

Abstract
Background
Chronic sputum production impacts on quality of life and is a feature of many respiratory diseases. Identification of the genetic variants associated with chronic sputum production in a disease agnostic sample could improve understanding of its causes and identify new molecular targets for treatment.
Methods
We conducted a genome-wide association study (GWAS) of chronic sputum production in UK Biobank. Signals meeting genome-wide significance (p<5×10−8) were investigated in additional independent studies, were fine-mapped and putative causal genes identified by gene expression analysis. GWASs of respiratory traits were interrogated to identify whether the signals were driven by existing respiratory disease among the cases and variants were further investigated for wider pleiotropic effects using phenome-wide association studies (PheWASs).
Results
From a GWAS of 9714 cases and 48 471 controls, we identified six novel genome-wide significant signals for chronic sputum production including signals in the human leukocyte antigen (HLA) locus, chromosome 11 mucin locus (containing MUC2, MUC5AC and MUC5B) and FUT2 locus. The four common variant associations were supported by independent studies with a combined sample size of up to 2203 cases and 17 627 controls. The mucin locus signal had previously been reported for association with moderate-to-severe asthma. The HLA signal was fine-mapped to an amino acid change of threonine to arginine (frequency 36.8%) in HLA-DRB1 (HLA-DRB1*03:147). The signal near FUT2 was associated with expression of several genes including FUT2, for which the direction of effect was tissue dependent. Our PheWAS identified a wide range of associations including blood cell traits, liver biomarkers, infections, gastrointestinal and thyroid-associated diseases, and respiratory disease.
Conclusions
Novel signals at the FUT2 and mucin loci suggest that mucin fucosylation may be a driver of chronic sputum production even in the absence of diagnosed respiratory disease and provide genetic support for this pathway as a target for therapeutic intervention.
Short abstract
Genome-wide association study in UK Biobank identifies six novel loci associated with chronic sputum production at genome-wide significance in a disease agnostic population. These include a FUT2 locus, highlighting a possible target for drug development. https://bit.ly/3IRVJeT
Introduction
Increased sputum production impacts on daily activities and quality of life, and is a shared feature of many respiratory diseases. Worldwide, 545 million people have chronic respiratory conditions, with those associated with chronic sputum production including COPD, asthma, bronchiectasis, chronic bronchitis and cystic fibrosis. Chronic respiratory disease is the third leading cause of death worldwide, with 3.91 million deaths in 2017 [1].
The determinants of chronic sputum production in disease are not completely understood [2]. Most studies of excess sputum production have been in subjects with chronic bronchitis and COPD where it has been associated with lower lung function [3, 4] and higher risk of both exacerbation and respiratory symptoms [5]. Risk factors for excess sputum production include smoking and occupational and environmental pollutants [4, 6–8]. Currently available drug treatments for those with chronic sputum production do not generally affect the rate of production of sputum, but act as mucolytics and expectorants [9–11].
Genome-wide association studies (GWASs) have highlighted pathways underlying a range of respiratory traits and diseases, and highlighted potentially relevant drug targets [12, 13]. Previous GWASs of sputum production [14–17] have not identified any genome-wide significant findings.
We hypothesised that identifying genetic variants that are associated with chronic sputum production in a large general population sample could improve understanding of its causes and identify new molecular targets for treatment. To test this hypothesis, we undertook a GWAS of risk of chronic sputum production in 9714 cases and 48 471 controls from UK Biobank, and sought replication of the association signals in five additional independent studies totalling 2203 cases and 17 627 controls. We performed phenome-wide association studies (PheWASs) and interrogation of gene expression data to characterise the association signals and determine which genes may be driving these signals.
Methods
Study population
Information about chronic sputum production was obtained from the online lifetime occupation survey that was e-mailed to 324 653 UK Biobank participants with existing e-mail addresses between June and September 2015 and achieved a response rate of 38% (31% of all of those contacted provided a full completion of the questionnaire [18]). For this study, we defined cases as those who answered “Yes” to the question “Do you bring up phlegm/sputum/mucus daily?” (UK Biobank data-field 22504; a total of 121 283 participants provided a “Yes” or “No” response). Controls were defined as those who answered “No” to this question. Cases and controls were further restricted to those of genetically determined European ancestry, as previously defined [19], with available smoking data (UK Biobank data-field 20160). Related individuals were removed, with cases preserved over controls when excluding one of a pair (or more) of related individuals (UK Biobank data-field 22021; “related” defined as a KING kinship coefficient ≥0.0884, equivalent to second-degree relatedness or closer). For related pairs within the cases or controls, the individual with the lowest genotype missingness (UK Biobank data-field 22005) was retained. From all available controls, we defined a subset of controls with a similar age (UK Biobank data-field 34) and sex (UK Biobank data-field 31) distribution to the cases at a 1:5 ratio with the cases.
Demographics and respiratory characteristics of the case and controls were derived using the following definitions: doctor-diagnosed asthma (UK Biobank data-field 22127), moderate-to-severe asthma (as previously described [20]), doctor-diagnosed chronic bronchitis (UK Biobank data-field 22129), cough on most days (UK Biobank data-field 22502), smoking status (UK Biobank data-field 20160), COPD Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage 1–4 and stage 2–4 (defined using baseline spirometry as previously described [19, 21]), and bronchiectasis and cystic fibrosis (supplementary tables S1 and S2).
UK Biobank has ethical approval from North West – Haydock Research Ethics Committee (21/NW/0157). Written informed consent was provided by all participants.
GWAS of chronic sputum production
Genetic data from the v3 March 2018 UK Biobank data release, imputed to the Haplotype Reference Consortium panel r1.1 2016, were used for the GWAS, giving 27 317 434 variants for analysis.
Association testing was performed using logistic regression under an additive genetic model in PLINK 2.0 [22] with age, sex, array version, never/ever-smoking status and the first 10 principal components of ancestry as covariates. Variants were excluded if they had an imputation quality INFO score <0.5 or a minor allele count (MAC) <20. Association signals were considered genome-wide significant at p<5×10−8. Independent signals were initially defined using a 1-Mb window (500 kb each side of the sentinel variant) and then using conditional analyses implemented in GCTA-COJO [23]. All variant coordinates are for genome build GRCh37. Region plots were created using LocusZoom [24].
Replication
We sought replication in five general population cohorts which surveyed participants for chronic sputum production: Generation Scotland [25], EXCEED Study [26], LifeLines 1, LifeLines 2 and Vlagtwedde-Vlaardingen [17]. Further details are provided in the supplementary material.
In addition, the overlap of primary care sputum codes with the chronic sputum production question (UK Biobank data-field 22504) was evaluated to identify whether primary care codes could be used to define an additional independent case–control dataset from those in UK Biobank who did not respond to the online lifetime occupation survey (supplementary material).
Fine-mapping
We undertook Bayesian fine-mapping for all genome-wide significant signals that were not in the human leukocyte antigen (HLA) region to define 99% credible sets of variants, i.e. sets of variants that are 99% probable to contain the true causal variant (assuming that it has been measured).
To fine-map signals within the HLA region (chromosome 6:29 607 078–33 267 103 (b37)) to a specific HLA gene allele or amino acid change, we re-imputed our discovery samples using IMPUTE2 v2.3.1 with a reference panel that enabled imputation of 424 classical HLA alleles and 1276 amino acid changes as described in Jia et al. [27]. We then repeated the association testing as described earlier.
