

Abstract
Background
Bipolar androgen therapy (BAT) results in rapid fluctuation of testosterone (T) between near-castrate and supraphysiological levels and has shown promise in metastatic castration-resistant prostate cancer (mCRPC). Its clinical effects may be mediated through induction of DNA damage, and preclinical studies suggest synergy with PARP inhibitors.
Patients and Methods
This was a single-center, Phase II trial testing olaparib plus BAT (T cypionate/enanthate 400 mg every 28 days) with ongoing androgen deprivation. Planned recruitment was 30 subjects (equal proportions with/without homologous recombination repair [HRR] gene mutations) with mCRPC post abiraterone and/or enzalutamide. The primary objective was to determine PSA50 response (PSA decline ≥50% from baseline) rate at 12-weeks. The primary analysis utilized the entire (intent-to-treat [ITT]) cohort, with those dropping out early counted as non-responders. Secondary/exploratory analyses were in those treated beyond 12-weeks (response-evaluable cohort).
Results
Thirty-six patients enrolled and 6 discontinued prior to response assessment. In the ITT cohort, PSA50 response rate at 12-weeks was 11/36 (31%; 95% CI 17%–48%), and 16/36 (44%, 95% CI 28%–62%) had a PSA50 response at any time on-study. After a median follow-up of 19 months, the median clinical/radiographic progression free survival in the ITT cohort was 13.0 months (95% CI 7–17). Clinical outcomes were similar regardless of HRR gene mutational status.
Conclusions:
BAT plus olaparib is associated with high response rates and long PFS. Clinical benefit was observed regardless of HRR gene mutational status.
Keywords: castration-resistant prostate cancer, DNA damage repair, homologous recombination, PARP inhibitor
Introduction
While androgen deprivation therapy (ADT) (i.e., medical/surgical castration) remains the backbone for treating metastatic prostate cancer, it has been long known that testosterone supplementation benefits some men with advanced prostate cancer. Contemporary studies have demonstrated that supraphysiological androgen (SPA) concentrations are associated with an antitumor effect in many preclinical models(1–3). Recently, high-dose testosterone has been translated into clinical use in the form of Bipolar Androgen Therapy (BAT), which involves rapid cycling between supraphysiological and near-castrate serum testosterone levels over a 28-day treatment cycle(2–4).
TRANSFORMER, the largest study evaluating BAT, randomized men with asymptomatic mCRPC who had previously progressed on abiraterone to BAT vs. enzalutamide(4). Overall, this study showed similar outcomes between treatment arms and consequently BAT has not been established as standard of care. TRANSFORMER did, however, provide important insights regarding the clinical utility of BAT. First, BAT appears to mitigate cross-resistance between AR-directed therapies, with response rates to enzalutamide being substantially higher when given post-BAT. Second, BAT appears to be associated with improvement in quality of life (QOL), which is an important consideration given the detrimental effects that ADT has on overall well-being. Thus, ongoing work to develop new approaches aimed at augmenting responses to BAT are justified.
It has been well documented that SPA leads to the induction of double-strand DNA (dsDNA) breaks and that this likely represent a key mechanism by which BAT exerts its clinical effects(1–3). SPA has also been shown to repress the expression of genes involved in DNA repair(3). Moreover, co-treatment with androgens plus the Poly (ADP-ribose) polymerase (PARP) inhibitor olaparib results in increased dsDNA damage and enhanced anti-tumor effects in preclinical models(3). We have also observed that response rates to BAT are higher in patients whose tumors harbor mutations in genes involved in homologous recombination repair (HRR)(3).
On the basis of the aforementioned data, we conducted a Phase II trial to evaluate the effects of BAT plus olaparib in men with mCRPC who had previously progressed on abiraterone and/or enzalutamide.
Subjects and Methods
Trial Design and Patient Population
This was an investigator-initiated, open-label, phase II study evaluating the clinical activity of BAT plus olaparib in patients with asymptomatic mCRPC who had progressed on abiraterone and/or enzalutamide. Our goal was to assess clinical efficacy in a biomarker unselected population. However, in order to explore whether outcomes associated with homologous recombination repair (HRR) gene mutational status, we required that all patients have baseline next-generation sequencing using any clinical grade assay, with the goal of enrolling an equal proportions of patients with/without at least one pathogenic alteration in an HRR gene. Patients with mCRPC harboring mutations in genes previously shown to associate with response to olaparib (e.g. BRCA2, ATM, FANCA, CHEK2, PALB2, CDK12) were assigned to the HRR-deficient group(5). Because genomic factors predictive of response to olaparib were poorly understood when this study was designed, additional alterations in genes associated directly or indirectly in HRR were considered qualifying for the HRR-deficient cohort at the primary investigator’s (MTS) discretion.
Upon enrollment, all patients continued on ADT. Eligible patients then initiated testosterone enanthate/cypionate 400 mg intramuscular every 28 days, which have identical pharmacokinetics and have been shown to result in rapid fluctuations between supraphysiologic and near-castrate serum testosterone levels in men maintained on ADT (2). Patients also received olaparib 300 mg by mouth twice daily. This study was approved by our center’s Institutional Review Board and registered with clinicaltrials.gov (NCT03516812). Informed consent was obtained from all subjects.
