

Abstract
Background
Genetic mutations of IKZF1 have been frequently delineated in B‐lineage acute leukaemia (B‐ALL) but rarely elucidated in acute myeloid leukaemia (AML). IKZF1 mutations confer a poor prognosis in AML, and hotspot mutations of IKZF1, N159Y and N159S tend to occur in B‐ALL and AML respectively. However, the pathogenesis of IKZF1 N159S in AML and IKZF1 lineage susceptibility are largely unknown.
Methods
The genetic and clinical characteristics of IKZF1‐mutated AML patients were evaluated. Multi‐omics analysis and functional assays were performed in vitro using IKZF1 mutations knock‐in AML cell lines.
Results
23 (4.84%) small sequence variants of IKZF1 were identified in 475 newly diagnosed AML (non‐M3) patients. Based on RNA sequencing, three classes of IKZF1‐related AML were defined, including 9 patients (39.13%) with IKZF1 N159S mutations, 10 (43.47%) with CEBPA mutations and 4 others (17.39%). IKZF1 N159S may define a unique subgroup with higher HOXA/B expression and native B‐cell immune fractions. Gene expression data of multiple knock‐in cell lines indicate that the lymphocyte differentiation‐related MME and CD44 kept high expression in IKZF1 N159Y but were downregulated in N159S. CUT&TAG sequencing showed that IKZF1 N159S reshaped the binding profiles of IKZF1. Integration analysis suggested that the pathogenesis of IKZF1 N159S may depend on the deregulation of several cofactors, such as oncogenic MYC and CPNE7 targets.
Conclusions
Collectively, we dissected the molecular spectrum and clinical features of IKZF1‐related AML, which may promote an in‐depth understanding of the pathogenesis, lineage susceptibility and clinical research of AML.
Keywords: acute myeloid leukaemia, DNA binding, gene expression profiles, IKZF1, sequence variants
Genetic mutations of IKZF1 bared unfavourable prognostic significance in the leukaemogenesis of AML, especially for the hotspot IKZF1 (p.N159S) point mutation.
IKZF1 (p.N159S) point mutation occurs preferentially in AML with poor prognosis and defines a rare molecular subtype with unique gene expression.
Recurrent IKZF1 (p.N159S) mutation exhibits a more aggressive disease progression potentially by deregulation of NOTCH signalling, VEGF, CPNE7 and MYC‐related factors.
1. INTRODUCTION
Acute myeloid leukaemia (AML) is a group of the most aggressive haematopoietic malignancies in adults associated with development blockage and over‐proliferation of myeloid lineage cells. 1 , 2 Recent advances in high‐throughput sequencing and large‐scale omics studies have significantly promoted the precise molecular classification in AML, as well as in other acute leukaemia (AL). It is widely appreciated that the chromosomal abnormalities (gene fusions) and sequence variants (multi‐amino acid sites) define the major subtypes of AML, 3 , 4 , 5 , 6 B‐progenitor acute lymphoblastic leukaemia (B‐ALL) 7 , 8 , 9 and T‐cell acute lymphoblastic leukaemia (T‐ALL). 10 , 11 , 12 , 13 Meanwhile, several entities with similar gene expression profiles (GEP) have been established in AL, including homologous markers, that is, fusion genes and hotspot mutations IKZF1 p.Asn159Tyr (N159Y), 8 , 9 heterogeneous genetic markers, that is, BCR::ABL1 and BCR::ABL1‐like pair. Recently, based on RNA‐sequencing (RNA‐Seq) data of 655 AML patients, the largest multi‐centre cohort in China, we have illuminated eight meta‐subgroups of AML, which provides a comprehensive framework to pinpoint rare molecular subtypes in AML, among which, recurrent IKZF1 mutations was discovered in this cohort. 14
IKZF1 encodes IKAROS, which is a critical transcription factor for lymphopoiesis and immune haematopoiesis, 15 and potentially works as a tumour suppressor by negatively regulating cell proliferation. 16 IKAROS contains the N‐terminal DNA‐binding domain (DBD) with four zinc fingers (ZFs) and two additional zinc fingers situated at the C‐terminal for homo‐ and heterodimerisation. 17 , 18 IKAROS mainly functions as a homodimer through ZFs, but it can also dimerise with other members of the IKAROS family or various transcriptional factors to exert multiple functions, 15 , 16 such as nucleosome remodelling and deacetylase (NuRD) and complex polycomb repressive complex 2 (PRC2). 19 Genetic deletions and mutations of IKZF1 are commonly involved in the pathogenesis of ALL, especially B‐ALL. Deletion of exons 4−7 of IKZF1 leads to the generation of Ik6 heterodimer, which interferes with normal B‐lymphocyte signalling and promotes B‐ALL development. 20 Somatic mutations of IKZF1 have even been more widely reported in the unfavourable molecular subtypes of B‐ALL, such as in nearly 40% of BCR::ABL1/‐like cases. 21 , 22 , 23 In an international collaborative study of B‐ALL, we reported that IKZF1 N159Y is a newly identified rare subtype of B‐ALL with unique gene expression profile, characterised by significant upregulation of the transcriptional coactivator YAP1, SALL1 and ARHGEF28B, and downregulation of the B‐cell receptor signalling and JAK‐STAT signalling pathways. 24 In addition, germline‐derived dominant negatives and haploinsufficient of IKZF1 mutations are predisposed to T, B, and myeloid cell combined immunodeficiency and AL. 15 , 25
Although a large number of studies have elucidated the important pathogenic role of IKZF1 in ALL, IKZF1 mutations have rarely been reported in AML. A recent study reported that the frequency of IKZF1 mutations in AML was 2.6% (5/193), which were significantly associated with SF3B1 and biCEBPA mutations. 26 Besides, IKZF1 mutations were retrospectively identified in OHSU (8/593, 1.35%), TCGA (1/200, 0.5%) and TARGET (4/95, 4.21%) cohorts. Among these 13 IKZF1 mutations, 5 were N195S mutation (5/13, 38.5%). 26 However, due to the small number of such cases and their sporadic occurrence, no targeted studies have been reported. In our previous work, we identified numerous small sequence variants of IKZF1 with unique gene expression profiles in AML. Among them, IKZF1 N159S was a recurrent hotspot mutation, which closely clustered in a subset of patients with myelodysplasia‐related mutations and upregulated gene expression of the HOXA/B family genes. 14 Notably, immunomodulatory drugs inducing IKAROS protein degradation showed potential therapeutic efficiency in the HOXA‐related AML, which suggests the emerging synergistic role of IKAROS in high‐risk AML. 27
To date, a few critical issues remain to be addressed to further explore the pathogenesis and improve the prognosis of AML patients with IKZF1 mutations. Firstly, genetic alterations of IKZF1 are common and widely reported in B‐ALL, however, the genomic landscape, molecular classification and clinical significance are scarcely investigated in a large AML cohort. Meanwhile, certain IKZF1 mutations tend to occur in B‐ALL (p.N159Y) or AML (p.N159S) have been reported but the potential mechanism of the IKZF1‐related lineage susceptibility is rarely studied. 14 Hence, in the present study, we outlined the mutational spectrum of IKZF1 in 383 AMLs that we previously published and 92 newly diagnosed primary AMLs excluding patients with PML::RARA (M3). In addition, via comparison of gene expression profiles as well as in vitro investigation, we proposed to understand the underlying leukaemogenesis of IKZF1 in AML. We hope our work will serve as a novel reference for future research on leukaemia‐lineage susceptibility, and as a starting point for a new, genetically based disease classification that will contribute to the improved risk stratification and ultimately better treatment outcomes.
