

Abstract
Simultaneous inhibition of tumor vasculature and the glycolysis pathway may be a targeted anti-tumor strategy to inhibit tumor nutrient supply. Flavonoids are natural products with strong biological activity, which inhibit hypoxia induction factor 1α (HIF-1α) regulating glycolysis and tumor angiogenesis, while salicylic acid can reduce the glycolysis level of tumor cells by inhibiting related rate-limiting enzymes. A series of salicylic acid-modified indole trimethoxy-flavone derivatives were designed and synthesized by introducing benzotrimethoxy-structure commonly used in blood vessel blockers, and their anti-tumor activities were evaluated. Among them, compound 8f exhibited significant anti-proliferative activity against two hepatoma cells, HepG-2 and SMMC-7721, with IC50 values of 4.63 ± 1.13 μM and 3.11 ± 0.35 μM, respectively. Colony formation experiments also further verified its excellent in vitro anti-tumor activity. In addition, compound 8f showed the ability to induce apoptosis in SMMC-7721 cells in a concentration-dependent manner. After treatment with compound 8f, the expressions of the rate-limiting enzymes PKM2, PFKM, HK2 and tumor angiogenesis-related vascular endothelial growth factor of the glycolytic pathway were all down-regulated, and the lactate level in the hepatoma cell SMMC-7721 was significantly reduced. The morphology of the nucleus and tubulin was also observed to disperse gradually with the increase of compound 8f concentration. And compound 8f showed strong binding ability to tubulin. Our results suggest that the strategy of synthesizing the salicylic acid-modified indole flavone derivative 8f is a way to obtain active anti-tumor candidate compounds that may be further developed as targeted agents to inhibit tumor vasculature and glycolytic pathways.
Simultaneous inhibition of tumor vasculature and the glycolysis pathway may be a targeted anti-tumor strategy to inhibit tumor nutrient supply.
1. Introduction
The tumor microenvironment is the internal environment for tumor growth, invasion and metastasis, which can promote tumor development through various pathways and mechanisms.1,2 Among them, hypoxia and abundant tumor vasculature are important features of solid tumors.3,4 The Warburg effect showed that tumors favored the glycolytic pathway for energy even in normoxia and was accompanied by an increase in lactate levels.5,6 In addition, the high expression of tumor vascular endothelial growth factor (VEGF) under hypoxia triggers tumor angiogenesis, which greatly promotes tumorigenesis and invasion.7,8 Meanwhile, existing blood vessels are also an important way for tumors to acquire capacity.9 Therefore, we hypothesized that simultaneous inhibition of tumor glycolysis and tumor vasculature might be an anti-tumor strategy targeting the tumor energy microenvironment.
In glycolysis, key metabolic enzymes phosphofructokinase (PFK), pyruvate kinase (PK), and hexokinase (HK) are regulated by transcription factors such as hypoxia-inducible factor 1α (HIF-1α) in the oncogenic factor signal transduction pathway.10,11 Flavonoid natural products have received extensive attention due to their effective and abundant biological activities.12,13 And their (e.g. chrysin, apigenin, acacetin) inhibitory ability on tumor HIF-1α and VEGF has been noted as a promising candidate for the development of anti-tumor drugs targeting glycolytic pathways and tumor neovasculars.14–16 In addition to inhibiting tumor angiogenesis, vascular blockers that block existing tumor vessels are also worthy of attention, including CA4P and its analogs OXi4503, AVE8062, colchicine analogs ZD6126, BNC-105, and CKD-516 (Fig. 1).17–21 In recent years, we have focused on the structure of flavonoids, introduced trimethoxy (Fig. 1), a common pharmacophore in vascular disrupting agents targeting tubulin, and synthesized and obtained trimethoxyflavonoid derivatives that inhibit tubulin, HIF-1α, and glycolysis rate-limiting enzymes with excellent anti-tumor activity (Fig. 2A).22–25 To further expand the exploration of this strategy, the anti-tumor activity of the classic NSAID acetylsalicylic acid (aspirin) was noted. Among them, salicylic acid and acetate salicylic acid have also been reported to reduce the level of glycolysis in tumor cells by inhibiting PFK in glycolysis.26 In addition, tubulin inhibitors based on indole skeletons have been extensively explored,27,28 and indole ring compounds with different substituents can enhance the antiproliferative activity against tumor cells.29
Fig. 1. The chemical structures of CA4P, OXi4503, AVE8062, ZD6126, BNC-105, and CKD-516.

Fig. 2. Design strategies for A-ring trimethoxy-flavone derivatives containing salicylic acid-modified indole substituents. (A) Chemical structures of compounds 7f and FS-7. (B) The structure of flavonoids, indoles and salicylic acids involved in the splicing principle, as well as the structure of two salicylic acid-modified indole trimethoxy-flavone derivatives containing an A-ring trimethoxy group.

Here, we expected to introduce salicylic acid-modified indoles and trimethoxy groups into the flavonoid structure to obtain prospective anti-tumor candidate compounds (Fig. 2B) Trimethoxy-flavone containing indolyl was synthesized from 3,4,5-trimethoxy-phenol, chloroacetyl chloride, indole-3-formaldehyde and indole-5-aldehyde. Subsequently, two indole-containing flavonoids reacted with chloroacetyl salicylic acid derivatives respectively to obtain the final compounds 8a–8g and 9a–9f. Compound 8f showed significant antiproliferative activity against human hepatoma cell lines HepG-2 and SMMC-7721 cells with IC50 values of 4.63 ± 1.13 μM and 3.11 ± 0.35 μM, respectively. In addition, compound 8f inhibited SMMC-7721 cell colony formation, induced cell apoptosis, and blocked G1 and S phase cell cycles in a concentration-dependent manner. After treatment with compound 8f, the expressions of VEGF and glycolytic rate-limiting enzymes PKM2, PFKM and HK2 were significantly down-regulated, and the formation of lactic acid was inhibited. The morphology of the nucleus and tubulin in SMMC-7721 cells was gradually dispersed under the action of compound 8f, and the molecular docking results showed the binding effect of compound 8f with tubulin. These results suggested that targeting tumor vasculature and glycolysis simultaneously might be a potential strategy for developing a new generation of anti-tumor drugs.
2. Results and discussion
2.1. Synthesis of salicylic acid modified indole flavonoid derivatives
According to our previous report,24,25,30 a series of B-ring trimethoxy flavonoids containing indole derivatives of salicylic acid were synthesized (Fig. 3). The important intermediate chlorophenone (5) was obtained from 3,4,5-trimethoxyphenol (1) as a starting material. Subsequently, two flavonoids containing indole structure (6, 7) were obtained by reacting with indole-3-formaldehyde and indole-5-aldehyde respectively. Salicylic acid and its derivatives (1) were methylated (2) and reacted with sulfoxide chloride to obtain acyl chloride salicylic acid derivatives (3). In the presence of a catalytic quantity of NaH, a DMF solution containing chloroacetyl salicylic acid derivatives was slowly dropped into DMF reaction solution dissolved 6 or 7, and allowed to continuously stir and react for about 24 h under the condition of an ice water bath. The final product was extracted with ethyl acetate and separated by silica gel column chromatography (Fig. S1–S35†).
Fig. 3. Synthesis of salicylic acid-modified indole trimethoxy-flavone derivatives containing an A-ring trimethoxy group 8a–8g and 9a–9f.

