

ABSTRACT
Eukaryotic stress granules (SGs) are highly dynamic assemblies of untranslated mRNAs and proteins that form through liquid-liquid phase separation (LLPS) under cellular stress. SG formation and elimination process is a conserved cellular strategy to promote cell survival, although the precise regulation of this process is poorly understood. Here, we screened six E3 ubiquitin ligases present in SGs and identified TRIM21 (tripartite motif containing 21) as a central regulator of SG homeostasis that is highly enriched in SGs of cells under arsenite-induced oxidative stress. Knockdown of TRIM21 promotes SG formation whereas overexpression of TRIM21 inhibits the formation of physiological and pathological SGs associated with neurodegenerative diseases. TRIM21 catalyzes K63-linked ubiquitination of the SG core protein, G3BP1 (G3BP stress granule assembly factor 1), and G3BP1 ubiquitination can effectively inhibit LLPS, in vitro. Recent reports suggested the involvement of macroautophagy/autophagy, as a stress response pathway, in the regulation of SG homeostasis. We systematically investigated well-defined autophagy receptors and identified SQSTM1/p62 (sequestosome 1) and CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) as the primary receptors that directly interact with G3BP1 during arsenite-induced stress. Endogenous SQSTM1 and CALCOCO2 localize to the periphery of SGs under oxidative stress and mediate SG elimination, as single knockout of each receptor causes accumulation of physiological and pathological SGs. Collectively, our study broadens the understanding in the regulation of SG homeostasis by showing that TRIM21 and autophagy receptors modulate SG formation and elimination respectively, suggesting the possibility of clinical targeting of these molecules in therapeutic strategies for neurodegenerative diseases.
Abbreviations: ACTB: actin beta; ALS: amyotrophic lateral sclerosis; BafA1: bafilomycin A1; BECN1: beclin 1; C9orf72: C9orf72-SMCR8 complex subunit; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; Co-IP: co-immunoprecipitation; DAPI: 4’,6-diamidino-2-phenylindole; FTD: frontotemporal dementia; FUS: FUS RNA binding protein; G3BP1: G3BP stress granule assembly factor 1; GFP: green fluorescent protein; LLPS: liquid-liquid phase separation; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; NBR1: NBR1 autophagy cargo receptor; NES: nuclear export signal; OPTN: optineurin; RFP: red fluorescent protein; SQSTM1/p62: sequestosome 1; SG: stress granule; TAX1BP1: Tax1 binding protein 1; TOLLIP: toll interacting protein; TRIM21: tripartite motif containing 21; TRIM56: tripartite motif containing 56; UB: ubiquitin; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type.
KEYWORDS: Autophagy receptor, CALCOCO2, G3BP1, SQSTM1, stress granule, TRIM21, ubiquitination
Introduction
Stress granules (SGs) are membraneless condensates of RNA and RNA-binding proteins that form in response to cellular stress [1]. Emerging evidence of a link between SGs and a range of neurodegenerative diseases has recently highlighted their physiological importance [2–4]. Earlier studies have shown that the key components of SGs include translation-related proteins, RNA binding proteins, and mRNAs [5,6], while more recent proteomics analyses suggest that the range of proteins in SGs is far more diverse than originally thought [7–9]. In particular, the prevalence of several quality control proteins suggests stringent monitoring of SGs. Recent findings have revealed that while the majority of these so-called physiological SGs (i.e., occurring in healthy cells) disassemble after stress is removed, a substantial proportion of SGs are eliminated by autophagy under prolonged exposure to stress conditions [10–12]. Several pathogenic protein variants, such as C9orf72 (C9orf72-SMCR8 complex subunit) and FUS (FUS RNA binding protein), that are genetically linked to Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) have been found to persist within cytoplasmic foci, forming irreversible clusters, referred to as pathological SGs [13–16]. The accumulation of these relatively solid RNA-protein condensates can cause gain-of-function toxicity that is highly linked to ALS-FTD and several studies have suggested that pathological SGs are also cleared by autophagy [17–22]. In addition, ubiquitination, as well as associated quality control proteins, has been shown to contribute to the recovery of cellular activities in response to heat shock stress [12,23], although it remains poorly understood how quality control proteins can precisely manage SG homeostasis.
Autophagy is a lysosomal degradation process responsible for the removal of protein aggregates, nonfunctional organelles, and intracellular pathogens [24–26]. Autophagy is highly conserved throughout eukaryotes, and is initiated by newly formed membranes (i.e., phagophores) that engulf targeted cargos, resulting in the formation of autophagosomes that fuse with the lysosome to deliver their cargo for degradation [27]. Phagophore-conjugated MAP1LC3 (microtubule associated protein 1 light chain 3) mediates recognition and binding to cargo receptors, and one or more specific autophagy receptors are required to initiate selective autophagy. There are currently six known cargo receptors, including SQSTM1, CALCOCO2, OPTN (optineurin), NBR1 (NBR1 autophagy cargo receptor), TOLLIP (toll interacting protein), and TAX1BP1 (Tax1 binding protein 1), that concurrently bind to specific cargos while recognizing MAP1LC3 through its “LC3-interacting region” [24,28,29]. Dysfunction of autophagy has been linked to several neurodegenerative diseases including ALS-FTD [17,18,22,30,31], and mutations in SQSTM1 and OPTN have been identified as causative factors of ALS-FTD [32–34]. However, the mechanisms by which autophagic dysfunction is linked to pathological SGs and ALS-FTD are poorly understood.
Tripartite motif (TRIM) proteins represent a large family of RING E3 ubiquitin ligases, many of which function in cellular processes such as protein quality control, autophagy, and innate immunity [35–37]. TRIMs have been shown to function as autophagy receptors and modulate selective autophagy [38–42]. Other studies have proposed that TRIMs can form platforms, or “TRIMosomes”, that facilitate coordination of core regulators of autophagosome formation, including ULK1 (unc-51 like autophagy activating kinase 1), BECN1 (beclin 1), SQSTM1 and Atg8 family members [38]. TRIMs can also act as specific receptors that target cargos for autophagic degradation [38]. In particular, TRIM21 was shown to induce autophagy and participate in the ubiquitin-mediated autophagic degradation of several innate immune responsive effectors such as IRF3 (interferon regulatory factor 3) under IFNG/IFN-γ (interferon gamma) treatment and IKBKB/IKK-β (inhibitor of nuclear factor kappa B kinase subunit beta) during virus infection [39,43]. TRIM21 was also reported to promote osteosarcoma cell autophagy [44]. However, no studies have investigated whether TRIM21 contributes to regulating stress granules.
In this study, we screened six E3 ligases present in SGs [7] (and unpublished data) and identified TRIM21 as a mediator of SG homeostasis. TRIM21 co-assembles with SGs under oxidative stress. TRIM21 knockdown decreases ubiquitination of G3BP1 and promotes SG formation whereas TRIM21 overexpression results in the ubiquitination of G3BP1 and inhibition of SG formation. In vitro assays showed that K63-linked ubiquitination of G3BP1 catalyzed by TRIM21 inhibits LLPS. We also found that TRIM21 overexpression inhibits the formation of FUS mutant protein and poly(GR) generated from the C9orf72 repeat expansion into pathological SGs. Furthermore, our results indicate that G3BP1 interacts with the autophagy proteins SQSTM1 and CALCOCO2, which serve as the major receptors for SG elimination. Taken together, our results show that SG homeostasis is modulated by TRIM21 and autophagy receptors which mediate SG formation and elimination respectively.
Results
TRIM21 is colocalized with SGs under oxidative stress and its overexpression inhibits stress granule formation
Previous reports have identified the presence of multiple E3 ligases in stress granules (SGs), including TRIM21, TRIM25 (tripartite motif containing 25), TRIM56 (tripartite motif containing 56), DTX3L (deltex E3 ubiquitin ligase 3L), NOSIP (nitric oxide synthase interacting protein) [7], as well as TRIM47 (tripartite motif containing 47) (unpublished data). The presence of these E3 ligases in SGs suggests a quality control system for protein translation and RNA homeostasis under stress. To investigate how these ligases participate in modulating SG dynamics, we generated FLAG-tagged constructs of all six E3 ligases and individually expressed each in U-2 OS cells subjected to arsenite-induced oxidative stress. Immunofluorescence staining of the SG component protein G3BP1, a marker for SG formation, showed that overexpression of TRIM25, TRIM47, DTX3L, or NOSIP had negligible or mild effects on SG formation under arsenite treatment compared to wild-type (WT) cells transfected with empty vector. By contrast, overexpression of TRIM21 or TRIM56 resulted in a remarkable decrease in SG number (Fig. S1A, quantified in S1B), suggesting that TRIM21 and TRIM56 may inhibit SG formation. Since TRIM21 overexpression led to a greater inhibitory effect, we focused on TRIM21 in subsequent analyses.
We first validated that the endogenous TRIM21 was localized to SGs under oxidative stress by immunofluorescence staining in U-2 OS cells (Figure 1A, quantified in 1B). To obtain more details for the localization pattern of TRIM21 and G3BP1, we resolved the distribution pattern of these two molecules in SGs, using the super-resolution structured illumination microscopy (SIM), and the results indicated that TRIM21 was non-uniformly intercalated into the G3BP1 clusters (Figure 1C). As a TRIM family protein, TRIM21 carries four characteristic domains, including an N-terminal RING domain; a B-box domain; a coiled-coil segment; and a C-terminal SPRY domain (Figure 1D). In TRIM21-overexpressing cells, we noted that TRIM21 itself formed fibers or aggregates in about 68% of the examined cells (Fig. S1C-D), in addition to its inhibition of SG formation, which was in line with a previous study that showed TRIMs can form homo- or hetero-oligomers through the coiled-coil domain [45]. To determine whether the coiled-coil domain was indeed responsible for this fiber formation, we exposed cells overexpressing TRIM21 or a TRIM21 variant with deletion for the coiled-coil domain (TRIM21ΔCC) to the cross-linker disuccinimidyl suberate (DSS). The results showed that overexpression of TRIM21, but not TRIM21ΔCC, resulted in oligomer formation, thus verifying the role of the coiled-coil domain in this phenomenon (Fig. S1E).
Figure 1.