Endpoints
PSA was measured each cycle prior to receiving testosterone and imaging studies occurred every 12 weeks. The primary objective was to determine the PSA50 response (i.e. PSA decline ≥50% from baseline) rate after 12 weeks of treatment. Patients were permitted to continue treatment until radiographic or clinical (i.e. increasing cancer-related pain) progression, whichever occurred first(6). Patients were not removed from study for PSA progression alone prior to completing 12 weeks of treatment; however, if a patient did not achieve a PSA50 response after 12 weeks, he had the option to discontinue treatment. Secondary endpoints were to determine the radiographic response rate (per RECIST v1.1), maximum PSA50 response rate (i.e. based on lowest PSA on treatment), clinical/radiographic progression-free survival (crPFS), PSA PFS, overall survival (OS), QOL changes, and safety (per CTCAE v4.0)(6, 7).
For those with a PSA decline on treatment, PSA progression was defined per Prostate Cancer Working Group 3 (PCWG3) criteria as a confirmed increase in PSA ≥25% and ≥2 ng/mL above the nadir(7). Because BAT can cause PSA to initially rise, subjects with no decline in PSA from baseline were considered to have PSA progression if PSA rose ≥25% and ≥2 ng/mL compared to the 12-week PSA value. Radiographic progression was defined as the time from the start of treatment until progression per RECIST v1.1 for soft tissue lesions or PCWG3 criteria for bone lesions(6, 7). Progression endpoints were defined as the time from the start of treatment until progression event or death, whichever occurred first. OS was the time from start of treatment until death from any cause. QOL was assessed using the FACT-P and IIEF surveys(8, 9). Exploratory biomarker analyses included whole exome sequencing to assess for genomic signatures associated with functional loss of HRR activity and immunohistochemical studies(10).
Statistical Considerations
The primary objective was to determine the PSA50 response rate after 12 weeks of therapy. Per protocol, subjects who dropped out of the study prior to 12 weeks were replaced. The primary analysis, safety assessments, and non-stratified survival analyses are reported for the intent-to-treat (ITT) cohort. Other secondary objectives and exploratory analyses were evaluated in the response-evaluable cohort (i.e. patients who did not drop out before 12 weeks). Response rates were compared between subgroups (e.g. HRR-deficient vs intact) using 2-sided exact binomial tests. Survival endpoints were calculated using Kaplan-Meier estimation, and comparisons between subgroups used log-rank tests. Changes in QOL relative to Day 1 were assessed using paired t-tests. Comparisons of functional HRR activity or baseline PSA levels used 2-sided Mann-Whitney rank sum tests.
Sample size
We assumed that BAT monotherapy would results in a PSA50 response rate of 25% in unselected patients with mCRPC(2, 4, 11). At the time of initial study design, clinical data indicated that >60% of men with a variety of HRR gene mutations would have a PSA50 response to olaparib(5). If there was no increased efficacy with combination therapy, we assumed olaparib would drive clinical responses for those with HRR gene mutations, while BAT would drive responses in those with intact HRR pathway. Therefore, given that we required half of the study cohort to have a HRR gene mutation, we hypothesized that the null rate (H0) for combination therapy to be approximately 50%. We sought to detect a 25% improvement over the null rate, corresponding to a PSA50 response rate of ≥75% (H1) for the entire study cohort. Based on these assumptions, a sample size of 30 patients provided 82% power based on a 1-sample test of proportions with a 2-sided α=5%.
Results
Thirty-six patients enrolled and 6 discontinued before 12-weeks. Patients discontinued early for progression (n=2), nausea (n=2), stroke (n=1), and myocardial infarction (n=1). Out of 30 response evaluable patients, 16 had tumors with mutations in genes involved in HRR (i.e. HRR-deficient), while 14 did not (i.e. HRR-intact). Demographic characteristics can be found in Table 1.
Table 1:
Patient Baseline Demographics.
| Characteristic | ITT Cohort (N = 36) |
HRR-Intact (N = 14) |
HRR-Deficient (N = 16) |
|---|---|---|---|
|
| |||
| Age, years (median [IQR]) | 70 (63, 76) | 75 (71, 81) | 67 (61, 72) |
| PSA, ng/mL (median [IQR]) | 26 (13, 114) | 22 (12, 66) | 25 (12, 114) |
| Race | |||
| Black or African American (n [%]) | 1 (2.8%) | 1 (7.1%) | 0 (0%) |
| Native Hawaiian or Other Pacific Islander (n [%]) | 1 (2.8%) | 0 (0%) | 1 (6.2%) |
| White (n [%]) | 25 (69%) | 9 (64%) | 12 (75%) |
| Unknown/Not Reported (n [%]) | 9 (25%) | 4 (29%) | 3 (19%) |
| ECOG performance status | |||
| 0 (n [%]) | 24 (67%) | 9 (64%) | 12 (75%) |
| 1 (n [%]) | 11 (31%) | 4 (29%) | 4 (25%) |
| 2 (n [%]) | 1 (2.8%) | 1 (7.1%) | 0 (0%) |
| Prior Docetaxel in hormone-sensitive setting (n [%]) | 9 (25%) | 2 (14%) | 5 (31%) |
| Prior Abiraterone (n [%])* | 14 (39%) | 5 (36%) | 7 (44%) |
| Prior Enzalutamide (n [%])* | 7 (19%) | 3 (21%) | 1 (6.2%) |
| Prior Abiraterone and Enzalutamide (n [%]) | 11 (31%) | 5 (36%) | 5 (31%) |
| Prior Radium-223 (n [%]) | 12 (33%) | 5 (36%) | 3 (19%) |
| Prior Sipuleucel-t (n [%]) | 13 (36%) | 5 (36%) | 6 (38%) |
Received only the single prior novel hormonal agent indicated (i.e. did not receive both abiraterone and enzalutamide)
Eleven out of 36 (31%, 95% CI 17%–48%) patients in the ITT cohort had a PSA50 response at 12 weeks, which corresponded to a 37% (95% CI 21%–56%) PSA50 response rate in the response-evaluable cohort (N=30) (Figure 1A). Sixteen out of 36 (44%, 95% CI 28%–62%) subjects achieved a PSA50 response as their maximum on-study PSA decline in the IIT cohort, which corresponded to a 53% (95% CI 35%–71%) PSA50 response rate in the response-evaluable cohort (Figure 1B). Thirteen subjects had measurable disease per RECIST v1.1 and were evaluable for a radiographic response. Overall, 7/13 (54%) had a radiographic response (i.e. complete or partial), including 2/13 (15%) complete responses. There was no significant difference in PSA50 or radiographic response rates between the HRR-deficient vs. HRR-intact cohorts (Figure 1A–C).