2.2. In vitro anti-tumor activity and cytotoxicity
The in vitro anti-tumor activity of salicylic acid-modified indole flavonoid derivatives was evaluated on human gastric cancer cells (HGC-27), human colon cancer cells (HCT-116), human liver cancer cells (HepG-2, SMMC-7721), and human breast cancer cells (MDA-MB-231) (Table 1). Combretastatin A4 (CA4), sodium citrate and 5-fluorouracil (5-Fu) were used as controls. Among them, these compounds generally showed low cell inhibitory activity against HCT-116 cells. We noted that flavonoid derivatives linked with an indole benzene ring (8a–8g) showed significantly better anti-tumor cell proliferation activity in vitro than those linked with an indole pyrrole ring (9a–9f). The absence of salicylic acid derivate-substituted compounds 6 and 7 showed a certain degree of inhibitory activity against normal hepatocytes (LO2), however, the activity was weakened in the presence of salicylic acid substituents, and little toxicity was observed against LO2 cells. This indicated that salicylic acid substituents on the indole ring could reduce the toxicity to normal cells. For compounds 8a–8g, the para-electron-withdrawing groups in the salicylic acid substituents decreased the activity of anti-cell proliferation, while the para-electron-donating groups enhanced it. Compound 8f showed excellent anti-proliferation activity on HGC-27, HepG-2, MDA MB-231 and SMMC-7721 cells, with IC50 values of 10.48 ± 0.44, 4.63 ± 1.13, 4.96 ± 0.91 and 3.11 ± 0.35 μM, respectively. Moreover, no toxicity of compound 8f was observed on normal liver cells, suggesting a certain selectivity. In contrast, although CA4 and 5-Fu were generally superior to salicylic acid-modified indole flavonoid derivatives in vitro for anti-tumor cell proliferation, their toxicity to normal liver cells could not be ignored. Based on the strongest anti-cell proliferation activity against SMMC-7721 cells, further evaluation was conducted.
The IC50 values of compounds 6, 7, 8a–8g and 9a–9f against six human tumor cell lines.
| Compounds | IC50 ± SD (μM) | |||||
|---|---|---|---|---|---|---|
| HGC-27 | HCT-116 | HepG-2 | MDA-MB-231 | SMMC-7721 | LO2 | |
| 8a | 66.37 ± 5.58 | >100 | 15.84 ± 0.44 | 27.32 ± 0.88 | 7.46 ± 0.46 | NDa |
| 8b | >100 | >100 | 32.11 ± 0.42 | 36.71 ± 1.47 | 16.51 ± 1.09 | >100 |
| 8c | 55.88 ± 7.47 | >100 | 29.04 ± 0.71 | 44.26 ± 1.22 | 19.48 ± 0.41 | >100 |
| 8d | >100 | >100 | 6.63 ± 1.29 | 38.05 ± 2.23 | 5.18 ± 0.46 | >100 |
| 8e | 41.54 ± 3.83 | >100 | >100 | 44.31 ± 0.88 | >100 | ND |
| 8f | 10.48 ± 0.44 | ND | 4.63 ± 1.13 | 4.96 ± 0.91 | 3.11 ± 0.35 | ND |
| 8g | 24.5 ± 1.67 | >100 | >100 | 59.86 ± 0.89 | 6.84 ± 0.65 | ND |
| 9a | ND | ND | 30.03 ± 1.65 | >100 | >100 | ND |
| 9b | >100 | >100 | >100 | ND | ND | >100 |
| 9c | 16.69 ± 2.33 | ND | ND | >100 | ND | >100 |
| 9d | >100 | >100 | 57.77 ± 0.96 | >100 | 16.67 ± 1.7 | ND |
| 9e | 20.83 ± 0.94 | >100 | 12.43 ± 1.42 | ND | 15.28 ± 1.78 | >100 |
| 9f | ND | ND | ND | ND | ND | ND |
| 6 | 3.93 ± 0.71 | 21.93 ± 0.48 | 1.3 ± 0.19 | 2.69 ± 0.58 | 1.22 ± 0.11 | 61.86 ± 1.48 |
| 7 | 15.16 ± 2.44 | 56.59 ± 4.25 | 5.44 ± 1.42 | 17.38 ± 1.04 | 4.72 ± 0.97 | 87.75 ± 0.71 |
| CA4 | 0.047 ± 0.033 | 0.29 ± 0.05 | 0.37 ± 0.18b | 1.5 ± 0.17b | 0.75 ± 0.01b | 81.83 ± 1.76 |
| Sodium citrate | >100 | >100 | >100 | >100 | >100 | >100 |
| 5-Fu | 0.5 ± 0.11 | 14.29 ± 1.22 | 0.37 ± 0.87 | 1.58 ± 0.38 | 1.59 ± 0.31 | 54.45 ± 1.97 |
ND: not detected.
nM.
2.3. Inhibition of colony formation
The inhibitory effect of compound 8f on colony formation of SMMC-7721 cells was investigated. As shown in Fig. 4, with increasing drug concentration, the crystal violet density in the pore showed a significant downward trend, indicating that the number of living cells in the pore also gradually decreased with increasing drug concentration. When the concentration of compound 8f was increased to 6 μM, the cell growth and colony formation were almost completely inhibited. The results indicated that compound 8f could inhibit the formation of SMMC-7721 colonies in a dose-dependent manner.
Fig. 4. Inhibitory effect of compound 8f on SMMC-7721 cell colony formation. Crystal violet-stained images of SMMC-7721 cell colonies treated with compound 8f at concentrations of 0, 1.5, 3, and 6 μM for 11 days.

2.4. Analysis of apoptosis and cell cycle assay
In order to investigate the apoptosis inducing ability of compound 8f, SMMC-7721 cells were treated with compound 8f at 0, 1.5, 3 and 6 μM concentrations for 48 h. Subsequently, the cells were analyzed by flow cytometry after annexin V-fluorescein isothiocyanate (FITC)/PI staining. The results showed that compared with the blank control group, the apoptosis of cells increased gradually with the increase of drug concentration, which were 7.9%, 18.07% and 24.3%, respectively (Fig. 5A and B). These results indicated that compound 8f could induce apoptosis of SMMC-7721 cells in a concentration-dependent manner.
Fig. 5. Effects of compound 8f on the apoptosis and cell cycle of SMMC-7721 cells. (A and B) Quantitative analysis of apoptotic SMMC-7721 cells treated with compound 8f (0, 1.5, 3, 6 μM) was performed with FITC-annexin V/PI flow cytometry. (C and D) Cell cycle distribution of SMMC-7721 cells treated with compound 8f (0, 1.5, 3, 6 μM) was determined using flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001, n = 3.

Disruption of the tumor cell cycle often leads to uncontrolled cell proliferation. Here, we further investigated the effect of compound 8f on the cell cycle of SMMC-7721 cells (Fig. 5C and D). The results showed that compared with the control group, for SMMC-7721 cells treated with compound 8f at concentrations of 0, 1.5, 3 and 6 μM, the proportion of G1 phase increased to a certain extent, which were 60.32%, 63.29% and 65.15%, respectively. The proportion of S phase cells was higher than that of the blank control group, 34.37%, 14.69%, 23.62%, respectively. The proportion of G2 phase in cells treated with compound 8f decreased. These results suggested that compound 8f could arrest the cell cycle in G1 and S phases to a certain extent.
2.5. Western blotting analysis
In order to further investigate the effects of compound 8f on the expression of VEGF and three rate-limiting enzymes PKM2, PFKM and HK2 in glycolysis, the Western blot assay was used to evaluate the expression of related enzymes in SMMC-7721 cells treated with compound 8f. The results showed that the expression of VEGF (Fig. 6A), PKM2 (Fig. 6B), PFKM (Fig. 6C) and HK2 (Fig. 6D) in SMMC-7721 cells treated with compound 8f decreased significantly with the increase of compound 8f concentration. The down-regulation of glycolytic rate-limiting enzyme expression may be one of the factors of glycolytic inhibition, and its effect on VEGF is conducive to blocking tumor angiogenesis, which may contribute to the excellent anti-tumor cell proliferation activity of compound 8fin vitro.
Fig. 6. Effect of compound 8f on the expression of VEGF (A) and three rate-limiting enzymes PKM2 (B), PFKM (C) and HK2 (D) in SMMC-7721 cells at the concentration of 0, 1.5, 3, 6 μM. *p < 0.05, **p < 0.01, ***p < 0.001, n = 3.