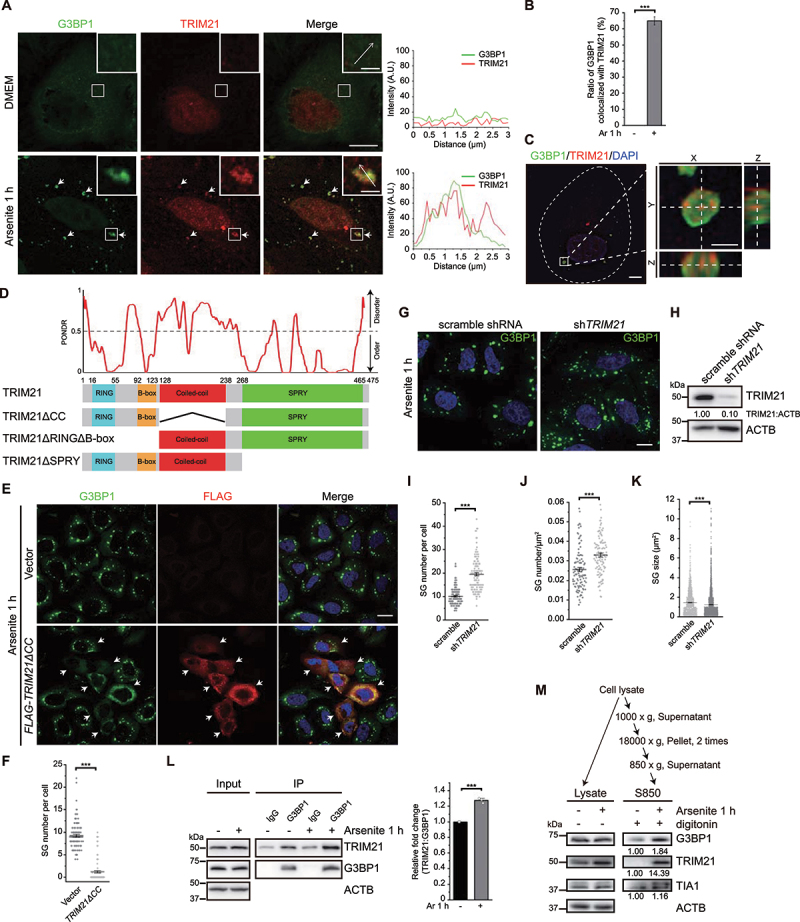
TRIM21 is localized to SGs under oxidative stress and its overexpression inhibits SG formation. (A) TRIM21 is recruited to SGs under oxidative stress. U-2 OS cells treated without or with arsenite were stained with TRIM21 (red) and G3BP1 (green). The arrows indicate colocalization of TRIM21 and G3BP1. Line graphs in the right panels show TRIM21 and G3BP1 signals along the indicated arrows in the insets. Scale bar: 10 μm. Inset scale bar: 2 μm. (B) Quantification of the percentage of SGs colocalized with TRIM21 from (A). Data are presented as means ± SEMs from three independent experiments with 100 cells counted. ***p < 0.001 (Student’s t-test). (C) Left, Z-stack projection of the representative image of TRIM21 and G3BP1 signals in U-2 OS cells under oxidative stress acquired by SIM. Cells were stained with G3BP1 (green), TRIM21 (red) and nuclei (DAPI, blue). Scale bar: 5 μm. Right, magnified orthogonal sectioning view of regions in the boxes; scale bar: 1 μm. (D) TRIM21 domain structures are shown and the TRIM21 truncations are made based on domain analysis: RING domain (16–55 aa), B-box domain (92–123 aa), coiled-coil (CC) domain (128–238 aa) and SPRY domain (268–465 aa). The level of structural disorder was predicted using the PONDR (predictor of natural disordered regions) VLXT algorithm (http://www.pondr.com/). (E) TRIM21ΔCC overexpression inhibits SG formation. U-2 OS cells were transfected with the empty vector or FLAG-TRIM21ΔCC and treated with arsenite before fixation. Cells were then stained with G3BP1 (green), FLAG (red) and nuclei (DAPI, blue). Scale bar: 20 μm. (F) Quantification of SG number per cell from (E). Data were pooled from three independent experiments with 120 cells counted. Error bars indicate SEM. ***p < 0.001 (Student’s t-test). (G) Knockdown of TRIM21 promotes the accumulation of SGs. A549 cells were infected with lentivirus-control shRNA or lentivirus-sh-TRIM21. Cells were then treated with arsenite before fixation. Cells were then stained with G3BP1 (green) and nuclei (DAPI, blue). Scale bar: 10 μm. (H) The knockdown efficiency of TRIM21. Quantifications of TRIM21 levels (normalized by ACTB) are shown. (I, J, K) Quantification of SG number per cell (I), SG number per cell area (J) and SG size (K) from (G). Data were pooled from three independent experiments with 120 cells counted. Error bars indicate SEM. ***p < 0.001 (Student’s t-test). (L) Immunoprecipitation analysis of the interaction between G3BP1 and TRIM21 in HEK293FT cells without or with arsenite treatment. Anti-G3BP1 antibody immunoprecipitated TRIM21 under normal and stress conditions. Relative fold change of immunoprecipitated TRIM21 is presented as mean of three replicate experiments ± SEM. ***p < 0.001 (Student’s t-test). (M) TRIM21 is enriched in SG fraction under oxidative stress. U-2 OS cells were treated without or with arsenite and SG fraction (S850) was extracted by serial centrifugations. The samples were analyzed by immunoblotting with the indicated antibodies. ACTB was used as the loading control. Protein levels of G3BP1, TRIM21 and TIA1 were quantified.
To eliminate potential artifacts caused by overexpression in phenotypic analyses of SGs, we therefore utilized TRIM21ΔCC instead of the full-length TRIM21. Cells transfected with FLAG-TRIM21ΔCC exhibited obvious inhibition of SG formation in immunofluorescence staining assays for FLAG and G3BP1, without detectable TRIM21 aggregate accumulation (Figure 1E, quantified in 1F). Immunofluorescence-based quantification and immunoblotting of total G3BP1 and TRIM21 levels in TRIM21ΔCC-overexpressing cells showed that overexpression of TRIM21ΔCC does not lead to changes in global G3BP1 levels under both normal and stress conditions (Fig. S1F, S2A-C). Furthermore, there is no obvious dose-dependence between TRIM21ΔCC expression level and SG number under our experimental conditions (Fig. S1G). Double staining of SGs with G3BP1 and an additional SG marker EIF3A revealed that both markers were always colocalized in SGs and concurrently decreased when EGFP-TRIM21ΔCC was overexpressed under arsenite treatment, indicating that rather than only the presence of G3BP1, SGs are reduced as a whole (Fig. S2D).
TRIM21 co-assembles with SGs under oxidative stress and its knockdown promotes SG formation
To further assess the role of TRIM21 in SG regulation at endogenous level, we stably suppressed the expression of TRIM21 in cells using lentivirus-mediated shRNA and this led to a significant increase in SG number (Figure 1G and 1H, quantified in 1I) and SG number per cell area (Figure 1J), but with a notable decrease in SG size (Figure 1K), as compared to the control cells under arsenite treatment. However, the ratio of cells with detectable SGs remains similar (~ 65%) in both TRIM21 knockdown and control cells under stress condition (Fig. S2E).
Next, we sort to determine which stage of the SG formation and dissolution process is affected by TRIM21. To this end, SG number in WT cells were compared with that in either TRIM21ΔCC-overexpressing cells or TRIM21 knockdown cells, at 4 time points under stress (0, 15, 30 and 60 min) and 2 time points during the recovery stage (60 and 90 min in TRIM21-overexpressing cells, or 15 and 30 min in TRIM21 knockdown cells). In TRIM21ΔCC-overexpressing cells, we observed marked decrease in SG number starting from 30 min of stress, as compared to the control cells (Fig. S2F, quantified in S2G). Whereas in TRIM21 knockdown cells, there was a significant increase in SG number at 60 min of stress, as compared to the control cells (Fig. S2H, quantified in S2I). Collectively, these results support that TRIM21 functions during the formation of SGs.
To then explore the mechanism underlying TRIM21 inhibition of SG formation, we first examined whether TRIM21 could interact with G3BP1, an essential core structural component of SGs, under arsenite treatment. To this end, we conducted co-immunoprecipitation (Co-IP) assays in HEK293 FT cells using anti-G3BP1 antibody. Western blots showed an obvious increase in interactions between G3BP1 and TRIM21 upon exposure to arsenite (Figure 1L). These data suggested that TRIM21 interacts with G3BP1, and hence SGs, under arsenite-induced oxidative stress, implying a role for TRIM21 in SG dynamics.
We then investigated how endogenous TRIM21 functions in response to arsenite treatment through SG fractionation experiments. U-2 OS cells were first treated with digitonin to release soluble cytosolic protein and the SG fraction was further enriched using a previously described method [46]. Western blots showed that the protein levels of the SG markers G3BP1 and TIA1 were enriched in the SG fraction of arsenite treated cells (Figure 1M). Notably, TRIM21 was highly accumulated in the SG fraction in response to arsenite treatment (Figure 1M). These findings suggested that TRIM21 could potentially affect SG dynamics through interaction with G3BP1 under oxidative stress conditions.
TRIM21 modifies G3BP1 with K63-linked ubiquitin chains to inhibit stress granule formation
In light of our above data, we next sought to determine whether TRIM21 could modulate SG formation through ubiquitin modification of granule components, based on its functional annotation as an E3 ligase. As a core SG structural protein, the depletion of G3BP1, along with its paralog G3BP2, abolishes SG accumulation in response to arsenite [47]. Therefore, we asked whether G3BP1 could serve as a substrate of TRIM21. In Co-IP assays using U-2 OS cells stably expressing EGFP-G3BP1, the G3BP1-conjugated ubiquitin signal trended higher in cells that also overexpressed TRIM21ΔCC treated with arsenite compared to that in cells without arsenite exposure (Fig. S3A). Furthermore, the conversion of three amino acids at the TRIM21ΔCC active site to disrupt ubiquitin ligase activity (i.e., TRIM21ΔCCC16A,C31A,H33W, or TRIM21ΔCC-LD hereafter) [48,49], abolished the signal for ubiquitin-conjugated G3BP1, suggesting that TRIM21 is required for G3BP1 ubiquitination (Fig. S3B). In line with these results, we observed ubiquitination of endogenous G3BP1 under oxidative stress, which is dependent on TRIM21, since overexpression of TRIM21ΔCC promoted and knockdown of TRIM21 inhibited ubiquitination of endogenous G3PB1 upon arsenite treatment (Figure 2A-B). In addition, G3BP1 appeared to be conjugated with K63-linked ubiquitin chains, since the expression of UBK63R, but not UBK48R, reduced the G3BP1-UB signal (Figure 2C). Interestingly, G3BP1 protein levels did not obviously change despite its ubiquitination by TRIM21ΔCC, implying that TRIM21 does not target G3BP1 for degradation. These results were also consistent with reports that showed G3BP1 undergoes K63-linked ubiquitination upon heat shock to weaken the stress granule–specific interaction network and induce SG disassembly [12].
Figure 2.
