

Abstract
Traditional radical-mediated ring-opening of bicyclo[1.1.0]butanes (BCBs) for cyclobutane synthesis suffers from poor diastereoselectivity. Although few reports on BCB ring-opening via polar mechanisms are available, the Lewis acid-catalyzed diastereoselective ring-opening of BCBs using carbon nucleophiles is still underdeveloped. Herein, we report a mild and diastereoselective Bi(OTf)3-catalyzed ring-opening of BCBs employing 2-naphthols. The anticipated carbofunctionalized trisubstituted cyclobutanes were obtained via a bicoordinated bismuth complex and the products are formed in good to excellent yields with high regio- and diastereoselectivity. The scope of the reaction was further extended using electron-rich phenols and naphthylamine. The functionalization of the synthesized trisubstituted cyclobutanes shows the synthetic utility of the present method.
A mild and diastereoselective Bi(OTf)3-catalyzed ring-opening of BCBs employing 2-naphthols leading to the synthesis of trisubstituted cyclobutanes is reported.
Introduction
Chemists have been captivated by the idea of molecular strain for decades.1 Numerous synthetically useful transformations have been developed because of research on the characteristics and reactivity of strained carbocyclic systems. In fact, the term “strain-release” reactions has become rooted into contemporary synthetic chemistry.2 Bicyclo[1.1.0]butanes (BCBs) are among the most structurally compact cyclic compounds known, owing to their small size and high strain energy. Because of this, they have received much attention from both theoretical and preparatory perspectives.3 BCBs can be utilized to build a variety of complex bicyclic scaffolds, primarily through annulation reactions.4 The complex bicyclic scaffolds can also be obtained via functionalization of BCBs without breaking the C–C bond.5 The development of heterolytic and homolytic ring-opening processes of BCBs has advanced significantly in recent years, enabling access to structurally complex cyclobutane derivatives.
Recently, cyclobutanes have been used as structural building blocks in medicinal chemistry due to their innovative structural design and advantageous electronic, steric, and conformational characteristics.6 Furthermore, since easily accessible ‘chemical space’ is poorly populated with cyclobutanes, the examination of such species potentially avoids patented synthetic methods or molecules. However, due to the difficulties in the synthesis of these four-membered rings, the methods for preparing cyclobutanes with a variety of synthetically relevant and pharmaceutically privileged functional groups are limited.7 Therefore, chemists have recently started looking into highly strained bicycles, such as BCBs, to identify innovative protocols for their synthesis to unravel efficient ways to prepare these important motifs. For instance, Baran and co-workers disclosed the synthesis of cyclobutyl amines utilizing sulfonyl BCBs as the electrophiles (Scheme 1, eqn (1)).8a The cyclobutylation of a modest number of medicinal substances and amino acids was also successfully tested using this strategy.8a,b However, this reaction was suitable only for heteroatom nucleophiles. Moreover, Aggarwal and co-workers investigated the nucleophilic reactivity of BCBs by in situ generation of highly strained BCB boronate complexes from the corresponding lithiated BCBs and boronic esters. These highly strained BCB boronate complexes can react with various types of nucleophiles for highly functionalized cyclobutane construction with very high diastereoselectivity (eqn (2)).9 It is worth noting that the generation of highly nucleophilic lithiated BCB required 1,1-dibromo-2-(chloromethyl)cyclopropane and tert-butyl lithium.
Scheme 1. Functionalized cyclobutane synthesis from bicyclobutanes.