2.6. Lactic acid analysis
Aerobic glycolysis is a typical feature of tumors. Even under normal oxygen conditions, tumor cells obtain energy mainly through glycolysis. As an important product of glycolysis, lactic acid plays a key role in shaping the acidic microenvironment of tumors and epigenetic modification. Therefore, based on the down-regulation of compound 8f on the expression of key rate limiting enzymes in the glycolysis pathway, the effect of compound 8f on cell lactic acid production was further investigated (Fig. 7). The results showed that the lactic acid content decreased significantly with the increase of compound 8f concentration, suggesting that compound 8f can effectively reduce the production of lactic acid, which may be related to the down-regulation of glycolytic rate-limiting enzyme expression.
Fig. 7. Effect of lactic acid content on SMMC-7721 cells treated with compound 8f for 48 h. ***p < 0.001, n = 3.

2.7. Microtubule disarrangement
Based on the common characteristics of the trimethoxy group of reported vascular blockers targeting tubulin, we expected that salicylic acid modified indole flavonoid derivatives containing the trimethoxy group could also play a certain role in inhibiting tubulin. The inhibition of tubulin is conducive to the arrest of mitosis of tumor cells, thus exerting the activity of anti-cell proliferation. As shown in Fig. 8, it was observed under a laser confocal microscope that after treatment with compound 8f, the round and full morphology of nuclei gradually shrunk and denaturated with the increase of drug concentration. At a concentration of 6 μM, nucleus rupture and dispersion were observed. Compared with the normal network structure displayed in the control group, the morphology of Cy3-stained tubulin became gradually dispersed with the increase of compound 8f concentration, and the skeleton morphology of microtubules gradually blurred and disappeared. When the concentration of compound 8f reached 6 μM, the network structure of tubulin almost disappeared completely. These results suggested that compound 8f showed an inhibitory effect on tubulin, which might contribute to its growth inhibitory activity against SMMC-7721 hepatoma cells.
Fig. 8. Immunofluorescence staining of SMMC-7721 cells treated with compound 8f at concentrations of 1.5, 3, and 6 μM for 48 h. The tubulins were labeled red with Cy3 dye, and the nuclei were labeled blue with DAPI.

2.8. Molecular docking studies
In order to further confirm the interaction between compound 8f and tubulin, the molecular docking simulation of compound 8f and colchicine was performed on tubulin (PDB: 1SA0) respectively. The results showed that the carbonyl group in the flavonoid structure of compound 8f and the carbonyl group in the substituted salicylic acid structure formed hydrogen bonds with Lys254 and Ala180 residues of tubulin, respectively (Fig. 9C and D). Colchicine in the colchicine–tubulin complex (PDB: 1SA0) was molecularly-docked with tubulin under the same conditions. Among them, colchicine formed hydrogen bonds with multiple amino acid residues in tubulin, including Met259, Val238, Ala180, Lys352, Thr179 and Ser178 (Fig. 9A and B). By molecular docking of compounds 8f and colchicine with tubulin, it was observed that the benzotrimethoxy groups of compounds 8f and colchicine were located in adjacent but different binding pockets. In addition, the substituent indole-salicylic acid of compound 8f extended into the colchicine binding pocket and shared a common hydrogen binding site Ala180 residue with colchicine (Fig. 9E). Binding energy was used to explain their ability to bind to tubulin. The binding energy of compound 8f to tubulin was lower than that of colchicine (Fig. 9F), indicating the stable binding ability of compound 8f to tubulin. These results suggested that the binding effect between compound 8f and tubulin might lead to inhibition of tubulin and significant anti-proliferation effects of tumor cells.
Fig. 9. Predicted docking modes of colchicine (A and B) and compound 8f (C and D) alone with tubulin, respectively. Prediction of the docking mode between colchicine–compound 8f mixing state and tubulin (E). Binding energy (kcal mol−1) of colchicine and compound 8f to tubulin (F).

In addition, in order to compare the protein interactions of compound 8f with known inhibitors of VEGF, PKM2, PFKM, and HK2, we performed molecular docking of compound 8f with lenalidomide (Fig. 10A), compound 10 (Fig. 10B), and lonidamine (Fig. 10D) in corresponding proteins, respectively. The results showed that compound 8f is similar to these inhibitors and can interact with the protein in the same binding pocket of the corresponding protein. Compound 8f was mainly its salicylic acid substituents in the same pockets as known inhibitors of these proteins and interacted with amino acid residues of those proteins. Moreover, the binding energy of compound 8f to VEGF, PKM2 and HK2 proteins was lower than that of the corresponding inhibitors (Table 2), suggesting that compound 8f had a more stable binding ability to these proteins. Although no suitable PFKM inhibitors were used, compound 8f interacted with Gly24, Ser23, Ser121, Arg88, and Arg301 of PFKM (Fig. 10C) and showed a low binding energy (−7.4585 kcal mol−1). These results suggested that the interaction of compound 8f with the above target proteins may be related to its inhibitory activity on the proteins and its anti-tumor cell activities.
Fig. 10. Predicted docking patterns of compound 8f/lenalidomide in VEGF (PDB: 4GLU) (A), compound 8f/compound 10 in PKM2 (PDB: 6TTQ) (B), compound 8f in PFKM (PDB: 4OMT) (C) and compound 8f/lonidamine in HK2 (PDB: 2NZT) (D).

Binding energy (kcal mol−1) of compound 8f, lenalidomide, compound 10 and lonidamine to VEGF, PKM2, PFKM, and HK2 protein, respectively.
| Compounds | VEGF | PKM2 | PFKM | HK2 |
|---|---|---|---|---|
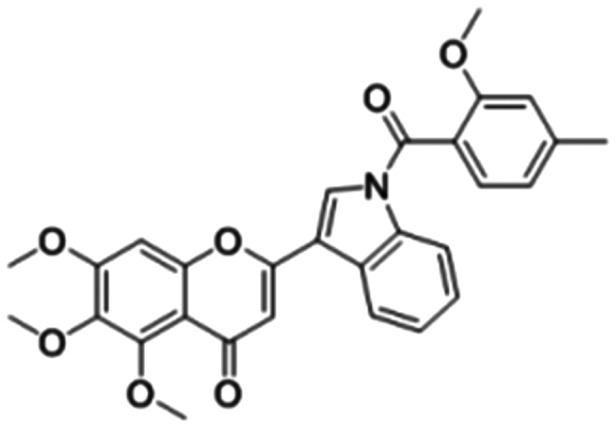 Compound 8f Compound 8f |
−7.9560 | −8.3930 | −7.4585 | −8.0091 |
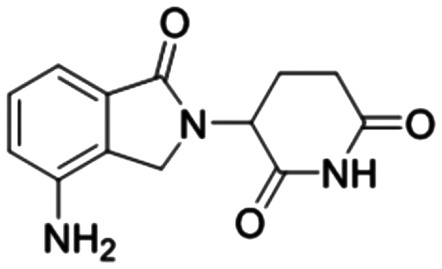 Lenalidomide Lenalidomide |
−5.8194 | |||
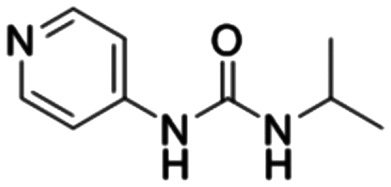 Compound 10 Compound 10 |
−4.9800 | |||
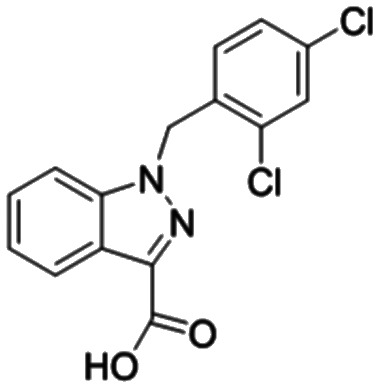 Lonidamine Lonidamine |
−5.6446 |
3. Conclusions
Simultaneously inhibiting tumor vasculature and glycolysis to cut off nutrient supply is an anti-tumor strategy worth considering. In our previous study, the synthesized flavonoid derivatives containing a trimethoxy group showed the expected target inhibition and anti-tumor activity in vivo and in vitro.22–25,30 Here, we further introduced bioactive indole and salicylic acid and synthesized two trimethoxy-flavone derivatives with salicylic acid-modified indole substituents. In the screening of several cell lines, compound 8f showed significant antiproliferative activity against human hepatoma cell lines HepG-2 and SMMC-7721, with IC50 values of 4.63 ± 1.13 μM and 3.11 ± 0.35 μM, respectively, while it showed low cytotoxicity against human normal hepatocyte LO2. Inhibition of SMMC-7721 cell colony formation also showed the antiproliferative activity of compound 8f. In addition, compound 8f induced apoptosis of SMMC-7721 cells and blocked the cell cycle in G1 and S phases in a concentration-dependent manner. After treatment with compound 8f, the expressions of VEGF and glycolytic rate-limiting enzyme PKM2, PFKM and HK2 were significantly down-regulated, suggesting that compound 8f could inhibit the formation of neovascularization and the glycolytic pathway to a certain extent. The inhibition of glycolysis was verified by the reduction of lactic acid formation in cells by compound 8f. For tubulin, the nucleus and tubulin morphology of treated SMMC-7721 cells gradually dispersed as the concentration of compound 8f increased. Molecular docking results also revealed the binding effect of compound 8f to tubulin. These results indicate that compound 8f is a promising candidate compound for the treatment of hepatocellular carcinoma, and suggest that simultaneous inhibition of tumor vasculature and glycolysis holds promise for the development of a new generation of targeted anti-tumor agents.
