

Abstract
The cGAS–STING axis plays an important role in protecting higher organisms against invading pathogens or cancer by promoting the production of cytokines and interferons. However, persistent or uncontrolled activation of this pathway could lead to inflamed environments, which is detrimental to the host in the long run. Persistent activation of STING is known to be the cause of STING-associated vasculopathy with onset in infancy (SAVI) and activated STING is believed to play important roles in worsening various diseased states, such as traumatic brain injury, diabetic kidney disease and colitis. Thus, antagonists of STING could play important roles in managing various inflammatory diseases. Herein, we report the discovery of small molecule STING inhibitors, HSD1077 and analogs, which are facilely synthesized via a Povarov–Doebner type three-component reaction involving an amine, ketone, and aldehyde. Structure–activity relationship, SAR, studies indicate that both the 3H-pyrazolo[4,3-f]quinoline and pyrazole moieties in HSD1077 are critical for STING binding. At concentrations as low as 20 nM, HSD1077 suppressed type-1 interferon expression in both murine RAW macrophages and human THP-1 monocytes upon treatment with 100 μM 2′-3′ cGAMP. Compounds containing the 3H-pyrazolo[4,3-f]quinoline moiety have the potential to be translated into anti-inflammatory compounds via STING inhibition.
The cGAS–STING axis plays an important role in protecting higher organisms against invading pathogens or cancer by promoting the production of cytokines and interferons.
Introduction
The activation of innate immunity is critical for mounting swift responses toward adverse events such as pathogenic infections or cellular damage.1 The sensing of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by pattern recognition receptors allows for downstream activation and induction of pro-inflammatory cytokine expression such as type 1 interferon, nuclear factor kappa-beta (NF-κβ) and other cytokines such as interleukins 1 and 6, which play vital roles in T cell priming and activation.1,2 As a signalling motif indicating cellular damage or an invading DNA-containing pathogen, cytosolic double stranded DNA is recognized by the DNA sensor cyclic GMP–AMP synthase (cGAS),3 initiating 2′-3′ cyclic GMP–AMP (cGAMP) production through the catalysis of phosphodiester bond formation between adenosine triphosphate (ATP) and guanosine triphosphate (GTP).4 Binding of cGAMP to the universal cyclic dinucleotide sensor protein, Stimulator of interferon genes (STING), results in the recruitment of tank binding kinase I (TBK1), which activates the transcription factor interferon regulatory factor 3 (IRF3) via phosphorylation.1,5,6 Following IRF3 activation and translocation to the nucleus, IRF3 serves as a transcriptional activator that promotes the potent induction of type 1 interferons, which are critical towards mounting appropriate immune responses against pathogenic invasion as illustrated6,7 (Fig. 1).
Fig. 1. cGAS–STING activation pathway. The cGAS pathway is activated by double-stranded DNA (dsDNA) from bacteria, viruses, cells undergoing apoptosis, damaged tumour cells and mitochondria. Activated cGAS catalyzes the formation of a second messenger, cGAMP using ATP and GTP as substrates. cGAMP binds to STING localized on the ER membrane and promotes STING translocation to the Golgi apparatus. The translocation recruits and activates TBK1, which in turn catalyzes the phosphorylation and nuclear translocation of IRF3. The phosphorylation and nuclear translocation activate interferon stimulation genes, which induces inflammatory genes and subsequently, cytokines. STING also induces nuclear factor kappa B (NF-kB) activation which induces proinflammatory cytokines upon nuclear translocation through binding of IκB kinase (IKK).

Despite the good side of the cGAS–STING activation, aberrant activation and dysfunction of this axis leads to chronic upregulation of cytokine expression which have been shown to play critical roles in the development of chronic autoimmune disorders.8–10 Constitutive activation of STING due to gain of function (GOF) mutations11 plays a critical role in the development of debilitating diseases such as STING associated vasculopathy with onset in infancy (SAVI), characterized by symptoms such as prominent vascular lesions and pulmonary inflammation.12–14
Alternatively, dysfunctions in cytosolic nucleic acid clearance mechanisms, as in the case of TREX1 exonuclease loss of function leading to DNA accumulation and chronic cGAS–STING activation,15 play major roles in the onset and development of other autoimmune disorders such as Aicardi–Goutières syndrome (AGS).16
With further mounting evidence suggesting implication of the cGAS–STING–TBK1 axis in various diseases where inflammation contributes towards disease onset,17 the cGAS–STING–TBK1 pathway has been viewed as an attractive target towards the amelioration of associated symptoms correlated with these autoimmune disorders and spurred discovery of therapeutic moieties seeking to modulate the activities of cGAS, TBK1 or STING.16–18 Solved crystal structures of human cGAS has since facilitated the recent identification of potent cGAS inhibitors such as compound S3 by Zhao et al.19 (IC50 = 4.9 μM against human cGAS) and CU-76 by Padilla-Salinas and colleagues20 (IC50 = 0.27 μM against murine cGAS) through respective in silico virtual screening methodologies and subsequent optimization. Similarly, the selective targeting of TBK1 has been reported by both academic and pharma scientists. The recent development of an efficacious TBK1 targeting PROTAC 3i by Crew et al.21 featuring an impressive half degradation constant (DC50) of 12 nM, or discovery of GSK8612 as a potent and highly selective small molecule TBK1 inhibitor by Thomson and co-workers22 are examples of work in the TBK1 inhibitor arena. Yet, with further discovery of possible STING activation and contribution towards immune responses via non-canonical means, independent of cGAS and TBK1, such as via DNA associated protein IFI16 pathway,23 direct modulators of STING could have immune modulating profiles that are distinct from inhibitors of cGAS or TBK1. To date, a small handful of small molecule-based inhibitors of STING have been discovered, primarily based on the use of cell-based quantification of cytokines downstream of the cGAS–STING pathway such as interferon-β. This is achieved either through reporter cell bioassays or strategies such as the quantification of cytokine mRNA expression via quantitative Polymerase chain reaction (qPCR) analyses, as with the discovery of previous STING antagonists such as C-176, H-151 and astin C (Fig. 2).24–27 While highly effective, the utility of cell-based screening can be complemented with other novel assays or in silico screening methodology as a first line strategy to improve efficiency towards the high-throughput screening of large library compounds. Indeed, the identification of antagonists such as SN-011 and C-18 were complemented by in silico virtual screening efforts or other screening strategies such as affinity selection-mass spectrometry (AS-MS) to reduce the number of compounds needed for downstream cellular-based based analyses (Fig. 2).28,29 Our group has a long standing interest in the identification of inhibitors of the cGAS–STING pathway and we reported earlier that the antiparasitic drug, suramin, is a potent inhibitor of cGAS.30 Herein, we report a small molecule inhibitor of STING, HSD1077, which is cell permeable and capable of suppressing type-1 interferon expression in both murine RAW macrophages and human THP-1 monocytes.
Fig. 2. Structures of reported small molecule STING antagonists.