Additionally, BCBs can also be opened with the addition of radicals, as demonstrated by Jui,10a Ernouf,10b Gryko,10c and Studer.10d Notably, all these reactions suffered from poor diastereoselectivity. Fox and co-workers disclosed the homoconjugate addition of organocuprate to BCBs, followed by the trapping of the enolate thus generated with an electrophile.11 While this chemistry exhibited a novel use of BCBs, the cyclobutane products often had low diastereoselectivity. Thus, the synthesis of cyclobutanes from bicyclobutanes either offered low diastereoselectivity of the products or the reaction procedures are not very straightforward. Inspired by Aggarwal's boronate transfer process, we hypothesized that Lewis acids may perform analogous to boronates. We speculated that a similar ordered transition state involving the nucleophile and BCBs can be attained by a strategic choice of substituents having distinct propensity to coordinate to a Lewis acid, thereby culminating in high diastereoselectivity. Notably, Leitch and co-workers recently uncovered the Lewis acid-catalyzed divergent synthesis of azabicyclohexanes and cyclobutenyl amines from BCBs.12 However, the use of such a strategy for Lewis acid catalyzed ring-opening carbofunctionalization of BCBs has not yet been reported. Our previous experience on the ability of Lewis acids to activate 2-naphthols motivated us to select it as the nucleophilic trigger.13 We envisioned that a Lewis acid can coordinate BCBs as well as naphthols and bring them closer, making the carbofunctionalization of BCBs possible, resulting in the formation of trisubstituted cyclobutanes (eqn (3)). It may be noted in this context that functionalized trisubstituted cyclobutanes are important building blocks used in medicinal chemistry.14
Results and discussion
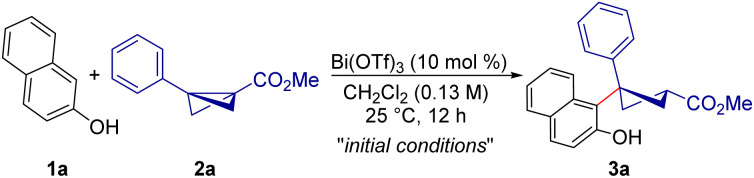
With the envisioned idea in mind, the present studies were initiated by the treatment of 2-naphthol 1a with BCB 2a in the presence of Bi(OTf)3 (10 mol%) in CH2Cl2 at 25 °C. Under these conditions, the anticipated carbofunctionalized product 3a was formed in 91% yield as a single diastereomer (Table 1, entry 1). Interestingly, the possible side products originating from the direct addition of the hydroxyl group of 2-naphthol to bicyclobutane were not observed under these conditions. Other Lewis acids such as Sc(OTf)3 and Yb(OTf)3 provided a reduced yield of 3a under the present conditions (entries 2 and 3). When triflic acid was used as a Brønsted acid catalyst, no desired product was obtained (entry 4). Other solvents were found to be ineffective for this desired transformation (entries 5 and 6). It is worth mentioning that, using MeCN as the solvent, the expected product was formed in 86% yield and with a 3 : 1 diastereomeric ratio (entry 7). Carrying out the reaction at 0 °C did not improve the yield of 3a (entry 8). Moreover, increasing the amount of 2a to 1.5 equiv. did not improve the yield of 3a considerably (entry 9). Lowering the amount of Lewis acid or performing the reaction under dilute conditions resulted in lower yield of the carbofunctionalized product (entries 10 and 11). Hence, entry 1 was chosen as the optimal condition for this diastereoselective carbofunctionalized cyclobutane synthesis.15
Optimization of the reaction conditionsa.
| ||
|---|---|---|
| Entry | Variation of the initial conditionsa | Yield of 3ab (%) |
| 1 | None | 91 |
| 2 | Sc(OTf)3 instead of Bi(OTf)3 | 28 |
| 3 | Yb(OTf)3 instead of Bi(OTf)3 | 15 |
| 4 | TfOH instead of Yb(OTf)3 | <5 |
| 5 | DCE instead of CH2Cl2 | 78 |
| 6 | THF instead of CH2Cl2 | 39 |
| 7cc | MeCN instead of CH2Cl2 | 86 |
| 8 | 0 °C instead of 25 °C | 84 |
| 9 | 1.5 equiv. of 2a | 92 |
| 10 | 5 mol% Bi(OTf)3 | 83 |
| 11 | 0.06 M CH2Cl2 | 81 |
Initial conditions: 1a (0.20 mmol), 2a (0.24 mmol), Bi(OTf)3 (10 mol%), CH2Cl2 (1.5 mL), 25 °C for 12 h.
Given are the yield of chromatographically purified 3a.
3.0 : 1 diastereomeric ratio was obtained in this case and the dr value was determined from 1H NMR of the crude reaction mixture.
