

Abstract
Although cyclopentanone (CPO) is a promising bio-derived fuel, thermodynamic data of its low-temperature oxidation under high-pressure conditions are lacking. In this work, the low-temperature oxidation mechanism of CPO is investigated in a flow reactor in the temperature range of 500–800 K and at a total pressure of 3 atm by a molecular beam sampling vacuum ultraviolet photoionization time-of-flight mass spectrometer. The electronic structure and pressure-dependent kinetic calculations are carried out at the UCCSD(T)-F12a/aug-cc-pVDZ//B3LYP/6-31+G(d,p) level to explore the combustion mechanism of CPO. Experimental and theoretical observations showed that the dominant product channel in the reaction of CPO radicals with O2 is HO2 elimination, yielding 2-cyclopentenone. The hydroperoxyalkyl radical (•QOOH) generated by 1,5-H-shifting is easily reacted with second O2 and forms ketohydroperoxide (KHP) intermediates. Unfortunately, the third O2 addition products are not detected. In addition, the decomposition pathways of KHP during the low-temperature oxidation of CPO are further assessed, and the unimolecular dissociation pathways of CPO radicals are confirmed. The results of this study can be used for future research on the kinetic combustion mechanisms of CPO under high pressure.
Introduction
Bio-derived fuels are considered environment-friendly, safe, and sustainable alternative fuels in practical combustion devices; thus, the potential of these fuels to replace fossil fuels and reduce greenhouse gas emissions has been proposed.1−4 Ketones are one of many types of biofuel molecules. Cyclopentanone (CPO) is a representative five-membered cyclic ketone that can be directly obtained by bio-processes such as cellulose breakdown by fungi5 and biomass pyrolysis.6 CPO can also be used as a precursor for renewable high-density fuels.7 Moreover, CPO has been recognized by the U.S. Department of Energy as one of the eight representative fuel blending components for optimizing gasoline engine performance, with excellent anti-knock properties.8 Given these characteristics, the ketone has received increased research attention in recent years. However, the oxidation mechanism of CPO under high-pressure conditions remains incompletely understood, and this knowledge gap limits its practical applications.
Research on CPO can be traced back to the 1950s. Johnson and co-workers9 found that the thermal decomposition products of CPO in the temperature range of 761–816 K at 1 atm include 2-cyclopentenone, H2, 1-butene, CO, and ethylene. Delles et al.10 studied the vapor-phase thermal decomposition of CPO over the temperature range of 805–854 K and observed 4-pentenal as a product for the first time; the related reaction pathways were then speculated. Zaras et al.11 calculated the unimolecular decomposition pathways of CPO by using the compound G3B3 method and calculated the relevant rate constants in the temperature range of 800–2000 K using RRKM theory. The fastest pathways were found to be keto–enol equilibrium and concerted ring-opening. Recently, Pastoors et al.12 used the imaging photoelectron photoion coincidence spectroscopy to investigate the pyrolysis of CPO at 800–1000 K and 1.1 atm in a microtubular reactor, and the pyrolysis products such as 1,3-butadiene, ketene, propyne, allene, and ethane were observed. Giri et al.13 monitored the thermal decomposition products CO of CPO behind reflected shock waves over 1150–1590 K and 750–1800 Torr by using quantum cascade laser absorption spectroscopy. Dong et al.14 studied the pyrolysis process of CPO at temperatures of 1156–1416 K and pressures of 8.53–10.06 atm using laser absorption spectra. The experimental results indicated that H radicals play a major role in the decomposition, and the whole decomposition process could be divided into three stages based on the H radical concentration. Li et al.15 investigated the thermal decomposition of CPO in a flow reactor at temperatures of 875–1428 K and pressures of 0.04 and 1 atm, and the keto–enol tautomers such as CPO, 1-cyclopentenol, 1,4-cyclopentadien-1-ol, 2-cyclopenten-1-one, and 3-cyclopenten-1-one were observed. However, O2 was not considered in the previous studies. Scheer et al.16 studied the low-temperature oxidation pathways of CPO in a flow tuber reactor between 550 and 700 K and at 4 or 8 Torr total pressure. The corresponding pathways were then analyzed by multiplex photoionization time-of-flight mass spectrometry (TOF-MS), and the experimental results were supported by quantum-chemical calculations at the CBS-QB3 level. Results indicated that the initial oxidation reactions are dominated by HO2 elimination to form 2-cyclopentenone. These findings are consistent with the results of homogeneous charge compression ignition conducted by Yang and Dec17 who found that the autoignition characteristics of CPO are essentially identical to those of ethanol, except that they have lower autoignition reactivity. Zhou et al.18 and Khanniche and Green19 carried out high-precision electronic structure calculations for the H atom abstraction and oxygenation processes of CPO, respectively. Abstraction from the β-H is the dominant process in the presence of OH. CPO oxidation in a jet-stirred reactor between 730 and 1280 K at 1 and 10 atm was studied by Thion et al.,20 who then established the related dynamics model. Reaction pathway analyses showed that H atom abstraction mainly occurs on β-CH2, yielding β-cyC5H7O; this result agrees well with the findings of Zhou.18 Zhang et al.21 investigated the oxidation of CPO by measuring ignition delay times and CO time histories over a wide range of conditions and then developed and validated a detailed kinetic model using the experimental data. The results revealed that the low-temperature reactivity of CPO is largely inhibited by competitive chain propagation processes, especially the elimination of olefin + HO2.