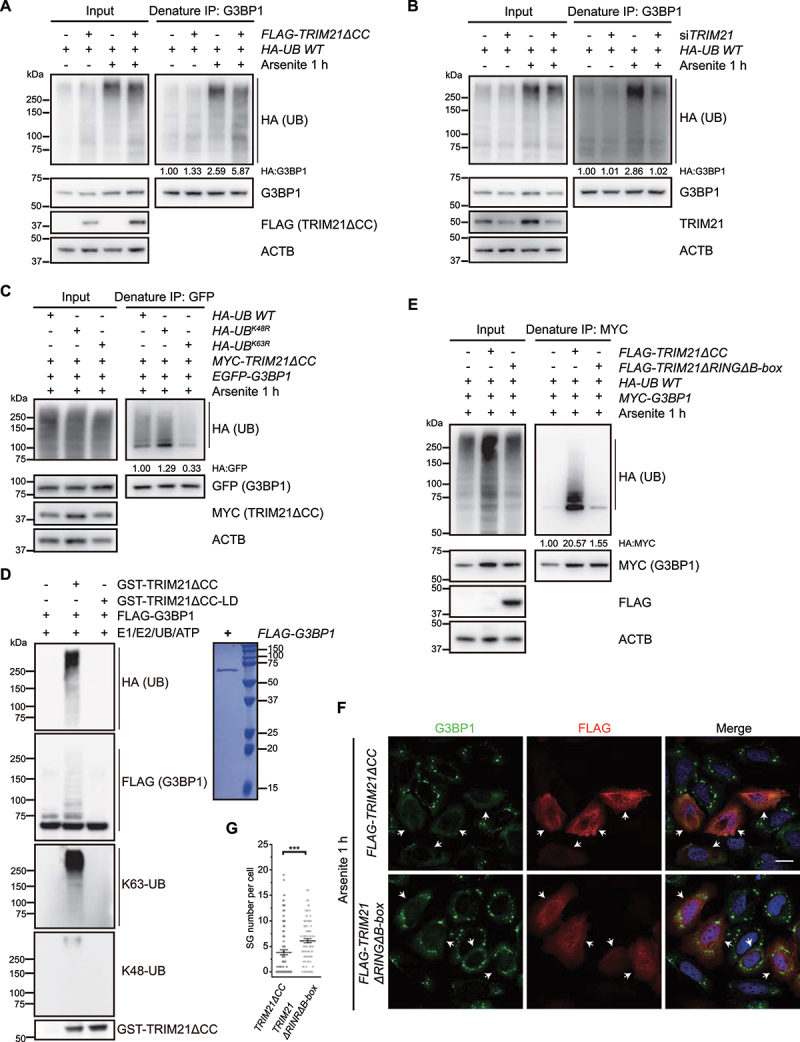
TRIM21 catalyzes K63-linked ubiquitination of G3BP1 to inhibit SG formation. (A) G3BP1 is ubiquitinated by TRIM21ΔCC. HEK293FT cells were transfected with the empty vector or FLAG-TRIM21ΔCC, along with HA-UB, and the cells were treated without or with arsenite. Proteins were immunoprecipitated with G3BP1 antibody under denaturing conditions and immunoblotted with the indicated antibodies. Ubiquitination levels (normalized by G3BP1 levels) were quantified (n = 2). (B) G3BP1 ubiquitination level is decreased in TRIM21 knockdown cells. HEK293FT cells were transfected with the scramble or TRIM21 siRNA, along with HA-UB, and the cells were treated without or with arsenite. Proteins were immunoprecipitated with G3BP1 antibody under denaturing conditions and immunoblotted with the indicated antibodies. Ubiquitination levels (normalized by G3BP1 levels) were quantified (n = 2). (C) G3BP1 is ubiquitinated by TRIM21 via K63-linked polyubiquitin chain. HEK293FT cells were transfected with EGFP-G3BP1 and MYC-TRIM21ΔCC, along with HA-UB, HA-UBK48R or HA-UBK63R, and cells were treated with arsenite. Ubiquitinated G3BP1 were immunoprecipitated with GFP antibody under denaturing conditions. Ubiquitination levels (normalized by GFP-G3BP1 levels) were quantified (n = 3). (D) In vitro ubiquitination of G3BP1 by TRIM21ΔCC shows K63-linked polyubiquitin chain. Mammalian purified FLAG-G3BP1 was incubated with GST-E1, His-UBE2D2 and HA-UB, together without or with GST-TRIM21ΔCC or GST-TRIM21ΔCC-LD, for 1 h at 37°C. The reaction samples were immunoblotted with the antibodies against HA, FLAG, GST, K63-linked and K48-linked ubiquitin. The Coomassie staining of the purified FLAG-G3BP1 are shown in the right. (E) TRIM21ΔRINGΔB-box is unable to form ubiquitin chains on G3BP1. HEK293FT cells were transfected with MYC-G3BP1 and HA-UB, along with the empty vector, FLAG-TRIM2ΔCC, or FLAG-TRIM21ΔRINGΔB-box, and cells were treated with arsenite. Ubiquitinated G3BP1 were immunoprecipitated with anti-MYC beads under denaturing conditions. Ubiquitination levels (normalized by MYC-G3BP1 levels) were quantified (n = 3). (F) TRIM21ΔRINGΔB-box is unable to inhibit stress granule formation. U-2 OS cells were transfected with FLAG-TRIM21ΔCC or FLAG-TRIM21ΔRINGΔB-box and cells were treated with arsenite. Cells were fixed and stained with G3BP1 (green), FLAG (red) and nuclei (DAPI, blue). Scale bar: 20 μm. (G) Quantification of SG number per cell from (F). Data were pooled from three independent experiments with 120 cells counted. Error bars indicate SEM. ***p< 0.001 (Student’s t-test).
Ubiquitination of G3BP1 by TRIM21 was further verified by in vitro ubiquitination assays in which purified mammalian FLAG-G3BP1 was incubated with E1, UBE2D2 (ubiquitin conjugating enzyme E2 D2), ubiquitin and ATP. The results indicated that ubiquitinated FLAG-G3BP1 accumulation markedly increased in the presence of GST-TRIM21ΔCC (Figure 2D). Moreover, the ubiquitin signal could only be detected by anti-K63-, but not anti-K48-linkage specific ubiquitin antibody, further supporting the formation of K63-linked ubiquitin chains on G3BP1, while the ligase dead mutant GST-TRIM21ΔCC-LD was unable to form ubiquitin chains on G3BP1 (Figure 2D).
These above data led us to propose that TRIM21 may inhibit SG formation through ubiquitination of G3BP1. In agreement with this potential function, we found that a RING domain and B-box deletion variant of TRIM21 (TRIM21ΔRINGΔB-box) could still bind to (Fig. S3C) but not ubiquitinate G3BP1 (Figure 2E), and overexpression of this variant in cells could no longer inhibit SG formation under oxidative stress condition (Figure 2F, quantified in 2G). This evidence further supported the ubiquitination of G3BP1 by TRIM21 via K63 linkage, likely inhibiting SG formation.
To better understand the mechanism of SG inhibition via TRIM21 ubiquitination of G3BP1, we performed in vitro LLPS assays. We first obtained high concentrations of FLAG-G3BP1 and ubiquitinated FLAG-G3BP1 (FLAG-G3BP1-UB) from mammalian cells. FLAG-G3BP1-UB was prepared by co-expressing FLAG-G3BP1 together with MYC-TRIM21ΔCC and HA-UB in HEK293FT cells. FLAG affinity isolation and FLAG peptide exchange were then used for protein purification. Immunoblotting confirmed the presence of purified proteins, and antibodies against HA showed an increase of G3BP1-UB levels under MYC-TRIM21ΔCC overexpression (Fig. S3D). We incubated FLAG-G3BP1 and FLAG-G3BP1-UB at four different concentrations with five different concentrations of PEG 8000 to examine LLPS-mediated granule formation in vitro. Notably, higher G3BP1-UB concentrations were required for granule formation under all examined PEG concentrations than those of the wild-type FLAG-G3BP1 (Fig. S3E-F). These data indicated that ubiquitination of G3BP1 by TRIM21 inhibits SG formation.
Both TRIM21 and G3BP1 interact with several autophagy machinery proteins upon arsenite treatment
In light of the above findings, we also considered that SGs are degraded by autophagy under prolonged stress [10–12]. TRIM21 is known to regulate the degradation of activated IRF3 and IKBKB via interactions with the autophagy factors ULK1, BECN1, SQSTM1, and MAP1LC3 [39,43]. We therefore investigated whether TRIM21 also participates in autophagic degradation of SGs using Co-IP experiments to examine whether TRIM21 interacts with these autophagy-related proteins under arsenite treatment. The results indeed showed strong interactions between TRIM21 and ULK1, BECN1 or MAP1LC3B in HEK293FT cells exposed to arsenite (Figure 3A-C). Interestingly, TRIM21 also exhibited strong interactions with the autophagy receptors SQSTM1, CALCOCO2, TOLLIP, and NBR1 under arsenite treatment, but interacted weakly with OPTN and TAX1BP1 (Figure 3D-E, S4A-D). These data suggest the involvement of autophagy in SG regulation.
Figure 3.
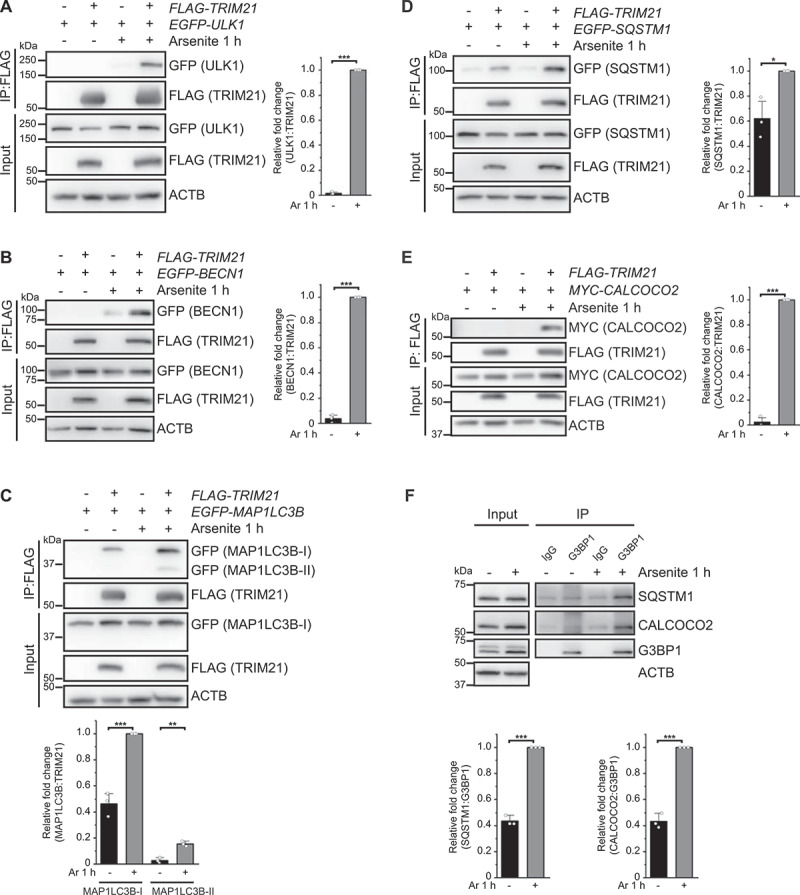
TRIM21 and G3BP1 interact with several autophagy machinery proteins upon arsenite treatment. (A-E) HEK293FT cells were transfected with EGFP-ULK1 (A), EGFP-BECN1 (B), EGFP-MAP1LC3B (C), EGFP-SQSTM1 (D) or MYC-CALCOCO2 (E), along with the empty vector or FLAG-TRIM21, and the cells were treated without or with arsenite. FLAG affinity isolation was performed with anti-FLAG beads, and the samples were analyzed by immunoblotting with the indicated antibodies. (F) HEK293FT cells were treated without or with arsenite for 1 h. Anti-G3PB1 immunoprecipitation was performed, and the samples were analyzed by immunoblotting with the indicated antibodies. Blots are representative of three biological replicates. Relative fold changes of the immunoprecipitated proteins are presented as means of three replicate experiments ± SEM. Significance was determined using Student’s t-test (A-F); *p < 0.05, **p < 0.01, ***p < 0.001.
