

Summary
Enzymatic hydrolysis holds great promise for plastic waste recycling and upcycling. The interfacial catalysis mode, and the variability of polymer specimen properties under different degradation conditions, add to the complexity and difficulty of understanding polymer cleavage and engineering better biocatalysts. We present a systemic approach to studying the enzyme-catalyzed surface erosion of poly(ethylene terephthalate) (PET) while monitoring/controlling operating conditions in real time with simultaneous detection of mass loss and changes in viscoelastic behavior. PET nanofilms placed on water showed a porous morphology and a thickness-dependent glass transition temperature (Tg) between 40°C and 44°C, which is >20°C lower than the Tg of bulk amorphous PET. Hydrolysis by a dual-enzyme system containing thermostabilized variants of Ideonella sakaiensis PETase and MHETase resulted in a maximum depolymerization of 70% in 1 h at 50°C. We demonstrate that increased accessible surface area, amorphization, and Tg reduction speed up PET degradation while simultaneously lowering the threshold for degradation-induced crystallization.
Keywords: catalytic degradation, plastic waste, enzymatic depolymerization, circular plastic economy, Langmuir monolayers, PET hydrolases, Ideonella sakaiensis, interfacial biocatalysis, film thickness, glass transition
Graphical abstract
Highlights
-
•
The Tg of PET in contact with water decreases to 40°C in nanometric films
-
•
PET turnover rate decreases above 50°C with a thermostable IsPETase variant
-
•
Incomplete PET degradation coincides with surface deposition of deactivated enzymes
-
•
Degradation-induced surface crystallization is not seen in thin films up to 60°C
The bigger picture
While the majority of plastic waste is landfilled, incinerated, or recycled—in that order—a large portion of ∼29% has an unknown fate: about 4% reaches the oceans, and other large amounts enter the biosphere as micro- and nanoplastics derived from environmental degradation. In a short period of time, microorganisms may have evolved various mechanisms to address these xenobiotics, such as biocatalytic depolymerization of plastics and assimilation of their monomers. Our research reveals that surface-layer degradation plays a crucial role in biocatalytic PET depolymerization. The change in physicochemical properties of a nanoscale thin film during biocatalytic degradation was monitored to mimic the surface-erosion process of bulk polymers. Our findings will strengthen the mechanistic understanding of biocatalytic polymer degradation in general and may serve as the basis for optimizing bioremediation strategies (e.g., for accumulated plastic pollution in wastewater treatment plants).
Enzymes can mostly access the surface layer of PET during depolymerization. We show that an amorphous PET thin film with comparable properties to the PET surface layer has a lower glass transition temperature (Tg) of 40°C than bulk PET’s Tg of 65°C–81°C. As a result, within a few hours at 40°C–50°, this PET thin film was rapidly depolymerized by an IsPETase variant. Our findings will be beneficial for industrial implementation of enzymatic plastic degradation using properly treated polymers.
Introduction
Seven types of plastics account for 80% of global plastic demand, with low- and high-density polyethylene (LDPE and HDPE, respectively), polypropylene (PP), and poly(ethylene terephthalate) (PET) being the top contributors.1,2 Due to the short lifetime of PET-based products used for packaging, approximately 95% of PET manufactured is discarded within a year.2 The waste processing industry relies on primary recycling—reprocessing to give a product that can be used for the same initial purpose—and secondary recycling (or mechanical “downcycling”) to produce lower-value materials for different uses from the original one. Current stringent regulations and legislative encouragement for recycling are opening new venues for the implementation of alternative and/or optimized schemes for the collection, sorting, and processing of PET waste to achieve a circular plastic economy while contributing to sustainable development.3,4,5 For instance, the EU directive (219/904) set a collection target of 65% recycling (excluding incineration) for single-use plastic bottles by 2025; moreover, these plastic bottles should at least contain 25% recycled plastic in their manufacture by 2025 (for PET bottles) and 30% by 2030 (for all bottles).3
As an emerging technology holding industrial promise, biocatalytic PET recycling targets the depolymerization of PET into monomers terephthalic acid (TPA) and ethylene glycol (EG) or low-molecular-weight oligomers such as mono-(2-hydroxyethyl) terephthalate (MHET). These small molecular degradation products can be either repolymerized to synthesize virgin-quality PET or transformed into products with significant added value (upscaling).6,7,8,9,10,11 Depolymerization can be achieved with genetically engineered microorganisms to produce polyester hydrolases and other key enzymes to metabolize PET building blocks as a carbon source.7,12,13,14,15 For a long time, PET was considered non-biodegradable (i.e., degradation by microorganisms), but Müller et al. changed that when they reported in 2005 the first PET hydrolase, a cutinase produced by the bacterium Thermobifida fusca.16 Since then, many PET hydrolases have been reported to degrade amorphous PET materials, including IsPETase, discovered in Ideonella sakaiensis 201-F6 bacterium, which also produces a MHETase that catalyzes the hydrolysis of MHET to TPA and EG but is not active on PET polymers.17 Despite this progress, most studies do not reach full degradation of PET specimens at given conditions, hindering the potential for technical implementation. The “passivation” or incomplete degradation of the polymer in response to enzymatic degradation has been attributed to either (1) the degradation-induced polymer crystallization or (2) the deactivation of enzymes through conformational changes.6,18,19 The latter could be induced either thermally or through interactions with the polymer and its degradation intermediates. Many publications in the field agree that pretreatment of PET improves the achivable efficiency and extent of degradation by engineered enzymes by reducing the polymer crystallinity (amorphization).6,20,21 A few publications have also suggested a relationship between the maximum turnover number and the particle size of micronized polymers.6,22,23 A recent paper published during the revision of this article suggests that while amorphization of PET is a necessary pretreatment step for degradation using a variant of the leaf compost-cutinase enzyme (LCC-ICCG), the particle size reduction has no appreciable effect on the overall degradation performance.23 Moreover, it is generally accepted that reaction temperatures, preferably above the glass transition temperature (Tg; 65°C–81°C reported for bulk PET in literature), can also increase catalytic rates.24,25,26 Although efforts in enzyme engineering succeeded in increasing the thermal stability of PET-hydrolyzing enzymes, enabling the use of a wider variety of enzymes at or above the Tg range of bulk PET,21,27 the mechanistic implications of amorphization, film thickness/particle size, temperature, and water as standard reaction media on the activity of these enzymes and on the passivation of the polymer are still not well understood. Thus, the selection conditions or rules for efficient enzymatic degradation of PET remain uncertain.
Since enzymes are confined to the polymer surface, and degradation proceeds as a surface erosion process, answering open questions on PET degradation might require a different perspective of interfacial biocatalysis to visualize the effect of the polymer surface amorphization, the plasticizing effect of water, and the Tg of the surface layer (Tgs) on the catalysis reaction. Given that Tgs can be lower than the one in bulk,28 due to the difference between surface and bulk chain entanglement and packing behaviors,29,30 the temperature range between Tgs and Tg could offer a sweet spot where the chains at the surface are mobile and degradable, while the chains in bulk are not yet able to crystallize, enabling enhanced reaction rates.