4. Experimental section
4.1. General experimental procedure
All chemicals and solvents used were obtained commercially. The progress of the reaction was monitored by thin-layer chromatography on silica gel 60 F254 plates under UV light at 254 nm or 365 nm. 1H and 13C NMR spectra were recorded on a Bruker 500 or AVANCE III 400 MHz spectrometer. The multiplicity of chemical shifts was represented by the following abbreviations: s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet, br = generalized. High-resolution mass spectral data for the compounds were recorded by a Waters GCT model mass spectrometer.
Human gastric cancer cells (HGC-27), human colon cancer cells (HCT-116), human liver cancer cells (HepG-2, SMMC-7721), human breast cancer cells (MDA-MB-231) and human normal liver cells (LO2) were purchased from the Institute of Cellular Biology of Chinese Academy of Sciences (China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was from Sigma-Aldrich (USA). Dulbecco's modified Eagle's medium (DMEM), trypsin and fetal bovine serum (FBS) were supplied by Gibco (USA). Cy3-labeled goat anti-mouse IgG, 4′,6-diamidino-2-phenylindole (DAPI), cell cycle kit, apoptosis kit and antibody for western blot were purchased from Beijing Solarbio Technology Co., LTD (China).
4.2. Chemistry
Synthesis of indole flavonoids, compounds 6 and 7
3,4,5-Trimethoxyphenol (4) was dissolved in 20 mL of dichloromethane, a catalytic amount of anhydrous aluminum chloride was added at room temperature, and then 0.5 mL of chloroacetyl chloride was slowly added dropwise. The reaction was heated under reflux at 60 °C for 1 h, the progress of the reaction was monitored by thin-layer chromatography, and a mixture of concentrated HCl and ice water (1 : 1, v/v) was added after the reaction was complete. The extracted dichloromethane phase product was purified by silica gel column chromatography to obtain compound 5. Indole-3-carbaldehyde and indole-5-carbaldehyde were respectively reacted with compound 5 in ethanol solution under base catalysis for 24 h, and then the pH was adjusted to about 7.0 by adding water to obtain a large number of bright yellow precipitates, which were intermediates 6 and 7.
2-(1H-Indol-3-yl)-5,6,7-trimethoxy-4H-chromen-4-one (6)
Yellow solid, yield: 72.1%, Mp: 236–237 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.71 (s, 1H, 9-H), 8.07 (d, J = 2.0 Hz, 1H, 10-H), 7.92 (dd, J = 6.9, 1.0 Hz, 1H, 12-H), 7.44 (dd, J = 6.9, 1.1 Hz, 1H, 15-H), 7.33–7.27 (m, 2H, 13-H, 14-H), 7.25 (s, 1H, 3-H), 6.55 (s, 1H, 8-H), 4.28 (s, 3H, 5-OCH3), 3.99 (s, 3H, 7-OCH3), 3.84 (s, 3H, 6-OCH3). 13C NMR (126 MHz, DMSO-d6) (ppm): δ 178.86 (s), 162.73 (s), 161.35 (s), 150.94 (s), 145.25 (s), 136.77 (s), 136.70 (s), 131.67 (s), 127.01 (s), 123.13 (s), 121.24 (s), 119.51 (s), 112.70 (s), 108.84 (s), 107.91 (s), 105.73 (s), 92.23 (s), 62.42 (s), 61.47 (s), 57.24 (s). HRMS (ESI) m/z calcd for C20H17NO5 [M + H]+, 352.1185; found, 352.1187.
2-(1H-Indol-5-yl)-5,6,7-trimethoxy-4H-chromen-4-one (7)
Yellow solid, yield: 69.8%, Mp: 186–187 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.34 (s, 1H, 12-H), 8.21 (s, 1H, 10-H), 7.76 (d, J = 8.5 Hz, 1H, 15-H), 7.44 (d, J = 8.6 Hz, 1H, 14-H), 6.95 (s, 1H, 3-H), 6.64 (s, 1H, 11-H), 6.60 (s, 1H, 8-H), 4.27 (s, 3H, 5-OCH3), 3.99 (s, 3H, 7-OCH3), 3.83 (s, 3H, 6-OCH3). 13C NMR (126 MHz, DMSO-d6) (ppm): δ 179.90 (s), 163.55 (s), 161.83 (s), 151.10 (s), 145.99 (s), 137.12 (s), 136.79 (s), 128.64 (s), 127.39 (s), 124.96 (s), 123.47 (s), 113.82 (s), 112.54 (s), 107.13 (s), 102.59 (s), 92.29 (s), 62.39 (s), 61.47 (s), 57.37 (s). HRMS (ESI) m/z calcd for C20H17NO5 [M + H]+, 352.1185; found, 352.1188.
Synthesis of compounds 8a–8h and 9a–9g
Dimethyl sulfate was slowly added dropwise into an alkaline aqueous solution of salicylic acid derivatives (1) and reacted at 40 °C, and then adjusted to pH 3.0 with 10% HCl to obtain the white precipitate of 2-methoxybenzoic acid derivatives (2), respectively. Thionyl chloride and a catalytic amount of DMF were added to the solution of 2-methoxybenzoic acid derivatives in dichloromethane, and allowed to react under reflux at 40 °C for 1 h to obtain 2-methoxybenzoyl chloride derivatives (3). Intermediates 6 or 7 dissolved in DMF were stirred for 30 min in an ice-water bath in the presence of a catalytic amount of sodium hydride, and then compound 7 was added to continue the reaction for 24 h. After monitoring the reaction, water was added to precipitate a precipitate, which was extracted with ethyl acetate, and the organic phase was dried with anhydrous Na2SO4. Finally, the crude product obtained by rotary evaporation under reduced pressure was purified by silica gel column chromatography to obtain compounds 8a–8h and 9a–9g.