Mapping association signals to putative causal genes
We used functional annotation and colocalisation with expression quantitative trait loci (eQTL) signals to identify putative causal genes at each signal.
Annotation of the variants in each credible set was performed using SIFT [28], PolyPhen-2 and CADD, all implemented using the Ensemble GRCh37 Variant Effect Predictor (VEP) [29], alongside FATHMM [30]. Variants were annotated as deleterious if they were labelled deleterious by SIFT, probably damaging or possibly damaging by PolyPhen-2, damaging by FATHMM (specifying the “Inherited Disease” option of the “Coding Variants” method and using the “Unweighted” prediction algorithm), or had a CADD scaled score ≥20.
We queried the sentinel variants in GTEx V8 [31] and BLUEPRINT [32] (see supplementary table S3 for list of tissues). We tested for colocalisation of GWAS and eQTL signals using coloc [33]; H4 >80% was used to define a shared causal variant for eQTL and GWAS signals.
Associations with other phenotypes
To investigate whether the signals of association with sputum production were driven by underlying respiratory phenotypes of the cases, a look-up for each signal was undertaken for 14 respiratory or respiratory-related traits from GWAS results: moderate-to-severe asthma (ncases=5135, ncontrols=25 675) [20], lung function (forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC), FEV1/FVC and peak expiratory flow (PEF)) (n=400 102) [19], respiratory infection (ncases=19 459, ncontrols=101 438) [34], chronic cough (ncases=15 213, ncontrols=94 731), chronic bronchitis (ncases=977, ncontrols=108 967), idiopathic pulmonary fibrosis (IPF) (ncases=2668, ncontrols=8591) [35], smoking traits (smoking age of onset (n=124 590), smoking cessation (ncases=141 649, ncontrols=27 321), smoking cigarettes per day (n=120 744) and smoking initiation (ncases=170 772, ncontrols=212 859)) and asthma (ncases=23 948, ncontrols=118 538) [36]. Smoking trait results were from the UK Biobank component of Jiang et al. [37]; chronic cough and chronic bronchitis were defined for this study using UK Biobank data (supplementary material). Where the sentinel variant was not available in the look-up dataset, we utilised an alternative variant from the credible set with the highest posterior probability of being causal. A Bonferroni adjustment for 84 association tests was applied requiring a p<5.95×10−4 for association to be classified as statistically significant. Imputed HLA gene allele or amino acid changes were used for signals in the HLA region.
To investigate associations of the chronic sputum-associated variants with a wider range of phenotypes, we performed a PheWAS for 2172 traits in UK Biobank (false discovery rate <0.01; supplementary material) and searched the Open Targets Genetics Portal (p<5×10−8, version 0.4.0 (bd664ca); accessed 16 April 2021 [38]). PheWAS for imputed HLA alleles was performed using DeepPheWAS (supplementary material) [39].
Sensitivity analyses
To further investigate whether the effects of the variants associated with risk of chronic sputum production differ between ever- and never-smokers, or between individuals with and without a history of chronic respiratory disease (spirometry-defined COPD GOLD 1–4, doctor-diagnosed asthma or doctor-diagnosed chronic bronchitis), we tested the association of sentinel variants in ever- and never-smokers and those with and without evidence of chronic respiratory disease separately. We additionally evaluated whether the associations differed between males and females or by the time of year of the survey (UK Biobank data-field 22500). Finally, we evaluated whether adjusting for current smoking (UK Biobank data-field 22506) (rather than ever- versus never-smoker status) affected the results.
Results
A total of 10 481 participants answered “Yes” to the question “Do you bring up phlegm/sputum/mucus daily?” and 110 802 answered “No” (supplementary table S4). After excluding those with missing genotype and essential covariate data, and those of genetically determined European ancestry, a total of 9714 cases and 48 471 controls (figure 1) were included in the GWAS. Ever-smoking and respiratory disease were more common in the cases than in the controls (table 1). The genomic control inflation factor (λ) was 1.026, so no adjustments to the test statistics were applied (supplementary figure S1). Six independent novel signals met the genome-wide significance threshold of p<5×10−8 (table 2 and supplementary figure S2). These were four common variant signals (minor allele frequency >5%) in or near MUC2, FUT2, HLA-DRB1 and NKX3-1, and two intronic rare variant signals (minor allele frequency <1%) in OCIAD1 and NELL1 (figure 2).
FIGURE 1.
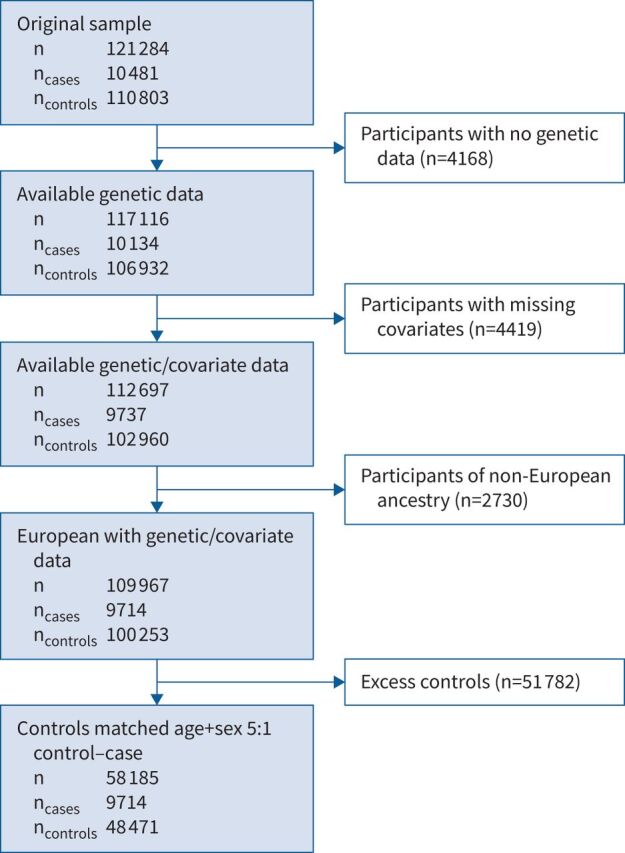
Study flowchart detailing case–control selection from the UK Biobank cohort.
TABLE 1.
Demographics and characteristics of cases and controls included in the genome-wide association study of chronic sputum production
| Cases (n=9714) | Controls (n=48 471) | |
| Mean age (years) | 57.7 | 57.7 |
| Female (%) | 42.5 | 42.5 |
| Smoking status | ||
| Ever-smoker | 5306 (54.6) | 20 912 (43.1) |
| Current smoker | 983 (10.2) | 1569 (3.2) |
| Doctor-diagnosed chronic bronchitis | 407 (4.2) | 416 (0.86) |
| Doctor-diagnosed asthma | 2630 (27.1) | 5251 (10.8) |
| Cough on most days | 7022 (72.3) | 3999 (8.3) |
| Moderate-to-severe asthma | 520 (5.4) | 521 (1.1) |
| Self-reported chronic sinusitis | 181 (1.9) | 1057 (2.2) |
| Meets spirometry criteria for GOLD 1–4 | 1511 (21.8)# | 4766 (13.1)# |
Data are presented as n (%), unless otherwise stated. COPD: Global Initiative for Chronic Obstructive Lung Disease. #: ncases=6942 and ncontrols=36 321 with available spirometry that passed quality control.