Results and discussion
Identification of a hit compound, HSD1077 as a STING binder
Taking advantage of the binding properties of STING towards cyclic dinucleotides, our group has previously reported the development of a STING-based fluorescence polarization assay for the efficient monitoring of the activities of cyclic dinucleotide metabolizing enzymes such as cyclases and phosphodiesterases.31 Binding of a commercially available fluorescein conjugated cyclic-di-GMP probe 2′-O-(6-[fluoresceinyl]aminohexylcarbamoyl)-cyclic-di-GMP (hereby known as fluorophore-c-di-GMP or F-c-di-GMP) towards STING was shown to induce increased fluorescence polarization, due to reduced rotation of the fluorophore when bound to STING. STING binders, such as c-di-GMP, c-di-AMP or cGAMP were shown to be capable of competing with fluorophore-c-di-GMP towards STING binding; displacement of fluorophore-c-di-GMP from STING resulted in decreased fluorescence polarization.31 We rationalized that the reported STING-based fluorescence polarization assay could also be repurposed for the identification of other small molecule (non-nucleotide) competitive binders of STING (Fig. 3). As a proof of concept, we utilized our proprietary compound library (synthesized via the Povarov–Doebner type multicomponent reaction)30–34 to screen for STING inhibitors. This yielded an initial hit compound, HSD1077 (see Fig. 4 for structure). HSD1077 could compete with 50 nM of fluorophore-c-di-GMP towards STING binding, with half maximal inhibitory concentration (IC50) of 10.65 μM (Fig. 4B).
Fig. 3. Probe displacement by STING binder. A) Structure of F-c-di-GMP probe. B) Schematic diagram depicting the repurposing of the developed FP assay for the potential screening of small molecule binders of STING, which competitively displace F-c-di-GMP.

Fig. 4. A) Synthesis of quinoline compounds using the Povarov–Doebner type multicomponent reaction and structure of hit compound HSD1077. B) Plot depicting anisotropy values versus concentration of HSD1077 shows dose-dependent competitive probe displacement. Concentrations of probe: 50 nM; STING: 10 μM.

SAR evaluation of HSD1077 analogues
With the quinoline HSD1077 identified as a novel scaffold that could bind to STING, resulting in competitive displacement of a fluorescent cyclic dinucleotide ligand, we sought to identify the salient motifs present in HSD1077 that could play critical roles towards STING binding. Binding efficiency of the compound towards STING was represented in the form of fraction of F-c-di-GMP bound (see Fig. 5B), where a low fraction of bound probe (fluorophore-c-di-GMP) suggests competitive binding of fluorophore-c-di-GMP with a competing small molecule, resulting in reduced fraction of fluorophore-c-di-GMP bound to STING.
Fig. 5. A) Analogues modified at ring A tested for STING binding through STING-FP assay. B) Fraction of probe bound to STING (Fbound) upon incubation with listed drug compounds. Fluorophore-c-di-GMP was used at a concentration of 50 nM, STING at 10 μM. All compounds (ADU-S100, DIABZI-3 and HSD1077) were used at 20 μM concentrations.

Initially, we sought to understand the importance of the 3H-pyrazolo[4,3-f]quinoline moiety, which contains ring A (see Fig. 4A), towards STING binding, and generated compounds that acted as isosteres of the hit compound with vital changes around ring A (Fig. 5A). As a preliminary screen, 20 μM of compounds and 50 nM of the probe was incubated with 10 μM of STING for 5 minutes prior to evaluation of fluorescence anisotropy. As positive controls, incubations of potent STING binding agonists ADU-S100 and DiABZI compound 3 (hereafter known as DIABZI-3) were also performed, resulting in expected high probe displacement from STING.
It was observed in general that the pyrazolo moiety of ring A was critical for STING binding. Deletion of the pyrazolo moiety present in HSD1077, as shown in analogue 1, resulted in an abrogation of STING binding. The direct replacement of the pyrazolo moiety with a dimethoxy or dioxolo moieties (compounds 2 and 3, Fig. 5) also resulted in a significant decrease in STING binding, compared to HSD1077. Fluorine substitution in the 3H-pyrazolo[4,3-f]quinoline moiety at position 5, (compound 4), was tolerated, with a modest reduction in STING binding compared to HSD1077. Changing the pyrazolo moiety in HSD1077 into pyrrolo (5) or imidazo (6), also led to a significant decrease in STING binding, highlighting the essentiality of the pyrazolo moiety for effective STING binding. Interestingly, another pyrazolo-containing compound 7 (1H-pyrazolo[4,3-h]quinoline) was a poor STING binder, compared to HSD1077, which is a 3H-pyrazolo[4,3-f]quinoline-containing compound. Compound 8, a 3-methyl-3H-pyrazolo[4,3-f]quinoline compound did not bind to STING as well as HSD1077, suggesting that the different functional group vectors in HSD1077 are important for STING binding.
HSD1077 contains a saturated six-membered ring (labelled ring B in Fig. 4A) and we sought to determine the essentiality of this moiety. The replacement of the cyclohexyl rings with a cyclopropyl group, in the case of 9 resulted in reduced STING binding. STING binding was restored with other ring systems (cyclopentyl or cycloheptyl rings), as observed in 10 and 11, with 12 harbouring the cyclopentyl ring being slightly less effective in binding to STING in comparison to HSD1077.
The incorporation of heteroatoms such as O and S, as seen in compounds 13 and 14, onto the saturated cyclohexyl ring decreased binding to STING binding.
Regarding substitution to the cyclohexyl ring, the incorporation of a non-polar substituent such as a methyl group in 14 did not affect STING binding but polar substituents such as amine, or alcohol moieties such as 15 and 16 were not well tolerated, with regards to STING binding (Fig. 6).
Fig. 6. A) Analogues modified at ring B tested for STING binding through STING-FP assay. B) Fraction of probe bound to STING (Fbound) upon incubation with listed drug compounds. Fluorophore-c-di-GMP was used at a concentration of 50 nM, STING at 10 μM. All compounds (ADU-S100, DIABZI-3 and HSD1077) were used at 20 μM concentrations.

Next, we explored modifications of the pyrazole ring C, where the pyrazole ring was replaced with other heterocycles (Fig. 7). The presence of the pyrazole ring was initially deemed to be critical in STING binding, as its deletion in compound 17 led to a significant decrease in STING binding. It was noted that C-3 alkylation of the pyrazole ring, in the form of methyl, t-butyl and trifluoromethyl substitutions as observed in 18, 19 and 20 led to reduced binding capabilities of the compounds towards STING. Nitrogen hopping within the pyrazole ring, as shown in 21 and 22, similarly negatively impacted STING binding. The replacement of a pyrazole to a triazole, as shown in 23 also led to a similar decrease in STING binding. The replacement of the pyrazole moiety with nitrogen containing aryl moieties such as pyridine and pyrimidine groups, as in the case of 24 and 25 also negatively affected STING binding. N1 methylation of the pyrazole ring, analogue compound 26, did not affect STING binding. These findings suggests that the overall electronic environment of ring C, especially the N-2 position, which is largely similar in both HSD1077 and 26, plays vital roles towards drug binding to STING. The tolerability of the N1 position towards modification opens the possibility of finding other analogs, which may inhibit STING. In future, we will explore the substitution of the N1 position of the pyrazole with other groups and investigate how such substitutions would impact STING binding and drug-like properties.
Fig. 7. A) Analogues modified at ring C tested for STING binding through STING-FP assay. B) Fraction of probe bound to STING (Fbound) upon incubation with listed drug compounds. Fluorophore-c-di-GMP was used at a concentration of 50 nM, STING at 10 μM. All compounds (ADU-S100, DIABZI-3 and HSD1077) were used at 20 μM concentrations.

HSD1077 attenuates type 1 interferon expression in murine RAW macrophages
Having identified HSD1077 as a STING binder that could competitively displace cyclic dinucleotides, we sought to determine if treatment of HSD1077 would modulate the cGAS–STING pathway through STING binding in cells.