5,6,7-Trimethoxy-2-(1-(2-methoxybenzoyl)-1H-indol-3-yl)-4H-chromen-4-one (8a)
Yellow solid, yield: 63.1%, Mp: 187–188 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.14 (s, 1H, 12-H), 7.96 (s, 1H, 10-H), 7.87 (d, J = 7.3 Hz, 1H, 15), 7.61 (t, J = 7.9 Hz, 1H, 20-H), 7.55 (d, J = 7.5 Hz, 1H, 21-H), 7.44–7.36 (m, 2H, 13-H, 14-H), 7.17 (t, J = 7.2 Hz, 1H, 19-H), 7.10 (d, J = 8.4 Hz, 1H, 18-H), 7.02 (s, 1H, 3-H), 6.33 (s, 1H, 8-H), 4.26 (s, 3H, 5-OCH3), 3.97 (s, 3H, 17-OCH3), 3.82 (s, 3H, 7-OCH3), 3.79 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.88 (s), 166.92 (s), 163.30 (s), 161.46 (s), 156.75 (s), 151.75 (s), 147.97 (s), 136.71 (s), 135.60 (s), 132.66 (s), 130.49 (s), 129.74 (s), 129.56 (s), 125.68 (s), 124.43 (s), 124.34 (s), 120.98 (s), 119.13 (s), 116.13 (s), 114.60 (s), 111.51 (s), 107.51 (s), 101.89 (s), 90.34 (s), 62.44 (s), 61.63 (s), 56.57 (s), 55.81 (s). HRMS (ESI) m/z calcd for C28H23NO7 [M + H]+, 486.1153; found, 486.1557.
2-(1-(4-Bromo-2-methoxybenzoyl)-1H-indol-3-yl)-5,6,7-trimethoxy-4H-chromen-4-one (8b)
Yellow solid, yield: 49.7%, Mp: 174–175 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.22 (s, 1H, 12-H), 7.87 (s, 1H, 10-H), 7.84 (dd, J = 5.9, 3.1 Hz, 1H, 15-H), 7.44–7.39 (m, 3H, 13-H, 14-H, 21-H), 7.33 (dd, J = 8.0, 1.3 Hz, 1H, 20-H), 7.25 (s, 1H, 18-H), 7.01 (s, 1H, 3-H), 6.32 (s, 1H, 8-H), 4.26 (s, 3H, 5-OCH3), 4.00 (s, 3H, 17-OCH3), 3.81 (s, 3H, 7-OCH3), 3.79 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.88 (s), 166.92 (s), 163.30 (s), 161.46 (s), 156.75 (s), 151.75 (s), 147.97 (s), 136.71 (s), 135.60 (s), 132.66 (s), 130.49 (s), 129.74 (s), 129.56 (s), 125.68 (s), 124.43 (s), 124.34 (s), 120.98 (s), 119.13 (s), 116.13 (s), 114.60 (s), 111.51 (s), 107.51 (s), 101.89 (s), 90.34 (s), 62.44 (s), 61.63 (s), 56.57 (s), 55.81 (s). HRMS (ESI) m/z calcd for C28H22BrNO7 [M + H]+, 564.0658; found, 564.0062.
2-(1-(5-Bromo-2-methoxybenzoyl)-1H-indol-3-yl)-5,6,7-trimethoxy-4H-chromen-4-one (8c)
Yellow solid, yield: 56.3%, Mp: 210–211 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.24 (s, 1H, 12-H), 7.90 (s, 1H, 10-H), 7.88–7.82 (m, 1H, 15-H), 7.73–7.66 (m, 2H, 19-H and 21-H), 7.46–7.39 (m, 2H, 13-H and 14-H), 7.01 (s, 1H, 3-H), 6.99 (d, J = 8.5 Hz, 1H, 18-H), 6.38 (s, 1H, 8-H), 4.26 (s, 3H, 5-OCH3), 3.99 (s, 3H, 17-OCH3), 3.82 (s, 3H, 7-OCH3), 3.79 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.83 (s), 165.00 (s), 163.34 (s), 161.58 (s), 155.98 (s), 151.78 (s), 148.23 (s), 136.75 (s), 135.52 (s), 135.24 (s), 132.08 (s), 129.98 (s), 129.76 (s), 125.97 (s), 125.94 (s), 124.74 (s), 119.18 (s), 116.28 (s), 115.05 (s), 113.49 (s), 112.86 (s), 107.44 (s), 101.52 (s), 90.44 (s), 62.45 (s), 61.64 (s), 56.69 (s), 56.19 (s). HRMS (ESI) m/z calcd for C28H22BrNO7 [M + H]+, 564.0658; found, 564.0662.
2-(1-(4-Chloro-2-methoxybenzoyl)-1H-indol-3-yl)-5,6,7-trimethoxy-4H-chromen-4-one (8d)
Yellow solid, yield: 47.7%, Mp: 174–175 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.20 (s, 1H, 12-H), 7.89 (s, 1H, 10-H), 7.87–7.81 (m, 1H, 15-H), 7.50 (d, J = 8.1 Hz, 1H, 21-H), 7.45–7.39 (m, 2H, 13-H, 14-H), 7.18 (dd, J = 8.1, 0.7 Hz, 1H, 20-H), 7.10 (s, 1H, 18-H), 7.01 (s, 1H, 3-H), 6.32 (s, 1H, 8-H), 4.26 (s, 3H, 5-OCH3), 3.99 (s, 3H, 17-OCH3), 3.82 (s, 3H, 7-OCH3), 3.79 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.81 (s), 165.86 (s), 163.24 (s), 161.57 (s), 157.53 (s), 151.79 (s), 148.14 (s), 138.44 (s), 136.74 (s), 135.50 (s), 130.56 (s), 130.26 (s), 129.74 (s), 125.84 (s), 124.63 (s), 122.93 (s), 121.21 (s), 119.14 (s), 116.14 (s), 114.87 (s), 112.31 (s), 107.43 (s), 101.60 (s), 90.26 (s), 62.44 (s), 61.64 (s), 56.62 (s), 56.19 (s). HRMS (ESI) m/z calcd for C28H22ClNO7 [M + H]+, 520.1163; found, 520.1168.
2-(1-(5-Chloro-2-methoxybenzoyl)-1H-indol-3-yl)-5,6,7-trimethoxy-4H-chromen-4-one (8e)
Yellow solid, yield: 57.8%, Mp: 200–201 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.22 (s, 1H, 12-H), 7.91 (s, 1H, 10-H), 7.88–7.83 (m, 1H, 15-H), 7.58–7.53 (m, 2H, 19-H and 21-H), 7.46–7.39 (m, 2H, 13-H and 14-H), 7.04 (d, J = 9.7 Hz, 1H, 18-H), 7.01 (s, 1H, 3-H), 6.37 (s, 1H, 8-H), 4.26 (s, 3H, 5-OCH3), 3.98 (s, 3H, 17-OCH3), 3.82 (s, 3H, 7-OCH3), 3.79 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.83 (s), 165.15 (s), 163.33 (s), 161.56 (s), 155.44 (s), 151.78 (s), 148.20 (s), 136.74 (s), 135.50 (s), 132.28 (s), 129.97 (s), 129.75 (s), 129.30 (s), 125.96 (s), 125.91 (s), 125.55 (s), 124.71 (s), 119.18 (s), 116.23 (s), 115.04 (s), 113.01 (s), 107.44 (s), 101.53 (s), 90.38 (s), 62.43 (s), 61.63 (s), 56.63 (s), 56.23 (s). HRMS (ESI) m/z calcd for C28H22ClNO7 [M + H]+, 520.1163; found, 520.1166.
5,6,7-Trimethoxy-2-(1-(2-methoxy-4-methylbenzoyl)-1H-indol-3-yl)-4H-chromen-4-one (8f)
Yellow solid, yield: 61.3%, Mp: 174–175 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.09 (d, J = 7.4 Hz, 1H, 12-H), 8.00 (s, 1H, 10-H), 7.90–7.84 (m, 1H, 15-H), 7.43 (d, J = 7.7 Hz, 1H, 21-H), 7.42–7.34 (m, 2H, 13-H and 14-H), 7.03 (s, 1H, 3-H), 6.97 (d, J = 7.8 Hz, 1H, 20-H), 6.89 (s, 1H, 18-H), 6.36 (s, 1H, 8-H), 4.27 (s, 3H, 5-OCH3), 3.98 (s, 3H, 17-OCH3), 3.82 (s, 3H, 7-OCH3), 3.76 (s, 3H, 6-OCH3), 2.50 (s, 3H, 19-CH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.89 (s), 167.04 (s), 163.29 (s), 161.41 (s), 156.91 (s), 151.76 (s), 147.89 (s), 143.61 (s), 136.69 (s), 135.64 (s), 130.79 (s), 129.69 (s), 125.54 (s), 124.28 (s), 121.64 (s), 121.50 (s), 119.13 (s), 116.03 (s), 114.36 (s), 112.24 (s), 107.55 (s), 102.11 (s), 90.35 (s), 62.45 (s), 61.63 (s), 56.52 (s), 55.72 (s), 22.06 (s). HRMS (ESI) m/z calcd for C29H25NO7 [M + H]+, 500.1709; found, 500.1712.
