

Abstract
Inhibiting PD-1:PD-L1 signaling has transformed therapeutic immune restoration. CD4+ T cells sustain immunity in chronic infections and cancer, yet little is known about how PD-1 signaling modulates CD4+ helper T (TH) cell responses or the ability to restore CD4+ TH-mediated immunity by checkpoint blockade. We demonstrate that PD-1:PD-L1 specifically suppressed CD4+ TH1 cell amplification, prevents CD4+ TH1 cytokine production and abolishes CD4+ cytotoxic killing capacity during chronic infection in mice. Inhibiting PD-L1 rapidly restored these functions, while simultaneously amplifying and activating TH1-like T regulatory cells, demonstrating a system-wide CD4–TH1 recalibration. This effect coincided with decreased T cell antigen receptor signaling, and re-directed type I interferon (IFN) signaling networks towards dominant IFN-γ-mediated responses. Mechanistically, PD-L1 blockade specifically targeted defined populations with pre-established, but actively suppressed proliferative potential, with limited impact on minimally cycling TCF-1+ follicular helper T cells, despite high PD-1 expression. Thus, CD4+ T cells require unique differentiation and functional states to be targets of PD-L1-directed suppression and therapeutic restoration.
Robust effector T cell responses are critical to the resolution of viral infection and when compromised, chronic infection ensues. At the onset of what will become a chronic viral infection, sustained antigen signaling remodels the immune environment, driving the production of immunosuppressive cytokines and the expression of multiple inhibitory receptors/ligands that further potentiate viral persistence (for example, IL-10, PD-1, PD-L1, Tim3). This immunosuppressive program diminishes CD8 T cell function (termed T cell exhaustion) and maintains virus-specific T cells in an attenuated functional and distinct epigenetic state that is unable to eliminate infection1. Characteristics of cellular and molecular exhaustion are also observed in T cells during cancer, indicating a conserved program of T cell dysfunction in response to chronic antigen stimulation. Further exacerbating cell-intrinsic CD8 T cell dysfunction in chronic infection is the progressive loss of virus-specific CD4+ type 1 helper T cells (TH1) and the reciprocal accumulation of B cell helping CD4+ follicular helper T cells (TFH) as virus persists2–5. Although the exhausted state of CD8 T cells is relatively well-defined, the cellular and molecular changes that comprise CD4+ T cell functional alterations in chronic infection and cancer are little understood. Further adding to this complexity is the multiple TH differentiation states that CD4+ T cells progress toward, leading to the unique requirement to consider functional changes in molecular programs within the context of specific TH differentiation states. CD4+ TH1 cell help is critical to sustain CD8 T cells during viral persistence, and the progressive CD4+ TH1 loss promotes CD8 T cell exhaustion and defective viral control6. Therapeutically, transfer of virus-specific CD4+ TH1 cells in the middle of an established chronic infection increased antiviral CD8 T cell numbers and function to enable viral control6. Thus, the loss of CD4+ TH1 cells is a critical mechanism promoting CD8 T cell dysfunction, and enhancing this distinct virus-specific CD4+ helper T cell subset can overcome CD8 T cell exhaustion to increase immune control of chronic infections.
Immunotherapy that blocks inhibitory receptors/ligands to reverse T cell exhaustion and enhance immune function has spurred a revolution in treating chronic disease. Among the most successful of these immunotherapies has been the blockade of the PD-1:PD-L1 (PD-1/L1) pathway. PD-1/L1 blockade enhances CD8 T cell function in many viral infections, including chronic lymphocytic choriomeningitis virus (LCMV), HIV, HCV; in numerous preclinical cancer models, and has been particularly successful in a subset of cancer patients1. Because CD8 T cells are critical effectors of viral and tumor clearance, the bulk of the work studying the effects of anti-PD-1/L1 therapy has focused on their role. Indeed, a distinct TCF-1+ memory-like CD8 T cell subset has been identified as the specific CD8 T cell population targeted by the PD-1/L1-blocking therapy7–13. Yet, in certain instances, patients respond to anti-PD-1/ L1 blockade independent of CD8 T cell responses14,15, indicating that many cell types are likely responding to and affecting the outcome of this therapy than previously recognized.
PD-1 can be highly expressed on activated CD4+ T cells, suggesting that these T cells may also be responsive to PD-1/L1-blocking therapies. Although blocking PD-1/L1 is not widely considered to target CD4+ T cells16, increasing evidence in patients suggests that, at least in some instances, CD4+ T cells can respond to this therapy14,17. The reason underlying these discrepancies in how or why CD4+ T cells may (or may not) respond to PD-1/L1-blocking immunotherapy is unclear, but may be explained by differences in precursor CD4+ helper T cell populations. For instance, if anti-PD-1/L1 specifically targets distinct types of CD4+ helper T cell subsets, the therapy will likely be effective only in cases in which these specific helper T cell subsets are present pretreatment. Yet to date, a precise role of PD-1/L1 blockade in restoring CD4+ T cell responses and the potential for differential helper T cell subset targeting remains unclear.