Previous research provided valuable insights into the oxidation mechanism of CPO. It is noted that only a few stable and high-content species were observed in the low-temperature oxidation of CPO. In previous experimental reports, only the first O2 addition products were reported in the low-temperature oxidation of CPO.16 Obviously, the introduction of the C=O group in CPO directly affects the overall reactivity compared to cyclopentane. To fully understand the combustion process of CPO, more combustion products need to be identified. Molecular-beam sampling photoionization time-of-flight mass spectrometery (MB-PI-TOFMS) has incomparable advantages in the diagnosis of microscopic molecular structure and has been widely used in combustion diagnosis. Recently, we have extended the pressure range of the reactor coupled with MB-PI-TOFMS to 3 atm.22 This will allow the detection conditions to more closely resemble the actual operating pressure of the combustion device.
In this work, the low-temperature oxidation of CPO between 500 and 800 K at 3 atm total pressure was investigated in a flow reactor. The products, including 2-cyclopentenone, cyclic ether, ketohydroperoxide (KHP), ethenone, and 2-propenal, were observed by MB-VUV-PI-TOFMS. The combustion mechanism of CPO, including the O2 addition, intra-H migration, second O2 addition, and unimolecular dissociation of the CPO radical, is explored theoretically. The temperature- and pressure-dependent kinetics of the low-temperature oxidation reactions of oxocyclopentyl + O2 are determined using master equation (ME) analysis. To the best of our knowledge, this work provides the first reported second O2 addition of CPO.
Experimental and Theoretical Methods
Experimental Method
The experiment was carried out on a homemade MB-VUV-PI-TOFMS instrument. The detailed descriptions about MB-VUV-PI-TOFMS have been introduced in our recent work;22 only a brief introduction will be provided here. The experimental apparatus includes four main components, i.e., a reaction furnace with a variable pressure flow reactor, a differentially pumped chamber with a molecular-beam sampling system, a MS chamber with a vacuum ultraviolet single-photon ionization source (VUV-SPI), and a homemade reflection time of flight mass spectrometry (RTOF-MS) system. The vacuum of each chamber of the MB-VUV-PI-TOFMS is mainly maintained by a combination of mechanical pump and molecular pump. The molecular beam is formed between the quartz reaction tube and the reaction furnace chamber. The parameters of the molecular beam can refer to our recent report.22 A nozzle of approximately 0.06 mm is horizontally drilled into the wall of the quartz tube by using a short-pulse laser. A cone-like skimmer with a 3 mm orifice is positioned at 2 mm behind the quartz reaction tube to collect the species produced by low-temperature oxidation reactions. Thereafter, the molecular beam enters the VUV-SPI region through a skimmer with a diameter of 2 mm and is ionized by a VUV lamp (L7293, Hamamatsu Photonics K. K., Japan) with 10.78 eV in the ionization region. The ions are introduced vertically into the acceleration region of the RTOF-MS and then detected by a microchannel plate detector.
The low-temperature oxidation reaction takes place in a 365 mm long quartz reaction tube with an inside diameter of 7 mm and an outside diameter of 10 mm, 210 mm of which is placed in a high-temperature furnace. The pressure in the reaction tube is controlled by adjusting the needle valve, and CPO (Sinopharm Chemical Reagent Co., Ltd. purity ≥99.9%) is accurately delivered into the carburetor by a feed pump. Ar (Fujian Yidong Gas Co., Ltd. 99.999%), as the carrier gas, is passed into the quartz reaction tube, and O2 (Fujian Yidong Gas Co., Ltd. 99.999%) is mixed with the reactants at the inlet of this tube. Ar and O2 are controlled by a mass flow controller to a flow rate of 100 standard cubic centimeters per minute (SCCM). The temperature distribution in the horizontal direction of the reactor is measured with a K-type thermocouple. The temperature distribution is directly referred to our previous report.22 Here, CPO oxidation is studied between 500 and 800 K, and the flow rates of Ar, O2, and reactants are 300 SCCM, 180 SCCM, and 0.1 mL/min, respectively. The pressures of the mass spectrometer chamber must be better than 2.0 × 10–4 Pa to ensure the safe operation of the time-of-flight mass spectrometer.
Computational Method
Theoretical calculations are performed to investigate the reaction pathways of the reactions. The geometries of the reactants and products, as well as transition states, are optimized using the hybrid density functional B3LYP23−26 together with the 6-31+G(d,p) basis set, followed by harmonic frequency calculations. Intrinsic reaction coordinates (IRC) are calculated to verify the connectivity between transition states and the corresponding reactants and products. The electronic structure calculations are performed by using the Gaussian 16 C01 software package.27 Single-point energies of stationaries are further calculated at the level of UCCSD(T)-F12a together with the aug-cc-pVDZ basis set, using the MOLPRO 2022 software package.28 Optimized geometries, rotational constants, and vibrational frequencies of all the stationary points are given in the Supporting Information. The single-point energy calculation at the aug-cc-pVDZ level is based on sufficient consideration of computational accuracy and efficiency. The energy differences calculated at different levels between CBS-QB3, aug-cc-pVDZ, and aug-cc-pVTZ are listed in Table S1. The maximum deviation of barrier height calculated by CCSD(T)-F12a/aug-cc-pVDZ and CCSD (T)-F12a/ug-cc-pVTZ was only 0.40 kcal/mol, while the using of the aug-cc-pVTZ method requires significantly more computational resources.