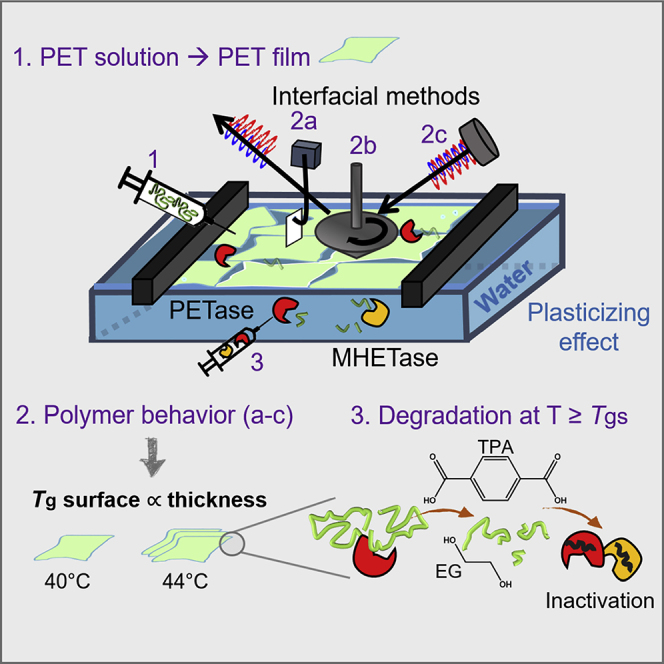
Langmuir-Blodgett monolayer degradation (LMD) systems present an in situ method to study interfacial hydrolysis of nanometric-thickness films, which resembles the surface erosion process of bulk polymers (Figure 1, considerations). This is possible by making use of detection systems to measure the properties of the thin layers before, during, and after degradation, including surface tension, rheological properties, infrared molecular fingerprints, microscopy, and area reduction.31 In this way, it is possible to identify the different stages of enzymatic catalysis (Figure 1, lateral view I–IV), as a function of area reduction versus time (Figure 1, characterization A), as well as the changes of the polymer physicochemical properties during depolymerization, e.g., viscoelasticity and crystallinity (Figure 1, characterization B).
Figure 1.

Overview of the interfacial degradation of PET by a dual-enzyme system
In bulk systems exposed to water, sorbed water molecules penetrate amorphous regions of the surface, lowering the glass transition temperature (Tg), due to a plasticizing effect (considerations).20,28,32 In a Langmuir degradation experiment, the polymer layer, which lies on top of the aqueous surface, forms a thin layer with a thickness of 2.5–7 nm, anticipating a further decrease in the polymer Tg and an increase in chains mobility at T = Tg.. At T = Tg, the binding of the enzymes, injected in the aqueous subphase under the formed layer, and the depolymerization mechanism (degradation, lateral view I–IV) can be monitored by interfacial rheology, infrared reflection absorption spectroscopy (IRRAS), and Brewster angle microscopy (BAM) (characterization). The dataset includes the decrease in the film area as a funtion of time (Characterization A), and the changes of storage and loss modulus thorughout the experiment (Characterization B).
Results and discussion
PET thin films form 2D non-organized porous-like structures
Thin films of PET were deposited on a liquid surface (air-water interface [A-W interface]) supported by a Langmuir trough and characterized as depicted in Figure 1 and explained in the experimental procedures. Similar to water-insoluble amphiphiles, PET surface pressure (π) versus mean molecular area (MMA; defined as area occupied per polymer repeating unit) compression isotherms (π-A isotherms) yield information on the film structure and the mechanism of its formation, as the number of contacts established between chains in the layer is linked to π.
The π-A isotherm and Brewster angle microscopy (BAM) analysis showed that PET generates free islands at the A-W interface upon spreading, which are observed at almost zero surface pressure by BAM (Figure 2). During compression by closing the barriers, the islands get into contact, increasing the π values (Figure 2A). As a result of the stress accumulation, a rearrangement occurred to form a continuous close-packed monolayer on the water surface. Such a scenario is verified by the analysis of the shear resistance of the layer. The storage modulus (G′), as an elastic part of the shear response, starts to increase once the islands are closely packed. The loss modulus (G′′), representing the viscous or dissipative part of the shear response, only increases slightly over the value of bare water since the compact PET layer behaves like an elastic solid (Figure 2A). Similar assemblies have been observed for nanoparticle Langmuir films.33 However, a complete filling of the layer was not possible due to the glassy state of the polymer (Figure 2B). The onset of the π increase in the π-A isotherm at 14 Å2 suggests the formation of dimer aggregates at the molecular level, assuming that one PET repeating unit lying flat atop water has a molecular area of 35 Å2 in a monolayer (10 Å for the repeating unit and 3.5 Å for the typical van der Waals distance between chains). PET is expected to be prone to aggregation at the A-W interface as it is in the case with complex amphiphiles for which the A-W interface is a poor solvent.34 This is most likely due to aromatic ring π–π stacking. Interfacial rheology data suggest a phase transition above 10 mN/m, as seen by an increase of both G′ and G″, which could be associated with the maximal lateral packing density (MLPD) of PET film (Figure 2A). Once the MLPD is reached, the film stiffens as it is forced out of the A-W interface to form a second layer, which is observed as considerably thicker structures on the BAM (Figure S1).
Figure 2.

Formation of two-dimensional (2D) PET thin films
(A) π-A isotherm (black line) and G″ and G′ as a function of mean molecular area (orange and pink lines) measured at the A-W interface.
(B) In situ BAM images taken at given points marked in the isotherm; 50 μm scale bar. See also Figure S1.
(C) AFM images of films transferred to Si wafers at point (c). From left to right: 1 × 5 (1 μm scale bar) and 1 × 1 μm scan (200 nm scale bar).
(D) Height count for AFM images.
The formation of a stable monolayer formed by compacted polymer islands was confirmed also by atomic force microscopy (AFM) studies of the films transferred onto solid substrates at stage π = 6 mN/m (Figure 2C). The film is approximately 2.5 nm thick (Figure 2D) and exhibits a porous-like appearance within the islands with heterogeneous micro- and nanoholes (50–250 nm diameter).
Furthermore, the chain conformations and orientations were assessed by polarization-modulated infrared reflection-absorbance spectroscopy (PM-IRRAS) at the interface (Figure S2). According to the PM-IRRAS selection rules at the AW interface, a positive peak from a group indicates that its dipole moment is oriented parallel to the interface, while a dip in the signal is a sign of a perpendicular orientation.35 The strong positive peak at 1,720 cm−1, which is attributed to the stretching vibration of the ester carbonyl group,30 with a vibrational dipole moment that is oriented nearly perpendicular to the main-chain axis of PET molecules and parallel to the plane of the phenyl ring, indicates that the polymer main chain is aligned parallel to the free surface, as reported by other authors.30,36 This orientation has been designated as oriented oblate ellipsoid conformation.30 The two bands at 1,342 and 1,370 cm−1 are assigned to the CH2 (in the O–CH2–CH2–O– moieties) wagging mode in trans and gauche conformations of the EG moiety.37,38 In semicrystalline and crystalline PET, the peak at 1,342 cm−1 is stronger than that at 1,370 cm−1. In our thin films at 21°C, a stronger signal of the trans conformer at 1,342 cm−1 was observed. However, we could not observe a notable degree of crystallinity of PET thin films at the A-W interface by electron diffraction and AFM, suggesting that the slightly higher signal of the trans conformation of the 2.5 nm film at the A-W interface is related to effects other than crystallization. For instance, the flattening of the polymer coils near the interface or a confinement effect of thin films could cause a higher number of trans conformers.30 Interestingly, others authors have reported hindered crystallization of PET surface layers (13.6 nm) at temperatures below 70°C, despite the Tg being detected around 48°C.28
The Tgs of PET decreases with decreasing thickness at the A-W interface
PET degradation through enzymatic hydrolysis has an interfacial nature and is promoted at temperatures close to or above the Tg of the PET material. The large surface-to-volume ratio in the thin films and the presence of a water-polymer-air interface enable us to explore the free surface and the role of the interface on the physicochemical parameters of PET. Using the Langmuir approach, PET films were formed at different packing densities ranging from MMA 15 to 12 Å2, equivalent to surface pressures between 2 and 9 mN/m, and the Tg of the films was measured in situ using an interfacial rheometer coupled to the Langmuir trough. The films at the A-W interface were subjected to a heating cycle from 21°C to ∼45°C, and the Tg was calculated as the intersection of two tangent lines from the plot of the logarithm of G′ versus linear temperature (Figure 3A). This analysis is similar to some ASTM methods, e.g., D 7028 for the onset Tg. Deposited on an aqueous subphase, the Tg of PET thin films (which refers to surface Tgs) was found to be between 40°C and 44°C, >20°C lower than that of the bulk. These results were also corroborated by the temperature at the peak of the tan(δ), 45°C ± 2°C, which is an alternative measurement to determine Tg using rheology (Figure S3). The low Tg of our film is associated with both the plasticizing effect of water molecules and the thickness of the films. The plasticizing effect of water has been evidenced for PET in a range of temperatures and humidity values,20,28,32,39 and its effect in lowering melting temperatures of semicrystalline polyesters on Langmuir films has been reported by our group.40 It has been also observed by several authors that Tg of amorphous PET films prepared on solid substrates (e.g., Si wafers) is lower than its bulk (between 65°C and 71°C) and decreases with the reduction of the film thickness, leading to a Tgs as low as 48°C for a surface thickness of 13.6 nm, as described by Shinotsuka et al.28
Figure 3.