We then examined whether these receptors also interact with G3BP1 under arsenite-induced oxidative stress in HEK293FT cells. Co-IP assays using anti-G3BP1 antibody to immunoprecipitate the receptors showed that G3BP1 could bind with SQSTM1, CALCOCO2, TOLLIP, and NBR1 under physiological conditions, and that these interactions were enhanced following arsenite treatment (Figure 3F, S4E). By contrast, G3BP1 exhibited no detectable binding with OPTN and TAX1BP1 (Fig. S4E). These results support that SGs can be regulated by the autophagy pathway [10], underscores the function of G3BP1 in these regulations.
SQSTM1 or CALCOCO2 knockout inhibits stress granule elimination during recovery from oxidative stress
Based on these results, we then investigated the function of autophagy pathway in SG formation/elimination through CRISPR-Cas9-mediated knockout of autophagy receptor genes individually in U-2 OS cells. When the cells were treated with arsenite for 1 h, we could detect a marginal difference in SG abundance between the WT control and SQSTM1 knockout cells (Figure 4A-C). Interestingly, when we examined SGs in these cells during the recovery stage at 1.5 h after stress removal, as reported in other studies of oxidative stress [50], we observed that SG number remained at a higher level in the SQSTM1 knockout cells than that in the WT controls (Figure 4A-C), thus indicating that SQSTM1 is required for the elimination of SGs.
Figure 4.
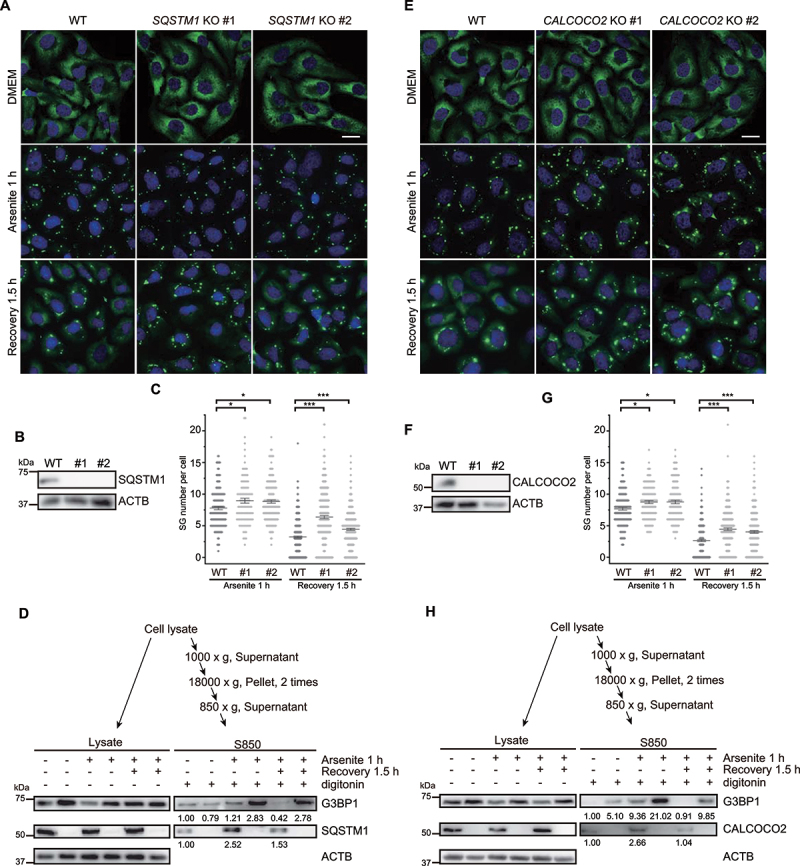
Knockout of SQSTM1 or CALCOCO2 prevents SG elimination during recovery from oxidative stress. (A and E) Two SQSTM1 knockout cell lines (#1 and #2) (A), or two CALCOCO2 knockout cell lines (#1 and #2) (E) were untreated, treated with arsenite for 1 h, or recovered for 1.5 h after removal of arsenite. Cells were fixed and stained with G3BP1 (green) and nuclei (DAPI, blue). Scale bar: 20 μm. (B and F) Immunoblots showed knockouts of SQSTM1 (B) or CALCOCO2 (F) from CRISPR-Cas9-generated U-2 OS cells. (C and G) Quantification of SG number per cell under oxidative stress and at the recovery stage from stress as represented in (A) and (E). Data were pooled from three independent experiments with 150 cells counted. Error bars indicate SEM. *p < 0.05, ***p < 0.001 (one-way ANOVA with Tukey’s test). (D and H) SQSTM1 (D) or CALCOCO2 (H) knockout results in the accumulation of G3BP1 under oxidative stress and in the recovery stage after arsenite treatment. SG fractions (S850) were obtained by serial centrifugations, and the samples were analyzed by immunoblotting with the indicated antibodies. Protein levels of G3BP1, SQSTM1 and CALCOCO2 were quantified.
We then conducted SG fraction enrichment in both WT and SQSTM1 knockout cells under the following three conditions: untreated, 1 h of arsenite treatment, and 1.5 h of recovery after arsenite treatment. We found that SQSTM1 knockout cells showed significantly higher level of G3BP1 accumulation in SG fractions compared to WT cells under both oxidative stress and recovery conditions, with especially obvious differences under the recovery condition (Figure 4D). Notably, SQSTM1 was also enriched in the SG fraction in WT cells in response to arsenite treatment and during the recovery stage after stress (Figure 4D). These results were consistent with previous reports that showed SQSTM1 serves as a key factor in the elimination of SGs by autophagy [19,51]. Interestingly, CALCOCO2 knockout resulted in a similar impairment of SG elimination under oxidative stress and during stress recovery as observed in SQSTM1 knockout cells (Figure 4E-H). Notably, immunofluorescence staining of G3BP1 revealed that knockout of other autophagy receptors such as OPTN, NBR1, TOLLIP, or TAX1BP1 respectively induced only a slight increase, no effect, or unexpectedly, a decrease in the number of SGs during recovery from oxidative stress (Fig. S5A-L). Collectively, these results demonstrate that SQSTM1 and CALCOCO2 function as specific receptors for SG cargo recognition and elimination.
The autophagy receptors SQSTM1 and CALCOCO2 are adjacent to SGs in response to arsenite treatment
Since the colocalization of autophagy receptor with cargo proteins can serve as an indicator of autophagic degradation of the cargos, we then co-stained endogenous G3BP1 and SQSTM1 or CALCOCO2 under different treatment conditions. Compared to the untreated cells, the 3D reconstituted confocal images of the SGs revealed that SQSTM1 and G3BP1 were closely apposed under arsenite treatment and during the recovery stage from oxidative stress (Figure 5A-B, quantified in 5C). Similarly, we found that CALCOCO2 was also adjacent to G3BP1-positive SGs under arsenite treatment as well as during recovery (Fig. 5D-E, quantified in 5F). EGFP-tagged full-length SQSTM1 or CALCOCO2 also exhibited adjacent localization with endogenous G3BP1 (Fig. S6A-B, quantified in S6C; Fig. S6D-E, quantified in S6F), which fully recapitulated the endogenous immunofluorescence results. Moreover, mapping results of individual domains revealed that the UBA domain of SQSTM1 and the UBZ domain, as well as the coiled-coil domain, of CALCOCO2, which mediate their interactions with G3BP1 under stress (Fig. S7A-B and Fig. S7D-E), are indispensable for these observed localizations (Fig. S7C and S7F), highlighting the specificity of interactions between G3BP1 and the two receptors.
Figure 5.
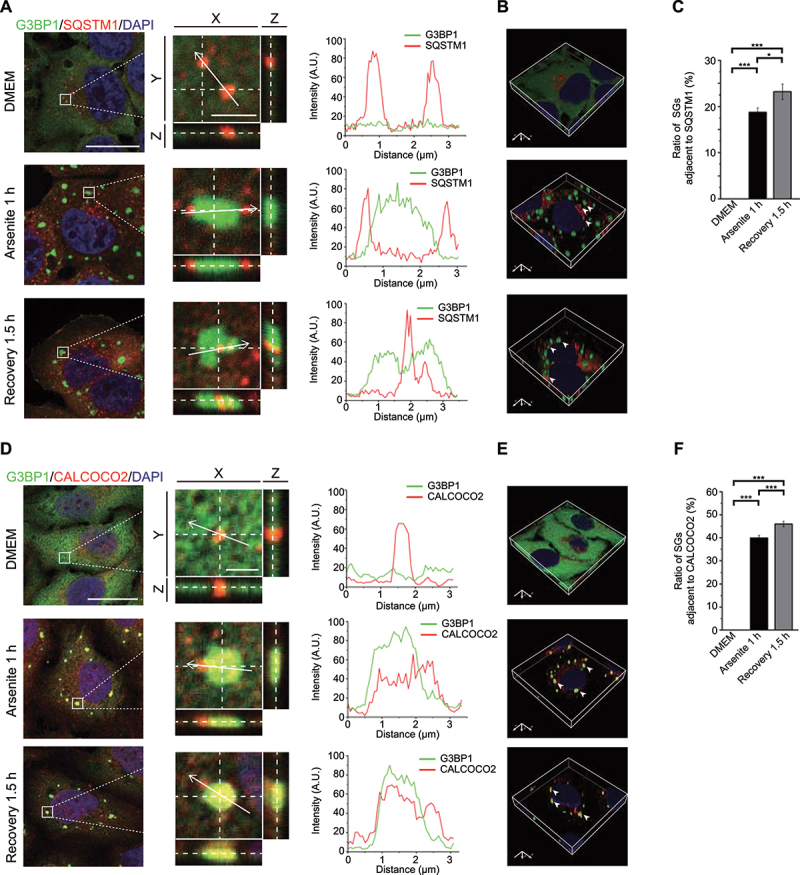
Endogenous SQSTM1 and CALCOCO2 are associated with SGs upon arsenite treatment and during recovery from oxidative stress. (A and D) HeLa cells were untreated, treated with arsenite for 1 h, or recovered for 1.5 h after removal of arsenite. Cells were fixed and stained with G3BP1 (green), nuclei (DAPI, blue) and SQSTM1 (red) (A) or CALCOCO2 (red) (D). Left, Z-stack projection of the images; scale bar: 20 μm. Middle, magnified orthogonal sectioning view of regions in the boxes; scale bar: 2 μm. Right, Line graphs show fluorescence plots along the white arrow. (B and E) 3D reconstructed images after Z-section confocal analysis. Arrows indicate adjacent localization of G3BP1 and SQSTM1 (B) or CALCOCO2 (E). (C and F) Percentage of SGs adjacent to SQSTM1 or CALCOCO2 as represented in (A) and (D). Data are presented as means ± SEMs from three independent experiments with 300 cells counted. *p < 0.05, ***p < 0.001 (one-way ANOVA with Tukey’s test).