The pressure- and temperature-dependent rate constants are calculated by the MESS29 code using Rice–Ramsberger–Kassel–Marcus/Master equation (RRKM/ME) theory.30 The quantum tunneling effect becomes significant at low temperatures, so in this study, the tunneling effects are considered by the incorporation of asymmetric Eckart corrections.31 The barrier elementary steps in the O2 addition process are studied by using the classical transition state theory (TST). For the barrier-less step of the initial O2 addition process, the variable-reaction-coordinate TST29,32 was used. Due to the change of spin multiplicity during the initial O2 addition process, potential energies along the reaction coordinate were calculated with the explicitly correlated multi-reference configuration interaction (MRCI + Q-F12)33 method realized with the MOLPRO 2022 software package. The MRCI + Q calculations are based on two states of CASSCF wave functions, with the active space chosen as (7e,5o), which includes six electrons in O2 as well as the radical electron in the CPO radical, together with the basis set of cc-pVDZ. For barrier-less channels of the R• + O2 and QOOH + O2, the whole potential energy curve along the reaction coordinate is further scaled by a factor of 0.95 and 1.10, respectively, to match the potential energy change of CCSD(T)-F12a results. Collision energy transfer probability model adopted the single-exponential-down model, ⟨Edown⟩ = 200 × (T/300)0.85 according to Zaras et al.11 The ME calculations are performed at the temperature range 300–1250 K, at pressure of 0.01, 0.1, 1, 3, 10, 100 atm. The collision frequency between the reactant and bath gas Ar is calculated using the Lennard-Jones (L-J) model. The L-J parameter values use in this study: Ar34 σ1 = 3.47 Å, ε1 = 79.2 cm–1, R•11 σ2 = 5.5 Å, ε2 = 650.75 cm–1 are from literature reports, while QOOH σ3 = 5.4 Å, ε3 = 569.30 cm–1 was calculated using the method of Chuang et al.35,36 The low-frequency torsional modes corresponding to internal rotations are simulated as one-dimensional (1-D) hindered rotors with hindrance potentials, which are obtained by a relaxed scan increment of 10° at the B3LYP/6-31+G(d,p) level.
Results and Discussion
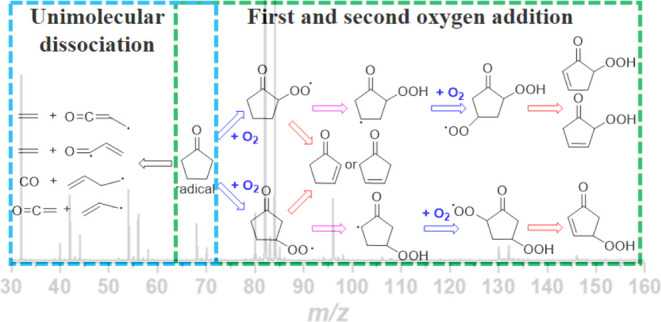
First and Second Oxygen Addition
As shown in Figure 1a, the structure of CPO presents an axis of symmetry centered on the ketone function, with H on both sides of this axis considered equivalent. For clarity, the carbon atom next to the carbonyl group is labeled α and the next carbon atom is labeled β. The C–H bond dissociation energies of CPO are presented at the corresponding sites and calculated at the UCCSD(T)-F12a/aug-cc-pVDZ level. The values obtained are in good agreement with the corresponding values of 90.3 and 97.3 kcal mol–1 computed by Scheer et al.16 via the CBS-QB3 method. The low-temperature oxidation process of the fuel is considered to involve reactions between fuel radical molecules and oxygen. In this study, CPO may collide with the background gas or reactor wall under certain temperature conditions and abstract H to form the first oxocyclopentyl radicals (2-oxocyclopentyl, CPO-2, Figure 1b or 3-oxocyclopentyl, CPO-3, Figure 1c). From an energy point of view, abstracting H from the α site is more dominant than abstracting from the β site. However, Zhou18 reported that abstraction from β-H is the dominant process when OH is involved, but the reverse holds true for the HO2 radical. These results are consistent with the findings of Thion et al.20 Both CPO radicals are expected in significant concentrations, as was also the case in studies of Cl reactions with acyclic ketones.37,38 To understand the low-temperature oxidation process of CPO in more detail, it is necessary to simultaneously consider the reaction process of CPO-2 and CPO-3 with O2.
Figure 1.

(a) Structure of CPO and corresponding C–H bond dissociation energies (kcal/mol); (b) structure of CPO-2; and (c) structure of CPO-3.
H-Shift and HO2-Elimination
Figure 2 shows the mass spectrum of CPO at 500–800 K and 3 atm total pressure. At temperatures below 650 K, only the peak of reactants at m/z = 84 (C5H8O) is observed, which means that the low-temperature oxidation reaction process of CPO has not started. When the temperature reaches 675 K, the dominant product peak is observed at m/z = 82 (Figure 2b), and the relative ion single appears to increase as the temperature increases.
Figure 2.

Product mass spectrum for elimination of CPO at 3 atm total pressure and (a) 500–650 K, (b) 675–800 K.