With the identified reaction conditions in hand, we evaluated the scope and drawbacks of this diastereoselective carbofunctionalization of BCBs (Scheme 2). The variation in the BCBs was examined first. A variety of bicyclo[1.1.0]butanes having electron-releasing, -neutral, or -withdrawing groups at the 4-position of the benzene ring underwent a smooth ring-opening reaction leading to the diastereoselective formation of the desired carbofunctionalized products in good yields (3a–e). The developed carbofunctionalization reaction was scalable, as evidenced by the isolation of 3a in 89% yield when the reaction was carried out on a 2.0 mmol scale. Furthermore, BCBs with substituents at the 3-position of the aryl ring were tolerated well under the current conditions, resulting in the formation of the expected cyclobutane products in high yields (3f, 3g). Disappointingly, 2-substituted aryl BCBs failed to give the desired product under the present reaction conditions, most likely for steric reasons. Moreover, this regio- and diastereoselective ring-opening reaction was not limited to methyl ester derived BCBs, but benzyl and isopropyl substituted BCBs afforded the target products in good yields (3h, 3i).
Scheme 2. Substrate scope of the reaction. Reaction conditions: 1 (0.20 mmol), 2 (0.24 mmol), Bi(OTf)3 (10 mol%), CH2Cl2 (1.5 mL), 25 °C for 12 h. Provided are isolated yields of the products. a Yield of the experiment conducted on a 2.0 mmol scale. b The dr value was determined from 1H NMR of the crude reaction mixture.

The scope of the reaction was then evaluated using variously substituted naphthols. The reaction conducted using 7-methoxynaphthalen-2-ol 1b as the nucleophilic trigger furnished the desired product 3j in 84% yield. 7-Bromo substituted 2-naphthol 1c also worked well and the anticipated product 3k was formed in 93% yield. In this case, the product structure was confirmed using X-ray analysis of the crystal.16 Moreover, 2-naphthols with various aryl and heteroaryl substitutions at the 7-position of the naphthol ring afforded good yields of carbofunctionalized cyclobutanes (3l–n). A wide range of 2-naphthols with electron-releasing, -withdrawing, or -neutral groups at the 6-position of the naphthol ring yielded the desired products in a single diastereomer (3o–s). Various 4-substituted 2-naphthols could also be used as substrates to obtain the corresponding carbofunctionalized cyclobutanes (3t–v) under the optimized reaction conditions. The reaction using 3-methoxy 2-naphthol afforded the target product 3w in 78% yield and 2 : 1 dr.
Interestingly, this carbofunctionalization reaction of BCB is not only limited to 2-naphthol as the nucleophilic trigger, but 1-naphthol also worked under the present reaction conditions and the target product 3x was formed in 48% yield. This ring-opening reaction can also be extended further towards electron-rich phenols as the nucleophile, and in that case, the products (3y and 3z) were obtained in moderate yields. Finally, naphthylamine could also be used as the nucleophile to open BCB and the reaction furnished the functionalized cyclobutane 3aa in 52% yield with 3 : 1 dr.
The reaction of monosubstituted ketone derived BCB 2j with 1a under the optimized conditions afforded the cyclobutyl ketone 3ab in 70% yield and an inseparable mixture of diastereomers in a 2 : 1 ratio (Scheme 3). Similar results are obtained with the 2-naphthyl-derived BCB 2k, and the product 3ac was formed in 68% yield and 2.3 : 1 dr. These experiments tend to indicate that the aryl group of BCB has a major role in this diastereoselective ring-opening reaction.17
Scheme 3. Reaction using unsubstituted ketone-derived BCBs.

A few mechanistic experiments were carried out to get insight into the mechanism of the present reaction. When the reaction was performed in the absence of Bi(OTf)3, no product formation was observed, indicating that the Lewis acid is crucial for this reaction. Moreover, the reaction conducted using methyl protected 2-naphthol 4 did not afford the desired product, shedding light on the role of the free –OH in this carbofunctionalization reaction. In the absence of the free –OH group, it is likely that the carbofunctionalization process was deemed unfavourable due to the lack of proper coordination between 2-naphthol and Bi(OTf)3 (Scheme 4, eqn (4)).
Scheme 4. Mechanistic studies.