2-(1-(2,4-Dimethoxybenzoyl)-1H-indol-3-yl)-5,6,7-trimethoxy-4H-chromen-4-one (8g)
Yellow solid, yield: 52.5%, Mp: 171–172 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.10 (d, J = 7.1 Hz, 1H, 12-H), 8.03 (s, 1H, 10-H), 7.86 (dd, J = 5.4, 2.2 Hz, 1H, 15-H), 7.53 (d, J = 8.3 Hz, 1H, 21-H), 7.43–7.33 (m, 2H, 13-H and 14-H), 7.04 (s, 1H, 3-H), 6.68 (d, J = 8.4 Hz, 1H, 20-H), 6.60 (s, 1H, 18-H), 6.40 (s, 1H, 8-H), 4.27 (s, 3H, 5-OCH3), 3.98 (s, 3H, 17-OCH3), 3.93 (s, 3H, 19-OCH3), 3.82 (s, 3H, 7-OCH3), 3.74 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 179.88 (s), 166.68 (s), 163.82 (s), 163.30 (s), 161.44 (s), 158.75 (s), 151.74 (s), 147.85 (s), 136.70 (s), 135.68 (s), 131.72 (s), 130.91 (s), 129.66 (s), 125.46 (s), 124.18 (s), 119.05 (s), 116.92 (s), 115.94 (s), 114.09 (s), 107.56 (s), 105.07 (s), 102.15 (s), 98.91 (s), 90.41 (s), 62.44 (s), 61.63 (s), 56.59 (s), 55.76 (s),55.68 (s). HRMS (ESI) m/z calcd for C29H25NO8 [M + H]+, 516.1658; found, 516.1664.
5,6,7-Trimethoxy-2-(1-(2-methoxybenzoyl)-1H-indol-5-yl)-4H-chromen-4-one (9a)
Yellow solid, yield: 34.2%, Mp: 228–229 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.46 (d, J = 7.8 Hz, 1H, 12-H), 8.08 (s, 1H, 10-H), 7.91 (d, J = 8.7 Hz, 1H, 15-H), 7.53 (t, J = 7.9 Hz, 1H, 19-H), 7.47 (d, J = 7.5 Hz, 1H, 21-H), 7.12 (s, 1H, 14-H), 7.10 (t, J = 7.5p Hz, 1H, 20-H), 7.04 (d, J = 8.4 Hz, 1H, 18-H), 6.90 (s, 1H, 3-H), 6.64–6.57 (m, 2H, 11-H, 8-H), 4.27 (s, 3H, 5-OCH3), 4.00 (s, 3H, 17-OCH3), 3.83 (s, 3H, 7-OCH3), 3.80 (s, 3H, 6-OCH3). HRMS (ESI) m/z calcd for C28H23NO7 [M + H]+, 486.1553; found, 486.1552.
2-(1-(4-Bromo-2-methoxybenzoyl)-1H-indol-5-yl)-5,6,7-trimethoxy-4H-chromen-4-one (9b)
Yellow solid, yield: 37.1%, Mp: 217–218 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.44 (d, J = 6.6 Hz, 1H, 12-H), 8.08 (s, 1H, 10-H), 7.92 (d, J = 8.7 Hz, 1H, 15-H), 7.35 (d, J = 8.0 Hz, 1H, 21-H), 7.29–7.27 (m, 1H, 20-H), 7.20 (s, 1H, 18-H), 7.10 (d, J = 3.3 Hz, 1H, 14-H), 6.90 (s, 1H, 3-H), 6.64 (d, J = 3.7 Hz, 1H, 11-H), 6.61 (s, 1H, 8-H), 4.28 (s, 3H, 5-OCH3), 4.01 (s, 3H, 17-OCH3), 3.84 (s, 3H, 7-OCH3), 3.80 (s, 3H, 6-OCH3). HRMS (ESI) m/z calcd for C28H22BrNO7 [M + H]+, 564.0658; found, 564.0657.
2-(1-(5-bromo-2-methoxybenzoyl)-1H-indol-5-yl)-5,6,7-trimethoxy-4H-chromen-4-one (9c)
Yellow solid, yield: 61.3%, Mp: 174–175 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.46 (s, 1H, 12-H), 8.08 (s, 1H, 10-H), 7.92 (d, J = 8.7 Hz, 1H, 15-H), 7.62 (dd, J = 8.8, 2.4 Hz, 1H, 19-H), 7.58 (d, J = 2.4 Hz, 1H, 21-H), 7.08 (d, J = 3.3 Hz, 1H, 14-H), 6.93 (d, J = 8.9 Hz, 1H, 18-H), 6.90 (s, 1H, 3-H), 6.64 (d, J = 3.8 Hz, 1H, 11-H), 6.60 (s, 1H, 8-H), 4.27 (s, 3H, 5-OCH3), 4.00 (s, 3H, 17-OCH3), 3.83 (s, 3H, 7-OCH3), 3.78 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 180.84 (s), 165.38 (s), 164.02 (s), 161.60 (s), 155.48 (s), 151.72 (s), 147.30 (s), 136.67 (s), 135.80 (s), 135.06 (s), 131.74 (s), 131.50 (s), 128.60 (s), 128.27 (s), 128.04 (s), 126.18 (s), 124.07 (s), 116.76 (s), 113.35 (s), 113.05 (s), 111.87 (s), 109.34 (s), 107.19 (s), 90.65 (s), 62.44 (s), 61.66 (s), 56.68 (s), 56.09 (s). HRMS (ESI) m/z calcd for C28H22BrNO7 [M + H]+, 564.0658; found, 564.0657.
2-(1-(4-Chloro-2-methoxybenzoyl)-1H-indol-5-yl)-5,6,7-trimethoxy-4H-chromen-4-one (9d)
Yellow solid, yield: 31.7%, Mp: 212–213 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.43 (d, J = 7.7 Hz, 1H, 12-H), 8.08 (s, 1H, 10-H), 7.91 (d, J = 8.8 Hz, 1H, 15-H), 7.41 (d, J = 8.1 Hz, 1H, 21-H), 7.14–7.07 (m, 1H, 20-H), 7.09 (s, 1H, indole 6-H), 7.04 (s, 1H, 18-H), 6.90 (s, 1H, 3-H), 6.63 (d, J = 3.7 Hz, 1H, 11-H), 6.61 (s, 1H, 8-H), 4.27 (s, 3H, 5-OCH3), 4.00 (s, 3H, 17-OCH3), 3.83 (s, 3H, 7-OCH3), 3.80 (s, 3H, 6-OCH3). HRMS (ESI) m/z calcd for C28H22ClNO7 [M + H]+, 520.1163; found, 520.1164.