Figure 1:
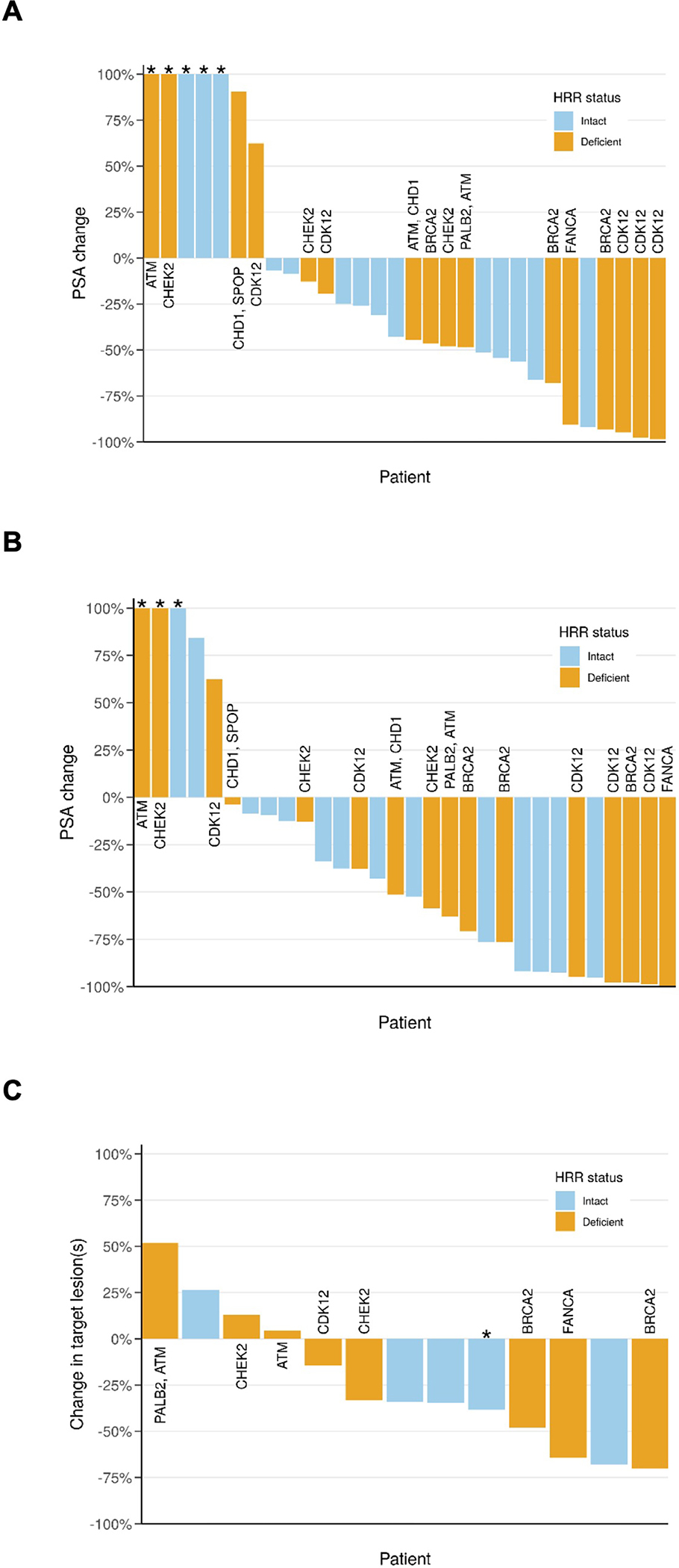
PSA and radiographic response waterfall plots.
(A) PSA change at 12 weeks. PSA50 responses at 12 weeks occurred in 6/16 (38%) patients with HRR-deficient tumors (orange) compared to 5/14 (36%) (P>0.9) patients with HRR-intact tumors (blue). *PSA change >100% was truncated at 100%.
