

Abstract
The ASXL1 and SRSF2 mutations in AML are frequently found in patients with preexisting myeloid malignancies and are individually associated with poor outcomes. In this multi-institutional retrospective analysis, we assessed the genetic features and clinical outcomes of 43 patients with ASXL1mutSRSF2mut AML and compared outcomes to patients with either ASXL1 (n = 57) or SRSF2 (n = 70) mutations. Twenty-six (60%) had secondary-AML (s-AML). Variant allele fractions suggested that SRSF2 mutations preceded ASXL1 mutational events. Median overall survival (OS) was 7.0 months (95% CI:3.8,15.3) and was significantly longer in patients with de novo vs s-AML (15.3 vs 6.4 months, respectively; P = .04 on adjusted analysis). Compared to ASXL1mutSRSF2wt and ASXL1wtSRSF2mut, co-mutated patients had a 1.4 and 1.6 times increase in the probability of death, respectively (P = .049), with a trend towards inferior OS (median OS = 7.0 vs 11.5 vs 10.9 months, respectively; P = .10). Multivariable analysis suggests this difference in OS is attributable to the high proportion of s-AML patients in the co-mutated cohort (60% vs 32% and 23%, respectively). Although this study is limited by the retrospective data collection and the relatively small sample size, these data suggest that ASXL1mutSRSF2mut AML is a distinct subgroup of AML frequently associated with s-AML and differs from ASXL1mutSRSF2wt/ASXL1wtSRSF2mut with respect to etiology and leukemogenesis.
1 |. INTRODUCTION
Acute myeloid leukemia (AML) is a hematologic malignancy of myeloid progenitors resulting in compromised hematopoiesis and bone marrow failure.1 Diverse and complex genetic alterations result in highly heterogeneous outcomes for patients. Prognosis has traditionally been determined using clinical characteristics and cytogenetic abnormalities. However, the recent widespread adoption of next generation sequencing (NGS) has allowed for the identification of several prognostically distinct genomic subgroups.2–6 The largest genomic classification of AML patients by Papaemmanuil et al. proposed 11 distinct subgroups based on patterns of leukemia-initiating mutations, comutations and genomic heterogeneity, which are hypothesized to better define the disease biology and predict outcomes.4
One proposed subgroup is defined by mutations in chromatin modifying and/or RNA-splicing genes.4 This chromatin-spliceosome subgroup includes patients with concomitant mutations in the chromatin modifying gene, ASXL1, and the splicesosome component, SRSF2, (ASXL1mutSRSF2mut AML). In the 15 patients with ASXL1mutSRSF2mut AML reported in this analysis, overall survival (OS) was dismal with no long-term survivors suggesting that these mutations may have multiplicative adverse effects.4 The small number of co-mutated (ASXL1mutSRSF2mut) patients in this study precluded the ability to determine how clinical factors such as type of treatment or a prior history of myeloid neoplasm influenced outcomes. Harboring an ASXL1 mutation, regardless of the presence or absence of a mutation in SRSF2, is a known poor prognostic indicator.3,4,7–10 SRSF2 is associated with poor outcomes for MDS patients, though the prognostic significance in AML is less well-defined.11–14 Determining the effect of a prior myeloid malignancy is of particular importance because ASXL1 and SRSF2 mutations are both common in myelodysplastic syndrome (MDS)15 and chronic myelomonocytic leukemia (CMML).16 We recently reported that ASXL1mutSRSF2mut AML, in contrast to ASXL1mutSRSF2wt AML, shares a mutational profile and immunophenotype with CMML suggesting that this genomic profile may identify patients with secondary AML (s-AML) from preexisting CMML.17 Further, Lindsley and colleagues identified ASXL1 and SRSF2, along with six other mutations related to chromatin and RNA-splicing (EZH2, BCOR, STAG2, SF3B1, U2AF1, and ZRSR2), to be highly specific (>95%) for predicting s-AML, an etiologic subtype of AML known to confer very poor outcomes..18,19
In this multi-institutional retrospective analysis, we sought to assess the genetic features and analyze the clinical outcomes of a larger cohort of patients with ASXL1mutSRSF2mut AML, and compare survival outcomes of this cohort to patients with ASXL1mutSRSF2wt AML and ASXL1wtSRSF2mut AML. We hypothesized that ASXL1mutSRSF2mut AML would be associated with worse outcomes and may represent a unique genomic footprint of s-AML irrespective of whether patients were diagnosed with a preexisting myeloid neoplasm.
2 |. METHODS
2.1 |. Study design
This is a multi-institutional retrospective cohort study involving patients who were identified through the electronic medical record review at the University of North Carolina in Chapel Hill, NC (UNC) and Moffitt Cancer Center in Tampa, FL (Moffitt) from 2011 to 2020. Patients were included in all analyses if they were ≥ 18 years old and had newly diagnosed AML with ASXL1 or SRSF2 mutations identified by NGS from 2011 to 2020. The 2016 WHO guidelines were used as diagnostic criteria for AML.6 In the primary analysis, outcomes of ASXL1mutSRSF2mut AML were described to evaluate the association between co-variates including etiology (de novo, s-AML, treatment-related AML [t-AML]) and survival. In a secondary analysis, survival outcomes of ASXL1mutSRSF2mut patients were compared to patients with ASXL1mutSRSF2wt AML and ASXL1wtSRSF2mut AML to evaluate the differences between these populations. This study was approved by the institutional review board at UNC and Moffitt according to the declaration of Helsinki.
2.2 |. Next-generation sequencing
Next generation sequencing was performed on DNA collected from diagnostic bone marrow or peripheral blood. Table S1 outlines the sequencing panels used for each patient with ASXL1mutSRSF2mut AML. A threshold of ≥5% variant allele fraction (VAF) for individual gene mutations was considered positive for most variants. However, for samples analyzed using the Illumina TruSight Myeloid 54-gene panel, ASXL1 c.1934dupG (p.Gly646fs) mutations required a minimum 10% VAF, given known potential false positive results at lower VAF on this platform.20
To be included in this study, patients were required to have ASXL1 or SRSF2 variants defined as pathogenic/likely pathogenic by the reporting laboratory. Germline control samples were not sequenced in parallel, but variants with high population allele frequencies in any subpopulation in the genome aggregation database (gnomAD, https://gnomad.broadinstitute.org) were excluded as presumed benign germline variants.
