

Abstract
Purpose:
The goal is to identify thermal exposures capable of reducing or eliminating cell survival on expanded polytetrafluoroethylene (ePTFE), in an effort to develop a mild hyperthermia treatment of neointimal hyperplasia in ePTFE vascular grafts.
Materials and methods:
Viable and dead bovine aortic endothelial cells were quantified following different thermal exposure conditions: cells on collagen-coated ePTFE sheets or tissue culture polystyrene dishes were heated at 42°and 45°C to determine their thermal sensitivity on different surfaces, and cells cultured on collagen-coated ePTFE sheets were heated at 43–50°C for various durations, followed by incubation at 37°C for 0 and 20 h, respectively. Significant cell death was set to be 50%. Two types of cell death, apoptosis and necrosis, were distinguished by cell morphology and membrane integrity assessments.
Results:
The attachment and survival of cells on ePTFE sheets were more sensitive to inhibition by mild heating than those on tissue culture dishes. Exposure to 45°C for 90 min and 50°C for 30 min caused significant necrotic cell death on ePTFE (65% and 75%, respectively). A 37°C/20-h incubation following 30-min exposures at 47°and 50°C increased total cell death (necrosis + apoptosis) from 20% to 50% and 75% to 100%, respectively.
Conclusion:
Cells grown on ePTFE were more susceptible to mild hyperthermia-induced death, compared to those on tissue culture dishes. Significant cell death on ePTFE mainly via apoptosis can be achieved by optimising temperature and duration of exposure.
Keywords: hyperthermia, apoptosis, necrosis, neointimal hyperplasia
Introduction
The vascular access is a veritable lifeline for the chronic haemodialysis patient. Vascular access dysfunction is the leading cause of morbidity and hospitalisation in these patients [1–2]. The three main types of vascular access are central venous catheters, native arteriovenous (AV) fistulas (where the patient’s vein is surgically connected directly to an artery) and synthetic AV grafts (where the patient’s vein is surgically connected to an artery through a synthetic graft). Central catheters are highly prone to infection and sepsis [3]. Although the fistula and graft are considered permanent vascular access, both have their respective limitations. Although the AV fistulas have a relatively low incidence of stenosis once they mature and become functional, it often takes months for a fistula to mature after surgical creation, and about half of native fistulas fail to mature to become a usable access [4–6].
Synthetic AV grafts made from expanded Teflon (expanded polytetrafluoroethylene, ePTFE) are used as vascular access in nearly a quarter of the chronic haemodialysis patients in the USA [3]. In contrast to AV fistulas, AV grafts do not require a maturation time and can usually be used within weeks after implantation. They also reliably allow high blood flow rates because of the pre-formed lumen [7]. Unfortunately, ePTFE grafts are prone to the development of neointimal hyperplasia (NH) on the graft’s luminal surface. NH occurs preferentially at and near the graft-venous anastamosis site and decreases the luminal area, thereby reducing the blood flow rate and increasing the risk of thrombosis. Stenosis as a result of NH accounts for 85% of dialysis AV graft failures. One-year and two-year primary patency rates for ePTFE grafts are only 50% and 25%, respectively [7, 8].
NH is comprised of smooth muscle cells (SMCs), extracellular matrices, and endothelial cells (ECs) that are crucial for angiogenesis. Currently, no effective pharmacotherapy or brachiotherapy is available to prevent the occurrence of NH. If the occurrence of NH can be prevented, reduced or significantly delayed, the synthetic graft would provide an excellent dialysis vascular access.
Several approaches have been proposed to inhibit NH. One approach is to selectively promote the growth of ECs on the luminal surface of the vascular graft [9, 10]. The expectation is that endothelialisation of the vascular graft will inhibit the growth of the underlying SMCs by the release of nitric oxide and other inhibitors. However, a recent study showed that the seeding of endothelial progenitor cells actually increased, rather than decreased, the stenosis of AV grafts in an animal model [11]. An alternative approach is to prevent the growth of all cells, including ECs, on the graft surface. This has been accomplished with potent anti-proliferative drugs, such as paclitaxel and sirolimus [12, 13]. While drug-eluting stents have greatly improved the outcome of coronary angioplasty, a disadvantage of this approach for AV grafts is that it requires an invasive procedure (angioplasty to deliver the stent) and implantation of a foreign material (the stent) which cannot be readily removed; further, the long-term results of this approach in maintaining the graft patency are unknown. Efforts have been made to achieve the sustained release of anti-proliferative drugs using hydrogel delivery systems in the vicinity of the graft anastomosis, the location that is most prone to the development of NH [14]. Catheterbased delivery of gamma-radiation is yet another method that has been investigated for preventing cell proliferation on graft lumens [15]. However, for synthetic vascular grafts, none of the above-mentioned strategies has yet proven effective in humans.
The long-term goal of this study is to investigate the use of mild hyperthermia to prevent excessive cell proliferation in vascular grafts. Mild hyperthermia has been shown to produce cell death of various mammalian cells in vitro [16–20]. The extent of cell death has been related to the thermal dose, a combination of temperature and duration of exposure [21]. Thermal exposure can produce apoptosis and necrosis [17, 22, 23], two different forms of cell death that can be readily distinguished by cell morphology and membrane integrity assessments [22–25]. Previous studies have reported mild hyperthermia of ECs cultured on Petri dishes [26–28]. However, to our knowledge, mild hyperthermia of ECs cultured on ePTFE, the material used in most AV grafts used in clinical dialysis, has not been investigated. This study examined (1) the differential thermal sensitivity of ECs on different surfaces (ePTFE versus tissue culture-treated polystyrene dish) and (2) the effects of various thermal exposures (through combinations of different temperatures and heating durations) on the death of ECs on ePTFE. The goal is to identify the optimal thermal exposures capable of inhibiting cell proliferation/survival on ePTFE to set the stage for the application of thermal therapy for vascular graft stenosis. The ranges of temperatures and heating durations in this study took into consideration this intended clinical application.
Materials and methods
Cell culture conditions
Bovine aortic endothelial cells, the model cells used in all in vitro experiments in this study, were purchased from Lonza (Basel, Switzerland) and cultured in Dulbecco’s Modified Eagle’s medium supplemented with 5% fetal bovine serum, 1% penicillin/streptomycin and 1% sodium pyruvate (collectively referred to as cDMEM). The cells were grown in 100-mm diameter tissue-culture grade polystyrene Petri dishes and kept in a 37°C, 5% CO2 humidified incubator. When the cells reached a confluent monolayer, they were detached from the Petri dish using EDTA/trypsin and seeded onto prepared ePTFE sheets (Zeus, Orangeburg, SC) (see protocols below). All experiments used cells of passages 8–11.
Preparation of collagen-coated ePTFE sheets and polystyrene dishes for cell culture
Inside a biosafety cabinet at room temperature, 0.4-mm thick square ePTFE sheets slightly larger than 5 mm × 5 mm (L × W) were wetted and sterilised by soaking in 70% ethanol until the sheets appeared translucent and were then washed in autoclaved nanopure water. The ePTFE sheets were soaked in 0.075 mg/mL type-I collagen and phosphate buffer saline (PBS) solution overnight to allow the adsorption of collagen to their surfaces. Next, the ePTFE sheets were fixed to the bottom of 35-mm diameter polystyrene Petri dishes using a silicone holder (Figure 1A). The holder sandwiched the ePTFE sheet between the solid silicone base on the bottom and a silicone gasket with a 5 mm×5 mm centre cut-out on the top to expose the ePTFE surface. The silicone base and gasket were 0.5 mm thick each. Next, cells were seeded onto the ePTFE sheets (Figure 1B), where they adhered to and grew on the ePTFE at 37°C until the thermal exposure experiments began.
Figure 1.