(B) Maximum PSA decline on study for the response evaluate cohort (i.e., patients completing at least 12 weeks of treatment). PSA50 responses at any time on study occurred in 10/16 (63%) patients with HRR-deficient tumors compared to 6/14 (43%) (P=0.3) with HRR intact-tumors. *PSA change >100% was truncated at 100%.
(C) Radiographic response for patients with measurable disease per RECIST v1.1.Radiographic response occurred in 4/8 (50%) patients with HRR-deficient tumors compared to 3/5 (60%) (P=0.8) patients with HRR-intact tumors. *Patient had a partial response in his lymph node metastases while also demonstrating progressive disease in bone lesions.
For the ITT cohort, median follow-up for PSA PFS was 20 months (IQR 6.2 to NR), for crPFS was 19 months (IQR 16 to 22), and for OS was 29 months (IQR 20 to 37). The median PSA PFS in the ITT cohort was 7 months (95% CI 4–11), median crPFS was 13 months (95% CI 7–17), and median OS was 26 months (95% CI 22-NR). In the response-evaluable cohort, there was no significant difference in PSA PFS (p=0.5) or crPFS (p=0.4) between the HRR-deficient vs. HRR-intact cohorts. There was a marginal difference in OS between subgroups (p=0.07) (Figure 2).
Figure 2:
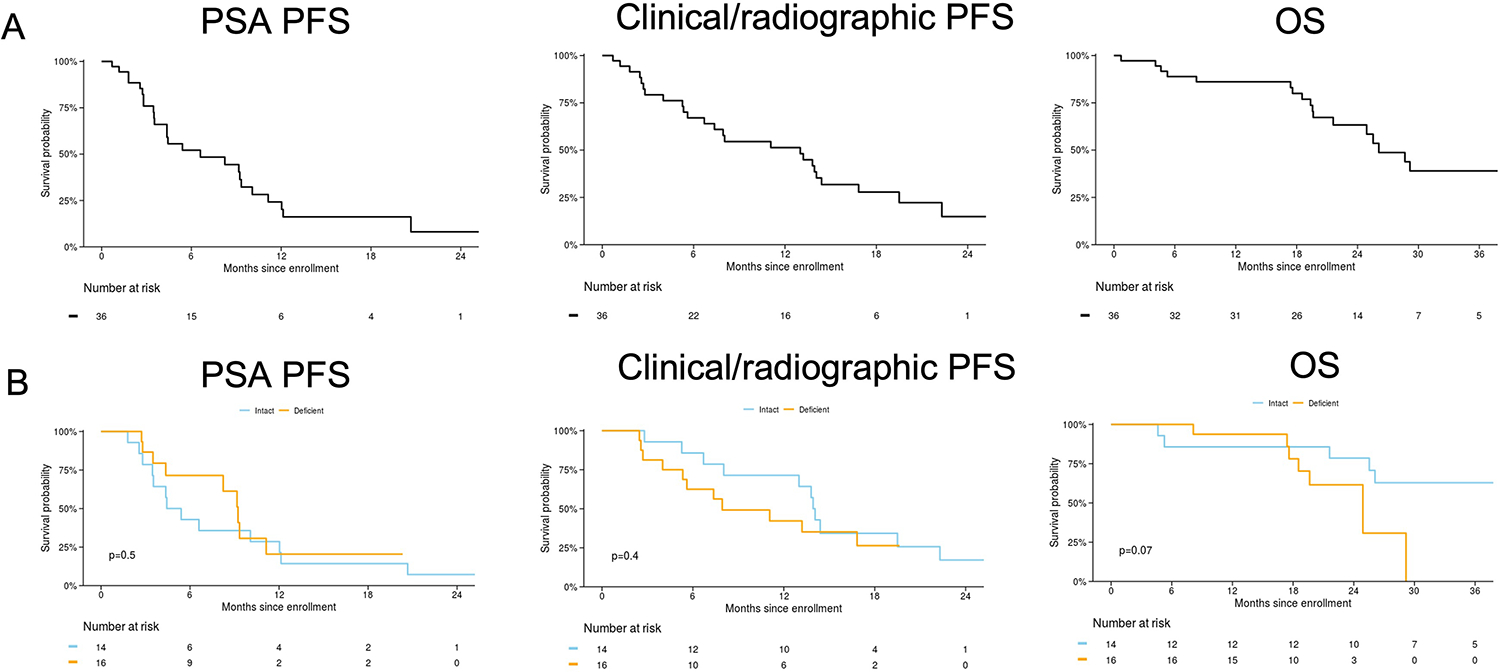
PSA progression-free survival (PSA PFS), clinical/radiographic PFS, and overall survival (OS). Data are presented for the intent-to-treat cohort (A) and for response-evaluable cohort (B) stratified by presence/absence of a homologous recombination repair (HRR) gene mutation (i.e. HRR deficient/intact). Median PSA PFS was 5 (95% CI 4–21) months for HRR-intact and 9 (95% CI 8-NR) months for HRR-deficient, median clinical/radiographic PFS was 14 (95% CI 13-NR) months for HRR intact and 8 (95% CI 5-NR) for HRR deficient, and median OS was NR (95% CI 26-NR) months for HRR-intact and 25 (95% CI 19-NR) for HRR-deficient.