2.3 |. Outcomes
The primary outcome was OS defined as time from diagnosis of AML to date of death or end of the study period (February 28, 2020). Patients who were alive at this time were censored. For patients with ASXL1mutSRSF2mut AML, the primary exposure was s-AML. S-AML was defined as having a documented history of a previous myeloid neoplasm. T-AML was defined as developing AML after receiving cytotoxic chemotherapy or radiation. Patients who developed a treatment-related myeloid neoplasm prior to the development of AML were classified as s-AML. Patients who did not have s-AML or t-AML were classified as having de novo AML. Both MDS and myeloproliferative neoplasms (MPNs) were defined according to current WHO criteria. The secondary outcome was rate of complete remission (CR) or complete remission with incomplete hematologic recovery (CRi) following initial chemotherapy defined by ELN guidelines.3 Exploratory analyses evaluated associations between treatment intensity, as well as serologic, genomic, and cytogenetic factors and outcomes. Patients receiving azacitidine or decitabine, regardless of the dose or schedule, were classified as receiving hypomethylating agents (HMAs). Patients receiving anthracycline-based chemotherapy (daunorubicin, idarubicin, or mitoxantrone) were considered to have received intensive induction chemotherapy (IC). Investigational agents given to patients as part of a clinical trial in addition to these regimens were not considered when determining treatment intensity.
2.4 |. Covariates
Covariates considered were based on literature review and included site, age, gender, race, performance status, cytogenetic risk group, splenomegaly, number of mutations, variant allele fraction of SRSF2 and ASXL1, and white blood cell count, hemoglobin, platelet count, and lactate dehydrogenase (LDH) at diagnosis.
2.5 |. Statistical methods
Patients’ baseline characteristics were summarized using descriptive statistics. Fisher’s exact test was used to compare categorical variables and the Wilcoxon two-group test and the Kruskal-Wallis test were used for to compare groupings of continuous variables. The Kaplan–Meier method was used to estimate the time-to-event function of OS. OS was calculated using the time from initial diagnosis (either by bone marrow biopsy or, if unavailable, flow cytometry of peripheral blood) to death or date of last contact (censored). Cox regression modeling was used to evaluate the association of OS and select patient covariates. Both univariable and adequately powered multivariable models were investigated. The OS curves were compared using log-rank and score tests. Logistic regression models were used to examine the association of select covariates to the dichotomized outcome response variable of CR/CRi to not CR/CRi. All reported P values are two-sided with P values less than .05 considered significant. Statistical analyses were done using Stata (Version 16.1, StataCorp, College Station, TX), SAS (version 9.4, Cary, NC) or R (2019, R Foundation, https://www.R-project.org).
3 |. RESULTS
3.1 |. Patient characteristics
Of 1564 AML patients screened, 43 (2.7%) were found to have ASXL1mutSRSF2mut AML. Baseline characteristics of the ASXL1mutSRSF2mut cohort are included in Table 1. The median age of patients was 71 years; most patients (81%) were white. Twenty-eight (64%) had normal cytogenetics; 35 (81%) had intermediate-risk cytogenetics by current ELN guidelines. Twenty-six patients (60%) were classified as having s-AML arising from MDS (n = 17), CMML (n = 7), and MPNs (n = 2) including one patient who developed t-MN prior to developing AML. Sixteen (37%) patients were classified as having de novo AML. One patient was classified as t-AML. Patients with s-AML had fewer blasts in the bone marrow as compared to de novo patients (median 30% vs 64%, P < .001).
TABLE 1.
Baseline characteristics of the cohort of patients with ASXL1mutSRSF2mut AML
