

Abstract
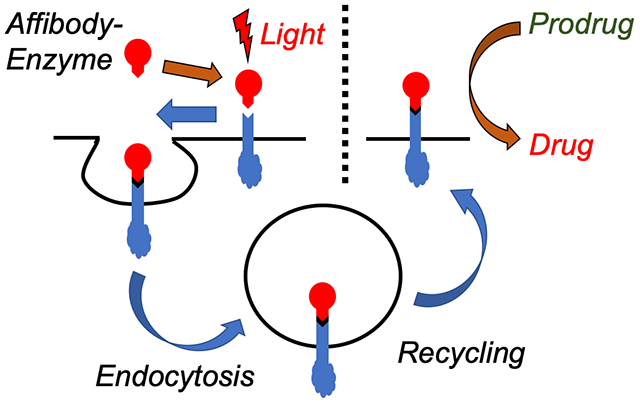
In this work, we show that a prodrug enzyme covalently photoconjugated to live cell receptors survive endosomal proteolysis and retain its catalytic activity over multiple days. Here, a fusion protein was designed with both an anti-epidermal growth factor receptor (EGFR) affibody and the prodrug enzyme cytosine deaminase, which can convert prodrug 5-fluorocytosine to the anticancer drug 5-fluorouracil. A benzophenone group was added at a site-specific mutation within the affibody, and the fusion protein was selectively photoconjugated to EGFR receptors expressed on membranes of MDA-MB-468 breast cancer cells. The fusion protein was next labeled with two dyes for tracking uptake: AlexaFluor 488 and pH-sensitive pHAb. Flow cytometry showed that fusion proteins photocrosslinked to EGFR first underwent receptor-mediated endocytosis within 12 h, followed by recycling back to the cell membrane within 24 h. These findings were also confirmed by confocal microscopy. The unique crosslinking of the affibody-enzyme fusion proteins were utilized for two anticancer treatments. First, the covalent linking of the protein to the EGFR led to inhibition of ERK signaling over a two-day period, whereas conventional antibody therapy only led to 6 h of inhibition. Second, when the affibody-CodA fusion proteins were photocrosslinked to EGFR overexpressed on MDA-MB-468 breast cancer cells, prodrug conversion was found even 48 h post-incubation without any apparent decrease in cell killing, while without photocrosslinking no cell killing was observed 8 h post-incubation. These studies show that affinity-mediated covalent conjugation of the affibody-enzymes to cell receptors allows for prolonged expression on membranes and retained enzymatic activity without genetic engineering.
Keywords: Affibody, Epidermal Growth Factor Receptor (EGFR), cytosine deaminase, enzyme prodrug therapy, receptor-mediated endocytosis, affinity-mediated
Graphical Abstract
INTRODUCTION
Because of the considerable negative side effects of anticancer drugs, many modern chemotherapy approaches seek to localize drug activity to cancerous tissues, for example by biasing drug accumulation through active targeting. Antibody-targeted enzyme prodrug therapy hones a few techniques into one approach1. First, an enzyme is conjugated to an antibody so that the conjugate localizes to a solid tumor environment. This step is followed by systemic administration of prodrugs, which are nontoxic molecules that are converted into toxic species by a chemical transformation in the body. By using a bacterial enzyme (e.g. beta-lactamase or cytosine deaminase), prodrug molecules may be converted to drug molecules without a competing background reaction in the human patient. Thus, although the prodrug itself has no inherent selectivity for the cancer, the overabundance of enzyme at the tumor causes selective conversion and toxicity, along with reduced systemic side effects.
However, one of the main roadblocks to clinical acceptance has been the need to administer enzymes multiple times per patient due to loss of activity. Because it is known that cancer cells begin DNA repair within 24 h of drug-induced strand breaks, the ability to administer multiple effective prodrug doses is critical for reducing tumor size1. Unfortunately, while the presence of the antibody on the enzyme conjugate allows homing to tumor tissue, the antibody also promotes receptor-mediated endocytosis, resulting in catabolic processes that degrade the enzyme2–6. Eventually, repeat dosings become ineffective because the body produces antibodies that clear the antibody-enzyme conjugate before it can reach the tumor1,7,8.
In this work, we utilize a method of affinity-mediated covalent photoconjugation to both confine an enzyme to a cancer cell and, significantly, maintain its expression and activity over multiple days by escaping proteolysis. A number of exquisite reactions have been shown to work both on live cell membranes and in live animals, but these tend to focus on small molecule labels rather than full proteins9–15. Here, we hypothesized that a covalent bond could mimic the neonatal Fc receptor (FcRn), which is responsible for prolonging the circulation of abundant proteins like albumin and IgG. Because the FcRn binds tightly to the internalized protein, the protein does not desorb from its receptor and is instead recycled out of the cell16,17. Thus, inducing covalent binding of a protein to a target receptor would similarly allow the protein to escape proteolysis and instead be recycled back to the membrane as a permanent fusion to the membrane protein. Thus, rather than losing activity, the enzyme remains presented on the membrane as a functional protein unit over longer time scales, where it can convert multiple prodrug doses into toxic drugs without the need for additional enzyme administrations. Finally, while the work shown here is aimed toward prodrug therapy applications, this method more generally describes a method to modify potentially any specific cell receptor with a unique biological tag without genetic engineering.
RESULTS AND DISCUSSION
In order to show retention of prodrug enzyme activity after photoconjugation to a cell receptor, a fusion protein was created consisting of cytosine deaminase (CodA) and an affibody capable of photocrosslinking to epidermal growth factor receptor (EGFR), its target ligand. In previous work, we showed that the EGFR-binding affibody ZEGFR:1907 (henceforth known as wildtype, or WT)18, once mutated specifically at its N23 position to a cysteine, could then be conjugated directly with benzophenone (BP). Upon photoirradiation with either long UV or NIR light in the presence of upconverting nanoparticles, the BP modified affibodies (N23BP) became covalently conjugated either to pure EGFR or to EGFR expressed on cells in both 2- and 3D models19 . In the work shown here, N23BP EGFR binding affibodies were fused with cytosine deaminase (CodA), a bacterial enzyme encoded by the CodA sequence of E. coli20 that can convert 5-fluorocytosine (5-FC) prodrug to toxic 5-fluorouracil (5-FU) and thus has been used frequently in ADEPT (Antibody Directed Enzyme Prodrug Therapy)21. As with the affibodies alone, the fusion proteins were expressed in the E. coli strain BL21(DE3) and purified from cell lysate by Ni-NTA bead capture. The purified proteins were then reacted with maleimide-benzophenone (BP), followed by removal of excess BP through 20 kDa MWCO dialysis cups (Thermo Fisher). Using similar protocols, proteins were expressed consisting of WT affibody fused CodA (WT-CodA), as well as CodA alone without affibody. Each of these proteins possessed expected molecular weights when analyzed by SDS-PAGE (Figure 1).
Figure 1.