Safety and Quality of Life
Treatment was generally well tolerated, and most adverse events (AEs) were low grade. The most frequently observed treatment-related AEs (TRAE) (i.e. at least possibly study drug-related) were gastrointestinal related and fatigue (Table S1). Five patients had grade ≥3 TRAEs, including one stroke (grade 4) and one myocardial infarction (MI) (grade 5). Following an amendment to exclude history of prior MI, no additional cardiovascular AEs were observed. Overall, there was no significant change in blood counts after 12 weeks (Figure S1). QOL was preserved on study, with erectile function score improving across multiple timepoints (Figure S2).
Exploratory Analyses
Functional defects in HRR pathways result in characteristic genome-wide alterations, which manifest as predictable genomic signatures(10). To evaluate if these signatures associated with outcomes, we performed paired germline-somatic whole exome sequencing as previously described on a subset of samples (N=10)(12–15). Several genomic HRR deficiency scores were evaluated, and while there was a general trend toward higher HRR deficiency score in those achieving a PSA50 response at any time, these results were not statistically significant (Table S2, Figure 3).
Figure 3:
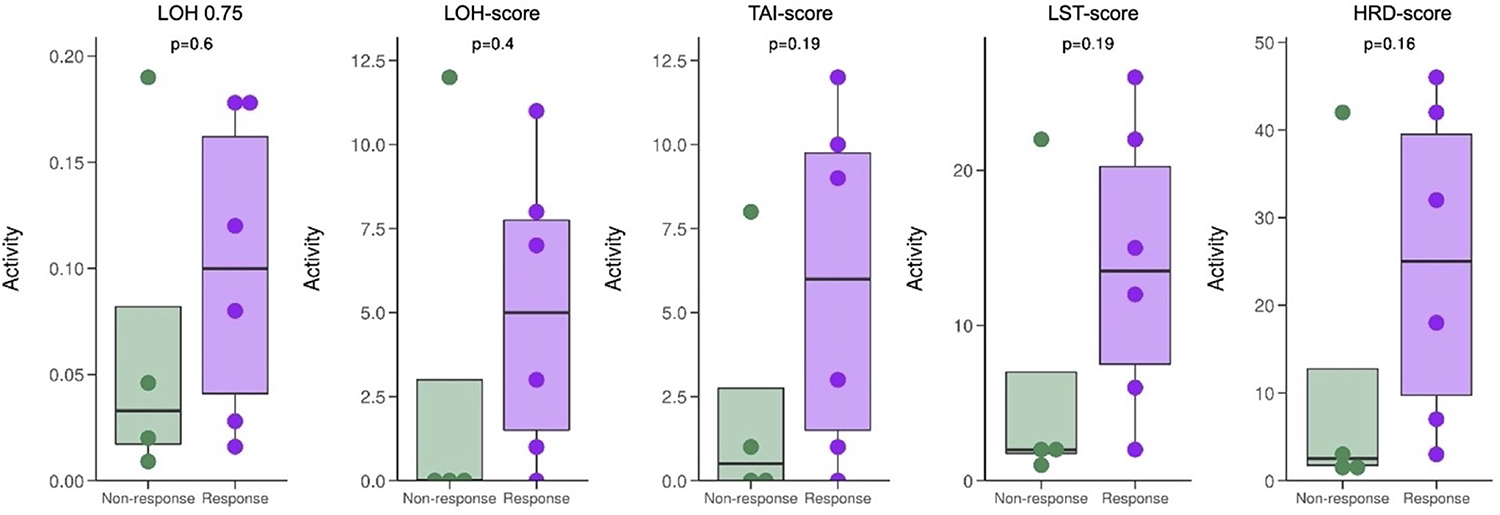
Associations between functional HRR deficiency scores and maximum PSA50 response.
LOH 0.75: Loss of heterozygosity (LOH) score, measuring the degree of LOH based on discontinuous SNP typing. LOH events larger than 75% of the respective chromosome arm length are excluded from the score analysis.
LOH-score: Number of LOH events assessed using the scarHRD tool. This score represents number of LOH regions exceeding 15 Mb that do not cover the whole chromosome.
TAI-score: Telomeric allelic imbalance (TAI) reflects the number of sub-chromosomal regions with allelic imbalance extending to the telomere.
LST-score: Number of large-scale state transitions (LST) assessed using the scarHRD tool. The LST score represents the number of chromosomal breaks between adjacent regions of at least 10 Mb length, with a distance between events not more than 3Mb.
HRD-score: Defined as sum of the burden of LOH-Scar, LST-Scar and TAI-scar (LOH-Scar score+ TAI-Scar score +LST-scar)
Because high AR expression has been shown to associate with sensitivity to androgen-induced cell death in preclinical models, we evaluated immunohistochemical staining patterns of AR in available metastatic tissue (N=6). AR expression was variable and the median crPFS for cases with AR expression above the median was 13.0 months (95% CI 11.1-NR) and 6.7 months (95% CI 2.5-NR) for those below the median (p=0.3) (Table S3, Figure S3). We also assessed if baseline PSA – an AR-regulated gene – was associated with outcomes. Consistent with findings from a prior study, higher baseline PSA level was associated with improved outcomes (Figure 4)(2). Similarly, there was weak evidence that crPFS was positively associated with baseline PSA.