| All Patients (n = 43) | De novo AML (n = 16) | Secondary AML (n = 26) | Treatment-related AML (n = 1) | P-value | |
|---|---|---|---|---|---|
| Median Age at Dx—yr. (range) | 71 (42–85) | 70 (42–83) | 72 (59–85) | 69 | .11 |
| Male sex—no. (%) | 30 (70%) | 11 (69%) | 19 (73%) | 1 | .43 |
| Race—no. (%) | .72 | ||||
| White | 34 (81%) | 11 (69%) | 22 (85%) | n/a | |
| African-American | 3 (7%) | 2 (13%) | 1 (4%) | ||
| Hispanic | 1 (2%) | 0 (0%) | 1 (4%) | ||
| Other | 4 (9%) | 2 (13%) | 2 (8%) | ||
| Unknown | 1 (2%) | 1 (6%) | 0 (0%) | ||
| ECOG—no. (%) | .49 | ||||
| 0–1 | 31 (72%) | 12 (75%) | 19 (73%) | ||
| 2–4 | 10 (23%) | 3 (19%) | 6 (23%) | ||
| Median WBC at Dx, 109/L (range) | 8.2 (0.6–157.5) | 10.1 (0.9–157.5) | 8.0 (0.6–94.3) | 86.1 | .3 |
| Median Hgb at Dx, g/dL (range) | 9.2 (6.1–13.8) | 9.9 (6.1–13.8) | 9.0 (7.2–13.4) | 10.7 | .75 |
| Median Plt at Dx, 109/L (range) | 63 (10–273) | 82 (12–199) | 57 (10–273) | 116 | .63 |
| Median LDH at Dx, U/L (range) | 406 (166–7960) | 958 (166–7960) | 370 (171–4435) | 3874 | .1 |
| Median peripheral blasts % at Dx (range) | 7% (0%−80%) | 12% (0%−80%) | 7% (0%−73%) | 35% | .41 |
| Median bone marrow blast % at Dx (range) | 36% (0.5%−93%) | 64% (24%−93%) | 30% (0.5%−85%) | n/a | <.001 |
| Splenomegaly | .07 | ||||
| Present | 12 (28%) | 3 (20%) | 9 (35%) | ||
| Absent | 16 (37%) | 10 (63%) | 6 (23%) | ||
| Total mutations (range) | 4 (2–10) | 5 (2–7) | 4 (2–9) | 6 | .27 |
| WHO classification | <.001 | ||||
| AML | 20 (47%) | 13 (81%) | 6 (23%) | 1 | |
| AML-MRC | 23 (53%) | 3 (19%) | 20 (76%) | ||
| Cytogenetics—no. (%) | .83 | ||||
| ELN Intermediate risk | 34 (79%) | 11 (67%) | 22 (85%) | 1 | |
| ELN Adverse risk | 6 (14%) | 4 (25%) | 2 (7%) | ||
| Unknown | 3 (7%) | 1 (6%) | 2 (7%) | ||
| Induction regimens—no. (%) | .058 | ||||
| Intensive induction | 20 (47%) | 9 (56%) | 11 (42%) | ||
| Hypomethylating agents | 16 (37%) | 7 (44%) | 9 (35%) | ||
| No AML directed chemotherapy | 7 (16%) | 0 (0%) | 6 (23%) | 1 (100%) | |
| Bone marrow transplant no. (%) | 6 (14%) | 4 (25%) | 2 (8%) | .29 |
Abbreviations: ANC, absolute neutrophil count; Dx, diagnosis; ECOG, Eastern Cooperative Oncology Group; ELN, European Leukemia Network; Hgb, Hemoglobin; LDH, Lactate dehydrogenase; MRC, myelodysplasia-related changes; Plt, Platelet count; WBC, white blood cell count;
3.2 |. Mutations and variant allele fraction
Figure 1 illustrates the identified co-mutations and clinical response to initial treatment among ASXL1mutSRSF2mut AML patients. The most common additional co-mutations seen in this cohort were TET2 (49%), RUNX1 (35%), STAG2 (25%), IDH1 (16%), NRAS (14%) and SETBP1 (14%). Although the small sample size precluded statistical analyses, IDH1, RUNX1, JAK2, NPM1 and EZH2 mutations appeared to be more common in de novo AML whereas ETV6, CEBPA and STAG2 mutations appeared to be more common in s-AML (Table S2).
FIGURE 1.
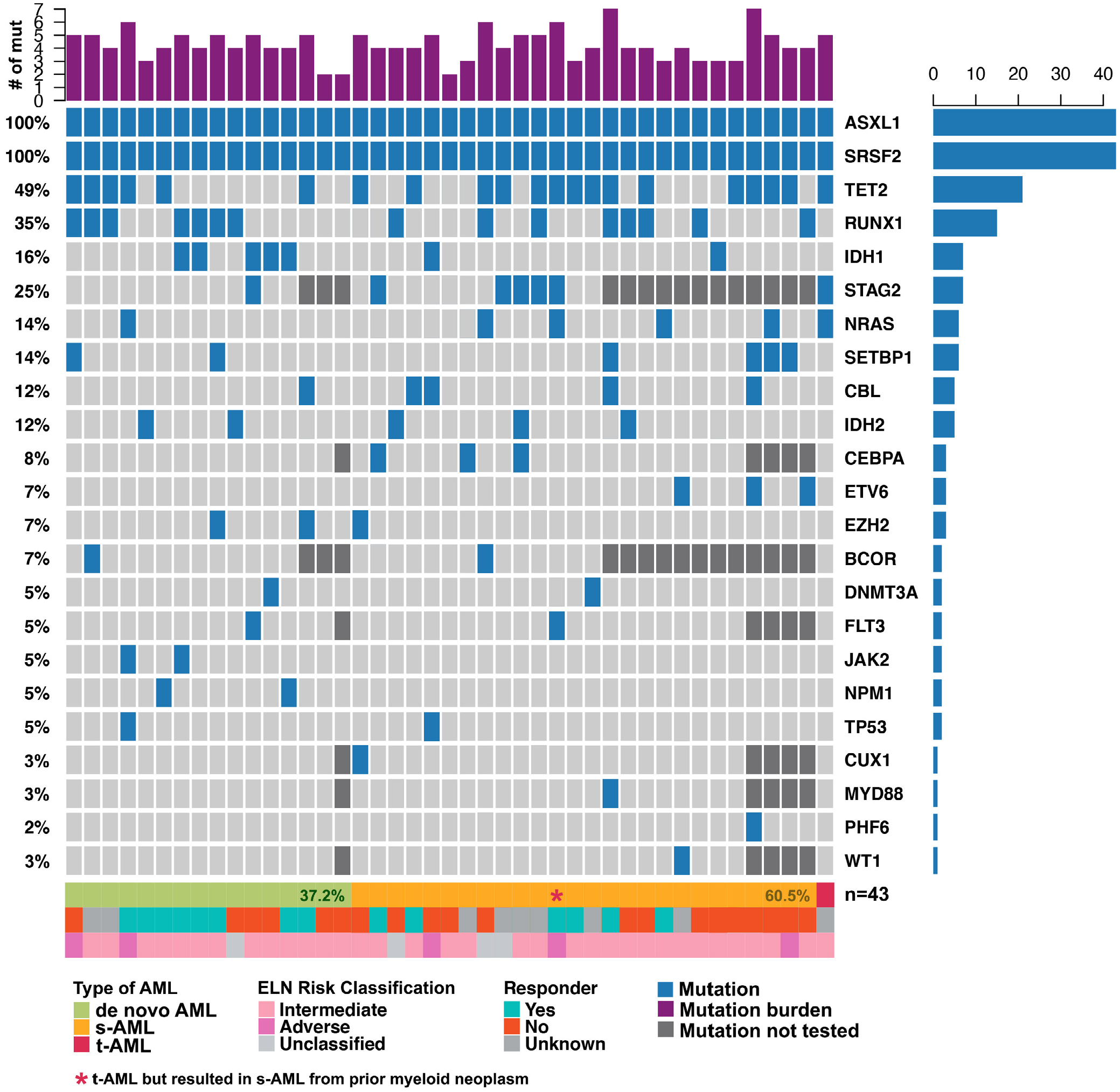
Co-mutations, ontogeny and responses of patients with co-mutated ASXL1/SRSF2. Patients are sorted left-to-right by type of AML (de novo, secondary-AML [s-AML], or treatment-related AML [t-AML]). Number of mutations includes all unique mutations identified including ASXL1 and SRSF2
Among co-mutated patients, all (43/43) ASXL1 variants resulted in protein truncation. In 29 cases, the pathogenic ASXL1 mutation was an insertion/deletion resulting in reading frameshift, and in 14 cases, the pathogenic ASXL1 mutation was a single nucleotide nonsense variant that introduced an early termination codon. Consistent with prior publications and public databases, ASXL1 c.1934dupG (p. Gly646TrpfsTer12) was the most common ASXL1 mutation, occurring in 21 of 43 cases (49%). All 43 SRSF2 mutations impacted codon 95, the canonical hotspot for mutations in this gene. Missense mutations impacting codon 95 were present in 40 cases (91%), with the four remaining variants (9%) representing in-frame deletions at this site. A list of all reported variants is included in Table S3.