2. METHODS
2.1. Ethics approval and consent to participate
Primary blasts were obtained according to the Declaration of Helsinki at disease onset from bone marrow of 475 AML patients with least 20% abnormal bone marrow blasts. RNA sequencing and targeted screening of 100 common leukaemia‐related genes was performed in all primary AML patients from Ruijin Hospital as previously described. 28 Clinical information of 23 AML with IKZF1 mutations is provided (Table S1). Informed consent was obtained according to procedures approved by the Institutional Review Board from Ruijin Hospital, affiliated to Shanghai Jiao Tong University School of Medicine. K562, U937, HEK‐293T and NCI‐H1299 cells were authenticated. Details are available in Supplemental Methods.
2.2. Other material and methods
FLAG‐tagged‐IKZF1 wild‐type or mutants were ectopically expressed in leukaemia cell line K562 and U937 using lentiviral‐mediate vector. Primers used for in vitro experimental assays are listed in Supplementary Methods. Bioinformatic/statistical analysis and other detailed information are described in Supplementary Methods.
2.3. Statistical analysis
Wilcoxon rank‐sum test was used for calculating the statistical significance when comparing of two groups while Kruskal‐Wallis test was used for more than two groups, paired‐samples two‐tailed Student's t‐test was used for estimating the statistical significance of RT‐qPCR. Data were presented as mean ± SD. All statistical analyses were performed using the R/Bioconductor statistical environment (https://www.r‐project.org/) and significance was defined as p < .05.
2.4. Role of the funding source
The funders have no role in the study design, data collection, data analysis, data interpretation or report writing.
Additional methods are outlined in Supplemental Methods.
3. RESULTS
3.1. Genomic classification of IKZF1‐positive AML
The similarity of the patient samples based on gene expression data was visualised in a t‐Distributed Stochastic Neighbour Embedding (t‐SNE) analysis, which showed an overall separation of AML samples according to different genetic abnormalities defined by the 5th edition of the World Health Organization (WHO) Classification of Hematolymphoid Tumors 29 (Figure 1A). We totally identified 23 small sequence variants of IKZF1, which comprised 4.84% (23/475) of newly diagnosed non‐M3 AML patients (Figure 1C and D; Table S1). Notably, hotspot mutations, IKZF1 N159S occurred in 1.89% (9/475) of AML, accounting for 39.13% (9/23) of all IKZF1 mutations in this cohort.
FIGURE 1.
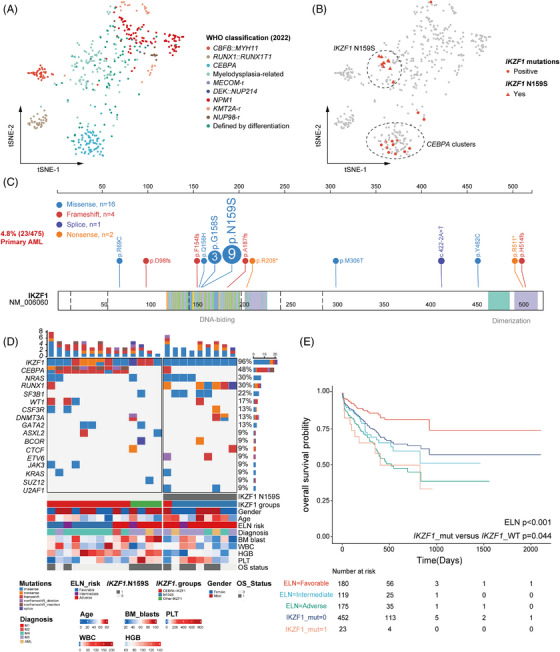
Genomic classification of IKZF1‐positive AML. (A) t‐SNE plots for the signatures of major somatic mutational WHO classes of our AML cohort. Each dot represents a cancer sample. Color of dots indicates cancer types or subtypes. (B) Expression clustering of representative markers for IKZF1 N159S, biCEBPA/‐like, and others AML are plotted onto the t‐SNE map. (C) Structure and mutation spectrum of IKZF1 in AML. All IKZF1 mutations (n = 23) are depicted in protein domains. Different colors represent different types of mutations. (D) Oncoplot and clinical features of patients with IKZF1 mutations. The heatmap shows the genomic landscape and clinical features of patients with IKZF1 mutations. (E) Overall survival of AML patients according to the ELN classes, Kaplan–Meier curves depicting the survival difference between IKZF1‐mutated patients and non‐IKZF1‐mutated patients.
Consistent with our recently published work, 14 through clustering of gene expression profiles, all cases with IKZF1 N159S were clustered into myelodysplasia‐related/‐like (G5) subgroup, while the majority of other IKZF1 mutant loci displayed a similar gene expression profile to biallelic CEBPA or ‐like (biCEBPA/‐like, G4) subgroup, which incorporated not only biCEBPA, but also moCEBPA mutations with loss of heterozygosity and several CEBPA WT cases. 14 Accordingly, three molecular subtypes with IKZF1 aberrations were recognised including IKZF1 N159S, biCEBPA/‐like and others (Figure 1D). Furthermore, patients harbouring IKZF1 mutations had a similar poor overall survival (OS) to those in the ELN adverse risk group (Figures 1E and S1A). Relevant clinical features suggested that haemoglobin (HGB) were significantly higher in biCEBPA/‐like IKZF1, while bone marrow blasts, platelet (PLT) and the variant allele frequency (VAF) of IKZF1 mutations appeared to be higher in IKZF1 N159S‐positive cases (Figure S1B). These factors might impact the development of normal immune cells and even dim the prognosis of IKZF1 N159S‐positive AML patients (Figure S1B and C).