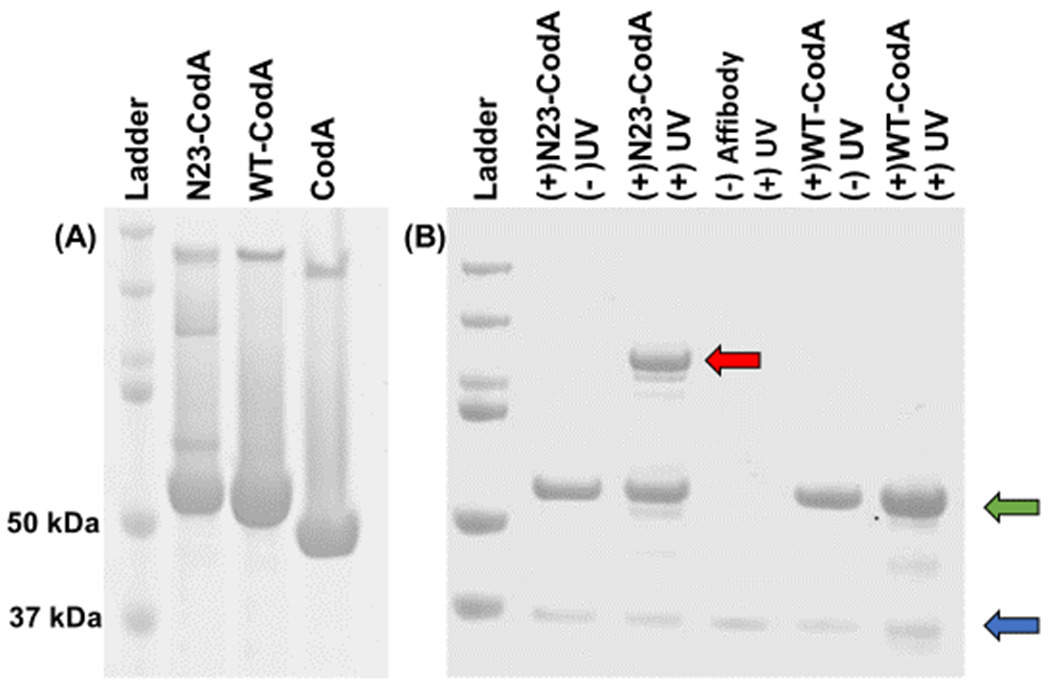
(A) SDS-PAGE of purified affibody-enzyme fusion proteins: (left to right) ladder, N23BP-CodA, WT-CodA, and free CodA. The molecular weights of the ladder proteins are (top to bottom): 200, 150, 100, 75, 50, and 37 kDa. (B) SDS-PAGE of affibody-enzyme fusion proteins photocrosslinked with the soluble extracellular fragment (ECF) of EGFR. Only the combination of N23BP-CodA and UV leads to the formation of a new band (red arrow). The affibody bands and the free EGFR ECF bands are shown in green and blue arrows, respectively. Left to right: Ladder, (+) N23BP-CodA (+) EGFR (−) UV, (+) N23BP-CodA (+) EGFR (+) UV, (−) affibody (+) EGFR (+) UV, (+) WT-CodA (+) EGFR (−) UV, and (+) WT-CodA (+) EGFR (+) UV. Ladder molecular weights are same as for (A).
The affibody-enzyme fusions were next characterized to ensure that they possessed EGFR-binding and photocrosslinking similar to free affibodies. First, non-covalent affinity for EGFR was tested by microscale thermophoresis measurements (MST). For this, the purified soluble fragment of EGFR was conjugated to AlexaFluor 647 (Thermo Fisher) and mixed with various concentrations of affibody-enzyme fusion proteins ranging from 10 μM to 100 pM. From these measurements, KD values were obtained showing KD = 25 nM ± 0.66 nm and 44 nM ± 1.02 nm for WT-CodA and N23BP-CodA, respectively (Figure S1). While these values are approximately an order of magnitude larger than those of the reported free affibody (KD ~ 2.8 nM)18 , they are still well within nanomolar range. Interestingly, the N23BP-CodA fusion proteins actually showed more favorable binding to EGFR than BP-substituted N23BP affibody alone19, which may be attributed to reduced solubility of the small affibodies after conjugation with benzophenone and dyes. Finally, temperature-dependent circular dichroism studies showed that the melting temperature of both affibody-enzyme fusion proteins (~80°C) were similar to the reported value for free CodA (85°C ± 2°C)22, which shows that the enzyme structure is not likely to be compromised as a result of affibody fusion (Figure S2).
Next, the affibody-CodA fusion proteins were tested for photocrosslinking to pure EGFR. For this, wild-type affibody-enzyme (WT-CodA), BP-modified mutant affibody-enzyme (N23BP-CodA), and the enzyme itself (CodA) were each mixed with purified soluble fragment of EGFR and irradiated with 890 μW/cm2 of 365 nm light for 30 min, which is within recommended limits23 and does not harm biological samples, as found in previous studies by us and others19,24,25. Following deglycosylation and thermal denaturation (see Methods), each mixture was run on a SDS-PAGE gel. While the WT-CodA reaction with EGFR yielded only individual bands corresponding to either the affibody-enzyme alone (57 kDa) or the soluble purified deglycoslyated EGFR fragment (40 kDa), the BP-containing N23BP-CodA fusion protein showed successful photocrosslinking to EGFR after irradiation as observed by the appearance of a new band >100 kDa which is the sum of the molecular weights of EGFR and N23BP-CodA. Free EGFR and free N23BP-CodA are also evident as separate bands on the gel, indicating that the reaction is not quantitative, which was also seen previously18. Thus, these results show that despite the affibody being fused to cytosine deaminase, the BP containing affibody was successfully photocrosslinked to EGFR (Figure 1B).
The next step was to track the fate of the fusion proteins once bound or crosslinked to receptors on live cells. Anticipating receptor-mediated endocytosis, the fusion proteins were conjugated with either of two dyes: pH-tolerant, green-fluorescent AlexaFluor 488 (AF488, Thermo Fisher), or the pH-sensitive dye pHAb (Promega), which shows strong emission in the red (580 nm) at pH < 6.5 (endosome) but has low photoluminescence at pH > 6.5 (cytosol/external fluid) (Figure S3)26. To attach the dyes, the affibody-enzyme fusion proteins were first reacted with either NHS-AF488 or NHS-pHAb at 10:1 molar ratios of dye:protein for 2 h at room temperature, followed by dialysis to remove any unreacted fluorophore. On average, this approach yielded a degree of labeling of ~1.2 moles of dye per mole protein as determined from UV-Vis spectra (Figure S4). Next, each form of affibody-CodA (WT, N23BP) was incubated with EGFR-positive MDA-MB-468 human breast carcinoma cells (~30,000 cells/well). After 3 h of incubation, the cells were irradiated with 890 μW/cm2 of 365 nm light for 30 min. Finally, cells were harvested at 2, 4, 6, 12, or 24 h (cells incubated for only 2 h were not irradiated) by washing three times to remove excess affibody-enzyme, trypsinized, and fixed with 4% paraformaldehyde. The cells were then transferred to PBS and the cell suspension was analyzed in a BDFACSCelesta flow cytometer (BD Biosciences) with simultaneous 488 laser and 561 laser excitations for tracking AF488 and pHAb, respectively.
While cells incubated with WT-CodA showed only a short increase in AF488 fluorescence at 2 h followed by a steady decrease (Figure 2), the cells that were treated with N23BP-CodA demonstrated a marked, progressive increase in fluorescence per cell over a time period as long as 24 h. These results are consistent with our previous results where N23BP affibodies were retained in 2- and 3D tumor models while WT affibodies showed a significant decrease in EGFR binding after 4 h19. The pHAb fluorescence of the WT-CodA proteins followed a similar trend, peaking at 2 h and decreasing thereafter. In direct contrast, however, the pHAb fluorescence of the N23BP-CodA shows that the photoconjugated N23BP-CodA proteins are first taken into endosomes – reaching a maximum fluorescence at 12 h incubation – followed by a decline at 24 h. To confirm both the specificity of EGFR targeting and that fusion protein uptake is an EGFR-mediated process, the above experiment was repeated with addition of equimolar free Epidermal Growth Factor (EGF), which led to reduced retention of N23BP-CodA in cells. This effect, which is attributed to competition of EGF and affibody-enzyme for EGFR binding sites27,28, was also observed for the accumulation of dye conjugated WT-CodA affibody-enzymes (Figure S5).
Figure 2.