TABLE 2.
Novel genome-wide significant signals of association with chronic sputum production
|
Chromosome:
position (GRCh37) |
rsID | Locus (distance from gene (bp))# |
Coded/
noncoded |
Coded allele frequency (% (count))¶ | OR (95 CI) | p-value | INFO+ | Variants in 99% credible set (n (highest posterior probability)) | |
| 4:48 854 355 | rs79998532 | OCIAD1 (intronic) | A/G | 0.2 (233) | Discovery | 2.36 (1.76–3.16) | 8.00×10−09 | 0.92 | 3 (0.86) |
| Replication | 3.3 (0.11–98.6) | 0.49 | |||||||
| Meta-analysis | 2.37 (1.77–3.17) | 6.36×10−09 | |||||||
| 6:32 496 534 | rs374248993 | HLA-DRB1 § | G/C | 57 (66 355) | Discovery | 1.12 (1.08–1.16) | 7.30×10−11 | 0.87 | HLA-DRB1*03:147§ |
| Replication | 1.01 (0.84–1.21) | 0.93 | |||||||
| Meta-analysis | 1.11 (1.08–1.15) | 1.31×10−10 | |||||||
| 8:23 480 686 | rs79401075 | NKX3-1 (59 765) | A/G | 10 (11 620) | Discovery | 1.18 (1.12–1.24) | 8.90×10−11 | 0.98 | 30 (0.32) |
| Replication | 1.20 (0.95–1.52) | 0.12 | |||||||
| Meta-analysis | 1.18 (1.12–1.24) | 2.65×10−11 | |||||||
| 11:1 116 931 | rs779167905 |
MUC2 (12 513) |
T/TTCTA | 67 (78 158) | Discovery | 1.12 (1.08–1.16) | 1.20×10−10 | 0.98 | 30 (0.15) |
| Replication | 1.09 (0.93–1.28) | 0.29 | |||||||
| Meta-analysis | 1.12 (1.08–1.15) | 6.99×10−11 | |||||||
| 11:20 887 601 | rs529240826 | NELL1 (intronic) | GC/G | 0.51 (588) | Discovery | 1.91 (1.52–2.4) | 2.50×10−08 | 0.67 | 2 (0.83) |
| Replication | 0.83 (0.38–1.78) | 0.63 | |||||||
| Meta-analysis | 1.79 (1.44–2.22) | 1.99×10−07 | |||||||
| 19:49 206 417 | rs492602 | FUT2 (exonic) | G/A | 51 (58 803) | Discovery | 1.11 (1.08–1.15) | 3.20×10−11 | 1 | 32 (0.07) |
| Replication | 1.06 (1–1.14) | 0.07 | |||||||
| Meta-analysis | 1.10 (1.07–1.13) | 1.21×10−11 |
#: start or end of nearest gene; ¶: values for discovery single nucleotide polymorphisms; +: INFO score (imputation quality) taken from discovery; §: amino acid change of threonine to arginine at codon 233 of exon 5 of HLA-DRB1 (HLA gene allele HLA-DRB1*03:147).
FIGURE 2.
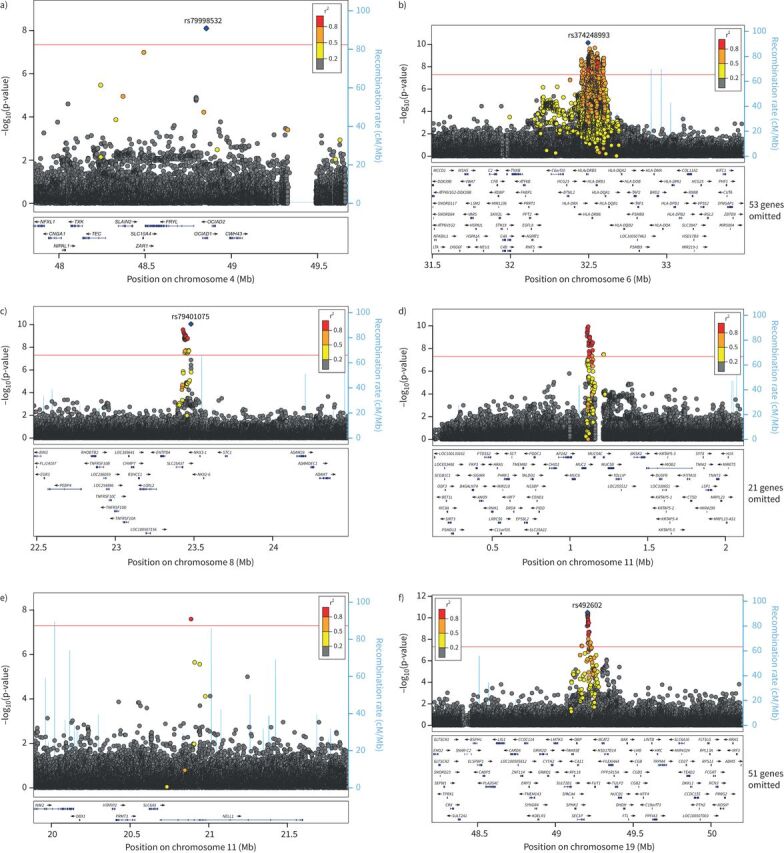
LocusZoom plots of the six sentinel signals: a) OCIAD1 signal (rs79998532), b) HLA-DRB1 signal (rs374248993), c) NKX3-1 signal (rs79401075), d) MUC2 signal (rs779167905), e) NELL1 signal (rs529240826) and f) FUT2 signal (rs492602).
No systematic differences were seen in effect sizes when stratifying by smoking status, by history of chronic respiratory disease, by sex, by time of year of survey or when including current smoking status as a covariate (supplementary table S5 and supplementary figures S3–S8) for the six sentinel variants. Through comparison of survey responses and linked primary care data we showed that primary care codes were not adequate proxies for the survey responses (supplementary material). We sought replication in five independent cohorts with a combined sample size of 1977 cases and 17 627 controls; data from all five replication cohorts were only available for the FUT2 locus. Although none of the signals met criteria for significance in a meta-analysis of the replication cohorts, the directions of effect were consistent with the discovery results for the signals in or near MUC2, FUT2, OCIAD1, HLA-DRB1 and NKX3-1, and all except the signals at NELL1 and HLA-DRB1 also increased in significance when the replication and discovery results were meta-analysed (table 2, supplementary table S13 and supplementary figure S9).
Novel associations with chronic sputum production
HLA locus
The HLA signal was fine-mapped to an amino acid change of threonine to arginine (frequency 36.8%) at codon 233 of exon 5 of HLA-DRB1 (HLA-DRB1*03:147) that was associated with decreased risk (OR 0.91 (95% CI 0.88–0.94)) of chronic sputum production (p=3.43×10−9). The amino acid change was in linkage disequilibrium with the GWAS sentinel variant rs374248993 (linkage disequilibrium r2=0.74 with HLA-DRB1*03:147) and the signal for rs374248993 was attenuated when conditioned on the amino acid change (supplementary figures S10 and S11).
HLA-DRB1*03:147 was significantly associated with FEV1, FEV1/FVC and PEF at genome-wide significance (p<5×10−8) (figure 3 and supplementary table S6). The amino acid associated with increased risk of chronic sputum production (threonine) was associated with increased lung function; this had not been previously reported. The HLA PheWAS identified multiple significant associations for the HLA allele associated with increased risk of chronic sputum production with a wide range of quantitative traits (e.g. blood cell traits and liver biomarkers) and diseases (including decreased risk of gastrointestinal and thyroid-associated diseases, and increased risk of bronchiectasis and asthma) (supplementary table S7).