Figure 3 shows the potential energy surfaces (PESs) for αR• + O2 and βR• + O2 calculated at the UCCSD(T)-F12a/aug-cc-pVDZ//B3LYP/6-31+G(d,p) level, which is generally consistent with previously reported results by Scheer et al.16 The process of O2 addition to α(β) R• and the formation of the corresponding alkylperoxy radicals (α(β) ROO•) is barrierless. The potential well depths for the initially formed adduct α(β) ROO• are 21.82 and 34.02 kcal/mol, respectively. The energy barrier of αR• is significantly lower than that of βR• owing to the presence of a conjugated system of free radical and carbonyl in the αR• radical, which reduces the energy of αR• + O2 and therefore has different reaction behaviors.39 For the αR• + O2 reaction, the energy barrier of the formation of 2-cyclopentenone P1 by the elimination of HO2 by the five-membered ring transition state TS1 of αROO• is 2.46 kcal/mol lower than that of the lowest isomerization pathway TS3. In the βR• + O2 reaction, there are two possible pathways for HO2 elimination: βROO• → P6/P1 + HO2 or βROO• → P6-1 + HO2, which, respectively, produce 2-cyclopentenone P6/P1 or 3-cyclopentenone P6-1. But the energy barrier for the former is more than 5 kcal/mol lower than that of the latter and slightly lower than the lowest isomerization reaction pathway TS9. Therefore, the elimination reaction pathway of α(β) ROO• tends to generate 2-cyclopentenone, which is more dominant than the isomerization reaction. As shown in Figure 2, the intensity of cyclopentenone m/z = 82 is higher than that of other product peaks. α(β)ROO• can form different structures of hydroperoxyalkyl through isomerization reactions: α•Q2OOH, α•Q3OOH, α•Q4OOH; β•Q7OOH, β•Q8OOH, β•Q9OOH. The energy of α•QOOH relative to the entrance αR• reactants is −9.33, −10.52, and −18.24 kcal/mol. And the energy of β•QOOH relative to the inlet βR• reactants is −25.25, −20.74, and −28.03 kcal/mol. According to the PESs of Figure 3, the order of isomerization energy barriers is: 1,5-H shift (TS3, TS9) < 1,5-H shift (TS4) < 1,4-H shift (TS2, TS7, TS8). Therefore, the dominant reaction pathway in the isomerization process starting from α(β)ROO• is to generate α•Q3OOH or β•Q9OOH through the six-membered ring transition state via the 1,5-H shift.
Figure 3.

Relative energy of stationary points on the PESs for (a) αR•+ O2 and (b) βR•+ O2.
Cyclic Ether Formation
Cyclic ether formation with OH elimination occurs through •QOOH via a concerted mechanism with O addition to C and O–O bond scission. Figure 3 shows multiple pathways through which •QOOH produces bicyclic structures with different numbers of atoms for cyclic ether. Although there is another channel for •QOOH to form cyclopentenone by eliminating HO2, the barrier is higher than that for cyclic ether. The barriers for the formation of 3-membered ring (P2, P7, and P8) are lower than those for the formation of 4-membered ring (P3, P9) and 5-membered ring (P4). For the αR• + O2, the energy barrier for the formation of the 3-membered ring P2 +•OH from α•Q2OOH is 10.42 kcal/mol, which is lower than the energy barriers of 22.79 and 29.41 kcal/mol for the formation of the 4-membered ring P3 + •OH and the 5-membered ring P4 +•OH, respectively. For the βR•+ O2, the energy barrier for the formation of the 3-membered ring P8 + OH from β•Q8OOH is 10.79 kcal/mol, lower than the energy barriers of 14.68 and 29.92 kcal/mol for the formation of the three-membered ring P7 + •OH and the four-membered ring P9 + •OH, respectively. Therefore, the •QOOH free radical is more likely to form the 3-membered ring ether bicyclic compound (P2 or P7). There is also a reaction pathway for α(β)ROO• to directly co-eliminate •OH to form the bicyclic tautomer of the cyclic ether (P5 or P10) as a diketone product. However, due to their relatively high reaction barriers, their contribution to the reaction products can be ignored.
Second O2 Addition and Ketohydroperoxide Formation/Decomposition
Although the energy barrier for α(β)ROO• to undergo 1,5-H shift isomerization to form α•Q3OOH and β•Q9OOH is relatively low, the dissociation pathway energy barrier for the subsequent formation of cyclic ether products is higher compared to other dissociation pathways. This makes it difficult to consume and results in its accumulation, leading to secondary oxygenation processes and chain branching reactions. The PESs for the primary reaction pathway of •QOOH with O2 at the CCSD(T)-F12a/aug-cc-pVDZ level are shown in Figure 4. The reaction of •QOOH with O2 is also a barrier-less process, forming adduct products •OOαQ3OOH and •OOβQ9OOH with potential well depths of 31.75 and 21.20 kcal/mol, respectively. For the α•Q3OOH + O2, there are two possible pathways for HO2 elimination from •OOαQ3OOH: •OOαQ3OOH → P11 + HO2, •OOαQ3OOH → P13 + HO2, forming two different structures of hydrogen peroxycyclopentenone P11 and P13, with energy barriers of 23.38 and 28.05 kcal/mol, respectively. The energy barrier of the former is 4.67 kcal/mol lower than that of the latter, and it is also 3.6 kcal/mol lower than the reaction pathway that produces diketohydroperoxide (DKHP) P12. Moreover, the energy of the transition states TS11, TS12, and TS13 is all lower than that of α•Q3OOH at the reaction entrance. From the perspective of reaction kinetics, all of these reaction pathways are favorable, and they tend to eliminate HO2 to generate P11 via a 4-membered ring transition state. •OOαQ3OOH can also undergo 1,4-H shift isomerization to generate HOOαQ14OOH and HOOαQ15OOH, but the energy barriers are higher than other reaction pathways, so their contribution to the secondary oxygenation process can be ignored. For the β•Q9OOH + O2, the energy of all reaction pathways is higher than that of the reaction entrance. The reaction barrier for •OOβQ9OOH to eliminate HO2 via a 5-membered ring transition state to generate P16 is the lowest at 23.53 kcal/mol, and other dissociation pathways are much higher than the HO2-elimination pathway. Therefore, in the secondary oxygenation process of ROO•, αROO• is more dominant and tends to generate P11 or P13 via the HO2-elimination pathway and P12 via the dissociation pathway. However, in the mass spectrum shown in Figure 2, only the product peak of P12 at m/z = 130 was detected, and no peak of hydrogen peroxycyclopentenone (P11 or P13) was detected at m/z = 114 because P11 and P13 have higher reactivity and further decompose. They can form ketone compounds through intramolecular dehydration reaction22 as shown in Figure 5 and the corresponding peak was detected at m/z = 96 in Figure 2. It is worth noting that some of the species in Figure 4 contain cis-trans isomers, and the energy comparison between them is shown in Table S2.