3.2. IKZF1 N159S defines a rare molecular subtype with unique gene expression profiles in AML
Unsupervised clustering using 1267 genes with the highest variance and p value less than .05 confirmed the genomic‐based classification in IKZF1‐positive AML (Figure 2A and B). At the gene expression level, patients with IKZF1 N159S and biCEBPA/‐like IKZF1 were mutually exclusive and pertained to two different hierarchical branches: high and low expression of HOXA/B family genes. In line with our previous work, 14 IKZF1 N159S was associated with myelodysplasia‐related changes, which frequently coexisted with RUNX1 and spliceosome mutations, and was characterised by the upregulation of haematopoietic stem progenitor cell signature (HSPC) and core markers including HOXA/B, MYCT1, PAWR and BEND4. 14 We hereby reproduced the above observations and extended more specific gene expression signatures of IKZF1 N159S hotspot mutations in this work (Figures 2B and S2), that is, cytoskeletal architecture‐related (SHROOM4), p53‐mediated apoptosis (CCDC8) and activator TGF‐beta pathway (RBPMS). We also provided a comparison of IKZF1‐positive gene expression profile with other non‐IKZF1 AML using the same 1267 genes expression, and further demonstrated that IKZF1 N159S may define a separate molecular subtype with unique GEP in all our AML cases, even within the subcluster of myelodysplasia‐related/‐like AML (Figure S3A). Oncogenic MYC contributes to the genesis of many human cancers, which strictly depends on its partner Max to regulate gene transcription. 30 GSEA analysis indicated that the MYCMAX, VEGF, B‐cell receptor, NOTCH, MAPK, WNT and TGF‐beta signalling pathways were significantly upregulated in the IKZF1 N159S‐positive AML, while DNA mismatch repair and nucleotide excision repair pathways were significantly downregulated when compared with biCEBPA/‐like IKZF1 (Figure 2C). The Gene Ontology (GO) enrichment analysis using differentially expressed genes (DEGs) cross validated the deregulation of extracellular matrix organisation and VEGF/VEGFA signalling pathways (Figure 2D). In addition, the Rap1 signalling pathway and RHO GTPase cycle were also enriched in the IKZF1 N159S‐positive AML based on DEGs enrichment analysis (Figure 2D). Meanwhile, the known oncogenic gene PDGFRB was also upregulated in the IKZF1 N159S‐related AML, which was associated with genomic translocations demonstrating gene expression signatures of BCR::ABL1‐like B‐ALL. 31 We noticed that the overall VAF distributions of IKZF1‐N159S tended to be slightly higher than that of RUNX1 mutations although there was no statistical significance, suggesting that IKZF1‐N159S might not be secondary events to RUNX1 mutations (Figure S3B). To evaluate the prognostic value of IKZF1 mutations under other concomitant factors, we constructed a multivariable Cox regression model incorporating age as well as IKZF1, CEBPA and RUNX1 mutations, and identified that mutant IKZF1, rather than RUNX1, could be an independent predictive factor for OS in AML patients (Figure S3C). Notably, the deconvolution of immune cells showed that the native B cells, T cells regulatory (Tregs), neutrophils and eosinophils were significantly higher in IKZF1 N159S‐positive AML, which may be associated with the blocked differentiation of B lymphocytes and specific regulation of microenvironment by IKZF1‐positive tumour cells (Figure S4).
FIGURE 2.
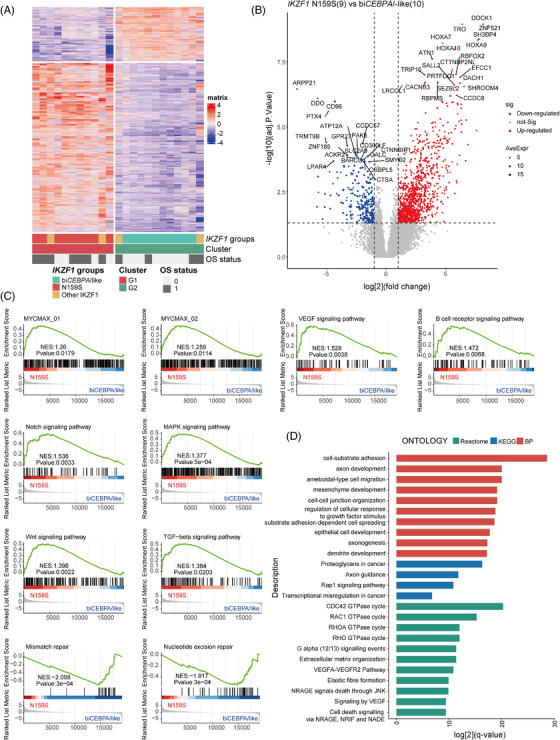
IKZF1 N159S defines a rare molecular subtype with unique gene expression profiles in AML. (A) Unsupervised clustering using the 1267 variance genes with p value less than 0.05 among three classes of classifications in IKZF1‐positive AML. Each column represents a patient and each row represent a gene. Up‐ and downregulated genes are shown in red and blue, genomic‐based classification in IKZF1‐positive AML patients demonstrated that IKZF1 N159S and biCEBPA/‐like were pertained to two different subgroups, which we defined as G1 and G2, respectively. (B) Volcano plot shows the differentially expressed genes (DEGs) between IKZF1 N159S and biCEBPA/‐like cases. The x axis represents log2‐transformed fold‐change values, while y axis shows –log10‐transformed p value. (C) Gene set enrichment analysis of IKZF1 N159S patients (versus biCEBPA/‐like). (D) Significant positive enrichment of terms in IKZF1 N159S‐positive AML.
3.3. Functional effects of IKZF1 variants G158S, N159Y and N159S in vitro
To evaluate the function of highly recurrent IKZF1 variants in a cellular context, we performed in vitro experiments to assess the impact of different variants on IKAROS activities and leukaemia cell phenotypes. Two most frequent point mutations in our cohort including IKZF1 N159S and G158S were chosen for further investigation, and IKZF1 N159Y was also selected as a positive control, which was extensively reported to affect the initiation and development of ALL and disturb the normal function of wild‐type (WT) IKAROS. 32 , 33 Ectopic expression of WT IKZF1 resulted in significant inhibition of cell growth and proliferation coinciding with its tumour‐suppression function, as measured by CCK‐8 assays in leukaemia cells K562 and U937. In contrast, cells with N159S and N159Y expression showed more aggressive cell proliferation; however, G158S expression cells showed a similar growth level to that of the WT IKZF1 cells, suggesting that G158S potentially retains the normal tumour‐suppression function of IKAROS, while N159S/Y could partially impair this function (Figure 3A). Results of GFP relative depletion further supported the CCK‐8 assays (Figure 3B). Flow cytometry of annexin V/PI staining was performed to evaluate the effects of various IKZF1 mutants on apoptosis of K562 and U937 leukaemia cells, which suggested that WT IKAROS and G158S could induce more apoptosis as compared with N159S and N159Y (Figure 3C and D). Cell cycle analysis by flow cytometry showed that in comparison with N159S/Y‐positive cells, more WT and G158S cells arrested in the G1 phase, resulting in fewer cells in the G2/M stage (Figure 3E). These results suggested that IKZF1 N159S might disturb the normal function of IKAROS and relieved the tumour‐suppression effects of wild‐type IKAROS.
