

Abstract
Recurrent de novo missense variants in H4 histone genes have recently been associated with a novel neurodevelopmental syndrome that is characterized by intellectual disability and developmental delay as well as more variable findings that include short stature, microcephaly, and facial dysmorphisms. A 4-year-old male with autism, developmental delay, microcephaly, and a happy demeanor underwent evaluation through the Undiagnosed Disease Network. He was clinically suspected to have Angelman syndrome, however molecular testing was negative. Genome sequencing identified the H4 histone gene variant H4C5 NM_003545.4: c.295T>C, p.Tyr99His, which parental testing confirmed to be de novo. The variant met criteria for a likely pathogenic classification and is one of seven known disease-causing missense variants in H4C5. A comparison of our proband’s findings to the initial description of the H4-associated neurodevelopmental syndrome demonstrates that his phenotype closely matches the spectrum of those reported among the 29 affected individuals. As such, this report corroborates the delineation of neurodevelopmental syndrome caused by de novo missense H4 gene variants. Moreover, it suggests that cases of clinically suspected Angelman syndrome without molecular confirmation should undergo exome or genome sequencing, as novel neurodevelopmental syndromes with phenotypes overlapping with Angelman continue to be discovered.
Keywords: H4C5, recurrent, missense, de novo, neurodevelopmental syndrome, microcephaly, tooth anomalies, digit anomalies, Angelman syndrome
Introduction
Neurodevelopmental disorders are a spectrum of conditions affecting brain development during childhood and leading to complex patterns of impairment across motor, cognitive, and neurobehavioral domains (Moreno-De-Luca et al., 2013). Current estimates suggest that 2-5% of children are affected with neurodevelopmental disease, and despite its heterogenous etiology, a monogenic cause is identified in approximately 40% of those who undergo exome sequencing (McRae et al., 2017; Srivastava et al., 2019). The ongoing discovery of disease-associated genes, now totaling approximately 1,500, continues to refine our understanding of distinct neurodevelopmental phenotypes (Leblond et al., 2021).
Deleterious germline variants in genes encoding histones have been found to cause neurodevelopmental syndromes. For instance, heterozygous missense variants in histone 3 family 3A and 3B have been identified to produce phenotype that includes progressive neurologic dysfunction, brain abnormalities and seizures, along with skeletal, genital, and cardiac anomalies (Bryant et al., 2020). Meanwhile, heterozygous truncating variants in HIST1H1E have been shown to underlie a syndrome of intellectual disability and overgrowth (Tatton-Brown et al., 2017).
In addition, a cohort of 29 affected individuals has provided compelling evidence that germline de novo missense variants in histone H4 genes produce a neurodevelopmental syndrome with intellectual disability, developmental delays along with a variable spectrum of growth impairments, craniofacial dysmorphisms, and skeletal abnormalities (Tessadori et al., 2022). These features can have significant phenotypic overlap with other well-established neurologic syndromes.
Here we describe a patient where Angelman syndrome was clinically suspected, however molecular testing was non-diagnostic, which is estimated to occur in approximately 10% of affected patients (Williams et al., 2010). Genome sequencing then revealed a pathogenic variant in H4C5 (HIST1H4E; MIM: 602830), which more closely matches his clinical findings and illustrates the need to pursue comprehensive testing in patients with complex neurodevelopmental disorders.
Materials and methods
Study Enrollment
The proband was evaluated at the University of Miami clinical site of the Undiagnosed Disease Network (UDN). Informed consent was obtained from the affected individual and parents under NIH-UDN protocol (15-HG-0130). The study was approved by the University of Miami Institutional Review Board. All clinical data and biological specimens collected were used and stored in accordance with the ethical standards described in the Declaration of Helsinki protocols.
Genome Sequencing
Genome sequencing was performed using DNA extracted from peripheral blood leukocytes. Specimens were obtained from the proband as well as both parents to facilitate trio-genome analysis. Sequencing was performed at Baylor Genetics as part of the UDN workflow. In brief, the KAPA Hyper Prep kit was used for library and sequence analysis was performed using the Illumina NovaSeq 6000 platform for 150 bp paired-end reads. Average sequenced coverage was greater than 40X across the genome and over 97.5% of target bases had a coverage greater 20X.