Figure 4.

Relative energy of stationary points of cis conformation on the PESs for (a) α•Q3OOH + O2; (b) β•Q9OOH + O2.
Figure 5.

(a) P11, (b) P13 further decomposition path energy calculated at UCCSD (T)—F12a/aug-cc-pVDZ level.
Unimolecular Dissociation
While numerous theoretical studies on the dissociation process of CPO have been conducted, few experimental reports on the dissociation products of CPO under high pressure are available. In this study, the CPO dissociation mass spectra (Figure 6) are obtained at 725 and 800 K and 3 atm total pressure. Figure 7 illustrates the main unimolecular decomposition pathways of CPO identified in this study. The CPO-2 and CPO-3 radicals initially formed following dissociation can undergo β scission reactions. CPO-2 radicals can ring-open via T20, T21, while CPO-3 ring-open via T22, T23. The energy barriers calculated in this work are consistent with the results reported by Zhou and Zaras et al.11,18 CPO-2 can be dissociated into ethylene (CH2=CH2) and propylene ketone radicals (CH2=CH–C=O) via T20, T20-1 and T21, T21-1, respectively. Therefore, the peak at m/z = 56.03 corresponds to propylene ketone (CH2=CH–CHO) dissociated from the CPO-2 radical. CPO-3 can be dissociated into ethenone (CH2=C=O) and allyl radical (CH2=CH–CH2•) by T22, T22-2 and T23, T23-1, respectively; it can also be decomposed into CO and 3-buten-1-yl radical (CH2=CH–CH2–CH2.) via the T22, T22-1 channel. Therefore, the peaks at m/z = 42.01, 42.05, and 56.06 correspond to ethenone (CH2=C=O), propylene (CH2=CH–CH3), and 1-butene (CH2=CH–CH2–CH3), respectively. 1-Butene (m/z = 56.06) can be observed at 725 K but disappeared at 800 K, whereas propylene (m/z = 42.05) is noted at 800 K but not at 725 K. This finding shows that CPO-3 radicals are easily decomposed to produce 1-butene and CO at low temperatures and ketene and propylene at high temperatures. This is consistent with the calculated dissociation PES in Figure 7b.
Figure 6.

Mass spectra of CPO dissociation products at atm (a) 725, (b) 800 K. The insets are respectively enlarged views of local species at corresponding temperatures.
Figure 7.

(a) CPO-2, (b) CPO-3 reaction energies constructed by the β site C–C fracture process obtained from UCCSD (T)-F12a/aug-cc-pVDZ level of potential energy together with ZPE corrections.
Rate Constants
The reaction process between CPO radicals with molecular oxygen dominates the entire process of low-temperature fuel oxidation, particularly the reaction process involving ROO• radicals, which determines the auto-ignition behavior of the fuel. Figure 8 shows the temperature-dependent rate constants for the main reaction channels during the addition of R• to O2. The reaction channel that forms ROO• through the addition of O2 dominates over the entire low-temperature range, regardless of whether it is the CPO free radical at the α or β position. As the temperature increases, the reaction channels for eliminating HO2 to form cyclopentene and undergoing 1,5-H shift isomerization will become increasingly important, and the competition between the two channels directly reflects the overall reactivity of CPO.
Figure 8.

Temperature-dependent rate constant for the CPO radical with O2 PES at a pressure of 3 atm. Reactions of (a) αR• + O2; (b) βR• + O2; (c) αROO•species; (d) α•QOOH species; (e) βROO•species; (f) β•QOOH species.
For αR• + O2, the reaction rate constant for αROO• eliminating HO2 to form 2-cyclopentenone P1 is greater than that for 1,5-H shift isomerization to form α•Q3OOH reaction channel, with a difference of 100 times at 700 K, and the two reaction rate constants are positively correlated with temperature. Other reaction channels have high reaction barriers and low reaction rates, so their contribution to the overall reaction can be ignored. For βR• + O2, the βROO• has two reaction channels that eliminate HO2 to form 2-cyclopentenone P6 and 3-cyclopentenone P6-1, respectively. At lower temperatures, the reaction channel for forming P6 is favorable, but as the temperature increases, the reaction channel for forming P6-1 will become increasingly important. Higher temperatures will further widen the gap in reaction rates between the reaction channel for HO2 formation of cyclopentenone and 1,5-H shift isomerization to form theα•Q9OOH reaction channel. The reaction rate constant for •QOOH further forming tricyclic ether products (P2, P7, and P8) is greater than that for other reaction channels because the molecular tension of the reaction transition state molecule during the formation process is minimized, which is favorable for the formation of tricyclic ether products. The Arrhenius parameter expanded for the temperature-dependent rate constant is listed in Table S3.
Figure 9 presents the pressure-dependent rate constants for the major reaction channels involved in the R• + O2 addition reaction process. Note that some rate coefficients are lost because at least one of the participating species is reacting faster than the slowest time scale corresponding to internal energy relaxation via collisions. In this case, it is not possible to establish a phenomenological rate coefficient,29,40 instead, the rapidly equilibrating species are merged automatically by MESS, preserving the validity of the rest of the rate coefficients.41 At temperatures below 600 K, the reaction channel that involves the consumption of ROO• by HO2 to form CPO shows almost no pressure dependence. However, at temperatures above 600 K, pressure dependence begins to appear for all reaction channels and shows a positive correlation with pressure. For the reaction channel that generates cyclic ethers, a significant pressure dependence effect exists throughout the entire temperature range.