Figure 4:
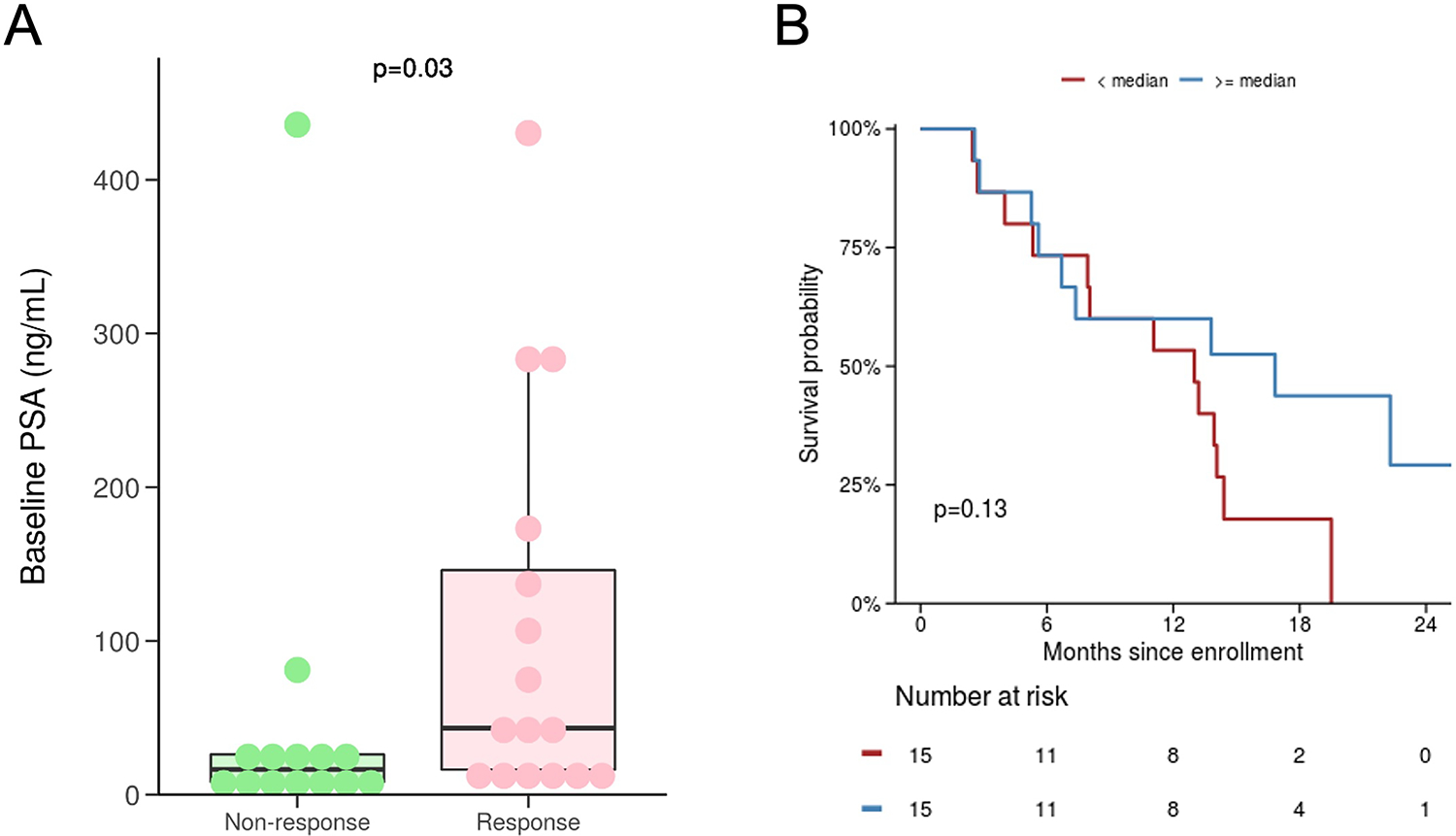
Associations between clinical outcomes and baseline PSA values. (A) Baseline PSA value is significantly higher in those achieving a PSA50 response anytime on study (i.e. responders vs. non-responders). The median baseline PSA in responders vs. non-responders was 43 vs. 16 ng/mL (P=0.03), respectively. (B) Baseline PSA value at or above vs. below the median PSA (22.3 ng/mL) is weakly associated with clinical/radiographic PFS.
Discussion
We observed high response rates and long crPFS with the combination of BAT plus olaparib. Although the PSA50 response rate did not exceed the hypothesized null rate, this study largely confirms our preclinical studies and provides evidence that the clinical activity of BAT can be augmented by co-treatment with olaparib. Importantly, we found that this combination was effective regardless of HRR gene mutational status. We also observed low rates of hematologic toxicity, which is likely due to the effects of testosterone and consistent with other studies showing that BAT resulted in increased hemoglobin(4). Finally, this regimen did not negatively impact QOL and patients reported improved sexual function while on study.
While cross-study comparisons should be interpreted with caution, it is important to put our findings into context given that this was a single-arm study. Prior trials have consistently shown that BAT monotherapy is associated with a PSA50 response rate of ~25% and a median PFS of <6 months in patients with mCRPC who have progressed on a novel hormonal agent (NHA)(2, 11, 16). Similarly, the PROFound study tested olaparib in patients with mCRPC who had progressed on one prior NHA and had at least one HRR gene mutation across 15 prespecified genes. That trial reported a PSA50 response rate of 30% and a median PFS of <6 months. This contemporary data suggests that a PSA50 response rate of 25% would reflect a more accurate historical control. Indeed, using this as a comparator, the PSA50 response rate of 53% (P=0.001) anytime on treatment with BAT plus olaparib compares quite favorably(5).