The SRSF2 mutations were consistently found at a high variant allele fraction (VAF), typically close to 50% (range 12% - 61%, median 45%), suggesting a heterozygous driver mutation present in most cases. In contrast, the VAF of ASXL1 mutations was more variable (range 6% – 49%, median 34%). To infer mutation ontogeny, SRSF2 and ASXL1 VAFs were compared by calculating a ratio of SRSF2 VAF: ASXL1 VAF. In 36/43 (84%) patients, the ratio was ≥1.0, suggesting that SRSF2 is the earlier mutational event in the majority of cases (Figure 2). Patients with s-AML had a non-significant increase in SRSF2:ASXL1 VAF compared with de novo AML (P = .10).
FIGURE 2.
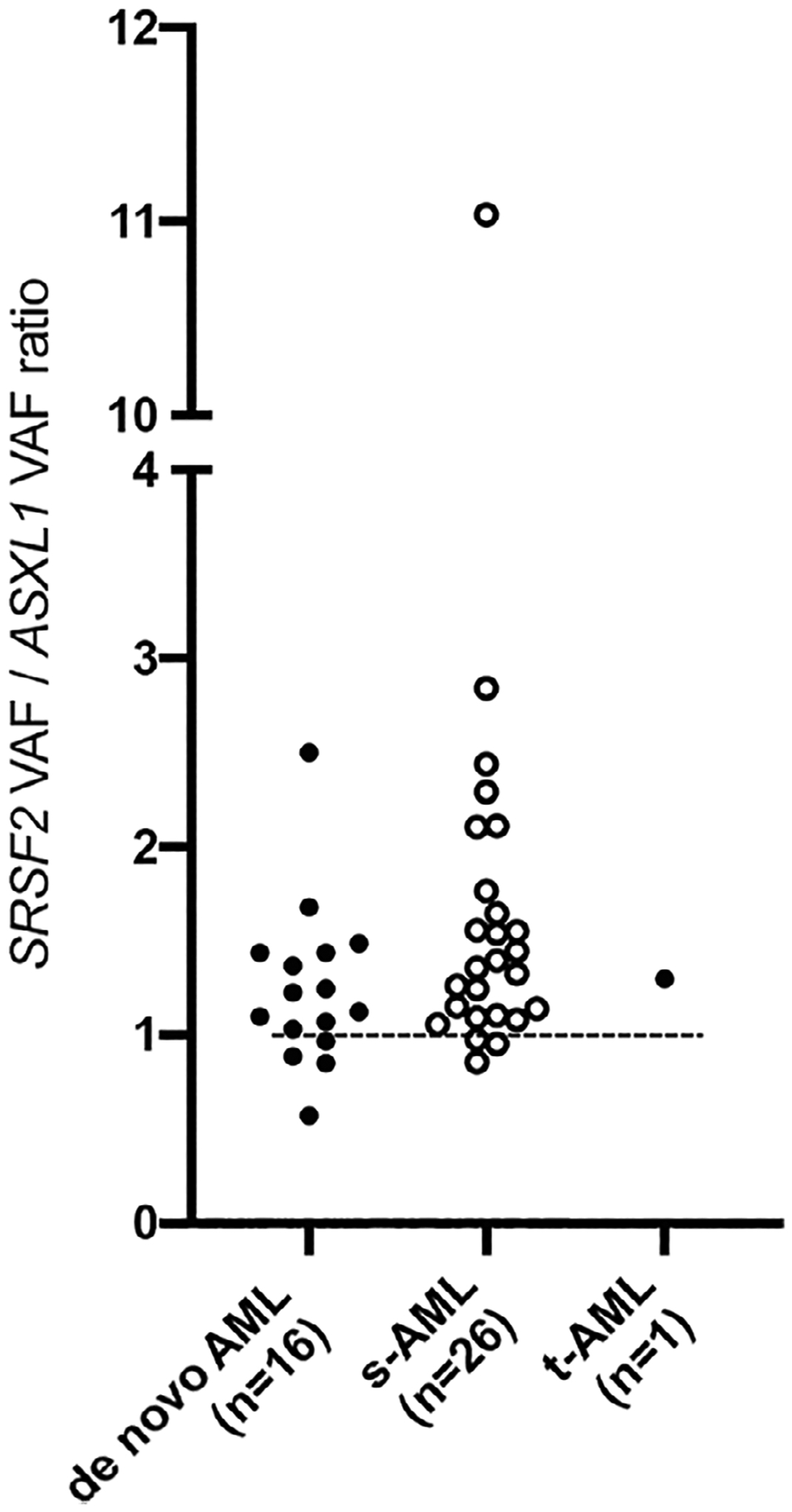
Ratio of variant allele fraction (VAF) of SRSF2 to ASXL1, stratified by de novo, secondary-AML (s-AML) and treatment-related AML (t-AML). Dashed line is one. Values above one indicate that the VAF of SRSF2 is higher than the VAF of ASXL1
3.3 |. Overall survival
The median follow-up for ASXL1mutSRSF2mut AML survivors was 11.2 months. The median OS was 7.0 months (95% CI: 3.8, 15.3; Figure 3(A)). Median OS for patients with s-AML (n = 26) and de novo AML (n = 16) was 6.4 months (95% CI: 2.5, 11.2) and 15.3 months (95% CI: 3.6, 24.2, P = .09), respectively (Figure 3(B)). The patient with t-AML survived 7 days. Median OS did not differ by age (P = .16) or total number of mutations (continuous, P = .40). Note, SWOG cytogenetic risk, splenomegaly, lLDH, hemoglobin, white blood cell count, blast count at diagnosis, type of secondary AML (MDS vs MPN vs CMML) and VAFs of ASXL1 or SRSF2 mutations were also not significantly associated with OS. Although site was significantly associated with OS on a univariable model (P = .04), adjusting for patients who did not receive treatment eliminated the significance of this association demonstrating that this association was due to variation in patient acuity. After adjusting for site on multivariable analysis, s-AML was significantly associated with having worse OS (P = .04) as compared to de novo AML. Adding additional covariates to the multivariable model did not significantly change this association, nor improve model fit.