Determination of the Tg of PET ultrathin films on an aqueous subphase
(A) G′ versus temperature plot at different surface pressures, indicating the first and second tangent lines for calculation of Tg. See also Figure S3.
(B and C) Influence of π and MMA on Tg of PET ultrathin layers at the A-W interface.
Interestingly, the onset of Tgs increased slightly as the layers were compressed to higher surface pressures and lower MMA following a linear relation (Figures 3B and 3C). This is most likely due to direction-oriented stress, which decreases the free volume and the flexibility of the polymer, introducing significant changes to the thermophysical properties of PET thin films. Pressure dependence of Tg is also observed in bulk, and this effect is typically on the order of 0.04 K/bar. The pressures in a Langmuir film can be calculated by dividing surface pressure through film thickness, so for a film with a thickness of 2.5 nm, a surface pressure of 1 mN/m corresponds to ca. 0.4 MPa = 4 bar. That leads to an estimate of 0.16 K/mN. The dependence of Tg on π observed for PET is 0.48 K/mN, which is somewhat higher but in the right order of magnitude.
The thicknesses of PET films prepared at 10°C, 21°C, and 42°C (all compressed to 6 mN/m), were measured by ellipsometer as 3.22 ± 0.2, 5.22 ± 0.43, and 6.58 ± 0.86 nm, respectively (Figure S4). These results are slightly higher than the AFM values because the bulk refractive index was used, which would need to be adjusted for ultrathin films. Overall, our results show a direct correlation between temperature, thickness and the onset of Tgs in ultrathin films solely in contact with an aqueous subphase. We also prove that much larger reductions in Tg are observed in the absence of interaction with solid substrates, as suggested by previous studies with other polymers and freely standing thin films (poly(methyl methacrylate) [PMMA]).41 With these results, we emphasize the benefits of using the Langmuir technique to infer changes in the dynamics of polymer thin films when an aqueous environment is intended for its application/degradation to avoid discrepancies from the polymer-subtract interaction.42
PET ultrathin porous layers do not crystallize at temperatures close to surface Tg
At temperatures close to Tg, depolymerization of PET is usually associated with surface crystallization, which consequently restricts enzymatic degradation. To evaluate the thermal behavior of thin layers at the A-W interface at their Tgs and the changes induced by “physical aging” (prolonged exposure to temperatures close to Tg), we used a combination of different analytical methods. Rheological monitoring was applied to PET ultrathin films (obtained at 6 mN/m) while undergoing 3 cycles of heating from 21°C to 45°C with following incubation duration times of 40, 80, and 100 min at 45°C (Figure 4A) at the A-W interface. The films before and after annealing at 45°C were transferred to Si wafers or copper grids for analysis by AFM and transmission electron microscopy (TEM), respectively (Figures 4B–4D). During annealing, the films lost about 5% of their initial area after each incubation cycle (MMA, black line in Figure 4A). G′ sharply decreased when Tg was reached, caused by activation of molecular segmental motions, yet similar values were obtained at the end of each cycle (orange line in Figure 4A) after the area lost was compensated by compression of the barriers. The AFM and TEM analyses of the film transferred at 21°C before heating (Figures 4B–4D) confirmed our previous observations that the layer is rather smooth with micro- and nanoholes. The ripples originate from the film transfer procedure. The large holes are caused by water condensation on the underlying substrate during transfer at elevated temperatures, resulting in parts of the film not being adsorbed to the substrate and being removed in the subsequent washing step.
Figure 4.

Characterization of PET ultrathin films at temperatures close to surface Tg
(A) Area variation (black lines) and G′ measurement (orange line) over time during heating and cooling cycles (red line). The layer was transferred at points (a)–(e).
(B and C) AFM of films transferred at points (a) and (e). From left to right: 1 × 5 and 1 × 1 μm scan. Scale bar, 3µm and 820-800 nm.
(D) Electron diffraction of films transferred to copper grids at points (a) and (e). Scale bar, 200nm.
It was also observed that the layer became rougher after annealing at up to 45°C (Figure 4C); however, no out-of-plane crystals were formed, and electron diffraction measurements of the annealed films on copper grids did not show a defined diffraction pattern (Figure 4D). While it is well established that cold crystallization of PET is inhibited in ultrathin films (<20 nm) that are confined between two solid walls,43 our results suggest PET crystallization in ultrathin films is inhibited even at the A-W interface. Still, as seen by the reduction of the film area and the change in film morphology, physical aging at Tg takes place.
Degradation of ultrathin films follows combined endo- and exo-mechanisms
The PETase from I. sakaiensis (IsPETase) is one of the most efficient PET hydrolases at ambient conditions. The IsPETase triple mutant (IsPETaseTM) used in this study with a Tm (mid-point of thermal denaturation) of 56.6°C44 showed a quite high affinity to the A-W interface, as indicated by an increase in π in the Langmuir trough (Figure S5) induced by low concentrations of enzyme, suggesting a remarkable affinity for hydrophobic PET and agreeing with recent findings.45
After the addition of IsPETaseTM beneath the polymer thin film at a final concentration of 0.25 μg/mL, four degradation stages could be defined by calculating the loss in area (A t/A0∗100), G′, and G″, as well as by microscopical characterization by BAM (Figures 1 and 5A). In stage I, a fast decrease in G′ and G″ after injection of the enzyme at constant area is observed, showing firstly a high affinity of IsPETaseTM to the thin PET film at the interface, most likely mediated by non-specific adsorption to the hydrophobic polymer surface, as suggested elsewhere.25 Dynamic-mechanical measurements by rheology offer valuable information on the molecular structure of polymers.46 At constant temperature, the decrease of G′ due to depolymerization in stage I at almost constant area (At = A 0) is consistent with a random-scission behavior (represented in Figure 1I).47,48,49,50 In random scission of polymers, the probability of exo-scission (cuts at chain ends) resulting in soluble monomers is inversely proportional to the chain length. For PET with a typical degree of polymerization of 200, the initial probability for exo-scission is much lower than for endo-scission. The fragmented chains generated by endo-scission remain at the A-W interface (Figure 1, I), given that only monomers (TPA and EG) and oligomers (MHET and BHET) are considered to be water soluble. While the chain length decreases close to exponentially during random scission of PET chains,51 clearly visible through the drop of G′, the number of chain ends available for exo-scissions increases drastically, which initiates stage II (mass loss phase) (see also Figure 1, II). The evolution of chain end formation can be observed by fitting a degradation curve using an analytical model and Monte Carlo simulations of polymer degradation52 (Figure S6). The softening of the film during stage I also enabled a rupture and compaction of the film by the externally applied compression as observed by BAM (Figure 5C). The dissolution of the small fragments from exo-scissions causes a decrease in surface pressure, which, under isobaric conditions, is compensated by an area reduction (At = A 0). Here, area loss is equivalent to mass loss in bulk degradation. As degradation accelerates, in stage III, the compression of the layer to compensate for the loss of surface pressure at the interface increases G′ and G″ until both surface pressure and moduli stabilize at stage IV due to the termination of degradation (Figure 1, IV). A compact film is again observed by BAM. It is worth noticing that at this point, when a maximum of 45% reduction of the film area is reached, the addition of more enzyme does not re-initiate degradation. In the following, we evaluate possible causes for the incomplete degradation of the PET monolayers, in striking similarity to the often-observed incomplete enzymatic degradation of bulk PET.