FIGURE 3.
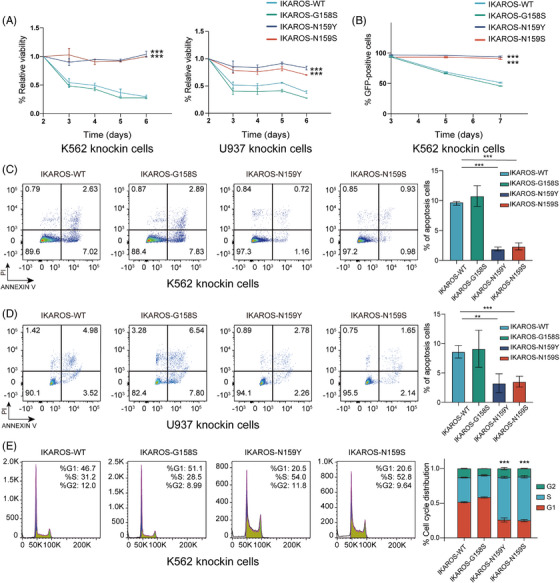
Functional effects of IKZF1 variants. (A) CCK8 assays demonstrating the proliferative capacity of K562 and U937 following transfection with various IKZF1 mutation, normalised to native K562 and U937. (B) Negative‐selection competition assays showing the percentage of various GFP+ IKZF1 mutants‐transduced K562 over time, as normalised to Day 3. (C) Apoptosis analysis in IKZF1 mutants‐expressing K562. IKZF1 WT and IKZF1 G158S induced apoptosis, as measured by annexin V/PI flow cytometric analyses, while IKZF1 N159S/Y defect the ability to induce apoptosis. The histogram depicts the quantification of total proportion of cells in early (annexin V+/PI–) and late apoptotic (annexin V+/PI+) stages. (D) Apoptosis analysis in IKZF1 mutants‐expressing U937 cells. (E) Cell cycle analysis of IKZF1 mutants‐expressing K562 cells. IKZF1 WT and IKZF1 G158S induce cell cycle arrested at G0/G1 phase, while IKZF1 N159S/Y defect the ability to arrest the cell cycle. The histogram depicts DNA content of cell cycle progression in IKZF1 mutants‐expressing K562 cells. All plots are representative of at least three independent experiments performed in duplicate and presented as the means ± standard deviation. (*p < .05, **p < .01 and ***p < .001).
3.4. Comparative analysis of IKZF1 mutant cell lines
According to the published genomic data from thousands of patients with acute leukaemia, we noticed that IKZF1 N159Y and N159S were almost mutually exclusive in B‐ALL and T‐ALL/AML. 24 , 26 To screen the potential lineage specific gene expression patterns following IKZF1 mutations, we then performed bulk RNA‐Seq of IKZF1 G158S, N159S and N159Y mutant human cell lines (Table S2). Unsupervised clustering of mutant and WT IKZF1 cell lines exhibited that different changes at the same amino acid position of IKZF1 or at different positions can produce distinct gene expression profiles (Figures 4A and S5). MME, a well‐known cell surface marker of B‐ALL, showed high expression in IKZF1 N159Y, which was close to WT phenotype, but downregulated in IKZF1 N159S/G158S (Figure 4B). Several specific downregulated genes in IKZF1 N159Y/S may also contribute to the lineage susceptibility, as exemplified by CD44, PAX6, RUNX2, IGSF9B and RUNX1T1 (Figure 4B). GSEA analysis indicated that NOTCH signalling, cell cycle, and MYC targets pathways were commonly upregulated in IKZF1 mutants, suggesting the tumour suppressor function of IKZF1. On the contrary, mitotic recombination and development of natural killer cell/mast cell were consistently downregulated in all three IKZF1 mutants (Figure 4C). Meanwhile, T‐cell immune regulation, antigen processing/presentation and mononuclear cell differentiation were downregulated in both IKZF1 N159S and N159Y. B‐cell differentiation and JAK‐STAT signalling were only downregulated in IKZF1 N159S mutant cell line (Figure 4C). When focusing on the DEGs of IKZF1 N159S (versus WT), the deregulated gene sets can be divided into six subclasses, namely, C1‐C6. These subclusters of genes were enriched in WT, WT/G158S, N159Y/S, N159S and N159Y, respectively. Among them, we identified at least 23 genes, that is, RELN, CXCR1, IGSF9B, ONECUT2, TRIB1 and MAP3K20, that were specifically upregulated in N159S mutant cell line (Figure 4E).
FIGURE 4.
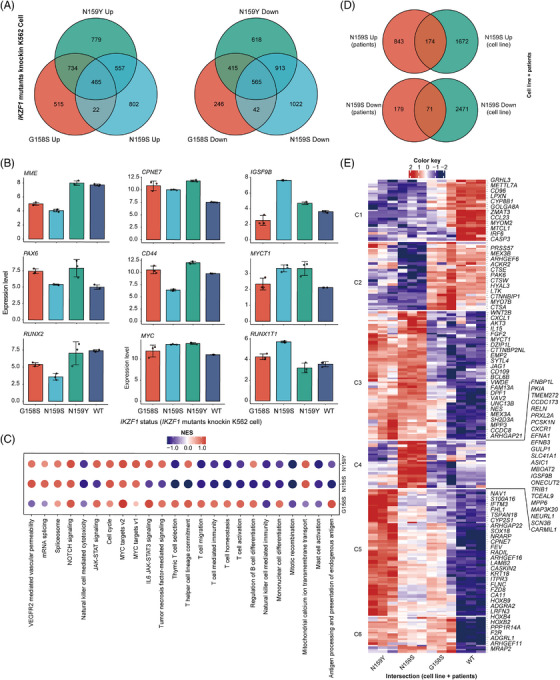
Bulk RNA‐Seq analysis of IKZF1 WT, G158S, N159S and N159Y knock‐in human cell lines define codrivers of IKZF1 N159S‐positive AML. (A) Venn diagram depicting the relationship of differentially up‐ and downregulated expressed genes (DEGs) between IKZF1 G158S, N159S and N159Y mutation knock‐in K562 cell line (versus WT). (B) Bar plots showing the expression level of 9 selected DEGs in the RNA‐Seq data. (C) Dot plot detailing the enriched pathways of IKZF1 G158S, N159S and N159Y mutation knock‐in K562 cell line. (D) Venn diagram depicting the relationship of differentially expressed genes (DEGs) between IKZF1 N159S‐positive AML and IKZF1 N159S knock‐in K562 cell line. (E) Heatmap showing the hierarchical clustering of IKZF1 G158S, N159S and N159Y mutation knock‐in K562 cell line (versus WT) based on DEGs intersection of IKZF1 N159S‐positive AML and IKZF1 N159S knock‐in cell. Gene signatures of IKZF1 N159S can be divided into six subclasses, namely, C1–C6. These subclusters of genes are enriched in WT, WT/G158S, N159Y/S, N159S and N159Y, respectively.