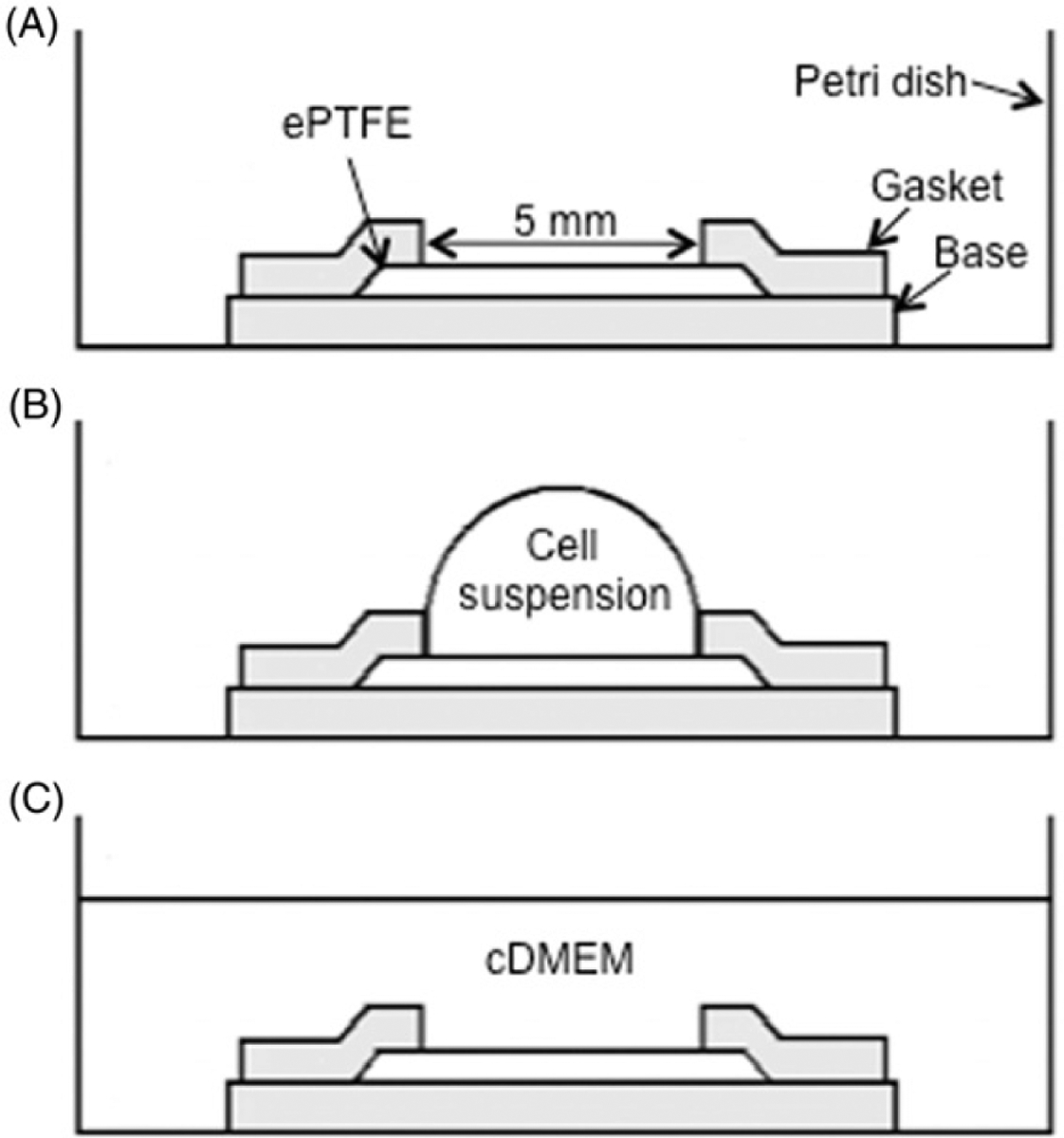
Sketch of the cell seeding process (side view). (A) The 5 mm×5 mm ePTFE sheet was held in place by a silicone holder’s base and gasket (shaded in the diagram) in a standard Petri dish for tissue culture. (B) Cell suspension was pipetted onto the ePTFE sheet. (C) 2.5 mL of cDMEM was added to the dish. Drawings not to scale.
In parallel experiments, tissue culture-treated (by vacuum gas plasma) polystyrene dish surfaces were coated with type-I collagen solution only (without ePTFE) overnight. Cells were seeded onto the collagen-coated polystyrene surface as described above for the ePTFE surface, and grown until confluence.
Thermal exposure experiments
Thermal exposures lasting 2 h or more were conducted in a 5% CO2 humidified, water-jacketed incubator (Thermo/Forma Scientific Model 3110). All other short-term (less than 2 h) exposures were performed using water baths (Fisher Scientific Model Isotemp 210, Pittsburgh, PA). The temperature in the incubator and water bath was monitored using mercury thermometers (14986 C, Fisher). In each water bath, a metal rack was used to hold the Petri dishes during the heating experiments. Immediately before a cell sample was placed in the water bath, the medium in the sample’s Petri dish was replaced with 2.5 mL of fresh cDMEM preheated to the temperature of the respective water bath. Three different groups of experiments were performed, as detailed below.
Protocol A: Differential thermal sensitivity
The objective of Protocol A was to determine the difference in thermal sensitivity of ECs cultured on ePTFE (vascular graft material) and those grown on tissue-culture treated polystyrene dishes (surrogate of native tissue). Both materials were coated with collagen to mimic the extracellular matrix environment in vivo and to promote cell adhesion. Two experiments were performed: (1) Survival: cells on collagen-coated ePTFE or polystyrene surfaces were cultured in a 37°C, 5% CO2 humidified incubator until they reached confluence. The confluent cells were exposed to 45°C for 2 or 4 h. The 45°C temperature was selected to ensure that cell death was induced, so that comparison of cell death on these two surfaces could be made; (2) Attachment: cell suspensions were added to collagen-coated ePTFE or polystyrene surfaces and kept in a 5% CO2, humidified incubator at 37° or 42°C for 24 h, which was sufficiently long to allow ECs to attach and spread on both surfaces under normal conditions (i.e. 37°C). The 42°C temperature was selected to show differential thermal sensitivity at a mild temperature that did not cause significant cell death. For both (1) and (2), cell viability/death was determined immediately following the thermal exposure, as described below.
Protocol B: Cell death detection immediately following heat exposure
The objective of Protocol B was to determine the percentage of cell death on ePTFE immediately after and over a range of thermal exposures. The cells (30 μL of cell suspension at a density of 60 cells/μL) were seeded onto the collagen-coated ePTFE surfaces (Figure 1B) and allowed to attach to the ePTFE surface for 30 min at 37°C in a 5% CO2 humidified incubator. Next, an aliquot of 2.5 mL of fresh cDMEM was added to the dish (Figure 1C) and the mixture was further cultured in the incubator overnight to allow for acclimatisation. The thermal exposure experiments were performed on the following day when the cells remained subconfluent. One set of samples was exposed to either 43° or 45°C for 30, 45 or 90 min; another set was exposed to either 47° or 50°C for 10, 20 or 30 min. Samples kept at 37°C served as a control. Cell viability/death was determined immediately following the thermal exposure, as described below (Figure 2).
Figure 2.
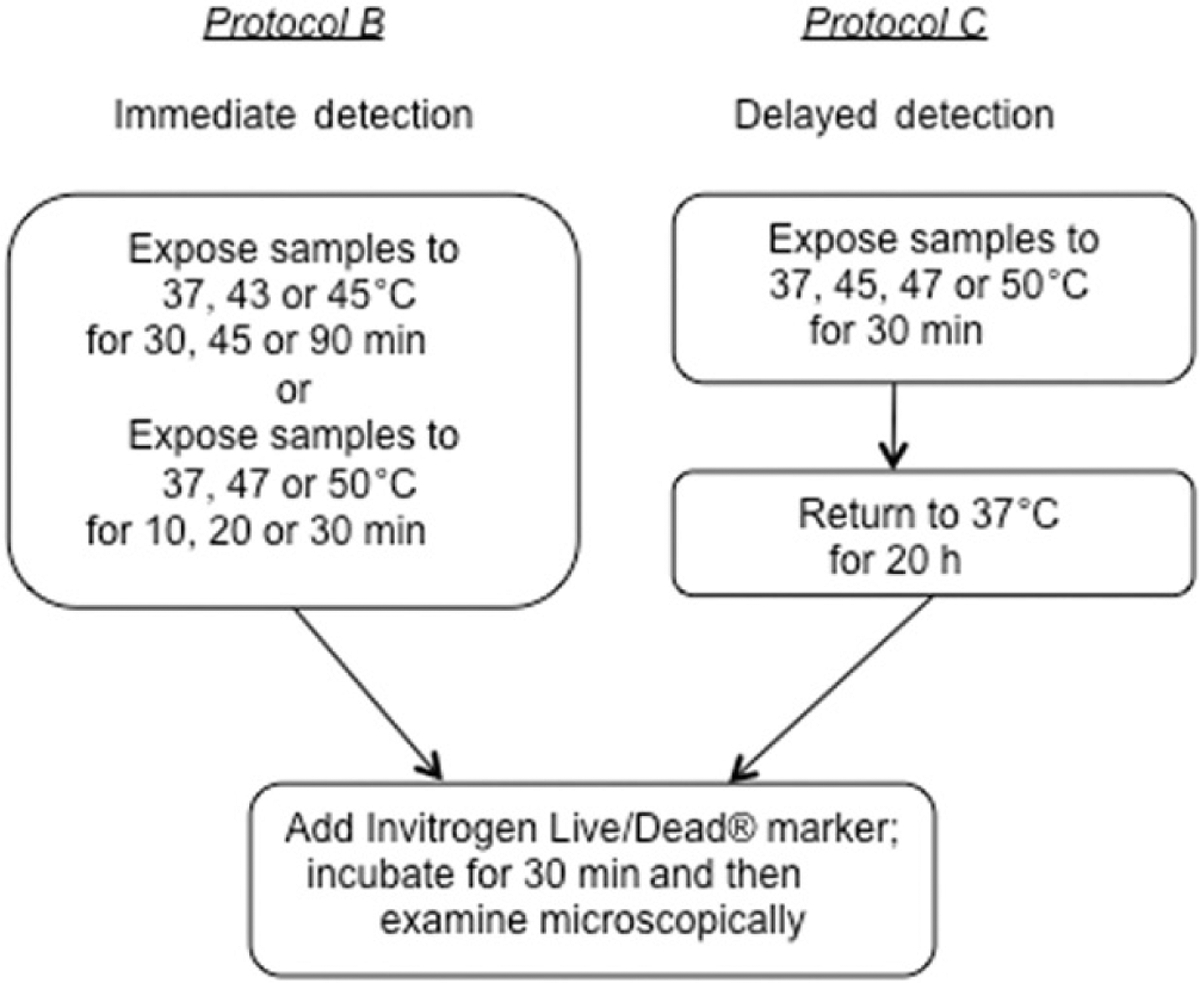
Experimental protocols for detecting immediate or delayed cell death on ePTFE after thermal exposure. Upper left panels, Protocol B: Cells were exposed to various temperatures for various durations, following which the cell viability/death was immediately assessed. Upper right panels, Protocol C: Cells were exposed to various temperatures for 30 min and returned to physiological temperature (37°C) for 20 h. The cell viability/death was then assessed.