Figure 9.

Pressure-dependent rate constant for the CPO radical with O2 PES. Reactions of (a) αROO• = P1 + HO2; (b) α•Q2OOH = P2 + •OH; (c) βROO• = P6 + HO2; (d) β•Q8OOH = P8 + •OH.
Figure 10 shows the high-pressure rate constant of the CPO radical secondary oxygenation reaction process. The reaction channel for eliminating HO2 and forming P11 from •OOαQ3OOH via TS11 dominates the entire temperature range. And the reaction channel for eliminating HO2 through TS13 at high temperatures has also become more important. Compared to the reaction channel that eliminates HO2, the rate constant of the reaction channel that forms cyclic ethers through 1,5-H migration isomerization is much lower, and its contribution to the entire reaction can be ignored. Similarly, the formation of P16 via TS16 from •OOβQOOH is dominant over the entire temperature range. Therefore, the generation of hydrogen peroxycyclopentenone is the most favorable path for the secondary oxygenation process of CPO. The Arrhenius parameter expanded for the high-pressure-limit rate constant is listed in Table S4.
Figure 10.

High-pressure-limit rate constant for the CPO radical with O2 PES. Reactions of (a) •OOαQ3OOH species; (b) •OOβQ9OOH species.
Figure 11 shows the rate constant of the unimolecular dissociation of CPO. The rate constants obtained at the UCCSD (T) - F12a/aug-cc-pVDZ level are consistent with the rate constants reported by Zhou et al.18 In the dissociation process of CPO radicals, the rate constant of scission of C1–C5 or C1–C2 bond is significantly higher than that of scission of the C3–C4 bond. It is noteworthy that the reaction rate constant of producing 3-buten-1-yl radial by eliminating CO in CPO-3 radicals is much higher than that of other pathways. This also means that this channel is the main dissociation channel, which is consistent with the reports of Dong et al.14 and Li et al.15 The Arrhenius parameter expanded for the high-pressure-limit rate constant is listed in Table S5.
Figure 11.

High-pressure-limit rate constant of the dissociation process of CPO. Reactions of (a) αR• = W20, αR• = W21, W20 = P20, W21 = P21; (b) βR• = W22, βR• = W23, W22 = P22, W23 = P23. And the CPO radical decomposition reaction channel from Zhou.18
Conclusions
In this work, the low-temperature oxidation of CPO was investigated between 525 and 800 K at 3 atm total pressure by MB-VUV-TOF-MS with a flow reactor. The PESs of the main reaction pathways were theoretically calculated at the UCCSD(T)-F12a/aug-cc-pVDZ//B3LYP/6-31+G(d,p) level. At the same time, the rate constants of each main reaction path in the process of primary oxygenation are calculated by using high-precision quantitative calculations combined with RRKM/ME theory. The primary conclusions included the following:
-
(1)
The initial oxidation reaction tends to eliminate HO2 from ROO• radicals to produce cyclopentenone, while •QOOH preferentially forms 3-membered cyclic ether products.
-
(2)
The •QOOH generated by 1,5-H-translocation has a high energy barrier for the subsequent formation of cyclic ether, which easily reacts with O2 and forms KHP intermediates. The mass spectral peaks of the related products are observed at m/z = 130. Some KHP intermediates with higher activity, such as cyclopentenone hydrogen peroxide (P11, P13, and P16), are not detected, but their related decomposition and conversion products are detected at m/z = 96, especially at high temperatures.
-
(3)
The second cycle of O2 addition to •QOOH preferentially produces ketone intermediates rather than ether intermediates.
-
(4)
The unimolecular dissociation products of CPO, such as ethylene (CH2=CH2), ethenone (CH2=C=O), propylene (CH2=CH–CH3), acrolein (CH2=CH–CHO), and 1-butene (CH2=CH–CH2–CH3), are experimentally and theoretically confirmed. And the dissociation of CPO to ethylene and CO is the main reaction channel.
The results of this study provide a reference for the combustion kinetics of CPO under high pressure. Further investigation of the combustion characteristics of CPO under different pressure conditions is recommended.
Acknowledgments
This work was financially supported by the National Major Scientific Research Instrument Development Project of the National Natural Science Foundation of China (grant no. 21827801) and the National Natural Science Foundation of China (grant nos. 91961107, 22171235, and 22173104). J.C. also thanks the financial support from the Fujian Science and Technology Innovation Laboratory for Optoelectronic Information of China (grant no. 2021ZR109).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02162.
Relative energies of various species in the αR•+O2 reaction with respect to the reactants calculated at the level of CCSD(T)-F12a together with the aug-cc-pVDZ or aug-cc-pVTZ basis set without ZPE level in comparison with the results by Scheer; energy difference between cis/trans isomerization in the second O2 addition; extended Arrhenius parameters for the rate coefficients of the dominant channels of first O2 addition; extended Arrhenius parameters for the rate coefficients of the dominant channels of second O2 addition; extended Arrhenius parameters for the rate coefficients of the dominant channels of unimolecular dissociation; and optimized geometries and frequencies of all the species on the PESs (PDF)
Author Contributions
Yihuang Jiang: conceptualization, methodology, investigation, formal analysis, data curation, writing—original draft, validation, and visualization. Zaifa Shi: conceptualization, methodology, data curation, writing—original draft, validation, and visualization. Jingxiong Yu: conceptualization, methodology, and formal analysis. Di Wu: investigation, data curation, and visualization. Jun Chen: conceptualization, resources, methodology, investigation, supervision, project administration, validation, visualization, writing—review and editing. Lansun Zheng and Zichao Tang: resources, conceptualization, methodology, supervision, and project administration.
