was measured using a microplate reader (Thermo Scientific, Varioskan Flash). Results were normalized to DMSO control group (DMSO vehicle; set as 100%).
Iron assay
Intracellular Fe2+ level was analyzed by FerroOrange staining. Cells were seeded at 1 × 105 cells per well into 12-well plates and incubated with the indicated treatments in an incubator of 5% CO2 at 37°C. During the last 30 min of incubation, 1 μM FerroOrange (DOJINDO, F347) probes were added. Then the cells were washed twice with HBSS and resuspended in HBSS. A green laser (ex. 532 nm) was used to excite FerroOrange and the fluorescent images were captured by using fluorescence microscopy (ZEISS, Axio Observer 5).
ELISA analysis
The concentrations or activity of AMY, MPO, and HMGB1 in the indicated samples were measured using ELISA kits according to the manufacturer’s guidelines. Data were normalized to protein or DNA concentration.
Animal model
We conducted all animal care and experiments in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org) and with approval from our institutional animal care and use committee. Mice were kept under standard pathogen-free conditions with an artificial 12-h light/dark cycle and constant 50%-60% humidity. Mice were allowed access to tap water and free (ad libitum) access to standard laboratory chow during the experimental period.
To generate a mouse xenograft model, 2.5 × 106 PANC-1 cells were injected into the flanks of 5-week-old male nude Balb/c mice [68]. After 1 week, the mice were treated with vehicle, IKE (20 mg/kg, i.p., once every other day), copper gluconate (0.15 mg/kg, i.g., once every day), or liproxstatin-1 (10 mg/kg, i.p., once every day) for 35 days. The length and width of tumors were measured every day, and volumes were calculated using the formula: π/6 × length × width2. Tumor xenografts were removed, weighed, stored, and fixed.
For L-arginine-induced pancreatitis, a sterile solution of L-arginine monohydrochloride (8%; Sigma-Aldrich, A5131) was prepared in normal saline and the pH was adjusted to 7.0 [69]. Mice received intraperitoneal injections of L-arginine (3 g/kg) for 3 days, while controls were administered saline. TM (0.7 mg per mouse per day, oral gavage) was used for 1 week before the first L-arginine treatment. The parameters of acute pancreatitis were assessed 12 h after the last L-arginine treatment. Animals were sacrificed at the indicated time by CO2 asphyxia, and a blood sample and tissue were collected.
Immunohistochemistry
Formalin-fixed tissues were embedded in paraffin and sectioned. Tissues sections (4 μm) were immunostained using the MaxVision Kit (Maixin Biol, KIT-5020) according to the manufacturer’s instructions. The primary antibodies were used as indicated. In all, 50 μl MaxVision reagent was applied to each slide. Color was developed with 0.05% diaminobenzidine and 0.03% H2O2 in 50 mM Tris-HCl (pH 7.6), and the slides were counterstained with hematoxylin. A negative control for every antibody was also included for each xenograft specimen by substituting the primary antibody with pre-immune rabbit serum. The slides were scanned using a digital pathology slide scanner (Aperio CS2).
Statistical analysis
GraphPad Prism 8.4.3 was used to collect and analyze data. Data are presented as mean ± SD. A one-way or two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used for comparison among the different groups. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82272660, 81972399), the Natural Science Foundation Research Team of Guangdong Province (2018B030312001), the Basic and Applied Basic Research Project of Guangzhou Basic Research Program (202201011411), the Open Fund of Key Laboratory of Minimally Invasive Techniques and Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province (21SZDSYS17) and the National Institutes of Health (GM131919).
Funding Statement
This work was supported by the National Natural Science Foundation of China [82272660, 81972399], the Natural Science Foundation Research Team of Guangdong Province [2018B030312001], the Basic and Applied Basic Research Project of Guangzhou Basic Research Program [202201011411], the Open Fund of Key Laboratory of Minimally Invasive Techniques and Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province [21SZDSYS17] and the National Institutes of Health [GM131919].