Protocol C: Detection of delayed cell death following heat exposure
The objective of Protocol C was to measure cell death on ePTFE after a delay following thermal exposure. Cells were prepared as described in Protocol B. After exposure to 37° (control), 45°, 47°or 50°C for 30 min, the cells were returned to the incubator and kept at the physiological temperature of 37°C for additional 20 h to allow apoptosis to progress. The samples were stained to determine cell viability/death as described below (Figure 2).
Cell viability/death staining
Cell death by apoptosis is a progression involving multiple steps, including cell shrinkage, chromatin compaction, blebbing, nuclear and cellular fragmentation. In contrast, necrosis results from acute cellular injury and is a form of traumatic cell death, not following organised biochemical and morphological criteria [29, 30]. The Invitrogen Live/Dead® viability/cytotoxicity kit (Invitrogen/Molecular Probes, Eugene, OR, USA) distinguishes and labels cells with compromised (necrotic and apoptotic) and uncompromised (viable) cellular membranes. Necrotic cells are readily detected using this kit, but cells undergoing apoptosis can only be detected at the later steps of the apoptotic death progress when the membrane becomes compromised [23, 25]. After exposure to various thermal exposures as described above, ECs on the ePTFE surfaces were stained using the Invitrogen Live/Dead® viability/cytotoxicity kit, according to the manufacturer’s instructions. In this procedure, the cytosols of living cells were labelled with calcein AM (appearing green) and the nuclei of necrotic and apoptotic cells were labelled with ethidium homodimer-1 (appearing red).
Microscopic image acquisition and analysis
After staining with the Invitrogen kit, the cells were examined under an Olympus IX70 inverted microscope using 10 × and 20 × objectives. The excitation/emission wavelengths for calcein AM and ethidium homodimer-1 are 494/517 nm and 517/617 nm, respectively. Microscopic fluorescent images were acquired with a cooled CCD camera (ORCA-ER, Hamamatsu, Bridgewater, NJ) and commercial imaging software IPLab (Scanalytics, Rockville, MD). The images of the live and dead cells were combined using ImageJ (National Institutes of Health), so that the viable and non-viable cells could be manually counted from the same image to avoid double counting. Ethidium homodimer-1-positive nuclei, appearing red, were considered dead (non-viable) and calcein AM-positive cytosols, appearing green, were considered live (viable). The percentage of dead cells was calculated as follows:
| (1) |
Morphological analysis (ImageJ software) was performed on the viable cells, whose cytosol appearing green, by manually tracing the cell perimeter. ImageJ software was then used to calculate the area, perimeter and roundness index of individual cells. The roundness index was calculated as the ratio of the cell area to the area of a circle with a diameter equal to the major axis of the best-fit ellipse:
| (2) |
By definition, the roundness index is in the range of 0 and 1, and a perfectly round cell corresponds to an index of 1.
Data presentation and statistical analysis
For each exposure condition (temperature and time), three to five independent experiments (N = 3–5) were conducted on separate days. Each independent experiment had duplicate or triplicate samples from which the average cell death percentage, cell area, cell perimeter and roundness index were calculated. The combined average of the independent experiments became the reported value for a specific exposure condition. The error bars in all figures represent standard error of the mean (SEM). Unequal variance t-test (Welch’s t-test) rather than the student’s t-test was used for comparison between groups [31]. P < 0.05 was considered statistically different.
Results
Differential thermal sensitivity
Figure 3 shows that the percentage of dead cells cultured on collagen-coated ePTFE sheets was approximately double that cultured on collagen-coated tissue culture dishes, after either 2 or 4 h of incubation at 45°C. Even after 4 h of thermal exposure at 45°C, the percentage of dead cells on dishes was quite low (~10%), while the percentage on ePTFE sheets had increased to almost 20%. It is worth noting that our percentage death of confluent endothelial cells on collagen-coated tissue culture dishes was lower than that reported by Ketis et al.[28]. Ketis et al. showed that the percentage death of confluent bovine aortic endothelial cells (the same cell type as ours) (both primary and passages 1–12) on tissue culture dishes was approximately 40% after 2 h of exposure to 45°C. However, Ketis et al. determined cell viability using a proliferation-based colony formation assay [32]. It is possible that thermal exposure may negatively impact EC proliferation, leading to detection of a higher death percentage.
Figure 3.
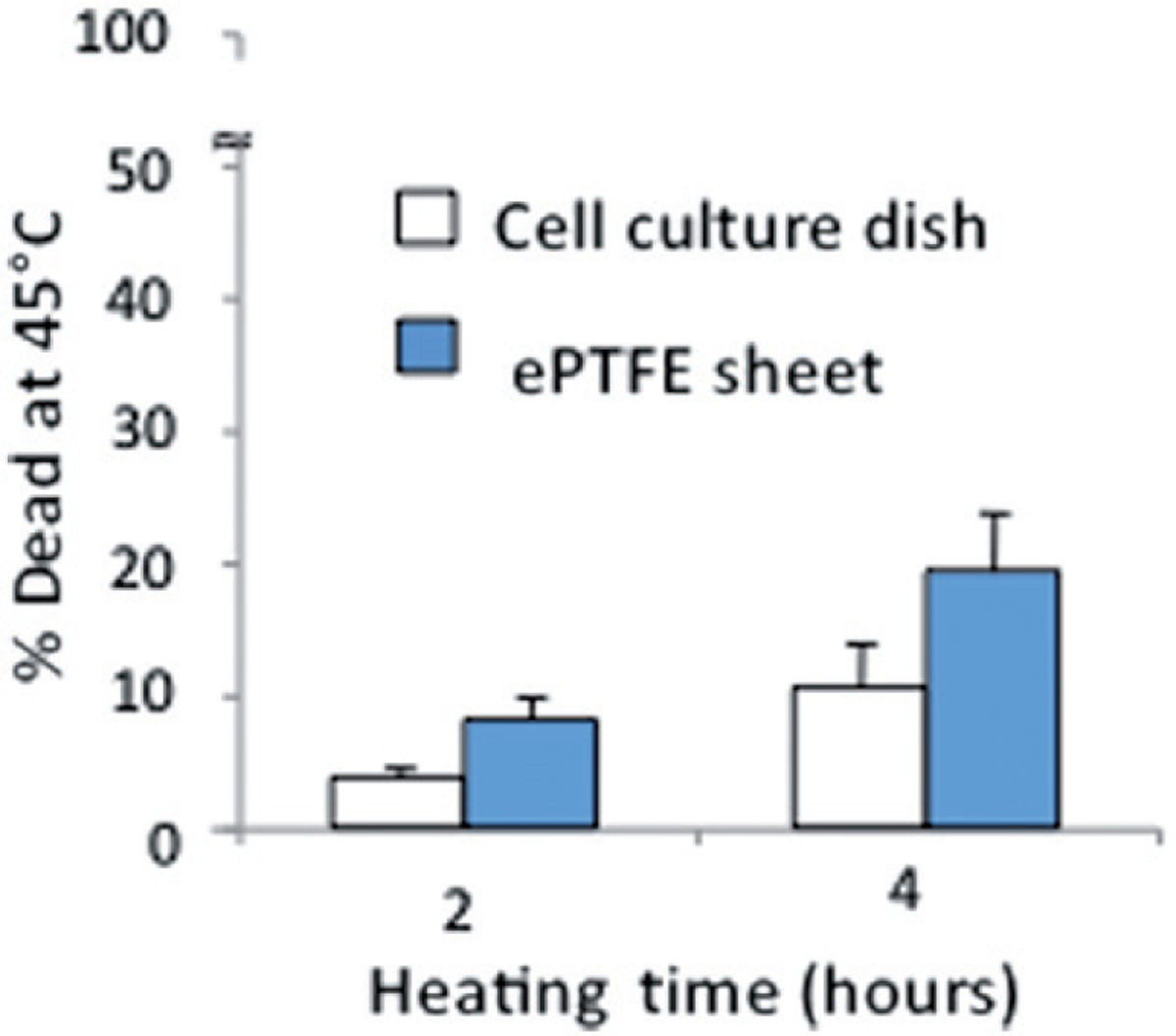
Differential survival of heated cells on collagen-coated ePTFE sheets and tissue culture dishes. Cells were cultured at 37°C until they reached confluence on either surface. The percentage of cell death detected after 2 or 4 h at 45°C was almost double on ePTFE sheets compared to tissue culture dishes. The percentage of cell death at 37°C was very low (near zero) on both surfaces.
Figure 4 shows representative images for cells attaching to collagen-coated ePTFE sheets or tissue culture dishes after 24 h of incubation at 37°and 42°C. As expected, cells attached to and formed a confluent monolayer on the tissue culture dishes at 37°C; cells on ePTFE surfaces exhibited similar attachment density and appearance at 37°C (Figures 4A–B). At 42°C, cells were able to form a confluent monolayer on the tissue culture dishes (Figure 4C). In contrast, fewer cells attached to ePTFE at 42°C and these cells failed to exhibit the spreading characteristic of healthy anchorage-dependent cells (Figure 4D).
Figure 4.