Figure 5.

Characterization of PET films upon degradation by IsPETaseTM
(A and B) Area variation (blue lines), G″ and G′ measurement (orange line), and compression speed (gray line) over time during degradation of PET by IsPETaseTM at a final concentration of 0.25 (A) and 0.5 μg/mL (B).
(C) In situ BAM images during stage I degradation. Time indicates the incubation time with the enzyme, and the arrowheads show selected locations on the film being degraded through time. Scale bar, 50 µm
(D) AFM of films transferred at the end of degradation (stage IV) at 40°C (left) and 50°C (right). From left to right: 1 × 5 and 1 × 1 μm scan. The dark regions represent holes in the film due to transfer at high temperatures.
Contact-angle measurements of films transferred after degradation showed that the layer becomes slightly more hydrophilic after degradation due to an increase in carboxylic acid and hydroxyl residues by hydrolysis and the presence of enzymes (Table S1). Moreover, we used a doubled excess concentration of enzyme in the subphase to assess if there was an enzyme saturation by an excess of substrate and obtained a longer reaction time in stage I but a similar maximum extent of depolymerization at 45% (Figure 5B). The longer reacting time in stage I, together with a fast decrease in G′, indicates that more cleavage sites were being attacked when a 2-fold concentration of enzyme was used, generating shorter PET chains with lower molecular weight.
At the termination of degradation, AFM of the films transferred to Si wafers (Figure 5D) showed a quite interesting film topography, with filament-like structures, contrary to the rough films observed when the films were transferred at 40°C in the absence of the enzymes (Figures 4B and 4C). As observed in the stepwise thermal annealing experiment (Figure 4A), the film does not crystallize (no diffraction pattern observed in TEM) but undergoes physical aging, which would certainly be accelerated by the mobility enabled by reduced average chain length and the external compression force. A denser packing of the chains can reduce the number of chain segments with enough conformational freedom to bind to the substrate binding pocket of the PET hydrolases. In addition to physical aging of the polymer, a change in the film topology can be caused by enzymes that undergo adsorption-induced denaturation and aggregation once they reached the interface and are compressed within the film. This (partly) enzymatic origin of the structures found on the film surface was confirmed by staining with the amine-reactive dye N-hydroxysuccinimide (NHS)-Fluorescein (Figure S7) and is also in line with their more pronounced appearance after degradation at 50°C (Figure 5D) when compared with 40°C. The denatured protein inhibits the adsorption of active enzymes, thereby preventing further degradation (Figure 1, IV). Along this line, we further investigated the role of product inhibition of IsPETaseTM by MHET (Figure 1, III) due to the lack of the helper enzyme MHETase as an alternative explanation for incomplete degradation.
Addition of IsMHETaseSM increases degradation by approximatley 20%
As the second key enzyme for the PET-assimilating activity of I. sakaiensis, IsMHETase can hydrolyze MHET to terephthalate and EG53 and is recently shown to have very limited activity on the chain-end esters in PET polymers.54 IsMHETase single mutation (IsMHETaseSM; single mutation: MHETaseW397A) used in this study showed no degradation performance on polymer chains without IsPETaseTM after 30 min of contact with the thin film at 40°C (Figure 6A). Following the subsequent addition of IsPETaseTM to the subphase (0.25 μg/mL), the above-mentioned degradation stages (I–IV) with only IsPETaseTM were observed. Degradation by both enzymes reached a maximum extent of 50%–60% area reduction at 60 min after the injection of IsPETaseTM. This time was shorter than the time for degradation using IsPETaseTM alone, confirming the synergistic relationship between both enzymes for degradation of PET. The AFM of the films transferred in the plateau region at the end of degradation showed similar structures as those detected for IsPETaseTM alone (Figure 6A). When the degradation temperature was increased to 50°C (Figure 6B), around 10% more area was lost from the interface, and degradation proceeded 5 times faster when compared with IsPETaseTM + IsMHETaseSM at 40°C (Figure 6B). In addition, the coverage of the Si wafer by the transferred polymer film was significantly lower, as observed by AFM (Figure 6D), consistent with the higher degradation extent; moreover, fewer aggregates were detected. At 60°C, the enzymes denatured rapidly, causing an increase in π and a considerably lower hydrolysis performance at the A-W interface than when incubated at lower temperatures (Figure 6B). We conclude that to maximize the mass loss, it is necessary to achieve a balance between the lifetime and the activity of the IsPETase, where the latter is clearly enhanced in the presence of IsMHETase. When transferring our findings to bulk PET, our findings support previous evidence on increasing the surface area of PET and reducing its crystallinity via preprocessing below a critical size to speed up degradation rates while reducing the threshold for degradation-induced crystallinity.
Figure 6.

Degradation of PET by IsMHETaseSM and IsPETaseTM enzymes
(A) Area variation (blue line) and G″ and G′ measurement (orange lines) over time during degradation of PET by IsPETaseTM and IsMHETaseSM at 40°C.
(B) Area reduction curves of PET degradation by IsPETaseTM and IsMHETaseSM at 40°C, 50°C, and 60°C.
(C) AFM images of films transferred after degradation in (A).
(D) AFM images of films transferred after degradation at 50°C in (B). AFM images from left to right: 1 × 18.8, 1 × 5, and 1 × 1 μm scan. The holes in the films are due to transfer at high temperatures. Confocal microscopy images of transferred films stained with amine reactive fluorescent probe N-hydroxysuccinimide (NHS)-Fluorescein suggests the presence of enzymes as the cause of the filament-like structures observed by AFM (see Figure S7).