The reaction of BCB 2a with Bi(OTf)3 in the absence of 2-naphthol 1a furnished the cyclobutene 5 in 51% yield under the optimized reaction conditions (eqn (5)). This study indicates that in the absence of 2-naphthol, BCB can directly coordinate with the Lewis acid, which results in the formation of 5.12 Most probably, 2-naphthol reduces the Lewis acidity of Bi(OTf)3 and therefore no cyclobutene product was obtained under our optimized reaction conditions. To rule out the possibility of carbofunctionalized product formation via the intermediacy of cyclobutene 5, 2-naphthol 1a and 5 were subjected to the present reaction conditions. However, the desired product 3a was not formed, thereby confirming that the reaction was not proceeding via the cyclobutene intermediate 5 (eqn (6)). When the reaction was performed using 1-bromo 2-naphthol 1s under the optimized conditions, the desired ring-opening product of BCB was not formed, and cyclobutene 5 was formed in 17% yield (eqn (7)).18 This indicates that the present reaction does not work with 2-naphthols with a substituent at the 1-position. Performing the reaction using sulfonyl group-containing BCB did not afford the desired ring-opening product, shedding light on the importance of the carbonyl moiety of BCB for effective Lewis acid binding in this diastereoselective ring-opening reaction (eqn (8)).
We also examined how the mode of reagent addition affects the reaction outcome. When 2-naphthol and Bi(OTf)3 were stirred for 30 min and BCB 2a was subsequently added, the expected product 3a was formed in 84% yield (Scheme 5, eqn (9)). It is noteworthy that reversing the order of reagent addition did not result in product formation (eqn (10)). These two reactions support the hypothesis about the intermediacy of a bicoordinated Bi-complex for a fruitful transformation. The pre-complexation of the Lewis acid with 2-naphthol (presumably) lowers the acidity of the Lewis acid, thereby effectively halting BCB decomposition; the soft coordination of a BCB ester with the precomplexed Bi(OTf)3 (with 2-naphthol) forms the bicoordinated Bi-complex which enables the diastereoselective ring-opening of activated BCB. Reversing the mode of addition leads to decomposition of the BCB, as the acidity of the Lewis acid could not be reduced by 2-naphthol coordination.
Scheme 5. Dependence of the reaction on the mode of addition.

Based on the mechanistic studies and DFT calculations,19 a catalytic cycle for this diastereoselective ring-opening reaction is presented in Scheme 6. Initially, the coordination between Bi(OTf)3 and 2-naphthol 1a forms the intermediate A. This intermediate A serves as the catalytically active species in this transformation. Following this, BCB 2a also coordinates with intermediate A displacing one of the triflate ions, and forms the cationic intermediate B. Interestingly, the BCB coordinates via C1, thereby generating a positive charge on C3 and further activating it towards the nucleophilic attack. Subsequently, the nucleophilic attack of the 2-naphthol moiety on BCB occurs through TS(B–C), leading to the formation of the intermediate C. This nucleophilic attack presents an activation free energy barrier of only 1.9 kcal mol−1 making it a highly facile process. This is followed by a 1,3-proton transfer that restores the aromaticity of 2-naphthol and generates the intermediate D. Further proton transfer results in the final product Fvia a proposed intermediate E, where the proton is envisioned to be transferred from a second molecule of 1a. Notably, the proton transfer from intermediate E can happen from either face of the substrate, resulting in the formation of two diastereomeric products. Thus, this step is most likely to be the diastereo-determining step of this transformation. The DFT studies indicated that the TSs for the direct proton transfer was of very high energy, thus suggesting that assisted proton transfer via triflate, 2-naphthol, etc., could take place. It was found that the observed diastereomer 3a is 1.7 kcal mol−1 more stable than the other diastereomer. Therefore, it is reasonable to assume that the diastereoselectivity determining step has the thermodynamic influence. We have also considered alternative pathways, the details of which along with the energetics of the unassisted proton transfer steps are provided in the ESI.†15
Scheme 6. Proposed catalytic cycle. Relative free energy values (ΔG) are given in kcal mol−1. The distance is in Å.

The trisubstituted cyclobutane 3a synthesized using the present method can be employed as a synthetically useful precursor for the synthesis of functionalized cyclobutanes (Scheme 7). Reduction of the ester group in 3a using LiAlH4 provided the primary alcohol 6 in 94% yield. Moreover, the methyl ester in 3a was hydrolysed to form the free carboxylic acid 7 in 78% yield. Methyl protection of the free –OH group was carried out and the expected product 8 was formed in 93% yield. In addition, the cyclobutane derivative 3a can be easily O-arylated under transition-metal-free conditions using arynes as the aryl source;20 thus, when 3a was treated with benzyne produced from the triflate precursor 9 using KF (with 18-crown-6 as an additive), the desired product 10 was obtained in 86% yield. Furthermore, treatment of the cyclobutane 3a with triflic anhydride in the presence of pyridine furnished the desired product 11 in 93% yield. Subsequent treatment of 11 with diphenylphosphine oxide in the presence of a catalytic amount of Pd(OAc)2 furnished the phosphine oxide 12 in 82% yield. Additionally, the α-allylation of the methyl protected methyl ester 8 using allyl bromide under basic conditions provided the desired allylation product 13 with two all-carbon quaternary stereocenters in 64% yield. The ester functionality in 8 could easily be converted to an amide moiety in two steps and the target amide 14 was formed in 79% yield. Subsequently, the amide was converted into the thioamide derivative 15 in 84% yield by the treatment with P2S5.