3.5. IKZF1 N159S reshapes its genome binding patterns
To explore the genomic binding profiles of the most significant hotspot mutation IKZF1 N159S in AML and recognise direct gene targets of IKZF1 N159S, CUT&TAG sequencing was performed in the IKZF1 N159S mutant cell line (Table S2). Compared with IKZF1 WT, the genome binding pattern was reshaped by IKZF1 N159S, which was in line with the variations of gene expression profiles (Figure 5A). These differential binding peaks may drive the common GEP of IKZF1 mutants, that is, CPNE7, VWDE; and specific signatures of IKZF1 N159Y/S, that is, HSH2D, LTK; and IKZF1 N159S, that is, IGSF9B, ONECUT2 and CARMIL1 (Figure 5B). When compared with DEGs of IKZF1 N159S‐positive AML, we screened 108 upregulation and 25 downregulation of IKZF1‐related DEGs, which showed consistent trend in RNA‐Seq data of patients and cell lines (Figure 5C). A protein interaction network was constructed to integrate and visualise these screened downstream factors of IKZF1 N159S (Figure S6), functional enrichment analysis indicated that the interacting proteins were significantly enriched in pathways related to cancer development, that is, P53 signalling pathways, and were also enriched in the cell cycle regulation‐related pathways, consistent with our previous function assays (Figure 5D). We finally focused on the genes that are differentially regulated by IKZF1 N159S both in patient cells, cell line RNA‐Seq and CUT&TAG data. CPNE7, a member of calcium‐dependent proteins family that was rarely reported in acute leukaemia, was commonly regulated by IKZF1 mutations and was significantly occupied by IKZF1 N159S rather than WT IKZF1. Hence, CPNE7 might be one of the common underlying factors possibly responsible for the high pathogenicity of IKZF1 mutations. Moreover, another member of the same gene family CPNE8 has been defined as the coexpression factor of HOXA/B, which could predict a significantly poor prognosis in AML. 14 To assess whether CPNE7 could be directly activated by IKZF1 N159S rather than WT IKZF1, we performed the luciferase experiments. The luciferase activity was higher in cells transfected with the IKZF1 N159S mimics and CPNE7 than in those transfected with the WT IKZF1 mimics and CPNE7 (Figure 5E, left panel). We also conducted the luciferase experiments focusing on MYC, the specific gene signature of IKZF1 N159Y/S (Figure 4B), which has been reported to be a target gene of IKZF1 and directly suppressed by WT IKZF1. 25 , 34 , 35 MYC was significantly activated by IKZF1 N159S rather than IKZF1 WT (Figure 5E, right panel), which further confirmed the leukaemogenic effect of IKZF1 N159S mutation.
FIGURE 5.
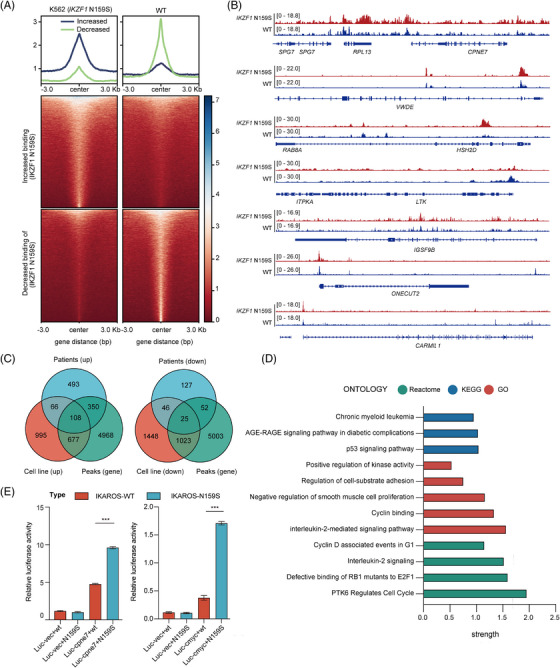
IKZF1 N159S reshapes its genome binding patterns. (A) Distribution of reads in differentially binding peaks in IKZF1 WT and IKZF1 N159S knock‐in K562 cell line. The upper panel shows a line plot depicting the intensity of IKZF1 WT and IKZF1 N159S binding signals centred at the peaks. The lower heatmap shows signals at and ± 3 kb the centre of the peaks. (B) Tracks showing the IKZF1 WT and IKZF1 N159S occupancy at genomic loci promoter and/or enhancer regions of selected target genes based on combination analysis of RNA‐Seq and CUT&TAG sequencing data. (C) Venn diagram depicting the intersection between differentially expressed genes (DEGs) of IKZF1 N159S‐positive patients, IKZF1 N159S knock‐in K562 cell line and CUT&TAG binding peaks. (D) Significant positive enrichment of terms in intersection gene sets based on Protein‐protein interaction (PPI) network analysis. (E) Luciferase activity assay detected the transcriptional regulation region of MYC and CPNE7 bound by IKZF1 WT and IKZF1 N159S. 1299 cells were transfected with the MYC and CPNE7 luciferase reporter plasmid and predicted IKZF1 WT and IKZF1 N159S mimics. The luciferase activity was detected 48 h after transfection in 1299 cells. The plots are representative of at least three independent experiments performed in duplicate and presented as the means ± standard deviation (*p < .05, **p < .01 and ***p < .001).