The authors declare no competing financial interest.
Supplementary Material
References
- Ma F.; Hanna M. A. Biodiesel production: a review1Journal Series #12109, Agricultural Research Division, Institute of Agriculture and Natural Resources, University of Nebraska–Lincoln.1. Bioresour. Technol. 1999, 70, 1–15. 10.1016/s0960-8524(99)00025-5. [DOI] [Google Scholar]
- Bhatti H.; Hanif M.; Qasim M.; Ataurrehman Biodiesel production from waste tallow. Fuel 2008, 87, 2961–2966. 10.1016/j.fuel.2008.04.016. [DOI] [Google Scholar]
- Jaecker-Voirol A.; Durand I.; Hillion G.; Delfort B.; Montagne X. Glycerin for New Biodiesel Formulation. Oil Gas Sci. Technol. 2008, 63, 395–404. 10.2516/ogst:2008033. [DOI] [Google Scholar]
- Quispe C. A. G.; Coronado C. J. R.; Carvalho J. A. Jr. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renewable Sustainable Energy Rev. 2013, 27, 475–493. 10.1016/j.rser.2013.06.017. [DOI] [Google Scholar]
- Strobel G. A.; Knighton B.; Kluck K.; Ren Y.; Livinghouse T.; Griffin M.; Spakowicz D.; Sears J. The production of myco-diesel hydrocarbons and their derivatives by the endophytic fungus Gliocladium roseum (NRRL 50072). Microbiology 2008, 154, 3319–3328. 10.1099/mic.0.2008/022186-0. [DOI] [PubMed] [Google Scholar]
- Demirbas A. The influence of temperature on the yields of compounds existing in bio-oils obtained from biomass samples via pyrolysis. Fuel Process. Technol. 2007, 88, 591–597. 10.1016/j.fuproc.2007.01.010. [DOI] [Google Scholar]
- Yang J.; Li N.; Li G.; Wang W.; Wang A.; Wang X.; Cong Y.; Zhang T. Synthesis of renewable high-density fuels using cyclopentanone derived from lignocellulose. Chem. Commun. 2014, 50, 2572–2574. 10.1039/c3cc46588h. [DOI] [PubMed] [Google Scholar]
- U.S. Department of Energy . Co-Optima FY17 Year in Review April, 2018; pp 1–44.
- Johnson E. R.; Walters W. D. The thermal decomposition of cyclopentanone. J. Am. Chem. Soc. 1954, 76, 6266–6271. 10.1021/ja01653a012. [DOI] [Google Scholar]
- Delles F. M.; Dodd L. T.; Lowden L. F.; Romano F. J.; Daignault L. G. The Pyrolysis of Cyclopentanone. J. Am. Chem. Soc. 1969, 91, 7645–7647. 10.1021/ja50001a021. [DOI] [Google Scholar]
- Zaras A. M.; Thion S.; Dagaut P. Computational Kinetic Study for the Unimolecular Decomposition of Cyclopentanone. Int. J. Chem. Kinet. 2015, 47, 439–446. 10.1002/kin.20921. [DOI] [Google Scholar]
- Pastoors J. I. M.; Bodi A.; Hemberger P.; Bouwman J. Dissociative Ionization and Thermal Decomposition of Cyclopentanone. Chemistry 2017, 23, 13131–13140. 10.1002/chem.201702376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri B. R.; AlAbbad M.; Barker J. R.; Farooq A. High temperature unimolecular decomposition of cyclopentanone. Proc. Combust. Inst. 2019, 37, 267–273. 10.1016/j.proci.2018.05.076. [DOI] [Google Scholar]
- Dong X.; Ninnemann E.; Ranasinghe D. S.; Laich A.; Greene R.; Vasu S. S.; Green W. H. Revealing the critical role of radical-involved pathways in high temperature cyclopentanone pyrolysis. Combust. Flame 2020, 216, 280–292. 10.1016/j.combustflame.2020.03.001. [DOI] [Google Scholar]
- Li W.; Ye L.; Fang Q.; Zou J.; Yang J.; Li Y. Exploration on Thermal Decomposition of Cyclopentanone: A Flow Reactor Pyrolysis and Kinetic Modeling Study. Energy Fuels 2021, 35, 14023–14034. 10.1021/acs.energyfuels.1c01672. [DOI] [Google Scholar]
- Scheer A. M.; Welz O.; Vasu S. S.; Osborn D. L.; Taatjes C. A. Low temperature (550-700 K) oxidation pathways of cyclic ketones: dominance of HO2-elimination channels yielding conjugated cyclic coproducts. Phys. Chem. Chem. Phys. 2015, 17, 12124–12134. 10.1039/c4cp06097k. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Dec J. E. Bio-Ketones: Autoignition Characteristics and Their Potential as Fuels for HCCI Engines. SAE Int. J. Fuels Lubr. 2013, 6, 713–728. 10.4271/2013-01-2627. [DOI] [Google Scholar]
- Zhou C. W.; Simmie J. M.; Pitz W. J.; Curran H. J. Toward the Development of a Fundamentally Based Chemical Model for Cyclopentanone: High-Pressure-Limit Rate Constants for H Atom Abstraction and Fuel Radical Decomposition. J. Phys. Chem. A 2016, 120, 7037–7044. 10.1021/acs.jpca.6b03994. [DOI] [PubMed] [Google Scholar]
- Khanniche S.; Green W. H. Reaction Pathways, Thermodynamics, and Kinetics of Cyclopentanone Oxidation Intermediates: A Theoretical Approach. J. Phys. Chem. A 2019, 123, 9644–9657. 10.1021/acs.jpca.9b05806. [DOI] [PubMed] [Google Scholar]
- Thion S.; Togbé C.; Dayma G.; Serinyel Z.; Dagaut P. Experimental and Detailed Kinetic Modeling Study of Cyclopentanone Oxidation in a Jet-Stirred Reactor at 1 and 10 atm. Energy Fuels 2016, 31, 2144–2155. 10.1021/acs.energyfuels.6b02061. [DOI] [Google Scholar]
- Zhang K.; Lokachari N.; Ninnemann E.; Khanniche S.; Green W. H.; Curran H. J.; Vasu S. S.; Pitz W. J. An experimental, theoretical, and modeling study of the ignition behavior of cyclopentanone. Proc. Combust. Inst. 2019, 37, 657–665. 10.1016/j.proci.2018.06.097. [DOI] [Google Scholar]
- Shi Z.; Jiang Y.; Yu J.; Chen S.; Chen J.; Tang Z.; Zheng L. Developing the Low-Temperature Oxidation Mechanism of Cyclopentane: An Experimental and Theoretical Study. Chem.—Eur. J. 2022, 28, e202200256 10.1002/chem.202200256. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16; Rev. C.01: Wallingford, CT, 2016;.