Scheme 7. Synthetic utility of trisubstituted cyclobutanes.

Conclusions
In conclusion, a Lewis acid-catalyzed diastereoselective carbofunctionalization of BCBs utilizing 2-naphthols as the nucleophilic trigger leading to the formation of tricyclic cyclobutanes has been realized. The desired transformation relies on the bicoordinated bismuth complex as the crucial intermediate. The present reaction is operationally simple, advances smoothly under mild conditions and can tolerate various functional groups. This reaction is not restricted to 2-naphthols as the nucleophilic trigger; electron-rich phenols and naphthylamine can also act as the nucleophile. Mechanistic experiments and DFT studies were carried out to get insight into the possible course of the reaction. To reveal synthetic handles for additional synthetic transformations, functional group interconversions were also performed.
Data availability
Details on experimental procedures, mechanistic experiments, characterization data of all the carbofunctionalized cyclobutanes, X-ray data of 3k, and DFT studies.
Author contributions
A. G. and A. T. B. conceived and designed the project. A. G. performed the optimization studies, substrate scope analysis and mechanistic studies. S. B. helped in the substrate scope studies. M. S. H. performed the DFT studies. A. G., S. B. and A. T. B. wrote the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support by the Science and Engineering Research Board (SERB), Government of India (File Number: CRG/2021/001803) is greatly acknowledged. A. G. thanks MHRD (for PMRF), and S. B. and M. S. H. thank CSIR (for SRF) for the research fellowship. We thank Prof. Garima Jindal for help in DFT studies, Mr. Kishorkumar Sindogi for collecting the X-ray data of 3k and Ms Shiksha Deswal, Mr Sayan Shee and Mr Sukriyo Chakraborty (OC, IISc) for helpful discussion.
Electronic supplementary information (ESI) available: Details on experimental procedures and characterization data of all compounds. CCDC 2233256. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01373a
Notes and references
- Wiberg K. B. Angew. Chem., Int. Ed. Engl. 1986;25:312. doi: 10.1002/anie.198603121. [DOI] [Google Scholar]
- (a) Turkowska J. Durkaab J. Gryko D. Chem. Commun. 2020;56:5718. doi: 10.1039/D0CC01771J. [DOI] [PubMed] [Google Scholar]; (b) Milligan J. A. Wipf P. Nat. Chem. 2016;8:296. doi: 10.1038/nchem.2485. [DOI] [PubMed] [Google Scholar]
- (a) Fawcett A. Pure Appl. Chem. 2020;92:751. doi: 10.1515/pac-2019-1007. [DOI] [Google Scholar]; (b) Walczak M. A. A. Krainz T. Wipf P. Acc. Chem. Res. 2015;48:1149. doi: 10.1021/ar500437h. [DOI] [PubMed] [Google Scholar]; (c) Kelly C. B. Milligan J. A. Tilley L. Sodano T. M. Chem. Sci. 2022;13:11721. doi: 10.1039/D2SC03948F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kleinmans R. Pinkert T. Dutta S. Paulisch T. O. Keum H. Daniliuc C. G. Glorius F. Nature. 2022;605:477. doi: 10.1038/s41586-022-04636-x. [DOI] [PubMed] [Google Scholar]; (b) Liang Y.-J. Kleinmans R. Daniliuc C. G. Glorius F. J. Am. Chem. Soc. 2022;144:20207. doi: 10.1021/jacs.2c09248. [DOI] [PubMed] [Google Scholar]; (c) Guo R. Chang Y. Herter L. Salome C. Braley S. E. Fessard T. C. Brown M. K. J. Am. Chem. Soc. 2022;144:7988. doi: 10.1021/jacs.2c02976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zheng Y. Huang W. Dhungana R. K. Granados A. Keess S. Makvandi M. Molander G. A. J. Am. Chem. Soc. 2022;144:23685. doi: 10.1021/jacs.2c11501. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Agasti S. Beltran F. Pye E. Kaltsoyannis N. Crisenza G. E. M. Procter D. J. Nat. Chem. 2023;15:535. doi: 10.1038/s41557-023-01135-y. [DOI] [PubMed] [Google Scholar]