3.6. IKZF1 N159S strengthen the expression of MYC and CPNE7 to promote carcinogenesis
According to the aforementioned analysis, consistently, a significant upregulation of MYC, CPNE7 and CPNE8 mRNA was observed in IKZF1 N159S‐expressing cells when compared to IKZF1 WT‐expressing cells (Figure 6A). In parallel, IKZF1 N159S was accompanied by increased expression levels of MYC, CPNE7 and CPNE8 proteins in contrast to that in IKZF1 WT (Figure 6B). MYC, a multifunctional transcription factor protein, behaved as a common gene signature upregulated by IKZF1 N159Y/S, 25 , 34 and contributed to the pathogenesis of various types of human cancers through different mechanisms. 36 , 37 Upregulation of MYC following IKZF1 N159S may herald underlying pathogenesis in this molecular subtype. In addition, the CPNE gene family are calcium‐dependent phospholipid‐binding proteins with intrinsic kinase activity, although the exact biological functions of CPNE family proteins remain unclear, an increasing number of studies have shown that CPNE family proteins may mediate various signalling pathways involved in tumourigenesis and progression. 38 Therefore, further investigation on the synergistic pathway of IKZF1 N159S‐CPNE7 may provide new insights into the pathogenesis and refine the treatment of AML. Based on the theoretical assumptions above, we designated the rescue experiment aiming at IKZF1 N159S‐expressing cells, and short hairpin RNAs (shRNAs) targeting MYC and CPNE7 were used to knockdown the expression level of MYC and CPNE7 in N159S‐expressing cells (Figure 6C). Decreased expression of MYC in IKZF1 N159S‐expressing cells was typically associated with a subsequent G1 cell cycle arrest (Figure 6D). Meanwhile, both MYC and CPNE7 depletion in IKZF1 N159S cells blocked cell growth detected by CCK‐8 assays (Figure 6E). To further exploit potential therapeutic options for patients with IKZF1 N159S mutation, 10058‐F4, an appealing inhibitor of MYC was applied in our research, which was reported to exert remarkable anti‐cancer capability in hepatocellular carcinoma, 39 prostate cancer 40 as well as various haematologic malignancies. 41 , 42 Western blot analysis of 10058‐F4‐treated IKZF1 N159S cells revealed dose‐dependent inhibition of MYC protein after 24 h (Figure 6F). A concentration‐dependent increase in the fraction of cells in the G1 phase was detected after 24‐h period of drug exposure (Figure 6G). CCK‐8 assays showed dose‐dependent inhibition of viability of IKZF1 N159S cells that in parallel with G1 cell cycle arrest (Figure 6H). These results suggested that IKZF1 missense mutation N159S may augment the biological function of a series of oncogenes, among which, the engagement of MYC and CPNE7 might play a pivotal role in IKZF1 N159S‐mediated tumourigenesis. Considering MYC is a proliferation‐related gene, a direct effect of inhibitor 10058‐F4 or MYC shRNA should not be neglected. Genetic ablation or pharmaceutical blockade of MYC or CPNE7 could potentially provide innovative treatment options for AML patients with IKZF1 N159S mutation.
FIGURE 6.
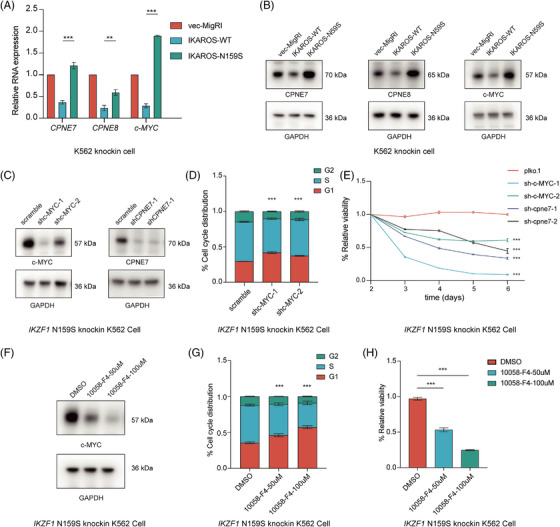
IKZF1 N159S mutation strengthen the expression of MYC and CPNE7 to promote carcinogenesis. (A) Qpcr validation of differential gene expressions. The graph depicting relative expression level of MYC, CPNE7 and CPNE8 mRNA in IKZF1 WT and IKZF1 N159S expressing K562 cells 5 days post lentiviral transduction, and gene expression level was normalised to the reference gene GAPDH. (B) Western blot of MYC, CPNE7 and CPNE8 in empty vector, IKZF1 WT and IKZF1 N159S transduced K562 cells 5 days post lentiviral transduction. (C) Western blot detection of MYC and CPNE7 in IKZF1 N159S expressing K562 cells after transducing with lentivirus expressing either a scramble (control) shRNA or two independent shRNAs targeting at MYC and CPNE7. Cells were selected and immunoblotted at 4 days post‐transduction. (D) Cell cycle distribution calculation of IKZF1 N159S expressing cells after knocking down MYC, a growing proportion of cells arrested in G1 phase after MYC knockdown. Cell cycle distribution assayed as described in the Methods section. (E) Cck8 proliferation assay of IKZF1 N159S expressing cells control versus MYC or CPNE7 knockdown, MYC or CPNE7 knockdown inhibit the proliferation ability of IKZF1 N159S expressing cells. The viability of cells was measured daily beginning 3 days post‐transduction of shRNAs. (F) Western blot analysis of MYC protein level in IKZF1 N159S expressing cells treated with 10058‐F4 for 24 h at the concentrations labelled, GAPDH served as a loading control. MYC protein expression is negatively regulated by 10058‐F4. (G) Flow‐cytometry‐based quantification of cell cycle distribution of IKZF1 N159S expressing cells treated with 10058‐F4, with increasing drug concentration, more and more cells are blocked in G1 phase. (H) Cck8 assay demonstrated the dose‐dependent effects of 10058‐F4 in the proliferation capability of IKZF1 N159S expressing cells, cell proliferation was inhibited with increasing drug concentration. All plots are representative of at least three independent experiments performed in duplicate and presented as the means ± standard deviation (*p < .05, **p < .01 and ***p < .001).
4. DISCUSSION
Acute myeloid leukaemia (AML) is a group of highly genetic heterogeneous diseases with various prognosis. Functionally, gene mutations could be classified into different types, as exemplified by the dysregulation of transcription factors (TFs), that is, CEBPA, RUNX1 and IKZF1 and epigenetic regulators, that is, DNMT3A, IDH1/2 and ASXL1, which are indispensable for the control of myeloid progenitor cell differentiation and lineage determination. 34 IKZF1, a well‐known tumour suppressor and transcription factor in haematopoiesis, plays a critical role in lymphoid differentiation, as well as in myeloid development. Several mouse models with constitutive or conditional IKZF1 knockout exhibited disturbance of normal lymphoid and myeloid differentiation, and developed aggressive diseases such as widespread haematopoiesis failure, thymic lymphoma or T‐cell malignancies.
With the advent of the era of high‐throughput sequencing and precision medicine of AL, genetic mutations of IKZF1 were firstly reported in aggressive B‐ALL subtypes, that is, BCR::ABL1/‐like, which were characterised by the high relapse rate and short survival duration. 8 , 16 , 25 However, IKZF1 mutations were poorly characterised in the research field of AML. Only a few studies have reported the rare IKZF1 mutations in acute myeloid leukaemia with a low frequency. 26 , 43 On the other hand, the lineage plasticity refers to the ability of transition from one committed developmental pathway to another, 44 relapse can be associated with a lineage switch from acute lymphoblastic to acute myeloid leukaemia, which may be resistant to chemo‐ and immunotherapies and result in poor clinical outcomes. 45 Previous studies have shown that the lineage switching is linked to a major rewiring of gene regulatory networks in patients with KMT2A translocations. 34 , 44 , 45 But the potential role of IKZF1 (IKAROS) in disease fate determination has been rarely explored.
Herein, we conducted a genomics/transcriptomics analysis in 475 newly diagnosed cases of non‐M3 AML. The genomic landscape of IKZF1 sequence variants (n = 23) in AML was elucidated. Three major classes of IKZF1 mutations were identified including IKZF1 N159S (n = 9, 39.13%), biCEBPA/‐like IKZF1 (n = 10, 43.47%) and others (n = 4). Most of them may result in abnormal genome binding patterns and gene expression regulations via impacting the DNA‐binding domain of IKZF1. Besides, the hotspot IKZF1 N159S mutations frequently coexisted with RUNX1 and spliceosome mutations. These comutations are common in the myelodysplasia‐related/‐like AML, particularly in elderly populations. Prognosis‐wise, AML patients with IKZF1 N159S had an extremely poor prognosis, even worse than TP53 mutation/complex karyotype AML in our cohort. According to the available drug response data, combination therapy of demethylation agents and Homoharringtonine (HHT)/all‐trans‐retinoic acid (ATRA) showed potential sensitivity in IKZF1 N159S‐positive AML (Table S1). Above all, incorporating IKZF1 into the prognostic stratification of AML may improve the diagnosis discrimination and facilitate the development of new tailored therapies for AML. Meanwhile, we also noticed that the IKZF1 mutations coexisted with biCEBPA are associated with more favourable prognosis, like DUX4 fusions in B‐ALL, which could reverse the negative effect of IKZF1 abnormalities. The related factors still warrant further exploration by more comparative functional studies.
Gene expression profiles indicated that compared with biCEBPA/‐like IKZF1, the HOXA/B, HSPC expression signature, proto‐oncogene pathways including VEGF, B‐cell receptor, NOTCH, MAPK, WNT, TGF‐beta, Rap1 signalling pathways were significantly upregulated in the IKZF1 N159S‐positive AML. Activated NOTCH signalling was also observed in T‐ALL with IKZF1 N159S. 14 Moreover, the known kinase signalling factor (PDGFRB) of BCR::ABL1‐like B‐ALL was also upregulated in IKZF1 N159S‐related AML. 31 In contrast, the DNA repair‐related pathways were significantly downregulated in patients with IKZF1 N159S. These specifically enriched pathways may partially interpret the unique immune microenvironment and clinical prognosis of IKZF1 N159S, providing a wealth of therapeutic targets/pathways for this high‐risk AML subtype.
Next, we performed in vitro functional experiments to investigate the cellular effects of IKZF1 genetic mutations. IKZF1 N159S exerted aberrant regulation of cell apoptosis, cell cycle and cell viability, which indicated the perturbation of the tumour‐suppression function of IKZF1. We then compared the gene expression profiles of hotspot mutations IKZF1 N159S, G158S and N159Y and found that B‐lymphoid development markers, such as MME and CD44, were highly expressed in N159Y‐positive but downregulated in N159S‐positive cell line. RUNX2 was also highly expressed in IKZF1 N159Y knock‐in AML cells, which was reported to be abundant in the haematopoietic stem/progenitor cells in mice, and the upregulation of RUNX2 led to the repression of myeloid differentiation. 46 Conversely, RUNX1T1, a well‐known factor for haematopoietic differentiation and myeloid development, was highly expressed in IKZF1 N159S knock‐in AML cells instead of IKZF1 N159Y. 47 These data suggested that the dysregulation of gene transcriptional profiles may underpin the fundamental lineage reprogramming. 45 We therefore propose that different regulation effect of genetic mutations at the same amino acid site of transcription factors may lead to different regulation effect and leukaemia lineage.
Based on gene expression profiling and CUT&TAG sequencing of knock‐in human cell lines, we identified several potential common and specific pathways, that is, JAK‐STAT, NOTCH signalling pathway and B‐cell development, and genes, that is, CPNE7, VWDE, LTK and IGSF9B, of IKZF1 N159S‐positive AML. Among these, we speculate that the inherent characteristics of recurrent IKZF1 N159S mutation might lead to a more deteriorative evolution of AML via upregulating MYC targets and CPNE7, which were directly regulated by IKZF1 N159S. Inhibitors of MYC have been widely exploited, that is, 10058‐F4, APTO‐253. Therefore, the employment of MYC inhibitors could bring new therapeutic ideas for patients with IKZF1 N159S mutations.
Taken together, our data identified the mutation spectrum of IKZF1 and defined the significant function of IKZF1 N159S in AML. The clinical and biological significance of IKZF1 variants were explored both in patients and cell line level, highlighting the key role of IKZF1 in the pathogenesis, treatment, and stratification of AML. Furthermore, it may appear as an important molecular target for tailored treatment, as exemplified by MYC inhibitor. These findings may further promote the accurate classification and improvement of prognosis for patients with IKZF1‐positive AML, which may also exert potential impact on other tumour researches involving immune microenvironment and lineage switching mechanism.
AUTHOR CONTRIBUTIONS
SY and LJF conceived and designed the study. WY and ZYY performed the experiments. WY, LJF and ZYL analysed and interpreted data. CWY, WY, STF, YW, ZJN and ZYM collected the clinical data; WY and LJF wrote the manuscript. CWY, ZYY and SY revised the paper. All authors reviewed the data and paper and approved the final version of the manuscript.
FUNDING
This work was supported by the National Natural Science Foundation of China (No. 82270116) and the Shanghai Municipal Education Commission‐Gaofeng Clinical Medicine Grant Support (No. 20161406).
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We wish to thank all our patients and their families for supporting this study.
Wang Y, Cheng W, Zhang Y, et al. Identification of IKZF1 genetic mutations as new molecular subtypes in acute myeloid leukaemia. Clin Transl Med. 2023;13:e1309. 10.1002/ctm2.1309
Yang Wang, Wenyan Cheng and Yvyin Zhang contributed equally to this work.
Contributor Information
Jianfeng Li, Email: ljf12500@rjh.com.cn.
Yang Shen, Email: yang_shen@sjtu.edu.cn.
DATA AVAILABILITY STATEMENT
Cell line sequencing data have been deposited into the GSA‐Human database (https://ngdc.cncb.ac.cn/gsa‐human/) under accession identification numbers subHRA004087. All data sets generated and/or analysed during the current study are available from the corresponding author on reasonable request. Please contact yang_shen@sjtu.edu.cn.