Contemporary data supports our hypothesis that combination BAT plus olaparib has at least additive clinical activity; however, our prespecified threshold for a clinically meaningful PSA50 response rate was not met. In large part, this was due to having limited data on the effects of olaparib in men with mCRPC at the time this study was designed. Key assumptions regarding the activity of olaparib were based on the TOPARP-A trial, which reported a PSA50 response rate of 63% in the small subset (N=16) of men with mCRPC that harbored alterations in HRR genes(5). On this basis, we assumed a high response rate would be observed within the group with HRR mutations. This is in direct contrast to the PROfound trial, which reported a PSA50 response rate of only 30% in men with a variety of HRR gene mutations receiving olaparib (N=243). Indeed, other studies have suggested that responses to PARP inhibitors are limited to patients with mutations in BRAC1/2 and a handful of other genes (e.g. PALB2) directly involved in HRR(17).
A key finding of our study is that the clinical activity of BAT plus olaparib appears independent of HRR gene mutational status, suggesting that biomarker selection is not necessary. This finding may be due to the ability of BAT to suppress HRR gene expression and thus sensitize tumors to PARP inhibition. We have previously reported that supraphysiological androgens (SPA) results in a substantial suppression of BRCA2 (4-fold reduction) and other DNA repair genes(3). Therefore, it seems plausible that BAT may induce an HRR-deficient phenotype and sensitize prostate cancer cells to olaparib. Similar to our observation that HRR gene mutational status was not associated with outcomes, we also did not see significantly enhanced efficacy in patients who had tumors demonstrating functional loss of HRR pathway as determined through genomic signature analysis. However, given that only a limited number of samples were available for this analysis, larger studies evaluating HRR signatures as a biomarker for response to BAT are still warranted.
PARP inhibitors have also been shown across multiple preclinical and clinical studies to augment the antitumor effects of DNA damaging cytotoxic agents as well as radiation(18–24). Given that prior preclinical studies have consistently shown that SPA can induce DNA damage, it is possible that the high response rates with BAT plus olaparib are due to an additive DNA damaging effect(1–3, 25). Importantly, while studies testing PARP inhibitors plus cytotoxic chemotherapy have reported high toxicity rates, the effects of BAT are restricted to AR-positive prostate cancer cells, which should limit off-target toxicity and may account for why this regimen was generally well tolerated.
When exploring biomarkers associated with response and resistance to combination treatment, there was a suggestion that increased AR activity may be associated with improved outcomes. Similar to our initial pilot study testing BAT, we observed an association between high PSA levels and improved outcomes(2). Consistent with this finding, we also estimated longer PFS in men with AR expression above the median as assessed by IHC. These confirmatory findings fit with preclinical models and recent clinical data showing that high AR transcriptional activity is associated with improved outcomes to BAT monotherapy(3, 26, 27). Larger studies incorporating metastatic tissue acquisition are needed to further assess whether baseline AR expression/activity is associated with improved outcomes.
There are several limitations to this study. As mentioned, key statistical assumptions were based on preliminary data showing that PARP inhibitors were highly active in patients with somatic alterations affecting an array of HRR-related genes. Subsequent studies have largely refuted this and in retrospect, the null PSA50 response rate was too high. The lack of a control arm also makes conclusions regarding activity of this regimen difficult. Another limitation is our small sample size, which limited our ability to draw conclusions in those with/without mutations in genes involved in HRR. In addition, we allowed for a permissive approach to defining HRR-deficiency given the uncertainty regarding which genes conferred functional loss of HRR activity when this study was designed. As such, the HRR-deficient cohort include patients with mutations in genes we now know are unlikely to predict response to PARP inhibition(17, 28–30). For example, one subject with CHD1 and SPOP mutations was included in the HRR-deficient cohort based on data suggesting that co-occurrence of these alterations may predict response to PARP inhibition – a finding yet to be confirmed clinically(30). Ultimately, it remains possible that we would have observed better outcomes had we enrolled more patients with BRCA1/2 mutations, although the lack of these patients does provide further evidence that this combination is active in biomarker unselected patients.
In conclusion, we observed encouraging signs of clinical activity with BAT plus olaparib. While the small size and lack of a control arm were key limitations of this study, these results do justify additional clinical trials. Future randomized studies evaluating BAT plus olaparib are warranted.
Supplementary Material
Acknowledgements:
This study was supported by a research grant from AstraZeneca. MTS was supported by US DOD Award W81XWH-16-1-0484 and a Prostate Cancer Foundation Young Investigator Award. RG was supported by NIH/NCI grants P50 CA097186 and R50 CA221836. HHC and PSN were supported by NIH/NCI Cancer Center Support Grant P30 CA015704. MCH was supported by the US DOD Awards W81XWH-20-1-0111 and W81XWH-21-1-0229, Grant 2021184 from the Doris Duke Charitable, and the V Foundation. NDS was supported by a Prostate Cancer Foundation Young Investigator Award. PSN was supported by NIH/NCI grants P50 CA097186, 5P01 CA163227, R01CA266452-01 and PC200262
Footnotes
Conflicts of Interest:
MTS: Paid consultant and/or received Honoria from Sanofi, AstraZeneca, PharmaIn and Resverlogix. He has received research funding to his institution from Zenith Epigenetics, Bristol Myers Squibb, Merck, Immunomedics, Janssen, AstraZeneca, Pfizer, Madison Vaccines, Hoffman-La Roche, Tmunity, SignalOne Bio and Ambrx, Inc.