TRIM21 inhibits the pathological stress granule formation
Hexanucleotide repeat expansions (HRE, [G4C2]n) in C9orf72 are a common genetic cause of familial ALS-FTD [52,53]. Translation of these HREs results in dipeptide repeat proteins (DPRs) (e.g., poly[GR]) that form pathological SGs through LLPS [54,55]. Given that TRIM21 overexpression inhibited physiological SG formation following oxidative stress, we then investigated whether TRIM21 could also inhibit pathological SG formation by the model DPR, EGFP-(GR)58 [56]. Since the vast majority of EGFP-(GR)58 was localized in the nucleus (data not shown), we sought to better monitor the cytosolic population of (GR)58 by inserting two nuclear export signal (NES) sequences into the N-terminus and the internal of EGFP-(GR)58 construct in order to enhance its cytosolic localization (Fig. S8A). The cytosolic population of 2xNES-(GR)58 appeared to form obvious puncta that colocalized with G3BP1 in the absence of arsenite-induced stress (Figure 6A, top row). Interestingly, TRIM21ΔCC overexpression resulted in a decreased number of (GR)58- and G3BP1-colocalized SGs (Figure 6A, quantified in 6B) as well as a reduced number of cells harboring these SGs (quantified in 6C). We further tested another ALS-related gene FUS [57,58], and found that FUS mutant protein FUSP525L also colocalized with G3BP1 to form SGs in HeLa cells under non-stress conditions, which was also inhibited under TRIM21ΔCC overexpression, suggesting a general inhibitory effect on SG formation by TRIM21 (Fig. S8B-C). Collectively, the above data show that TRIM21 can inhibit formation of pathological SGs, in addition to stress-induced physiological SGs.
Figure 6.
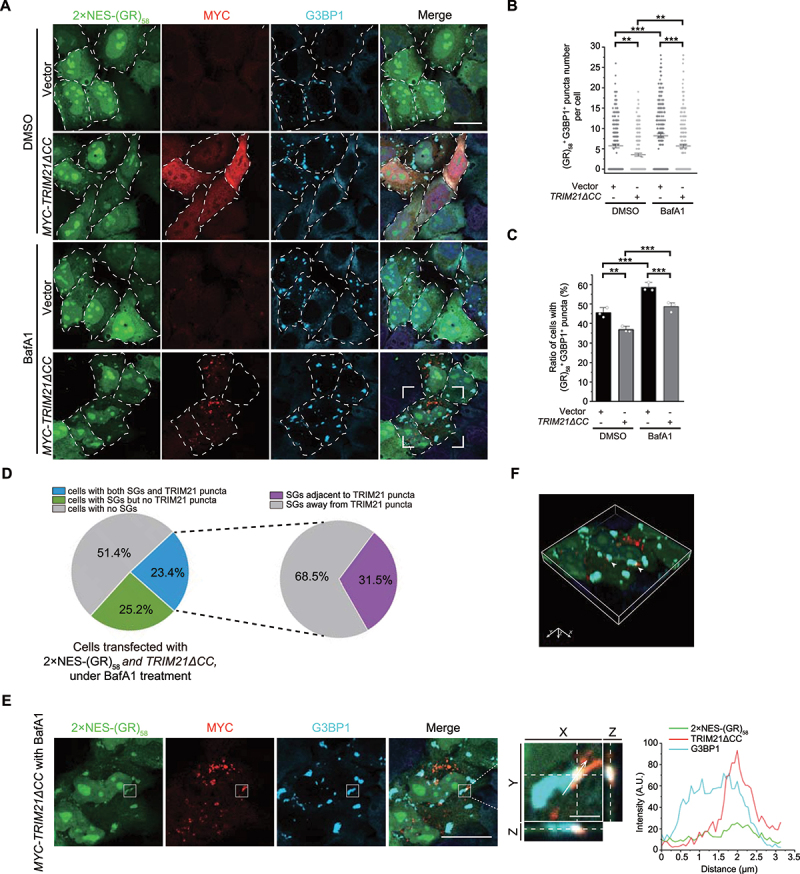
TRIM21 inhibits the pathological SG formation. (A) HeLa cells were transfected with 2xNES-(GR)58, along with the empty vector or MYC-TRIM21ΔCC, and treated without or with 100 nM BafA1 for 4 h. Cells were fixed and stained with MYC (red), G3BP1 (cyan) and nuclei (DAPI, blue). Scale bar: 20 μm. (B) Quantification of the number of (GR)58- and G3BP1-colocalized SGs per cell from (A). Data were pooled from three independent experiments with 200 cells counted. Error bars indicate SEM. **p < 0.01, ***p < 0.001 (one-way ANOVA with Tukey’s test). (C) Quantification of the percentage of cells with SGs from (A). Data are presented as means ± SEMs from three independent experiments with 200 cells counted. **p < 0.01, ***p < 0.001 (one-way ANOVA with Tukey’s test). (D) HeLa cells were transfected with 2×NES-(GR)58 and TRIM21ΔCC and then treated with 100 nM BafA1. 51.4% of cells had no SGs, 25.2% of cells had SGs but no TRIM21 puncta, and 23.4% of cells had both 2xNES-(GR)58 SGs and TRIM21 puncta. In the 23.4% of cells with both 2xNES-(GR)58 SGs and TRIM21 puncta, 31.5% of 2xNES-(GR)58-positive SGs were adjacent to TRIM21 puncta. (E) The enlarged image of the white square drawn in the bottom row in (A). TRIM21 is localized to the periphery of G3BP1- and 2×NES-(GR)58-colocalized puncta. Left, Z-stack projection of the images; scale bar: 20 μm. Middle, magnified orthogonal sectioning view of regions in the boxes; scale bar: 2 μm. Right, Line graphs show fluorescence plots along the white arrow. (F) A 3D reconstructed image after Z-section confocal analysis. Arrows indicate the colocalized G3BP1, 2×NES-(GR)58 and TRIM21.
Next, we investigated whether pathological SGs are subjected to autophagic degradation. We observed significant accumulation of 2xNES-(GR)58 and an increased number of cells containing pathological SGs under treatment with the autophagy blocker, bafilomycin A1 (BafA1) (Figure 6A, quantified in 6B and 6C). Notably, TRIM21 reduced SG numbers to similar extents in both the presence and absence of BafA1 (Figure 6B), suggesting that TRIM21-mediated SG regulation may not depend on the autophagy pathway. However, further analysis of TRIM21ΔCC-overexpressing cells under BafA1 treatment showed that among the 23.4% of cells harboring both 2xNES-(GR)58 SGs and TRIM21 puncta, 31.5% of 2xNES-(GR)58-positive SGs were localized to the periphery of TRIM21 puncta (Figure 6D), as exemplified by 3D reconstituted confocal images (Figure 6E-F). These data, as well as the above results, indicated that TRIM21 may function in preventing over-accumulation of the pathological SGs.
Knockout of SQSTM1 or CALCOCO2 prevents the elimination of (GR)58
In light of these results, we finally sought to examine whether the SQSTM1 and CALCOCO2 autophagy receptors function in the elimination of pathological SGs as well as physiological SGs. Therefore, we examined the fate of pathological (GR)58 SGs by transiently expressing 2xNES-(GR)58 in SQSTM1- or CALCOCO2-knockout HeLa cells. Immunofluorescence staining indicated that knockout of SQSTM1 or CALCOCO2 resulted in significant accumulation of (GR)58- and G3BP1-colocalized SGs (Figure 7A-B and 7D-E, quantified in 7C and 7F), suggesting that these proteins contribute to the elimination of pathological (GR)58 SGs. Taken together, these data, as well as the BafA1 treatment results shown above, demonstrate that pathological SGs are cleared by autophagy through autophagy receptors SQSTM1 and CALCOCO2.
Figure 7.
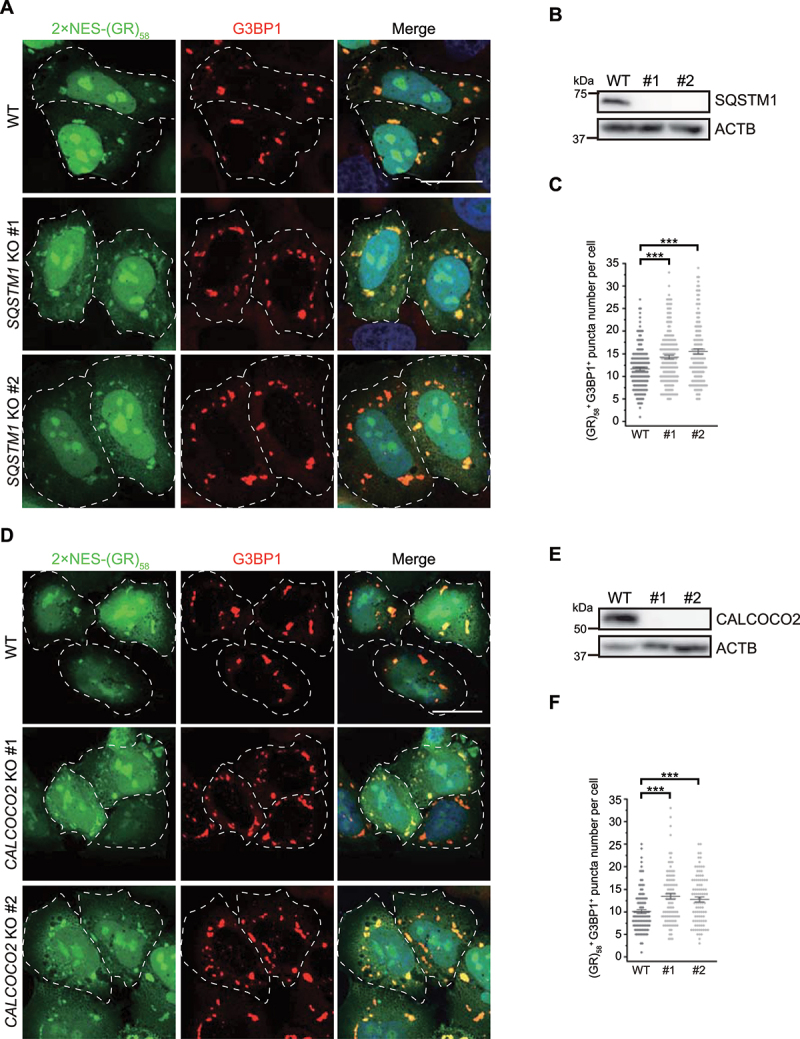
Knockout of SQSTM1 or CALCOCO2 inhibits the elimination of pathological (GR)58. (A and D) SQSTM1 (A) or CALCOCO2 (D) knockout resulted in the accumulation of 2xNES-(GR)58. Numbers indicate different single clones. Cells were fixed and stained with G3BP1 (red) and nuclei (DAPI, blue). Scale bar: 20 μm. (B and E) Immunoblots showing knockout of SQSTM1 (B) or CALCOCO2 (E) from CRISPR-Cas9-generated HeLa cells. (C and F) Quantification of the SG number per cell as represented in (A) and (D). Data were pooled from three independent experiments with 150 cells counted. Error bars indicate SEM. ***p < 0.001 (one-way ANOVA with Tukey’s test).
Discussion
In this report, we provide evidence to show that SG homeostasis is regulated by TRIM21-mediated ubiquitination of G3BP1 and autophagy-dependent elimination of SGs. Our data demonstrate that TRIM21 is enriched in SGs and its knockdown promotes SG formation in cells. Furthermore, we found that autophagy also participates in eliminating SGs by degrading both physiological and pathological SGs. Interestingly, the autophagy receptors localize to the area adjacent to SGs and interact with the SG structural protein G3BP1 under arsenite treatment, providing a direct link between SGs and autophagy. Based on these findings, we proposed a working model for TRIM21 and autophagy machinery functioning together to maintain SG homeostasis (Figure 8). TRIM21 inhibits SG formation by catalyzing the ubiquitination of G3BP1 under stress or pathological conditions. Meanwhile, G3BP1 interacts with the autophagy receptors under stress and during the recovery stage after stress removal to mediate the elimination of physiological or pathological SGs.