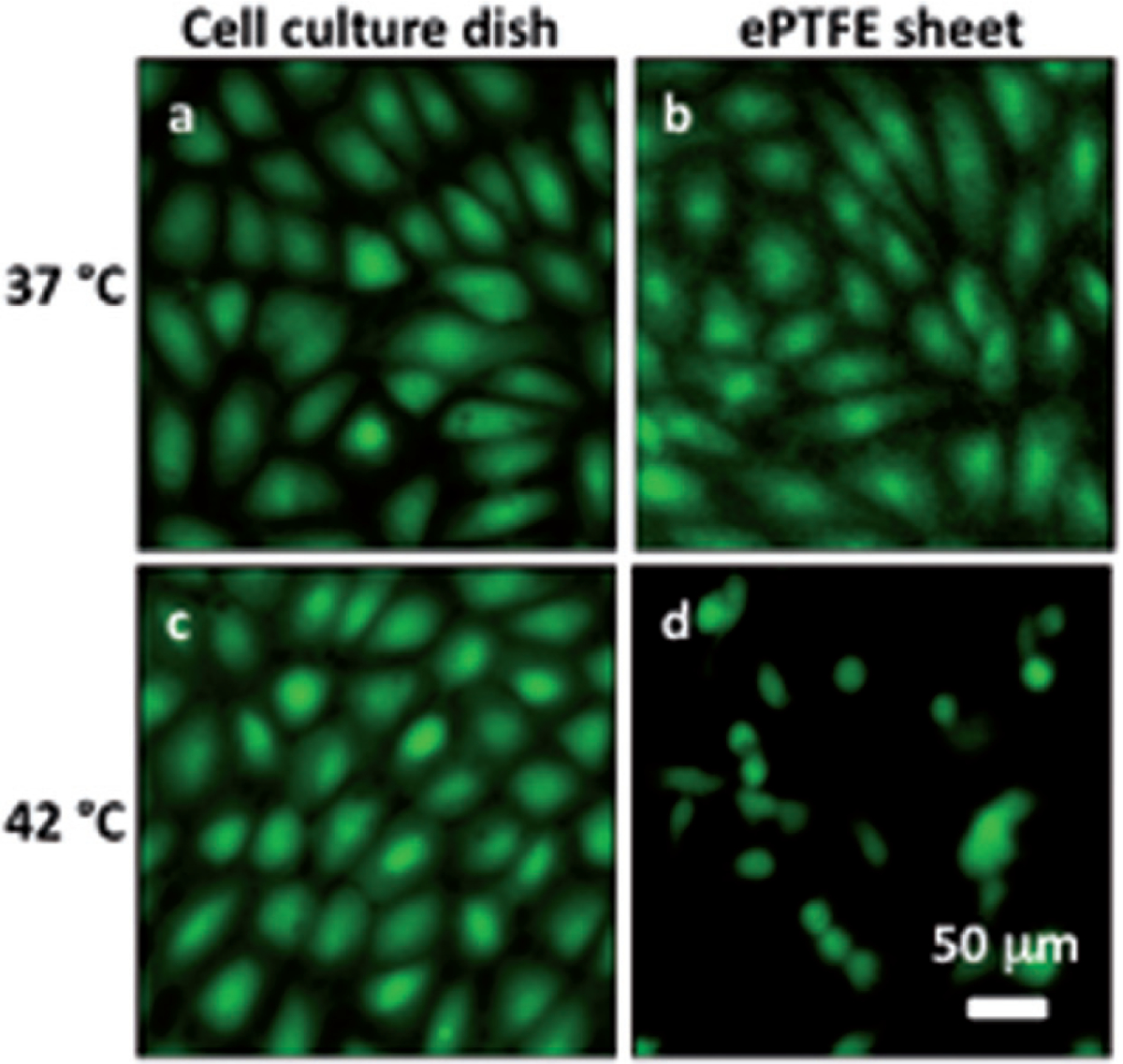
Differential adhesion of heated cells onto collagen-coated ePTFE sheets and tissue culture dishes. Cells in suspension were allowed to attach to either surface for 24 h at 37°or 42°C for 24 h. The cells were then stained with calcein AM (which labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (which labels the nuclei of dead cells, appearing red) and then imaged. At 37°C, endothelial cells formed confluent monolayers and displayed typical cobblestone morphology on both ePTFE sheets (B) and dishes (A). At 42°C, however, the cell density on ePTFE sheets (D) was significantly lower than that on dishes (C). Further, the cells on ePTFE (D) exhibited a condensed cytoplasm, an early indication of apoptosis, though the cells were negative of ethidium homodimer-1 staining.
Temperatures exceeding 47°C can cause epidermal and dermal injury [33]. Thus, for our intended clinical application (i.e. using mild hyperthermia to prevent cell proliferation on ePTFE vascular grafts), our target temperature should be 47°C or lower. Figures 3 and 4 show that 42°and 45°C damaged cells on ePTFE sheets while the damage to cells on the tissue culture dishes (used as surragate of native tissue) was minimal when exposed to thesetemperatures for multiple hours. Therefore, the following experiments (Protocols B and C) were focused on behaviours of cells on ePTFE at various temperatures higher than 42°C and up to 47°C. Exposure to the extreme temperature of 50°C was conducted as a positive control, and significant cell death was set to be 50% or higher. Inasmuch as subconfluent cells appear to be more sensitive to heat inhibition (see results in the next section), our ultimate clinical objective is to prevent cells from proliferating and forming a confluent monolayer on the ePTFE graft surface. With this objective in mind, the following experiments focused on subconfluent cells.
Necrotic cell death determined by immediate staining
Figure 5A shows the percentage of dead (necrotic) cells after cells had been exposed to the lower heating temperatures of 43°and 45°C for 30, 45 or 90 min. In this group, only 45°C/90-min exposure caused significant cell death (specifically, 65%). Figure 5B shows results of cell samples that were exposed to 47°or 50°C for 10, 20 or 30 min. In this group, the maximum percentage of cell death, 75%, occured after 30 min of heating at 50°C. Other than these two thermal exposures, all cell deaths were less than 30%. Values for the 37°C controls were always lower than 10%.
Figure 5.
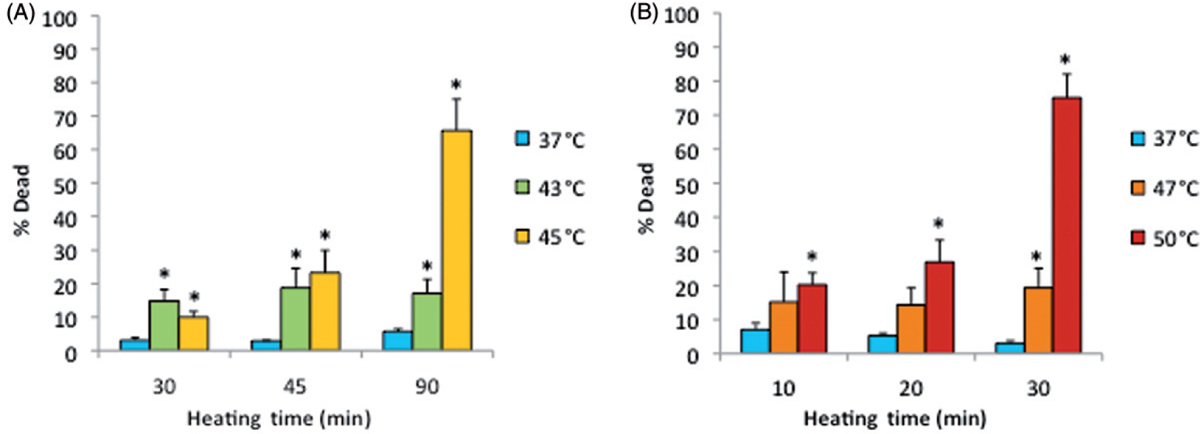
Death of cells on ePTFE as determined immediately after various thermal exposures. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to 43° and 45°C (A) or 47° and 50°C (B) for various durations. After the heat exposure, the cell viability/death was immediately assessed, and the percentage of dead (necrotic) cells were reported. Significant cell death (more than 50% of cell death) was detected only after long heating durations or exposure at high temperature. Exposure to 37°C served as the control. N = 3–4. Error bars represent SEM. *P < 0.05 compared to the control with the same exposure duration.
Note that the percentage of dead cells was 65% (Figure 5A) for subconfluent ECs exposed at 45°C for 90 min, but was only 10% for confluent ECs exposed at the same temperature for 120 min (Figure 3). Therefore, these data suggest that thermal sensitivity may be influenced by cell density. While the underlying mechanism is beyond the scope of this study, our finding that confluent ECs are more resistant to heat stress maybe explained by the published reports, showing that cell-to-cell contacts enhance the survival of ECs [34, 35]. Figure 6 shows representative fluorescent images of the morphological changes induced by thermal exposure of 30 min at 37°, 45°, 47°and 50°C. Quantitative morphological analyses (area, perimeter and roundness index) of the viable (green) cells that did not have ethidium homodimer-1-positive (red) nuclei are presented in Figures 7, 8 and 9, respectively. At 45°, 47°and 50°C, fewer cells spread on the ePTFE and many viable (green) cells exhibited morphological rounding rather than spreading, an early indication of apoptosis (indicated by arrows in Figures 6B–D). In contrast, viable cells on ePTFE maintained at 37°C (Figure 6A) displayed large area and large perimeter but a low roundness index, characteristics of the typical spread out, fan shape of subconfluent ECs on tissue culture dishes. In general, the average cell size (area and perimeter) decreased and the cell roundness increased when the temperatures and/or heating exposures increased. For example, within the 30-min heating exposure, the cell roundness index increased from 0.55 at 37°C to 0.66 at 50°C, whereas the average cell area and perimeter decreased from 871 μm2 and 158 μm at 37°C to 488 μm2 and 97 μm at 50°C, respectively (Table I). Because the decrease in size and increase in roundness index are indicators of apoptosis, our results suggested that these ‘viable (green) cells’ at 45°, 47°and 50°C may be undergoing the early steps of apoptosis and have not yet progressed to having ethidium homodimer-1-positive (red) nuclei. Apoptosis is irreversible [23, 25], so will continue even after the heating ceases. In the following experiments, cells were exposed to 45°, 47° and 50°C for 30 min, and then kept at 37°C for 20 h for apoptosis to continue and complete. If our hypothesis was correct, we expected to see an increase in cell death after the 20-h incubation at 37°C.
Figure 6.
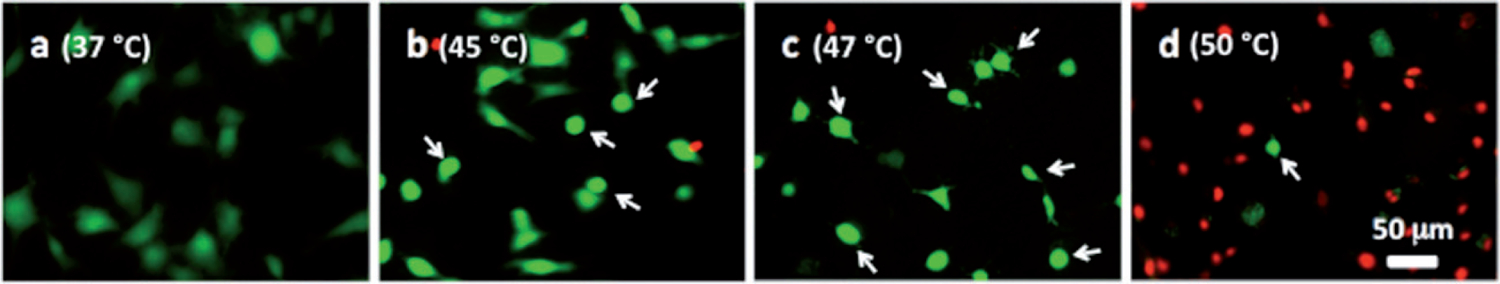
Representative microscopic fluorescent images of subconfluent cells on ePTFE immediately after various thermal exposures. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to (A) 37, (B) 45, (C) 47 and (D) 50°C for 30 min. Next, they were immediately stained with calcein AM (which labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (which labels the nuclei of dead cells, appearing red) and then imaged. Arrows in B–D point to cells that were viable (green) but exhibited a condensed cytoplasm, an early indication of apoptosis, though these cells were negative of ethidium homodimer-1. Most cells at 50°C were necrotic.
Figure 7.
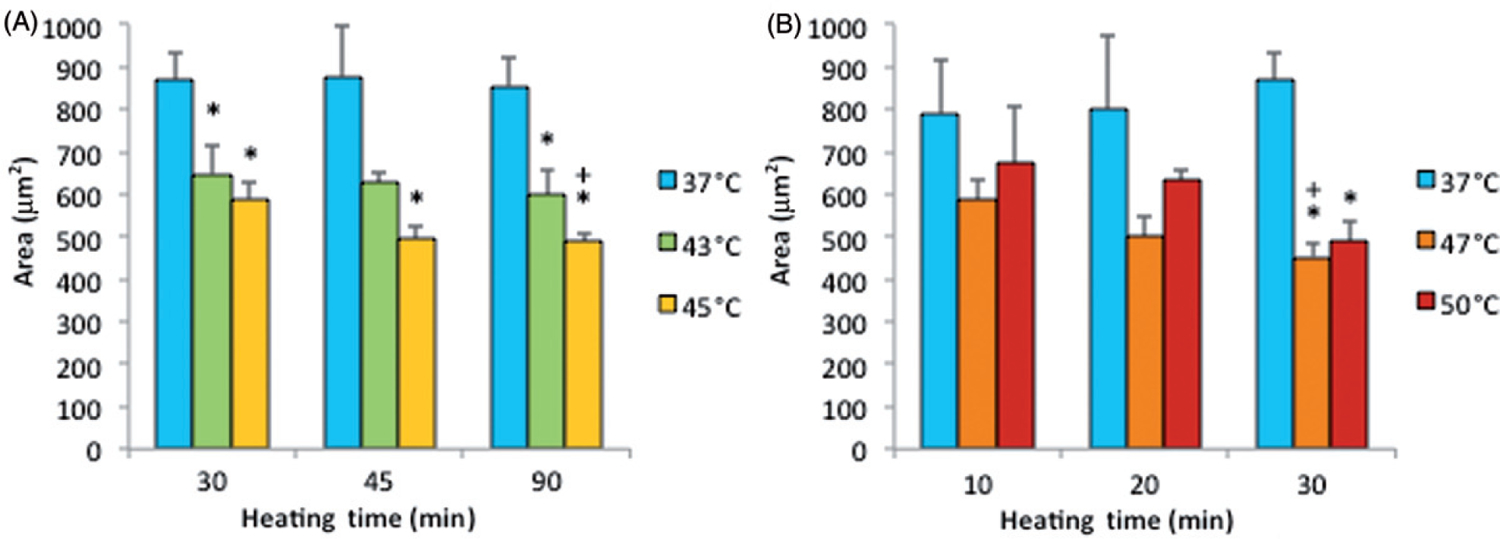
Cell area on ePTFE as determined immediately after various thermal exposures. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to 43° and 45°C (A) or 47°and 50°C (B) for various durations. Next, they were immediately stained with calcein AM (which labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (which labels the nuclei of dead cells, appearing red), and the area of viable cells was measured and reported as averaged ‘area (μm2)’ per cell. While cell area decreased for all exposures above 37°C, the cell area was the smallest (500 μm2 or less) following the 47° and 50°C/30-min exposures and the 45°C/45-min and 90-min exposures. N = 3–5. Error bars represent SEM. *P < 0.05 compared to 37°C with the same exposure duration. +P < 0.05 compared to the same temperature at the minimum exposure duration.
Figure 8.
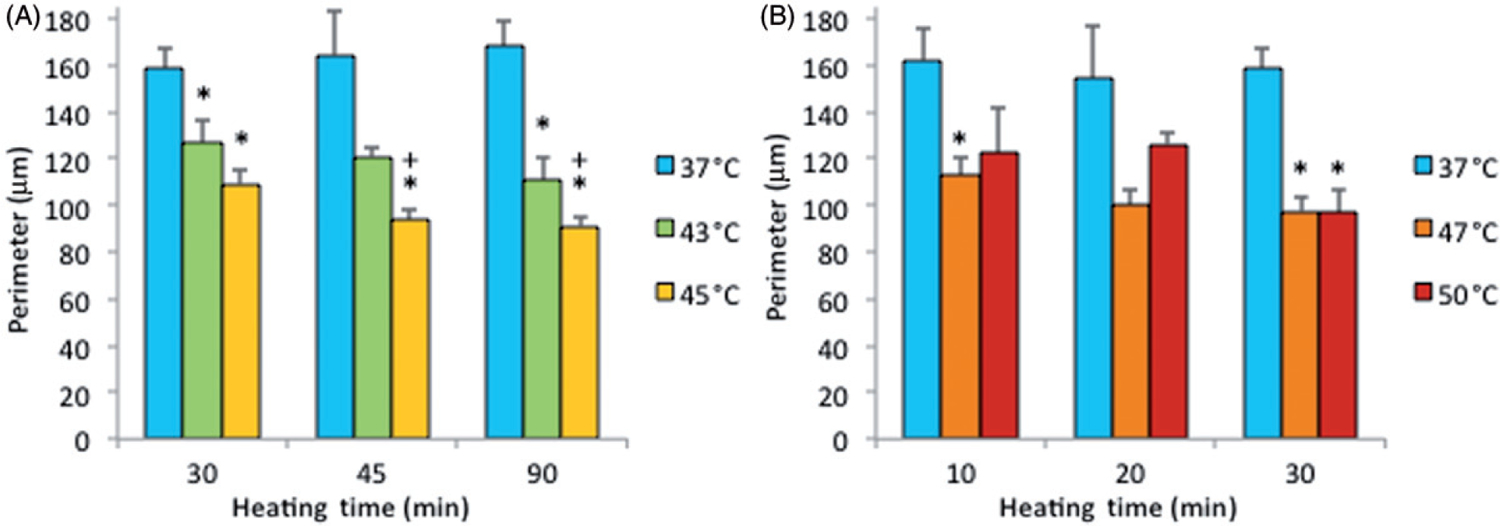
Cell perimeter on ePTFE as determined immediately after various thermal exposures. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to 43° and 45°C (A) or 47° and 50°C (B) for various durations. Next, they were immediately stained with calcein AM (which labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (which labels the nuclei of dead cells, appearing red), and the perimeter of viable cells was measured and reported as averaged ‘perimeter (μm)’ per cell. While cell perimeter decreased for all exposures above 37°C, the cell perimeter was the smallest (100 μm or less) following the 47° and 50°C/30-min exposures and the 45°C/45 and 90-min exposures. N = 3–5. Error bars represent SEM. *P < 0.05 compared to 37°C with the same exposure duration. + P < 0.05 compared to same temperature at the minimum exposure duration.
Figure 9.
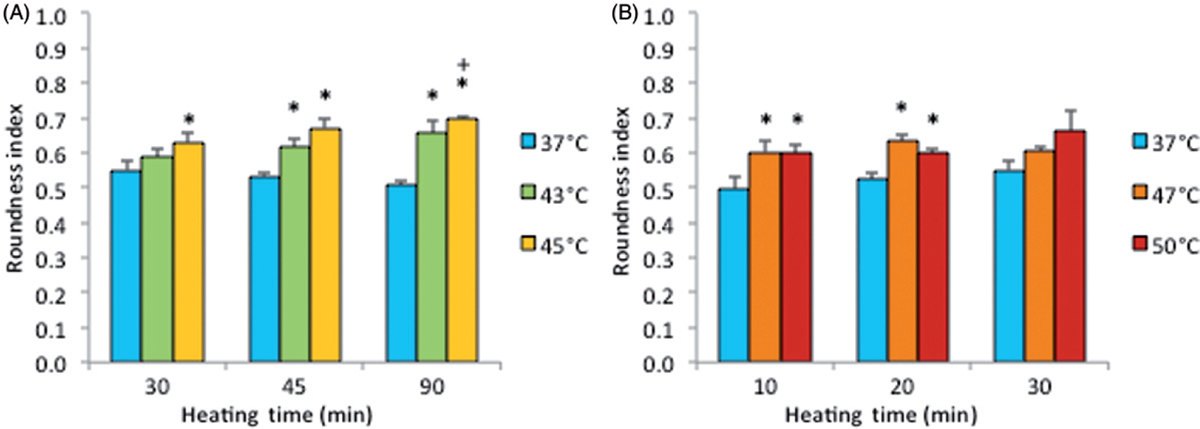
Cell roundness index on ePTFE as determined immediately following various thermal exposures. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to 43° and 45°C (A) or 47° and 50°C (B) for various durations. Next, they were immediately stained with calcein AM (which labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (which labels the nuclei of dead cells, appearing red), and the roundness index (in the range of 0 and 1) of viable cells was measured and reported as averaged ‘roundness index’ per cell. A perfectly round cell corresponds to an index of 1. Index increased with temperature and exposure duration. N = 3–5. Error bars represent SEM. *P < 0.05 compared to 37°C with the same exposure duration. +P < 0.05 compared to the same temperature at the minimum exposure duration.
Table I.
Cell morphology and death on ePTFE immediately after 30-min thermal exposures. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to 37°(control), 45°, 47°and 50°C for 30 min. Next, they were stained with calcein AM (which labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (which labels the nuclei of dead cells, appearing red) for determining the morphology of viable cells and the percentage of dead cells. In general, as the temperature increased, the size (area and perimeter) of viable cells decreased and the percentage of dead cells increased.
| Temperature (°C) | Area (μm2) | Perimeter (μm) | Roundness index | Cell death |
|---|---|---|---|---|
| 37 | 871 | 158 | 0.55 | 3% |
| 45 | 589* | 109* | 0.63* | 10%* |
| 47 | 452* | 98* | 0.60 | 19%* |
| 50 | 488* | 97* | 0.66 | 75%* |
P < 0.05 with respect to the 37°C/30-min exposure in the same column.
Increase in total cell death (apoptosis and necrosis) with incubation delay
Figure 10 displays the effect of a post-heating incubation period on the death of cells grown on ePTFE, presumably due to apoptosis. These samples were heated for 30 min at 37°, 45°, 47°or 50°C and incubated for an additional 0 or 20 h at 37°C to allow for the progression of apoptosis before staining. No significant apoptotic cell death was observed following the 45°C exposure. In contrast, there was a substantial increase in apoptotic cell death after 20 h of incubation at 37°C following the 47°C exposure. A thermal exposure at 47°C for 30 min eventually killed, through both apoptosis and necrosis, almost 50% of the cells, although only less than 20% were initially labelled as dead immediately after the heat exposure. These data suggested that apoptosis contributed to most of the cell death for the 47°C/30 min exposure. Exposure to 50°C caused mostly necrotic death, since cell death was 75% immediately after the heat exposure and reached 100% after an additional 20-h incubation at 37°C. A small but statistically significant increase in apoptosis (from 3% to 7%) after 30 min plus 20 h of incubation at 37°C was also observed, which might be attributed to the change, albeit modest, of the environment (i.e. from the water bath to the incubator). Representative figures of the cells stained after 20 h of post-heating incubation at 37°C are shown in Figure 11.
Figure 10.
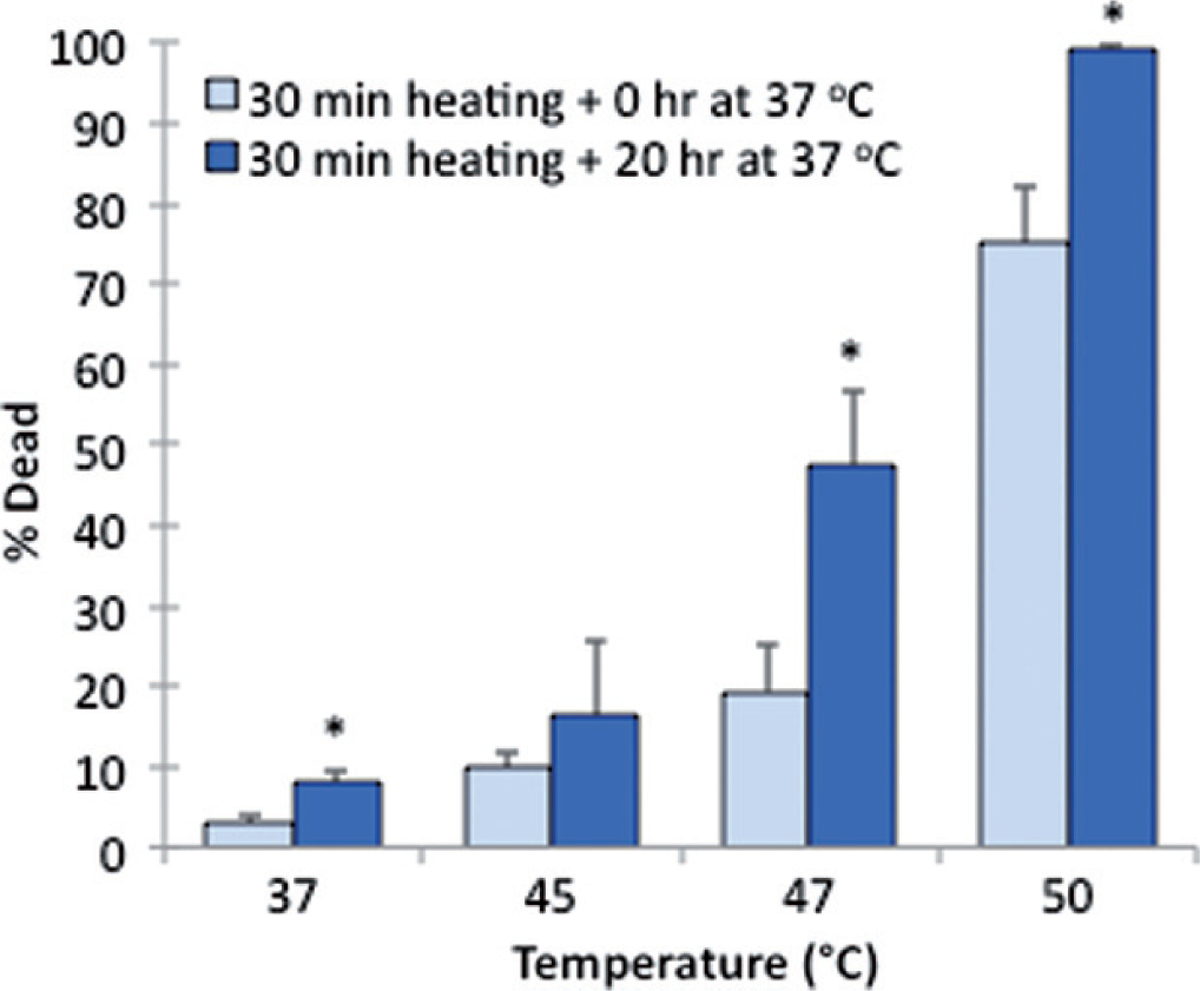
Delayed cell death on ePTFE upon various thermal exposures. Percentage of cell death was determined following one exposure for 30 min at 37°, 45°, 47° or 50°C with a subsequent 0-h (control) or 20-h incubation period at 37°C. The additional incubation period at 37°C allowed for the progression of apoptosis prior to staining. N = 3–4. Error bars represent SEM. *P < 0.05, compared to the control (zero incubation time after heating) at the same temperature.
Figure 11.