FIGURE 3.

Results for association of sentinel variant risk alleles with respiratory traits. Results are aligned to the risk allele for chronic sputum production; effect direction “Increasing” can be read as increasing risk for binary traits and increasing values in quantitative traits. Chronic bronchitis and smoking age of onset, cigarettes per day and cessation phenotype look-ups were omitted as no associations with p<0.05 found. *: p<5.95×10−4 (Bonferroni adjustment for 84 association tests); **: p<5×10−8. Note that the idiopathic pulmonary fibrosis (IPF) association for rs779167905 (using proxy single nucleotide polymorphism (SNP) rs10902094) was attenuated when conditioned on rs35705950 (OR 0.99; p=0.784). FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; PEF: peak expiratory flow.
MUC2 locus
For the mucin locus signal (rs779167905 allele T), the allele associated with risk of chronic sputum production was also significantly associated with increased risk of asthma (OR 1.06; p=0.0027) and moderate-to-severe asthma (OR 1.13; p=6.3×10−7), increased FVC (β=0.0087; p=6×10−4) and decreased risk of IPF (OR 0.84; p=7.5×10−6) (figure 3 and supplementary table S6). There were no associations with gene expression for rs779167905 in GTEx or BLUEPRINT. However, we have previously shown that a proxy of rs779167905 (rs11602802, r2=0.94) was associated with mRNA levels of MUC5AC in bronchial epithelial brush samples collected from asthma patients, with the risk allele being associated with elevated MUC5AC expression [20].
Genome-wide significant associations with IPF [40] and moderate-to-severe asthma [20] have previously been reported at this chromosome 11 locus, so we undertook a conditional analysis to identify whether the chronic sputum production signal was independent of these previous signals. Repeating the association testing for this variant conditioning on the previously reported variants (rs35705950 [40] and rs11603634 [20]) identified that the chronic sputum production GWAS signal was independent of the IPF signal (rs779167905, conditional p=1.18×10−10) but was not independent of the previously reported moderate-to-severe asthma signal (rs779167905, conditional p=0.0039) (supplementary figures S12 and S13). Furthermore, the IPF association for rs779167905 (using proxy single nucleotide polymorphism (SNP) rs10902094) was also attenuated when conditioned on rs35705950 (OR 0.99; p=0.784).
Our PheWAS and Open Targets Genetics Portal analysis identified that the MUC2 locus signal (rs779167905) allele that was associated with increased risk of chronic sputum production (allele T) was associated with higher risk of asthma and asthma-related traits in other studies [41–43] and with lower risk of gall bladder disease (supplementary tables S7 and S8).
FUT2 locus
The FUT2 credible set included two variants that were annotated as functional using VEP. This included a stop-gain variant in FUT2 (rs601338, linkage disequilibrium r2=0.992 with sentinel rs492602) and a nearby missense variant (rs602662, r2=0.882 with sentinel rs492602) that resulted in a glycine to serine amino acid change for the allele positively correlated with the chronic sputum production risk allele (supplementary tables S9 and S10).
Sentinel variant rs492602 at the FUT2 locus was associated with gene expression for FUT2, NTN5, RASIP1, SEC1P and MAMSTR, for which there was support for colocalisation of eQTL and GWAS signals in multiple tissues from GTEx V8 (figure 4 and supplementary table S11). Increased risk of chronic sputum production was consistently correlated with increased expression of NTN5 and MAMSTR across a range of tissues. In contrast, the direction of the FUT2 expression signal varied by tissue, with increased risk of chronic sputum production correlated with decreased expression of FUT2 in brain tissues and with increased expression in gastrointestinal tissue. There were no associations in lung tissue and upper airway tissues were not available.
FIGURE 4.

Results for expression quantitative trait loci colocalisation for the FUT2 locus using variant rs492602. The numbers within the grid are the posterior probability of colocalisation (H4), with results aligned to the risk allele G for the rs492602 variant. Missing numbers indicate no data were available for the respective gene and tissue. GWAS: genome-wide association study; EBV: Epstein–Barr virus.
The sentinel variant for the FUT2 region signal on chromosome 19 (rs492602) was associated with lung function measures FEV1/FVC and PEF (p=2.2×10−6 and p=1.1×10−6, respectively), with the chronic sputum production risk allele (G) associated with decreased lung function (figure 3 and supplementary table S6).
Our PheWAS and Open Targets Genetics Portal analysis for this variant identified 141 associations spanning multiple disease areas, phenotypes and biomarkers (supplementary tables S7 and S8). In summary, the allele associated with increased risk of chronic sputum production was associated with increased risk of gallstones [42, 44, 45], type 1 diabetes [46] and Crohn's disease [47–50], elevated vitamin B12 [51–54] and cholesterol and fat metabolites [41, 42, 55–59], hypertension/cardiovascular disease [41, 42, 44], excess alcohol with associated sequelae [44, 60–62], and increased risk of mumps and lower risk of childhood ear infections [63]. Higher risk of chronic sputum production was also associated with higher levels of γ-glutamyl transferase, total bilirubin and aspartate amino transferase, and lower levels of alanine aminotransferase and alkaline phosphatase.
Other novel loci
Using functional annotation of variants and eQTL analysis, no putative causal genes could be assigned to the signals in or near OCIAD1 and NELL1. There was a single colocalising eQTL for SLC25A37 in the NKX3-1 locus with increased risk of chronic sputum production associated with a reduced expression of SLC25A37 in brain cortex (supplementary table S11 and supplementary figure S14).
Discussion
We describe a GWAS of chronic sputum production to identify genome-wide significant signals, and our novel findings implicate genes involved in mucin production and fucosylation, as well as the HLA class II histocompatibility antigen, HLA-DRB1. We provide functional evidence that the SNP signals we identify are associated with gene expression of FUT2, MUC5AC and SLC5A37.
Smoking is believed to be the main cause of excess sputum production, and is also associated with chronic infections, reduced lung function and susceptibility to chronic respiratory disease. Through identification of genetic association signals that are independent of smoking and history of chronic respiratory disease, our study demonstrates the value in studying a disease-relevant phenotype in a very large population that is agnostic to respiratory disease or smoking status.