Figure 8.
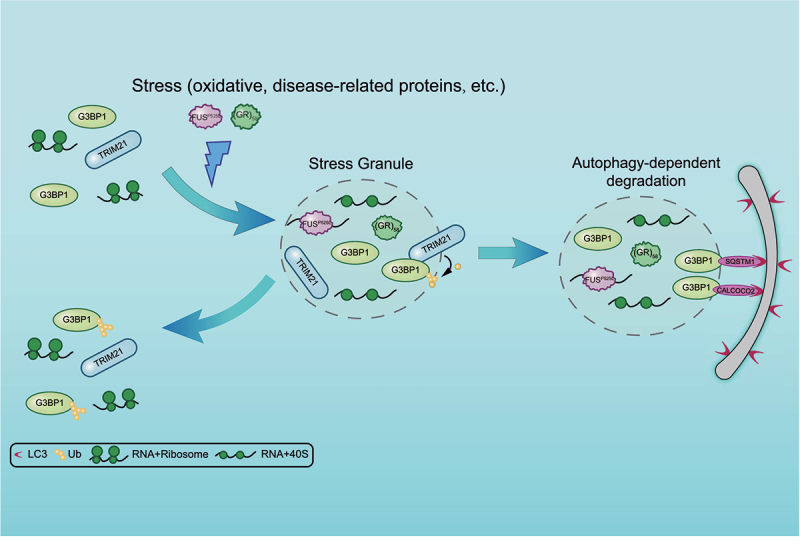
A proposed model for TRIM21 and autophagy machinery in regulating SG homeostasis. TRIM21 is highly enriched in SGs under arsenite-induced oxidative stress. It inhibits SG formation by catalyzing K63-linked ubiquitination of G3BP1. Meanwhile, G3BP1 recruits the autophagy receptors SQSTM1 and CALCOCO2 for the elimination of physiological or pathological SGs.
Previous studies have shown that TRIM21 plays an important role in immune regulation during pathogen infection through its function as an E3 ligase. More specifically, it can either enhance the innate immune response to viral infection by catalyzing K27-linked ubiquitination of MAVS (mitochondrial antiviral signaling protein) [59], or it can downregulate innate immunity by targeting and degrading various proteins in the immune-response pathway [60–62]. Interestingly, TRIM21 also mediates autophagic degradation of some cytoplasmic regulators of innate immunity [39,43]. Furthermore, recent reports suggest that TRIM21 can promote autophagy in osteosarcoma cells [44]. However, it remains unclear whether TRIM21 also participates in other signal pathways. The presence of multiple E3 ligases, including TRIM21, in SG components suggests that these ligases could contribute to the regulation of SG homeostasis. In this study, we discovered a previously unrecognized function of TRIM21 in regulating SG dynamics by ubiquitinating G3BP1 to inhibit SG formation.
Consistent with this hypothetical function, a recent study reported that G3BP1 ubiquitination via K63 linkage is a critical step in SG elimination, either by disassembly when stress is retracted or by autophagy-dependent degradation under prolonged stress treatment or pathological mutations [12]. Moreover, various TRIM proteins have been reported to form complexes with different autophagy receptors to mediate autophagic degradation of target substrates. For example, TRIM44 (tripartite motif containing 44) expression stimulates SQSTM1 oligomerization, thereby promoting autophagic clearance of aggregates [42]. TRIM32 (tripartite motif containing 32) interacts with TAX1BP1, subsequently increasing the selective autophagic degradation of TICAM1/TRIF (TIR domain containing adaptor molecule1) to suppress TLR3 (toll like receptor 3)- or TLR4 (toll like receptor 4)-mediated innate immune responses [63]. Furthermore, previous studies also demonstrated that TRIMs form a “TRIMosome” structural platform to facilitate autophagosome formation and cargo degradation [38]. In this work, though treatment with the autophagy blocker did not fully restore SG formation in TRIM21-overexpressing cells, our data demonstrate that TRIM21 interacts with several autophagy machinery proteins. This is consistent with the previously proposed formation of a “TRIMosome”, which could provide additional interactions to help connect SGs with the autophagy-related proteins. These findings broaden the scope of our understanding of TRIM21 function beyond its well-established role in inflammatory response.
In line with our data showing that TRIM21 overexpression inhibits SG formation via K63-linked ubiquitination of G3BP1 (Figure 2C-D), other studies have shown that TRIM21 catalyzes K63 ubiquitination to modulate intracellular signal transduction [49,64–66]. For instance, TRIM21 can suppress SQSTM1 oligomerization and protein sequestration for autophagic degradation by K63-linked ubiquitination of SQSTM1 [49,66]. Similarly, TRIM21 also catalyzes K63-linked ubiquitination of NMI (N-myc and STAT interactor) to promote its interaction with IFI35 (interferon induced protein 35), consequently inhibiting innate antiviral response [64]. Conversely, it is well known that TRIM21 functions as an antibody receptor and catalyzes the formation of free K63-linked ubiquitin chains to initiate cellular innate immune response against viral infection [67]. Therefore, it is highly possible that ubiquitination of G3BP1 may block self-interactions in the intrinsically disordered region of G3BP1 [47] consequently disrupting SG formation, which aligns well with our in vitro LLPS results. It is worth noting that because the full-length TRIM21 was aggregation-prone, the coiled-coil deletion mutant was used for the phenotypic characterization and G3BP1 ubiquitination investigation. The coiled-coil region is mainly involved in mediating the multimerization of TRIMs to increase their catalytic activity [68–70]. Accordingly, TRIM21ΔCC may have a reduced ligase activity compared to the full-length TRIM21, which may account for the small amount of ubiquitinated G3BP1 purified from the mammalian cells (Fig. S3D) and the moderate difference of the LLPS results between G3BP1 and ubiquitinated G3BP1 (Fig. S3E-F).
Among the six known autophagy receptors, SQSTM1 was identified here as a key factor responsible for autophagic degradation of SGs. First, knockout of SQSTM1 led to substantial accumulation of SGs during the recovery stage after stress treatment (Figure 4A-D). Second, SQSTM1 exhibited a remarkably high adjacent signal with G3BP1 under both oxidative stress and afterward in the recovery stage (Figure 5A-C, S6A-C). We also found that CALCOCO2 performs a comparable role to that of SQSTM1 (Fig. 4E-H, 5D-F, S6D-F), while OPTN knockout results in a slight increase in SG accumulation. By contrast, knockout of NBR1, TOLLIP, or TAX1BP1 has no obvious effect on SG degradation or even (in the case of TOLLIP and TAX1BP1) results in detection of fewer SGs. The Co-IP analysis further supported the increased interaction between G3BP1 and SQSTM1 and/or CALCOCO2 upon arsenite treatment (Figure 3F). Accordingly, SQSTM1 and CALCOCO2 were recently reported to promote the clearance of SGs via selective autophagy [19,51]. Furthermore, a recent study reported that SQSTM1, the major driver of ubiquitin condensate, sequentially recruits NBR1 and TAX1BP1 to mediate the autophagic degradation of SQSTM1-ubiquitin condensates [71]. Collectively, our data suggest that SQSTM1 and CALCOCO2 mediate the autophagic clearance of SGs.
Taken together, our findings reveal that SG homeostasis is modulated by TRIM21-catalyzed K63-linked ubiquitination of G3BP1 to inhibit SG formation and the autophagy receptors SQSTM1 and CALCOCO2 mediated elimination of SGs.
Materials and methods
Cell culture
HEK293FT (PTA-5077), HeLa (CRM-CCL-2), U-2 OS (HTB-96) and A549 (CCL-185) cells from ATCC were cultured in DMEM (Thermo Fisher Scientific, 12800017), supplemented with 10% fetal bovine serum (Lonsera, S711-001S) and 100 U/ml penicillin G and 100 μg/ml streptomycin (Thermo Fisher Scientific, 15140148) at 37°C under 5% CO2. For SG assembly experiments, cells were subject to 0.5 mM sodium arsenite (Sigma-Aldrich, S7400) for 1 h; for SG recovery experiments, cells were first treated with 0.5 mM sodium arsenite for 1 h and then recovered for 1.5 h from oxidative stress. A moderate amount of SGs remained in U-2 OS cells after 1.5 h recovery.
Generating knockout cell lines
All knockout cell lines were generated with CRISPR-Cas9 systems. The guide RNAs sequences were designed from the website http://chopchop.cbu.uib.no/. The guide RNAs sequences were inserted into pGL3-U6-sgRNA-PGK-puromycin (Addgene 51133; deposited by Xingxu Huang) plasmid. U-2 OS or HeLa Cells were co-transfected with sgRNA plasmids and pST1374-NLS-FLAG-linker-Cas9 (Addgene 44758; deposited by Xingxu Huang). After 24 h, cells were selected with 5 μg/ml puromycin (Sigma-Aldrich, 540411) and 10 μg/ml blasticidin (Thermo Fisher Scientific, R21001) for 7 days. Cells were confirmed for the knockout of the targeted gene by immunoblotting. After confirmation, single cell colonies were individually sorted by flow cytometry into 96-well plates. Single cell colonies were screened by immunoblotting. The target sequences are listed in Table S1.
Plasmids
FLAG-TRIM21 and FLAG-G3BP1 were generated by cloning the corresponding cDNA into pRK5-FLAG [72]. MYC-G3BP1 and MYC-CALCOCO2 were generated by inserting the corresponding cDNA into pRK5-MYC. FLAG-TRIM21ΔCC, FLAG-TRIM21ΔRINGΔB-box and MYC-TRIM21ΔCC were generated by overlap extension of PCR-mediated deletion on pRK5-FLAG-TRIM21 and pRK5-MYC-TRIM21. EGFP-G3BP1, EGFP-ULK1, EGFP-BECN1, EGFP-SQSTM1 and EGFP-CALCOCO2 were generated by inserting the corresponding cDNA into pEGFP.C1 (Clontech, 6084–1). EGFP-MAP1LC3B was constructed by inserting EGFP-MAP1LC3B fragment into pcDNA3.0 (Invitrogen, A-150228). GST-TRIM21ΔCC was generated by inserting the corresponding fragment into pGEX-6P-1 (GE Healthcare Life Sciences, 28–9546-48). GST-TRIM21ΔCC-LD was generated by using pRK5-MYC-TRIM21ΔCC-LD as a template. pRK5-HA-UB was described previously [73]. HA-UBK48R and HA-UBK63R were generated based on PCR-mediated site-directed mutagenesis on pRK5-HA-UB. All mutations were confirmed by DNA sequencing. The construct for expression of His-tagged UBE2D2 was kindly provided by Dr. Cynthia Wolberger (John Hopkins University, Baltimore, MD, USA). The constructs pT7-7-His-HA-UB for expressing the recombinant HA-UB were kindly provided by Professor Kazuhiro Iwai (Kyoto University, Kyoto, Japan). EGFP-(GR)58 was provided by Dr. Lei Li (ShanghaiTech University, Shanghai, China). 2xNES-(GR)58 was generated by inserting two NES fragments into the EGFP-(GR)58 plasmid, with the first NES fragment obtained from HIV-1 rev [74] inserted at the N-terminus of EGFP-(GR)58, and the second NES fragment from TARDBP/TDP-43 (TAR DNA binding protein) inserted in between EGFP and (GR)58. For generation of EGFP-G3BP1 stable cell line, EGFP-G3BP1 was constructed by inserting EGFP-G3BP1 fragment into pCDH-CMV-MCS-EF1-Puro (System Biosciences, CD510B-1).