- (a) McNamee R. E. Thompson A. L. Anderson E. A. J. Am. Chem. Soc. 2021;143:21246. doi: 10.1021/jacs.1c11244. [DOI] [PubMed] [Google Scholar]; (b) McNamee R. E. Haugland M. M. Nugent J. Chan R. Christensen K. E. Anderson E. A. Chem. Sci. 2021;12:7480. doi: 10.1039/D1SC01836A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lovering F. Bikker J. Humblet C. J. Med. Chem. 2009;52:6752. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; (b) Lovering F. MedChemComm. 2013;4:515. doi: 10.1039/C2MD20347B. [DOI] [Google Scholar]; (c) Wrobleski M. Reichard G. Paliwal S. Shah S. Tsui H. Duffy R. Lachowicz J. Morgan C. Varty G. Shih N. Bioorg. Med. Chem. Lett. 2006;16:3859. doi: 10.1016/j.bmcl.2006.04.031. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou K. C. Vourloumis D. Totokotsopoulos S. Papakyriakou A. Karsunky H. Fernando H. Gavrilyuk J. Webb D. Stepan A. F. ChemMedChem. 2016;11:31. doi: 10.1002/cmdc.201500510. [DOI] [PubMed] [Google Scholar]
- (a) Namyslo J. C. Kaufmann D. E. Chem. Rev. 2003;103:1485. doi: 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]; (b) Seiser T. Saget T. Tran D. N. Cramer N. Angew. Chem., Int. Ed. 2011;50:7740. doi: 10.1002/anie.201101053. [DOI] [PubMed] [Google Scholar]; (c) Poplata S. Troster A. Zou Y. Q. Bach T. Chem. Rev. 2016;116:9748. doi: 10.1021/acs.chemrev.5b00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Gianatassio R. Lopchuk J. M. Wang J. Pan C.-M. Malins L. R. Prieto L. Brandt T. A. Collins M. R. Gallego G. M. Sach N. W. Spangler J. E. Zhu H. Zhu J. Baran P. S. Science. 2016;351:241. doi: 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lopchuk J. M. Fjelbye K. Kawamata Y. Malins L. R. Pan C.-M. Gianatassio R. Wang J. Prieto L. Bradow J. Brandt T. A. Collins M. R. Elleraas J. Ewanicki J. Farrell W. Fadeyi O. O. Gallego G. M. Mousseau J. J. Oliver R. Sach N. W. Smith J. K. Spangler J. E. Zhu H. Zhu J. Baran P. S. J. Am. Chem. Soc. 2017;139:3209. doi: 10.1021/jacs.6b13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Bennett S. H. Fawcett A. Denton E. H. Biberger T. Fasano V. Winter N. Aggarwal V. K. J. Am. Chem. Soc. 2020;142:16766. doi: 10.1021/jacs.0c07357. [DOI] [PubMed] [Google Scholar]; (b) Fawcett A. Biberger T. Aggarwal V. K. Nat. Chem. 2019;11:117. doi: 10.1038/s41557-018-0181-x. [DOI] [PubMed] [Google Scholar]; (c) Silvi M. Aggarwal V. K. J. Am. Chem. Soc. 2019;141:9511. doi: 10.1021/jacs.9b03653. [DOI] [PubMed] [Google Scholar]
- (a) Pratt C. J. Aycock R. A. King M. D. Jui N. T. Synlett. 2020;31:51. doi: 10.1055/s-0039-1690197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ernouf G. Chirkin E. Rhyman L. Ramasami P. Cintrat J. Angew. Chem., Int. Ed. 2020;59:2618. doi: 10.1002/anie.201908951. [DOI] [PubMed] [Google Scholar]; (c) Ociepa M. Wierzba A. J. Turkowska J. Gryko D. J. Am. Chem. Soc. 2020;142:5355. doi: 10.1021/jacs.0c00245. [DOI] [PubMed] [Google Scholar]; (d) Yu X. Lübbesmeyer M. Studer A. Angew. Chem., Int. Ed. 2021;60:675. doi: 10.1002/anie.202011738. [DOI] [PubMed] [Google Scholar]