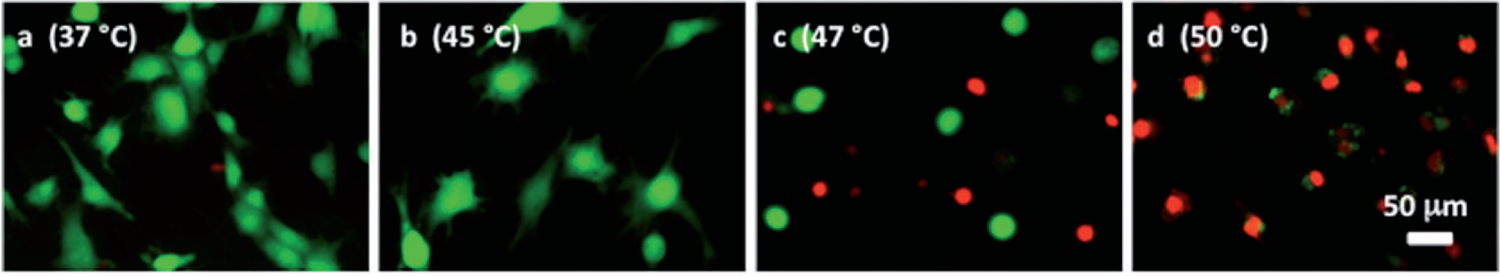
Representative microscopic fluorescent images of subconfluent cells on ePTFE after various thermal exposures and a delay. Cells were cultured on collagen-coated ePTFE at 37°C for a day, and then exposed to (A) 37°, (B) 45°, (C) 47° and (D) 50°C for 30 min, followed by a 20-h incubation at 37°C that allowed for apoptosis to progress. Next, they were stained with calcein AM (that labels the cytosol of viable cells, appearing green) and ethidium homodimer-1 (that labels the nuclei of dead cells, appearing red) and then imaged. When compared to cells of the same high temperature exposure in Figure 6, especially the 47°and 50°C exposures (Figures 6C and 6D, respectively), there is an increase in the number of dead cells because the 37°C/24-h incubation allowed for the progression of apoptosis.
Discussion
Most previous in vitro experiments have been conducted using standard tissue culture ware in which cells are exposed to surfaces such as polystyrene. Little is known whether cell responses to mild hyperthermia treatment vary with different substrates. To our knowledge, this study is the first to report differential thermal sensitivity of ECs on different surfaces. A major new finding of this study was that ECs were more sensitive to hyperthermia-induced death when cultured on ePTFE sheets than on polystyrene dishes that were pretreated for tissue culture and used as surrogate of native tissue in this study. We extrapolate, with cautions, that in the in vivo setting cells found on an ePTFE graft’s luminal surface might be more sensitive to thermal exposure than cells in native tissues. The reason ePTFE renders ECs more sensitive to thermal exposure is not understood and remains unexplored. Nevertheless, we can take advantage of this effect to devise a strategy of using thermotherapy for preventing neointimal hyperplasia on ePTFE grafts without damaging native tissues.
Our experiments were also designed to investigate the effects of various thermal exposures (through combination of different temperatures and heating durations) on the death of ECs on ePTFE. The ranges of temperatures and heating durations in this study took into consideration our intended clinical application (i.e. using mild hyperthermia to prevent cell proliferation on ePTFE vascular grafts). Temperatures exceeding 47°C can cause epidermal and dermal injury [33]. Thus, our target temperature should be 47°C or lower (in other words, 47°C is the highest temperature to use). We found that the lowest temperature needs to be higher than 42°C, because 24 h of heating at 42°C did not cause significant cell death (Figure 4). Therefore, most of the experiments were conducted using 43–47°C; exposure to the high temperature of 50°C was conducted as a positive control. After the range of the temeratures was determined, we next decided the heating durations accordingly. The relationship between the percentage of dead cells and the heating duration is not linear. Henle and Dethlefsen analysed published results of hyperthermia of several mammalian cell types and reported they all exhibited an exponential relationship between ‘exposure temperature and duration’ and ‘cell death’ [49]. While an exponential trend is also suggested in the 50°C treatments of the present study (Figure 5B), more importantly our results show that two combinations of exposure temperature and duration produced greater than 50% necrotic cell death (45°C/90-min and 50°C/30-min exposures). Causing significent cell death through different combinations of temperatures and heating durations provides flexibility in tailoring the thermal treatment so that damage to nearby normal tissue (i.e. native tissues adjacent to the graft) can be minimised while achieving the desired cell death on the graft wall. Treatment flexibility increases the probability of designing a successful therapy to prevent neointimal hyperplasia using mild hyperthermia.
For survival and proliferation, anchorage-dependent cells such as ECs require cytoskeletal forces against the extracellular matrix to form the flattened, spread shape common to anchorage-dependent cells [37–39]. Separation of a cell from its neighbours and morphological rounding are features of apoptosis [40–41]. Welch et al. [42–43] found that hyperthermia treatment (incubation for 3 h at 42–43°C) changed cell morphology and cytoskeleton organisation significantly in rat fibroblasts, particularly a collapse and aggregation of the vimentin-containing intermediate filaments around the nucleus. We found that viable endothelial cells maintained at 37°C displayed a spread shape, the normal morphology for anchored ECs (Figure 6A). At the higher temperatures of 45°, 47°and 50°C, fewer cells spread on the ePTFE and more cells displayed a condensed round shape (Figures 6B–D).
Both necrotic cells and late-stage apoptotic cells have ethidium homodimer-1-positive (red) nuclei. By only performing morphological analyses of viable (green) cells that did not have ethidium homodimer-1-positive (red) nuclei, the influence of necrosis was eliminated from the morphology results and changes in cell morphology reported here (i.e. the decrease in cell size (area and perimeter) and the increase in roundness, as reported in Figures 7–9 and Table I) were caused by only one form of cell death, apoptosis, at the early stage. Our results suggest apoptosis occurs with the 43°and 45°C/30-min exposure, but is more prevalent during 47°C and 50°C/30-min exposure. This interpretation is consistent with significant increases in the percentage of cell death detected after the 20-h post-heating incubation at 37°C: for 47°C, the percentage death increased from ~20% immediately after 30-min heating to ~50% after 30-min heating + 20-h incubation at 37°C; for 50°C, the increase was from ~75 % to ~100%.
We found that necrosis is the prominent course of cell death at 50°C and that, based on the 30-min exposure results, the mechanism of cell death changes from apoptosis to necrosis between 47° and 50°C. Research using Jurkat and murine mastocytoma cells indicates a change in cell death mechanisms between 44° and 46°C. Samali et al. [20] exposed suspended Jurkat cells to temperatures ranging from 37° to 46°C for 60 min. By analysing membrane integrity and cell morphology to detect necrotic and apoptotic death, they reported more than 90% apoptosis for 44°C exposures and almost 100% necrosis for 46°C exposures, suggesting a turning point from apoptosis to necrosis at 45°C. Harmon et al. [17] reached a similar conclusion about a 45°C turning point by treating suspended murine mastocytoma cells for 30 min to temperatures between 37° and 47°C. Our research suggests a turning point slightly higher than 45°C. The small difference in turning point temperature could be caused by the difference between suspended and adhered cells (implying adhered cells are more resistant to heating than suspended cells) and/or the difference in cell origin and species (implying that endothelial cells are more resistant to heating than Jurkat or murine mastocytoma cells). It has been reported that cellular response to heat differs with cell origin and species [28].
We found that cell death percentage increased, likely due to apoptosis, after 20 h of incubation at 37°C following the 47°C/30-min exposure and the 50°C/30-min exposure, where the total cell death reached almost 50% and 100%, respectively. However, 50°C exposure caused 75% death due to necrosis, but 47°C exposures only caused 20% death due to necrosis. By applying heat at a lower temperature, the native healthy tissue surrounding the graft will experience less heating, while in the graft lumen the cells are sufficiently heated to cause significant death via apoptosis. Our data showed that applying temperatures greater than 47°C increases the risk of necrotic death. In fact, high temperatures have actually been shown to increase neointimal growth [33]. Necrotic death is characterised by cellular membrane rupture and a release of cytosolic contents. This leads to inflammation and the attraction of macrophages which promote cellular growth and proliferation. Therefore, despite causing nearly 100% cell death (~75% by necrosis), the 50°C/30-min exposure should be avoided to reduce inflammatory responses in the graft region. Consequently, the 47°C/30-min exposure would be preferred for graft heating because it generated 50% cell death (~30% via apoptosis) without extreme temperature. The 47°C/30-min exposure may be able to limit cell proliferation in a graft lumen and extend the vascular access lifetime.
Conclusion
In summary, we report here that (1) ECs were more sensitive to hyperthermia-induced death when cultured on ePTFE sheets than on polystyrene dishes that were used as surrogate of native tissue; and (2) when mild hyperthermia was used to cause endothelial cell death on ePTFE, different combinations of temperature and exposure time produced cell death of 50% or more. Two thermal doses (50°C/30 min and 45°C/90 min) produced greater than 50% necrotic cell death. However, since necrosis may stimulate re-growth of cells and paradoxically predispose to neointimal hyperplasia, apoptotic cell death is preferable. A 30-min heating at 47°C generated 50% cell death, over half of which was through apoptosis. Therefore, by inducing apoptosis at the luminal surface of ePTFE grafts, repeated exposures around 47°C could reduce hyperplasia and hence stenosis. In vitro studies to examine the thermal sensitivity of other cell types involved in neointimal hyperplasia (such as smooth muscle cells) and in vivo animal studies are warranted to examine this treatment strategy.
Footnotes
Declaration of interest: A.K. Cheung received support from the National Heart, Lung and Blood Institute (RO1HL67646) and the Veterans Affairs Merit Review Program. D.A. Christenson received support from the University of Utah Research Foundation. Y.E. Shiu received support from the American Heart Association (0970307 N and 0765113 Y). The authors alone are responsible for the content and writing of the paper.