Lentivirus production and generation of TRIM21 knockdown cell line
To generate lentiviral particles, HEK293FT cells were co-transfected with the lentiviral vector pLKO.1-Puro (Addgene 8453; deposited by Bob Weinberg) encoding a scramble shRNA or TRIM21 shRNA, the lentivirus packaging plasmids psPAX2 (Addgene 12260; deposited by Didier Trono) and pMD2.G (Addgene 12259; deposited by Didier Trono) using Lipofectamine® 2000 (Thermo Fisher Scientific, 11668019). Lentiviral particles were harvested 48 h post-transfection, centrifuged to remove cell debris. A549 cells were plated in 6-well plates prior to being infected with viral particles for 24 h, then virus particle-containing medium were exchange to fresh medium. Forty-eight h after infection, cells were selected with 2 μg/ml puromycin. Knockdown of the target protein was detected by immunoblotting. The target sequences are listed in Table S1.
Antibodies and reagents
Mouse anti-MYC (2276), rabbit anti-HA (3724), rabbit anti-TAX1BP1 (5105S) and rabbit anti-CALCOCO2 (60732, for immunoblotting) were purchased from Cell Signaling Technology. Rabbit anti-FLAG (F3165), mouse anti-FLAG (F7425), rabbit anti-TRIM21 (HPA005673, for immunofluorescence) and rabbit anti-MAP1LC3B (L7543) were purchased from Sigma-Aldrich. Rabbit anti-TRIM21 (12108-1-AP), mouse anti-TRIM21 (67136-1-Ig), rabbit anti-G3BP1 (67136-2-AP for immunoprecipitation and immunoblotting), mouse anti-G3BP1 (66486-1-Ig, for immunoprecipitation and immunoblotting) and rabbit anti-NBR1 (16004-1-AP) were purchased from Proteintech. Rabbit anti-G3BP1 (A3968, for immunofluorescence), rabbit anti-SQSTM1 (A11247), rabbit anti-TOLLIP (A2202) and mouse anti-HA (AE008) were purchased from ABclonal, Inc. Mouse anti-TIA1 (sc-166247) was purchased from Santa Cruz Biotechnology. Rabbit anti-OPTN (23666), rabbit anti-CALCOCO2 (ab68588, for immunofluorescence) and rabbit anti-K48-linkage specific ubiquitin (ab140601) were purchased from Abcam. Rabbit anti-GFP was made against a recombinant GFP from Aequorea victoria. Anti-ACTB (HRP-DirecT; PM053-7) was purchased from Medical & Biological Laboratories Co., LTD. Mouse anti-K63-linkage specific ubiquitin (BML-PW0600-0025) was purchased from Enzo Life Sciences. The secondary antibodies goat anti-mouse IgG (H+L), HRP (111–035-146), goat anti-rabbit IgG (H+L), HRP (111–035-144) and Cy5-AffiniPure Donkey Anti-Rabbit IgG (H+L) (711–175-152) were purchased from Jackson ImmunoResearch Inc. The secondary antibodies goat anti-rabbit IgG (H+L), Alexa Fluor 488 (A-11034), goat anti-mouse IgG (H+L), Alexa Fluor 488 (A-11029), goat anti-rabbit IgG (H+L), Alexa Fluor 568 (A-11036) and goat anti-mouse IgG (H+L), Alexa Fluor 568 (A-11031) were purchased from Invitrogen.
Immunoprecipitation and immunoblotting
For immunoprecipitation, 24 h post-transfection, HEK293FT cells were lysed in NP40 lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5% NP40 [Sangon Biotech, A600385-0100]) containing EDTA-free protease inhibitor cocktail (Bimake, B14002) for 30 min at 4°C. The lysates were centrifuged at 17,000 x g for 10 min at 4°C, and the soluble supernatant fractions were subjected to immunoprecipitation with either protein A beads (Cell Signaling Technology, 9863S) pre-bound with G3BP1 or GFP antibody, FLAG (Bimake, B26102) or MYC (Bimake, B26302) magnetic beads. After incubation for 2 h at 4°C, beads were washed three times with NP40 wash buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.1% NP40), and the samples were immunoblotted with the indicated antibodies. For immunoprecipitation under denaturing condition, harvested cells were lysed in a buffer with 1% SDS (Sinopharm Chemical Reagent Co., 30166480) and 5 mM DTT (MDBio, Inc., D023) in PBS (Sangon Biotech, B548117-0500). The samples were then heated at 95°C for 5 min and diluted with NP40 lysis buffer. The soluble supernatant fractions were harvested and subjected to immunoprecipitation as described above. Immunoblotting was performed using polyvinylidene fluoride membrane (Bio-Rad, 1620177) and the indicated antibodies. Protein signals were detected by ECL western blotting detection reagents (PerkinElemer, NEL105001EA). The chemiluminescence bands were imaged under Amersham Imager 680 (GE Healthcare Life Sciences, USA). The bands were adjusted within the linear range, and quantified by ImageJ2 (v2.3.0, NIH).
Immunofluorescence microscopy
Cells grown on cover glasses were fixed with 4% paraformaldehyde (Thermo Fisher Scientific, 43368) in PBS for 15 min at room temperature. Cells were then permeabilized and incubated with the blocking buffer (4% FBS, 0.5% Triton X-100 [Sangon Biotech, A110694-0100] in PBS) for 10 min at room temperature. Cells were further incubated with indicated primary antibodies in blocking buffer for 1 h at room temperature, and then incubated with fluorescent dye-conjugated secondary antibodies and DAPI in blocking buffer for 30 min at room temperature. Finally, cells were mounted with Anti-Fade Fluorescence Mounting Medium (Abcam, ab104135). Images were acquired on Zeiss LSM800 microscope (Zeiss, Germany) with a 63×, 1.4 NA oil objective. Same acquisition parameters were used for a specific set of experiments. Z-stacks with a thickness of 1.7 μm made up of 9 images were collected by the 358 nm, 488 nm, 561 nm or 633 nm laser at 1% power, respectively. The parameter settings are: pixel size = 0.05 µm; dwell time per pixel = 2.06 µs; line average = 2; distance between slices = 0.19 µm. Z series are displayed as maximum Z-projections processed with ImageJ2. 3D images were processed with ICY software (Institut Pasteur and France-BioImaging version 2.4.2.0). The ratio of colocalization was determined considering a total of 200 cells from three independent experiments. SIM images were obtained with the Zeiss Lattice SIM microscope (Zeiss, Germany) using a Plan-Apochromat oil objective lens 63x, 1.4 NA. Images were captured using PCO edge sCMOS camera (Acquisition pixel size, 63 nm at 63x objective) and G6 gride (23 μm). Data was reconstructed using a 3D default method in Zen Black.
Protein purification
pT7-7-His-HA-UB, His-UBE2D2, GST-TRIM21ΔCC and GST-TRIM21ΔCC-LD were purified from E. coli BL21 (DE3; Transgen Biotech, CD601–01), according to a previously described method [75]. Protein eluted from Glutathione Sepharose (GE Healthcare Life Sciences, 17–5132-02) or Ni-NTA (GE Healthcare Life Sciences, 17–5318-02) were fractionated on a Superdex 200 HR (10/300) column (GE Healthcare Life Sciences, 28–9909-44) in a buffer containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl and 2 mM DTT.
FLAG-G3BP1 and FLAG-G3BP1-UB were purified from HEK293FT cells. FLAG-G3BP1 and HA-UB, together with the empty vector or MYC-TRIM21ΔCC, were transfected into HEK293FT cells. Cells were harvested and lysed with NP40 lysis buffer, followed by centrifugation at 17,000 x g for 30 min at 4°C. The supernatants were incubated with FLAG magnetic beads for 2 h at 4°C. The beads were then washed with NP40 wash buffer for three times and once with LLPS buffer (20 mM Tris-HCl, pH 7.5, 85 mM KCl, 1 mM MgCl2, and 0.5 mM DTT). FLAG-tagged proteins were eluted with 3xFLAG peptide (Sigma-Aldrich, F4799) with a final concentration of 0.2 mg/ml in LLPS buffer. The eluted proteins and their conjugated-ubiquitin were validated by western blot with indicated antibodies.
In vitro ubiquitination assay
For in vitro ubiquitination assays, the mammalian purified substrate FLAG-G3BP1 (4.0 µM) was incubated with GST-E1 (140 nM; BostonBiochem, E-306), His-UBE2D2 (2.0 µM), GST-TRIM21ΔCC (2.0 µM) or GST-TRIM21ΔCC-LD (2.0 µM), and HA-UB (10 µM) in the reaction buffer (25 mM Tris-HCl, pH 7.4, 2 mM MgCl2, ATP [Sigma-Aldrich, A2383], and 1 mM DTT) at 37°C for 1 h.
Stress granule fractionation and enrichment
Stress granule enrichment was performed according to a previously described method [46]. Briefly, U-2 OS in 15 cm tissue-culture dishes was treated with 0.5 mM sodium arsenite for 1 h or recovered for 1.5 h after removal of arsenite. 90% of the cells were then treated with 0.029% digitonin (Abcam, ab141501) for 30 s to permeabilize the plasma membrane and then wash once by PBS. Cells were harvested and flash frozen with liquid N2 and the cell pellets were stored at −80°C. Then the pellets were thawed on ice for 10 min and re-suspended in 200 µl stress granule lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM DTT, 50 µg/mL heparin [Yeasen, 60351ES03], 0.5% NP40, 1:5000 Antifoam B [Sigma-Aldrich, A5757], EDTA-free protease inhibitor cocktail and RNase inhibitor [Thermo Fisher Scientific, EO0381] in DEPC H2O). Syringe lysis was also performed with a 25G, 5/8 needle by 8 passages through the needle on ice to lyse the cells sufficiently. After lysis, spin the samples at 1000 x g for 5 min at 4°C to pellet cell debris, and the supernatants were transferred to new tubes and centrifuged at 18,000 x g for 20 min at 4°C. After that, the supernatants were discarded and the pellets were re-suspended with 1 ml stress granule lysis buffer. The samples were again centrifuged at 18,000 x g for 20 min at 4°C. The pellets were re-suspended in 90 µl stress granule lysis buffer followed by spin at 850 x g for 2 min at 4°C, and the supernatants (S850) represent the stress granule core-enriched fractions. The input lysate was prepared by directly lysing the remaining 10% of the cells without digitonin treatment.