Genome sequencing data was processed and aligned to GRCh37/hg19, with filtering and prioritization of variants based on inheritance models, allele frequency and predicted functional impact for the proband using Genesis 2.0 genome analysis software in accordance with previously described protocols (DePristo et al., 2011; McKenna et al., 2010). Copy number variant detection was performed with CNVnator.
Results
Phenotype of the proband
The proband is a 4-year-old male with congenital hypotonia and gross motor delays first observed at 6 months of age. A follow-up assessment performed when he was 1 year old also revealed fine motor and speech delays. By age 3, he was diagnosed with autism spectrum disorder due to lack of verbal communication, social disinhibition, and engagement in stereotypical behaviors. He was also observed to have frequent staring spells, however evaluation with electroencephalographic studies and brain MRI, both of which were interpreted as normal.
On physical evaluation, his height and weight were at the 22nd and 28th percentiles for his age and sex. His head circumference met criteria for microcephaly (z = −2.05), and right-sided plagiocephaly was also noted. Craniofacial dysmorphisms included mild hypertelorism, periorbital fullness, protruding ears, broad nasal tip, deep philtrum, and thin upper lip. Dental anomalies included small, pointed teeth with a gap between upper central incisors and a bifid lateral lower incisor (Figure 1A (i) (ii)). He also exhibited skeletal abnormalities such as tapering fingers, mild genu varus of the right foot and short toes (Figure 1A (iii) (iv)). Neurological assessment was remarkable for the presence of truncal hypotonia and a wide-based gait. He exhibited a happy demeanor.
FIG 1.
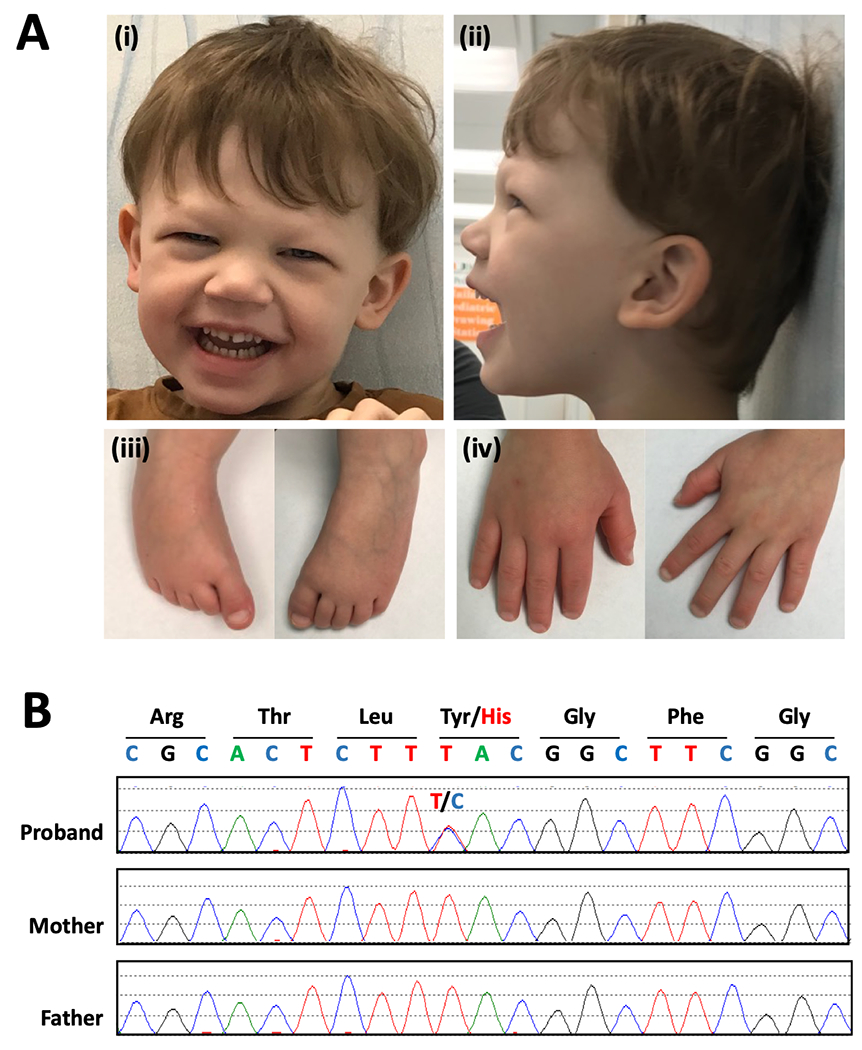
A: Clinical features. (i) (ii): Craniofacial features of the patient at age 4. Note the microcephaly with right-sided plagiocephaly, mild hypertelorism, and broad nasal tip. Dental abnormalities include small, pointed teeth with a gap between upper central incisors, and a bifid lateral lower incisor. Also observed to have periorbital fullness and deep philtrum (iii): Feet of the patient. Right foot with mild genu varus deformity and toes are short bilaterally. (iv): Hands of the patient. B: Molecular findings. Chromatogram of Sanger sequencing in proband and parents, which confirms the proband is heterozygous for the missense variant c.295T>C and that neither parent harbors this variant.
The patient was clinically suspected to have a diagnosis of Angelman syndrome, however molecular genetic testing, which included chromosomal microarray, Fragile X analysis, methylation-specific MLPA for Prader-Willi and Angelman syndromes, exome and mitochondrial sequencing was all negative. Biochemical testing was also non-diagnostic.
He is the second liveborn to a nonconsanguineous couple of Eastern European and Ashkenazi Jewish descent. Both of his parents, his older sister and his younger brother are all healthy.
Molecular analysis of H4C5 variant
Trio genome sequencing of the proband and his unaffected parents revealed that he was heterozygous for a missense variant in H4C5 (NM_003545.4: c.295T>C, NP_003536.1: p.Tyr99His). Neither of his parents harbored the variant, confirming that it occurred de novo in our proband (Figure 1B). The variant is absent in Genome Aggregation Database (gnomAD v2.1.1) (Karczewski et al., 2020). Analysis of the missense variant using in silico prediction tools yielded conflicting assessments. MutationTaster predicted the variant was disease causing with an accuracy of 0.99, however its REVEL score predicted to alteration to be benign with a score of 0.317 (Ioannidis et al., 2016; Schwarz et al., 2014).
Review of the literature demonstrated that the missense variant has previously been reported in two unrelated individuals (Tessadori et al., 2022). In both individuals, the H4C5 p.Tyr99His variant was reported to occur de novo and lead to a neurodevelopmental phenotype with manifestations that overlapped with our proband.
Using the ACMG guidelines for the interpretation of variants, we determined that our proband’s H4C5 missense variant meets criteria for a pathogenic classification (Richards et al., 2015). This was based on our findings that the variant was confirmed to be de novo (PS2), functional studies of the variant in zebrafish show a deleterious effect (PS3), and the variant is absent in population databases (PM2).
Evaluation of our proband’s phenotype compared to the initial H4 cohort
We examined the first and only published report of individuals with neurodevelopmental impairments found to have de novo missense variants in H4 genes (Tessadori et al., 2022). The authors documented the phenotypic spectrum among the 29 individuals in their cohort and noted that all these patients had intellectual disability and developmental delay. Other frequently observed features included microcephaly (69%), visual impairment (61%), short stature and failure to thrive (38%), hypotonia (34%), as well as craniofacial features such as broad nasal tip (38%) and teeth anomalies (21%). We reviewed the complete characteristics of this cohort and identified which of these findings were also seen in our proband (Table 1). Of note, all of our proband’s clinical characteristics were described in the initial H4 cohort of patients. We then compared our proband’s features to the previously described patients with identical p.Tyr99His missense variants. Among all three of affected individuals with p.Tyr99His variants, shared features included intellectual disability, developmental delay, and microcephaly. The remaining characteristics, however, were found to be present in one or two of the individuals, but not all three, emphasizing the syndrome’s variable expressivity.
TABLE 1.
Comparison of our proband’s phenotype to the initial H4 cohort
| H4 COHORT CHARACTERISTICS (Percent affected) | PROBAND FINDINGS | P23 p.Tyr99His (Tessadori et al.)a | P24 p.Tyr99His (Tessadori et al.)a |
|---|---|---|---|
| Neurodevelopment | |||
| Intellectual disability (100%) | Present | Present | Present |
| Developmental delay (100%) | Present | Present | Present |
| Hypotonia (34%) | Present | Present | - |
| Seizures (17%) | - | Present | - |
| Autism (17%) | Present | Present | - |
| Ataxia (14%) | Present | - | - |
| Growth/Skeletal features | |||
| Microcephaly (69%) | Present | Present | Present |
| Short stature (38%) | - | - | Present |
| Failure to thrive (38%) | - | - | Present |
| Craniosynostosis (7%) | - | - | - |
| Digit anomalies (14%) | Present | - | Present |
| Vertebral anomalies (15%) | - | - | Present |
| Facial Features | |||
| Hypertelorism (17%) | Present | - | - |
| Upslanting palpebral fissures (10%) | - | - | - |
| Broad nasal tip (38%) | Present | Present | - |
| Thin upper lip/vermillion (14%) | Present | - | - |
| Teeth anomalies (21%) | Present | Present | - |
| Other Features | |||
| Recurrent infections (14%) | - | - | - |
| Visual impairment (61%) | - | Present | Present |
| Hearing impairment (24%) | - | Present | - |
The p.Tyr99His variant has been previously described as p.Tyr98His in keeping with the convention of not including the first post-translationally removed methionine when numbering amino acid residues, however, here HGSV nomenclature is used.
DISCUSSION
We report here a 4-year-old male with language impairment, developmental delay, autism, hypotonia, wide-based gait, post-natal microcephaly, and a happy demeanor as well as craniofacial dysmorphisms including a gap in upper central incisors. He was clinically suspected of Angelman syndrome; however, his molecular testing was non-diagnostic.
Trio genome sequencing revealed him to be heterozygous for a de novo missense variant in H4C5. Moreover, the proband’s features closely match the initial delineation of the H4-associated neurodevelopmental syndrome based on a cohort of twenty-nine affected individuals, though among them there was considerable variability in phenotype (Tessadori et al., 2022).
This patient illustrates the value of pursuing comprehensive molecular testing with exome or genome sequencing in a patient with a complex neurodevelopmental disorder, even when clinical diagnosis has been made or is highly suspected, as broad phenotypic overlap exists across many neurodevelopmental disorders. And in fact, ACMG guidelines now recommend the use of exome and genome sequencing as a 1st- or 2nd-line test for pediatric patients with developmental delay or intellectual disability due to its high diagnostic yield in this context (Manickam et al., 2021). It should be noted that a previously performed exome sequencing was unrevealing as the causal role of H4C5 variants in developmental disorders was discovered after the exome analysis, which highlights the value of reanalysis for improving the diagnostic yield in patients for whom initial testing is inconclusive (Ewans et al., 2018)
Furthermore, this case independently corroborates the delineation of a novel, H4-associated neurodevelopmental syndrome, as the proband in this report exhibits many of the specific characteristics observed in the initial H4 cohort and harbors the recurrent H4C5 c.295T>C variant, which is one of only seven pathogenic variants of the gene reported in the literature.
Interestingly, the variant produces an amino acid substitution in the C-terminal domain, which is buried in the nucleosome core and believed affect the contact between histone H3-H4 subunits and histone chaperones (Tessarz & Kouzarides, 2014). Missense variants appear to cluster in this region, as well as in and around the core H4 globular domain (Tessadori et al., 2022). And although the mechanism of disease is currently unclear, a dominant mechanism is proving likely, as heterozygous loss-of-function variants have not been associated with the phenotype but are present in the healthy population (Karczewski et al., 2020).
Future studies seeking to clarify this mechanism may reveal the commonalities among the spectrum of neurodevelopmental disorders associated with histones and histone lysine methyltransferases as well as demethylases (Ojaimi et al., 2022). We expect that better understanding the role of histones in the control of human development will be essential to discovering effective therapies.
Acknowledgements
We deeply appreciate our patients, their families, and the staff at the UDN and University of Miami for participating. The findings reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number [1U01HG010230]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
The authors have no conflicts of interest to report.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Bryant L, Li D, Cox SG, Marchione D, Joiner EF, Wilson K, Janssen K, Lee P, March ME, Nair D, Sherr E, Fregeau B, Wierenga KJ, Wadley A, Mancini GMS, Powell-Hamilton N, Kamp J. van de, Grebe T, Dean J, … Bhoj EJ (2020). Histone H3.3 beyond cancer: Germline mutations in Histone 3 Family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients. Science Advances, 6(49), eabc9207. 10.1126/sciadv.abc9207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Angel G. del, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, & Daly MJ. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewans LJ, Schofield D, Shrestha R, Zhu Y, Gayevskiy V, Ying K, Walsh C, Lee E, Kirk EP, Colley A, Ellaway C, Turner A, Mowat D, Worgan L, Freckmann M-L, Lipke M, Sachdev R, Miller D, Field M, … Roscioli T. (2018). Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genetics in Medicine, 20(12), 1564–1574. 10.1038/gim.2018.39 [DOI] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, … Sieh W (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. The American Journal of Human Genetics, 99(4), 877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Le T-L, Malesys S, Cliquet F, Tabet A-C, Delorme R, Rolland T, & Bourgeron T (2021). Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Molecular and Cellular Neuroscience, 113, 103623. 10.1016/j.mcn.2021.103623 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, & DePristo MA (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, Alvi M, Ambridge K, Barrett DM, Bayzetinova T, Jones P, Jones WD, King D, Krishnappa N, Mason LE, Singh T, … Hurles ME (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, & Ledbetter DH (2013). Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. The Lancet Neurology, 12(4), 406–414. 10.1016/s1474-4422(13)70011-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaimi MA, Banimortada BJ, Othman A, Riedhammer KM, Almannai M, & El-Hattab AW (2022). Disorders of histone methylation: Molecular basis and clinical syndromes. Clinical Genetics, 102(3), 169–181. 10.1111/cge.14181 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, & Committee ALQA (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, Firth HV, Frazier T, Hansen RL, Prock L, Brunner H, Hoang N, Scherer SW, Sahin M, Miller DT, & Group, N. E. S. R. W. (2019). Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genetics in Medicine, 21(11), 2413–2421. 10.1038/s41436-019-0554-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, Elliott A, Wylie H, Ardissone A, Rittinger O, Stewart F, Temple IK, Cole T, Collaboration CO, Mahamdallie S, Seal S, Ruark E, & Rahman N (2017). Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. The American Journal of Human Genetics, 100(5), 725–736. 10.1016/j.ajhg.2017.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessadori F, Duran K, Knapp K, Fellner M, Study DDD, Smithson S, Meireles AB, Elting MW, Waisfisz Q, O’Donnell-Luria A, Nowak C, Douglas J, Ronan A, Brunet T, Kotzaeridou U, Svihovec S, Saenz MS, Thiffault I, Viso FD, … Haaften G. van. (2022). Recurrent de novo missense variants across multiple histone H4 genes underlie a neurodevelopmental syndrome. The American Journal of Human Genetics, 109(4), 750–758. 10.1016/j.ajhg.2022.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, & Kouzarides T (2014). Histone core modifications regulating nucleosome structure and dynamics. Nature Reviews Molecular Cell Biology, 15(11), 703–708. 10.1038/nrm3890 [DOI] [PubMed] [Google Scholar]
- Williams CA, Driscoll DJ, & Dagli AI (2010). Clinical and genetic aspects of Angelman syndrome. Genetics in Medicine, 12(7), 385–395. 10.1097/gim.0b013e3181def138 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.