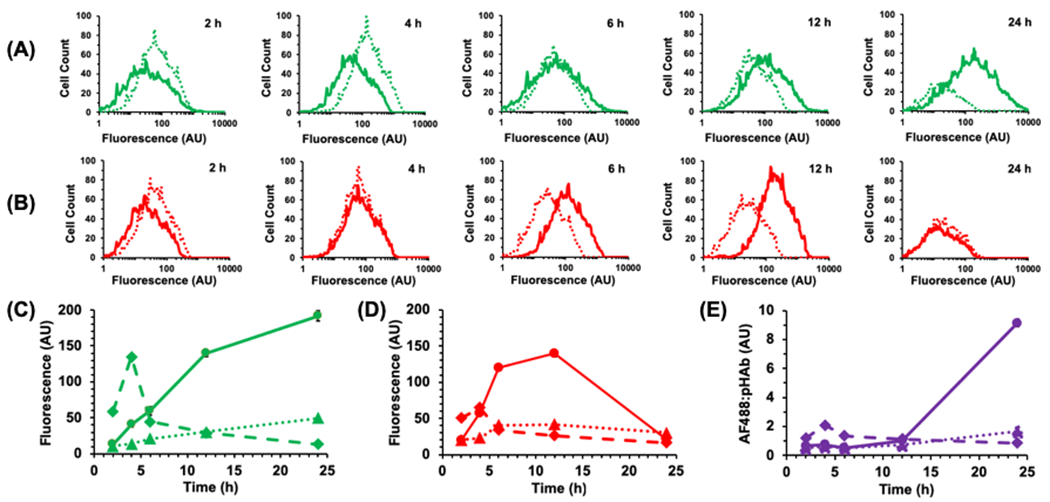
Representative flow cytometry histograms of MDA-MB-468 cells showing the change in fluorescence intensity of (A) AF488 and (B) pHAb conjugated to N23BP-CodA (solid line) and WT-CodA (dotted line) at various times after incubation. Samples were irradiated at the 3 h mark; 2 h samples were not irradiated. Samples were run in triplicate (n=3), but representative plots are shown for clarity. (C, D) Change of mean fluorescence intensity with time of (C) AF488 and (D) pHAb conjugated to N23BP-CodA (solid line) and WT-CodA (dashed line). Dotted line shows fluorescence from N23BP-CodA with EGF added. (E) Ratio of AF488:pHAb fluorescence for N23BP-CodA (solid), WT-CodA (dashed), and N23BP-CodA + EGF (dotted) as function of time. For C-E, error bars represent standard error; in some cases, the error range is smaller than the data marker.
In addition to flow cytometry, confocal microscopy was utilized to examine the fate of affibody-enzymes in MDA-MB-468 cells. The cells were grown on poly(D-lysine) coated glass coverslips (Corning BioCoat) and incubated with either AF488-pHAb-N23BP-CodA or AF488-pHAb-WT-CodA. After 3 h, the cells were irradiated for 30 min as described above. At 4, 12, and 24 h total incubation, the cells were washed and incubated with the nuclear stain 4’,6-diamidino-2-phenylindole (DAPI) for 10 min. The cells were washed an additional three times with PBS before mounting the cells with Prolong Gold antifade mountant (Thermo Fisher) and then imaging with a Nikon A1R Laser Confocal Microscope. For cells with WT-CodA, significant fluorescence was observed in both the green and red channels at 4 h, corresponding to AF488 and pHAb, respectively (Figure 3). However, both channels showed marked decrease in fluorescence at 12 and 24 h. For the cells with N23BP-CodA, bright fluorescence was observed in both the green and red channels at 4 h and 12 h. At 24 h, however, a strong green fluorescence was observed with a dim red fluorescence, which is consistent with the theory that the photocrosslinked affibody-enzyme is being recycled back to the cell membrane. To quantify the relationship between AF488 and pHAb fluorescence, colocalization analysis was performed on the images from the green and red channels without thresholding. A heat map of normalized pixel intensities is shown in Figure 3, along with a Pearson Correlation Coefficient to provide a measure of colocalization. For the WT-CodA, a PCC of 0.601 was found at 4 h, which is consistent with considerable pixel overlap. At 12 and 24 h, however, the PCC decreases to 0.494 and 0.313, with likely capture of green AF488 emission in the red channel. For N23BP-CodA, high PCC values of 0.720 and 0.814 were obtained at 4 h and 12 h. At 24 h, when green fluorescence is mostly present, the PCC drops to 0.409. Thus, the N23BP-CodA appears to retain its structure at 12 h when non-covalently bound proteins would otherwise be degraded, while the red fluorescence drops away at 24 h as was observed in the flow cytometry studies, indicating that the N23BP-CodA leaves the endosome intact.
Figure 3.
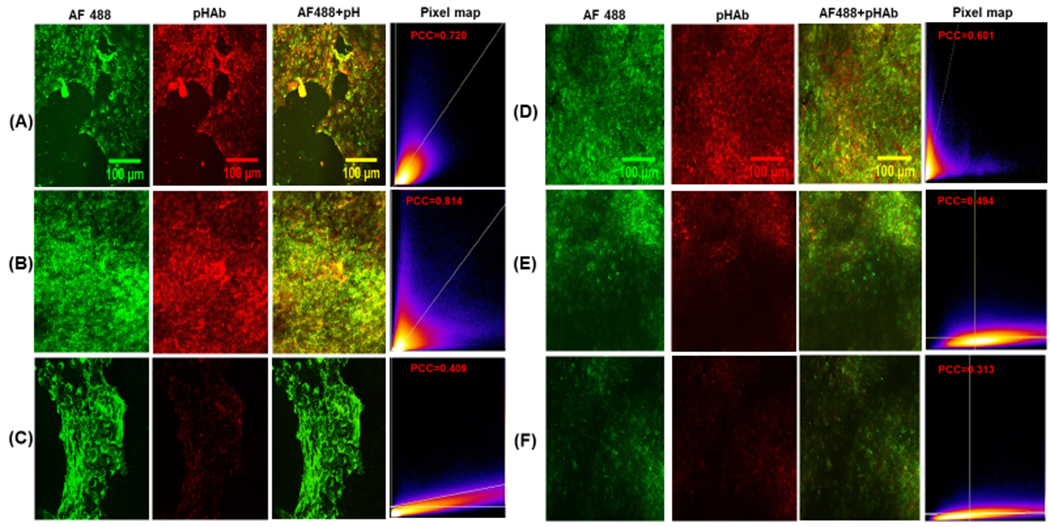
(A-C) Confocal microscopy images of MDA-MB-468 breast cancer cells treated with AF488-pHAb-N23BP-CodA for (A) 4 h, (B) 12 h, and (C) 24 h. The pixel map shows the colocalization of the two dyes (x: green, y: red) with the reported Pearson Correlation Coefficient (PCC). (D-F) Confocal microscopy images of MDA-MB-468 breast cancer cells treated with AF488-pHAb-WT-CodA for (D) 4 h, (E) 12 h, and (F) 24 h, with pixel map showing colocalization and PCC.
Having shown that the affibody-enzyme remains attached to the EGFR receptor after irradiation, we next sought to determine the intracellular signaling effect of covalently attaching the affibody fusions to EGFR, as EGFR inhibition is a potential method for treating certain cancers, including lung, colorectal, and head and neck29. While extracellular signal-regulated kinases (ERK) signaling can be detected using Western blot30 , this technique provides only a static snapshot of cellular activities; instead, to measure ERK activity in living cells in real-time, a genetically encoded Forster Resonance Energy Transfer (FRET)-based sensor named EKAR (Extracellular Signal Regulated Kinase Activity Reporter) was used31,32. Upon initiation of ERK activity, the CFP-YFP-containing EKAR sensor undergoes a conformational change to reduce the intramolecular distance between the donor-acceptor dye pair, thereby leading to an increase in FRET mediated fluorescence. Thus, in order to monitor ERK signaling in MDA-MB-468 cells, the cells were first transfected with the EKAR sensor (see Materials and Methods). The transfected cells (Figure S6) were next seeded at a density of 10000 cells/well and incubated with 100 nM of N23BP-CodA or Cetuximab (a clinically approved anti-EGFR monoclonal antibody) followed by 30 min UV photocrosslinking after 3 h. The cells were imaged at 0.5, 2, 6, 12 and 24 h. After 24 h, the old media was removed and 100 nM EGF in fresh media was added to the cells, and FRET fluorescence was imaged again at 0.5, 2, 6, 12, and 24 h post-addition. To obtain a background EKAR FRET intensity, two comparison studies without N23BP-CodA were performed: one control received a simple media change after 24 h while the other received 100 nM EGF in fresh media. At each time point, the cells were imaged using Molecular Devices ImageXpress. Fluorescence was measured and converted into a quantitative signal via a custom MATLAB software.