References
- 1.K-DOQI (Kidney Disease Outcomes Quality Initiative) Clinical practice guidelines for vascular access. Am J Kidney Dis 2006;48:S177–S276. [Google Scholar]
- 2.Hemodialysis Access. New York: National Kidney Foundation. Available from: http://www.kidney.org/atoz/content/hemoaccess.cfm (cited November 2009). [Google Scholar]
- 3.DOPPS. 2009. Annual Report of the Dialysis Outcomes and Practice Patterns Study: Hemodialysis Data 1999–2008. Ann Arbor, MI: Arbor Research Collaborative for Health. [Google Scholar]
- 4.Hodges T, Fillinger M, Zwolak R, Walsh D, Bech F, Cronenwett J. Longitudinal comparison of dialysis access methods: Risk factors for failure. J Vasc Surg 1997;26:1009–1019. [DOI] [PubMed] [Google Scholar]
- 5.Leapman S, Boyle M, Pescovitz M, Milgrom M, Jindal R, Filo R. The arteriovenous fistula for hemodialysis access: Gold standard or archaic relic? Am Surg 1996;62:652–656. [PubMed] [Google Scholar]
- 6.Miller P, Tolwani A, Luscy C, Deierhoi M, Bailey R, Redden D, et al. Predictors of adequacy of arteriovenous fistulas in hemodialysis patients. Kidney Int 1999;56:275–280. [DOI] [PubMed] [Google Scholar]
- 7.Pisoni R, Young E, Dykstra D, Greenwood R, Hecking E, Gillespie B, et al. Vascular access use in Europe and United States: Results from the DOPPS. Kidney Int 2002;61:305–316. [DOI] [PubMed] [Google Scholar]
- 8.Pisoni R, Young E, Mapes D, Keen M, Port F. Vascular access use and outcomes in the US, Europe, and Japan: Results from the Dialysis Outcomes and Practice Patterns Study. Nephrol News Issues 2003;17:38–43,47. [PubMed] [Google Scholar]
- 9.Bordenave L, Fernandez P, Remy-Zolghadri M, Villars S, Daculsi R, Midy D. In vitro endothelialized ePTFE prostheses: Clinical update 20 years after the first realization. Clin Hemorheol Microcirc 2005;33:227–234. [PubMed] [Google Scholar]
- 10.Pawlowski K, Rittgers S, Schmidt S, Bowlin G. Endothelial cell seeding of polymeric vascular grafts. Front Biosci 2004;9:1412–1421. [DOI] [PubMed] [Google Scholar]
- 11.Rotmans J, Heyligers J, Verhagen H, Velema E, Nagtegaal M, de Kleijn D, et al. In vivo cell seeding with anti-CD34 antibodies successfully accelerates endothelialization but stimulates intimal hyperplasia in porcine arteriovenous expanded polytetrafluoroethylene grafts. Circulation 2005; 112:12–18. [DOI] [PubMed] [Google Scholar]
- 12.Colombo A, Drzewiecki J, Banning A, Grube E, Hauptmann K, Silber S, et al. Randomized study to assess the effectiveness of slow-and moderate-release polymer-based paclitaxel-eluting stents for coronary artery lesions. Circulation 2003;108:788–794. [DOI] [PubMed] [Google Scholar]
- 13.Regar E, Serruys P, Bode C, Holubarsch C, Guermonprez J, Wijns W, et al. Angiographic findings of the multicenter Randomized Study with the Sirolimus-Eluting Bx Velocity Balloon-Expandable Stent (RAVEL): Sirolimus-eluting stents inhibit restenosis irrespective of the vessel size. Circulation 2002;106:1949–1956. [DOI] [PubMed] [Google Scholar]
- 14.Kopecek J Smart and genetically engineered biomaterials and drug delivery systems. Eur J Pharm Sci 2003;20:1–16. [DOI] [PubMed] [Google Scholar]
- 15.Sun S, Beitler J, Ohki T, Calderon T, Schechner R, Yaparpalvi R, et al. Inhibitory effect of brachytherapy on intimal hyperplasia in arteriovenous fistula. J Surg Res 2003;115:200–208. [DOI] [PubMed] [Google Scholar]
- 16.Gorman A, Heavey B, Creagh E, Cotter T, Samali A. Antioxidant-mediated inhibition of the heat shock response leads to apoptosis. FEBS Lett 1999;445:98–102. [DOI] [PubMed] [Google Scholar]
- 17.Harmon B, Corder A, Collins R, Gobé G, Allen J, Allan D, Kerr J. Cell death induced in a murine mastocytoma by 42–47°C heating in vitro: Evidence that the form of death changes from apoptosis to necrosis above a critical heat load. Int J Radiat Biol 1990;58:845–858. [DOI] [PubMed] [Google Scholar]
- 18.Qian L, Xueli S, Huirong R, Jingbo G, Suqi C. Mitochonrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress Chaperones 2004;9:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roti J Cellular responses to hyperthermia (40–46°C): Cell killing and molecular events. Int J Hyperthermia 2008;24:3–15. [DOI] [PubMed] [Google Scholar]
- 20.Samali A, Holmberg C, Sistonen L, Orrenius S. Thermotolerance and cell death are distinct cellular responses to stress: Dependence on heat shock proteins. FEBS Lett 1999;461:306–310. [DOI] [PubMed] [Google Scholar]
- 21.Dewey W, Sapareto S. Thermal dose determination in cancer therapy. J Radiat Oncol Biol Phys 1984;10:787–800. [DOI] [PubMed] [Google Scholar]
- 22.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 23.Ziegler U, Groscurth P. Morphological features of cell death. News Physiol Sci 2004;19:124–128. [DOI] [PubMed] [Google Scholar]
- 24.Lodish H, Berk A, Kaiser C, Krieger M, Scott M, Bretscher A, et al. Cell Biology, 6th edn. New York: Freeman, 2008. [Google Scholar]
- 25.Razvi E, Welsh R. Apoptosis in viral infections. Adv Virus Res 1995;49:1–8. [DOI] [PubMed] [Google Scholar]
- 26.Fajardo L, Schreiber A, Kelly N, Hahn G. Thermal sensitivity of endothelial cells. Radiat Res 1985;103:276–285. [PubMed] [Google Scholar]
- 27.Fajardo L, Prionas S. Endothelial cells and hyperthermia. Int J Hyperthermia 1994;10:347–353. [DOI] [PubMed] [Google Scholar]
- 28.Ketis N, Hoover R, Karnovsky M. Effects of hyperthermia on cell survival and patterns of protein synthesis in endothelial cells from different origins. Cancer Res 1988;48: 2101–2106. [PubMed] [Google Scholar]
- 29.Kerr J Shrinkage Necrosis: A distinct mode of cellular death. J Path 1971;105:13–20. [DOI] [PubMed] [Google Scholar]
- 30.Kerr J, Wyllie A, Currie A. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruxton G The unequal variance t-test is an underused alternative to Student’s ttest and the Mann-Whitney U test. Behav Ecol 2006;17:688–690. [Google Scholar]
- 32.Rockwell S Effects of clumps and clusters on survival measurements with clonogenic assays. Cancer Res 1985; 45:1601–1607. [PubMed] [Google Scholar]
- 33.Hehrlein C, Chuang C, Tuntelder J, Tatsis G, Littmann L, Svenson R. Effects of vascular runoff on myointimal hyperplasia after mechanical balloon or thermal laser arterial injury in dogs. Circulation 1991;84: 884–890. [DOI] [PubMed] [Google Scholar]
- 34.Herren B, Levkau B, Raines EW, Ross R. Cleavage of betacatenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: Evidence for a role for caspases and metalloproteinases. Mol Biol Cell 1998;9:1589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallat Z, Tedgui A. Apoptosis in the vasculature: Mechanisms and functional importance. Br J Pharmacol 2000;130:947–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henle K, Dethlefsen L. Time-temperature relationships for heat-induced killing of mammalian cells. Ann N Y Acad Sci 1980;335:234–253. [DOI] [PubMed] [Google Scholar]
- 37.Chicurel M, Chen C, Ingber D. Cellular control lies in the balance of forces. Curr Opin Cell Biol 1998;10:232–239. [DOI] [PubMed] [Google Scholar]
- 38.Galbraith C, Sheetz M. Forces on adhesive contacts affect cell function. Curr Opin Cell Biol 1998;10:566–571. [DOI] [PubMed] [Google Scholar]
- 39.Katsumi A, Orr A, Tzima E, Schwartz M. Integrins in mechanotransduction. J Biol Chem 2004;279:12001–12004. [DOI] [PubMed] [Google Scholar]
- 40.Kampinga H Thermotolerance in mammalian cells. Protein denaturation and aggregation, and stress proteins. J Cell Sci 1993;104:11–17. [DOI] [PubMed] [Google Scholar]
- 41.Champagne M, Dumas P, Orlov S, Bennett M, Hamet P, Tremblay J. Protection against necrosis but not apoptosis by heat-stress proteins in vascular smooth muscle cells: Evidence for distinct modes of cell death. Hypertension 1999; 33:906–913. [DOI] [PubMed] [Google Scholar]
- 42.Welch W, Mizzen L. Characterization of the thermotolerant cell. II. Effects on the intracellular distribution of heat-shock protein 70, intermediate filaments, and small nuclear ribonucleoprotein complexes. J Cell Biol 1988;106: 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welch W, Suhan J. Morphological study of the mammalian stress response: Characterization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance of intranuclear actin filaments in rat fibroblasts after heat-shock treatment. J Cell Biol 1985;101:1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]