The most significant signal implicated the gene FUT2 which has been widely studied for its role in blood group antigen expression and association with gastric and respiratory infection. FUT2 encodes fucosyltransferase 2 which mediates the transfer of fucose to the terminal galactose on glycan chains of cell surface glycoproteins and glycolipids. FUT2 creates a soluble precursor oligosaccharide called the H antigen, FuC-α((1,2)Galβ-), which is an essential substrate for the final step in the soluble ABO blood group antigen synthesis pathway. The FUT2 locus allele associated with increased risk of chronic sputum production in this study is correlated with a nonsense allele that leads to inactivated FUT2 which results in a nonsecretory phenotype of ABO(H) blood group antigens [64] for homozygous carriers. This nonsense allele (rs601338 allele A) has frequencies of 25–50% in South Asian, European and African populations, but is rare (<1%) in East Asian populations [65]. Candidate gene studies of this locus have identified that nonsecretors (at increased risk of chronic sputum production according to our study) have a lower risk of Helicobacter pylori infection [66], rotavirus A infection [67, 68], norovirus infection [69–71], infant (12–24 months) respiratory illness [72], asthma exacerbations [73], otitis media [74], exacerbation in non-cystic fibrosis bronchiectasis and Pseudomonas aeruginosa airway infection in the same group [75], some evidence of slower HIV progression [71], and a higher risk of pneumococcal and meningococcal infection [76]. The T allele of another variant in high linkage disequilibrium at this locus (rs681343, r2=0.996 with rs492602), associated with increased risk of chronic sputum production in our study, was recently reported to be associated with increased risk of human polyomavirus 1 (BKV) virus infection, as measured by antibody response [77]. A recent GWAS of critically ill cases of coronavirus disease 2019 (COVID-19) (ncases=7491) showed that the risk allele for chronic mucus production (G) of rs492602 was protective against life-threating COVID-19 (OR 0.88 (95% CI 0.87–0.90); p=4.55×10−9) [78]. However, this finding was not replicated in the latest COVID-19 Host Genetics Initiative results for a similar phenotype [79]. The differing directions of effect of this signal on different phenotypes may be explained by the SNP effects on FUT2 expression which differ across cell and tissue types. Further targeted experiments in relevant cell and tissue types would be needed to elucidate this and define the likely effects of targeting FUT2 directly or indirectly.
Epitopes that are fucosylated by FUT2 play a role in cell–cell interaction, including host–microbe interaction [80, 81], and mediate interaction with intestinal microbiota, thereby influencing its composition [82–85]. While there has been no direct evidence of host–pathogen binding on the FUT2 generated epitopes for nongastrointestinal infection, there is evidence that FUT2 can influence nonbinding ligands such as sialic acid [86]. Sialic acid binding has been shown to be important for adenovirus binding in cell models [87] and modulating this binding has been implicated as a possible mechanism for increasing risk of mumps infection [63].
FUT2 may also be key to the function of mucins, including those encoded by genes at our other significant locus (i.e. MUC2, MUC5AC and MUC5B). Mucins are a major constituent of airway mucus and MUC5AC is major gel-forming mucin secreted by airway epithelial cells. FUT2 may play a key role in MUC5AC regulation leading to excess mucus production or its increased viscosity, a common characteristic observed in patients with airway obstructive diseases including asthma, bronchitis and COPD. Analysis of oligosaccharides released from insoluble colonic mucins, largely Muc2, by mass spectrometry shows complete lack of terminal fucosylation of O-linked oligosaccharides in Fut2-LacZ-null mice [88]. FUT2 has also been shown to determine the O-glycosylation pattern of Muc5ac in mice [89]. The significant signal at MUC2 in our analysis was not independent of the previously reported moderate-to-severe asthma signal [20] for which MUC5AC was implicated as the most likely causal gene using gene expression data from bronchial epithelial cells. In that study we went on to show that the signal (rs11602802 used as proxy) was associated with mRNA levels of MUC5AC in bronchial epithelial brush samples collected from asthma patients, with the risk allele being associated with elevated MUC5AC. There was also a nonsignificant trend for MUC5B to have a reduced mRNA level in the presence of the moderate-to-severe asthma risk allele. These ex vivo observations have recently been replicated in nasal epithelial cell brush samples in an independent cohort and extended to show this signal (rs12788104 within the credible set of MUC2 signal) regulates MUC5AC protein levels in vitro using nasal epithelial cells from genotyped subjects in the air–liquid interface model [90]. Although our analysis did not identify an association at the MUC2 locus with COPD-related traits (FEV1 and FEV1/FVC), a recent study has also highlighted MUC5AC as a potential biomarker for COPD prognosis [91].
The particular allele that was found to explain the association signal in the HLA region (HLA-DRB1*03:147 [92]) has only recently been reported and so there is limited information about functionality. Associations of this allele with other GWAS traits should be interpreted with caution given the high linkage disequilibrium across the region. Furthermore, the association of this allele with increased sputum production and increased lung function reminds us that increased sputum production is part of the adaptive immune response to environmental insult and approaches to target mucus production must also consider potential negative effects of reducing sputum production.
We only report overlap of chronic sputum production association signals with association signals for gene expression regulation where there is statistical support that these signals share a causal variant. In addition to a comprehensive PheWAS, we provide a deeper assessment of associations with relevant respiratory phenotypes that highlights previously unreported associations with lung function for the HLA-DRB1 and FUT2 signals.
As only a subset of UK Biobank participants provided answers to the sputum production question, we expected that we might be able to define a replication case–control dataset from the remaining >300 000 participants using primary care data. However, evaluation of the positive predictive value of primary care codes for sputum production, when compared with the questionnaire data, was very low (supplementary material). This could reflect a low utilisation of sputum codes in primary care or that participants have not reported this symptom to their general practitioner. We obtained supportive evidence for four of the signals utilising data from five general population cohorts. The limited sample size (the case sample size for replication was 23% of the size available for discovery) impacted our ability to show statistically significant replication. Furthermore, we note that, for three of the replication cohorts (LifeLines 1, LifeLines 2 and Vlagtwedde-Vlaardingen), the sputum production question asked specifically about winter symptoms, while the UK Biobank survey did not restrict to any specific season. However, given the strong evidence summarised earlier for the involvement of the probable causal genes in control of pathways relevant to mucus production, we believe the associations identified are highly likely to be real. Due to very low numbers, we were unable to evaluate the effects of these signals in individuals of non-European ancestry, thereby limiting the generalisability of our findings to non-European ancestry groups. Efforts are urgently needed to improve diversity in genomics research [93] such as the planned Our Future Health initiative in the UK. In summary, the HLA, MUC2 and FUT2 loci show strong candidacy for a role in sputum production, with overlap with infection and related phenotypes and known mechanistic interactions between the genes at the FUT2 and MUC2 loci, suggesting that these signals are likely to be robust. The large number of associations of the FUT2 locus with a broad array of phenotypes, tissue-dependent expression of FUT2 and association with expression of other genes in the region may have implications for drug targeting guided by this locus. Experimental studies to characterise the specific interplay between FUT2 activity and mucin genes expressed in the airways are warranted.
Conclusions
Chronic sputum production is a phenotype characteristic of several respiratory diseases, as well as being a common cause for referrals in the absence of overt disease, and is of interest for pharmaceutical intervention. We report novel genetic factors which influence chronic sputum production and these signals highlight fucosylation of mucin as a driving factor of chronic sputum production. These signals could provide insight into the molecular pathways of sputum production and represent potential future targets for drug development [94].
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary text, figures and table captions ERJ-01667-2022.Supplement (1.5MB, pdf)
STREGA reporting recommendations ERJ-01667-2022.STREGA (176.6KB, pdf)
Supplementary tables ERJ-01667-2022.Tables (104.1KB, xlsx)
Shareable PDF
Footnotes
Data availability: Genome-wide association statistics from the case–control analysis of chronic sputum production will be made available via GWAS Catalog (GCP000629).