As shown in Figure 4, photocrosslinking affibody-enzyme to MDA-MB-468 cells resulted in EGFR inhibition that was prolonged past 24 h. In contrast, addition of Cetuximab led an initial decrease in ERK activity that was dissipated after only 6 h. When fresh EGF was added after 24 h to cells photocrosslinked to N23BP-CodA, only mild increases in ERK activity were observed due to the presence of other growth factors in the media, similar to a control sample without EGF. In contrast, cells treated with Cetuximab follwed by fresh EGF at 24 h showed a larger gain in ERK activity similar to untreated cells with fresh EGF. This data further supports the result that photocrosslinking fusion proteins to EGFR prevents detachment and catabolism of the targeting ligands such that EGF cannot bind and minimal increases in ERK activity. As a result, EGFR inhibition lasts much longer than for current antibody therapies.
Figure 4.
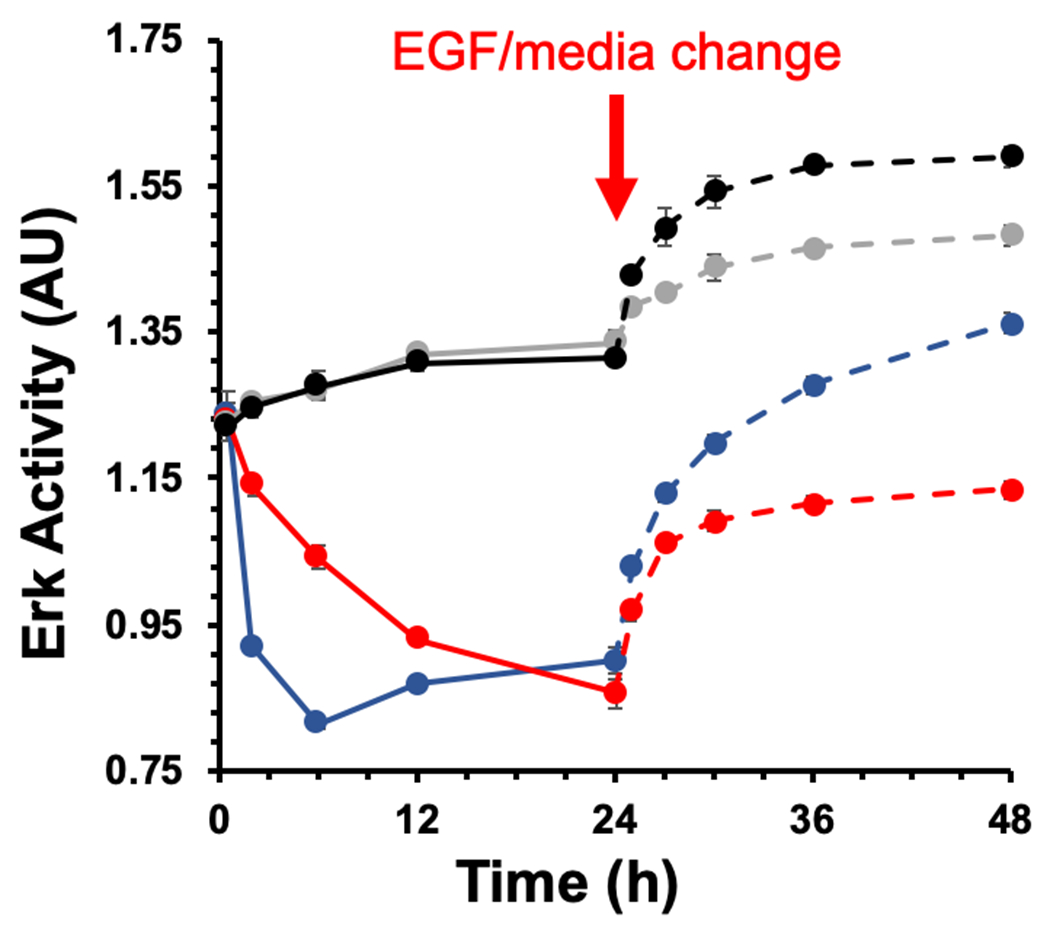
ERK activity inhibition in the presence of N23BP-CodA compared with Cetuximab. The time points used for the experiments are 0.5, 2, 6, 12, and 24 h, with UV irradiation after 3 h. Red: (+) N23BP-CodA; Blue: (+) Cetuximab; Gray: (−) treatment, (−) EGF; Black: (−) treatment, (+) EGF. The solid lines represent the time points before EGF addition and the dotted lines indicate time points after EGF addition or media change (gray only). Each sample was run in quadruplicate and the error bars indicate standard error (n=4).
Finally, having established both the photocrosslinking of the N23BP-CodA affibody-enzyme to EGFR on living cells and retention of protein expression before and after endocytosis, the enzymatic activity of the fusion proteins was then leveraged for cell killing by prodrug conversion. First, as determined by measuring cytosine conversion to uracil by UV-Vis spectroscopy, the KM and kcat of the affibody-enzyme fusion proteins (N23BP and WT) were within 70% of those displayed by the native enzyme (Figure S7, Table S1)33 . Next, prodrug therapy experiments were run against MDA-MB-468 cells in vitro. In 96-well plates, cells were incubated with 1 μM of WT-CodA, N23BP-CodA, or CodA alone. As before, after 3 h the cells were irradiated with 365 nm light for 30 min followed by further incubation to a total time of 8, 24, or 48 h. When each time point was reached, the excess affibody-enzymes were removed by washing the cells three times with PBS, and 2 mM of 5-FC was added. Three control samples were also included, one without 5-FC, one without UV, and one with neither 5-FC nor UV. A few wells also contained just 5-FC (prodrug) and 5-FU (drug) to establish baseline toxicities. After 12 h incubation with 5-FC, excess prodrug was removed, cells were washed with PBS, and viability was measured by MTT assay. Cells treated with WT-CodA or free CodA showed ~70-80% ±1-4% viability (Figure 5A, slightly less than the 5-FC baseline viability of 85% ± 2%. However, photocrosslinked N23BP-CodA proteins incubated with 5-FC showed a marked decrease in viability to ~50-55% ± 2.5%, similar to the baseline 5-FU toxicity (47% ± 2.6%). The differences in cell viability between N23BP-CodA and WT-CodA were found to be statistically significant, with p < 0.05 by Student’s t-test for all samples (N=3). Without either UV irradiation or 5-FC addition, little toxicity was observed. Interestingly, this same trend was found at all the enzyme incubation time points, even 48 h after adding the affibody-enzymes to the MDA-MB-468 cells. In order to increase the cytotoxic effect of the affibody-enzyme system, the cells were incubated with the affibody-enzymes for 3 h, followed by UV irradiation for 30 min and incubated further for a total of 8 h. Next, the cells were dosed with 2 mM 5-FC for 24, 48, and 72 h with fresh 5-FC added every 12 h. In this assay, cells treated with WT-CodA or free CodA showed a viability of 66-74% ± 1.5-4% (Figure 5B) while the baseline viability for 5-FC for all these time points was 77-84% ± 1-3%. The cells incubated with N23BP-CodA affibody-enzymes showed a much lower viability of 35-50% ± 1-3%, close to the 5-FU viability for these timepoints (30-45% ± 0.5-4%). The differences in cell viability between N23BP-CodA and WT-CodA were found to be statistically significant, with p < 0.05 by Student’s t-test for all samples (N=3). Thus, this affibody-enzyme photocrosslinking strategy holds promise for longer-term retention of prodrug activation by prolonging the expression, lifetime, and activity of the affibody-enzymes on the cell membrane.
Figure 5.
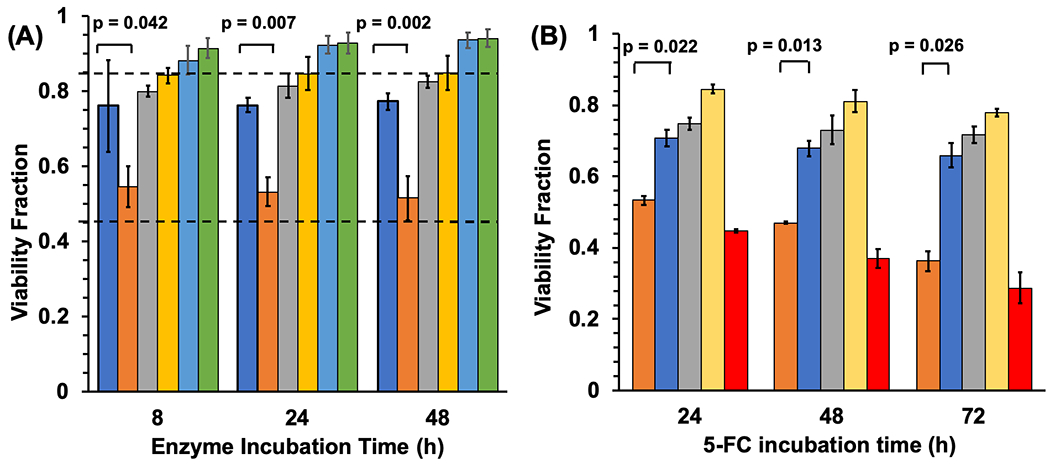
(A) Cell viability fraction of MDA-MB-468 cells in combination with affibody-enzyme fusion proteins, 5-fluorocytosine prodrug, and UV light, as measured by MTT assay for different affibody-enzyme incubation times. Top dotted line: baseline viability of cells incubated with 5-FC prodrug (0.854). Middle dotted line: baseline viability of cells incubated with 5-FU drug (0.467). Left to right at each timepoint: (+)WT-CodA (+)5-FC (+)UV, (+)N23BP-CodA (+)5-FC (+)UV, (+)CodA (+)5-FC (+)UV, (+)N23BP-CodA (+)5-FC (−)UV, (+)N23BP-CodA (−)5-FC (+)UV, and (+)N23BP-CodA (−)5-FC (−)UV. Error bars represent standard error. (B) Cell viability fraction of MDA-MB-468 cells in combination with affibody-enzyme fusion proteins, 5-fluorocytosine prodrug, and UV light, as measured by MTT assay for different 5-fluorocytosine incubation time points. Left to right at each timepoint: (+) N23BP-CodA, (+) WT-CodA, (+) CodA, (+) 5-FC, (+) 5-FU (no 5-FC). Error bars represent standard error. All experiments were performed in triplicate (n=3).
CONCLUSION
This work shows the possibility of using a direct, gene-free affinity-mediated covalent chemistry approach to tag receptors on live cells with unique protein segments while retaining expression over multiple days. This result was shown by first synthesizing an affibody-CodA construct that could form covalent photocrosslinks with EGFR through coupling of an attached benzophenone. Flow cytometry measurements showed that affibody-enzymes photocrosslinked to EGFR on live MDA-MB-468 cells were first internalized, but then were recycled outside of endosomal compartment. Without photoconjugation, WT affibody-enzymes showed internalization followed by rapid proteolysis. In addition, confocal microscopy images revealed high colocalization of dually-dye-labeled affibody-enzyme fusion proteins, showing the fidelity of the protein structure during and after internalization by the cell. These photocrosslinkable affibodies were also found to inhibit ERK activity caused by EGF addition for a longer time period than Cetuximab, a commercial anti-EGFR antibody used for cancer therapy. Finally, photocrosslinkable affibody-enzyme proteins were found to function as effective agents for prodrug therapy even two days after crosslinking, as shown by continued prodrug activation and cell killing. In contrast, the WT affibody-enzyme proteins showed minimal cell killing even after only 8 h of incubation with cells. Thus, while non-photocrosslinked fusion proteins are internalized and eventually proteolyzed by the cell, covalently conjugating the N23BP-CodA to EGFR enables the fusion protein to successfully evade degradation.
The reported work may both improve upon targeted cancer therapies and expand the cell bioconjugation toolkit. First, a major limitation of antibody-directed enzyme prodrug therapy (ADEPT) is that enzyme activity is lost at the tumor site after endocytosis and proteolysis by the cancer cells. In this work, by covalently conjugating the enzyme to a cell receptor to prevent dissociation, the enzyme could avoid lysosomal degradation and could either return to the cell surface or maintain viable inside the cell as a result of receptor recycling, and prodrug conversion was shown days after adding the targeted enzyme to cells. As opposed to non-covalent association, we believe that covalently conjugating the fusion protein masks its presence to the cell. Thus, as the EGFR is recycled out of the endosome, the fusion protein goes along for the ride, escaping the harsh environment of the endosome and lysosome. We are still investigating the exact location of the photoconjugated affibody post-uptake. In addition, this work shows how photocrosslinking expressed proteins to membrane proteins results in their display on cell surfaces for any application requiring modification of cell receptors, all without modifying genetic machinery of the cell.
MATERIALS AND METHODS
Amplification of CodA Gene.
E. coli strain BL21(DE3) was used as the template for amplifying CodA gene encoded for cytosine deaminase. The sequence for the forward and the reverse primers (NEB) were 5’-aagcttGGCGGTGGCTCGAATAACGCTTTACAAACAATTATTAAC-3’ and 3’-ctcgagACGTTTGTAATCGATGGCTTCT-5’, where the bold regions are the annealing nucleotides. The primers incorporate a HindIII site at the 5’ position and an XhoI site at the 3’ position of the amplified sequence. The amplification was done using a standard PCR technique (Applied Biosystems, Gene Amp, PCR systems 9700) where primers were annealed at 65°C (Figure S8).
Expression, Synthesis, and Purification of Affibody-enzyme Fusion Proteins.
The codon sequence for WT and N23BPC affibody (in vector Pet21b+) was digested for 1 h at 37°C using the restriction enzymes HindIII and XhoI. The two plasmids (amplified CodA and the digested WT and N23BPC) were ligated using a standard ligation mixture supplied by NEB. E. coli BL21 (DE3) was made competent using an Eppendorf Eporator (4309000019) at 1300 V, followed by transformation of the ligated plasmid. The transformed E. coli were then grown on an ampicillin (selection marker) coated agar plate overnight.
A single colony from the overnight plate was pricked and suspended in 5 mL Luria Broth (LB) supplemented with 5 μL ampicillin and allowed to grow for 12 h. 500 μL of this culture was then transferred to 50 mL of LB supplemented with 50 μL of ampicillin and allowed to grow for 2 h, after which the OD600 of the culture was measured. At OD 0.7, 50 μL of the inducer solution 1 M isopropyl β-D-1 thiogalactopyranoside (IPTG, GoldBio) was added. The culture was allowed to grow further for 3 more hours, after which the culture was centrifuged (10000g for 5 min), and the supernatant discarded. The bacterial pellet from this culture was then resuspended in 25 mL equilibration buffer (5 mM imidazole in water) and sonicated at 25% power (1 min and 1 min off, total on: 4 min) to lyse the pellets (Branson Ultrasonic Cell Disruptor). The lysed cells were then centrifuged (12000g for 20 min) and 200 μL of HisPur Ni-NTA suspension (Thermo Fisher) was added to the lysate and incubated for 1 h at RT on a rotisserie shaker. The Ni-NTA beads were then centrifuged (700g for 2 min) and washed 5x with a wash buffer (25 mM imidazole in water) and finally the adsorbed fusion affibodies were eluted by incubating the elution buffer (250 mM imidazole in water) with the beads for 10-15 min on a rotisserie shaker.
The concentrations of the newly purified affibody-enzyme fusions were measured using a Nanodrop Lite Spectrophotometer (Thermo Fisher), then reacted with 20:1 molar excess of 4-N-maleimido benzophenone (VWR) overnight in the dark at RT. Excess benzophenone and imidazole were removed using 20k MWCO Zeba spin desalting columns (Life Technologies) and the final affibody-enzyme fusion concentration was measured by UV-Vis spectroscopy. The purified fusion affibodie-enzymes were then stored at −20°C in 10 μL aliquots with 5% glycerol. 10 μL of the purified fusion affibody-enzymes were then denatured with 2% w/w SDS and heating for 10 min at 70°C in a thermocycler (GeneAmp PCR Systems 9700, Applied Biosystem). The samples were then loaded into polyacrylamide gel (Thermo Fisher) and electrophoresed for 35 min at 200V in MES running buffer (50 mM MES, 50 mM Tris base, 1% SDS, 1mM EDTA). The gel was washed thrice in boiling water, followed by staining with Coomassie (Simply Blue Stain, Thermo Fisher). The gels were then imaged with a Typhoon FLA 9500 scanner to check for sample purity.
Microscale Thermophoresis (MST) Measurements.
Extracellular domain of purified human EGFR (Sino Biological, 10001-H08H) was conjugated to NHS- AF647 (Thermo Fisher) in 10:1 ratio. The excess dye was removed using 20k MWCO dialysis cups (Thermo Fisher), leading to a final degree of labeling of 1:1. Labeled AF647-EGFR was then mixed with sixteen different concentrations (50 μM to 100 pM) of unlabeled N23BP-CodA and WT-CodA affibody-enzymes in PBS with 0.05% Tween in Low-Adhesion Microcentrifuge Tubes (Genemate). The mixtures were then loaded onto micro-capillaries (NanoTemper, Monolith NT.115 Series) and the signal from each were measured using the MST instrument (NanoTemper). The signals were then fit to a binding model using the instrument software to obtain respective Kd values.
Photocrosslinking of Affibody-enzyme fusions with EGFR.
200 nM of purified human extracellular EGFR (Sino Biological, 70 kDa) was mixed with 5 μM of N23BP-CodA and WT-CodA affibody-enzymes in a microcentrifuge tube and irradiated with 890 μW/cm2 365 nm light for 30 min. The mixtures were then deglycosylated with PNGase F (NEB, this removes all the N-linked oligosaccharides, which contribute about 30 kDa of the EGFR molecular weight34) for 2 h at 37°C. The samples were denatured with 2% w/v SDS and heated at 70°C for 10 min. The samples were then loaded onto 4-12% Bis-Tris polyacrylamide gels (Thermo Fisher) and electrophoresed for 35 min at 200 V in MES running buffer (50 mM MES, 50 mM Tris base, 1% SDS, 1 mM EDTA). The gel was washed thrice in boiling water, followed by staining with Coomassie (Simply Blue Stain, Thermo Fisher). The gels were then imaged with Typhoon FLA 9500 scanner to check for the photo-crosslinked products.
Circular Dichroism (CD) Measurements.
Affibody-enzymes were diluted to 10 μM in PBS and measurements were obtained at 222 nm as temperature was raised from 20 to 90°C at 1 °C/min in a Applied Photophysics Chirascan Plus CD instrument. Temperature inside the cuvette was monitored using a thermocouple probe.
Affibody-enzyme Kinetic Activity Assays with Cytosine.
The enzymatic assay for N23BP-CodA, WT-CodA, and CodA were performed by varying substrate concentration and measuring the absorbance using a UV-VIS spectrophotometer (Agilent Technologies, Cary Series 100). A stock of 10 mM cytosine was prepared in 50 mM Tris-HCl at pH 7.5. Varying concentrations (250, 200, 150 and 100 μM) of cytosine solution were prepared from the stock and mixed with 50 nM of the enzyme and enzyme-affibodies. The reaction was run for 15 min with OD267 (cytosine) and OD260 (uracil) readings taken every 12 s. The concentration of cytosine was calculated for each time point and the kinetic parameters KM and kcat were calculated using the methods of initial rates and double reciprocal plots.
Conjugation of AlexaFluor 488 (AF488) and pHAb to Affibody-Enzymes.
The affibody-enzymes (N23BP-CodA and WT-CodA) were conjugated to 1:10 molar excess NHS-AlexaFluor 488 (Thermo Fisher) and the pH-sensitive NHS-pHAb (Promega) in separate tubes for 2 h at RT in the dark in PBS. The mixtures were then transferred to a 20k MWCO dialysis cups (Thermo Fisher) and excess dye was removed by dialysis. After 36 h dialysis, the degree of labeling for both the dye-conjugated affibody fusions were calculated using UV-Vis spectrophotometry (Agilent Technologies, Cary Series 100), by using the absorbance at 545 nm for the endotracker pHAb and 495 nm for AF488 as compared to the absorbance at 280 nm for both affibody-fusions.
Flow Cytometry Measurements.
MDA-MB-468 cells were grown to confluence at 37°C under 5% CO2. The cells were trypsinized with 0.025% Trypsin-EDTA (Gibco) and 500 μL of the cell suspension was seeded at 25,000 cells per well in 24-well cell culture plates (Corning). The cells were then allowed to grow in the wells for 36 h at 37°C and 5% CO2. The media was then removed and the cells were washed with PBS. The cells were then incubated with 1 μM AF488-conjugated affibody-enzymes and pHAb-conjugated affibody-enzymes separately in media for 2, 4, 6, 12, and 24 h. After 3 h, the wells were irradiated with 890 μW/cm2 365 nm light for 30 min, after which the wells were placed back in the incubator. At each timepoint, the media was removed and the cells were washed thrice to remove excess dye-conjugated affibody-enzymes. The cells were then trypsinized with 0.025% Trypsin-EDTA (Gibco) and fixed with 4% paraformaldehyde. Before flow cytometry, the paraformaldehyde solution was removed and the cells were resuspended in DPBS. The fluorescence from the cells was measured using BD FACSCelesta instrument (BD Biosciences). For measurement of AF488 fluorescence, a 488 nm laser was used, while for measuring the phAb fluorescence a 561 nm laser was used. The raw data was then analyzed using Flowing Software (OmicX).
Culture of MDA-MB-468 Breast Cancer Cells.
MDA-MB-468 breast carcinoma cells were procured from the American Type Culture Collection (ATCC) and grown in 75 cm2 culture flasks at 37°C under 5% CO2 in the presence of Dulbecco’s Modified Eagle Media (DMEM, with glucose and glutamine, phenol red but no sodium pyruvate, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Thermo Fisher).
Confocal Microscopy.
MDA-MB-468 cells were grown to confluence at 37°C under 5% CO2. The cells were trypsinized with 0.025% Trypsin-EDTA (Gibco) and 500 μL of the cell suspension was seeded at 20,000 cells per well in 24-well cell culture plates (Corning) with a poly-L-lysine coated glass coverslip (Corning BioCoat) already placed in each well. The cells were then allowed to grow in the wells for 36 h at 37°C and 5% CO2, after which the media was removed and the cells were washed with PBS. The cells were then incubated with 1 μM AF488-conjugated affibody-enzymes (both N23BP-CodA and WT-CodA) and pHAb-conjugated affibody-enzymes (N23BP-CodA and WT-CodA) separately in media for 4, 12 and 24 h. After 3 h, the wells were irradiated with 890 μW/cm2 365 nm light for 30 min, after which the wells were placed back in the incubator. After each time point, the media was removed, and the cells were washed with PBS to remove excess dye-affibody enzymes. The cells were then incubated with 1 μg/mL of 4’,6-diamidino-2-phenylindole (DAPI) for 10 min to stain the cell nucleus. The cells were again washed thrice with PBS. The cells were then mounted with ProLong Antifade agent (Thermo Fisher) on a glass slide and imaged with a Nikon A1R Laser Confocal Microscope at 20X magnification.
Transfection of the MDA-MB-468 cells with the EKAR Sensor.
The EKAR sensor contains three critical domains. The first is a phospho-binding domain that upon phosporalation induces a conformational change that reduces the distance bettween a CFP (donor) and a YFP (acceptor) FRET pair on either ends of their peptide sequence. It also has a ERK docking domain, and thirdly it includes a peptide sequence that mimics the domain of Cdc25C, a common target of ERK (Figure S9)35.
500,000 MDA-MB-468 cells were seeded in a standard 6-well plate (Corning) in 2.5 mL of media. 7.5 μL of Lipofectamine Reagent (Thermo) was dissolved in 125 μL of Opti-MEM (with HEPES, 2.4 g/L of Sodium Bicarbonate and Glutamine) in Tube 1 and 5 μL of P3000 reagent (Thermo) and 2.5 μg of the EKAR plasmid was dissolved in 125 μL of Opti-MEM in Tube 2. The contents of the two tubes were then mixed at RT for 10-15 min. 250 μL of the mixture was then added to the cells and this was replaced with fresh media after 24 h. The cells were then imaged for DNA expression by fluorescence microscopy, 48 h post-transfection. The cells were then trypsinized and sorted in a BDFACS Aria Fusion into Fluorobrite Dulbecco’s Modified Eagle Media (DMEM, with glucose, glutamine and sodium bicarbonate but no phenol red) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin and streptomycin (Thermo).
Monitoring of ERK Activity.
The MDA-MB-468 cells were grown to confluence, after which the cells were washed once with DPBS (Gibco) and then trypsinized with 0.025% trypsin-EDTA (Gibco). 200 μL of the cell suspension in media was seeded in 96-well cell 3603 imaging (Corning) at density of 10,000 cells/well. The cells were then grown for 24 h at 37°C under 5% CO2. The N23BP-CodA affibody-enzyme, Cetuximab (ThermoFisher) and EGF (Sino Biological) were diluted in Fluorobrite DMEM to a final conc of 100nM. After removal of old media from the wells, 100 μL of the appropriate protein solution (N23BP-CodA and Cetuximab) was added and the wells were imaged after 0.5, 2, 6, 12, and 24 h by a Molecular Devices ImageXpress microscope. After 3 h, cells were irradiated with 890 μW/cm2 of 365 nm light for 30 min. After 24 h, the old media was removed and 100 nM of EGF in fresh media was added and the wells were imaged after 0.5, 2, 6, 12 and 24 h post-EGF addition. As controls, there were wells which did not receive any sort of treatment and wells in which only EGF was added. Both CFP and FRET measurements from all the wells with the samples and also a blank well, with cell-free media for background correction were recorded. Briefly, the following filter sets were used for FRET measurements: FRET (Semrock MOLE 0189), excitation: 438/25, dichroic, emission: 542/25. CFP (Semrock 2432B NTE ZERO), excitation: 438/25, dichroic, emission 483/25. The images were imported into the program, after which the background fluorescence was subtracted from each image. Pixels with an intensity above 800 units were classified as cells. FRET ratio was calculated as FRET intensity over CFP intensity by averaging all the relevant pixels, and reported as ERK activity. Each image was recorded at 10X magnification and measurements were done in quadruplicates. Here, the normalized FRET signal was used rather than raw FRET to ensure that overexpression of CFP fluorescence was not causing excess fluorescence in the FRET images. The signal from the images were converted into quantitative signal via a custom MATLAB software36.
Prodrug Cytotoxicity (MTT) Assays.
The MDA-MB-468 cells were grown to confluence, after which the cells were washed once with DPBS (Gibco) and then trypsinized with 0.025% trypsin-EDTA (Gibco). 200 μL of the cell suspension in media was seeded in 96-well cell culture plates (Corning) at density of 2000 cells/well. The cells were then grown for 24 h at 37°C under 5% CO2. The N23BP-CodA and WT-CodA affibody-enzymes and CodA enzyme itself were diluted in DMEM to a final conc. of 1 μM. After removal of old media from the wells, 100 μL of the appropriate protein solution (N23BP-CodA, WT-CodA, CodA, or no protein) was added. The cells were then incubated at 37°C under 5% CO2 with the enzyme/affibody-enzyme. After 3 h, cells were irradiated with 890 μW/cm2 of 365 nm light for 30 min. For the (−)UV controls, cells were not irradiated with UV. When the timepoint was reached (8, 24, or 48 h after introducing the protein), the cells were washed thrice with PBS to remove any excess enzyme/affibody-enzyme. In a separate experiment, cells were incubated with the affibody-enzymes just for 8 h. Next, 100 μL of 2 mM 5-FC was added to each well, while for control wells 100 μL was added instead. To measure the effect of 5-FC and 5-FU on cells, some wells were treated with only 5-FC and just 5-FU, without any protein. Excess 5-FC was removed after 12 h and the cells were washed with PBS and 100 μL of 1 mg/mL (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (MTT) in media was added to each of the wells.
In the other viability experiment, after incubation of 5-FC for 12 h, another dose of 2mM 5-FC was added instead of adding MTT. The wells were incubated with 5-FC for a total of 24, 48, and 72 h, with fresh dose of 5-FC after every 12 h. After 4 h of MTT addition, the media was removed from each well carefully to avoid disturbing the formazan crystals at the bottom. The crystals were dissolved in 100 μL isopropyl alcohol (IPA), and absorbance from the wells was measured at 570 nm in a Tecan Safire II Multimode Plate reader. The readings were then normalized to the cells that were treated with neither enzyme, nor enzyme-affibody, nor 5-FC/5-FU to calculate the fractional viability. All experiments were performed in triplicate.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank NIH DP2EB020401 and NSF DMR 1420736 for support of this research. The Nanotemper Microscale Thermophoresis instrumentation was supported by the instrumentation grant NIH S10OD21603, and the University of Colorado Biochemistry Instrument Core provided the Applied Photophysics Chirascan Plus CD instrument. The imaging work was performed at the BioFrontiers Institute Advanced Light Microscopy Core. Laser scanning confocal microscopy was performed on a Nikon A1R microscope supported by NIST-CU Cooperative Agreement award number 70NANB15H226. Dr. Joe Dragavon provided invaluable guidance for the confocal micrscopy and David Bull helped with colocalization anaylsis. Finally, we would like to thank Prof. Jerome Fox for use of his Tecan Safire II Multimode Plate Reader.
Footnotes
Supporting Information. MST spectra, CD spectra, fluorescence of AF488 and pHAb vs. pH, UV-Vis absorbance spectra of affibody-enzymes with dyes, kinetic data, additional EGF competition flow cytometry experiments.
DATA AVAILABILITY
Raw and processed data not included in the supplementary information will be supplied upon request to the corresponding author.
REFERENCES
- (1).Sharma SK, and Bagshawe KD (2017) Antibody Directed Enzyme Prodrug Therapy (ADEPT): Trials and Tribulations. Adv. Drug Deliv. Rev 118, 2–7. [DOI] [PubMed] [Google Scholar]
- (2).Sharma SK, Pedley RB, Bhatia J, Boxer GM, El-Emir E, Qureshi U, Tolner B, Lowe H, Michael NP, Minton N, et al. (2005) Sustained Tumor Regression of Human Colorectal Cancer Xenografts Using a Multifunctional Mannosylated Fusion Protein in Antibody-Directed Enzyme Prodrug Therapy. Clin. Cancer Res 11 (2), 814–825. [PubMed] [Google Scholar]
- (3).Kamath AV (2016) Translational Pharmacokinetics and Pharmacodynamics of Monoclonal Antibodies. Drug Disc. Today: Technol. 21–22, 75–83. [DOI] [PubMed] [Google Scholar]
- (4).Alley SC, Okeley NM, and Senter PD (2010) Antibody–Drug Conjugates: Targeted Drug Delivery for Cancer. Curr. Opin. Chem. Biol 14, 529–537. [DOI] [PubMed] [Google Scholar]
- (5).Chari RVJ, Miller ML, and Widdison WC (2014) Antibody–Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed 53, 3796–3827. [DOI] [PubMed] [Google Scholar]
- (6).Ryman JT, and Meibohm B (2017) Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst. Pharmacol. 6 (9), 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sharma SK, and Bagshawe KD (2017) Translating Antibody Directed Enzyme Prodrug Therapy (ADEPT) and Prospects for Combination. Expert Opin. Biol. Ther 17, 1–13. [DOI] [PubMed] [Google Scholar]
- (8).Schmidt SR (2009) Fusion-Proteins as Biopharmaceuticals -- Applications and Challenges. Curr. Opin. Drug Discov. Devel 12, 284–295. [PubMed] [Google Scholar]
- (9).Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, and Bertozzi CR (2007) Copper-Free Click Chemistry for Dynamic in Vivo Imaging. Proc. Natl. Acad. Sci. U.S.A 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, and Bertozzi CR (2010) Copper-Free Click Chemistry in Living Animals. Proc. Natl. Acad. Sci. U.S.A 107, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Prescher JA, Dube DH, and Bertozzi CR (2004) Chemical Remodelling of Cell Surfaces in Living Animals. Nature 430, 873. [DOI] [PubMed] [Google Scholar]
- (12).Chin JW, and Schultz PG (2002) In Vivo Photocrosslinking with Unnatural Amino Acid Mutagenesis. ChemBioChem 3, 1135–1137. [DOI] [PubMed] [Google Scholar]
- (13).Hayashi T, Yasueda Y, Tamura T, Takaoka Y, and Hamachi I (2015) Analysis of Cell-Surface Receptor Dynamics through Covalent Labeling by Catalyst-Tethered Antibody. J. Am. Chem. Soc 137, 5372–5380. [DOI] [PubMed] [Google Scholar]
- (14).Matsuo K, Nishikawa Y, Masuda M, and Hamachi I (2018) Live-Cell Protein Sulfonylation Based on Proximity-Driven N-Sulfonyl Pyridone Chemistry. Angew. Chem. Int. Ed 130, 667–670. [DOI] [PubMed] [Google Scholar]
- (15).Tamura T, and Hamachi I (2019) Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems. J. Am. Chem. Soc 141, 2782–2799. [DOI] [PubMed] [Google Scholar]
- (16).Grevys A, Nilsen J, Sand KMK, Daba MB, Øynebråten I, Bern M, McAdam MB, Foss S, Schlothauer T, Michaelsen TE, et al. (2018) A Human Endothelial Cell-Based Recycling Assay for Screening of FcRn Targeted Molecules. Nature Commun. 9, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tzaban S, Massol RH, Yen E, Hamman W, Frank SR, Lapierre LA, Hansen SH, Goldenring JR, Blumberg RS, and Lencer WI (2009) The Recycling and Transcytotic Pathways for IgG Transport by FcRn Are Distinct and Display an Inherent Polarity. J. Cell Biol 185, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brasino M, Roy S, Erbse AH, He L, Mao C, Park W, Cha JN, and Goodwin AP (2018) Anti-EGFR Affibodies with Site-Specific Photo-Cross-Linker Incorporation Show Both Directed Target-Specific Photoconjugation and Increased Retention in Tumors. J. Am. Chem. Soc 140, 11820–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Friedman M, Orlova A, Johansson E, Eriksson TLJ, Höidén-Guthenberg I, Tolmachev V, Nilsson FY, and Ståhl S (2008) Directed Evolution to Low Nanomolar Affinity of a Tumor-Targeting Epidermal Growth Factor Receptor-Binding Affibody Molecule. J. Mol. Biol 376, 1388–1402. [DOI] [PubMed] [Google Scholar]
- (20).Hall RS, Fedorov AA, Xu C, Fedorov EV, Almo SC, and Raushel FM (2011) Three-Dimensional Structure and Catalytic Mechanism of Cytosine Deaminase. Biochemistry 50, 5077–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Deckert PM, Renner C, Cohen LS, Jungbluth A, Ritter G, Bertino JR, Old LJ, and Welt S (2003) A33scFV-Cytosine Deaminase: A Recombinant Protein Construct for Antibody-Directed Enzyme-Prodrug Therapy. Brit. J. Cancer 88, 937–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mahan SD, Ireton GC, Knoeber C, Stoddard BL, and Black ME (2004) Random Mutagenesis and Selection of Escherichia Coli Cytosine Deaminase for Cancer Gene Therapy. Protein Eng., Des. Sel 17, 625–633. [DOI] [PubMed] [Google Scholar]
- (23).National Research Council. (2011) Prudent Practices in the Laboratory: Handling and Management of Chemical Hazards, Updated Version. Washington, DC: The National Academies Press. [PubMed] [Google Scholar]
- (24).Brasino M, and Cha JN (2017) Real-Time Femtomolar Detection of Cancer Biomarkers from Photoconjugated Antibody-Phage Constructs. Analyst 142, 91–97. [DOI] [PubMed] [Google Scholar]
- (25).Grunbeck A, Huber T, Sachdev P, and Sakmar TP (2011) Mapping the Ligand-Binding Site on a G Protein-Coupled Receptor (GPCR) Using Genetically Encoded Photocrosslinkers. Biochemistry 50, 3411–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nath N, Godat B, Zimprich C, Dwight SJ, Corona C, McDougall M, and Urh M (2016) Homogeneous Plate Based Antibody Internalization Assay Using PH Sensor Fluorescent Dye. J. Immunol. Methods 431, 11–21. [DOI] [PubMed] [Google Scholar]
- (27).Nordberg E, Friedman M, Göstring L, Adams GP, Brismar H, Nilsson FY, Ståhl S, Glimelius B, and Carlsson J (2007) Cellular Studies of Binding, Internalization and Retention of a Radiolabeled EGFR-Binding Affibody Molecule. Nucl. Med. Biol 34, 609–618. [DOI] [PubMed] [Google Scholar]
- (28).Nordberg E, Ekerljung L, Sahlberg SH, Carlsson J, Lennartsson J, and Glimelius B (2010) Effects of an EGFR-Binding Affibody Molecule on Intracellular Signaling Pathways. Int. J. Oncol 36, 967–972. [DOI] [PubMed] [Google Scholar]
- (29).Tomas A, Futter CE, and Eden ER (2014) EGF Receptor Trafficking: Consequences for Signaling and Cancer. Trends Cell Biol. 24, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yung Y, Dolginov Y, Zhong Y, Rubinfeld H, Michael D, Hanoch T, Roubini E, Lando Z, Zharhary D, and Seger R (1997) Detection of ERK Activation by a Novel Monoclonal Antibody. FEBS Lett. 408, 292–296. [DOI] [PubMed] [Google Scholar]
- (31).Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, and Svoboda K (2008) A Genetically Encoded Fluorescent Sensor of ERK Activity. Proc. Natl. Acad. Sci. U.S.A 105, 19264–19269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ting AY, Kain KH, Klemke RL, and Tsien RY (2001) Genetically Encoded Fluorescent Reporters of Protein Tyrosine Kinase Activities in Living Cells. Proc. Natl. Acad. Sci. U.S.A 98, 15003–15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fuchita M, Ardiani A, Zhao L, Serve K, Stoddard BL, and Black ME (2009) Bacterial Cytosine Deaminase Mutants Created by Molecular Engineering Show Improved 5-Fluorocytosine-Mediated Cell Killing In Vitro and In Vivo. Cancer Res. 69, 4791–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang XQ, Sun P, Gorman OM, Tai T, and Paller AS (2001) Epidermal Growth Factor Receptor Glycosylation Is Required for Ganglioside GM3 Binding and GM3-Mediated Suppresion of Activation. Glycobiology 11, 515–522. [DOI] [PubMed] [Google Scholar]
- (35).Wang Z, Wang M, Lazo JS, and Carr BI (2002) Identification of Epidermal Growth Factor Receptor as a Target of Cdc25A Protein Phosphatase. J. Biol. Chem 277, 19470–19475. [DOI] [PubMed] [Google Scholar]
- (36).Chapnick DA, Bunker E, and Liu X (2015) A Biosensor for the Activity of the “Sheddase” TACE (ADAM17) Reveals Novel and Cell Type-Specific Mechanisms of TACE Activation. Sci. Signal 8, rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data not included in the supplementary information will be supplied upon request to the corresponding author.