Kinetic analysis of PET-degradation process
Having excluded surface crystallization as a major cause for the incomplete degradation of the PET ultrathin film, we infer that the enzymes have been deactivated during our experiments (stage IV). Deactivation could be induced either thermally or through interaction with the polymer and its fragments. In a previous study,55 we observed a deactivated enzymatic degradation process of lactide/glycolide copolyesters and derived an appropriate kinetic model based on the assumption that the activity of enzymes decreases exponentially either by thermal deactivation or through the catalytic activity. The model was used to extract the reaction and deactivation rate constants from the monolayer dissolution curves (Table S2). For the biocatalysis with IsPETaseTM at 30°C to 50°C, the deactivation rate constant was greater than that for the polymer degradation. The degradation rate constant went through a maximum at 40°C, whereas the deactivation rate constant was in reasonable agreement with Arrhenius’ law and an Arrhenius energy barrier of about 100 kJ/mol. This is a rather low deactivation energy barrier for enzymes, but the unfolding energy of cutinases with PET degrading activity is even lower.56 Based on the model, the optimum temperature for PET degradation with IsPETaseTM is 40°C, which coincides with the onset of the Tg of the ultrathin PET Langmuir films. In that regard, the greater segmental mobility of the molecules in the surface layer, due to lower intermolecular interaction forces57 in combination with the plasticizing effect of water, seems to be an important factor enabling the in vitro degradation of PET by mesophilic hydrolases. When IsPETaseTM is combined with IsMHETaseSM, the highest degradation performed was achieved at 50°C, where the reaction and deactivation rates are higher than that of the IsPETaseTM alone. The current work shows that the Langmuir technique can help to gain the necessary fundamental insights also for polyesters that have very little amphiphilicity, hinting at its applicability to other classes of hydrolysable polymers such as polyamides or polyurethanes.
Conclusions and outlook
Our study highlights that the highest enzymatic activity of the mesophilic IsPETase coincides with the glass transition of the thin films and polymer surfaces plasticized by water (between 40°C and 48°C). IsPETase has a high affinity for the PET-water interface, initiating a rapid endo-scission or random depolmerization (Figure 1, I) that evolves into an exo-scission where soluble fragments from the chain-end cuts are dissolved in the water (Figure 1, II). The degradation products lower the reaction rate, which is mitigated by the second enzyme MHETase (Figure 1, III∗). Finally, thermal deactivation, which requires a low activation energy, leads to formation of protein deposits on the PET surface that block further degradation and induce a “passivation” of the surface (Figure 1, IV), even when the polymer remains amorphous. With these results, we provide insights on the main factors lowering the depolymerization rate of PET surfaces, which are named surface Tg, product inhibition, and surface deactivation of enzymes. Moreover, the high relevance of the thermal transitions of the polymer surface and its impact on the enzymatic degradation underlines the necessity for a holistic approach to polymer degradation. We advise that the combination of a rational design of the enzymes targeting the state in which the polymer is most degradable, suitable polymer pretreatments, and a continuous depolymerization process where small fragments and inactivated enzymes are removed/recovered could lead to improved industrial applicability of plastic recycling. This is imperative given the exponential accumulation of plastic waste, which has raised official awareness at the political, social, and scientific levels and is a clear threat to the UN’s Sustainable Development Goals.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Natalia A. Tarazona (natalia.tarazona@hereon.de).
Materials availability
This study did not generate new unique reagents.
Materials
Amorphous PET sheets of 2 mm thickness, sized 150 × 150 mm, were obtained from Goodfellow (product number GF21713734). Polymer solutions for the Langmuir experiments were prepared using hexafluoro-2-propanol ≥99% (HFIP) from Sigma-Aldrich (CAS 920-66-1). Thermophysical properties of PET were characterized by differential scanning calorimetry on a Netzsch DSC 204 Phoenix (Selb, Germany) at heating and cooling rates of 10 K min−1. Samples were weighed directly into hermetic aluminum pans. Measurements were taken during the first cooling and second heating run in a temperature range from −100°C to +200°C to determine the thermal behavior of bulk PET (Figure S8). Given the amorphous state of PET, only a quite smooth Tg was observed around 60°C.
Expression and purification of IsPETaseTM and IsMHETaseSM enzyme mutants
Plasmids harboring genes encoding IsPETaseS121E/D186H/R280A (IsPETaseTM)58 and MHETaseW397A (IsMHETaseSM)17 were transformed into chemically competent E. coli SHuffle T7 Express cells (New England Biolabs, Frankfurt am Main, Germany) and spread onto Luria-Bertani (LB) agar plates containing 100 μg mL−1 ampicillin. The plates were incubated overnight at 30°C. One colony was picked and used to inoculate an overnight culture containing 100 μg mL−1 ampicillin. Cultivation was performed in LB medium containing 100 μg mL−1 ampicillin. The LB medium was inoculated with overnight culture and incubated at 30°C and 160 RPM until an optical density (OD600) of 1 was reached. Expression of both enzymes was then induced by the addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). Cultivation was then continued for IsPETaseTM at 16°C and 160 RPM overnight. For the expression of the MHETase mutant, the temperature was reduced to 16°C when an OD600 of 2.5 was reached. Cultivation was then continued overnight. Cells were harvested by centrifugation at 3,500 × g and 4°C for 50 min. The cell pellet was washed with 50 mM TRIS-HCl buffer (pH 7.5) and subsequently stored at −20°C until cells were lysed. Cells were resuspended in buffer A (50 mM TRIS-HCl [pH 7.5], 150 mM NaCl, 10 mM imidazole) and disrupted by ultrasonication. Cell debris was then removed by centrifugation at 10,000 × g and 4°C for 20 min. Purification was performed with the ÄktaPure chromatography system (GE Healthcare Europe, Freiburg, Germany) using a HisTrapTM FF crude 5 mL column (Cytiva Europe, Freiburg, Germany). Undesired proteins were first washed away with buffer A and then with buffer A supplemented with 100 mM imidazole. Elution of IsPETase and MHETase mutants was then performed with buffer A supplemented with 200 mM imidazole. Elution fractions containing the protein of interest were pooled and concentrated using Vivaspin 6 centrifugal concentrator (10 kDa MWCO, Sartorius, Göttingen, Germany). PD-10 Desalting Columns (Cytiva Europe, Freiburg, Germany) were used to exchange the elution buffer to 50 mM TRIS-HCl buffer (pH 7.5). Protein content was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA).
PET thin film formation
PET Langmuir ultrathin films were prepared on a polytetrafluoroethylene Langmuir trough with a maximum surface area of A = 550 cm2 and two Delrin barriers for symmetric compression (KSV NIMA, Espoo, Finland). π-A isotherms were recorded with an electrobalance and a platinum Wilhelmy plate (perimeter 3.94 cm) as a surface pressure sensor. Milli-Q deionized water or PBS buffer (pH 7.4) were used as the subphase. As PET is badly soluble in common solvents except for HFIP, which cannot be used for Langmuir experiments alone, the working spreading solution was prepared mixing 5 μL of a PET:HFIP solution (10 mg/mL) with 250 μL chloroform:DCM (3:1). The PET-HFIP solution was sonicated at 40°C before use. After mixing, the solution was immediately spread on the aqueous surface to achieve a final concentration of 0.16 mg/mL PET. After spreading, the solvents were allowed to evaporate for 10 min before the measurements. For π-A isotherms, the layers were laterally compressed at constant rates of 10 mm/min. The area per molecule (MMA) for PET was calculated based on the average weight of the repeating units (one TG and one EG molecule) and the surface area of the trough during compression. π-A isotherms were repeated at least three times. All presented isotherm data correspond to individual experiment data reproducible with a random measurement error of ≈5% concerning the π or the MMA values for the independently repeated experiments.