Disclosure statement
The authors declare no potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2165323
References
- [1].Mizushima N, Levine B.. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang L, Chen X, Yang Q, et al. Broad Spectrum Deubiquitinase Inhibition Induces Both Apoptosis and Ferroptosis in Cancer Cells. Front Oncol. 2020;10:949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu Y, Wang Y, Liu J, et al. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2021;28(1–2):55–63. [DOI] [PubMed] [Google Scholar]
- [9].Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and Open Issues. Antioxid Redox Signal. 2018;29(1):61–74. [DOI] [PubMed] [Google Scholar]
- [10].Denoyer D, Masaldan S, La Fontaine S, et al. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics. 2015;7(11):1459–1476. [DOI] [PubMed] [Google Scholar]
- [11].Ge EJ, Bush AI, Casini A, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22(2):102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jiang Y, Huo Z, Qi X, et al. Copper-induced tumor cell death mechanisms and antitumor theragnostic applications of copper complexes. Nanomedicine (Lond). 2022;17(5):303–324. [DOI] [PubMed] [Google Scholar]
- [13].Chen X, Dou QP, Liu J, et al. Targeting ubiquitin-proteasome system with copper complexes for cancer therapy. Front Mol Biosci. 2021;8:649151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saporito-Magrina CM, Musacco-Sebio RN, Andrieux G, et al. Copper-induced cell death and the protective role of glutathione: the implication of impaired protein folding rather than oxidative stress. Metallomics. 2018;10(12):1743–1754. [DOI] [PubMed] [Google Scholar]
- [15].Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu J, Liu Y, Wang Y, et al. HMGB1 is a mediator of cuproptosis-related sterile inflammation. Front Cell Dev Biol. 2022;10:996307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen X, Yu C, Kang R, et al. Iron metabolism in Ferroptosis. Front Cell Dev Biol. 2020;8:590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen X, Zeh HJ, Kang R, et al. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. 2021;18(11):804–823. [DOI] [PubMed] [Google Scholar]
- [20].Liu J, Liu Y, Wang Y, et al. TMEM164 is anew determinant of autophagy-dependent ferroptosis. Autophagy. 2022. p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dai E, Han L, Liu J, et al. Autophagy-Dependent Ferroptosis Drives Tumor-Associated Macrophage Polarization via Release and Uptake of Oncogenic KRAS Protein. Autophagy. 2020;16:2069–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kuang F, Liu J, Xie Y, et al. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol. 2021;28(6):765–75 e5. [DOI] [PubMed] [Google Scholar]
- [23].Huang C, Santofimia-Castaño P, Liu X, et al. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner. Cell Death Discov. 2021;7(1):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nawrocki ST, Carew JS, Dunner K Jr., et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65(24):11510–11519. [DOI] [PubMed] [Google Scholar]
- [25].Xie Y, Zhu S, Zhong M, et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology. 2017;153(5):1429–43 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Song X, Zhu S, Xie Y, et al. JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology. 2018;154(5):1480–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ding Y, Chen X, Liu C, Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al . Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(8):2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. 4th. Autophagy. 2021;1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zellner S, Behrends C. Autophagosome content profiling reveals receptor-specific cargo candidates. Autophagy. 2021;17(5):1281–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang Z, Klionsky DJ. CCT2, a newly identified aggrephagy receptor in mammals, specifically mediates the autophagic clearance of solid protein aggregates. Autophagy. 2022;18(7):1483–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vargas JNS, Hamasaki M, Kawabata T, et al. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2022. DOI: 10.1038/s41580-022-00542-2 [DOI] [PubMed] [Google Scholar]
- [33].Harrison MD, Jones CE, Dameron CT. Copper chaperones: function, structure and copper-binding properties. J Biol Inorg Chem. 1999;4(2):145–153. [DOI] [PubMed] [Google Scholar]
- [34].Scheerer P, Borchert A, Krauss N, et al. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry. 2007;46(31):9041–9049. [DOI] [PubMed] [Google Scholar]
- [35].Chen X, Kang R, Kroemer G, et al. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6):e20210518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu K, Liu J, Zou B, et al. Trypsin-Mediated Sensitization to Ferroptosis Increases the Severity of Pancreatitis in Mice. Cell Mol Gastroenterol Hepatol. 2022;13(2):483–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. [DOI] [PubMed] [Google Scholar]
- [40].Chen X, Song X, Li J, et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. In: Autophagy. 2022. p. 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kang R, Tang D. Autophagy and ferroptosis - What’s the Connection? Curr Pathobiol Rep. 2017;5(2):153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu K, Huang J, Liu J, et al. Induction of autophagy-dependent ferroptosis to eliminate drug-tolerant human retinoblastoma cells. Cell Death Dis. 2022;13(6):521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. In: Autophagy. 2021;17(11):3361–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003. [DOI] [PubMed] [Google Scholar]
- [47].Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu J, Yang M, Kang R, et al. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15(11):2033–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shang M, Weng L, Xu G, et al. TRIM11 suppresses ferritinophagy and gemcitabine sensitivity through UBE2N/TAX1BP1 signaling in pancreatic ductal adenocarcinoma. J Cell Physiol. 2021;236(10):6868–6883. [DOI] [PubMed] [Google Scholar]
- [50].Sarraf SA, Shah HV, Kanfer G, et al. Loss of TAX1BP1-Directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. 2020;80(5):779–95 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dai C, Chen X, Li J, et al. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020;27(9):645–656. [DOI] [PubMed] [Google Scholar]
- [53].Han L, Bai L, Fang X, et al. SMG9 drives ferroptosis by directly inhibiting GPX4 degradation. Biochem Biophys Res Commun. 2021;567:92–98. [DOI] [PubMed] [Google Scholar]
- [54].Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77(8):2064–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang J, Qiu Q, Wang H, et al. TRIM46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating GPX4 ubiquitination. Exp Cell Res. 2021;407(2):112800. [DOI] [PubMed] [Google Scholar]
- [56].Kang R, Zeng L, Zhu S, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe. 2018;24(1):97–108 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu L, Liu B, Guan G, et al. Cyclophosphamide-induced GPX4 degradation triggers parthanatos by activating AIFM1. Biochem Biophys Res Commun. 2022;606:68–74. [DOI] [PubMed] [Google Scholar]
- [58].Canli Ö, Alankus YB, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu J, Zhu S, Zeng L, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. In: Autophagy. 2021. p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yang L, Ye F, Liu J, et al. Extracellular SQSTM1 exacerbates acute pancreatitis by activating autophagy-dependent ferroptosis. In: Autophagy. 2022. p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–296. [DOI] [PubMed] [Google Scholar]
- [63].Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang W, Green M, Choi JE, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang R, He Z, Cai P, et al. Surface-functionalized modified copper sulfide nanoparticles enhance checkpoint blockade tumor immunotherapy by photothermal therapy and antigen capturing. ACS Appl Mater Interfaces. 2019;11(15):13964–13972. [DOI] [PubMed] [Google Scholar]
- [66].Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chen X, Huang J, Yu C, et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun. 2022;13(1):6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liu J, Song X, Kuang F, et al. NUPR1 is a critical repressor of ferroptosis. Nat Commun. 2021;12(1):647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146(4):1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.