Liquid-liquid phase separation (LLPS)
The in vitro LLPS experiments were performed at room temperature. The LLPS of different concentration of FLAG-G3BP1 (0, 11, 22, 44 μM) and FLAG-G3BP1-UB (0, 11, 22, 44 μM) was induced by addition of indicated concentration of PEG 8000 (Yeasen, 60304ES76). The proteins and PEG 8000 were mixed in EP tubes and the samples were transferred to a glass bottom dish (Cellvis, D29-20-1-N) for observations. Samples were observed under a DIC microscope using Zeiss Axio Observer Z1 inverted phase contrast microscope (Zeiss, Germany) with a 40×, 0.95 NA objective.
Quantification and statistical analysis
All experiments were independently repeated at least three times. The automatic counting of SG numbers and SG colocalized with other signals were conducted in ImageJ2. The image processing steps included background subtraction, smoothing, and converting to binary masks via thresholding. Then the numbers of SG in different groups were measured using the analyze particle function with particle size and circularity parameters fixed. Data were pooled from three independent experiments with at least 100 cells counted. Error bars indicate SEM. Quantifications of the colocalization signals were presented as the mean of three replicates ± SEM, with at least 200 cells per condition from three biological replicates analyzed. Mean fluorescent intensity in each cell was quantified by tracing a region of interest (ROI) manually according to the cell boundary. Backgrounds were subtracted using the subtract background function of ImageJ2. The Pearson’s coefficient (r) is used to quantify the degree of correlation between two different fluorescent channels. All statistical analysis were performed using OriginPro 2021. The significance between two groups was obtained using the Student’s t-test. The significance among multiple groups was obtained using one-way ANOVA followed by Tukey’s multiple comparisons test. n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Acknowledgments
We thank Professor Cynthia Wolberger (John Hopkins University, Baltimore, MD, USA), Professor Kazuhiro Iwai (Kyoto University, Kyoto, Japan), Dr. Lei Li and Dr. Haopeng Wang (ShanghaiTech University, Shanghai, China) and Dr. Cong Liu (Chinese Academy of Sciences, Shanghai, China) for providing the various constructs. We thank the Image Core Facility and the Molecular and Cell Biology Core Facility in the School of Life Science and Technology at ShanghaiTech University for assisting with confocal microscopy and technical support.
Funding Statement
This work was supported by the National Natural Science Foundation of China [32070697 to Y.F.L.]; the Young Scientists Fund of the National Natural Science Foundation of China [32100624 to Y.J.K]; National Natural Science Foundation of China [31770831 to Y.F.L.]; National Natural Science Foundation of China [31670756 to Y.B.]; ShanghaiTech University start-up fund [2015F0202-000 to Y.F.L.].
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2164427
References
- [1].Ivanov P, Kedersha N, Anderson P.. Stress granules and processing bodies in translational control. Cold Spring Harb Perspect Biol. 2019;11(5):a032813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20(11):649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154(4):727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dobra I, Pankivskyi S, Samsonova A, et al. Relation between stress granules and cytoplasmic protein aggregates linked to neurodegenerative diseases. Curr Neurol Neurosci Rep. 2018;18(12):107. [DOI] [PubMed] [Google Scholar]
- [5].Riggs CL, Kedersha N, Ivanov P, et al. Mammalian stress granules and P bodies at a glance. J Cell Sci. 2020;133(16):jcs242487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Panas MD, Ivanov P, Anderson P. Mechanistic insights into mammalian stress granule dynamics. J Cell Biol. 2016;215(3):313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jain S, Wheeler JR, Walters RW, et al. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164(3):487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Markmiller S, Soltanieh S, Server KL, et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018;172(3):590–604 e513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Youn JY, Dunham WH, Hong SJ, et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol Cell. 2018;69(3):517–532 e511. [DOI] [PubMed] [Google Scholar]
- [10].Buchan JR, Kolaitis R-M, Taylor JP, et al. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153(7):1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Seguin SJ, Morelli FF, Vinet J, et al. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 2014;21(12):1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gwon Y, Maxwell BA, Kolaitis R-M, et al. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science. 2021;372(6549):abf6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li YR, King OD, Shorter J, et al. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mahboubi H, Stochaj U. Cytoplasmic stress granules: dynamic modulators of cell signaling and disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(4):884–895. [DOI] [PubMed] [Google Scholar]
- [15].Baradaran-Heravi Y, Van Broeckhoven C, van der Zee J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol Dis. 2020;134:104639. [DOI] [PubMed] [Google Scholar]
- [16].Dudman J, Qi X. Stress granule dysregulation in amyotrophic lateral sclerosis. Front Cell Neurosci. 2020;14:598517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Deng Z, Sheehan P, Chen S, et al. Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Mol Neurodegener. 2017;12(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nguyen DKH, Thombre R, Wang J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci Lett. 2019;697:34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guo H, Chitiprolu M, Gagnon D, et al. Autophagy supports genomic stability by degrading retrotransposon RNA. Nat Commun. 2014;5:5276. [DOI] [PubMed] [Google Scholar]
- [20].Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Donde A, Sun M, Jeong YH, et al. Upregulation of ATG7 attenuates motor neuron dysfunction associated with depletion of TARDBP/TDP-43. Autophagy. 2020;16(4):672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y, Gu J, Sun Q. Aberrant stress granule dynamics and aggrephagy in ALS pathogenesis. Cells. 2021;10(9):2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Maxwell BA, Gwon Y, Mishra A, et al. Ubiquitination is essential for recovery of cellular activities after heat shock. Science. 2021;372(6549):eabc3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol. 2019;35:453–475. [DOI] [PubMed] [Google Scholar]
- [26].Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85:685–713. [DOI] [PubMed] [Google Scholar]
- [27].Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes … wait, I’m confused. Autophagy. 2014;10(4):549–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vainshtein A, Grumati P. Selective autophagy by close encounters of the ubiquitin kind. Cells. 2020;9(11):2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126(Pt 15):3237–3247. [DOI] [PubMed] [Google Scholar]
- [30].Wu JJ, Cai A, Greenslade JE, et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci U S A. 2020;117(26):15230–15241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Deng Z, Lim J, Wang Q, et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2020;16(5):917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Deng Z, Purtell K, Lachance V, et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017;27(7):491–504. [DOI] [PubMed] [Google Scholar]
- [33].Teyssou E, Takeda T, Lebon V, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–522. [DOI] [PubMed] [Google Scholar]
- [34].Maruyama H, Kawakami H. Optineurin and amyotrophic lateral sclerosis. Geriatr Gerontol Int. 2013;13(3):528–532. [DOI] [PubMed] [Google Scholar]
- [35].Hatakeyama S. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci. 2017;42(4):297–311. [DOI] [PubMed] [Google Scholar]
- [36].Koepke L, Gack MU, Sparrer KM. The antiviral activities of TRIM proteins. Curr Opin Microbiol. 2021;59:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Di Rienzo M, Romagnoli A, Antonioli M, et al. TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020;27(3):887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30(4):394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015;210(6):973–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mandell MA, Kimura T, Jain A, et al. TRIM proteins regulate autophagy: TRIM5 is a selective autophagy receptor mediating HIV-1 restriction. Autophagy. 2014;10(12):2387–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jena KK, Kolapalli SP, Mehto S, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018;37(18):e98358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lyu L, Chen Z, McCarty N. TRIM44 links the UPS to SQSTM1/p62-dependent aggrephagy and removing misfolded proteins. Autophagy. 2022;18(4):783-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Niida M, Tanaka M, Kamitani T. Downregulation of active IKK beta by Ro52-mediated autophagy. Mol Immunol. 2010;47(14):2378–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang HT, Zeng Q, Wu B, et al. TRIM21-regulated Annexin A2 plasma membrane trafficking facilitates osteosarcoma cell differentiation through the TFEB-mediated autophagy. Cell Death Dis. 2021;12(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Esposito D, Koliopoulos MG, Rittinger K. Structural determinants of TRIM protein function. Biochem Soc Trans. 2017;45(1):183–191. [DOI] [PubMed] [Google Scholar]
- [46].Wheeler JR, Jain S, Khong A, et al. Isolation of yeast and mammalian stress granule cores. Methods. 2017;126:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang P, Mathieu C, Kolaitis RM, et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell. 2020;181(2):325–345 e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wada K, Kamitani T. Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun. 2006;339(1):415–421. [DOI] [PubMed] [Google Scholar]
- [49].Pan JA, Sun Y, Jiang YP, et al. TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. 2016;62(1):149–151. [DOI] [PubMed] [Google Scholar]
- [50].Turakhiya A, Meyer SR, Marincola G, et al. ZFAND1 recruits p97 and the 26S proteasome to promote The clearance of arsenite-induced stress granules. Mol Cell. 2018;70(5):906–919 e907. [DOI] [PubMed] [Google Scholar]
- [51].Chitiprolu M, Jagow C, Tremblay V, et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun. 2018;9(1):2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang YJ, Gendron TF, Ebbert MTW, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018;24(8):1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lee KH, Zhang P, Kim HJ, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167(3):774–788 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Freibaum BD, Lu Y, Lopez-Gonzalez R, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. [DOI] [PubMed] [Google Scholar]
- [59].Xue B, Li H, Guo M, et al. TRIM21 promotes innate immune response to RNA viral infection through Lys27-linked polyubiquitination of MAVS. J Virol. 2018;92(14):e00321-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang Z, Bao M, Lu N, et al. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol. 2013;14(2):172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Higgs R, Ni Gabhann J, Ben Larbi N, et al. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. 2008;181(3):1780–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li Z, Huan C, Wang H, et al. TRIM21-mediated proteasomal degradation of SAMHD1 regulates its antiviral activity. EMBO Rep. 2020;21(1):e47528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yang Q, Liu TT, Lin H, et al. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 2017;13(9):e1006600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Das A, Dinh PX, Pattnaik AK. Trim21 regulates Nmi-IFI35 complex-mediated inhibition of innate antiviral response. Virology. 2015;485:383–392. [DOI] [PubMed] [Google Scholar]
- [65].Yang L, Jin L, Ke Y, et al. E3 ligase Trim21 ubiquitylates and stabilizes keratin 17 to induce STAT3 activation in psoriasis. J Invest Dermatol. 2018;138(12):2568–2577. [DOI] [PubMed] [Google Scholar]
- [66].Jin J, Meng X, Huo Y, et al. Induced TRIM21 ISGylation by IFN-beta enhances p62 ubiquitination to prevent its autophagosome targeting. Cell Death Dis. 2021;12(7):697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].McEwan WA, Tam JC, Watkinson RE, et al. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat Immunol. 2013;14(4):327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Koliopoulos MG, Esposito D, Christodoulou E, et al. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J. 2016;35(11):1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sanchez JG, Okreglicka K, Chandrasekaran V, et al. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc Natl Acad Sci U S A. 2014;111(7):2494–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Li Y, Wu H, Wu W, et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014;24(6):762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Turco E, Savova A, Gere F, et al. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat Commun. 2021;12(1):5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Liu Y, Soetandyo N, Lee JG, et al. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. Elife. 2014;3:e01369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhao Y, Feng Z, Zou Y, et al. The E3 ubiquitin ligase SYVN1 ubiquitinates atlastins to remodel the endoplasmic reticulum network. iScience. 2020;23(9):101494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Aligeti M, Behrens RT, Pocock GM, et al. Cooperativity among Rev-associated nuclear export signals regulates HIV-1 gene expression and is a determinant of virus species tropism. J Virol. 2014;88(24):14207–14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.