PET thin film characterization
Rheology, IRRAS, and BAM measurements at the A-W interface were achieved by coupling the instruments to the same Langmuir trough setup described above. These measurements track macroscopic behavior stimulated by the changes of the atomic and/or molecular order at micro- or nanoscale. Rheology experiments were carried out with an Interfacial Shear Rheometer (IRS, model MCR502) from Anton Paar (Graz, Austria), which consists of a biconical disk coupled to a driving motor and to a torque and normal force transducer unit. Measurements were carried out at a defined strain of 1% and an oscillation frequency of ω = 6.2 rad s−1 s (1 Hz). The detailed protocol has been described before.50 The storage modulus (G′) accounts for the elastic component, and the loss modulus (G″) for the viscous component of the response to oscillatory shear. BAM measurements were performed with an Ellipsometer Accurion Nanofilm EP3 (EP3, Accurion, Göttingen, Germany). The instrument was equipped with a 658 nm class IIIB laser source and a high-performance CCD camera (1,392 × 1,040 pixels). Images were taken simultaneously during π-A isotherms and degradation experiments with a resolution limit of 1 μm. IRRAS measurements were carried out with a PM-IRRAS spectrometer (Biolin Scientific, Espoo, Finland) The photoelastic modulator was set to achieve a phase shift between p- and s-polarized light of at 2,900 cm−1. The angle of incidence was set to 74°. The integration time was 500 s. For ellipsometry, TEM, and AFM analyses, the monolayers were transferred onto a Si wafer directly from the A-W interface using the Langmuir-Schäfer technique.
Determination of PET Tg on an aqueous surface
Similar to dynamic mechanical analysis, the Tg of a polymer can be determined via rheology, where the temperature of the specimen is increased while applying a small-amplitude shear deformation to measure the dynamic moduli G′, G″, and tan(δ). Rheological and dynamic mechanical analysis (DMA) techniques are particularly sensitive to the glass transition compared with differential scanning calorimetry (DSC) and thermomechanical analysis (TMA). PET films prepared at different surface pressures were subjected to a heating cycle from 21°C to ∼45 °C at a heating rate of approximately 2.5°C/min. The Tg of the films was determined as the intersection of two tangent lines from the plot of the logarithm of G′ versus linear temperature. The first tangent line is manually selected at the temperature before the transition, and the second tangent line is constructed at the inflection to mid-point of the modulus drop.
Ellipsometry measurements
AFM was used to examine the surface topography and nanoroughness of the films transferred to solid substrates using an AFM from Asylum Research (Santa Barbara, CA, USA) equipped with a silicon cantilever (OLYMPUS OMCL AC160TS-R3). The scans were evaluated with ImageJ software.
Degradation experiments on the Langmuir trough
Before degradation experiments, all enzymes used were tested for their surface activity to determine the concentration limit, above which the enzymes have an inherent affinity for the interface. The surface activity of IsPETaseTM, a catalytically improved variant of I. sakaiensis PETase in Glycine + NaOH 50 mM (pH 9) was about 3 times higher than PBS buffer, the latter being quite favorable for interfacial studies. The concentration limit for IsPETaseTM was detected at 1 μg/mL in PBS buffer at 40°C (Figure S5). The IsMHETaseSM surface activity limit was detected at 0.5 μg/mL. To evaluate the enzymatic degradation of PET, ultrathin layers were formed at the A-W interface on PBS buffer (pH 7.4) at 21°C (approximately 50 μg of PET), followed by a layer compression step to reach 6 mN/m and a further increase in temperature to 40°C (or 50°C). After stabilization for about 30 min at constant surface pressure, the enzymes are injected under the films. During degradation, the surface pressure is maintained constant, therefore the loss in surface pressure due to degradation is compensated by the reduction in the area using the movable barriers in the through. The data are presented as area reduction versus time (At/A0)∗100. A = 100% corresponds to the area at which the enzyme was inserted.
Confocal microscopic imaging
Films transferred to Si wafers were subjected to NHS-ester labeling using amine reactive fluorescent probe NHS-Fluorescein (Thermo Fisher Scientific, 46409). Briefly, 1 mg NHS-Fluorescein was dissolved in 100 μL DMSO according to manufacturer instructions (stock solution). Si wafers were incubated with with 2 mL working solution prepared with 10 μL stock solution and 10 mL PBS buffer. The reaction took place at room temperature for 30 min followed by three washing cycles. Fluorescence was analyzed by Zeiss LSM 780 confocal microscopy, 20× lens, Zeiss ZEN Software (black edition). Raw images were exported as czi image files.
Acknowledgments
The authors thank Ing. Manuela Keller for her support in the monolayer degradation laboratory. This work was financially supported by the Helmholtz Association through program-oriented funding. The authors R.W. and U.T.B. acknowledge the financial support of the MixUp project, which has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 870294.
Author contributions
Conceptualization, N.A.T., R.M., R.W., and A.L.; methodology and investigation, N.A.T. and R.M.; resources, R.W., S.B., and L.P.; writing – original draft, N.A.T. and R.M.; writing – review and editing, R.M., A.L., R.W., and U.T.B.; supervision, R.M. and A.L.; funding acquisition, A.L., R.W., and U.T.B.
Declaration of interests
The authors declare no competing interests.
Published: November 23, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.checat.2022.11.004.
Contributor Information
Natalia A. Tarazona, Email: natalia.tarazona@hereon.de.
Ren Wei, Email: ren.wei@uni-greifswald.de.
Rainhard Machatschek, Email: rainhard.machatschek@hereon.de.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.PlasticsEurope . 2020. Plastics - the Facts 2020.https://www.plasticseurope.org/en/resources/publications [Google Scholar]
- 2.Martín A.J., Mondelli C., Jaydev S.D., Pérez-Ramírez J. Catalytic processing of plastic waste on the rise. Chem. 2021;7:1487–1533. doi: 10.1016/j.chempr.2020.12.006. [DOI] [Google Scholar]
- 3.Directive of the European Parliament and of the Council on the Reduction of the Impact of Certain Plastic Products on the Environment. (2019). https://eur-lex.europa.eu/eli/dir/2019/904/oj.