To understand possible modulatory effects of HSD1077 quantification of type 1 interferon expression in-cellulo upon HSD1077 treatment via a commercially available murine macrophage cell line RAW ISG Blue, which express secreted embryonic alkaline phosphatase (SEAP) under the control of an interferon stimulated gene 54, (ISG54), inducible promoter. Upon the stimulation of pathways, such as cGAS–STING which activate type 1 interferon production, SEAP would be expressed and can be quantified using a chromogenic detection substrate Quantiblue™. Treatment of RAW ISG-blue cells with HSD1077 did not induce type 1 interferon production, suggesting that HSD1077 did not act as an agonist in the cGAS–STING pathway (Fig. 8A).
Fig. 8. HSD1077 inhibits interferon regulatory factor activation. A) Treatment of HSD1077 to RAW ISG Blue (blue) for 24 hours showed non-significant changes in interferon expression levels. Pre-treatment of HSD1077, followed by cGAMP induction results in attenuated expression of type 1 interferon in a dose dependent manner. B) RAW ISG cells were pre-treated with either HSD1077 or H-151 as a positive control for 6 hours, and subsequently stimulated with 100 μM of cGAMP for 3 hours for induction of interferon-β. mRNA levels was quantified by RT-PCR. Gene expression was normalized with β-actin. Experiments were performed in two biological replicates. Error bars indicate error of the mean of two independent experiments.

Moreover, pre-treatment of HSD1077 (1 μM or lower, tolerated doses by macrophage) prior to STING activation with the natural ligand cGAMP was shown to result in a dose dependent attenuation in type 1 interferon expression, suggesting that HSD1077 modulates the cGAS–STING axis as a STING antagonist. Excitedly, concentrations as low as 20 nM caused noticeable decrease in interferon expression (Fig. 8).
As a secondary analysis, mRNA levels of murine interferon-β were also quantified via quantitative PCR (qPCR). HSD1077 pre-treatment, followed by cGAMP stimulation on RAW macrophages similarly results in a dose dependent reduction in murine interferon-β levels as compared to DMSO treated cell samples stimulated with cGAMP, confirming our findings through the Quantiblue assay (Fig. 8B).
HSD1077 treatment leads to decreased IRF3 phosphorylation in RAW macrophages
Given that the phosphorylation of IRF3 represents a highlight of the STING–TBK1–IRF3 axis, we also investigated and compared the phosphorylation of STING and IRF3 in HSD1077 treated and untreated samples of RAW ISG cells. The results showed that pre-treatment of HSD1077 at 5 μM for 4 hours prior to cGAMP stimulation showed decreased levels of phospho-STING and phospho-IRF3 when compared to samples pre-treated with DMSO prior to cGAMP stimulation. The results obtained confirms previous studies that showed that the inhibition of the STING pathway represents a mode of action for attenuating type 1 interferon (Fig. 9).
Fig. 9. HSD1077 attenuates STING and IRF3 phosphorylation in Raw ISG cells. Raw ISG cells were treated with 5 μM HSD1077 or 1 μM H151 for 4 h, followed by 100 μM cGAMP treatment for 3 h. Control cells were treated with DMSO and sterile water. p-STING and p-IRF3 levels were analysed using western blotting. The experiment was done in two biological replicates.

HSD1077 attenuates type 1 interferon expression in human THP-1 monocytes
Given that murine STING (mSTING) and human STING (hSTING) have only 61% amino acid identity in the ligand binding domain,35 this could lead to differences between mSTING and hSTING in terms of drug binding and interactions to STING. This was exemplified in the case of the mSTING agonist DMXAA, which was unable to illicit agonistic effects in hSTING.11 As such, we sought to investigate if HSD1077 would similarly result in an inhibition of type 1 interferon expression upon cGAMP stimulation in human derived THP-1 monocytes. Using THP-1 dual cell lines (Invivogen) featuring a luciferase gene under the control of an ISG54 inducible promoter, relative quantifications of type 1 interferon expression could be achieved through luciferase detection upon cGAS–STING pathway induction. Our results showed that pre-treatment of THP-1 monocytes with HSD1077 prior to cGAMP stimulation showed reduced type 1 interferon expression in a dose dependent manner similar to results obtained with murine RAW macrophage pre-treatment (Fig. 10A). This suggests that HSD1077 administration to macrophages results in the attenuation of type 1 interferon in both murine and human cell lines and can target both mSTING and hSTING.
Fig. 10. HSD1077 attenuates interferon induction in human THP-1 cells A) pre-treatment of HSD1077 to THP-1 dual monocytes for 2 hours, prior to induction of interferon by 2′-3′ cGAMP stimulation for 24 hours results in attenuated expression of interferon in a dose dependent manner. B) Treatment of HSD1077 to THP-1 dual (KI STING N154S) with a point mutation resulting in a gain of function showed a decrease in interferon expression levels upon 24 hours incubation.

In autoimmune diseases featuring chronic inflammation such as SAVI, plausible contributory factors include the expression of constitutionally active STING mutants, such as N154S or V155M STING isoforms. This results in chronically upregulated expression of type 1 interferons without the need for ligand activation, leading to undesired inflammatory symptoms.35 Hence, we sought to also identify if HSD1077 could reduce type 1 interferon expression in cell models expressing constitutively active STING isoforms. Upon treatment of HSD1077 to THP-1 dual cells expressing a knock in constitutionally active STING (N154S) isoform and detection of expressed luciferase corresponding to, it was observed that upon a 24 hour treatment of THP-1 dual monocytes expressing a knock-in STING (N154S) phenotype with HSD1077 and quantification of Lucia luciferase expressed using the luminescence reagent QuantiLuc™, a 42% reduction in luminescence was observed when compared to DMSO control (Fig. 10B), suggesting that HSD1077 was indeed able to modulate type 1 interferon expression through STING inhibition, even in cell lines with constitutive STING activation.
Conclusions
With the implication of the cGAS–STING pathway in inflammatory diseases, such as autoimmune and neurodegenerative diseases, there is a need for new therapeutic agents for the modulation of the cGAS–STING pathway. Herein, we describe the identification of a novel quinoline-containing compound capable of binding to STING and resulting in competitive displacement of cyclic dinucleotides. Initial SAR efforts have shown that the 3H-pyrazolo[4,3-f] ring moiety and pyrazole heterocycle present on the hit compound HSD1077 are important for STING binding. However, N-methyl substitution on the pyrazole ring was shown to be tolerated and future studies will explore the limit of the pyrazole substitution, with an eye toward which substitution would afford a good balance between drug-like properties and STING antagonism. Cellular assays have determined HSD1077 to act as a STING antagonist, suppressing type 1 interferon expression and downregulating IRF3 phosphorylation upon treatment in murine RAW macrophage. A potential limitation of HSD1077 is that while the compound is effective at inhibiting STING signaling at tolerable concentrations of 1 μM and lower (see Fig. 8), at high micromolar concentrations, we observed some cell death after 24 h. Ongoing work is therefore aimed at developing analogs that retain STING inhibition but possess better therapeutic windows than HSD1077. These privileged quinoline compounds could serve as a basis for the potential development of inhibitors of the cGAS–STING axis for the management and treatment of chronic autoimmune diseases such as SAVI and AGS, in which the cGAS–STING pathway is implicated.
Experimental
Expression and affinity purification of hSTING
The cloned plasmid harbouring hSTING gene (PET28a, SUMO) was obtained from Dr. Li Pingwei (Texas A&M University) as a gift and transformed into E. coli Rosetta™ 2(pLysS) cells. Kanamycin (50 μg mL−1) and chloramphenicol (32 μg mL−1) was employed as selection factors. A single colony was picked and grown in 10 mL of LB media with selection factors incorporated and incubated at 37 °C overnight. The culture was then inoculated into 1 L of terrific broth with kanamycin and chloramphenicol supplementation and grown at 37 °C till exponential phase was reached, where OD600 = 0.6. Protein expression was induced through the addition of isopropyl-b-d-thiogalactopyranoside to a 1 mM final concentration. The culture was incubated at 25 °C for 18 hours. Obtained cells were pelleted via centrifugation for 30 minutes at 5000 rpm. The obtained bacteria pellet was then resuspended in 25 mL of lysis buffer (50 mM Na3PO4, 300 mM NaCl, 20 mM imidazole, 5 mM 2-mercaptoethanol, 10% glycerol and 1× complete protein inhibitor cocktail (Roche, Basel, Switzerland)). Cells were lysed through sonication and the lysate centrifuged at 22 000 rpm for 25 minutes, followed by collection of the supernatant. The supernatant containing hSTING was purified by passing through a HisTrap-HP column (Cytiva) and pure hSTING eluted with elution buffer containing 50 mM Na3PO4 (pH = 7.4), 300 mM NaCl, 300 mM imidazole, 5 mM 2-mercaptoethanol, 10% glycerol. The purified hSTING was dialyzed for 24 hours in a dialysis buffer containing 50 mM Na3PO4 (pH = 7.4), 300 mM NaCl, 5 mM β-mercaptoethanol and 10% glycerol for the removal of imidazole, and quantified via absorbance measurements at λ = 280 nm with ε = 47 955 M−1 cm−1.
STING based fluorescence polarization assay
50 nM of 2′-Fluo-AHC-c-diGMP (Biolog) was incubated with 10 μM hSTING and 20 μM of screening compounds in dimethyl sulfoxide (DMSO) for 5 minutes in 1× phosphate buffered saline at room temperature. Fluorescence polarization (λex/em = 485/528 nm) was then quantified via Biotek Cytation 5 multi-mode reader, with anisotropy calculated using Gen5 microplate reader and imaging software. Anisotropy was normalized via equating measurements with 0 μM hSTING to zero. Experiments was performed in triplicates using 384 Greiner-Bio 384 fluorometric plate (flat plate). Anisotropy was converted to fraction bound by F-c-di-GMP using the following equation: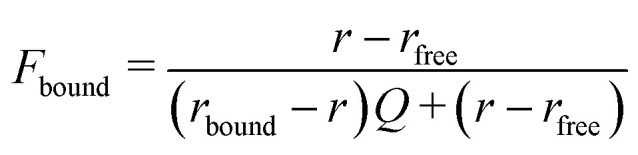 where r refers to the anisotropy value at 10 μM hSTING concentration upon compound addition, rfree refers to the anisotropy value of unbound fluorophore, and Q refers to ratio of fluorescence intensities between bound and free fluorophore. Anisotropy changes with increasing concentration of HSD1077 was plotted, with a 4-parameter dose–response fit applied to obtain the reported IC50via GraphPad Prism (San Diego, CA, USA).
where r refers to the anisotropy value at 10 μM hSTING concentration upon compound addition, rfree refers to the anisotropy value of unbound fluorophore, and Q refers to ratio of fluorescence intensities between bound and free fluorophore. Anisotropy changes with increasing concentration of HSD1077 was plotted, with a 4-parameter dose–response fit applied to obtain the reported IC50via GraphPad Prism (San Diego, CA, USA).
General synthesis of library compounds
General considerations
Solvents and reagents were obtained from commercial sources and utilized without further purification. 1H and 13C NMR spectra presented were obtained in methanol-d4 or DMSO-d6 using a Bruker AV500 (500 MHz) or AV800 (800 MHz) spectrometer with tetramethylsilane as an internal standard. 1H NMR data were reported as shown: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Chemical shifts were reported in downfield order in parts per million (δ ppm). Multiplicities are reported as follows: s = singlet, brs = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or combinations thereof. Electron spray ionization (ESI) technique and TOF mass analysis were used to record high-resolution mass spectra (HRMS). All the synthesized compounds were characterized using 1H, 13C and HRMS.
Synthesis of HSD1077 and analogues
The quinoline library was prepared through a previously established methodology, unless otherwise mentioned.26 Briefly, in a 20 mL screw capped glass vial, the corresponding amine (1 mmol) and aldehyde (1 mmol) were refluxed in absolute ethanol (5 mL) for 2 hours. After that the reaction was cooled to room temperature followed by addition of corresponding ketone (2.5 mmol) and a catalytic amount of conc. hydrogen chloride. Further reaction was allowed to reflux for an additional 6 to 12 hours. Upon completion, the reaction mixture was concentrated and purified using silica gel column chromatography (hexanes : ethyl acetate 50 : 50 to 0 : 100) or ethyl acetate/methanol (99 : 01 to 80 : 20).
7-(1H-Pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (HSD1077)
See reference for experimental details.32
6-(1H-Pyrazol-4-yl)-7,8,9,10-tetrahydrophenanthridine (1)
Yellow solid (99 mg, 40%) 1H NMR (500 MHz, DMSO-d6) δ 8.22 (brs, 1H), 8.08 (brs, 1H) 7.96 (d, J = 4.2 Hz, 1H), 7.89 (d, J = 3.9 Hz, 1H), 7.26 (td, J = 7.1, 1.2 Hz, 1H), 7.50 (td, J = 5.7, 1.2 Hz, 1H), 3.13 (t, J = 6.3 Hz, 2H), 2.94 (t, J = 6.0 Hz, 2H), 1.90–1.87 (m, 2H), 1.81–1.78 (m, 2H); 13C NMR (125 MHz, DMSO-d6): 153.06, 145.73, 141.85, 140.20, 129.46, 128.67, 128.24, 126.22, 126.02, 123.19, 121.61, 28.40, 25.75, 22.86, 22.01. HRMS (ESI) m/z calcd for C16H15N3 [M + H]+ 250.1344, found 250.1343.
2,3-Dimethoxy-6-(1H-pyrazol-4-yl)-7,8,9,10-tetrahydrophenanthridine (2)
Off-white solid (133 mg, 43%) 1H NMR (500 MHz, DMSO-d6): δ 8.16 (s, 1H), 7.99 (s, 1H), 7.25 (s, 1H), 7.17 (s, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.06 (t, J = 6.4 Hz, 2H), 2.89 (t, J = 6.0 Hz, 2H), 1.90–1.85 (m, 2H), 1.80–1.77 (m, 2H); 13C NMR (125 MHz, DMSO-d6): 151.46, 150.46, 149.18, 142.51, 140.87, 140.40, 126.09, 121.83, 121.25, 108.41, 101.66, 55.94, 55.37, 28.26, 26.05, 23.05, 22.16. HRMS (ESI) m/z calcd for C18H19N3O2 [M + H]+ 310.1555, found 310.1557.
5-(1H-Pyrazol-4-yl)-1,2,3,4-tetrahydro-[1,3]dioxolo[4,5-b]phenanthridine (3)
Brown solid (58 mg, 20%) 1H NMR (500 MHz, DMSO-d6) δ 8.15 (brs, 1H), 7.98 (brs, 1H), 7.30 (s, 1H), 7.22 (s, 1H), 6.15 (s, 2H), 2.99 (t, J = 5.9 Hz, 2H), 2.87 (t, J = 5.5 Hz, 2H), 1.86–1.83 (m, 2H), 1.78–1.77 (m, 2H); 13C NMR (125 MHz, DMSO-d6): 150.61, 149.58, 147.48, 143.65, 141.04, 139.81, 129.39, 126.41, 122.72, 121.63, 105.62, 102.08, 99.10, 28.17, 26.22, 22.93, 22.17 HRMS (ESI) m/z calcd for C17H15N3O2 [M + H]+ 294.1242, found 294.1244.
5-Fluoro-7-(1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (4)
See reference for experimental details.34
7-(1H-Pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrrolo[3,2-a]phenanthridine (5)
Yellow solid (147 mg, 51%). 1H NMR (500 MHz, DMSO-d6) δ 12.38 (s, 1H), 8.37 (s, 2H), 8.13–7.93 (m, 2H), 7.72–7.54 (m, 1H), 7.15 (s, 1H), 3.39 (t, J = 6.4 Hz, 2H), 2.97 (t, J = 6.2 Hz, 2H), 2.02–1.93 (m, 2H), 1.82–1.76 (m, 2H); 13C NMR (200 MHz, DMSO-d6) δ 150.92, 144.49, 140.42, 135.86, 134.05, 133.38, 131.51, 128.59, 126.08, 122.16, 119.80, 115.93, 114.52, 106.42, 31.00, 28.12, 21.98, 21.78; HRMS (ESI) m/z calcd for C18H17N4 [M + H]+ 289.1453, found 289.1451.
7-(1H-Pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-imidazo[4,5-a]phenanthridine (6)
Light brown solid (80 mg, 27%). 1H NMR (800 MHz, DMSO-d6) δ 8.59–8.47 (m, 3H), 8.27 (d, J = 9.2 Hz, 1H), 8.07 (d, J = 9.3 Hz, 1H), 3.78–3.59 (m, 2H), 3.03–2.90 (m, 2H), 1.99–1.87 (m, 2H), 1.83–1.75 (m, 2H); 13C NMR (200 MHz, DMSO-d6) δ 152.88, 145.51, 142.11, 140.01, 136.34, 134.55, 132.16, 129.47, 119.54, 116.19, 112.73, 31.20, 27.90, 21.68, 21.45. HRMS (ESI) m/z calcd for C17H16N5 [M + H]+ 290.1405, found 290.1405.
5-(1H-Pyrazol-4-yl)-6,7,8,9-tetrahydro-3H-pyrazolo[4,3-c]phenanthridine (7)
Pale brown solid (84 mg, 29%) 1H NMR (500 MHz, DMSO-d6): δ 8.54 (s, 1H), 8.23 (brs, 2H), 7.86 (d, J = 4.2 Hz, 1H), 7.63 (d, J = 4.5 Hz, 1H), 2.97 (t, J = 7.7 Hz, 2H), 2.96 (t, J = 5.2 Hz, 2H), 1.90 (d, J = 2.5 Hz, 2H), 1.82 (d, J = 2.5 Hz, 2H); 13C NMR (125 MHz, DMSO-d6): 152.12, 142.89, 141.10, 135.54, 133.91, 126.39, 122.26, 121.94, 121.03, 119.96, 111.37, 28.34, 26.56, 22.90, 22.24. HRMS (ESI) m/z calcd for C17H15N5 [M + H]+ 304.1562, found 304.1565.
3-Methyl-7-(1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a] phenanthridine (8)
Off-white solid (81 mg, 27%) 1H NMR (500 MHz, DMSO-d6): δ 8.45 (s, 1H), 8.22 (brs, 1H), 8.06 (brs, 1H), 7.93 (d, J = 4.7 Hz, 1H), 7.83 (d, J = 4.6 Hz, 1H), 4.15 (s, 3H), 2.99 (t, J = 6.0 Hz, 2H), 2.48 (t, J = 1.6 Hz, 2H), 1.97 (quin, J = 2.3 Hz, 2H), 1.84 (quin, J = 2.4 Hz, 2H); 13C NMR (125 MHz, DMSO-d6): δ 149.95, 143.57, 142.13, 140.96, 139.98, 138.04, 134.85, 129.57, 129.12, 121.60, 120.82, 117.30, 113.68, 36.24, 29.82, 22.42. HRMS (ESI) m/z calcd for C18H17N5 [M + H]+ 304.1562, found 304.1565.
9-Cyclopropyl-7-(1H-pyrazol-4-yl)-3H-pyrazolo[4,3-f]quinoline (9)
Brown solid (66 mg, 24%) 1H NMR (500 MHz, methanol-d4): δ 8.79 (s, 1H), 8.31 (s, 2H), 7.94–7.86 (m, 2H), 7.67 (s, 1H), 1.35–1.28 (m, 2H), 1.02–1.00 (m, 2H); 13C NMR (125 MHz, methanol-d4): 149.76, 149.20, 143.05, 128.51, 123.68, 122.54, 119.50, 118.99, 117.33, 114.66, 110.48, 99.65, 39.01, 29.34, 15.86. HRMS (ESI) m/z calcd for C16H13N5 [M + H]+ 276.1249, found 276.1248.
7-(1H-Pyrazol-4-yl)-3,8,9,10-tetrahydrocyclopenta[c]pyrazolo[4,3-f]quinoline (10)
Off-white solid (115 mg, 42%) 1H NMR (500 MHz, DMSO-d6): δ 8.40 (s, 1H), 8.28 (brs, 1H), 8.15 (brs, 1H), 7.85–7.79 (m, 2H), 3.47 (2H, brs), 3.27 (t, J = 7.4 Hz, 2H), 2.32 (quin, J = 7.6 Hz, 2H); 13C NMR (125 MHz, DMSO-d6): 149.00146.75, 144.64, 138.80, 137.96, 134.86, 129.01, 128.48, 122.38, 118.35, 116.99, 114.36, 33.41, 32.78, 24.09. HRMS (ESI) m/z calcd for C16H13N5 [M + H]+ 276.1249, found 276.1252.
7-(1H-Pyrazol-4-yl)-3,8,9,10,11,12-hexahydrocyclohepta[c]pyrazolo[4,3-f]quinoline (11)
Brown solid (39 mg, 15%) 1H NMR (methanol-d4) δ 7.96 (s, 1H), 7.56 (s, 2H), 7.19 (d, J = 4.35 Hz, 1H) 6.86 (d, J = 4.4 Hz, 1H), 4.04 (d, J = 4.0 Hz, 1H), 3.03 (d, J = 4.3 Hz, 1H), 2.85 (d, J = 1.6 Hz, 1H), 2.23 (t, J = 8.3 Hz, 1H), 1.74–1.72 (m, 2H), 1.65–1.60 (m, 2H), 1.38–1.36 (m, 1H), 1.10 (d, J = 5.0 Hz, 1H); 13C NMR (125 MHz, methanol-d4): δ 149.73, 143.70, 142.13, 139.91, 138.49, 136.15, 129.62, 129.17, 128.90, 121.64, 121.00, 116.55, 114.16, 49.06, 29.85, 28.66, 22.64, 22.48. HRMS (ESI) m/z calcd for C18H17N5 [M + H]+ 304.1562, found 304.1563.
7-(1H-Pyrazol-4-yl)-3,8,10,11-tetrahydropyrano[3,4-c]pyrazolo[4,3-f]quinoline (12)
White solid (75 mg, 26%) 1H NMR (500 MHz, methanol-d4): δ 8.55 (s, 1H), 8.07 (s, 2H), 7.91–7.84 (m, 2H), 4.99 (s, 2H), 4.22 (t, J = 4.4 Hz, 2H) 3.41 (brs, 2H); 13C NMR (125 MHz, methanol-d4): δ 149.87, 146.49, 145.79, 143.68, 139.77, 136.12, 132.18, 128.68, 128.06, 127.39, 120.99, 119.91, 76.08, 66.82, 64.04, 54.83, 31.66, 29.03, 29.00, 28.61. HRMS (ESI) m/z calcd for C16H13N5O [M + H]+ 292.1198, found 292.1200.
7-(1H-Pyrazol-4-yl)-3,8,10,11-tetrahydropyrazolo[4,3-f]thiopyrano[3,4-c]quinoline (13)
Pale brown solid (120 mg, 38%) 1H NMR (500 MHz, DMSO-d6): δ 8.56 (s, 1H), 8.22 (brs, 1H), 8.11 (brs, 1H), 7.87–7.81 (m, 2H), 4.11 (s, 2H), 3.59 (brs, 2H), 3.13 (t, J = 6.0 Hz, 2H); 13C NMR (125 MHz, DMSO-d6): 148.93, 144.06, 141.91, 138.64, 136.30, 129.61, 126.76, 121.38, 121.01, 116.25, 114.86, 31.44, 29.04, 25.66. HRMS (ESI) m/z calcd for C16H13N5S [M + H]+ 308.0969, found 308.0974.
9-Methyl-7-(1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (14)
Yellow solid (118 mg, 39%) 1H NMR (500 MHz, DMSO-d6): δ 8.49 (s, 1H), 8.17 (brs, 1H), 8.05 (brs, 1H), 7.81 (s, 2H), 3.27–3.21 (m, 1H), 3.00 (dd, J = 8.3 Hz, 3.7 Hz, 1H), 2.68–2.63 (m, 1H), 2.09 (brs, 1H), 1.82 (brs, 1H), 1.55 (hept, J = 6.0 Hz, 1H), 1.11 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 149.72, 143.71, 141.80, 140.03, 138.49, 136.14, 129.56, 128.63, 121.63, 120.82, 116.58, 114.19, 36.89, 30.43, 29.79, 28.58, 22.08. HRMS (ESI) m/z calcd for C18H17N5 [M + H]+ 304.1562, found 304.1560.
2-(7-(1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-9-yl)isoindoline-1,3-dione (S-1)
Off-white solid (191 mg, 44%). 1H NMR (800 MHz, DMSO-d6) δ 8.78–8.66(m, 1H), 8.43–8.21 (m, 3H), 8.16 (s, 1H), 7.96–7.77 (m, 4H), 4.65–4.50 (m, 1H), 3.78–3.65 (m, 2H), 3.60 (s, 1H), 3.24 (dd, J = 16.3, 5.1 Hz, 1H), 2.86–2.66 (m, 1H), 2.43–2.26 (m, 1H); 13C NMR (200 MHz, DMSO-d6) δ 168.37, 145.76, 138.71, 136.17, 134.70, 132.15, 128.66, 123.46, 123.34, 121.47, 118.25, 115.45, 46.51, 31.19, 30.76, 25.36; HRMS (ESI) m/z calcd for C25H19N6O2 [M + H]+ 435.1569, found 435.1565.
7-(1H-Pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-9-amine (15)
Synthesized from substrate S-1. In a 20 mL reaction vial substrate S-1 (150 mg, 0.34 mmol) was dissolved in methanol (4 mL) followed by addition of hydrazine monohydrate (0.5 mmol). Reaction was refluxed for 5 hours. After completion, the reaction was concentrated to dryness and purified via silica gel chromatography to get the desired deprotected compound. Off-white solid (79 mg, 75%). 1H NMR (800 MHz, DMSO-d6) δ 8.67–8.51 (m, 3H), 8.22 (s, 2H), 7.93 (s, 2H), 3.57 (s, 1H), 3.52–3.40 (m, 2H), 3.36–3.29 (m, 1H), 3.23–3.11(m, 1H), 2.49–2.40 (m, 1H), 2.12–2.01 (m, 1H); 13C NMR (200 MHz, DMSO-d6) δ 147.96, 146.45, 143.58, 141.84, 140.30, 138.61, 136.00, 131.28, 125.57, 120.64, 118.77, 115.85, 46.27, 32.26, 28.23, 25.82. HRMS (ESI) m/z calcd for C17H17N6 [M + H]+ 305.1514, found 305.1512.
7-(1H-Pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridin-9-ol (16)
Off-white solid (100 mg, 26%). 1H NMR (500 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.12 (brs, 2H), 7.88–7.68 (m, 2H), 4.92 (d, J = 3.3 Hz, 1H), 4.06 (td, J = 7.2, 3.6 Hz, 1H), 3.47–3.26 (m, 2H), 3.19 (td, J = 15.5, 14.4, 3.7 Hz, 1H), 2.9 1(dd, J = 16.5, 7.1 Hz, 1H), 2.14 (q, J = 6.5 Hz, 1H), 1.93 (dq, J = 12.7, 6.7, 5.9 Hz, 1H); 13C NMR (125 MHz, DMSO) δ 149.91, 143.78, 141.67, 138.53, 136.18, 129.47, 127.02, 121.51, 120.60, 116.53, 114.35, 64.71, 37.54, 30.26, 27.71. HRMS (ESI) m/z calcd for C17H16N5O [M + H]+ 306.1354, found 306.1356.
8,9,10,11-Tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (17)
See reference for experimental details.33
7-(3-Methyl-1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (18)
See reference for experimental details.32
7-(3-(tert-Butyl)-1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (19)
See reference for experimental details.32
7-(3-(Trifluoromethyl)-1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (20)
See reference for experimental details.32
7-(1H-Pyrazol-3-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (21)
Off-white solid (135 mg, 47%). 1H NMR (500 MHz, DMSO-d6) δ 8.61 (s, 1H), 8.03–7.94 (m, 2H), 7.91–7.77 (m, 1H), 6.92 (s, 1H), 3.31 (t, J = 6.5 Hz, 2H), 3.08 (t, J = 6.2 Hz, 2H), 1.96 (q, J = 6.2 Hz, 2H), 1.85–1.71 (m, 2H); 13C NMR (200 MHz, DMSO-d6) δ 147.10, 145.88, 138.36, 137.58, 136.14, 135.75, 130.88, 130.30, 122.28, 121.82, 117.39, 115.57, 107.43, 30.32, 28.20, 21.97, 21.86; HRMS (ESI) m/z calcd for C17H16N5 [M + H]+ 290.1405, found 290.1403.
7-(1H-Imidazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (22)
See reference for experimental details.38
7-(1H-1,2,3-triazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (23)
See reference for experimental details.32
7-(Pyridin-3-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (24)
See reference for experimental details.31
7-(Pyrimidin-5-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (25)
See reference for experimental details.30
7-(1-Methyl-1H-pyrazol-4-yl)-8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a]phenanthridine (26)
Off-white solid (64 mg, 21%). 1H NMR (800 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.44 (d, J = 5.3 Hz, 1H), 8.12 (d, J = 5.4 Hz, 1H), 8.03 (t, J = 7.3 Hz, 1H), 7.96 (t, J = 7.2 Hz, 1H), 3.98 (s, 3H), 3.25–3.08 (m, 2H), 2.94–2.83 (m, 2H), 1.99–1.89 (m, 2H), 1.83–1.75 (m, 2H); 13C NMR (200 MHz, DMSO-d6) δ 148.71, 145.80, 140.35, 138.14, 137.32, 135.56, 133.22, 129.96, 123.44, 121.49, 117.17, 116.65, 115.23, 39.25, 30.37, 28.12, 21.82, 21.74. HRMS (ESI) m/z calcd for C18H18N5 [M + H]+ 304.1562, found 304.1565.