2-(1-(5-Chloro-2-methoxybenzoyl)-1H-indol-5-yl)-5,6,7-trimethoxy-4H-chromen-4-one (9e)
Yellow solid, yield: 56.3%, Mp: 253–254 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.46 (s, 1H, 12-H), 8.08 (s, 1H, 10-H), 7.92 (d, J = 8.7 Hz, 1H, 15-H), 7.48 (dd, J = 8.8, 2.5 Hz, 1H, 19-H), 7.45 (d, J = 2.5 Hz, 1H, 21-H), 7.08 (d, J = 3.2 Hz, 1H, 14-H), 6.98 (d, J = 8.8 Hz, 1H, 18-H), 6.90 (s, 1H, 3-H), 6.64 (d, J = 3.7 Hz, 1H, 11-H), 6.60 (s, 1H, 8-H), 4.27 (s, 3H, 5-OCH3), 4.00 (s, 3H, 17-OCH3), 3.83 (s, 3H, 7-OCH3), 3.79 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 180.84 (s), 165.51 (s), 164.02 (s), 161.59 (s), 154.98 (s), 151.73 (s), 147.30 (s), 136.67 (s), 135.81 (s), 132.11 (s), 131.50 (s), 128.95 (s), 128.60 (s), 128.27 (s), 128.05 (s), 126.08 (s), 125.74 (s), 124.07 (s), 116.76 (s), 112.91 (s), 111.86 (s), 109.33 (s), 107.19 (s), 90.65 (s), 62.44 (s), 61.66 (s), 56.68 (s), 56.15 (s). HRMS (ESI) m/z calcd for C28H22ClNO7 [M + H]+, 520.1163; found, 520.1163.
2-(1-(2,4-Dimethoxybenzoyl)-1H-indol-5-yl)-5,6,7-trimethoxy-4H-chromen-4-one (9f)
Yellow solid, yield: 42.3%, Mp: 191–192 °C; 1H NMR (400 MHz, CDCl3) (ppm): δ 8.40 (d, J = 8.6 Hz, 1H, 12-H), 8.07 (s, 1H, 10-H), 7.89 (d, J = 8.8 Hz, 1H, 15-H), 7.44 (d, J = 8.4 Hz, 1H, 21-H), 7.20 (d, J = 3.7 Hz, 1H, 14-H), 6.90 (s, 1H, 3-H), 6.63–6.58 (m, 3H, 11-H, 18-H and 20-H), 6.55 (s, 1H 8-H), 4.27 (s, 3H, 5-OCH3), 4.00 (s, 3H, 17-OCH3), 3.90 (s, 3H, 19-OCH3), 3.83 (s, 3H, 7-OCH3), 3.76 (s, 3H, 6-OCH3). 13C NMR (126 MHz, CDCl3) (ppm): δ 180.87 (s), 167.01 (s), 164.01 (s), 163.47 (s), 161.54 (s), 158.36 (s), 151.70 (s), 147.16 (s), 136.64 (s), 136.03 (s), 131.48 (s), 131.18 (s), 128.72 (s), 128.07 (s), 127.91 (s), 124.04 (s), 117.03 (s), 116.61 (s), 112.20 (s), 108.30 (s), 107.24 (s), 105.02 (s), 98.89 (s), 90.66 (s), 62.44 (s), 61.66 (s), 56.67 (s), 55.72 (s), 55.64 (s). HRMS (ESI) m/z calcd for C29H25NO8 [M + H]+, 516.1658; found, 516.1663.
4.3. Anti-proliferation activity in vitro
The in vitro antiproliferative activity of the compounds on tumor cells (HGC-27, HCT-116, HepG-2, MDA-MB-231, SMMC-7721) and the cytotoxicity on normal liver cells (LO2) were detected by methyl thiazolyl tetrazolium (MTT) colorimetry. Cells were seeded in 96-well plates at a density of 5 × 103 cells per well, and the medium was replaced with fresh medium containing 128, 64, 32, 16, 8, 4, 2, and 1 μM drug concentrations after overnight incubation. Combretastatin A4 (CA4), sodium citrate, and 5-fluorouracil (5-FU) were used as positive controls. Incubation was continued for 48 h at 37 °C in an incubator containing 5% CO2. After MTT staining, the absorbance of each well was measured at a wavelength of 490 nm by a Wellscan MK-2 microplate reader and IC50 values were calculated.
4.4. Colony formation assay
SMMC-7721 cell suspensions were seeded in 6-well plates at a density of 1.5 × 103 cells per well, incubated in an incubator (37 °C, 5% CO2) for 24 h, and then the medium was replaced with fresh medium containing compound 8f (0, 1.5 μM, 3 μM, 6 μM). The culture was continued for 10 days, the medium containing the same concentration of compound 8f was changed every 48 h, then the medium was removed on day 11 and washed three times with phosphate buffer saline (PBS). Cells were fixed with paraformaldehyde solution (4%) for 15 min, then washed with PBS to remove excess fixative and stained with crystal violet solution (0.1%) for 15 min. Finally, the excess dye was gently washed three times with PBS, then dried at room temperature and photographed.
4.5. Cell cycle and apoptosis assay
SMMC-7721 cells were seeded in a six-well plate at a density of 1 × 104 cells per well, cultured in an incubator (37 °C, 5% CO2) for 24 h, and then treated with media containing different concentrations (0, 1.5 μM, 3 μM, 6 μM) of compound 8f for 48 h. Cells were harvested after trypsinization, washed twice with PBS by centrifugation, and fixed with pre-cooled 70% ethanol overnight. The fixative was removed and washed again with PBS, then RNaseA was added to resuspend the cells and treated at 37 °C for 30 min. Finally, pyridine iodide (PI) dye was added and incubated for 30 min in the dark at 4 °C, and the cell cycle progression was tested by flow cytometry at 488 nm wavelength. The cells collected as above were washed with PBS, then incubated with Annexin V-FITC and PI dyes in sequence in the dark, and then cell apoptosis was measured by flow cytometry.
4.6. Western blotting analysis
The SMMC-7721 cells treated with different concentrations of compound 8f were washed twice with pre-cooled PBS, and then RIPA buffer was added to lyse the cells. The lysate was centrifuged at 4 °C and 12 000 rpm, and the total protein concentration in the supernatant was determined by the BCA method. The proteins were transferred to PVDF membranes by the semi-dry method, and then the membranes were blocked with 5% nonfat milk at room temperature for 2 h with slow shaking. Subsequently, the cells were incubated with the primary antibody overnight at 4 °C, washed three times with TBST solution and incubated with the secondary antibody at room temperature. Finally, the PVDF membrane was taken out and washed with TBST solution again and imaged by a chemiluminescence imager (Tanon 5200, China). Quantitative analysis was performed by means of Image J software to record grayscale values.
4.7. Determination of lactic acid content
SMMC-7721 cell suspensions were seeded in 6-well plates at a density of 1 × 103 cells per well and incubated in an incubator (37 °C, 5% CO2) for 24 h. Cell-free medium was obtained after continued treatment with fresh medium containing various concentrations (0, 1.5 μM, 3 μM, 6 μM) of compound 8f for 48 h. Then, the enzyme working solution and the developer were added in sequence, mixed well and incubated in a water bath at 37 °C for 10 min. After the addition of 2 mL of stop solution, the absorbance values of the samples at wavelength of 530 nm were measured, and the lactic acid concentrations were calculated as follows:Lactic acid concentration (mM) = (SampleOD − BlankOD)/(StandardOD − BlankOD) × concentration of standard × sample dilution
4.8. Immunofluorescence assay
SMMC-7721 cells were seeded into 6-well plates plated with coverslips, treated with compound 8f at concentrations of 0, 1.5, 3, and 6 μM for 48 h and washed with PBS. Cells in each well were fixed with 4% paraformaldehyde and the excess fixative was washed away. Subsequently, the above cells blocked with bovine serum albumin were sequentially treated with β-action antibody, Cy3-labeled goat anti-mouse IgG and DAPI, and then observed under a laser scanning confocal microscope and images were collected.
4.9. Molecular docking
Molecular Operating Environment (MOE 2010.06; Chemical Computing Group, Canada) was used to perform molecular docking to further investigate the interaction of compound 8f with tubulin. ChemDraw was used to draw the chemical structure of the compound which was converted into 3D form in the MOE program window. The colchicine–tubulin complex (PDB: 1SA0) was downloaded from the Protein Data Bank (PDB) (https://www.rcsb.org/). All tests were performed on Windows 10 with an Intel Core i5 2.5 GHz processor, 4 GB of memory. The colchicine co-crystallization sites (PDB: 1SA0) in tubulin were used as docking sites. And co-crystallized colchicine was redocked into the tubulin binding pocket. After molecular docking of tubulin with compound 8f and colchicine, two-dimensional visual analysis was performed by MOE. The binding energy (kcal mol−1) of molecular docking was calculated as the total score to compare the difference in binding of the known inhibitor colchicine and compound 8f to the target protein. The same test was also used for molecular docking studies of compound 8f with lenalidomide, compound 10 and lonidamine in VEGF (PDB: 4GLU), PKM2 (PDB: 6TTQ, compound 10–PKM2 complex), PFKM (PDB: 4OMT) and HK2 (PDB: 2NZT), respectively.