FIGURE 3.
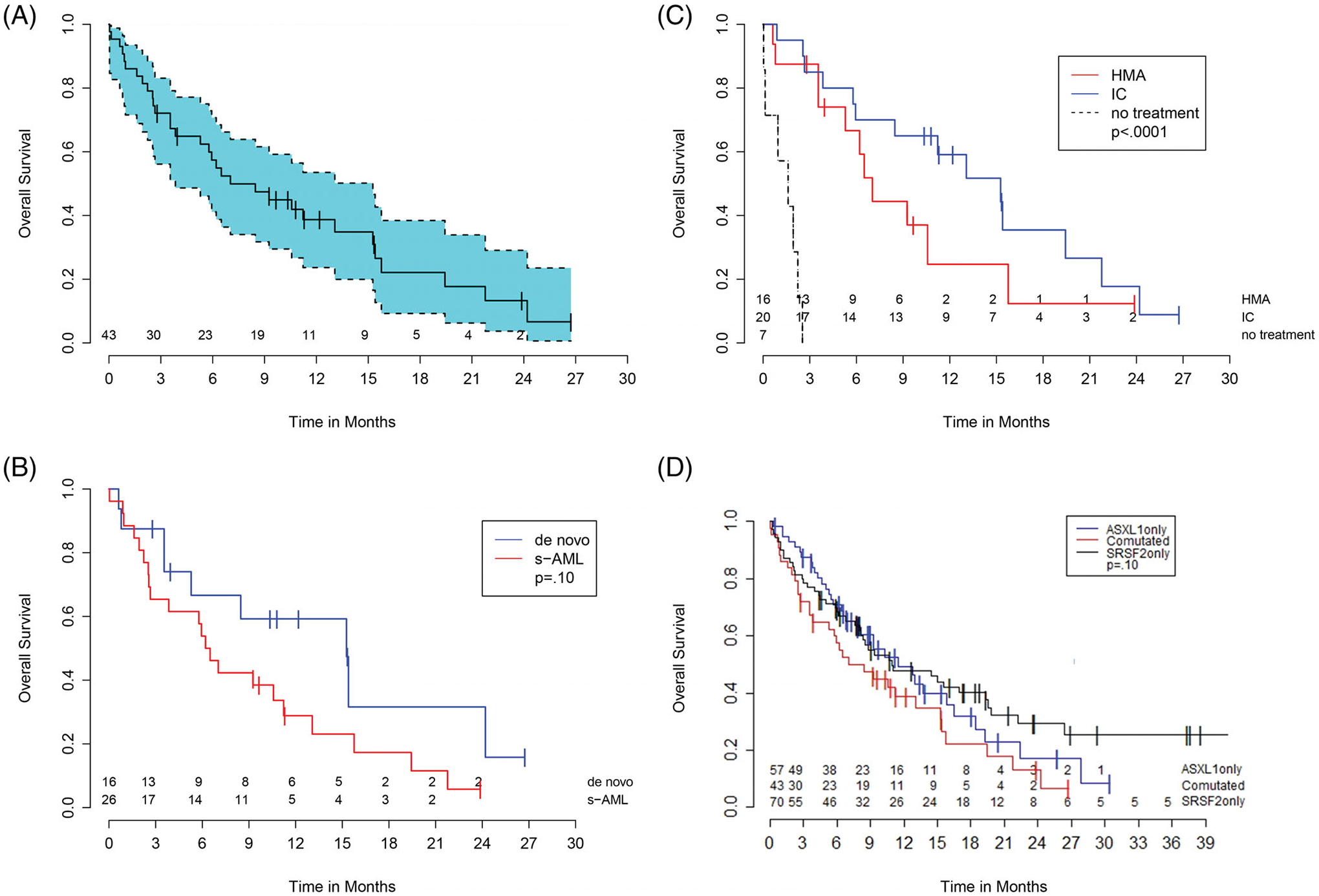
Kaplan–Meier curves of overall survival of ASXL1mutSRSF2mut AML patients. (A), Entire cohort of ASXL1mutSRSF2mut AML patients. Blue shading represents the confidence interval. (B), ASXL1mutSRSF2mut AML cohort stratified by secondary-AML (s-AML) vs de novo AML. (D), ASXL1mutSRSF2mut AML cohort stratified by treatment intensity (intensive induction chemotherapy [IC] vs hypomethylating agent [HMA] vs no-treatment). (D), ASXL1mutSRSF2mut AML (comutated) vs ASXL1mutSRSF2wt AML (ASXL1 only) vs ASXL1wtSRSF2mut AML (SRSF2 only)
Thirty-six patients received either IC (n = 20) or an HMA (n = 16) (Table S4). OS varied significantly by receipt of chemotherapy (no treatment vs IC vs HMAs, P < .001, Figure 3(C), Table S4) mostly due to the very poor outcomes of the seven patients who received no chemotherapy. Median OS for these patients was only 1.6 months compared to 10.6 months for those who received chemotherapy (P < .001). As compared to those who received HMAs (n = 16), patients who received IC (n = 20) appeared to have a longer median OS although this was not statistically significant (15.3 months vs 6.6 months, P = .20). Patients achieving a CR or CRi (n = 11) on first induction had a median OS of 15.3 months (95% CI: 5.3, 24.2). Six patients (four de novo and two s-AML) underwent allogeneic stem cell transplant (allo-SCT) with a median OS not reached (median follow-up 15.6 months). Five of the six (83%) received IC as initial treatment; four (67%) were alive at last follow-up including the two patients with s-AML.