- 4.Vollmer I., Jenks M.J.F., Roelands M.C.P., White R.J., van Harmelen T., de Wild P., van der Laan G.P., Meirer F., Keurentjes J.T.F., Weckhuysen B.M. Beyond mechanical recycling: giving new life to plastic waste. Angew. Chem. Int. Ed. Engl. 2020;59:15402–15423. doi: 10.1002/anie.201915651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarazona N.A., Machatschek R., Balcucho J., Castro-Mayorga J.L., Saldarriaga J.F., Lendlein A. Opportunities and challenges for integrating the development of sustainable polymer materials within an international circular (bio)economy concept. MRS Energy & Sustainability. 2022;9:28–34. doi: 10.1557/s43581-021-00015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tournier V., Topham C.M., Gilles A., David B., Folgoas C., Moya-Leclair E., Kamionka E., Desrousseaux M.L., Texier H., Gavalda S., et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature. 2020;580:216–219. doi: 10.1038/s41586-020-2149-4. doi: 10.1038/s41586-020-2149-4. [DOI] [PubMed] [Google Scholar]
- 7.Tiso T., Narancic T., Wei R., Pollet E., Beagan N., Schröder K., Honak A., Jiang M., Kenny S.T., Wierckx N., et al. Towards bio-upcycling of polyethylene terephthalate. Metab. Eng. 2021;66:167–178. doi: 10.1016/j.ymben.2021.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Werner A.Z., Clare R., Mand T.D., Pardo I., Ramirez K.J., Haugen S.J., Bratti F., Dexter G.N., Elmore J.R., Huenemann J.D., et al. Tandem chemical deconstruction and biological upcycling of poly(ethylene terephthalate) to β-ketoadipic acid by Pseudomonas putida KT2440. Metab. Eng. 2021;67:250–261. doi: 10.1016/j.ymben.2021.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Kim H.T., Hee Ryu M., Jung Y.J., Lim S., Song H.M., Park J., Hwang S.Y., Lee H.-S., Yeon Y.J., Sung B.H., et al. Chemo-biological upcycling of poly(ethylene terephthalate) to multifunctional coating materials. ChemSusChem. 2021;14:4251–4259. doi: 10.1002/cssc.202100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu B., Wang D., Wei N. Enzyme discovery and engineering for sustainable plastic recycling. Trends Biotechnol. 2022;40:22–37. doi: 10.1016/j.tibtech.2021.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Wei R., Tiso T., Bertling J., O’Connor K., Blank L.M., Bornscheuer U.T. Possibilities and limitations of biotechnological plastic degradation and recycling. Nat. Catal. 2020;3:867–871. doi: 10.1038/s41929-020-00521-w. [DOI] [Google Scholar]
- 12.Kim H.T., Kim J.K., Cha H.G., Kang M.J., Lee H.S., Khang T.U., Yun E.J., Lee D.-H., Song B.K., Park S.J., et al. Biological valorization of poly(ethylene terephthalate) monomers for upcycling waste PET. ACS Sustain. Chem. Eng. 2019;7:19396–19406. doi: 10.1021/acssuschemeng.9b03908. [DOI] [Google Scholar]
- 13.Ballerstedt H., Tiso T., Wierckx N., Wei R., Averous L., Bornscheuer U., O’Connor K., Floehr T., Jupke A., Klankermayer J., et al. MIXed plastics biodegradation and UPcycling using microbial communities: EU Horizon 2020 project MIX-UP started January 2020. Environ. Sci. Eur. 2021;33:99. doi: 10.1186/s12302-021-00536-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei R., Zimmermann W. Biocatalysis as a green route for recycling the recalcitrant plastic polyethylene terephthalate. Microb. Biotechnol. 2017;10:1302–1307. doi: 10.1111/1751-7915.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan F., Wei R., Cui Q., Bornscheuer U.T., Liu Y.-J. Thermophilic whole-cell degradation of polyethylene terephthalate using engineered Clostridium thermocellum. Microb. Biotechnol. 2021;14:374–385. doi: 10.1111/1751-7915.13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller R.J., Schrader H., Profe J., Dresler K., Deckwer W.D. Enzymatic degradation of poly (ethylene terephthalate): rapid hydrolyse using a hydrolase from. Macromol. Rapid Commun. 2005;26:1400–1405. [Google Scholar]
- 17.Palm G.J., Reisky L., Böttcher D., Müller H., Michels E.A.P., Walczak M.C., Berndt L., Weiss M.S., Bornscheuer U.T., Weber G. Structure of the plastic-degrading Ideonella sakaiensis MHETase bound to a substrate. Nat. Commun. 2019;10:1717. doi: 10.1038/s41467-019-09326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei R., Breite D., Song C., Gräsing D., Ploss T., Hille P., Schwerdtfeger R., Matysik J., Schulze A., Zimmermann W. Biocatalytic degradation efficiency of postconsumer polyethylene terephthalate packaging determined by their polymer microstructures. Adv. Sci. 2019;6:1900491. doi: 10.1002/advs.201900491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa M., Kawakami N., Tomizawa A., Miyamoto K. Efficient degradation of poly (ethylene terephthalate) with Thermobifida fusca cutinase exhibiting improved catalytic activity generated using mutagenesis and additive-based approaches. Sci. Rep. 2019;9:16038–16039. doi: 10.1038/s41598-019-52379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomsen T.B., Hunt C.J., Meyer A.S. Influence of substrate crystallinity and glass transition temperature on enzymatic degradation of polyethylene terephthalate (PET) N. Biotechnol. 2022;69:28–35. doi: 10.1016/j.nbt.2022.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Wei R., von Haugwitz G., Pfaff L., Mican J., Badenhorst C.P.S., Liu W., Weber G., Austin H.P., Bednar D., Damborsky J., Bornscheuer U.T. Mechanism-based design of efficient PET hydrolases. ACS Catal. 2022;12:3382–3396. doi: 10.1021/acscatal.1c05856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gamerith C., Zartl B., Pellis A., Guillamot F., Marty A., Acero E.H., Guebitz G.M. Enzymatic recovery of polyester building blocks from polymer blends. Process Biochem. 2017;59:58–64. doi: 10.1016/j.procbio.2017.01.004. [DOI] [Google Scholar]
- 23.Brizendine R.K., Erickson E., Haugen S.J., Ramirez K.J., Miscall J., Salvachúa D., Pickford A.R., Sobkowicz M.J., McGeehan J.E., Beckham G.T. Particle size reduction of poly(ethylene terephthalate) increases the rate of enzymatic depolymerization but does not increase the overall conversion extent. ACS Sustain. Chem. Eng. 2022;10:9131–9140. doi: 10.1021/acssuschemeng.2c01961. [DOI] [Google Scholar]
- 24.Kawai F., Kawabata T., Oda M. Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl. Microbiol. Biotechnol. 2019;103:4253–4268. doi: 10.1007/s00253-019-09717-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bååth J.A., Borch K., Jensen K., Brask J., Westh P. Comparative biochemistry of four polyester (PET) hydrolases. Chembiochem. 2021;22:1627–1637. doi: 10.1002/cbic.202000793. [DOI] [PubMed] [Google Scholar]
- 26.Pfaff L., Gao J., Li Z., Jäckering A., Weber G., Mican J., Chen Y., Dong W., Han X., Feiler C.G., et al. Multiple substrate binding mode-guided engineering of a thermophilic PET hydrolase. ACS Catal. 2022;12:9790–9800. doi: 10.1021/acscatal.2c02275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Then J., Wei R., Oeser T., Barth M., Belisário-Ferrari M.R., Schmidt J., Zimmermann W. Ca2+ and Mg2+ binding site engineering increases the degradation of polyethylene terephthalate films by polyester hydrolases from Thermobifida fusca. Biotechnol. J. 2015;10:592–598. doi: 10.1002/biot.201400620. [DOI] [PubMed] [Google Scholar]
- 28.Shinotsuka K., Bliznyuk V.N., Assender H.E. Near-surface crystallization of PET. Polymer. 2012;53:5554–5559. doi: 10.1016/j.polymer.2012.09.048. [DOI] [Google Scholar]
- 29.Brown H.R., Russell T.P. Entanglements at polymer surfaces and interfaces. Macromolecules. 1996;29:798–800. doi: 10.1021/ma951123k. [DOI] [Google Scholar]