Conflict of interest: L.V. Wain, M.D. Tobin, I. Sayers and I.P. Hall report collaborative research funding from GSK to undertake the submitted work. L.V. Wain, M.D. Tobin, C. John, A.L. Guyatt and R.J. Packer report funding from Orion Pharma, outside of the submitted work. L.V. Wain reports consultancy for Galapagos. J. Davitte, E. Hessel, D. Michalovich, J.C. Betts and A. Yeo were employees of GSK at the time of this study. D. Michalovich is an employee of Benevolent AI. C.A. Melbourne is an employee of Mirador Analytics. N. Shrine, R. Hall, R. Thompson, A.T. Williams, M.L. Paynton, P.H. Lee, A. Campbell, C. Hayward, M. de Vries, R.J. Allen and J.M. Vonk report no competing interests.
Support statement: L.V. Wain holds a GSK/Asthma and Lung UK Chair in Respiratory Research (C17-1). M.D. Tobin is supported by a Wellcome Trust Investigator Award (WT202849/Z/16/Z). M.D. Tobin and I.P. Hall hold NIHR Senior Investigator Awards. C. John held a Medical Research Council Clinical Research Training Fellowship (MR/P00167X/1). L.V. Wain, M.D. Tobin, I. Sayers and I.P. Hall report collaborative research funding from GSK to undertake the submitted work. The research was partially supported by the NIHR Leicester Biomedical Research Centre and the NIHR Nottingham Biomedical Research Centre; the views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. This research was funded in part by the Wellcome Trust. We acknowledge the support of the Health Data Research UK BREATHE Digital Innovation Hub (UKRI Award MC_PC_19004). This research was conducted under UK Biobank application 45243. This research used the SPECTRE and ALICE High Performance Computing Facility at the University of Leicester. Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates (CZD/16/6) and the Scottish Funding Council (HR03006) and is currently supported by the Wellcome Trust (216767/Z/19/Z). Genotyping of the GS:SFHS samples was carried out by the Genetics Core Laboratory at the Edinburgh Clinical Research Facility, University of Edinburgh, Scotland and was funded by the Medical Research Council UK and the Wellcome Trust (Wellcome Trust Strategic Award “STratifying Resilience and Depression Longitudinally” (STRADL) Reference 104036/Z/14/Z). C. Hayward is supported by a Medical Research Council University Unit Programme grant MC_UU_00007/10 (QTL in Health and Disease). Recruitment to the Generation Scotland CovidLife study was facilitated by SHARE (Scottish Health Research Register and Biobank). SHARE is supported by NHS Research Scotland, the Universities of Scotland and the Chief Scientist Office of the Scottish Government. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Li X, Cao X, Guo M, et al. . Trends and risk factors of mortality and disability adjusted life years for chronic respiratory diseases from 1990 to 2017: systematic analysis for the Global Burden of Disease Study 2017. BMJ 2020; 368: m234. doi: 10.1136/bmj.m234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 187: 228–237. doi: 10.1164/rccm.201210-1843CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim V, Zhao H, Boriek AM, et al. . Persistent and newly developed chronic bronchitis are associated with worse outcomes in chronic obstructive pulmonary disease. Ann Am Thorac Soc 2016; 13: 1016–1025. doi: 10.1513/AnnalsATS.201512-800OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelkonen M, Notkola I-L, Nissinen A, et al. . Thirty-year cumulative incidence of chronic bronchitis and COPD in relation to 30-year pulmonary function and 40-year mortality: a follow-up in middle-aged rural men. Chest 2006; 130: 1129–1137. doi: 10.1378/chest.130.4.1129 [DOI] [PubMed] [Google Scholar]
- 5.Kim V, Han MK, Vance GB, et al. . The chronic bronchitic phenotype of COPD: an analysis of the COPDGene Study. Chest 2011; 140: 626–633. doi: 10.1378/chest.10-2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dijkstra AE, de Jong K, Boezen HM, et al. . Risk factors for chronic mucus hypersecretion in individuals with and without COPD: influence of smoking and job exposure on CMH. Occup Environ Med 2014; 71: 346–352. doi: 10.1136/oemed-2013-101654 [DOI] [PubMed] [Google Scholar]
- 7.Trupin L, Earnest G, San Pedro M, et al. . The occupational burden of chronic obstructive pulmonary disease. Eur Respir J 2003; 22: 462–469. doi: 10.1183/09031936.03.00094203 [DOI] [PubMed] [Google Scholar]
- 8.Matheson MC, Benke G, Raven J, et al. . Biological dust exposure in the workplace is a risk factor for chronic obstructive pulmonary disease. Thorax 2005; 60: 645–651. doi: 10.1136/thx.2004.035170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarrant BJ, Le Maitre C, Romero L, et al. . Mucoactive agents for chronic, non-cystic fibrosis lung disease: a systematic review and meta-analysis: mucoactive agents in chronic non-CF management. Respirology 2017; 22: 1084–1092. doi: 10.1111/resp.13047 [DOI] [PubMed] [Google Scholar]
- 10.Rubin BK. Mucolytics, expectorants, and mucokinetic medications. Respir Care 2007; 52: 859–865. [PubMed] [Google Scholar]
- 11.Shen Y, Huang S, Kang J, et al. . Management of airway mucus hypersecretion in chronic airway inflammatory disease: Chinese expert consensus (English edition). Int J Chron Obstruct Pulmon Dis 2018; 13: 399–407. doi: 10.2147/COPD.S144312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wain LV, Shrine N, Artigas MS, et al. . Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 2017; 49: 416–425. doi: 10.1038/ng.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Husseini ZW, Gosens R, Dekker F, et al. . The genetics of asthma and the promise of genomics-guided drug target discovery. Lancet Respir Med 2020; 8: 1045–1056. doi: 10.1016/S2213-2600(20)30363-5 [DOI] [PubMed] [Google Scholar]
- 14.Dijkstra AE, Boezen HM, van den Berge M, et al. . Dissecting the genetics of chronic mucus hypersecretion in smokers with and without COPD. Eur Respir J 2015; 45: 60–75. doi: 10.1183/09031936.00093314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dijkstra AE, Smolonska J, van den Berge M, et al. . Susceptibility to chronic mucus hypersecretion, a genome wide association study. PLoS One 2014; 9: e91621. doi: 10.1371/journal.pone.0091621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JH, Cho MH, Hersh CP, et al. . Genetic susceptibility for chronic bronchitis in chronic obstructive pulmonary disease. Respir Res 2014; 15: 113. doi: 10.1186/s12931-014-0113-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng X, Vonk JM, de Jong K, et al. . No convincing association between genetic markers and respiratory symptoms: results of a GWA study. Respir Res 2017; 18: 11. doi: 10.1186/s12931-016-0495-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Matteis S, Jarvis D, Young H, et al. . Occupational self-coding and automatic recording (OSCAR): a novel web-based tool to collect and code lifetime job histories in large population-based studies. Scand J Work Environ Health 2017; 43: 181–186. doi: 10.5271/sjweh.3613 [DOI] [PubMed] [Google Scholar]
- 19.Shrine N, Guyatt AL, Erzurumluoglu AM, et al. . New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 2019; 51: 481–493. doi: 10.1038/s41588-018-0321-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shrine N, Portelli MA, John C, et al. . Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med 2019; 7: 20–34. doi: 10.1016/S2213-2600(18)30389-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Global Initiative for Chronic Obstructive Lung Disease (GOLD) . Pocket guide to COPD diagnosis, management, and prevention. A guide for health care professionals. 2019. Available from: http://goldcopd.org/
- 22.Purcell S, Chang C. Plink 2.0. 2022. www.cog-genomics.org/plink/2.0 Date last accessed: 28 February 2023.