Although not statistically significant, the rates of CR/CRi were higher following IC (8/20: 40%) compared with HMAs (3/16: 19%, P = .17). Of the 16 patients with de novo AML, seven achieved CR/CRi (44%): five of nine patients receiving IC (56%) and two of seven patients receiving HMAs (28%). Of the 26 patients with s-AML, four achieved CR/CRi (15%): three of 11 patients receiving IC (27%) and one of 10 patients receiving HMAs (10%).
Notably, two co-mutated patients had NPM1 mutations, both of whom had a normal karyotype and were classified as de novo AML. Both received IC, achieved a CR/CRi, and did not undergo allo-SCT. They are both alive without relapse at over 2 years follow-up.
3.4 |. OS comparison among co-mutated and ASXL1mutSRSF2wt and ASXL1wtSRSF2mut patients
Fifty-seven patients were identified with ASXL1mutSRSF2wt AML and 70 patients were identified with ASXL1wtSRSF2mut AML. The median age of patients did not differ between co-mutated patients (70.3 years) and ASXL1mutSRSF2wt (69.5 years) or ASXL1wtSRSF2mut patients (70.2 years, P = .95). Race, gender, and performance status also did not differ among the groups. Co-mutated patients were more likely to be classified as s-AML (60% vs 32% vs 23%, P = .001). Patients in the ASXL1mutSRSF2wt and ASXL1wtSRSF2mut subgroups had roughly equivalent probability of death. However, co-mutated patients had a 1.4 times increase in the probability of death compared to ASXL1mutSRSF2wt patients, and 1.6 times the probability of death when compared to ASXL1wtSRSF2mut patients (P = .049). Co-mutated patients appeared to have worse median OS compared to ASXL1mutSRSF2wt and ASXL1wtSRSF2mut patients (7.0 months, [95% CI 3.8, 15.3] vs 11.5 months [95% CI 7.8, 16.4] vs 10.9 months [95% CI 8.1, 19.4], respectively, P = .10; Figure 3(D)), though this did not reach statistical significance. The multivariable model for OS using s-AML status alone was significantly better than the model using mutation status alone (P = .001) suggesting that the difference in survival outcomes is due to differences in the proportion of s-AML patients. The OS by mutational status and etiology is shown in Table S5.
4 |. DISCUSSION
Over the last decade, advances in the understanding of the genetic determinants of AML and the widespread adoption of NGS in routine care has allowed for refinement of prognostically distinct genetic subgroups. Here, we report the largest cohort to date of patients with ASXL1mutSRSF2mut AML and compare survival outcomes to patients with either ASXL1 or SRSF2 mutations. The overall prevalence of ASXL1mutSRSF2mut AML was 2.7% which is consistent with data available from The Cancer Genome Atlas (13/672, 1.9%).21
The cohort of patients with ASXL1mutSRSF2mut AML was enriched for s-AML (60%) and harbored a relatively high number of overall mutations (median = 4). Although comparisons of number of mutations across datasets are prone to bias due to differential effects of sensitivity and breadth of sequencing, the high number of mutations illustrates the complexity of the mutational ontogeny in these patients. Other concomitant mutations frequently seen in this cohort of patients include those associated with preexisting myeloid neoplasms (such as RUNX1, STAG2, NRAS, SETBP1, and CBL) supporting the work of others suggesting that this genomic profile may be a footprint for preexisting MDS/MPNs.18 In most patients (84%), SRSF2 VAF was higher than ASXL1 VAF, suggesting that these patients had a heterozygous SRSF2 driver mutation and that SRSF2 mutations preceded ASXL1 mutational events. The ratio of SRSF2 to ASXL1 appeared higher among s-AML patients (P = .10) which might suggest differences in leukemogenesis between groups though larger studies are needed to validate this result.
Both ASXL1 and SRSF2 mutations are common in MDS and CMML and are frequently found in patients with s-AML.9,13,15 Our group has previously shown that ASXL1mutSRSF2mut AML has mutational and immunophenotypic features overlapping with CMML.17 Thus, we hypothesized those patients who are classified as having de novo ASXL1mutSRSF2mut AML may have actually harbored a preexisting undiagnosed myeloid neoplasm, and are biologically and clinically similar to patients with documented s-AML. Our findings confirm that ASXL1mutSRSF2mut AML patients have a poor prognosis (median OS: 7.0 months), consistent with recent studies.4, 22 This poor prognosis was shared among patients regardless of age, number of mutations, or cytogenetic risk category, which are typically strong predictors of OS. However, OS was worse in patients with s-AML when compared with de novo AML (6.1 months vs 15.4 months), after adjusting using multivariable analysis. While this does not disprove our hypothesis as larger samples are needed to confidently control for multiple covariates, it does highlight the continued clinical importance of a preexisting myeloid neoplasm. Additional studies, such as single cell sequencing, are needed to definitively determine whether clinical de novo ASXL1mutSRSF2mut AML biologically differs from ASXL1mutSRSF2mut s-AML and to describe the outcome of these clones after different treatment regimens.