- 30.Xu J., Zhang H., Li J., Zhang L., Zuo B., Tsui O.K.C., Wang X. Conformation-sensitive surface dynamics in thin poly(ethylene terephthalate) film. Macromolecules. 2019;52:2580–2588. doi: 10.1021/acs.macromol.8b02284. [DOI] [Google Scholar]
- 31.Tarazona N.A., Machatschek R., Lendlein A. Influence of depolymerases and lipases on the degradation of polyhydroxyalkanoates determined in Langmuir degradation studies. Adv. Mater. Interfaces. 2020;7:2000872. doi: 10.1002/admi.202000872. [DOI] [Google Scholar]
- 32.Zhang W., Yan Q., Ye K., Zhang Q., Chen W., Meng L., Chen X., Wang D., Li L. The effect of water absorption on stretch-induced crystallization of poly(ethylene terephthalate): an in-situ synchrotron radiation wide angle X-ray scattering study. Polymer. 2019;162:91–99. doi: 10.1016/j.polymer.2018.12.029. [DOI] [Google Scholar]
- 33.Vegso K., Siffalovic P., Majkova E., Jergel M., Benkovicova M., Kocsis T., Weis M., Luby S., Nygård K., Konovalov O. Nonequilibrium phases of nanoparticle Langmuir films. Langmuir. 2012;28:10409–10414. doi: 10.1021/la301764t. [DOI] [PubMed] [Google Scholar]
- 34.Tarazona N.A., Machatschek R., Schulz B., Prieto M.A., Lendlein A. Molecular insights into the physical adsorption of amphiphilic protein PhaF onto copolyester surfaces. Biomacromolecules. 2019;20:3242–3252. doi: 10.1021/acs.biomac.9b00069. [DOI] [PubMed] [Google Scholar]
- 35.Blaudez D., Turlet J.-M., Dufourcq J., Bard D., Buffeteau T., Desbat B. Investigations at the air/water interface using polarization modulation IR spectroscopy. Faraday Trans. 1996;92:525–530. doi: 10.1039/FT9969200525. [DOI] [Google Scholar]
- 36.Loch C.L., Ahn D., Chen C., Wang J., Chen Z. Sum frequency generation studies at poly (ethylene terephthalate)/silane interfaces: hydrogen bond formation and molecular conformation determination. Langmuir. 2004;20:5467–5473. doi: 10.1021/la0494526. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y., Lu Y., Yan S., Shen D. Orientation study of poly (ethylene terephthlate) ultrathin films during crystallization. Polym. J. 2005;37:133–136. [Google Scholar]
- 38.Zhang Y., Zhang J., Lu Y., Duan Y., Yan S., Shen D. Glass Transition Temperature Determination of Poly(ethylene terephthalate) thin films using reflection−absorption FTIR. Macromolecules. 2004;37:2532–2537. doi: 10.1021/ma035709f. [DOI] [Google Scholar]
- 39.Dubelley F., Planes E., Bas C., Pons E., Yrieix B., Flandin L. Water vapor sorption properties of polyethylene terephthalate over a wide range of humidity and temperature. J. Phys. Chem. B. 2017;121:1953–1962. doi: 10.1021/acs.jpcb.6b11700. [DOI] [PubMed] [Google Scholar]
- 40.Saretia S., Machatschek R., Bhuvanesh T., Lendlein A. Effect of water on crystallization and melting of telechelic oligo(ε-caprolactone)s in ultrathin films. Adv. Mater. Interfaces. 2021;8 doi: 10.1002/admi.202001940. [DOI] [Google Scholar]
- 41.Kim J.H., Jang J., Zin W.-C. Thickness dependence of the glass transition temperature in thin polymer films. Langmuir. 2001;17:2703–2710. doi: 10.1021/la001125k. [DOI] [Google Scholar]
- 42.Zhang W., Douglas J.F., Starr F.W. Why we need to look beyond the glass transition temperature to characterize the dynamics of thin supported polymer films. Proc. Natl. Acad. Sci. USA. 2018;115:5641–5646. doi: 10.1073/pnas.1722024115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanroy B., Wübbenhorst M., Napolitano S. Crystallization of thin polymer layers confined between two adsorbing walls. ACS Macro Lett. 2013;2:168–172. doi: 10.1021/mz300641x. [DOI] [PubMed] [Google Scholar]
- 44.Brott S., Pfaff L., Schuricht J., Schwarz J.-N., Böttcher D., Badenhorst C.P.S., Wei R., Bornscheuer U.T. Engineering and evaluation of thermostable IsPETase variants for PET degradation. Eng. Life Sci. 2022;22:192–203. doi: 10.1002/elsc.202100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Badino S.F., Bååth J.A., Borch K., Jensen K., Westh P. Adsorption of enzymes with hydrolytic activity on polyethylene terephthalate. Enzyme Microb. Technol. 2021;152 doi: 10.1016/j.enzmictec.2021.109937. [DOI] [PubMed] [Google Scholar]
- 46.Münstedt H. Rheological measurements and structural analysis of polymeric materials. Polymers. 2021;13:1123. doi: 10.3390/polym13071123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarazona N.A., Machatschek R., Lendlein A. Relation between surface area and surface potential change during (co)polyesters degradation as Langmuir monolayer. MRS Adv. 2020;5:667–677. doi: 10.1557/adv.2019.458. [DOI] [Google Scholar]
- 48.Eberl A., Heumann S., Brückner T., Araujo R., Cavaco-Paulo A., Kaufmann F., Kroutil W., Guebitz G.M. Enzymatic surface hydrolysis of poly(ethylene terephthalate) and bis(benzoyloxyethyl) terephthalate by lipase and cutinase in the presence of surface active molecules. J. Biotechnol. 2009;143:207–212. doi: 10.1016/j.jbiotec.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Gigli M., Quartinello F., Soccio M., Pellis A., Lotti N., Guebitz G.M., Licoccia S., Munari A. Enzymatic hydrolysis of poly(1, 4-butylene 2, 5-thiophenedicarboxylate) (PBTF) and poly(1, 4-butylene 2, 5-furandicarboxylate) (PBF) films: a comparison of mechanisms. Environ. Int. 2019;130 doi: 10.1016/j.envint.2019.05.046. [DOI] [PubMed] [Google Scholar]
- 50.Tarazona N.A., Machatschek R., Lendlein A. Unraveling the interplay between abiotic hydrolytic degradation and crystallization of bacterial polyesters comprising short and medium side-chain-length polyhydroxyalkanoates. Biomacromolecules. 2020;21:761–771. doi: 10.1021/acs.biomac.9b01458. [DOI] [PubMed] [Google Scholar]
- 51.Lyu S., Untereker D. Degradability of polymers for implantable biomedical devices. Int. J. Mol. Sci. 2009;10:4033–4065. doi: 10.3390/ijms10094033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffmann F., Machatschek R., Lendlein A. Analytical model and Monte Carlo simulations of polymer degradation with improved chain cut statistics. J. Mater. Res. 2022;37:1093–1101. doi: 10.1557/s43578-022-00495-4. [DOI] [Google Scholar]
- 53.Yoshida S., Hiraga K., Takehana T., Taniguchi I., Yamaji H., Maeda Y., Toyohara K., Miyamoto K., Kimura Y., Oda K. A bacterium that degrades and assimilates poly (ethylene terephthalate) Science. 2016;351:1196–1199. doi: 10.1126/science.aad6359. [DOI] [PubMed] [Google Scholar]
- 54.Sagong H.-Y., Seo H., Kim T., Son H.F., Joo S., Lee S.H., Kim S., Woo J.-S., Hwang S.Y., Kim K.-J. Decomposition of the PET film by MHETase using exo-PETase function. ACS Catal. 2020;10:4805–4812. doi: 10.1021/acscatal.9b05604. [DOI] [Google Scholar]
- 55.Machatschek R., Lendlein A. Fundamental insights in PLGA degradation from thin film studies. J. Control. Release. 2020;319:276–284. doi: 10.1016/j.jconrel.2019.12.044. [DOI] [PubMed] [Google Scholar]
- 56.Sulaiman S., You D.J., Kanaya E., Koga Y., Kanaya S. Crystal structure and thermodynamic and kinetic stability of metagenome-derived LC-cutinase. Biochemistry. 2014;53:1858–1869. doi: 10.1021/bi401561p. [DOI] [PubMed] [Google Scholar]
- 57.Lebedev D.V., Marikhin V.A., Myasnikova L.P., Yakushev P.N., Ivankova E.M. Segmental mobility in polyethylene near-surface layers. J. Macromol. Sci. Part B. 2013;52(12):1770–1783. doi: 10.1080/00222348.2013.808558. [DOI] [Google Scholar]
- 58.Son H.F., Cho I.J., Joo S., Seo H., Sagong H.-Y., Choi S.Y., Lee S.Y., Kim K.-J. Rational Protein Engineering of thermo-stable PETase from Ideonella sakaiensis for highly efficient PET degradation. ACS Catal. 2019;9:3519–3526. doi: 10.1021/acscatal.9b00568. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.