Cell viability of RAW ISG Blue reporter cells upon HSD1077 treatment
RAW ISG Blue macrophage reporter cells (Invivogen) were cultured in DMEM containing 10% heat inactivated foetal bovine serum and 1× penicillin/streptomycin in 37 °C, 5% CO2. 2 × 103 cells were seeded in 96 well plates and incubated for 24 hours to allow for adherence. Cells were then treated with increasing concentrations of HSD1077 for 24 hours. After which, CellTiter-Blue cell viability assay reagent (Promega) was added based on manufacturer recommendations and incubated for 3 hours. Fluorescence (λex/em = 560/590 nm) of each well was quantified via Biotek Cytation 5 multi-mode reader. Experiments were performed in biological triplicates, with data reported as the mean and standard deviation of 3 data points. Readings from cell samples treated with DMSO was normalized to 100%.
Detection of IRF activation in RAW ISG Blue reporter cells
RAW ISG Blue macrophage reporter cells (Invivogen) were cultured in DMEM containing 10% heat inactivated foetal bovine serum and 1× penicillin/streptomycin in 37 °C, 5% CO2. 1 × 105 cells were seeded in 96 well plates and incubated for 24 hours to allow for adherence. After which, cells were pre-treated with the drug compound for 2 hours, followed by the addition of 100 μM of 2′-3′ cGAMP for induction of cGAS–STING pathway and incubated for 24 hours. After which, media from each respective well was collected and for estimation of IRF activation via SEAP colorimetric assay via QUANTI-blue reagent (Invivogen) as per manufacturer protocols, using a clear 96 well flat bottom plate. 20 μL of the media obtained in each individual well was incubated with 180 μL of QUANTI-Blue™ Solution for 6 hours at 37 °C. Absorbance (630 nm) of each well was quantified via Biotek Cytation 5 multi-mode reader. Experiments were performed in biological triplicates, with data reported as the mean and standard deviation of 3 data points.
Detection of IRF activation in THP-1 dual reporter cells
THP-1 dual or THP-1 (STING N154S) dual reporter cells (Invivogen) were cultured in RPMI media containing 10% heat inactivated foetal bovine serum and 1× penicillin/streptomycin in 37 °C, 5% CO2. 1 × 105 cells were seeded in 96 well plates and incubated for 24 hours. After which, cells were pre-treated with the drug compound for 1 hour, followed by 100 μM of 2′-3′ cGAMP for induction of cGAS–STING pathway and incubated for 24 hours. After which, cell suspension from each respective well was collected and for relative quantification of IRF activation via Lucia luciferase based luminescent assay via QUANTI-Luc™ reagent (Invivogen). 10 μL of the cell suspension obtained in each individual well was added to 50 μL of QUANTI-Luc™ Solution. Endpoint luminescence measurements was then obtained via Biotek Cytation 5 multi-mode reader with a 4 second start time, and 0.1 second reading time. Experiments were performed in biological triplicates, with data reported as the mean and standard deviation of 3 data points.
Evaluation of interferon-β mRNA levels in RAW cells via qPCR
RAW ISG Blue macrophages were cultured in DMEM containing 10% heat inactivated foetal bovine serum. 1 × 106 cells were seeded in 6 well plates and incubated for 24 hours to allow for adherence. After 24 hours, cells were pre-treated with HSD1077 or H-151 for 6 hours, followed by 100 μM of 2′-3′ cGAMP for 3 more hours. Cells were subsequently harvested, and RNA extraction was performed via the use of TRIzol reagent (Thermo-fisher). 1 μg of RNA harvested from each sample was used for cDNA synthesis using random hexamers, dNTPs, and superscript II reverse transcriptase (Thermo-fisher). For the qPCR analysis, 2× Quanti-tect SYBR green master-mix (Qiagen) was used as per manufacturer recommendations. The following forward and reverse primers was used for the relative quantification of interferon-β and β-actin.36,37
| Gene | Sequence | |
|---|---|---|
| mIFN-β | Forward | 5′ – GAG TTA CAC TGC CTT TGC CAT CC – 3′ |
| Reverse | 5′ – ACT GTC TGC TGG TGG AGT TCA T – 3′ | |
| β-Actin | Forward | 5′ – TCA TGA AGT GTG ACG TTG ACA TCC GT – 3′ |
| Reverse | 5′ – CCT AGA AGC ATT TGC GGT GCA CGA TG – 3′ | |
Evaluation of pIRF3 levels in RAW cells via western blotting
0.5–1 × 106 Raw ISG cells were seeded in 6 well plates. Post 24 h, cells were treated with HSD1077 or H151 for 4 h, which was followed by 100 μM 2′,3′-cGAMP treatment for 3 h. Cells were then harvested in RIPA lysis buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate) with protease inhibitor cocktail (Roche) and 1 mM phenylmethylsulfonyl fluoride (Thermo-Fisher Scientific). Post sonication, cells were centrifuged at 14 000 × g for 15 min and the supernatant was collected. Protein quantification was done using a Pierce rapid gold BCA protein assay kit (Thermo-Fisher). Next, 10% SDS polyacrylamide gel electrophoresis was performed, and then proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was probed with pSTING (CST #50907), STING (CST, #13647), pTBK1 (CST #5483), TBK1 (CST #3013) pIRF3 (CST #E7J8G), IRF3 (CST #4302) and β-actin (CST #8457) antibodies overnight at 2–8 °C. After overnight incubation, the membrane was further incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at 37 °C for 2 h. SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo-Fisher) was used for signal detection on Azure 300 imaging system (Azure Biosystems).
Author contributions
HOS oversaw the project. ND and WWSO synthesized the compounds. WWSO performed in vitro STING binding assay. WWSO, JL and RC performed cellular assays. All authors contributed to the writing of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Supplementary Material
Acknowledgments
We thank Purdue University for the funding and support. This work was supported in part by the Research Instrumentation Centre in the Department of Chemistry at Purdue University.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00061c
References
- Ritchie C. Carozza J. A. Li L. Annu. Rev. Biochem. 2022;91:599–628. doi: 10.1146/annurev-biochem-040320-101629. [DOI] [PubMed] [Google Scholar]
- Pimkova Polidarova M. Vanekova L. Brehova P. Dejmek M. Vavrina Z. Birkus G. Brazdova A. ACS Infect. Dis. 2023;9:23–32. doi: 10.1021/acsinfecdis.2c00424. [DOI] [PubMed] [Google Scholar]
- Li X. Shu C. Yi G. Chaton C. T. Shelton C. L. Diao J. Zuo X. Kao C. C. Herr A. B. Li P. Immunity. 2013;39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. Ralph E. C. Shanker S. Wang H. Byrnes L. J. Horst R. Wong J. Brault A. Dumlao D. Smith J. F. Dakin L. A. Schmitt D. C. Trujillo J. Vincent F. Griffor M. Aulabaugh A. E. Protein Sci. 2017;26:2367–2380. doi: 10.1002/pro.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Bai X. C. Chen Z. J. Immunity. 2020;53:43–53.6. doi: 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- Yum S. Li M. Fang Y. Chen Z. J. Proc. Natl. Acad. Sci. U. S. A. 2021;118(14):e2100225118. doi: 10.1073/pnas.2100225118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalskov L. Narita R. Andersen L. L. Jensen N. Assil S. Kristensen K. H. Mikkelsen J. G. Fujita T. Mogensen T. H. Paludan S. R. Hartmann R. Nucleic Acids Res. 2020;48:11421–11433. doi: 10.1093/nar/gkaa873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thim-uam A. Prabakaran T. Tansakul M. Makjaroen J. Wongkongkathep P. Chantaravisoot N. Saethang T. Leelahavanichkul A. Benjachat T. Paludan S. Pisitkun T. Pisitkun P. iScience. 2020;23:101530. doi: 10.1016/j.isci.2020.101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Jesus A. A. Marrero B. Yang D. Ramsey S. E. Montealegre Sanchez G. A. Tenbrock K. Wittkowski H. Jones O. Y. Kuehn H. S. Lee C.-C. R. DiMattia M. A. Cowen E. W. Gonzalez B. Palmer I. DiGiovanna J. J. Biancotto A. Kim H. et al. . N. Engl. J. Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena K. K. Mehto S. Nath P. Chauhan N. R. Sahu R. Dhar K. Das S. K. Kolapalli S. P. Murmu K. C. Jain A. Krishna S. Sahoo B. S. Chattopadhyay S. Rusten T. E. Prasad P. Chauhan S. Chauhan S. EMBO Rep. 2020;21:e50051. doi: 10.15252/embr.202050051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. Jin L. Genes Immun. 2019;20:82–89. doi: 10.1038/s41435-018-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedel H. Roers A. Rösen-Wolff A. Luksch H. Clin. Immunol. 2020;216:108466. doi: 10.1016/j.clim.2020.108466. [DOI] [PubMed] [Google Scholar]
- Luksch H. Stinson W. A. Platt D. J. Qian W. Kalugotla G. Miner C. A. Bennion B. G. Gerbaulet A. Rösen-Wolff A. Miner J. J. J. Allergy Clin. Immunol. 2019;144:254–266.e8. doi: 10.1016/j.jaci.2019.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szego E. M. Malz L. Bernhardt N. Rösen-Wolff A. Falkenburger B. H. Luksch H. eLife. 2022;11:e81943. doi: 10.7554/eLife.81943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. Du J. Goodier J. L. Hou J. Kang J. Kazazian H. H. Zhao K. Yu X. F. Nucleic Acids Res. 2017;45:4619–4631. doi: 10.1093/nar/gkx178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decout A. Katz J. D. Venkatraman S. Ablasser A. Nat. Rev. Immunol. 2021;21:548–569. doi: 10.1038/s41577-021-00524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X. Zhang R. Cen S. Zhou J. Eur. J. Med. Chem. 2019;182:111591. doi: 10.1016/j.ejmech.2019.111591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. Zou Y. Zhou X. Zhou J. Front. Immunol. 2022;13:5232. doi: 10.3389/fimmu.2022.954129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W. Xiong M. Yuan X. Li M. Sun H. Xu Y. J. Chem. Inf. Model. 2020;60:3265–3276. doi: 10.1021/acs.jcim.0c00171. [DOI] [PubMed] [Google Scholar]
- Padilla-Salinas R. Sun L. Anderson R. Yang X. Zhang S. Chen Z. J. et al. . J. Org. Chem. 2020;85:1579–1600. doi: 10.1021/acs.joc.9b02666. [DOI] [PubMed] [Google Scholar]
- Crew P. Raina K. Dong H. Qian Y. Wang J. Vigil D. Serebrenik Y. V. Hamman B. D. Morgan A. Ferraro C. Siu K. Neklesa T. K. Winkler J. D. Coleman K. G. Crews C. M. J. Med. Chem. 2018;61:583–598. doi: 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Thomson D. W. Poecker D. Zinn N. Rau C. Stronmer K. Wagner A. J. Graves A. P. Perrin J. Bantscheff M. Duempelfeld B. Kasparcova V. Ramanjulu J. M. Pesiridis G. S. Muelbaier M. ACS Med. Chem. Lett. 2019;10(5):780–785. doi: 10.1021/acsmedchemlett.9b00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy G. Flannery S. M. Almine J. F. Connolly D. J. Paulus C. Jonsson K. L. Jakobsen M. R. Nevels M. M. Bowie A. G. Unterholzner L. Mol. Cell. 2018;71:745–760. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag S. M. Gulen M. F. Reymond L. Gibelin A. Abrami L. Decout A. Heymann M. Van Der Goot F. G. Turcatti G. Behrendt R. Ablasser A. Nature. 2018;559:269–273. doi: 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- Li S. Hong Z. Wang Z. Li F. Mei J. Huang L. Lou X. Zhao S. Song L. Chen W. Wang Q. Liu H. Cai Y. Yu H. Xu H. Zeng G. Wang Q. Zhu J. et al. . Cell Rep. 2018;25:3405–3421.e7. doi: 10.1016/j.celrep.2018.11.097. [DOI] [PubMed] [Google Scholar]
- Hong Z. Mei J. Li C. Bai G. Maimaiti M. Hu H. Yu W. Sun L. Zhang L. Cheng D. Liao Y. Li S. You Y. Sun H. Huang J. Liu X. Lieberman J. Wang C. Proc. Natl. Acad. Sci. U. S. A. 2021;118(24):e2105465118. doi: 10.1073/pnas.2105465118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu T. Altman M. D. Baltus G. A. Childers M. Ellis J. M. Gunaydin H. Hatch H. Ho T. Jewell J. Lacey B. M. Lesburg C. A. Pan B. S. Sauvagnat B. Schroeder G. K. Xu S. ACS Med. Chem. Lett. 2019;10:92–97. doi: 10.1021/acsmedchemlett.8b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. Sooreshjani M. A. Mikek C. Opoku-Temeng C. Sintim H. O. Future Med. Chem. 2018;10(11):1301–1317. doi: 10.4155/fmc-2017-0322. [DOI] [PubMed] [Google Scholar]
- Karanja C. W. Yeboah K. S. Ong W. W. S. Sintim H. O. RSC Chem. Biol. 2020;2:206–214. doi: 10.1039/D0CB00187B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opoku-Temeng C. Dayal N. Hernandez D. E. Naganna N. Sintim H. O. Chem. Commun. 2018;54:4521–4524. doi: 10.1039/C8CC01154K. [DOI] [PubMed] [Google Scholar]
- Opoku-Temeng C. Dayal N. Sooreshjani M. A. Sintim H. O. Bioorg. Chem. 2018;78:418–426. doi: 10.1016/j.bioorg.2018.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal N. Opoku-Temeng C. Hernandez D. E. Sooreshjani M. A. Carter-Cooper B. A. Lapidus R. G. Sintim H. O. Future Med. Chem. 2018;10:823–835. doi: 10.4155/fmc-2017-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal N. Mikek C. G. Hernandez D. Naclerio G. A. Yin Chu E. F. Carter-Cooper B. A. Lapidus R. G. Sintim H. O. Eur. J. Med. Chem. 2019;180:449–456. doi: 10.1016/j.ejmech.2019.06.089. [DOI] [PubMed] [Google Scholar]
- Dayal N. Řezníčková E. Hernandez D. E. Peřina M. Torregrosa-Allen S. Elzey B. D. Škerlová J. Ajani H. Djukic S. Vojáčková V. Lepšík M. Řezáčová P. Kryštof V. Jorda R. Sintim H. O. J. Med. Chem. 2021;64:10981–10996. doi: 10.1021/acs.jmedchem.1c00330. [DOI] [PubMed] [Google Scholar]
- Ding C. Song Z. Shen A. Chen T. Zhang A. Acta Pharm. Sin. B. 2020;10:2272–2298. doi: 10.1016/j.apsb.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiser C. Kim B. Vincent J. Ascano M. Sci. Rep. 2020;10:1–11. doi: 10.1038/s41598-019-56847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. Chaudhuri R. Ong W. W. S. Sintim H. O. ACS Chem. Biol. 2021;16:1663–1670. doi: 10.1021/acschembio.1c00342. [DOI] [PubMed] [Google Scholar]
- Sintim H. O., Dayal N. and Opoku-Temeng C., Inhibitors of kinase networks and uses therof, PCT Int. Appl., 1e190, WO2018183586A1, 2018
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.