4.10. Statistical analysis
The quantitative values were presented as mean ± SD. Student's t-test between two groups was performed using GraphPad Prism 6.0 software. P value < 0.05 was considered statistically significant.
Conflicts of interest
The authors declare no competing financial interest.
Supplementary Material
Acknowledgments
This work was financially supported by Hunan Provincial Key Laboratory of Tumor Microenvironment Responsive Drug Research (Approval number: 2019-56), the Natural Science Foundation of Hunan Province (Award Number: 2020JJ4534), Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study, and Tumor Microenvironment Responsive to Natural Product R&D Graduate Innovation Base.
Electronic supplementary information (ESI) available: NMR spectra and HRMS spectra of compounds 6, 7, 8a–8g, 9a–9f. See DOI: https://doi.org/10.1039/d3md00128h
References
- Xiao Y. Yu D. Pharmacol. Ther. 2021;221:107753. doi: 10.1016/j.pharmthera.2020.107753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T. Dai Y. Cancer Lett. 2017;387:61–68. doi: 10.1016/j.canlet.2016.01.043. [DOI] [PubMed] [Google Scholar]
- Wei X. Chen Y. Jiang X. Peng M. Liu Y. Mo Y. Ren D. Hua Y. Yu B. Zhou Y. Liao Q. Wang H. Xiang B. Zhou M. Li X. Li G. Li Y. Xiong W. Zeng Z. Mol. Cancer. 2021;20:7. doi: 10.1186/s12943-020-01288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.-X. Chen S.-Y. Yi N.-B. Li X. Chen S.-L. Lei Z. Cheng D.-B. Sun T. J. Controlled Release. 2022;350:829–840. doi: 10.1016/j.jconrel.2022.09.003. [DOI] [PubMed] [Google Scholar]
- Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G. Cantley L. C. Thompson C. B. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G. Benjamin L. E. Nat. Rev. Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- Claesson-Welsh L. Welsh M. J. Intern. Med. 2013;273:114–127. doi: 10.1111/joim.12019. [DOI] [PubMed] [Google Scholar]
- Siemann D. W. Cancer Treat. Rev. 2011;37:63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W. Chen Z. Zhang W. Cheng Y. Zhang B. Wu F. Wang Q. Wang S. Rong D. Reiter F. P. De Toni E. N. Wang X. Signal Transduction Targeted Ther. 2020;5:87. doi: 10.1038/s41392-020-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J. Li J. Wu L. Yu Q. Ji J. Wu J. Dai W. Guo C. J. Exp. Clin. Cancer Res. 2020;39:126. doi: 10.1186/s13046-020-01629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel L. Routaboul J. M. Cheynier V. Lepiniec L. Debeaujon I. Trends Plant Sci. 2007;12:29–36. doi: 10.1016/j.tplants.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Wen K. Fang X. Yang J. Yao Y. Nandakumar S. K. Salem L. M. Cheng K. Curr. Med. Chem. 2021;28:1042–1066. doi: 10.2174/0929867327666200713184138. [DOI] [PubMed] [Google Scholar]
- Fu B. Xue J. Li Z. Shi X. Jiang B.-H. Fang J. Mol. Cancer Ther. 2007;6:220–226. doi: 10.1158/1535-7163.MCT-06-0526. [DOI] [PubMed] [Google Scholar]
- Liu L.-Z. Jing Y. Jiang L. L. Jiang X.-E. Jiang Y. Rojanasakul Y. Jiang B.-H. Biochem. Biophys. Res. Commun. 2011;413:299–305. doi: 10.1016/j.bbrc.2011.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva S. Kim N. D. Chiu K. Franzen C. A. Bergan R. C. Pelling J. C. Mol. Carcinog. 2008;47:686–700. doi: 10.1002/mc.20421. [DOI] [PubMed] [Google Scholar]
- Kremmidiotis G. Leske A. F. Lavranos T. C. Beaumont D. Gasic J. Hall A. O'Callaghan M. Matthews C. A. Flynn B. Mol. Cancer Ther. 2010;9:1562–1573. doi: 10.1158/1535-7163.MCT-09-0815. [DOI] [PubMed] [Google Scholar]
- Lee J. Kim S. J. Choi H. Kim Y. H. Lim I. T. Yang H.-m. Lee C. S. Kang H. R. Ahn S. K. Moon S. K. Kim D.-H. Lee S. Choi N. S. Lee K. J. J. Med. Chem. 2010;53:6337–6354. doi: 10.1021/jm1002414. [DOI] [PubMed] [Google Scholar]
- Micheletti G. Poli M. Borsotti P. Martinelli M. Imberti B. Taraboletti G. Giavazzi R. Cancer Res. 2003;63:1534–1537. [PubMed] [Google Scholar]
- Pettit G. R. Lippert J. W. Herald D. L. Hamel E. Pettit R. K. J. Nat. Prod. 2000;63:969–974. doi: 10.1021/np0000623. [DOI] [PubMed] [Google Scholar]
- Rustin G. J. S. Galbraith S. M. Anderson H. Stratford M. Folkes L. K. Sena L. Gumbrell L. Price P. M. J. Clin. Oncol. 2003;21:2815–2822. doi: 10.1200/JCO.2003.05.185. [DOI] [PubMed] [Google Scholar]
- Deng X. Li Z. Xiong R. Liu J. Liu R. Peng J. Chen Y. Lei X. Cao X. Zheng X. Xie Z. Tang G. Biomed. Pharmacother. 2019;109:1659–1669. doi: 10.1016/j.biopha.2018.11.001. [DOI] [PubMed] [Google Scholar]
- Deng X. Pi Y. Li Z. Xiong R. Liu J. Zhao J. Xie Z. Lei X. Tang G. Chem.-Biol. Interact. 2020;327:109186. doi: 10.1016/j.cbi.2020.109186. [DOI] [PubMed] [Google Scholar]
- Liu R. Deng X. Peng Y. Feng W. Xiong R. Zou Y. Lei X. Zheng X. Xie Z. Tang G. Bioorg. Chem. 2020;96:103652. doi: 10.1016/j.bioorg.2020.103652. [DOI] [PubMed] [Google Scholar]
- Sun X. Zhao Y. Zhao J. Xie Z. Lei X. Liu X. Li Y. Huang S. Wang Z. Tang G. Drug Dev. Res. 2023:1–17. doi: 10.1002/ddr.22035. [DOI] [PubMed] [Google Scholar]
- Spitz G. A. Furtado C. M. Sola-Penna M. Zancan P. Biochem. Pharmacol. 2009;77:46–53. doi: 10.1016/j.bcp.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Naaz F. Neha K. Haider M. R. Shafi S. Future Med. Chem. 2021;13:1795–1828. doi: 10.4155/fmc-2020-0385. [DOI] [PubMed] [Google Scholar]
- Sang Y. L. Zhang W. M. Lv P. C. Zhu H. L. Curr. Top. Med. Chem. 2017;17:120–137. doi: 10.2174/1568026616666160530154812. [DOI] [PubMed] [Google Scholar]
- Wan Y. Li Y. Yan C. Yan M. Tang Z. Eur. J. Med. Chem. 2019;183:111691. doi: 10.1016/j.ejmech.2019.111691. [DOI] [PubMed] [Google Scholar]
- Wang Z. Deng X. Xiong R. Xiong S. Liu J. Cao X. Lei X. Chen Y. Zheng X. Tang G. MedChemComm. 2018;9:305–315. doi: 10.1039/C7MD00578D. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.