Given our initial hypothesis, we speculated that characteristics and outcomes of co-mutated patients would be different than ASXL1mutSRSF2wt patients. This is clinically relevant as there is an established association between ASXL1 mutations and adverse-risk disease.(4, 7–10) Analysis of VAF ratios in our data suggest that, among co-mutated patients, SRSF2 mutational events preceded ASXL1 mutations. Together with our previous findings that co-mutated patients harbor a unique immunophenotype, and mutational profile similar to patients with CMML, and differing significantly from ASXL1mutSRSF2wt AML,17 these data suggest that ASXL1mutSRSF2mut AML is a unique subgroup of AML with respect to etiology and leukemogenesis as compared to ASXL1mutSRSF2wt AML. Survival outcomes also appeared to differ between groups with worse outcomes in co-mutated patients (Figure 3(D)). Although we demonstrate that survival differences are attributable to the larger proportion of s-AML patients in the co-mutated group, the striking discrepancy in proportion of s-AML patients (60% vs 32%) serves only to further illustrate the difference between ASXL1mutSRSF2mut AML and ASXL1mutSRSF2wt AML.
There have been substantial therapeutic advances in the treatment of AML since the inception of this data. Liposomal cytarabine and daunorubicin (CPX-351) was FDA-approved for AML with myelodysplasia-related changes (MRC) in 2017, and venetoclax was FDA-approved in addition to HMAs or low dose cytarabine in 2018 for patients who are not candidates for IC.23–26 Because ASXL1mutSRSF2mut AML is more prevalent in elderly patients and those with MRC, further studies are needed to investigate the use of these novel regimens in this population. Only two patients in this cohort received CPX-351 and no one received venetoclax. Monocytic phenotypes have been shown to up-regulate MCL-1 and thus confer resistance to venetoclax-based regimens.27 Further investigation is warranted to determine whether ASXL1mutSRSF2mut AML may have a less favorable response to venetoclax-based regimens due to its association with monocytic phenotypes.17 Recent studies suggest that ASXL1 mutations may predict for response to azacitidine and anti-PD-1 combinations.28 However, there is a lack of data analyzing outcomes of patients with concomitant mutations in ASXL1 and SRSF2 receiving immune-based therapies. Given the relatively high mutational burden and poor outcomes with conventional chemotherapy regimens in this cohort, immune-based investigational strategies should be further explored in future trials.
The NPM1 mutations are associated with de novo AML and typically confer a favorable response to therapy and improved clinical outcomes.29–32 Some suggest that NPM1 and ASXL1 mutations may actually represent different routes of leukemogenesis given substantial differences in outcomes and other factors between groups.33 In this cohort, two patients with NPM1 mutations have survived over 2 years without allo-SCT or relapse suggesting that NPM1 mutations may continue to confer favorable outcomes even with these concomitant pathogenic mutations. This is consistent with current ELN guidelines that recommend against classifying patients with concomitant ASXL1 and NPM1 as adverse-risk based on the presence of ASXL1 alone.3
This study has several important limitations. First, while this is the largest cohort to date of ASXL1mutSRSF2mut AML, the small effective sample size limited our ability to detect statistically significant associations to only large effect sizes. This is particularly relevant in our comparison of survival outcomes between de novo vs. s-AML and IC vs. HMA therapy. Comparing outcomes by treatment regimen in a retrospective review is complicated by both known and unknown confounders. Although we attempted to account for known confounders through multivariable analysis, the small sample size limited exploratory models to only those with two or three covariates. Additionally, our data are taken from two large academic referral centers and are therefore susceptible to selection bias, which limits generalizability. We believe that the effect of this selection bias is minimal because most AML patients who are candidates for chemotherapy are treated at similar centers.
Nonetheless, these findings have several practical implications. First, although this study contributes to the body of evidence on the importance of ASXL1/SRSF2 mutational status on prognosis, our findings highlight the critical importance of clinically-defining s-AML, especially for patients who are candidates for intensive therapies and allo-SCT. Though there are currently no FDA-approved agents that specifically target ASXL1 or SRSF2, fit patients may be considered for early allo-SCT and/or clinical trials given poor outcomes with conventional chemotherapy agents. Further, frail patients who are not eligible for intensive induction should be prioritized for novel approaches given dismal outcomes with HMA therapy. These data add to the accumulating evidence for utilizing NGS at diagnosis in AML to inform not only prognosis but also treatment regimen and intensity of therapy. Awaiting full molecular and cytogenetic results prior to initiating induction chemotherapy has been shown to be feasible and is currently being utilized in clinical trials such as BEAT AML to guide therapeutic decisions.34 This strategy may become the standard of care as rapid NGS panels are becoming more available and more molecularly targeted agents are evaluated during induction.35–37 Importantly, the results of this study need to be replicated with other data sets from different institutions and in different practice settings. Needless to say, integration of these data into risk models will be highly valuable. Given the poor prognosis seen among patients in this cohort, investigational trials with novel therapeutic agents are sorely needed.
In conclusion, we have provided further support to existing evidence for the classification of a unique subset of AML patients with co-mutated ASXL1 and SRSF2. Eligible patients may benefit from IC and early allo-SCT though future studies are warranted to validate these data and identify preferred treatment regimens for this patient population.
Supplementary Material
FUNDING INFORMATION
This work was supported in part through a National Research Service Award Post-Doctoral Traineeship from the Agency for Healthcare Research and Quality (Grant No. 5 T32 HS000032-28) (D.R.R).