- Werner H.-J.; Knowles P. J.; Knizia G.; Manby F. R.; Schütz M. Molpro: a general-purpose quantum chemistry program package. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 242–253. 10.1002/wcms.82. [DOI] [Google Scholar]
- Georgievskii Y.; Miller J. A.; Burke M. P.; Klippenstein S. J. Reformulation and solution of the master equation for multiple-well chemical reactions. J. Phys. Chem. A 2013, 117, 12146–12154. 10.1021/jp4060704. [DOI] [PubMed] [Google Scholar]
- Klippenstein S. J.; Miller J. A. From the Time-Dependent, Multiple-Well Master Equation to Phenomenological Rate Coefficients. J. Phys. Chem. A 2002, 106, 9267–9277. 10.1021/jp021175t. [DOI] [PubMed] [Google Scholar]
- Eckart C. The Penetration of a Potential Barrier by Electrons. Phys. Rev. 1930, 35, 1303–1309. 10.1103/physrev.35.1303. [DOI] [Google Scholar]
- Georgievskii Y.; Klippenstein S. J. Transition State Theory for Multichannel Addition Reactions: Multifaceted Dividing Surfaces. J. Phys. Chem. A 2003, 107, 9776–9781. 10.1021/jp034564b. [DOI] [Google Scholar]
- Shiozaki T.; Knizia G.; Werner H. J. Explicitly correlated multireference configuration interaction: MRCI-F12. J. Chem. Phys. 2011, 134, 034113. 10.1063/1.3528720. [DOI] [PubMed] [Google Scholar]
- Hippler H.; Troe J.; Wendelken H. J. Collisional deactivation of vibrationally highly excited polyatomic molecules. II. Direct observations for excited toluene. J. Chem. Phys. 1983, 78, 6709–6717. 10.1063/1.444670. [DOI] [Google Scholar]
- Chung T. H.; Lee L. L.; Starling K. E. Applications of Kinetic Gas Theories and Multiparameter Correlation for Prediction of Dilute Gas Viscosity and Thermal Conductivity. Ind. Eng. Chem. Res. 1984, 23, 8–13. 10.1021/i100013a002. [DOI] [Google Scholar]
- Chung T. H.; Ajlan M.; Lee L. L.; Starling K. E. Generalized multiparameter correlation for nonpolar and polar fluid transport properties. Ind. Eng. Chem. Res. 1988, 27, 671–679. 10.1021/ie00076a024. [DOI] [Google Scholar]
- Allen J. W.; Scheer A. M.; Gao C. W.; Merchant S. S.; Vasu S. S.; Welz O.; Savee J. D.; Osborn D. L.; Lee C.; Vranckx S.; Wang Z.; Qi F.; Fernandes R. X.; Green W. H.; Hadi M. Z.; Taatjes C. A. A coordinated investigation of the combustion chemistry of diisopropyl ketone, a prototype for biofuels produced by endophytic fungi. Combust. Flame 2014, 161, 711–724. 10.1016/j.combustflame.2013.10.019. [DOI] [Google Scholar]
- Scheer A. M.; Welz O.; Zador J.; Osborn D. L.; Taatjes C. A. Low-temperature combustion chemistry of novel biofuels: resonance-stabilized QOOH in the oxidation of diethyl ketone. Phys. Chem. Chem. Phys. 2014, 16, 13027–13040. 10.1039/c3cp55468f. [DOI] [PubMed] [Google Scholar]
- Meng Q.; Zhao X.; Zhang L.; Zhang P.; Sheng L. A theoretical kinetics study on low-temperature reactions of methyl acetate radicals with molecular oxygen. Combust. Flame 2018, 196, 45–53. 10.1016/j.combustflame.2018.05.023. [DOI] [Google Scholar]
- Miller J. A.; Klippenstein S. J. Master Equation Methods in Gas Phase Chemical Kinetics. J. Phys. Chem. A 2006, 110, 10528–10544. 10.1021/jp062693x. [DOI] [PubMed] [Google Scholar]
- Al Rashidi M. J.; Mehl M.; Pitz W. J.; Mohamed S.; Sarathy S. M. Cyclopentane combustion chemistry. Part I: Mechanism development and computational kinetics. Combust. Flame 2017, 183, 358–371. 10.1016/j.combustflame.2017.05.018. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.