HHC: Paid consultant for AstraZeneca. She received royalties from UpToDate. Research funds to her institution were from Clovis Oncology, Color Genomics, Janssen, Medivation, Promontory Therapeutics (formerly Phosplatin) and Sanofi.
PSN: Paid consultant for Janssen and Pfizer. He received royalties from UpToDate, Research funds to his institution were from Janssen.
Ethics Approval and Consent
This clinical study was approved by the Fred Hutchinson Cancer Center institutional review board. All research subjects provided informed consent prior to participating in this study.
Statement on Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials.
References
- 1.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42(8):668–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Science translational medicine. 2015;7(269):269ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chatterjee P, Schweizer MT, Lucas JM, Coleman I, Nyquist MD, Frank SB, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. The Journal of clinical investigation. 2019;129(10):4245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denmeade SR, Wang H, Agarwal N, Smith DC, Schweizer MT, Stein MN, et al. TRANSFORMER: A Randomized Phase II Study Comparing Bipolar Androgen Therapy Versus Enzalutamide in Asymptomatic Men With Castration-Resistant Metastatic Prostate Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2021:Jco2002759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. The New England journal of medicine. 2015;373(18):1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European journal of cancer (Oxford, England : 1990). 2009;45(2):228–47. [DOI] [PubMed] [Google Scholar]
- 7.Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cella D, Nichol MB, Eton D, Nelson JB, Mulani P. Estimating clinically meaningful changes for the Functional Assessment of Cancer Therapy--Prostate: results from a clinical trial of patients with metastatic hormone-refractory prostate cancer. Value Health. 2009;12(1):124–9. [DOI] [PubMed] [Google Scholar]
- 9.Rosen RC, Allen KR, Ni X, Araujo AB. Minimal clinically important differences in the erectile function domain of the International Index of Erectile Function scale. European urology. 2011;60(5):1010–6. [DOI] [PubMed] [Google Scholar]
- 10.Lord CJ, Ashworth A. BRCAness revisited. Nature reviews Cancer. 2016;16(2):110–20. [DOI] [PubMed] [Google Scholar]
- 11.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. The Lancet Oncology. 2018;19(1):76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Sarkar N, Dasgupta S, Chatterjee P, Coleman I, Ha G, Ang LS, et al. Genomic attributes of homology-directed DNA repair deficiency in metastatic prostate cancer. JCI Insight. 2021;6(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sztupinszki Z, Diossy M, Krzystanek M, Reiniger L, Csabai I, Favero F, et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer. 2018;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takaya H, Nakai H, Takamatsu S, Mandai M, Matsumura N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Scientific reports. 2020;10(1):2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Z S. scarHRD R package Manual 2020. [Available from: https://github.com/sztup/scarHRD.
- 16.Markowski MC, Wang H, Sullivan R, Rifkind I, Sinibaldi V, Schweizer MT, et al. A Multicohort Open-label Phase II Trial of Bipolar Androgen Therapy in Men with Metastatic Castration-resistant Prostate Cancer (RESTORE): A Comparison of Post-abiraterone Versus Post-enzalutamide Cohorts. European urology. 2021;79(5):692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schweizer MT, Cheng HH, Nelson PS, Montgomery RB. Two Steps Forward and One Step Back for Precision in Prostate Cancer Treatment. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2020;38(32):3740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(9):2728–37. [DOI] [PubMed] [Google Scholar]
- 19.Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6(7):2860–7. [PubMed] [Google Scholar]
- 20.Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(12):3916–25. [DOI] [PubMed] [Google Scholar]
- 21.Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, et al. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Molecular cancer therapeutics. 2003;2(4):371–82. [PubMed] [Google Scholar]
- 22.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(44):17079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo L, Keyomarsi K. PARP inhibitors as single agents and in combination therapy: the most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin Investig Drugs. 2022;31(6):607–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matulonis UA, Monk BJ. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: does a development pathway forward exist? Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2017;28(3):443–7. [DOI] [PubMed] [Google Scholar]
- 25.Denmeade SR, Isaacs JT. Bipolar androgen therapy: the rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate. 2010;70(14):1600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam HM, Nguyen HM, Labrecque MP, Brown LG, Coleman IM, Gulati R, et al. Durable Response of Enzalutamide-resistant Prostate Cancer to Supraphysiological Testosterone Is Associated with a Multifaceted Growth Suppression and Impaired DNA Damage Response Transcriptomic Program in Patient-derived Xenografts. European urology. 2020;77(2):144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sena LA, Kumar R, Sanin DE, Thompson EA, Rosen DM, Dalrymple SL, et al. Prostate cancer androgen receptor activity dictates efficacy of bipolar androgen therapy through MYC. The Journal of clinical investigation. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016;17(11):1609–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2017;28(7):1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Y, Wen J, Huang G, Mittlesteadt J, Wen X, Lu X. CHD1 and SPOP synergistically protect prostate epithelial cells from DNA damage. The Prostate. 2021;81(1):81–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials.