

Graphical abstract

Keywords: Hepatotoxicity, Hepatotoxicity decision tree, Chemical categories, SAR read-across, Mode of action
Highlights
-
•
More than 1100 chemicals were evaluated in order to construct a decision tree, and included chemicals that are directly reactive, that require metabolic activation, or that exert toxicity by receptor binding.
Abstract
The liver is the most common target organ in toxicology studies. The development of chemical structural alerts for identifying hepatotoxicity will play an important role in in silico model prediction and help strengthen the identification of analogs used in structure activity relationship (SAR)- based read-across. The aim of the current study is development of an SAR-based expert-system decision tree for screening of hepatotoxicants across a wide range of chemistry space and proposed modes of action for clustering of chemicals using defined core chemical categories based on receptor-binding or bioactivation. The decision tree is based on ∼ 1180 different chemicals that were reviewed for hepatotoxicity information. Knowledge of chemical receptor binding, metabolism and mechanistic information were used to group these chemicals into 16 different categories and 102 subcategories: four categories describe binders to 9 different receptors, 11 categories are associated with possible reactive metabolites (RMs) and there is one miscellaneous category. Each chemical subcategory has been associated with possible modes of action (MOAs) or similar key structural features. This decision tree can help to screen potential liver toxicants associated with core structural alerts of receptor binding and/or RMs and be used as a component of weight of evidence decisions based on SAR read-across, and to fill data gaps.
1. Introduction
Most xenobiotics that enter systemic circulation are transported to the liver; therefore, this organ is one of the first and most exposed to chemical damage. In addition, because of its significant biotransformation of xenobiotics the liver is also the site of formation of toxic metabolites. Thus, liver-related adverse effects are important in understanding the safety of chemicals. Due to the complexity and diversity of xenobiotics, as well as the type and number of pathways the liver uses to process these xenobiotics, developing predictive systems for potential hepatotoxicity currently remains a significant challenge. In silico screening and prioritization of compounds developed for focused chemical classes have been widely used for many years in the pharmaceutical industry, but not for the chemical industry in general. Nonetheless, there are several reports that apply in silico QSAR models to predict liver toxicity (Mulliner et al., 2016, Low et al., 2011), most of them focused on drug candidates. Liu et al. proposed the use of ToxCast in vitro biological activity data and chemical structure to predict hepatotoxicity. The authors demonstrated the utility of data from high-throughput assays and the benefit of using hybrid representations that integrate biological activity and chemical structure for characterizing rodent hepatotoxicants (Liu et al., 2015a). In addition to the QSAR models, several research groups have successfully developed drug-related chemical structural alerts which could potentially be used in the screening of compounds to highlight potential hepatotoxicity. Many mechanistically supported structural alerts and some molecular fragments associated with drug-induced hepatotoxicity have also been proposed (Hewitt et al., 2013, Mellor et al., 2016, Pizzo et al., 2015, Liu et al., 2015b).
Our research group has undertaken a systematic approach for identifying and evaluating analogs for read-across assessments based upon chemical and biochemical principles (Wu et al., 2010, Blackburn et al., 2011). As support for these read-across methods, we have developed a decision tree for identifying potential developmental and reproductive toxicants (DART; Wu et al., 2013) based on chemical features that are present in these chemicals. The DART decision tree framework was used to define the appropriate magnitude of an uncertainty factor to account for missing reproductive and/or developmental toxicity data for a specific chemical when repeat dose data are available for this chemical (Blackburn et al., 2015).
Continuing our efforts to develop more robust SAR read-across methods for systemic toxicity, here we describe a decision tree to identify chemicals with structural features associated with hepatotoxicity. The decision framework is based on liver toxicity data for approximately 1180 different chemicals in humans or relevant animal models. Some of these chemicals produce adverse effects by interaction with receptors, others by reactive chemistry, either as the parent compound or by formation of reactive metabolites (RMs). In both cases we identify structural alerts for these chemicals and rules for defining the boundaries for each category of chemicals in the decision tree.
2. Methods
1. Data sources. The decision tree for hepatotoxicity flags was based on primary evaluation of several compiled datasets: a. NIH LiverTox data set (https://www.ncbi.nlm.nih.gov/books/NBK547852/), containing more than 1000 drugs; b. Liver QSAR dataset, a set of 951 compounds reported to produce a wide range of effects in liver of humans, rodents, and nonrodents (Fourches et al., 2010); c. Drugs and chemicals from review articles on hepatotoxicity (Hewitt et al., 2013, Stepan et al., 2011); and d. Cosmetics chemicals from the Scientific Committee on Consumer Safety (SCCS)/Scientific Committee on Consumer Products and Non-Food Products (SCCNFP) (Vinken et al., 2012). After eliminating duplicates (same CAS#) we collected 1180 compounds. Chemicals lacking hepatotoxicity data, negative for hepatotoxicity or not classifiable within a subcategory definition in the dataset were eliminated from further consideration, with the exception of a small set of chemicals with structural features closely related to hepatotoxicants. Our intention in choosing the SCCS data set, as well as much of the literature review, was to broaden the chemical coverage as much as possible beyond pharmaceutical compounds, but we acknowledge that the overall data set is biased towards drugs. We also searched metabolism reports using a metabolism data base (Biovia Metabolite, Dassault Systemes, San Diego, CA) and the primary literature. The metabolic pathways which could generate RMs relevant to identifying the proximate hepatotoxicants were flagged Supplementary Information.
2. Chemical class categorization. We binned chemicals based on expert judgement about possible receptor binding activity or reactive metabolite formation. All chemicals were analyzed by their core structural features (e.g. acyclic alkyl chain, cyclic/heterocyclic and aromatic/heteroaromatic rings, etc.), key functional groups (e.g. halogenated hydrocarbons, esters, aldehydes, acids, amides, alcohols, amines, urea, etc.) and common structural fragments within molecules as well as the potential to form RMs. Chemical groupings were also influenced by what is known from the literature about receptor interactions and/or known metabolic reactions to ensure that groupings made sense from a biological as well as a chemical perspective. For the interaction with receptors, we focused on the nuclear receptor family and grouped them into four categories: a. steroid hormone receptors (ER (estrogen receptor), GR (glucocorticoid receptor), AR (androgen receptor),); b. regulation of CYPs and transporter gene expression (PXR (pregnane X receptor), CAR (constitutive androstane receptor), AhR (aryl hydrocarbon receptor)); c. PPA7R (peroxisome proliferator-activated receptor) and d. RAR (retinoic acid receptor). Because many chemicals are metabolized extensively in the liver, one of the major tasks for the categorization of chemicals in the decision tree was to search possible RM generation pathways that may be associated with hepatotoxicity. We identified 11 different categories of RMs and their precursors as well as one miscellaneous category: a. p-, o-iminoquinones; b. p-, o-quinones; c. p-, o-quinone methides; d. αβ-unsaturated carbonyl chemicals; e. alkylating agents and carbon cation; f. alkyl & aryl radical formation: g. epoxides; h. acyl halides, acyl carbon cations β-lactams; i. aryl/heteroaryl carboxylic acid, acetic acid j. S-S bond formations; k. nitroso formation from aromatic and aliphatic amine; l. hepatotoxicity induced by miscellaneous mechanisms and metabolic pathways. It is important to note that because of the complexity of hepatotoxicity as an endpoint, the chemical grouping represents only one of several possible mechanistic explanations.
3. Decision tree construction and organization. Categories and subcategories of the tree consist of groups of chemicals that share similar core structural features or undergo similar bioactivation. We also ensured that adding or removing less toxic (e.g., alkyl) substituents, switching position of functional groups would not affect the pathway of bioactivation or receptor interaction. The selection of subcategories is biased by the frequency of occurrence of similar analogs or homologs which exhibit hepatotoxic effects. The definition of core structural features, especially the cut-off values (for example, chain length and certain aryl substituents) are set broadly unless data are available to clearly define the limits. This becomes more apparent as the structures of the chemicals become more complex (e.g. containing multiple reactive moieties). In addition, there are situations where more complex chemicals fit into more than one subcategory. For example,17-β-estradiol (E2 CAS# 50–28-2) derivatives belong to subcategories of estrogen receptor binders and p-, o-quinone, catechol, p-diphenol and precursors. In these cases, we grouped the chemicals based on known receptor binding affinities first, then on RM-induced mechanism. Chemicals with known hepatotoxic effects but with insufficient mechanistic or bioactivation information to form a group were grouped with the chemical category that shares similar structural alerts. Otherwise, they were placed into a miscellaneous chemicals group. In these cases, one must be very cautious of expanding the core structural or alert structural features to flag similar compounds due to the potential for highly specific modes of action. We would anticipate that, as we learn more about mode of action, these miscellaneous chemicals will be placed with other chemicals in either new groups or be incorporated with expanded rules in existing groups.
Overall, the decision tree includes 4 categories of chemicals associated with receptor binders (include 9 different nuclear receptors), 11 categories of chemicals associated with potential reactive chemicals/metabolites, and one miscellaneous category. We further divided the 16 main categories into multiple subcategories based on structural features.
3. Results and discussion
3.1. Categorization of chemicals and the expert system decision tree
Identification of structural alerts is useful in the prediction of toxicity and grouping of chemicals with similar features and can directly facilitate SAR read-across. In this study, we developed categories (and subcategories) and arranged them into a decision tree based on the following criteria: reactive chemicals (including RMs) which induced liver injury (e.g. enlarged livers, jaundice, hepatitis, hepatic hypertrophy, elevation of serum transaminase etc.) or liver tumors or chemicals that elicit hepatotoxicity through interaction with receptors. Each category contains a similar core structural fragment/bioactivation pathway. Chemical classes without clear structural alerts and RM formation pathways were grouped in a miscellaneous category. The domains covered by the categories/subcategories are driven by the chemistry of substances in the open literature that was evaluated; there was no attempt to broadly define the chemical space. The decision tree is based on organic chemicals only and organized into two major groupings: possible receptor binders and possible RMs that appear to be responsible for hepatotoxicity. Compounds that fit into multiple categories are grouped based on the following priorities: receptor binders > RM-related alerts > miscellaneous structural fragments. Fig. 1 outlines the simplified decision tree and Table 1 provides a short description of the core structural features for each category (see Appendix II for details). The largest categories are: p-, o-quinone-imines, p-, o-aromatic-diimines and precursors (1 5 8); p-, o-quinone, catechol, p-dihydroxyphenol and precursors (69); p-, o-quinone, imine methides (30); Michael acceptors and precursors (51); alkylation and potential alkylating reagents (82); alkyl or aryl radicals (23); epoxides and precursors (90); isocyanate, acetyl carbonium ion and acetyl chloride-like (or ketene) electrophiles (52); arylacetic acid, aryl/heteroaryl carboxylic acid and precursors (28); S-S bond formation (from -S-OH) or metal chelation (29); nitroso formation from aromatic and alphatic amines (1 1 2).
Fig. 1.

Overall process of the simplified decision tree for screening hepatotoxicity (detail decision tree see Appendix II).
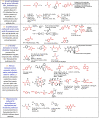
Table 1.
Decision Tree Categories with Representative Core Structures (core structural features are shown in “red”)*.
 |
 |
 |
If chemicals land in the bin of “known possible receptor binderRM induced hepatotoxic potential” in the decision tree based on the definition of each subcategory (see Appendix II for details), they are flagged as potential hepatotoxicants. Chemicals landing in the “no known possible receptor binder/RM induced hepatotoxic potential” bin either lack hepatotoxic potential or their potential receptor biding activity or major metabolites are outside of the evaluation domain of the decision tree and require further investigation to define their potential to elicit hepatotoxicity. It is important to keep in mind that the structures shown are intended to be used as guidelines to demonstrate the types of chemicals associated with hepatotoxic activity and are not intended to be rigid rules. The decision tree does not presume to be accurately predictive for every individual chemical but can serve as a component of a screening system to identify chemicals of potential concern to support SAR read-across.
3.2. Examples of category information (see Appendix II for detailed category information)
The collected reference information for each category member’s hepatotoxicity is summarized online in Appendix II where we have discussed what is known about the structural features and possible mechanism associated with hepatotoxicity for each category member. It is important to note that the mechanisms of hepatotoxicity may be more complicated than a single receptor binding or bioactivation pathway being affected by chemical exposure. Due to the length of this summary material, our discussion is primarily focused on selected examples of chemical categories associated with nuclear receptor binding, reactive chemicals/metabolites to illustrate to the reader the information contained in Appendix II.
3.2.1. Examples of chemical categories associated with receptor binding
Example 1
Steroid nucleus derived receptor binders: Androgenic and anabolic steroid (AR)-like derivatives (section 3.2.1. (a.3) in Appendix II).
Androgenic steroids include naturally occurring (e.g. testosterone) and synthetic anabolic steroids (Gao et al., 2005). As a group, these chemicals are capable of inducing several distinct forms of liver toxicity in humans, including temporary increases in hepatic serum enzymes; acute cholestatic syndrome; chronic vascular injury to the liver and hepatic tumors. Although the exact mechanism of liver injury is unclear, it is known that anabolic steroids interfere with bile acid-dependent flow (BADF) and bile acid-independent flow (BAIF) to cause cholestatic effects (Zimmerman, 1999, Becker, 2001) and/or an increase in oxidative stress leading to changes in bile salt transporter proteins or impairment of function of the bile salt export pump (LiverTox; Kafouni et al., 2007). The anabolic steroids in Fig. 2 contain a wide variety of substituents associated with these adverse effects. Generally, these compounds contain a carbonyl group at position C-3 and double bond between carbon atoms C-4 and C-5 as well as may be a double bond between C-1 and C-2 on the A ring. The R5 at position C-11 on the C-ring is hydrogen for a majority of these compounds. For the D-ring, the substituent R2 is a 17β-hydroxyl group and R3 is a methyl or other small alkyl group, ethynyl as well as R2 and R3 can form a spirolactone in a few cases and R4 is typically hydrogen. The 17α-alkyl group (R3) and 17β hydroxyl groups play an important role in causing hepatotoxicity (Kicman, 2008). Almost all orally active androgens are 17α-alkylated derivatives of testosterone and have possible serious effects on the liver (cholestasis, peliosis hepatitis, benign and malignant liver tumors). Androgens lacking a 17α-alkyl group or17β-hydroxyl group have weak or no hepatotoxicity. The liver toxicity is not found in “replacement dosages” of parenteral 17β-hydroxyl esters of testosterone (Becker. 2001). This suggests that 17β-hydroxyl is blocked by the acetyl moiety, abolishing hepatotoxicity. In addition to the general structural features described above, some anabolic steroids shown in (1b, 1c and 1d) are hepatotoxic. For example, conversion of the 3-keto to a 1H-pyrazole fused to the A ring (1b), addition of an ethanol group at the 2 position of the A ring (1c) or conversion of the A ring to a lactone (1d) are also favorable for hepatotoxicity. Due to the structural similarity in the steroid skeleton of steroidal AR ligands, complete separation of androgenic and anabolic activity has not been accomplished with synthetic steroids. Furthermore, steroidal AR tends to cross-react with other steroid receptors, which might cause adverse effects as well.
Example 2
Pregnane X Receptor (PXR) binders: Cholic acid and azole fungicide derivatives (section 3.2.2-1. (a) & (c) in Appendix II).
Fig. 2.

The scope of structural features of androgenic and anabolic steroid (AR) like derivatives.
The pregnane X receptor (PXR) is associated with the regulation of multiple genes involved in chemical metabolism and transport. The activity of PXR can be modulated by a variety of structurally diverse chemicals, which act as agonists or antagonists (Chen and Nie, 2009), and result in multiple biological effects, some physiologically necessary (e.g. PXR activation plays an important role in cholesterol and bile acid homeostasis), but other effects are pathological (e.g. hepatotoxicity). The PXR pathway is important for preventing bile acid toxicity under cholestatic conditions with pathological accumulation of bile acids (Rezen et al., 2011). Some chemicals interact with PXR and elicit an increase in the expression of various CYP enzymes (e.g. various isoforms of CYP3A; Schuetz et al., 1998). However, hepatotoxicity has not been directly linked to interaction with PXR. It has been suggested that the PXR-dependent induction of CYP3A4, UDP glucuronosyltransferase family 1 member A1 (UGT1A1), multidrug resistance-associated protein 2 (MRP2) and organic solute transporter beta (OSTβ), could contribute to the anticholestatic effect elicited by some chemicals, resulting in persistent hepatocellular secretory failure and subsequent liver damage (Van Dijk et al., 2015). Further, growing evidence indicates that some PXR agonists lead to different clinicopathological subtypes of hepatotoxicity. Wang et al. (2014) proposed that at least two types of mechanisms could explain PXR-mediated liver damage: 1. PXR agonist activates PXR-regulated expression of drug-metabolizing enzymes and transporters, contributing to the formation of toxic metabolites; and 2. PXR agonists activate the expression of critical liver enzymes in major metabolic pathways that may alter the balance of endobiotic formation and clearance, leading to accumulation of endogenous toxicants. As indicated, PXR has a key role in regulating the metabolism and transport of structurally diverse endogenous and exogenous compounds. Activation of PXR has the potential to initiate adverse effects, such as hepatic steatosis in mice (Zhou et al., 2008), enhancing drug metabolism which might cause unwanted drug–drug interactions and perturbing normal physiological functions (Dybdahl et al., 2012).
Cholic acid is one of the two major bile acids produced by the liver, where it is synthesized from cholesterol. Bile acids are relevant ligands of PXR, the main form being lithocholic acid (LCA) and its oxidized 3-keto form which activate PXR (Staudinger et al., 2001). LCA is a cytotoxic bile acid (Vogel et al., 2012) and induces cholestasis that results in extensive liver damage. Based on the degree of hydroxylation, the bile acids include tri-, di- and monohydroxylated bile acids, namely cholic acid (CA), deoxycholic acid (DCA) and lithocholic acid. DCA is the most hepatotoxic, while CA is the least hepatotoxic and cholestatic compound, indicating that hepatotoxicity of bile acids does not depend on their degree of hydroxylation (Delzenne et al., 1992). The representative active bile acids with general core structures are shown in (2a-1) in Fig. 3 where the R and R2 groups can be hydrogen or hydroxyl groups. R1 can be a hydroxyl group or amino alkyl acid and amino alkyl sulfonic acid groups.
Fig. 3.

The scope of structural features of cholic acid derivatives.
Azole fungicides are considered to cause liver toxicity, including hepatocellular steatosis and hypertrophy by a mechanism involving PXR (Knebel et al., 2019). In general, triazoles activate PXR but may also have agonist potential for multiple receptors. Structurally, these triazoles contain core features of halogenated phenethyl triazole or halogenated phenoxyl triazole moiety (e.g. the Y can be a tertiary or quaternary carbon or oxygen atom) as shown in (2b-1), exemplified by several azole fungicides (e.g. cyproconazole, epoxiconazole, propiconazole, tebuconazole, myclobutanil and triadimefon etc. as shown in (2c) to (2i) in Fig. 4). Cyproconazole has shown the most pronounced effects on increasing liver weight of animals (Heise et al., 2015). Some of these chemicals (e.g. cyproconazole, epoxiconazole, propiconazole etc.) may also have hepatocarcinogenic potential. It has also been reported that the common set of toxicological effects altered by these conazoles include hepatomegaly, hepatocellular hypertrophy, decreased serum cholesterol, decreased hepatic levels of all-trans retinoic acid, and increased hepatic cell proliferation (Hester et al., 2012). The hepatotoxicity and the structural features of other triazole related chemicals are discussed in section 3.2.2-1c of Appendix II.
Example 3
Constitutive androstane receptor (CAR) activators (section 3.2.2-2. (a) in Appendix II).
Fig. 4.

The scope of structural features of azole fungicide -like chemicals.
CAR is predominantly expressed in the liver. It interacts with CAR activators and translocates to the nucleus to form functional heterodimers with RXR to induce the expression of CAR target genes. Similar to PXR, CAR regulates the expression of various CYPs (e.g., CYP 2B10) and is primarily associated with chemical metabolism (including regulation of cholesterol and bile acid metabolism). For example, phenobarbital (PB),a non-genotoxic indirect CAR activator, induces cytochrome P450 (CYP) and other xenobiotic metabolizing enzymes and is known to produce liver foci/tumors in mice and rats. Epidermal growth factor receptor (EGFR) has been identified as a PB-responsive receptor, and PB activates CAR by inhibiting EGFR signaling (Kobayashi et al., 2015). One of the key events in the mechanism of the PB-induced liver tumors may be activation of CAR and subsequent changes in gene expression that increase cell proliferation, formation of hepatic foci and ultimately the development of liver tumors. Other events, such as epigenetic changes, induction of hepatic CYP2B enzymes, liver hypertrophy and decreased apoptosis and inhibition of gap junctional intercellular communication, are also associated with the MOA (Elcombe et al., 2014). However, the relevance of this MOA for humans is unclear (Elcombe et al., 2014, Qatanani and Moore, 2005). We placed CAR activators in the decision tree for completeness but the results may not be applicable for human risk assessment. Ten of 14 barbiturate derivatives in this dataset are hepatotoxic; the other 4 have no data. As shown in (3a-1) in Fig. 4, the R group can be phenyl, isopropyl, isopentyl, pentan-2-yl, sec-butyl, allyl or cyclohexenyl. The R1 group can be ethyl or allyl and R2 can be hydrogen, methyl or ethyl. The literature also indicated that some barbiturates blocked NADH oxidation, interrupting mitochondrial electron transfer (Chan et al., 2005).
Example 4
PPARα activation and peroxisome proliferator (PP) chemicals (section 3.2.3. (a), (c), and (d) in Appendix II).
The peroxisome proliferator-activated receptors (PPARs) are composed of three family members: PPARα, PPARδ and PPARγ. PPARs function as transcription factors regulating the expression of specific genes and play essential roles in the regulation of cellular differentiation, development, metabolism, as well as tumorigenesis (Michalik et al., 2006, Belfiore et al., 2009). PPARα is highly expressed in the liver and its function is to induce hepatic peroxisomal fatty acid oxidation during periods of fasting. In general, peroxisome proliferators induce the synthesis of peroxisomes in the liver and induce neoplastic lesions as well as cause liver cancer following chronic, high exposures, along with liver enlargement, elevated serum transaminase levels, hepatocellular hypertrophy, hyperplasia and changes in apoptosis (Corton et al., 2014, Gonzalez, 2002, Cohen and Grasso, 1981). The MOA of PPARα appears to be species dependent. Rats and mice are most sensitive to PPARα-induced toxicity while other species, such as humans, primates and hamsters are much less sensitive (Corton et al., 2014). Even though a variety of chemical classes may be involved in activating PPARα, these compounds (both the parents and their ester hydrolysis/oxidation metabolites) contain two key structural features: a. they all have a hydrophilic group (e.g. a carboxylic acid) and a nonpolar moiety (aryl or aryloxy ring, alkyl chain etc.) connected to the carboxylic acid; b. most of the compounds have substituents (e.g. chlorine, fluorine vs hydrogen or branched alkyl vs non-branched alkyl) which could resist metabolic detoxification.
a. Phthalate derivatives.
The first class of PPARα activators included in our data set is represented by phthalate derivatives as shown in (4a-1) in Fig. 6. Many phthalate diesters are metabolized to active species by esterases. These esterases cleave one of the two side chains from the parent diester phthalate producing an active monoester phthalate which increases hepatocyte peroxisome and cellular proliferation and leads to predictable adaptations in the liver consisting of hepatocellular hypertrophy and hyperplasia (Lock et al., 1989) but after chronic exposure may lead to liver tumors in male and female mice and rats (Klaunig et al., 2003). As shown in (4a-1) in Fig. 5, the R and R1 groups can be C4-C12 nonbranched/branched alkyl, allyl, ethoxylated alkyl, benzyl and cyclic ring groups. The R and R1 groups can be the same or different. The chemicals are normally inactive when the R and R1 groups are methyl or ethyl. In addition to activating PPARα, the hepatotoxicity of some phthalates may involve other factors. For example, literature reports indicated that the generation of allyl alcohol (AA) or acrolein (the active metabolite of AA, (Eigenberg et al., 1986) from diallyl phthalate could partially contribute to its hepatotoxicity. Di-(2-ethylhexyl) phthalate (DEHP) induces apoptosis in hepatocytes via the activation of the ERK/NF-κB signaling pathway, in which calcium ions and hydrogen peroxide act as the pivotal mediators of the apoptotic signaling (Ghosh et al., 2010). However, there are data generated in PPARα KO mice where DEHP exposure elicits hepatocellular tumors, suggesting that another mechanism is responsible (Ito et al., 2007).Fig. 6
Fig. 6.

The scope of structural features of phthalate derivatives.
Fig. 5.

The scope of structural features of phenobarbital -like chemicals.
b. Nitro diphenyl ether derivatives.
Several structurally related nitro diphenyl ether pesticides such as fomesafen, bifenox, nitrofen, lactofen, acifluorfen and oxyfluorfen are hepatocarcinogenic in rodents. These chemicals have a nitro substituted diphenyl ether core (shown in (4b-1) and (4b-2) in Fig. 7). The R group can be hydrogen, small alkoxy and acid or precursors of the acid. The R1 group is normally chlorine while the R2 group is normally chlorine or trifluoromethyl. In addition to the nitrophenyl toxicophore, the hepatocarcinogenesis of these chemicals may be partially associated with PPARα-mediated peroxisome proliferation. There is evidence that the induction of liver tumors by lactofen requires PPARα activation (Williams 1997). SAR studies indicate that an acidic functional group (e.g., carboxylic, sulfonic) either in the parent compound or a metabolite play a major role for most peroxisome proliferators (Woo and Lai, 2003). For the nitrofen and oxyfluorfen shown in (4 g-2), hepatocarcinogenic potential may involve the nitrophenyl moiety directly.
Fig. 7.

The scope of structural features of nitro diphenyl ether derivatives.
c. Perfluorooctanoate (PFOA) derivatives.
Perfluorooctanoate (PFOA) exposure results in peroxisome proliferation and benign liver tumors in rats, events associated with activation of PPARα (Vanden Heuvel et al., 2006). The core structural features for PFOA and related chemicals are indicated in (4c-1) in Fig. 8 where R is OH, F (or another halogen), or OR1. R1 are alkyl groups with 1–4 carbons. SAR study indicated that there is an existence of “active cliff” of chain length which can lead to a large in biological response. It is believed that the alkyl chain length can range from 8 to 14 carbons (n = 4–10) and a chain length greater than seven carbons is required to induce peroxisomal enzyme activity (Goecke-Flora and Reo, 1996). It is also believed that activity of perfluorooctanoic acid (PFOA) is partially associated with resistance of metabolism because of the strength of the C-F bonds.
Fig. 8.

The scope of structural features of perfluorooctanoate (PFOA) derivatives.
3.2.2. Examples of chemical categories associated with reactive chemicals and metabolites
In these categories, we classified chemicals based on possible covalent bond formation by electrophilic or radical reactions between the chemicalsor their metabolites (electrophiles) and protein/DNA (nucleophiles). The RMs that generate idiosyncratic drug toxicity were comprehensively reviewed by Stepan et al. (Stepan et al., 2011). CYP3A4 is the predominant isoform of P450 in liver. CYP3A4 metabolizes many chemicals commonly used by humans and is also responsible for metabolic activation of chemicals resulting in liver injury (Mizuno et al., 2009). Many chemicals have the potential to generate RMs. The subsequent molecular damage can cause hepatotoxicity through a number of downstream events. Given the complexity of metabolism for each chemical, attempting to develop specific rules for hepatotoxicity via specific RMs is outside the scope of this paper. We have tried to cast a wide net with the design of category rules such that any chemical in the broad group with the potential to form RMs will be flagged. This is a crucial aspect for supporting SAR read-across to increase relevance and reliability. It is also important to keep in mind that using RMs and/or structural alerts as standalone predictors of hepatotoxicity may overpredict toxicity potential and their evaluation requires expert judgment and potentially some experimental work for verification.
Example 5
p-, o-quinone-imines, p-, o-aromatic-diimines and precursors (section 3.3.1. (RM-1) in Appendix II).
a. quinone-imine and aromatic-diimine-related compounds (see section 3.3.1. (a) in Appendix II).
The formation of quinone-imine and aromatic diimine RMs has been implicated as an important factors in hepatotoxicity for many chemicals. It is believed that quinone-imine and aromatic diimine metabolites can bind covalently to macromolecules to cause cell damage or trigger an immune response leading to cell death (Wen and Moore, 2011, Kamimura et al., 2015). We have identified several different core structural fragments in our collected chemicals, as shown in (5a-1) to (5a-8) in Fig. 9, which could potentially form a quinone-imine or aromatic-diimine reactive metabolite. The key structural pattern of these chemicals is that the p-, or o-position of amine/substituted amine groups have either no substituent or substituents with the metabolic potential to be a hydroxyl or amino group. For (5a-3), X can be hydrogen, hydroxyl, nitro, methoxy, diazo or fluoro/chloro groups; Y can be hydrogen, alkyl, aryl, heteroaryl, acetyl groups; R1 can be hydrogen, alkyl or aryl groups. Most of the p- and o-hydroxyl aryl amines and N-acetyl/N-aryl substituted aryl amines have been reported to form corresponding quinone-imine/protein adducts. The metabolic conversion of p-, or o-alkoxyl and p-, o-C-hydroxylation, as well as cleavage of diazo bond of aryl amines and N-acetyl/N-aryl substituted aryl amines or reduction of nitro group, will form similar adducts. In addition, the chemicals having a p-halogen (e.g. F, Cl) with an electron withdrawing group (e.g. CF3) at the o-position of may be converted to the corresponding p-hydroxyl metabolite and then form adducts. These metabolic reactions are P450-mediated, with generation of p-, o- hydroxyl or p-, o- amino aryl amines and N-acetyl/N-aryl substituted aryl amines as the initial step. Both the p-, and o- quinone-imine and p-, and o-aromatic-diimine are reactive metabolites which are capable of binding macromolecules (e.g. proteins and DNA). However, the formation of p- and o-aromatic hydroxyl derivatives from corresponding aromatic alkoxyl precursors need case by case analysis.
Fig. 9.

p-, o-quinone-imines, p-, o-aromatic-diimines and precursors.
In the case of (5a-4), R group can be aryl, alkyl substituents and n can be 5, 6 or even 7 membered rings. For (5a-5), the Y group can be a hydrogen, hydroxyl, amino or methoxy; the R group can be hydrogen, amino, aryl or alkyl; the X group is sulfur, the hetero aryl ring size is 5 (n = 0); however, when X is carbon or nitrogen, the hetero aryl ring size can be 5–6 (n = 0, 1). In the case of (5a-6), the Y group can be a hydrogen, hydroxyl, amino or methoxy; R, R1 and R2 can be hydrogen and alkyl respectively. In the case of (5a-7), the Y group can be a hydrogen, hydroxyl, amino or methoxy; the substituent R can be chlorine or other substituents; R1 can be alkyl amine substituents; the X can be NH, S and n can be 0, or 1. In the case of (5a-8), R may be hydrogen, chlorine or small alkyl while R1 may be piperazine or 4-substituted piperazine (substituents can be a small alkyl (C1-C3) group or alkyl substituted ethylene glycol chain); X can be N or O. The general mechanisms to generate the corresponding reactive metabolites (RM-1a to RM-1r) via p-,o-quinone-imines, p-,o-aromatic-diimines formation of the representative chemicals are shown in Fig. 10 (Stepan et al., 2011, MacAllister et al., 2013, Calder et al., 1981, Madsen et al., 2008, Jamieson et al., 2011, Li et al., 2009a, Teo et al., 2015, Walker et al., 2008, Srivastava et al., 2014).
Fig. 10.

The formation of quinone-imine and aromatic-diimine RMs.
b. Benzimidazole related derivatives (see section 3.3.1. (d) in Appendix II).
The benzimidazole fungicides have been reported to disrupt cell division and cause hepatotoxicity. They may induce oxidative stress or hypersensitivity via RM formation and result in hepatotoxicity to cause clinical liver injury. Structurally, two imidazole nitrogen atoms in the benzimidazole may contribute to increase electron density in the phenyl ringand lead to oxidation to form reactive p- quinone-imine reactive metabolites as shown in (RM-1 s or RM-1 t) in Fig. 11. As indicated in (5b-1) and (5b-2) in Fig. 11, the core structural alert for the majority of the assembled compounds is the amino benzimidazole where substituents R on the benzimidazole ring can be hydrogen, or a phenylthio-, propylthio-, phenylsulfinyl-, benzoyl-, fluorobenzoyl-, thiophene-2-carbonyl- or thiazol-4-yl group. The R1 group of these chemicals can be hydrogen, N-butylcarbamoyl or benzyl but majority is hydrogen. R2 and R3 groups in these structures can be hydrogen, alkyl or R2 is a hydrogen while R3 is ester moiety. The simple analog of (5b-1) is albendazole which contains a benzimidazole core structure feature. Albendazole caused hepatotoxicity, testicular toxicity and activation of the immune system (Committee for veterinary medicinal products: Albendazole Sulpoxide summary (1) The European Agency for the Evaluation of Medicinal Product EMEA/MRL/094/96-final June 1996). Limited data indicate that replacement of the -N(R2R3) group in (5b-1) with a thiazol-4-yl group or alkylthio group on the benzimidazole ring (e.g. (5b-2)) and the metabolic precursors of (5b-1) such as (5c), (5d) and (5e) may show hepatotoxic activity.
Fig. 11.


Benzimidazole related derivatives and RM formation.
Another class of benzimidazole derivatives (5b-3) shown in Fig. 11 which have core structural feature of benzimidazol-2-ylthioacetylpiperazine exhibited hepatotoxicity comparable to the benzimidazole derived carbamate (e.g. albendazole) (Mavrova et al., 2006). For these chemicals, the substituent R can be hydrogen, methyl or nitro groups. The R1 can be di-phenylmethyl, methylphenyl, or nitrophenyl groups. The role of substituent R for the pattern of hepatotoxic effects is not clear to date. However, most derivatives possess hepatotoxicity comparable or less than that of albendazole.
Example 6
p-, o-quinone, catechol, p-diphenol and precursors (section 3.3.2. (RM-2) in Appendix II).
a. quinone, catechol, p-diphenol related compounds (see section 3.3.2. (a) in Appendix II).
RMs of p-, o-quinone also generate hepatotoxicity (Leeming et al., 2015). The catechol or p-dihydroxyphenol moiety can be metabolized to semiquinone radicals or p-, o-quinone RMs (Walgren et al., 2005). In addition, o-quinones undergo nonenzymatic redox cycling with the concomitant production of ROS which can in turn induce the oxidative stress (Penning et al., 1999). Many catechol or p-dihydroxyphenol precursors, such as o-methylendioxyphenols, p-, o-hydroxyl methoxyphenols and p-, o-dimethoxybenzenes, can undergo metabolic transformation to form catechol or p-dihydroxyphenol and then be converted to quinone RMs. The general core moieties are shown in (6a-1) to (6a-6) and representative examples of reactive metabolites (RM-2a to RM-2 k) in Fig. 12. In the cases of (6a-3) and (6a-4), X and Y are a hydroxy or precursor of hydroxy group and in (6a-5), X, Y and Z can all be a hydroxy or precursors of hydroxy group. Most chemicals with these features are converted to the corresponding catechol or p-dihydroxyphenol. For example, most o-methylendioxyphenols can undergo O-demethylenenation to form the corresponding catechol. Catechol or p-dihydroxyphenol may also undergo o-, p-C-hydroxylation of the phenolic ring and methoxyphenyl or alkoxyphenyl moiety of the compounds (e.g. in (6a-3 to 6a-5)). One of many examples is that hepatotoxicity of Atomoxetine (ATX) may be induced by a RM, p-toluquinone (RM-2j), which is mediated via hydroxylation and O-dealkylation/oxidation pathways (You et al., 2021). Furthermore, converting the benzofuran ring (e.g. in (6a-6)) to corresponding o-quinones via hydroxylation/oxidation pathways has been proposed to cause liver damage (McDonald and Rettie, 2007). For example, amiodarone, benzarone, benzbromarone and benzidarone, which contain a 3-benzoyl-1-benzofuran moiety, induce hepatotoxicity by generating o-quinones RMs (e.g. RM-2 h). Some other categorized chemicals which could undergo the o-, p-quinone intermediate are shown in Fig. 12 (Dietz and Bolton, 2007, Lambert et al., 2002, Mete et al., 2012, Ratziu et al., 1991, Carvalho et al., 1997, Tasaki et al., 2013, Hildebrand et al., 2010, McDonald and Rettie, 2007, Yu et al., 2004). The 14-membered phyto-macrocyclic lactones, zearalanone and zearalanol derivatives, caused liver effects including decrease in the levels of alkaline phosphatase, lactate dehydrogenase, alanine and aspartate aminotransferases to severe histological changes in the liver. The quinone RM (RM-2 k) formation may be responsible for increasing lipid peroxidation and inducing free radical production as well as forming DNA adducts in liver (Salah-Abbès et al., 2009, Zinedine et al., 2007, Creppy, 2002).
Fig. 12.


p-, o-quinone, catechol, p-diphenol RMs formation and precursors.
b. Anthraquinone and macrocyclic amide antibiotics (e.g. rifamycin S, rifabutin) related compounds (see section 3.3.2. (c) and (d) in Appendix II).
The anthraquinone derived tetracyclic compounds, such as anthracyclines (6b-1) in Fig. 13 (e.g. doxorubicin, epirubicin and idarubicin), show hepatotoxic effects. They are cancer chemotherapeutics that act by forming complexes between topoisomerase II and DNA, preventing rejoining of DNA strands (Tsao and Stewart, 2009). This group of molecules is planar, and some have been shown to intercalate into DNA, alter membrane function, and form free radicals causing direct damage to the hepatocytes. Doxorubicin, an anthracycline derivative, has been reported to cause hepatotoxicity through the formation of a semiquinone free radical (RM-2 l) via single-electron transfer process. This suggests that free radicals produced during the metabolic activation of doxorubicin-derived quinone–semiquinone are responsible for hepatotoxic effects (Kalender et al., 2005). It also could be associated with enzymatic biotransformation of the amine moiety of desosamine to a reactive nitroso species (will be further discussed in example 9 (b) for RM-5b formation) (Pessayre et al., 1985). The majority of anthracyclines in the dataset contain a hydroxyl R group. However, one compound has a methoxy R group with remaining substituents listed in Fig. 13. Other anthraquinones, such as hydroxyl, amine, nitro and other p substituted anthraquinone derivatives (as shown in 6b-2) also show hepatotoxic potential. These chemicals may be associated with the formation of RM-2 m and then induce superoxide radical anion (O2–) as the beginning of a cascade that generates hydrogen peroxide (H2O2) and hydroxyl radical (HO.) to initiate lipid peroxidation and be toxic to mitochondria. The chemicals with hydroxy or carboxyl groups at the beta position of the anthraquinone ring also show uncoupling activity and inhibitory effects on mitochondrial respiration (Vinken et al., 2012, Bironait and Ollingerbj, 1997, Kawal et al., 1986, Kågedal et al., 1999). The core structural alert for these chemicals is anthraquinone and the substituents R and R1 on the anthraquinone ring can be hydroxyl, amine/alkyl amines, nitro, carbolic acid, alkyl or halogen groups.
Fig. 13.


Tetracycline, anthraquinone derivatives and semiquinone free radical RMs formation.
Similar to the anthracyclines, the macrocyclic amide antibiotics, such as rifabutin (6c), rifamycin (6d), rifampin and rifapentine, have been associated with hepatotoxicity (Rao and Cederbaum, 1996, Nakajima et al., 2011). The MOA for hepatotoxicity by these rifamycins may be the formation of reactive oxygen species during redox-cycling (e.g. quinonimine (RM-2n) for rifabutin and quinone (RM-2o) for rifamycin in Fig. 14) with the subsequent production of hydroxyl radicals when iron complexes are present (Rao and Cederbaum, 1996). As shown in (6c) in Fig. 14, the core structural feature is a 25-membered macrocyclic amide.
Example 7
p-, o-Quinone, imine methides formation (see section 3.3.3. (RM-3) in Appendix II).
Fig. 14.

Rifamycin S, rifabutin related antibiotics and quinonimine, quinone RM formation.
Phenolic compounds (7a-1, Fig. 15) containing a p-alkyl substituent with at least one benzylic hydrogen can be oxidized in experimental animals to p-quinone methides (RM-3a) (DeVito, 1996, Sharma et al., 2012, Minet et al., 2012). p-Quinone methides are electrophilic, with positive charge density centered mainly on the exocyclic methylene carbon. The methylene carbon is conjugated to carbonyl moiety and is characterized by Michael addition of cellular nucleophiles to form benzylic adducts. p-Quinone methides have also been described as resonance-stabilized carbocations, which are capable of formation of covalent bonds with cellular nucleophiles to initiate a variety of cytotoxic and/or genotoxic responses. Therefore, many p-alkylphenols produce hepatotoxicity or lung toxicity and promote tumor formation. Although the enhanced reactivity of the quinone methide intermediate (e.g. electron-withdrawing groups on the ring) and rate of formation of the quinone methide may play a role, the stability of the quinone methide metabolite is a major determinant of the toxicity of alkylphenols (Thompson et al., 1995a). Normally, the stability of quinone methides increases with increasing substitution of electron-donating groups on the aromatic ring as well as with increasing length of the alkyl moiety (Thompson et al., 1996b). For example, increasing the length or branching of the p-alkyl substituent (e.g. from p-methyl to p-ethyl and to p-isopropyl) increases the rate of quinone methide formation and hepatotoxicity (Thompson et al., 1995b). The reactive o-quinone methides (RM-3b) in Fig. 15 are known to form from o-alkyl-phenols. However, reduction by quinone reductases has been shown to occur more readily than for the para isomer (Kucera et al., 2013) which indicates that the o-quinone methides are less active than the corresponding p-quinone methides. Some examples of active p-quinone methides are listed in (RM-3a) to (RM-3 g) in Fig. 15.
Fig. 15.


p-Quinone methides formation and examples.
The iminoquinone methides are also toxic (Damsten et al., 2008, Stepan et al., 2011). For example, it has been reported that indole derivatives, such as 3-methyl indole and related derivatives may be metabolized to the corresponding Michael acceptors (e.g. RM-3 h) and react with nucleophilic sites of DNA and proteins to form Michael adducts (Fig. 15) (Kalgutkar et al., 2005, Regal et al., 2001, Kassahun et al., 2005).
Example 8
Michael acceptors and precursors (section 3.3.4. (RM-4) in Appendix II).
Michael acceptors share structural features of unsaturated C = C moieties directly connected to an electron withdrawing group such as carbonyl (aldehyde, ketone), ester, amide, nitrile, nitro and sulfonyl groups. These chemicals contain an activated alpha–beta unsaturated carbon–carbon bond and the electron-deficient beta-carbon atom can function as an electrophile to react with biological nucleophiles (DNA, proteins). These chemicals include alpha–beta-unsaturated carbonyls and their precursors.
a. Vinyl amide, aldehyde and ester derivatives and some precursors (see section 3.3.4. (a) in Appendix II).
These chemicals belong to the group of vinyl amides, aldehydes and esters represented by the general core structure (8a-1), (8a-2) and (8a-3) shown in Fig. 16. The double bonds in these compounds can be unsubstituted or substituted by small alkyl groups (C1-C2, e.g. R = Me or Et), and the electron withdrawing groups attached to the double bond can be amide, N-alkyl amide, aldehyde, or ester groups (e.g. -CONHR1, (R1 = H, alkyl (C1-C3), OH, –CH2-NHCOCH = CH2); -COOR2, (R2 = alkyl (C1-C10); COH). Although the mechanism of hepatotoxicity is not clear, it has been proposed that one of the mechanisms is adduction of macromolecules and proteins by Michael addition (Shearn et al., 2014, Chung et al., 2012).
Fig. 16.

Vinyl amide, aldehyde and ester derivatives and active Michael acceptors (RM-4a) to (RM-4d) formation.
Additionally, allyl acetate and some allyl alcohol esters (allyl acetate, allyl cinnamate, allyl phenylacetate) are suggested as prohepatotoxicants due to formation of highly reactive acrolein once metabolized (RM-4a) (Amada et al., 2013, Silver and Murphy, 1978, Auerbach et al., 2008). In contrast, felbamate (CAS# 25451–15-4), valproic acid (CAS# 99–66-1) and terbinafine (CAS# 91161–71-6), have core structural features that do not belong to this class but have demonstrated hepatotoxic effects in animals or humans. The literature indicates that these chemicals share a common metabolic pathway forming Michael acceptors (RM-4b) to (RM-4d) (Iverson and Uetrecht, 2001, Thompson et al., 1996a). Other investigators (Pandit et al., 2012; Fromenty and Pessayre, 1995) conclude that the hepatotoxicity caused by valproic acid (VPA) may be due to interference with the β–oxidation of endogenous lipids, and the formation of an ester conjugate with carnitine that leads to secondary carnitine deficiency. Several lines of indirect evidence and in vitro studies indicate that the thioester derivative (RM-4a) of VPA and coenzyme A may exist as a metabolic intermediate in liver tissue. Depletion of coenzyme A or the VPA CoA ester itself may inhibit mitochondrial metabolism, which can lead to cell death.
b. Thiophene derivatives (see section 3.3.4. (b) in Appendix II).
Several compounds in this class cause hepatotoxicity. The thiophene ring (as shown in (8b-1) to (8b-3) in Fig. 17) appears to be converted to electrophilic intermediates ((RM-4e) to (RM-4 h) in Scheme 17), via cytochrome P450-mediated bioactivation (Shimizu et al., 2011). The proposed mechanism for the induction of hepatotoxicity involves the formation of S-oxide metabolites, which react rapidly with various nucleophiles by a Michael-type addition resulting in covalent binding to proteins at the thiophene ring (Silverman, 2004, Valadon et al., 1996). In addition to the sulfoxidation pathway, thiophene can also generate a reactive metabolite via an epoxide pathway (O'Donnell et al., 2003). A variety of substituents, such as acetyl, alkyl, amine, fused cyclic, aromatic ring, on the thiophene may generate S-oxide metabolites via a S-oxidation pathway. Interestingly, from these identified chemicals, duloxetine (CAS# 116539–59-4) has a naphthalene, rather than a thiophene moiety, as the preferred site of bioactivation (RM-4i) (Chan et al., 2011). The SAR of substitution at each position of the thiophene ring has not been studied. Steric and/or electronic effects may reduce the affinity towards the P450 enzyme that metabolizes the unsubstituted thiophene. A computational approach may be helpful to identify this reactive metabolite (Dang et al., 2017).
Example 9
Nitroso formation from aromatic and aliphatic amines (section 3.3.11. (RM-11) in Appendix II).
Fig. 17.

Thiophene derivatives and thiophene RMs (RM-4e) to (RM-4i) formation.
a. p-aminophenyl substituted sulfonamides (see section 3.3.11. (a) in Appendix II).
The core structures of p-aminophenyl substituted sulfonamides are shown in (9a-1), (9b) in Fig. 18. These chemicals contain a variety of aromatic or heteroaromatic sulfonyl moieties. For the 4-amino (or amino precursor) substituted phenylsulfonamide derivatives (9a-1), most of the N-substituents are heteroaryl groups (e.g. pyrimidinyl, pyridazinyl, isoxazolyl, thiazolyl or pyridinyl group) with some small alkyl or alkoxy groups (e.g. methyl, methoxy) on the nitrogen which are associated with the hepatotoxicity. These chemicals cause a characteristic idiosyncratic liver injury through drug allergy or hypersensitivity (Khalili et al., 2011). Some of these chemicals can also cause mild and transient ALT elevations. One proposed mechanism of inducing allergy or hypersensitivity involves the metabolism of aromatic amines, generating toxic reactive or antigenic metabolites. For example, the aromatic amine moiety may undergo hydroxylation, or be metabolized through oxidation to form the nitroso derivative (RM-5a) in Fig. 18 (Kalgutkar et al., 2010) that could form protein adducts. All the hepatotoxic chemicals shown in (9a-1) in Fig. 18, have R groups with hydrogen, acetyl, benzoyl, or heteroaryl groups such as pyrimidinyl, methyl- or small alkyl or methoxy pyrimidinyl, pyridazinyl, isoxazolyl or thiazoyl. Additionally, one class of proteinomimetic protease inhibitor, exemplified by amprenavir (9b), has an aromatic sulfonamide moiety. Amprenavir induced hepatotoxicity (Chang and Schiano, 2007, Pandit et al., 2012) but the mechanism is unclear..
Fig. 18.

p-aminophenyl substituted sulfonamides and reactive metabolites (RM-5a) formation.
b. Nitroso formation from alphatic amines in phyto-macrocyclic lactones (see section 3.3.11. (c) in Appendix II).
The group of 14-membered macrocyclic lactones with ketone group at the 10 position of the ring (9c-1) and 15-membered macrocyclic lactones with a nitrogen atom in the ring (9d) include the antibacterial agents erythromycin, clarithromycin, telithromycin and azithromycin (Fig. 19). They are reported to cause cholestatic hepatitis, fulminant liver failure and acute toxic cholestatic reaction and severe hepatotoxicity although these effects are rare (LiverTox; Braun, 1969, McCormack et al., 1977, Johnson and Hall, 1961, Gaeta et al., 1985). The different substituents of these compounds are listed in Fig. 19. Structurally, this class of chemicals share in common a single desosamine unit which is connected to a central macrocyclic lactone core. One of proposed mechanisms of causing idiosyncratic hepatotoxicity may be associated with enzymatic biotransformation of the amine moiety of desosamine to a reactive nitroso species (RM-5b) which is capable of interacting with proteins at thiol-incorporating residue (Pessayre et al., 1985, Moseley, 2013).
Fig. 19.

Erythromycin, azithromycin related derivatives and RM (RM-11c) formation.
3.3. Application of hepatotoxicity decision tree
It is important to point out that the hepatotoxicity decision tree is not intended to be used as a standalone tool. It can be used for several purposes: a. to evaluate the potential hepatotoxicity of new chemicals as one piece of a weight of evidence assessment for SAR read-across to fill data gaps; b. to screen chemicals to determine whether they fall into structural categories with known hepatotoxicity effects induced via possible receptor binding/or bioactivation. c. as part of chemoinformatic efforts to map and build common structural alerts for hepatotoxicity.
3.3.1. Screening chemicals with potential hepatotoxicity effects induced by receptor binding/or bioactivation
We demonstrate the use of the hepatotoxicity decision tree with six chemicals (Fig. 20). Detailed descriptions of the groups/sub-groups in the decision tree are available in Appendix II.
Fig. 20.

Flow diagram illustration of 6 examples taken through out the simplified decision tree (detailed description in Appendix II).
Chemical 1 (CAS# 58–18-4) is identified as having features in common with known precedents for hepatotoxicity via 4 steps (1, 2, 3, 4) to androgen receptor (AR) binding compounds (category 1).
Chemical 2 (CAS# 51–66-1) and Chemical 5 (CAS# 55142–85-3) are identified as having features in common with known precedents for hepatotoxicity via 6 steps (1, 2, 3, 4, 5, 6) to p-, o-quinone-imine formation (category 5) and Michael acceptor formation (category 8), respectively.
Chemical 3 (CAS# 60–56-0) and Chemical 4 (CAS# 3847–29-8) are identified as having features in common with known precedents for hepatotoxicity via 9 steps (1, 2, 3, 4, 5, 6, 7, 8, 9) to S-S bond formation (category 14) and Nitroso formation from aromatic and aliphatic amines (category 15), respectively.
Chemical 6 (CAS# 295–17-0) is identified as having features in common with chemicals that do not have known hepatotoxic potential via 10 steps (1, 2, 3, 4, 5, 6, 7, 8, 9,10) to “No known precedent for hepatotoxic potential”. This chemical would require further evaluation by experts.
3.3.2. Screening chemicals with structural/bioactivation features similar to the chemicals with precedent hepatotoxicity effects to support SAR read-across.
One of the applications of the decision tree is to screen chemicals to see if they fit into a subcategory that has precedent receptor binding/or bioactivation induced hepatotoxicity. For example, although no direct hepatotoxicity data have been identified for several 4-amino (or amino precursor) substituted phenylsulfonamide derivatives (10a), (10b), (10c), and (10d), these chemicals share similar core structural features, bioreactivity,or possible major reactive metabolites and physicochemical properties with a class of hepatotoxicants (9a-1) (Fig. 21). These chemicals belong to the same chemical subcategory and can be flagged as having hepatotoxic potential through SAR read-across. Chemical (10d) is a diazo-derived aryl sulfonamide which has core structural features outside of the core structural coverage of this subcategory. However, it may undergo diazo bond cleavage to generate amino aryl substituted sulfonamide that could converge to RM similar to that of (9a-1). Therefore, it is expected that chemical (10d) would have similar hepatotoxicity potential as the other chemicals in this group.
Fig. 21.

4-amino substituted phenylsulfonamide analogs that would be identified by the decision tree as having hepatotoxic potential.
Anabolic steroids provide another example. SAR evaluation of the anabolic steroids indicated that the 17β-hydroxyl group appears to be crucial for interaction with the receptor and a 17α-alkyl group can diminish first-pass metabolism and increase liver toxicity. Several anabolic steroids (1a-1–1 to 1a-1–5 and 11a-1, 11b) as shown in Fig. 22 share similar core structural features. The only differences for these anabolic steroids are substituents around the core steroid structure; thus, these chemicals are expected to be in the same category. However, chemicals (11a-1 and 11b) have acetyl and alkyl ester substitutions at the key C17β-hydroxy position which would block the 17β-hydroxyl and alter the interaction with androgen receptors, greatly reducing or abolishing hepatotoxicity potential. Therefore, chemicals (1a-1–1 to 1a-1–5) are not suitable to SAR read-across for chemicals (11a-1 and 11b) despite their similar core structural features.
Fig. 22.

Evaluation of common structural features/MoA of androgenic steroid to improve SAR read-across.
We also evaluated the performance of the decision tree for a set of fifteen chemicals that were not used in its construction (Table 2) but have been reported to be hepatotoxic (Rikans, 1987, Mulliner et al., 2016, Wu et al., 2017, Thakkar et al., 2020, Kalgutkar, 2020, Liu et al., 2020; Ding et al., 2022, Li et al., 2022, Lim et al., 2022, Zhang et al., 2022, Chen, 2023). All fifteen could be categorized as having precedent structures in the decision tree, including epoxides/epoxide precursors (category 12); o,p-quinone-imines and diimines (category 5); o,p-quinones, catechols and p-diphenols (category 6); αβ-unsaturated amides, aldehydes, ketones, esters (category 8); and arylacetic acids (category 13). One of the chemicals could be considered to belong to two categories.
Table 2.
Chemicals used to determine performance of the decision tree.
| Name | CAS Number | Decision tree category |
|---|---|---|
| furan | 110–00-9 | 12 |
| perampanel | 380917–97-5 | 12 |
| marrubiin | 465–92-9 | 12 |
| nomilin | 1063–77-0 | 12 |
| rutaevin | 33237–37-5 | 12 |
| 2-aminothiazole | 96-50-4 | 12 |
| benzamide,2-chloro-5-[[(2,2-dimethyl-1-oxopropyl)amino]methyl]-N-1H-imidazol-2-yl- |
1381846–21-4 | 12 |
| aniline | 62–53-3 | 5 |
| cyamemazine | 3546-03-0 | 5 |
| frovatriptan | 158747-02-5 | 5 |
| tadalafil | 171596-29-5 | 5 |
| yohimbine | 146-48-5 | 5 and 6 |
| 1-propanone, 1-(2-hydroxyphenyl)-3-phenyl- | 3516–95-8 | 6 |
| acrolein | 107–02-8 | 8 |
| ((3S)-6-((3-(4-(3-methanesulfonylpropoxy)-2,6-dimethylphenyl]phenyl}methoxy)-2,3-dihydro-1-benzofuran-3-yl)acetic acid | 1000413–72-8 | 13 |
3.3.3. Screening chemical categories with both hepatotoxicity and DART potential
Many chemicals that reach systemic circulation are capable of eliciting toxic effects in multiple organs. It is not surprising that many hepatotoxic chemicals also cause DART effects because the embryo expresses many of the same receptors, and reactive chemicals are as disruptive to embryonic cells as much as to adult cells. By comparing chemical categories in the hepatotoxicity and DART decision trees (Wu et al., 2013), we determined that there is a great deal of overlap in chemical categories which exhibit both hepatotoxic and DART potential. This comparison provides a good opportunity to understand mechanistic connections between both endpoints.
An example is retinoic acid and acitretin-related retinoids (see section 3.2.4. in Appendix II) which have RAR and RXR receptor binding potential. Retinoids can induce hepatotoxicity through altering glycoprotein synthesis or gene expression and inducing non-specific damage to hepatocellular membranes (Hewitt et al., 2013). These retinoids are also capable of interfering with morphogenesis in the embryo to induce developmental toxicity (Degitz et al., 2000, Nau, 1993).
Another example is the azole fungicides category. These azole derivatives are considered to cause liver toxicity, such as hepatocellular steatosis by a mechanism involving PXR (Knebel et al., 2019). The decreased hepatic levels of retinoic acid caused by azole chemicals (Hester et al., 2012) may result in hepatotoxic effects. Furthermore, these azole fungicides also induce teratogenesis by inhibiting CYP26 which plays a crucial role in maintaining proper gradients/concentrations of retinoic acid in embryonic tissue (Marotta and Tiboni, 2010).
One of the advantages of categorizing chemicals as done in this paper is that it could be used both as a component of SAR read-across to identify structural alerts and as a component of a screening system to identify chemicals of potential concern. For example, we have identified several benzimidazoles and their precursors as shown in Fig. 23. Among these, 8 compounds ((5b-1–1) to (5b-1–7) were reported to have both hepatotoxicity and DART potential. Three precursors (5c), (5d), (5e) have DART potential but lack hepatotoxicity data and one compound (5b-1–8) has hepatotoxic data only. Compound (5b-1–8) which lacks DART data could be considered to have DART potential via category read-across based on DART potential of analogs (5b-1–1) to (5b-1–7)). On the other hand, compounds (5c), (5d) and (5e) could undergo metabolism to form compounds (5b-1–1), (5b-1–6) and (5b-1–5), respectively (Virkel et al., 2004, Cristofol et al., 1997, Klausz et al., 2015). Therefore, these three chemicals would be flagged as a concern of having hepatotoxicity potential. Therefore, the common core structural features of these chemicals would suggest that amino benzimidazole may be a structural alert for both hepatotoxicity and DART, even though it lacks an unambiguous mode of action. One possible mechanism is the formation of quinone-imine RM (RM-1u or RM-1v) via a potential bioactivation pathway (Srivastava et al., 2014) as shown in Fig. 11. The quinone-imine RM is capable of interacting with macromolecules to either cause direct cell damage or trigger an immune response leading to cell death.
Fig. 23.

Benzimidazoles with common structural alerts for hepatotoxicity and DART potential.
3.3.4. Screening common structural features in different hepatotoxic chemical categories
The hepatotoxicity of some cross-category chemicals may be enhanced by common structural features. Of the 12 categories of potential RMs, the most prevalent RMs appear to be the formation of p, o-quinone-imine, aryl di-imine and nitroso RMs (Fig. 24). The potential structural alerts linked to these RMs are associated with p, o-amino-phenol (and precursors), anilines and nitroaromatic compounds.Fig. 25.
Fig. 24.

RM categories related to hepatotoxicity.
Fig. 25.

Common structural features leading to quinone-imines.
In addition to these structural alerts, some common structural features across many different classes of hepatotoxic chemicals were related to quinone-imines formation.
1. Anilide moiety: Anilide related moieties are common structural features of multiple chemicals (greater than40 chemicals) across classes identified in this data set. Anilide moieties can undergo bioactivation pathways such as hydroxylation/oxidation to generate alkyl amide iminone RMs (e.g. RM-1 h, RM-1p).
2. Ar-NH-Ar moiety: Masubuchi Y et. al. studied the diphenylamine associated NSAID drugs (e.g. flufenamic acid, mefenamic acid, tolfenamic acid and glafenine) and proposed a “structure dependent” mechanism that diphenylamine containing compounds induce hepatotoxicity by the uncoupling of mitochondrial oxidative phosphorylation (Masubuchi et al., 1998). However, careful analysis of structural features of these NSAIDs indicates that it is also possible for some chemicals to form quinone-imine RMs (e.g. RM-1b, RM-1 g) via hydroxylation at the para position of the phenyl group to induce hepatotoxicity. Some tyrosine kinase inhibitors such as gefitinib, erlotinib, lapatinib etc. also can form p-quinone-imine which is sufficiently reactive to bind to the cysteine groups of proteins (Li et al., 2009b, Wen and Moore, 2011).
3. Phenothiazine moiety: More than 19 chemicals contain this structural feature and majority of them showed hepatotoxic potential. The two Ar groups of Ar-NH-Ar are connected by a S atom. Most induced cholestatic hepatic injury. It was found that phenothiazine (CAS# 92–84-2), which does not contain the substituents, maintains hepatotoxicity effects suggesting that the two aryl ring-fused thiomorpholine moiety of phenothiazine derivatives may be essential for hepatotoxicity. Some instances of liver injury may be caused by production of a reactive intermediate such as quinone-imines (RM-1d) via a bioactivation sequence involving P450-catalyzed oxidation (Wen and Zhou, 2009).
4. Limitations of the decision tree
The hepatotoxicity decision tree was constructed based on our understanding of structural features, bioactivation and modes of action (where available) of the different chemical categories. It is not intended to be used as a stand-alone tool. By design, it is expected and intended to broadly capture chemicals with receptor binding activity, bioactivation (RMs formation) that are similar to chemicals with precedent for hepatotoxic effects. Each step is formatted to allow expansion of chemical coverage, when sufficient related structures with liver toxicity data are available. A major limitation is the uncertainty in predicting reactive metabolites and inadequate relationship between the structural alerts in some cases. This could increase the difficulty in screening hepatotoxicants due to differences in biotransformation which in turn have different toxic profiles. One other limitation is that the boundaries (or cut-off values) for any given subcategory may be poorly defined, mostly due to the fact that the decision tree is based primarily on grouping chemicals that show toxicity, with fewer inactive chemicals included in the same subcategory. This limitation will be reduced in the future by iterative addition of positive and negative chemicals within each subcategory and associated refinement of the corresponding rules. The decision tree does not quantify the hepatotoxic potential of any chemical (e.g. dose–response relationship) since the chemical sub-categories are not built on quantitative data. This limitation can also be reduced by defining the hepatotoxic potency of each chemical within a subcategory.
5. Conclusions
Chemical induced hepatotoxicity (including liver carcinogenicity) occurs through different mechanisms and involves a variety of chemical classes and reactive metabolites. This complexity makes it very difficult to predict this endpoint by a single computational model. The current study provides an update on our understanding of hepatotoxicity of commonly used chemical classes. The hepatotoxicity decision tree is based on the compiled ∼ 1180 different chemicals and associated literature with possible modes of action of chemicals which are associated with receptor binding and chemical structural alerts/reactive metabolites to define chemical categories. The advantage of this approach is that it allows to quickly screen chemical categories with precedent hepatotoxicity associated with receptor binding/bioactivation in the literature. It can be used in the initial assessment of a chemical of interest with data gaps, to determine its potential to elicit hepatotoxicity, and at the same time to identify high quality suitable analogs with toxicity data usable in SAR read-across evaluations. Also, it can be used to integrate core chemical structural features/RMs formation and mechanistic explanations to improve the relevance, consistency and transparency of SAR read-across and as one part of new approaches method (NAM) to support SAR-based toxicological assessments or define testing needs based on an increased level of concern for the chemicals of interest..
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
We gratefully acknowledge Dr. Donald Bjerke for his valuable scientific comments and suggestions. We would also like to thank Andrea Larsonpeters, Tanya FitzGerald and Greg Dameron as well as the CREG group for support.
Funding
This study was part of internal research program of the Procter & Gamble Company and partially funded by a grant from the Cosmetics Europe Long-range Science Strategy.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crtox.2023.100108.
Contributor Information
Shengde Wu, Email: wu.s.3@pg.com.
George Daston, Email: daston.gp@pg.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
No data was used for the research described in the article.
References
- Amada T., Tanaka Y., Hasegawa R., Sakuratani Y., Yamada J., Kamata E., Ono A., Hirose A., Yamazoe Y., Mekenyan O., Hayashi M. A category approach to predicting the repeated-dose hepatotoxicity of allyl esters. Regul Toxicol. Pharmacol. 2013;65:189–195. doi: 10.1016/j.yrtph.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Auerbach S.S., Mahler J., Travlos G.S., Irwin R.D. A comparative 90-day toxicity study of allyl acetate, allyl alcohol and acrolein. Toxicology. 2008;253:79–88. doi: 10.1016/j.tox.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, K. L. (2001) Principles and Practice of Endocrinology and Metabolism, Ed by Becker, K. L. Lippincott Williams & Wilkins, 2001- Medical-2477 pages.
- Belfiore A., Genua M., Malaguarnera R. PPAR-gamma agonists and their effects on IGF-I receptor signaling: Implications for cancer. PPAR Res. 2009;2009:1–18. doi: 10.1155/2009/830501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bironait D., Ollingerbj K. The hepatotoxicity of rhein involves impairment of mitochondrial functions. Chemico-Biological Interactions. 1997;103:35–50. doi: 10.1016/s0009-2797(96)03747-7. [DOI] [PubMed] [Google Scholar]
- Blackburn K., Bjerke D., Daston G., Felter S., Mahony C., Naciff J., Robison S., Wu S. Case studies to test: A framework for using structural, reactivity, metabolic and physicochemical similarity to evaluate the suitability of analogs for SAR-based toxicological assessments Regul. Toxicol. Pharmacol. 2011;60:120–135. doi: 10.1016/j.yrtph.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Blackburn K., Daston G., Fisher F., Lester C., Naciff J.M., Stuard S.B. A strategy for safety assessment of chemicals with data gaps for developmental and/or reproductive toxicity. Regul. Toxicol. Pharmacol. 2015;72:202–215. doi: 10.1016/j.yrtph.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Braun P. Hepatotoxicity of erythromycin. J. Infect. Dis. 1969;119:300–306. doi: 10.1093/infdis/119.3.300. [DOI] [PubMed] [Google Scholar]
- Calder I.C., Hart S.J., Smail M.C., Tange J.D. Hepatotoxicity of phenacetin and paracetamol in the Gunn rat. Pathology. 1981;13:757–762. doi: 10.3109/00313028109086649. [DOI] [PubMed] [Google Scholar]
- Carvalho F., Remiao F., Soares M.E., Catarino R., Queiroz G., Bastos M.L. d-Amphetamine-induced hepatotoxicity: possible contribution of catecholamines and hyperthermia to the effect studied in isolated rat hepatocytes. Arch. Toxicol. 1997;74:429–436. doi: 10.1007/s002040050407. [DOI] [PubMed] [Google Scholar]
- Chan C.Y., New L.S., Ho H.K., Chan E.C.Y. Reversible time-dependent inhibition of cytochrome P450 enzymes by duloxetine and inertness of its thiophene ring towards bioactivation. Toxicol. Lett. 2011;206:314–324. doi: 10.1016/j.toxlet.2011.07.019. [DOI] [PubMed] [Google Scholar]
- Chan K., Truong D., Shangari N., O’Brien P.J. Drug-induced mitochondrial toxicity. Exp. Opin. Drug Metab. toxicol. 2005;1:655–669. doi: 10.1517/17425255.1.4.655. [DOI] [PubMed] [Google Scholar]
- Chang C., Schiano T. Review article: drug hepatotoxicity Aliment Pharmacol Ther. 2007;25:1135–1151. doi: 10.1111/j.1365-2036.2007.03307.x. [DOI] [PubMed] [Google Scholar]
- Chen H.J.C. Mass spectrometry analysis of DNA and protein adducts as biomarkers in human exposure to cigarette smoking: acrolein as an example. Chem. Res. Toxicol. 2023;36:132–140. doi: 10.1021/acs.chemrestox.2c00354. [DOI] [PubMed] [Google Scholar]
- Chen Y., Nie D. Pregnane X receptor and its potential role in drug resistance in cancer treatment. Recent Pat. Anticancer Drug Discov. 2009;4:19–27. doi: 10.2174/157489209787002498. [DOI] [PubMed] [Google Scholar]
- Chung F.L., Wu M.Y., Basudan A., Dyba M., Nath R.G. Regioselective formation of acrolein-derived cyclic 1, N-(2)-propanodeoxyguanosine adducts mediated by amino acids, proteins, and cell lysates. Chem. Res. Toxicol. 2012;25:1921–1928. doi: 10.1021/tx3002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A.G., Grasso P. Review of the hepatic response to hypolipidaemic drugs in rodents and assessment of its-toxicological significance to man. Food Chem. Toxicol. 1981;19:585–605. doi: 10.1016/0015-6264(81)90509-5. [DOI] [PubMed] [Google Scholar]
- Committee for veterinary medicinal products: Albendazole Sulpoxide summary (1) The European Agency for the Evaluation of Medicinal Product EMEA/MRL/094/96-final June 1996.
- Corton J.C., Cunningham M.L., Hummer B.T., Lau C., Meek B., Peters J.M., Popp J.A., Rhomberg L., Seed J., Klaunig J.E. Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPAR) as a case study. Crit. Rev. Toxicol. 2014;44:1–49. doi: 10.3109/10408444.2013.835784. [DOI] [PubMed] [Google Scholar]
- Creppy E.E. Update of survey, regulation and toxic effects of mycotoxins in. Eur. Toxicol. Lett. 2002;127:19–28. doi: 10.1016/s0378-4274(01)00479-9. [DOI] [PubMed] [Google Scholar]
- Cristofol C., Navarro M., Franquelo C., Valladares J.E., Carretero A., Ruberte J., Arboix M. Disposition of Netobimin, Albendazole, and Its Metabolites in the Pregnant Rat: Developmental Toxicity. Toxicol Appl Pharmacol. 1997;144:56–61. doi: 10.1006/taap.1997.8114. [DOI] [PubMed] [Google Scholar]
- Damsten M.C., de Vlieger J.S., Niessen W.M., Irth H., Vermeulen N.P., Commandeur J.N. Trimethoprim: novel reactive intermediates and bioactivation pathways by cytochrome p450s. Chem. Res. Toxicol. 2008;21:2181–2187. doi: 10.1021/tx8002593. [DOI] [PubMed] [Google Scholar]
- Dang N.L., Hughes T.B., Miller G.P., Swamidass S.J. Computational approach to structural alerts: furans, phenols, nitroaromatics, and thiophens Chem. Res. Toxicol. 2017;30:1046–1059. doi: 10.1021/acs.chemrestox.6b00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degitz S.J., Kosian P.A., Makynen E.A., Jensen K.M., Ankley G.T. Stage- and Species-Specific Developmental Toxicity of All-Trans Retinoic Acid in Four Native North American Ranids and Xenopus laevis. Toxicol Sci. 2000;57:264–274. doi: 10.1093/toxsci/57.2.264. [DOI] [PubMed] [Google Scholar]
- Delzenne N.M., Calderon P.B., Taper H.S., Roberfroid M.B. Comparative hepatotoxicity of cholic acid, deoxycholic acid and lithocholic acid in the rat: in vivo and in vitro studies. Toxicol. Lett. 1992;61:291–304. doi: 10.1016/0378-4274(92)90156-e. [DOI] [PubMed] [Google Scholar]
- Dietz B., Bolton J.L. Botanical Dietary Supplements Gone Bad Chem. Res. Toxicol. 2007;20:586–590. doi: 10.1021/tx7000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z., Wang X., Zhang N., Sun C., Zhao G., Peng Y., Zheng J. Metabolic activation of perampanel mediated by CYP1A2. Chem. Res. Toxicol. 2022;35:490–498. doi: 10.1021/acs.chemrestox.1c00396. [DOI] [PubMed] [Google Scholar]
- Dybdahl M., Nikolov N.G., Wedebye E.B., Jónsdóttir S., Niemelä J.R. QSAR model for human pregnane X receptor (PXR) binding: Screening of environmental chemicals and correlations with genotoxicity, endocrine disruption and teratogenicity. Toxicol. Appl. Pharmacol. 2012;262:301–309. doi: 10.1016/j.taap.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Eigenberg D.A., Carter D.E., Schram K.H., Sipes I.G. Examination of the differential hepatotoxicity of diallyl phthalate in rats and mice. Toxicol. Appl. Pharmacol. 1986;86:12–21. doi: 10.1016/0041-008x(86)90395-9. [DOI] [PubMed] [Google Scholar]
- Elcombe C.R., Peffer R.C., Wolf D.C., Bailey J., Bars R., Bell D., Cattley R.C., Ferguson S.S., Geter D., Goetz A., Goodman J.I., Hester S., Jacobs A., Omiecinski C.J., Schoeny R., Xie W., Brian G., Lake B.G. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit. Rev. Toxicol. 2014;44:64–82. doi: 10.3109/10408444.2013.835786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourches D., Barnes J.C., Day N.C., Bradley P., Reed J.Z., Tropsha A. Cheminformatics analysis of assertions mined from literature that describe drug-induced liver injury in different species. Chem. Res. Toxicol. 2010;23:171–183. doi: 10.1021/tx900326k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromenty B., Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacology & Therapeutics. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- Gaeta G.B., Utili R., Adinolfi L.E., Abernathy C.O., Giusti G. Characterization of the effects of erythromycin estolate and erythromycin base on the excretory function of the isolated rat liver. Toxicol. Appl. 1985;pharmacol. 80:185-I 92. doi: 10.1016/0041-008x(85)90074-2. [DOI] [PubMed] [Google Scholar]
- Gao W., Casey E., Bohl C.E., Dalton J.T. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005;105:3352–3370. doi: 10.1021/cr020456u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh J., Das J.J., Manna P., Sil P.C. Hepatotoxicity of di-(2-ethylhexyl)phthalate is attributed to calcium aggravation, ROS-mediated mitochondrial depolarization, and ERK/NF-κB pathway activation. Free Radic. Biol. Med. 2010;49:1779–1791. doi: 10.1016/j.freeradbiomed.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Goecke-Flora, C. M., Reo, N. V. (1996) Influence of Carbon Chain Length on the Hepatic Effects of Perfluorinated Fatty Acids. A 19F- and 31P-NMR Investigation. Chem. Res. Toxicol. 9, 689-695). [DOI] [PubMed]
- Gonzalez F.J. The peroxisome proliferator-activated receptor alpha (PPAR): Role in hepatocarcinogenesis. Molecular and Cellular Endocrinology. 2002;193:71–79. doi: 10.1016/s0303-7207(02)00098-9. [DOI] [PubMed] [Google Scholar]
- Heise T., Schmidt F., Knebel C., Rieke S., Haider W., Pfeil R., Kneuer C., Niemann L., Marx-Stoelting P. Hepatotoxic effects of (tri)azole fungicides in a broad dose range. Arch. Toxicol. 2015;89:2105–2117. doi: 10.1007/s00204-014-1336-1. [DOI] [PubMed] [Google Scholar]
- Hester S., Moore T., Padgett W.T., Murphy L., Wood C.E., Nesnow S. The hepatocarcinogenic conazoles: cyproconazole, epoxiconazole, and propiconazole induce a common set of toxicological and transcriptional responses. Toxicol. Sci. 2012;127:54–65. doi: 10.1093/toxsci/kfs086. [DOI] [PubMed] [Google Scholar]
- Hewitt M., Enoch S.J., Madden J.C., Przybylak K.R., Cronin M.T. Hepatotoxicity: A scheme for generating chemical categories for read-across, structural alerts and insights into mechanism(s) of action. Crit. Rev. Toxicol. 2013;43:537–558. doi: 10.3109/10408444.2013.811215. [DOI] [PubMed] [Google Scholar]
- Hildebrand A., Pfeiffer E., Metzler M. Aromatic hydroxylation and catechol formation: A novel metabolic pathway of the growth promotor zeranol. Toxicol. Lett. 2010;192:379–386. doi: 10.1016/j.toxlet.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Ito Y., Yamanoshita O., Asaeda N., Tagawa Y., Lee C.H., Aoyama T., Ichihara G., Furuhashi K., Kamijima M., Gonzalez F.J., Nakajima T. Di(2-ethylhexyl)phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor alpha-independent pathway. J. Occup. Health. 2007;49:172–182. doi: 10.1539/joh.49.172. [DOI] [PubMed] [Google Scholar]
- Iverson S.L., Uetrecht J.P. Identification of a reactive metabolite of terbinafine: Insights into terbinafine-induced hepatotoxicity. Chem. Res. Toxicol. 2001;14:175–181. doi: 10.1021/tx0002029. [DOI] [PubMed] [Google Scholar]
- Jamieson J.D., Smith E.B., Dalvie D.K., Stevens G.J., Yanochko G.M. Myeloperoxidase-mediated bioactivation of 5-hydroxythiabendazole: A possible mechanism of thiabendazole toxicity. Toxicology In Vitro. 2011;25:1061–1066. doi: 10.1016/j.tiv.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Johnson D.F., Jr., Hall W.H. Allergic hepatitis caused by propionyl erythromycin ester of lauryl sulfate. N. Engl. J. Med. 1961;265:1200–1202. doi: 10.1056/NEJM196112142652407. [DOI] [PubMed] [Google Scholar]
- Kafouni M.I., Anders R.A., Verma S. hepatotoxicity associated with dietary supplements containing anabolic steroids Clin Gastroenterol hepatol. 2007;5:809–812. doi: 10.1016/j.cgh.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Kågedal K., Bironaite D., Öllinger K. Anthraquinone Cytotoxicity and Apoptosis in Primary Cultures of Rat Hepatocytes. Free Rad. Res. 1999;31:419–428. doi: 10.1080/10715769900300981. [DOI] [PubMed] [Google Scholar]
- Kalender Y., Yel M., Kalender S. Doxorubicin hepatotoxicity and hepatic free radical metabolism in rats: The effects of vitamin E and catechin. Toxicology. 2005;209:39–45. doi: 10.1016/j.tox.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A.S. Designing around structural alerts in drug discovery. J. Med. Chem. 2020;63:6276–6302. doi: 10.1021/acs.jmedchem.9b00917. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A.S., Gardner I., Obach R.S., Shaffer C.L., Callegari E., Henne K.R., Mutlib A.E., Dalvie D.K., Lee J.S., Nakai Y., O'Donnell J.P., Boer J., Harriman S.P. A comprehensive listing of bioactivation pathways of organic functional groups. Curr. Drug Metab. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- Kamimura H., Ito S., Nozawa K., Nakamura S., Chijiwa H., Nagatsuka S., Kuronuma M., Ohnishi Y., Suemizu H., Ninomiya S. Formation of the accumulative human metabolite and human-specific glutathione conjugate of diclofenac in TK-NOG chimeric mice with humanized livers. Drug Metab. Dispos. 2015;43:309–316. doi: 10.1124/dmd.114.061689. [DOI] [PubMed] [Google Scholar]
- Kassahun K., Skordos K., McIntosh I., Slaughter D., Doss G.A., Baillie T.A., Yost G.S. Zafirlukast metabolism by cytochrome P450 3A4 produces an electrophilic alpha, beta-unsaturated iminium species that results in the selective mechanism-based inactivation of the enzyme. Chem. Res. Toxicol. 2005;18:1427–1437. doi: 10.1021/tx050092b. [DOI] [PubMed] [Google Scholar]
- Kawal K., Mori H., Sugie S., Yoshimi N., Inoue T., Nakamaru T., Nozawa Y., Matsushima T. Genotoxicity in the hepatocyte/DNA repair test and toxicity to liver mitochondria of 1-hydroxyanthraquinone and several dihydroxyanthraquinones. Cell Biology and Toxicology. 1986;2:457–467. doi: 10.1007/BF00117848. [DOI] [PubMed] [Google Scholar]
- Khalili H., Soudbakhsh A., Hajhossein T.A. Severe hepatotoxicity and probable hepatorenal syndrome associated with sulfadiazine. Am. J. Health-Syst Ph. 2011;68:888–892. doi: 10.2146/ajhp100516. [DOI] [PubMed] [Google Scholar]
- Kicman A.T. Pharmacology of anabolic steroids. Br. J Pharmacol. 2008;154:502–521. doi: 10.1038/bjp.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig J.E., Babich M.A., Baetcke K.P., Cook J.C., Corton J.C., David R.M., DeLuca J.G., Lai D.Y., McKee R.H., Peters J.M., Roberts R.A., Fenner-Crisp P.A. PPARα agonist-induced rodent tumors: modes of action and human relevance. Crit. Rev. Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Klausz, G., Keller, E., Sara, Z., Szekely-Koermoeczy, P., Laczay, P., Ary, K., Sotonyi, P., Rona, K. (2015) Simultaneous determination of praziquantel, pyrantel embonate, febantel and its active metabolites, oxfendazole and fenbendazole, in dog plasma by liquid chromatography/mass spectrometry Biomed Chromatogr 29, 1859-1865. [DOI] [PubMed]
- Knebel C., Buhrke T., Süssmuth R., Lampen A., Marx-Stoelting P., Braeuning A. Pregnane X receptor mediates steatotic efects of propiconazole and tebuconazole in human liver cell lines. Archives of Toxicology. 2019;93:1311–1322. doi: 10.1007/s00204-019-02445-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Hashimoto M., Honkakoski P., Negishi M. Regulation of gene expression by CAR: an update. Arch. Toxicol. 2015;89:1045–1055. doi: 10.1007/s00204-015-1522-9. [DOI] [PubMed] [Google Scholar]
- Kucera H.R., Livingstone M., Moscoso C.G., Gaikwad N.W. Evidence for NQO1 and NQO2 catalyzed reduction of ortho- and para-quinone methides. Free Radical Research. 2013;47:1016–1026. doi: 10.3109/10715762.2013.847527. [DOI] [PubMed] [Google Scholar]
- Lambert J.D., Zhao D., Meyers R.O., Kuester R.K., Timmermann B.N., Dorr R.T. Nordihydroguaiaretic acid: hepatotoxicity and detoxification in the mouse. Toxicon: official journal of the International Society on Toxinology. 2002;40:1701–1708. doi: 10.1016/s0041-0101(02)00203-9. [DOI] [PubMed] [Google Scholar]
- Leeming M.G., Gamon L.F., Wille U., Donald W.A., O'Hair R.A. What are the potential sites of protein arylation by N-acetyl-p-benzoquinone imine (NAPQI)? Chem. Res. Toxicol. 2015;28:2224–2233. doi: 10.1021/acs.chemrestox.5b00373. [DOI] [PubMed] [Google Scholar]
- Li F., Chordia M.D., Huang T., Macdonald T.L. In vitro nimesulide studies toward understanding idiosyncratic hepatotoxicity: Diiminoquinone formation and conjugation. Chem. Res. Toxicol. 2009;22:72–80. doi: 10.1021/tx800152r. [DOI] [PubMed] [Google Scholar]
- Li X., Kamenecka T.M., Cameron M.D. Bioactivation of the Epidermal Growth Factor Receptor Inhibitor Gefitinib: Implications for Pulmonary and Hepatic Toxicities. Chem. Res. Toxicol. 2009;22:1736–1742. doi: 10.1021/tx900256y. [DOI] [PubMed] [Google Scholar]
- Li J., Shi J., Jia C., Li W., Peng Y., Zheng J. Metabolic activation and cytotoxicity of propafenone mediated by CYP2D6. Chem. Res. Toxicol. 2022;35:829–839. doi: 10.1021/acs.chemrestox.2c00013. [DOI] [PubMed] [Google Scholar]
- Lim S., Kim Y., Gu J., Lee S., Shin W., Kim S. Supervised chemical graph mining improves drug-induced liver injury prediction. iScience. 2022;26 doi: 10.1016/j.isci.2022.105677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Liu C., Liu Y., Ge Q., Sun C. Cytochrome P450 mediated bioactivation of rutaevin, a bioactive and potentially hepatotoxic component of Evodia rutaecarpa Chem. Res. Toxicol. 2020;33:3054–3060. doi: 10.1021/acs.chemrestox.0c00475. [DOI] [PubMed] [Google Scholar]
- Liu J., Mansouri K., Judson R.S., Martin M.T., Hong H., Chen M., Xu X., Thomas R., Shah I. Predicting hepatotoxicity using ToxCast in vitro bioactivity and chemical structure. Chem. Res. Toxicol. 2015;28:738–751. doi: 10.1021/tx500501h. [DOI] [PubMed] [Google Scholar]
- Liu R., Yu X., Wallqvist A. Data-driven identification of structural alerts for mitigating the risk of drug-induced human liver injuries. Journal of Cheminformatics. 2015:7–12. doi: 10.1186/s13321-015-0053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock E.A., Mitchell A.M., Elcombe C.R. Biochemical mechanisms of induction of hepatic peroxisome proliferation. Annu. Rev. Pharmacol. Toxicol. 1989;29:145–163. doi: 10.1146/annurev.pa.29.040189.001045. [DOI] [PubMed] [Google Scholar]
- Low Y., Uehara T., Minowa Y., Yamada H., Ohno Y., Urushidani T., Sedykh A., Muratov E., Kuz’min, V., Fourches, D., Zhu, H., Rusyn, I., Tropsha, A. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 2011;24:1251–1262. doi: 10.1021/tx200148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAllister S.L., Young C., Guzdek A., Zhidkov N., O'Brien P.J. Molecular cytotoxic mechanisms of chlorpromazine in isolated rat hepatocytes. Canadian Journal of Physiology and Pharmacology. 2013;91:56–63. doi: 10.1139/cjpp-2012-0223. [DOI] [PubMed] [Google Scholar]
- Madsen K.G., Skonberg C., Jurva U., Cornett C., Hansen S.H., Johansen T.N., Olsen J. Bioactivation of diclofenac in vitro and in vivo: Correlation to electrochemical studies. Chem. Res. Toxicol. 2008;21:1107–1119. doi: 10.1021/tx700419d. [DOI] [PubMed] [Google Scholar]
- Marotta, F., Tiboni, G. M. (2010) Molecular aspects of azoles-induced teratogenesis Expert Opin. Drug Metab. Toxicol. (2010) 6(4) 10.1517/17425251003592111. [DOI] [PubMed]
- Masubuchi Y., Saito H., Horie T. Structural requirements for the hepatotoxicity of nonsteroidal anti-inflammatory drugs in isolated rat hepatocytes. J. Pharmacol. Exp. Ther. 1998;287:208–213. [PubMed] [Google Scholar]
- Mavrova A.T., Anichina K.K., Vuchev D.I., Tsenov J.A., Denkova P.S., Kondeva M.S., Micheva M.K. Antihelminthic activity of some newly synthesized 5(6)-(un)substituted-1H-benzimidazol-2-ylthioacetylpiperazine derivatives. Eur. J. Med. Chem. 2006;41:1412–1420. doi: 10.1016/j.ejmech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- McCormack W.M., George H., Donner A., Kodgis L.F., Alpert S., Lowe E.W., Kass E.H. Hepatotoxicity of erythromycin estolate during pregnancy. Antimicrobil Agents and Chemotherapy. 1977;12:630–636. doi: 10.1128/aac.12.5.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald M.G., Rettie A.E. Sequential Metabolism and Bioactivation of the hepatotoxin benzbromarone: Formation of glutathione adducts from a catechol intermediate. Chem. Res. Toxicol. 2007;20:1833–1842. doi: 10.1021/tx7001228. [DOI] [PubMed] [Google Scholar]
- Mellor C.L., Steinmetz F.P., Cronin M.T. Using molecular initiating events to develop a structural alert based screening workflow for nuclear receptor ligands associated with hepatic steatosis. Chem. Res. Toxicol. 2016;29:203–212. doi: 10.1021/acs.chemrestox.5b00480. [DOI] [PubMed] [Google Scholar]
- Mete A., Mehmet I., Altug S., Gokhan A., Cem K., Yildiran S. Paroxetine induced toxic hepatitis: case report. Turkiye Klinikleri Tip Bilimleri Dergisi. 2012;32:264–266. [Google Scholar]
- Michalik L., Auwerx J., Berger J.P., Chatterjee V.K., Glass C.K., Gonzalez F.J., Grimaldi P.A., Kadowaki T., Lazar M.A., O'Rahilly S., Palmer C.N., Plutzky J., Reddy J.K., Spiegelman B.M., Staels B., Wahli W. International union of pharmacology. LXI. Peroxisome proliferator-activated receptors“. Pharmacol. Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- Minet E.F., Daniela G., Meredith C., Massey E.D. A comparative in vitro kinetic study of [14C]-eugenol and [14C]-methyleugenol activation and detoxification in human, mouse, and rat liver and lung fractions. Xenobiotica. 2012;42:429–441. doi: 10.3109/00498254.2011.637582. [DOI] [PubMed] [Google Scholar]
- Mizuno K., Katoh M., Okumura H., Nakagawa N., Negishi T., Hashizume T., Nakajima M., Yokoi T. Metabolic activation of benzodiazepines by CYP3A4. Drug Metab. Dispos. 2009;37:345–351. doi: 10.1124/dmd.108.024521. [DOI] [PubMed] [Google Scholar]
- Moseley, R. H. (2013) Chapter 26 - Hepatotoxicity of Antimicrobials and Antifungal Agents, InDrug-Induced Liver Disease (Third Edition) (Kaplowitz, N., and DeLeve, L. D., Eds.) pp 463-481, Academic Press, Boston, USA.
- Mulliner D., Schmidt F., Stolte M., Spirkl H.-P., Czich A., Amberg A. Computational models for human and animal hepatotoxicity with a global application scope. Chem. Res. Toxicol. 2016;29:757–767. doi: 10.1021/acs.chemrestox.5b00465. [DOI] [PubMed] [Google Scholar]
- Nakajima A., Fukami T., Kobayashi Y., Watanabe A., Nakajima M., Yokoi T. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: rifampicin, rifabutin, and rifapentine. Biochemical pharmacology. 2011;82:1747–1756. doi: 10.1016/j.bcp.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Nau H. Embryotoxicity and Teratogenicity of Topical Retinoic Acid. Skin Pharmacol. 1993;6:35–44. doi: 10.1159/000211162. [DOI] [PubMed] [Google Scholar]
- O'Donnell J.P., Dalvie D.K., Kalgutkar A.S., Obach R.S. Mechanism-based inactivation of human recombinant P450 2C9 by the nonsteroidal anti-inflammatory drug suprofen. Drug Metab. Dispos. 2003;31:1369–1377. doi: 10.1124/dmd.31.11.1369. [DOI] [PubMed] [Google Scholar]
- Pandit A., Sachdeva T., Bafna P. Drug-Induced Hepatotoxicity: A Review. J. Appl. Pharmaceutical Sci. 2012;2:233–243. [Google Scholar]
- Penning T.M., Burczynski M.E., Hung C.-F., McCoull K.D., Palackal N.T., Tsuruda L.S. Dihydrodiol Dehydrogenases and Polycyclic Aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chem. Res. Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- Pessayre D., Larrey D., Funck-Brentano C., Benhamou J.P. Drug interactions and hepatitis produced by some macrolide antibiotics. J Antimicrob Chemother. 1985;16 Suppl A:181–194. doi: 10.1093/jac/16.suppl_a.181. [DOI] [PubMed] [Google Scholar]
- Pizzo F., Gadaleta D., Lombardo A., Nicolotti O., Benfenati E. Identification of structural alerts for liver and kidney toxicity using repeated dose toxicity data. Chemistry Central Journal. 2015;9:62–73. doi: 10.1186/s13065-015-0139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qatanani M., Moore D.D. CAR, the continuously advancing receptor, in drug metabolism and disease. Curr. Drug Metab. 2005;6:329–339. doi: 10.2174/1389200054633899. [DOI] [PubMed] [Google Scholar]
- Rao D.N.R., Cederbaum A.I. A comparative study of the redox-cycling of a quinone (rifamycin S) and a quinonimine (rifabutin) antibiotic by rat liver microsomes. Free Radical Biology & Medicine. 1996;22:439–446. doi: 10.1016/s0891-5849(96)00335-8. [DOI] [PubMed] [Google Scholar]
- Ratziu V., Poynard T., Sabah H., Naveau S., Chaput J.C. Hepatotoxicity of moxisylyte chlorhydrate: a new case. Gastroenterologie Clinique et biologique. 1991;15:980. [PubMed] [Google Scholar]
- Regal K.A., Laws G.M., Yuan C., Yost G.S., Skiles G.L. Detection and characterization of DNA adducts of 3-methylindole. Chem. Res. Toxicol. 2001;14:1014–1024. doi: 10.1021/tx0100237. [DOI] [PubMed] [Google Scholar]
- Rezen T., Rozman D., Pascussi J.-M., Monostory K. Interply between cholesterol and grug metabolism. Biochimica et Biophysica Acta. 2011;1814:146–160. doi: 10.1016/j.bbapap.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Rikans L.E. The oxidation of acrolein by rat liver aldehyde dehydrogenases. Relation to allyl alcohol hepatotoxicity. Drug Metab. Dispos. 1987;15:356–362. [PubMed] [Google Scholar]
- Salah-Abbès J.B., Abbès S., Haous Z., Oueslati R. Raphanus sativus extract prevents and ameliorates zearalenone-induced peroxidative hepatic damage in Balb/c mice. JPP. 2009;61:1545–1554. doi: 10.1211/jpp/61.11.0015. [DOI] [PubMed] [Google Scholar]
- Schuetz E.G., Brimer C., Schuetz J.D. Environmental xenobiotics and the antihormones cyproterone acetate and spironolactone use the nuclear hormone pregnenolone X receptor to activate the CYP3A23 hormone response element. Molecular Pharmacology. 1998;54:1113–1117. doi: 10.1124/mol.54.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.M., Li Y., Novalen M., Hayes M.A., Uetrecht J. Bioactivation of nevirapine to a reactive quinone methide: implications for liver injury. Chem. Res. Toxicol. 2012;25:1708–1719. doi: 10.1021/tx300172s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn C.T., Backos D.S., Orlicky D.J., Smathers-McCullough R.L., Petersen D.R. Identification of 5' AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J. Biol. Chem. 2014;289:15449–15462. doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S., Atsumi R., Nakazawa T., Izumi T., Sudo K., Okazaki O., Saji H. Ticlopidine-induced hepatotoxicity in a GSH-depleted rat model. Arch. Toxicol. 2011;85:347–353. doi: 10.1007/s00204-010-0594-9. [DOI] [PubMed] [Google Scholar]
- Silver E.H., Murphy S.D. Effect of carboxylesterase inhibitors on the acute hepatotoxicity of esters of ally1 alcohol. Toxicol. Appl. Pharmacol. 1978;45:377–389. doi: 10.1016/0041-008x(78)90102-3. [DOI] [PubMed] [Google Scholar]
- Silverman R.B. second edition. Elsevier academic press; 2004. The Organic Chemistry of Drug Design and Drug Action; p. 445. [Google Scholar]
- Srivastava A., Ramachandran S., Hameed S.P., Ahuja V., Hosagrahara V.P. Identification and mitigation of a reactive metabolite liability associated with aminoimidazoles Chem. Res. Toxicol. 2014;27:1586–1597. doi: 10.1021/tx500212c. [DOI] [PubMed] [Google Scholar]
- Staudinger J.L., Goodwin B., Jones S.A., Diane Hawkins-Brown D., MacKenzie K.I., LaTour A., Liu Y., Klaassen C.D., Brown K.K., Reinhard J., Willson T.M., Koller B.H., B. H., Steven A. Kliewer, S. A. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. PNAS. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepan A., Walker D., Bauman J., Price D., Baillie T., Kalgutkar A., Aleo M. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- Tasaki M., Kuroiwa Y., Inoue T., Hibi D., Matsushita K., Ishii Y., Maruyama S., Nohmi T., Nishikawa A., Umemura T. Oxidative DNA damage and in vivo mutagenicity caused by reactive oxygen species generated in the livers of p53-proficient or -deficient gpt delta mice treated with non-genotoxic hepatocarcinogens. J. Appl. Toxicol. 2013;33:1433–1441. doi: 10.1002/jat.2807. [DOI] [PubMed] [Google Scholar]
- Teo Y.L., Ho H.K., Chan A. Formation of reactive metabolites and management of tyrosine kinase inhibitor-induced hepatotoxicity: a literature review. Expert Opin Drug Metab. Toxicol. 2015;11:231–242. doi: 10.1517/17425255.2015.983075. [DOI] [PubMed] [Google Scholar]
- Thakkar S., Li T., Liu Z., Wu L., Roberts R., Tong W. Drug-induced liver injury severity and toxicity (DILIst): binary classification of 1279 drugs by human hepatotoxicity. Drug Discov Today. 2020;25:201–208. doi: 10.1016/j.drudis.2019.09.022. [DOI] [PubMed] [Google Scholar]
- Thompson C.D., Kinter M.T., Macdonald T.L. Synthesis and in vitro reactivity of 3-carbamoyl-2-phenylpropionaldehyde and 2-phenylpropenal: Putative reactive metabolites of felbamate. Chem. Res. Toxicol. 1996;9:1225–1229. doi: 10.1021/tx9601566. [DOI] [PubMed] [Google Scholar]
- Thompson D.C., Perera K., Bolton J.L. Studies on the mechanism of hepatotoxicity of 4-methylphenol (p-cresol): effects of deuterium labeling and ring substitution. Chemico-Biological Interactions. 1996;101:1–11. doi: 10.1016/0009-2797(96)03707-6. [DOI] [PubMed] [Google Scholar]
- Thompson D.C., Perera K., Krol E.S., Bolton J.L. o-Methoxy & alkylphenols that form quinone methides of intermediate reactivity are the most toxic in rat liver slices. Chem. Res. Toxicol. 1995;8:323–327. doi: 10.1021/tx00045a001. [DOI] [PubMed] [Google Scholar]
- Thompson D.C., Perera K., London R. Quinone methide formation from para isomers of methylphenol (Cresol), ethylphenol, and isopropylphenol: Relationship to toxicity. Chem. Res. Toxicol. 1995;8:55–60. doi: 10.1021/tx00043a007. [DOI] [PubMed] [Google Scholar]
- Tsao, A., Stewart, D. (2009) Collateral Damage Associated with Chemotherapy pp 18-32 IN: Yeung, S-C., Escalante, C., Gagel, R. Medical care of cancer patients. BC Decker Inc.
- Valadon P., Dansette P.M., Girault J.-P., Amar C., Mansuy D. Thiophene sulfoxides as reactive metabolites: Formation upon microsomal oxidation of a 3-aroylthiophene and fate in the presence of nucleophiles in vitro and in vivo. Chem. Res. Toxicol. 1996;9:1403–1413. doi: 10.1021/tx9601622. [DOI] [PubMed] [Google Scholar]
- Van Dijk R., Kremer A.E., Smit W., van den Elzen B., van Gulik T., Gouma D., Lameris J.S., Bikker H., Enemuo V., Stokkers P.C., Feist M., Bosma P., Jansen P.L., Beuers U. Characterization and treatment of persistent hepatocellular secretory failure. Liver Int. 2015;35:1478–1488. doi: 10.1111/liv.12603. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel J.P., Thompson J.T., Frame S.R., Gillies P.J. Differential Activation of Nuclear Receptors by Perfluorinated Fatty Acid Analogs and Natural Fatty Acids: A Comparison of Human, Mouse, and Rat Peroxisome Proliferator-Activated Receptor-, -, and -, Liver X Receptor-, and Retinoid X Receptor- Toxicol. Sci. 2006;92:476–489. doi: 10.1093/toxsci/kfl014. [DOI] [PubMed] [Google Scholar]
- Vinken M., Pauwels M., Ates G., Vivier M., Vanhaecke T., Rogiers V. Screening of repeated dose toxicity data present in SCC(NF)P/SCCS safety evaluations of cosmetic ingredients. Arch. Toxicol. 2012;86:405–412. doi: 10.1007/s00204-011-0769-z. [DOI] [PubMed] [Google Scholar]
- Virkel G., Lifschitz A., Sallovitz J., Inza G., Lanusse C. Effect of the ionophore antibiotic monensin on the ruminal biotransformation of benzimidazole anthelmintics. Vet J. 2004;167:265–271. doi: 10.1016/S1090-0233(03)00079-0. [DOI] [PubMed] [Google Scholar]
- Vogel S.M., Bauer M.R., Joerger A.C., Wilcken R., Brandt T., Veprintsev D.B., Rutherford T.J., Fersht A.R., Boeckler F.M. Lithocholic acid is an endogenous inhibitor of MDM4 and MDM2. Proc. Natl Acad. Sci. USA. 2012;109:16906–16910. doi: 10.1073/pnas.1215060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walgren J.L., Mitchell M.D., Thom D.C. Role of metabolism in drug-induced idiosyncratic hepatotoxicity. Crit. Rev. Toxicol. 2005;35:325–361. doi: 10.1080/10408440590935620. [DOI] [PubMed] [Google Scholar]
- Walker, D. P., Bi, F. C., Kalgutkar, A. S., Bauman, J. N., Zhao, S. X., Soglia, J. R., Aspnes, G. E., Kung, D. W., Klug-McLeod, J., Zawistoski, M. P., McGlynn, M. A., Oliver, R., Dunn, M., Li, J-C., Richter, D. T., Cooper, B. A., Kath, J. C., Hulford, C. A., Autry, C. L., Luzzio, M. J., Ung, E. J., Roberts, W. G., Bonnette, P. C., Buckbinder, L., Mistry, A., Griffor, M. C., Han, S., Guzman-Perez, A. (2008) Trifluoromethylpyrimidine-based inhibitors of proline-rich tyrosine kinase 2 (PYK2): Structure–activity relationships and strategies for the elimination of reactive metabolite formation Bioorganic & Medicinal Chemistry Letters 18 6071–6077. [DOI] [PubMed]
- Wang Y.M., Chai S.C., Brewer C.T., Chen T. Associate member pregnane X receptor and drug-induced liver injury. Expert Opin. Drug Metab. Toxicol. 10, 1521–1532. Williams, G. M. (1997) Chemicals with carcinogenic activity in the rodent liver; mechanistic evaluation of human risk. Cancer Lett. 2014;117:175–188. doi: 10.1016/s0304-3835(97)00229-2. [DOI] [PubMed] [Google Scholar]
- Wen B., Moore D.J. Bioactivation of glafenine by human liver microsomes and peroxidases: identification of electrophilic iminoquinone species and GSH conjugates. Drug Metab. Dispos. 2011;39:1511–1521. doi: 10.1124/dmd.111.039396. [DOI] [PubMed] [Google Scholar]
- Wen B., Zhou M. Metabolic activation of the phenothiazine antipsychotics chlorpromazine and thioridazine to electrophilic iminoquinone species in human liver microsomes and recombinant P450s. Chem Biol Interact. 2009;181:220–226. doi: 10.1016/j.cbi.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Williams G.M. Chemicals with carcinogenic activity in the rodent liver; mechanistic evaluation of human risk. Cancer Lett. 1997;117:175–188. doi: 10.1016/s0304-3835(97)00229-2. [DOI] [PubMed] [Google Scholar]
- Woo Y.T., Lai D.Y. In: Quantitative structure-activity relationship (QSAR) analysis of mutagens and carcinogens. Benigni R., editor. CRC Press; 2003. Mechanism of action of chemical carcinogens and their role in structure-activity relationships analysis and risk assessment; pp. 41–80. [Google Scholar]
- Wu S., Blackburn K., Amburgey J., Jaworska J., Federle T. A framework for using structural, reactivity, metabolic and physicochemical similarity to evaluate the suitability of analogs for sar-based toxicological assessments. Regul. Toxicol. Pharmacol. 2010;56:67–81. doi: 10.1016/j.yrtph.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Wu S., Fisher J., Naciff J., Laufersweiler M., Lester C., Daston G., Blackburn K. Framework for identifying chemicals with structural features associated with the potential to act as developmental or reproductive toxicants. Chem. Res. Toxicol. 2013;26:1840–1861. doi: 10.1021/tx400226u. [DOI] [PubMed] [Google Scholar]
- Wu L., Liu Z., Auerbach S., Huang R., Chen M., McEuen K., Xu J., Fang H., Tong W. Integrating drug's mode of action into quantitative structure-activity relationships for improved prediction of drug-Induced liver injury. J Chem Inf Model. 2017;57:1000–1006. doi: 10.1021/acs.jcim.6b00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito, S. C. (1996) ACS Symposium Series 640, Chapter 2, 16.
- You Y., Wang X., Ma K., Li J., Peng Y., Zheng J. Metabolic Activation of Atomoxetine Mediated by Cytochrome P450 2D6 Chem. Res. Toxicol. 2021;34:2135–2144. doi: 10.1021/acs.chemrestox.1c00216. [DOI] [PubMed] [Google Scholar]
- Yu L., Liu H., Li W., Zhang F., Luckie C., van Breemen R.B., Thatcher G.R.J., Bolton J.L. Oxidation of raloxifene to quinoids: Potential toxic pathways via a diquinone methide and o-quinones. Chem. Res. Toxicol. 2004;17:879–888. doi: 10.1021/tx0342722. [DOI] [PubMed] [Google Scholar]
- Zhang, N., Yang., Y., Li, W., Zhou, S., Li, W., Peng, Y. and Zheng, J. (2022) . Chem. Res. Toxicol. 25: 1821-1830. [DOI] [PubMed]
- Zhou J., Febbraio M., Wada T., Zhai Y., Kuruba R., He J., Lee J.H., Khadem S., Ren S., Li S., Silverstein R.L., Xie W. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology. 2008;134:556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Zimmerman, H. J. (1999) Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver By Hyman J. Zimmerman Lippincott Williams & Wilkins, 1999 - Medical - 789 pages.
- Zinedine A., Soriano J.M., Moltó J.C., Mañes J. Review on the toxicity, occurrence, metabolism, detoxification, regulations and intake of zearalenone: An oestrogenic mycotoxin. Food Chem. Toxicol. 2007;45:1–18. doi: 10.1016/j.fct.2006.07.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.