REFERENCES
- 1. Newell LF, Cook RJ. Advances in acute myeloid leukemia. BMJ. 2021;375:n2026. [DOI] [PubMed] [Google Scholar]
- 2. Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol. 2016;34(5):525‐527. [DOI] [PubMed] [Google Scholar]
- 3. Taube F, Georgi JA, Kramer M, et al. CEBPA mutations in 4708 patients with acute myeloid leukemia: differential impact of bZIP and TAD mutations on outcome. Blood. 2022;139(1):87‐103. [DOI] [PubMed] [Google Scholar]
- 4. Rio‐Machin A, Vulliamy T, Hug N, et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun. 2020;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bolouri H, Farrar JE, Triche T, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age‐specific mutational interactions. Nat Med. 2018;24(1):103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burd A, Levine RL, Ruppert AS, et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med. 2020;26(12):1852‐1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu YF, Wang BY, Zhang WN, et al. Genomic profiling of adult and pediatric B‐cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stanulla M, Dagdan E, Zaliova M, et al. IKZF1(plus) defines a new minimal residual disease‐dependent very‐poor prognostic profile in pediatric B‐cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36(12):1240. [DOI] [PubMed] [Google Scholar]
- 9. Gu Z, Churchman ML, Roberts KG, et al. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seki M, Kimura S, Isobe T, et al. Recurrent SPI1 (PU.1) fusions in high‐risk pediatric T cell acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1274‐1281. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T‐cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T‐lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen B, Jiang L, Zhong ML, et al. Identification of fusion genes and characterization of transcriptome features in T‐cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2018;115(2):373‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng WY, Li JF, Zhu YM, et al. Transcriptome‐based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia. Proc Natl Acad Sci U S A. 2022;119(49):e2211429119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boutboul D, Kuehn HS, Van de Wyngaert Z, et al. Dominant‐negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. 2018;128(7):3071‐3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. John LB, Ward AC. The Ikaros gene family: transcriptional regulators of hematopoiesis and immunity. Mol Immunol. 2011;48(9‐10):1272‐1278. [DOI] [PubMed] [Google Scholar]
- 17. Stanulla M, Cavé H, Moorman AV. IKZF1 deletions in pediatric acute lymphoblastic leukemia: still a poor prognostic marker? Blood. 2020;135(4):252‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoshino A, Boutboul D, Zhang Y, et al. Gain‐of‐function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci Immunol. 2022;7(69):eabi7160. [DOI] [PubMed] [Google Scholar]
- 19. Heizmann B, Kastner P, Chan S. The Ikaros family in lymphocyte development. Curr Opin Immunol. 2018;51:14‐23. [DOI] [PubMed] [Google Scholar]
- 20. Kaya‐Okur HS, Wu SJ, Codomo CA, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10(1):1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stanulla M, Dagdan E, Zaliova M, et al. IKZF1(plus) defines a new minimal residual disease‐dependent very‐poor prognostic profile in pediatric B‐cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36(12):1240‐1249. [DOI] [PubMed] [Google Scholar]
- 22. Roberts KG, Li Y, Payne‐Turner D, et al. Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mullighan CG, Su X, Zhang J, Radtke I, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li JF, Dai YT, Lilljebjorn H, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A. 2018;115(50):E11711‐E20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Churchman ML, Qian M, Te Kronnie G, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. 2018;33(5):937‐948. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X, Zhang X, Li X, et al. The specific distribution pattern of IKZF1 mutation in acute myeloid leukemia. J Hematol Oncol. 2020;13(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aubrey BJ, Cutler JA, Bourgeois W, et al. IKAROS and MENIN coordinate therapeutically actionable leukemogenic gene expression in MLL‐r acute myeloid leukemia. Nat Cancer. 2022;3(5):595‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jin P, Jin Q, Wang X, et al. Large‐scale in vitro and in vivo CRISPR‐Cas9 knockout screens identify a 16‐gene fitness score for improved risk assessment in acute myeloid leukemia. Clin Cancer Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome‐like acute lymphoblastic leukemia. Blood. 2017;130(19):2064‐2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21(2):122‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Churchman ML, Low J, Qu C, et al. Efficacy of retinoids in IKZF1‐mutated BCR‐ABL1 acute lymphoblastic leukemia. Cancer Cell. 2015;28(3):343‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Assi SA, Imperato MR, Coleman DJL, et al. Subtype‐specific regulatory network rewiring in acute myeloid leukemia. Nat Genet. 2019;51(1):151‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huo Q, Ge C, Tian H, et al. Dysfunction of IKZF1/MYC/MDIG axis contributes to liver cancer progression through regulating H3K9me3/p21 activity. Cell Death Dis. 2017;8(5):e2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Madden SK, de Araujo AD, Gerhardt M, Fairlie DP, Mason JM. Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c‐Myc. Mol Cancer. 2021;20(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carter JL, Hege K, Yang J, et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther. 2020;5(1):288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang H, Pang P, Qin Z, et al. The CPNE family and their role in cancers. Front Genet. 2021;12:689097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin CP, Liu JD, Chow JM, Liu CR, Liu HE. Small‐molecule c‐Myc inhibitor, 10058‐F4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anti‐Cancer Drugs. 2007;18(2):161‐170. [DOI] [PubMed] [Google Scholar]
- 40. Carabet LA, Lallous N, Leblanc E, et al. Computer‐aided drug discovery of Myc‐Max inhibitors as potential therapeutics for prostate cancer. Euro J Med Chem. 2018;160:108‐119. [DOI] [PubMed] [Google Scholar]
- 41. Sheikh‐Zeineddini N, Bashash D, Safaroghli‐Azar A, et al. Suppression of c‐Myc using 10058‐F4 exerts caspase‐3‐dependent apoptosis and intensifies the antileukemic effect of vincristine in pre‐B acute lymphoblastic leukemia cells. J Cell Biochem. 2019;120(8):14004‐14016. [DOI] [PubMed] [Google Scholar]
- 42. Bashash D, Sayyadi M, Safaroghli‐Azar A, Sheikh‐Zeineddini N, Riyahi N, Momeny M. Small molecule inhibitor of c‐Myc 10058‐F4 inhibits proliferation and induces apoptosis in acute leukemia cells, irrespective of PTEN status. Int J Biochem Cell Biol. 2019;108:7‐16. [DOI] [PubMed] [Google Scholar]
- 43. de Rooij JD, Beuling E, van den Heuvel‐Eibrink MM, et al. Recurrent deletions of IKZF1 in pediatric acute myeloid leukemia. Haematologica. 2015;100(9):1151‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Quintanal‐Villalonga A, Chan JM, Yu HA, et al. Publisher correction: lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. 2020;17(6):382. [DOI] [PubMed] [Google Scholar]
- 45. Tirtakusuma R, Szoltysek K, Milne P, et al. Epigenetic regulator genes direct lineage switching in MLL/AF4 leukaemia. Blood. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuo YH, Zaidi SK, Gornostaeva S, Komori T, Stein GS, Castilla LH. Runx2 induces acute myeloid leukemia in cooperation with Cbfbeta‐SMMHC in mice. Blood. 2009;113(14):3323‐3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Al‐Harbi S, Aljurf M, Mohty M, Almohareb F, Ahmed SOA. An update on the molecular pathogenesis and potential therapeutic targeting of AML with t(8;21)(q22;q22.1);RUNX1‐RUNX1T1. Blood Adv. 2020;4(1):229‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
Cell line sequencing data have been deposited into the GSA‐Human database (https://ngdc.cncb.ac.cn/gsa‐human/) under accession identification numbers subHRA004087. All data sets generated and/or analysed during the current study are available from the corresponding author on reasonable request. Please contact yang_shen@sjtu.edu.cn.