- Panish R. Chintala S. R. Boruta D. T. Fang Y. Taylor M. T. Fox J. M. J. Am. Chem. Soc. 2013;135:9283. doi: 10.1021/ja403811t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhake K. Woelk K. J. Becia J. Un A. Jenny S. E. Leitch D. C. Angew. Chem., Int. Ed. 2022;61:e202204719. doi: 10.1002/anie.202204719. [DOI] [PubMed] [Google Scholar]
- Kaicharla T. Roy T. Thangaraj M. Gonnade R. G. Biju A. T. Angew. Chem., Int. Ed. 2016;55:10061. doi: 10.1002/anie.201604373. [DOI] [PubMed] [Google Scholar]
- (a) Tkachenko A. N. Radchenko D. S. Mykhailiuk P. K. Afonin S. Ulrich A. S. Komarov I. V. Angew. Chem., Int. Ed. 2013;52:6504. doi: 10.1002/anie.201301344. [DOI] [PubMed] [Google Scholar]; (b) Wager T. Pettersen B. A. Schmidt A. W. Spracklin D. K. Mente S. Butler T. W. Howard H. Lettiere D. J. Rubitski D. M. Wong D. F. Nedza F. M. Nelson F. R. Rollema H. Raggon J. W. Aubrecht J. Freeman J. K. Marcek J. M. Cianfrogna J. Cook K. W. James L. C. Chatman L. A. Iredale P. A. Banker M. J. Homiski M. L. Munzner J. B. Chandrasekaran R. Y. J. Med. Chem. 2011;54:7602. doi: 10.1021/jm200939b. [DOI] [PubMed] [Google Scholar]; (c) Homon A. A. V Shynder L. Demchuk O. P. V Hryshchuk O. Kondratov I. S. Gerus I. I. Grygorenko O. O. J. Fluorine Chem. 2022;263:110041. doi: 10.1016/j.jfluchem.2022.110041. [DOI] [Google Scholar]
- See the ESI† for details
- CCDC 2233256 (3k) contains the supplementary crystallographic data for this paper.†
- It may be noted that the reaction performed using 3-unsubstituted BCB with ester at the 1-position failed to afford the desired ring-opened product under the optimized conditions
- Hazra A. Kanji T. Banerjee P. J. Org. Chem. 2022;88:960. doi: 10.1021/acs.joc.2c02378. [DOI] [PubMed] [Google Scholar]
- For reports on dual coordination of bismuth, see: ; (a) Kawai N. Abe R. Matsuda M. Uenishi J. J. Org. Chem. 2011;76:2102. doi: 10.1021/jo102452d. [DOI] [PubMed] [Google Scholar]; (b) Kawai N. Abe R. Uenishi J. Tetrahedron Lett. 2009;50:6580. doi: 10.1016/j.tetlet.2009.09.053. [DOI] [Google Scholar]; (c) Komeyama K. Saigo N. Miyagi M. Takaki K. Angew. Chem., Int. Ed. 2009;48:9875. doi: 10.1002/anie.200904610. [DOI] [PubMed] [Google Scholar]; (d) Kargbo R. B. Hashemi Z. S. Roy S. Jin X. Herr R. J. Tetrahedron Lett. 2013;54:2018. doi: 10.1016/j.tetlet.2013.02.012. [DOI] [Google Scholar]; (e) Komeyama K. Takahashi K. Takaki K. Org. Lett. 2008;10:5119. doi: 10.1021/ol8019567. [DOI] [PubMed] [Google Scholar]
- (a) Shi J. Li L. Li Y. Chem. Rev. 2021;121:3892. doi: 10.1021/acs.chemrev.0c01011. [DOI] [PubMed] [Google Scholar]; (b) Fluegel L. L. Hoye T. R. Chem. Rev. 2021;121:2413. doi: 10.1021/acs.chemrev.0c00825. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Werz D. B. Biju A. T. Angew. Chem., Int. Ed. 2020;59:3385. doi: 10.1002/anie.201909213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Biju A. T., Modern Aryne Chemistry, Wiley-VCH Verlag, Weinheim, Germany, 2021 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Details on experimental procedures, mechanistic experiments, characterization data of all the carbofunctionalized cyclobutanes, X-ray data of 3k, and DFT studies.