- 23.Yang J, Lee SH, Goddard ME, et al. . GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82. doi: 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruim RJ, Welch RP, Sanna S, et al. . LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–2337. doi: 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith BH, Campbell H, Blackwood D, et al. . Generation Scotland: the Scottish Family Health Study; a new resource for researching genes and heritability. BMC Med Genet 2006; 7: 74. doi: 10.1186/1471-2350-7-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.John C, Reeve NF, Free RC, et al. . Cohort profile: Extended Cohort for E-health, Environment and DNA (EXCEED). Int J Epidemiol 2019; 48: 1734. doi: 10.1093/ije/dyz073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia X, Han B, Onengut-Gumuscu S, et al. . Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 2013; 8: e64683. doi: 10.1371/journal.pone.0064683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sim N-L, Kumar P, Hu J, et al. . SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012; 40: W452–W457. doi: 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaren W, Gil L, Hunt SE, et al. . The Ensembl Variant Effect Predictor. Genome Biol 2016; 17: 122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shihab HA, Gough J, Cooper DN, et al. . Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 2013; 34: 57–65. doi: 10.1002/humu.22225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GTEx Consortium . Genetic effects on gene expression across human tissues. Nature 2017; 550: 204–213. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Ge B, Casale FP, et al. . Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 2016; 167: 1398–1414. doi: 10.1016/j.cell.2016.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giambartolomei C, Vukcevic D, Schadt EE, et al. . Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 2014; 10: e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams AT, Shrine N, Naghra-van Gijzel H, et al. . Genome-wide association study of susceptibility to hospitalised respiratory infections. Wellcome Open Res 2021; 6: 290. doi: 10.12688/wellcomeopenres.17230.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen RJ, Guillen-Guio B, Oldham JM, et al. . Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2020; 201: 564–574. doi: 10.1164/rccm.201905-1017OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demenais F, Bisgaard H, Barnes KC, et al. . Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet 2018; 50: 42–53. doi: 10.1038/s41588-017-0014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Y, Li Y, Brazel DM, et al. . Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019; 51: 237–244. doi: 10.1038/s41588-019-0530-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghoussaini M, Mountjoy E, Carmona M, et al. . Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res 2021; 49: D1311–D1320. doi: 10.1093/nar/gkaa840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Packer RJ, Williams AT, Hennah W, et al. . DeepPheWAS: an R package for phenotype generation and association analysis for phenome-wide association studies. Bioinformatics 2023; 38: btad073. doi: 10.1093/bioinformatics/btad073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seibold MA, Wise AL, Speer MC, et al. . A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503–1512. doi: 10.1056/NEJMoa1013660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Y, Byrne EM, Zheng Z, et al. . Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat Commun 2019; 10: 1891. doi: 10.1038/s41467-019-09572-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.UK Biobank . GWAS V2 results. 2018. www.nealelab.is/uk-biobank Date last accessed: 3 August 2022.
- 43.Pividori M, Schoettler N, Nicolae DL, et al. . Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: genome-wide and transcriptome-wide studies. Lancet Respir Med 2019; 7: 509–522. doi: 10.1016/S2213-2600(19)30055-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou W, Nielsen JB, Fritsche LG, et al. . Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 2018; 50: 1335–1341. doi: 10.1038/s41588-018-0184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferkingstad E, Oddsson A, Gretarsdottir S, et al. . Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun 2018; 9: 5101. doi: 10.1038/s41467-018-07460-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Onengut-Gumuscu S, Chen W-M, Burren O, et al. . Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 2015; 47: 381–386. doi: 10.1038/ng.3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu JZ, van Sommeren S, Huang H, et al. . Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47: 979–986. doi: 10.1038/ng.3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Lange KM, Moutsianas L, Lee JC, et al. . Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017; 49: 256–261. doi: 10.1038/ng.3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franke A, McGovern DPB, Barrett JC, et al. . Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010; 42: 1118–1125. doi: 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jostins L, Ripke S, Weersma RK, et al. . Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124. doi: 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hazra A, Kraft P, Selhub J, et al. . Common variants of FUT2 are associated with plasma vitamin B12 levels. Nat Genet 2008; 40: 1160–1162. doi: 10.1038/ng.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nongmaithem SS, Joglekar CV, Krishnaveni GV, et al. . GWAS identifies population-specific new regulatory variants in FUT6 associated with plasma B12 concentrations in Indians. Hum Mol Genet 2017; 26: 2551–2564. doi: 10.1093/hmg/ddx071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanaka T, Scheet P, Giusti B, et al. . Genome-wide association study of vitamin B6, vitamin B12, folate, and homocysteine blood concentrations. Am J Hum Genet 2009; 84: 477–482. doi: 10.1016/j.ajhg.2009.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hazra A, Kraft P, Lazarus R, et al. . Genome-wide significant predictors of metabolites in the one-carbon metabolism pathway. Hum Mol Genet 2009; 18: 4677–4687. doi: 10.1093/hmg/ddp428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klarin D, Damrauer SM, Cho K, et al. . Genetics of blood lipids among ∼300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 2018; 50: 1514–1523. doi: 10.1038/s41588-018-0222-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willer CJ, Schmidt EM, Sengupta S, et al. . Discovery and refinement of loci associated with lipid levels. Nat Genet 2013; 45: 1274–1283. doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teslovich TM, Musunuru K, Smith AV, et al. . Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466: 707–713. doi: 10.1038/nature09270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiss FU, Schurmann C, Guenther A, et al. . Fucosyltransferase 2 (FUT2) non-secretor status and blood group B are associated with elevated serum lipase activity in asymptomatic subjects, and an increased risk for chronic pancreatitis: a genetic association study. Gut 2015; 64: 646–656. doi: 10.1136/gutjnl-2014-306930 [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann TJ, Theusch E, Haldar T, et al. . A large electronic-health-record-based genome-wide study of serum lipids. Nat Genet 2018; 50: 401–413. doi: 10.1038/s41588-018-0064-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M, Jiang Y, Wedow R, et al. . Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019; 51: 237–244. doi: 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chambers JC, Zhang W, Sehmi J, et al. . Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 2011; 43: 1131–1138. doi: 10.1038/ng.970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanchez-Roige S, Palmer AA, Fontanillas P, et al. . Genome-wide association study meta-analysis of the Alcohol Use Disorders Identification Test (AUDIT) in two population-based cohorts. Am J Psychiatry 2019; 176: 107–118. doi: 10.1176/appi.ajp.2018.18040369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tian C, Hromatka BS, Kiefer AK, et al. . Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun 2017; 8: 599. doi: 10.1038/s41467-017-00257-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelly RJ, Rouquier S, Giorgi D, et al. . Sequence and expression of a candidate for the human Secretor blood group α(1,2)fucosyltransferase gene (FUT2): homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the nonsecretor phenotype. J Biol Chem 1995; 270: 4640–4649. doi: 10.1074/jbc.270.9.4640 [DOI] [PubMed] [Google Scholar]