CONFLICT OF INTEREST
D.R.R., D.M.S., D.T.M, S.M.J, O.C., J.G., S.E., H.V.D., and N.D.M report no conflicting financial interests. M.A.H has served as a consultant for Adaptive Technologies, Amgen, Decibio, Guidepoint, Stemline, Jannsen. M.C.F has received grant funding from Bellicum Pharmaceulticals, Celgene, and Macrogenomics; and has received consulting fees from Abbvie, Novartis, and Shire. C.C.C has served as a consultant for Abbvie and Covance; has received honoraria from Abbvie, AstraZeneca, LOXO, MEI Pharma, Octapharma; and has received institutional research funding from Gilead, Incyte, H3 Biomedicine, and LOXO. D.A.S. has served on the advisory board for Agios, BMS, Celyad, Intellia, Kite, Syndax; has been a consultant for Incyte; has received research funding from Celgene and Jazz; and has been on the speakers bureau for AbbVie, Agios, Incyte, and Novartis. J.F.Z has received honoraria from AbbVie, Agios, Bristol Myers Squibb/Celgene, Daiichi-Sankyo, Genentech, Pfizer, and Takeda; has received consultancy fees from AsystBio Laboratories, Celgene and Takeda; has received research funding from AROG, Celgene, Forty Seven, Merck, Sumitomo Dainippon Pharma, and Takeda.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1136–1152. [DOI] [PubMed] [Google Scholar]
- 2.Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a southwest oncology group/eastern cooperative oncology group study. Blood. 2000;96(13):4075–4083. [PubMed] [Google Scholar]
- 3.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374 (23):2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley TJ, Miller C, Ding L, et al. Genomic and Epigenomic landscapes of adult De novo acute myeloid leukemia. N Engl J Med. 2013;368(22): 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 7.Pratcorona M, Abbas S, Sanders MA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012;97(3):388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnittger S, Eder C, Jeromin S, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27(1): 82–91. [DOI] [PubMed] [Google Scholar]
- 9.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paschka P, Schlenk RF, Gaidzik VI, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian acute myeloid leukemia study group. Haematologica. 2015; 100(3):324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arbab Jafari P, Ayatollahi H, Sadeghi R, Sheikhi M, Asghari A. Prognostic significance of SRSF2 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: a meta-analysis. Hematology. 2018;23(10):778–784. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Yao D-M, Ma J-C, et al. The prognostic implication of SRSF2 mutations in Chinese patients with acute myeloid leukemia. Tumour Biol. 2016;37(8):10107–10 114. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S-J, Rampal R, Manshouri T, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119(19):4480–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bamopoulos SA, Batcha AMN, Jurinovic V, et al. Clinical presentation and differential splicing of SRSF2, U2AF1 and SF3B1 mutations in patients with acute myeloid leukemia. Leukemia. 2020;34(10):2621–2634. [DOI] [PubMed] [Google Scholar]
- 15.Yoshizato T, Nannya Y, Atsuta Y, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129(17):2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elena C, Gallì A, Such E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128(10):1408–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson SM, Richardson DR, Galeotti J, et al. Acute myeloid leukemia with co-mutated ASXL1 and SRSF2 exhibits monocytic differentiation and has a mutational profile overlapping with chronic myelomonocytic leukemia. Hemasphere 2019;3(4):e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9): 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone RM, Mazzola E, Neuberg D, et al. Phase III open-label randomized study of cytarabine in combination with amonafide L-malate or daunorubicin as induction therapy for patients with secondary acute myeloid leukemia. J Clin Oncol. 2015;33(11):1252–1257. [DOI] [PubMed] [Google Scholar]
- 20.Yannakou CK, Jones K, McBean M, et al. ASXL1 c.1934dup;p. Gly646Trpfs*12—a true somatic alteration requiring a new approach. Blood Cancer Journal. 2017;7(12):656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Cancer Genome Atlas [Internet]. [cited 2020 Apr 27]. Available from: https://www.cancer.gov/tcga
- 22.Fleming S, Tsai C-H, Döhner H, et al. Use of machine learning in 2074 cases of acute myeloid leukemia for genetic risk profiling. Blood. 2019;134(suppl. 1):1392–1392. [Google Scholar]
- 23.Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional Cytarabine plus Daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36(26):2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alfayez M, Kantarjian H, Kadia T, Ravandi-Kashani F, Daver N. CPX-351 (vyxeos) in AML. Leuk Lymphoma. 2020;61(2):288–297. [DOI] [PubMed] [Google Scholar]
- 25.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei AH, Strickland SA, Hou J-Z, et al. Venetoclax combined with low-dose Cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019; 37(15):1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pei S, Pollyea DA, Gustafson A, et al. Monocytic subclones confer resistance to Venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discovery. 2020;10(4):536–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daver N, Garcia-Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to Azacitidine and Nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discovery. 2019;9(3):370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnittger S, Schoch C, Kern W, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106(12):3733–3739. [DOI] [PubMed] [Google Scholar]
- 30.Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107(10):4011–4020. [DOI] [PubMed] [Google Scholar]
- 31.Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106(12):3740–3746. [DOI] [PubMed] [Google Scholar]
- 32.Patel SS, Ho C, Ptashkin RN, et al. Clinicopathologic and genetic characterization of nonacute NPM1-mutated myeloid neoplasms. Blood Adv. 2019;3(9):1540–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbuccia N, Trouplin V, Gelsi-Boyer V, et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia. 2010;24(2):469–473. [DOI] [PubMed] [Google Scholar]
- 34.Burd A, Levine RL, Ruppert AS, et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML master trial. Nat Med. 2020;26:1852–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roboz GJ, DiNardo CD, Stein EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020;135(7):463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pollyea DA, Tallman MS, de Botton S, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33 (11):2575–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winer ES, Stone RM. Novel therapy in acute myeloid leukemia (AML): moving toward targeted approaches. Ther Adv Hematol. 2019;10: 2040620719860645. 10.1177/2040620719860645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.