- 65.The 1000 Genomes Project Consortium . A global reference for human genetic variation. Nature 2015; 526: 68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ikehara Y, Nishihara S, Yasutomi H, et al. . Polymorphisms of two fucosyltransferase genes (Lewis and Secretor genes) involving type I Lewis antigens are associated with the presence of anti-Helicobacter pylori IgG antibody. Cancer Epidemiol Biomarkers Prev 2001; 10: 971–977. [PubMed] [Google Scholar]
- 67.Imbert-Marcille B-M, Barbé L, Dupé M, et al. . A FUT2 gene common polymorphism determines resistance to rotavirus A of the P[8] genotype. J Infect Dis 2014; 209: 1227–1230. doi: 10.1093/infdis/jit655 [DOI] [PubMed] [Google Scholar]
- 68.Payne DC, Currier RL, Staat MA, et al. . Epidemiologic association between FUT2 secretor status and severe rotavirus gastroenteritis in children in the United States. JAMA Pediatr 2015; 169: 1040. doi: 10.1001/jamapediatrics.2015.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larsson MM, Rydell GEP, Grahn A, et al. . Antibody prevalence and titer to norovirus (Genogroup II) correlate with secretor (FUT2) but not with ABO phenotype or Lewis (FUT3) genotype. J Infect Dis 2006; 194: 1422–1427. doi: 10.1086/508430 [DOI] [PubMed] [Google Scholar]
- 70.Ruvoën-Clouet N, Belliot G, Le Pendu J. Noroviruses and histo-blood groups: the impact of common host genetic polymorphisms on virus transmission and evolution: noroviruses and herd innate protection. Rev Med Virol 2013; 23: 355–366. doi: 10.1002/rmv.1757 [DOI] [PubMed] [Google Scholar]
- 71.Carlsson B, Kindberg E, Buesa J, et al. . The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 norovirus infection. PLoS One 2009; 4: e5593. doi: 10.1371/journal.pone.0005593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barton SJ, Murray R, Lillycrop KA, et al. . FUT2 genetic variants and reported respiratory and gastrointestinal illnesses during infancy. J Infect Dis 2019; 219: 836–843. doi: 10.1093/infdis/jiy582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Innes AL, McGrath KW, Dougherty RH, et al. . The H antigen at epithelial surfaces is associated with susceptibility to asthma exacerbation. Am J Respir Crit Care Med 2011; 183: 189–194. doi: 10.1164/rccm.201003-0488OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Santos-Cortez RLP, Chiong CM, Frank DN, et al. . FUT2 variants confer susceptibility to familial otitis media. Am J Hum Genet 2018; 103: 679–690. doi: 10.1016/j.ajhg.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taylor SL, Woodman RJ, Chen AC, et al. . FUT2 genotype influences lung function, exacerbation frequency and airway microbiota in non-CF bronchiectasis. Thorax 2017; 72: 304–310. doi: 10.1136/thoraxjnl-2016-208775 [DOI] [PubMed] [Google Scholar]
- 76.Blackwell CC, Jónsdóttir K, Hanson M, et al. . Non-secretion of ABO antigens predisposing to infection by Neisseria meningitidis and Streptococcus pneumoniae. Lancet 1986; 328: 284–285. doi: 10.1016/S0140-6736(86)92103-3 [DOI] [PubMed] [Google Scholar]
- 77.Kachuri L, Francis SS, Morrison M, et al. . The landscape of host genetic factors involved in immune response to common viral infections. Genomic Med 2020; 12: 93. doi: 10.1186/s13073-020-00790-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kousathanas A, Pairo-Castineira E, Rawlik K, et al. . Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 2022; 607: 97–103. doi: 10.1038/s41586-022-04576-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.COVID-19 Host Genetics Initiative . Mapping the human genetic architecture of COVID-19. Nature 2021; 600: 472–477. doi: 10.1038/s41586-021-03767-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lindesmith L, Moe C, Marionneau S, et al. . Human susceptibility and resistance to Norwalk virus infection. Nat Med 2003; 9: 548–553. doi: 10.1038/nm860 [DOI] [PubMed] [Google Scholar]
- 81.Borén T, Falk P, Roth KA, et al. . Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993; 262: 1892–1895. doi: 10.1126/science.8018146 [DOI] [PubMed] [Google Scholar]
- 82.Wacklin P, Mäkivuokko H, Alakulppi N, et al. . Secretor genotype (FUT2 gene) is strongly associated with the composition of Bifidobacteria in the human intestine. PLoS One 2011; 6: e20113. doi: 10.1371/journal.pone.0020113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wacklin P, Tuimala J, Nikkilä J, et al. . Faecal microbiota composition in adults is associated with the FUT2 gene determining the secretor status. PLoS One 2014; 9: e94863. doi: 10.1371/journal.pone.0094863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rausch P, Rehman A, Künzel S, et al. . Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (secretor) genotype. Proc Natl Acad Sci USA 2011; 108: 19030–19035. doi: 10.1073/pnas.1106408108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galeev A, Suwandi A, Cepic A, et al. . The role of the blood group-related glycosyltransferases FUT2 and B4GALNT2 in susceptibility to infectious disease. Int J Med Microbiol 2021; 311: 151487. doi: 10.1016/j.ijmm.2021.151487 [DOI] [PubMed] [Google Scholar]
- 86.Cohen M, Hurtado-Ziola N, Varki A. ABO blood group glycans modulate sialic acid recognition on erythrocytes. Blood 2009; 114: 3668–3676. doi: 10.1182/blood-2009-06-227041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walters RW, Pilewski JM, Chiorini JA, et al. . Secreted and transmembrane mucins inhibit gene transfer with AAV4 more efficiently than AAV5. J Biol Chem 2002; 277: 23709–23713. doi: 10.1074/jbc.M200292200 [DOI] [PubMed] [Google Scholar]
- 88.Hurd EA, Holmén JM, Hansson GC, et al. . Gastrointestinal mucins of Fut2-null mice lack terminal fucosylation without affecting colonization by Candida albicans. Glycobiology 2005; 15: 1002–1007. doi: 10.1093/glycob/cwi089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Magalhães A, Rossez Y, Robbe-Masselot C, et al. . Muc5ac gastric mucin glycosylation is shaped by FUT2 activity and functionally impacts Helicobacter pylori binding. Sci Rep 2016; 6: 25575. doi: 10.1038/srep25575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sajuthi SP, Everman JL, Jackson ND, et al. . Nasal airway transcriptome-wide association study of asthma reveals genetically driven mucus pathobiology. Nat Commun 2022; 13: 1632. doi: 10.1038/s41467-022-28973-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Radicioni G, Ceppe A, Ford AA, et al. . Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort. Lancet Respir Med 2021; 9: 1241–1254. doi: 10.1016/S2213-2600(21)00079-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ralazamahaleo M, Elsermans V, Top I, et al. . Characterization of the novel HLA-DRB1*03:147 allele by sequencing-based typing. HLA 2019; 93: 53–54. doi: 10.1111/tan.13419 [DOI] [PubMed] [Google Scholar]
- 93.Tobin MD, Izquierdo AG. Improving ethnic diversity in respiratory genomics research. Eur Respir J 2021; 58: 2101615. doi: 10.1183/13993003.01615-2021 [DOI] [PubMed] [Google Scholar]
- 94.Okeley NM, Alley SC, Anderson ME, et al. . Development of orally active inhibitors of protein and cellular fucosylation. Proc Natl Acad Sci USA 2013; 110: 5404–5409. doi: 10.1073/pnas.1222263110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary text, figures and table captions ERJ-01667-2022.Supplement (1.5MB, pdf)
STREGA reporting recommendations ERJ-01667-2022.STREGA (176.6KB, pdf)
Supplementary tables ERJ-01667-2022.Tables (104.1KB, xlsx)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01667-2022.Shareable (787.6KB, pdf)