Results
Virus-specific CD4+ T cell heterogeneity during chronic infection.
To investigate how exhausted CD4+ T cells are affected by anti-PD-L1 therapy during chronic viral infection, LCMV-glycoprotein (GP)61–80-specific CD4+ T cell antigen receptor (TCR) transgenic (SMARTA) T cells were adoptively transferred into naïve mice that were subsequently infected 1 day later with LCMV clone 13 (Cl13) to generate a chronic infection. The SMARTA T cells, all expressing the same TCR, functionally and phenotypically recapitulate host-derived endogenous GP61–80-specific CD4+ T cells2,18,19, and are used to control for inherent differences in strength of TCR signaling in the total population. Twenty-five days after infection, mice were treated with either anti-PD-L1-blocking antibody or isotype control antibody and were continued on this treatment regimen every 3 days. SMARTA cells were analyzed after the first and third treatments and co-clustered based on time-of-flight mass cytometric (CyTOF) staining (Extended Data Fig. 1a and Supplementary Tables 1 and 2). PD-L1 blockade did not alter viral titers following one treatment, but in this experiment decreased viral titers were observed after three treatments (Extended Data Fig. 2a). All the CD4+ SMARTA T cells are primed early after infection20, are activated throughout infection and exhibited high PD-1 expression at the start of the treatment. Analysis revealed that the SMARTA T cells formed ten distinct clusters that could be broadly grouped into those that expressed the transcription factor TCF-1 (c1–5, c7 and c10) and those that did not (c6, c8 and c9) (Fig. 1a,b). TCF-1 drives expression of the TFH transcriptional regulator Bcl-6 (refs. 21,22), and is absent in TH1 cells. Consistent with this, TCF-1-negative c6, c8 and c9 had very low expression of Bcl-6 and expressed multiple TH1 cell-associated proteins. In c8 and 9, these included GzmB, Eomes, as well as Tim3 and CD39 (inhibitory receptors that are highly expressed on TH1 cells), whereas c6 had high expression of T-bet (a transcription factor driving TH1 differentiation) and SLAMF1 (an activation marker defining TH1 cells in LCMV infection). Of the TH1 clusters, c8 was unique in that it expressed the TH1 chemokine receptor CX3CR1 and appeared to be more terminally differentiated with a higher proportion of cells expressing Tim3 and GzmB, and the highest expression of CD80 and Lag3 (Fig. 1a). Thus, c6, c8 and c9 were residual TH1 clusters sustained during chronic viral infection, with c6 a potentially less-differentiated TH1 population and c8 the most terminally differentiated.
Fig. 1 |. PD-L1 blockade specifically amplifies and functionally enhances CD4+ TH1 cells.
Virus-specific CD4+ SMARTA T cells were transferred into naïve mice that were then infected with LCMV-Cl13 one day later. Twenty-five days after infection mice were treated with either isotype (n = 5) or anti-PD-L1 (n = 5) antibody and subsequently every 3 days for a total of three treatments. a, UMAP plots of CyTOF data showing high-dimensional distribution of PhenoGraph clusters of SMARTA T cells showing (left) the combined data of one and three isotype and anti-PD-L1 treatments) and (right) 60 h after the first isotype or anti-PD-L1 treatment. Clusters that are significantly increased or decreased in abundance (P < 0.05 calculated by the edgeR test in the diffcyt R package) following one anti-PD-L1 treatment are colored red and blue, respectively. The heatmap depicts the column normalized z-scores of the arcsinh-transformed MSI of the indicated protein in each cluster. b, UMAP plots show the single-cell expression of the indicated protein. c, Total number of SMARTA T cells in each cluster 60 h following the first isotype or anti-PD-L1 treatment. d, Graph depicts the PD-L1/Iso fold change between TFH and TH1 SMARTA cell numbers in the same mice. e, Total number of IFN-γ, TNF, IL-10 and geometric mean fluorescence intensity (GMFI) of IFN-γ producing SMARTA T cells at 60 h following the first isotype or anti-PD-L1 treatment. f, Flow plots depict SLAMF1 and CXCR5 expression by wild-type (WT; n = 5) and PD-1 knockout (KO; n = 5) SMARTA cells 9 days after LCMV-Cl13 infection. Box plots indicate the percentage and total number of TH1 (SLAMF1hi CXCR5lo) and TFH (SLAMF1lo CXCR5hi) wild-type and knockout SMARTA cells, the number of IFN-γ producing SMARTA T cells and the GMFI of IFN-γ expression. g, Total number of IFN-γ, TNF and IL-10 producing SMARTA T cells following three treatments. Data are representative of n = 5 mice per group examined over two (f) or four (a–d), and two (e,g) experiments totaling n = 10 SMARTA and n = 10 PD1−/− SMARTA (f), n = 19 isotype and n = 20 anti-PD-L1 (a–d) and n = 10 mice per group (e,g). Bar graphs show the average and error bars indicate s.d.. Box plots indicate the median, upper and lower quartile, and the whiskers show the high and low value. *P < 0.05; unpaired, two-tailed Student’s t-test (c,e–g) or paired t-test (d).
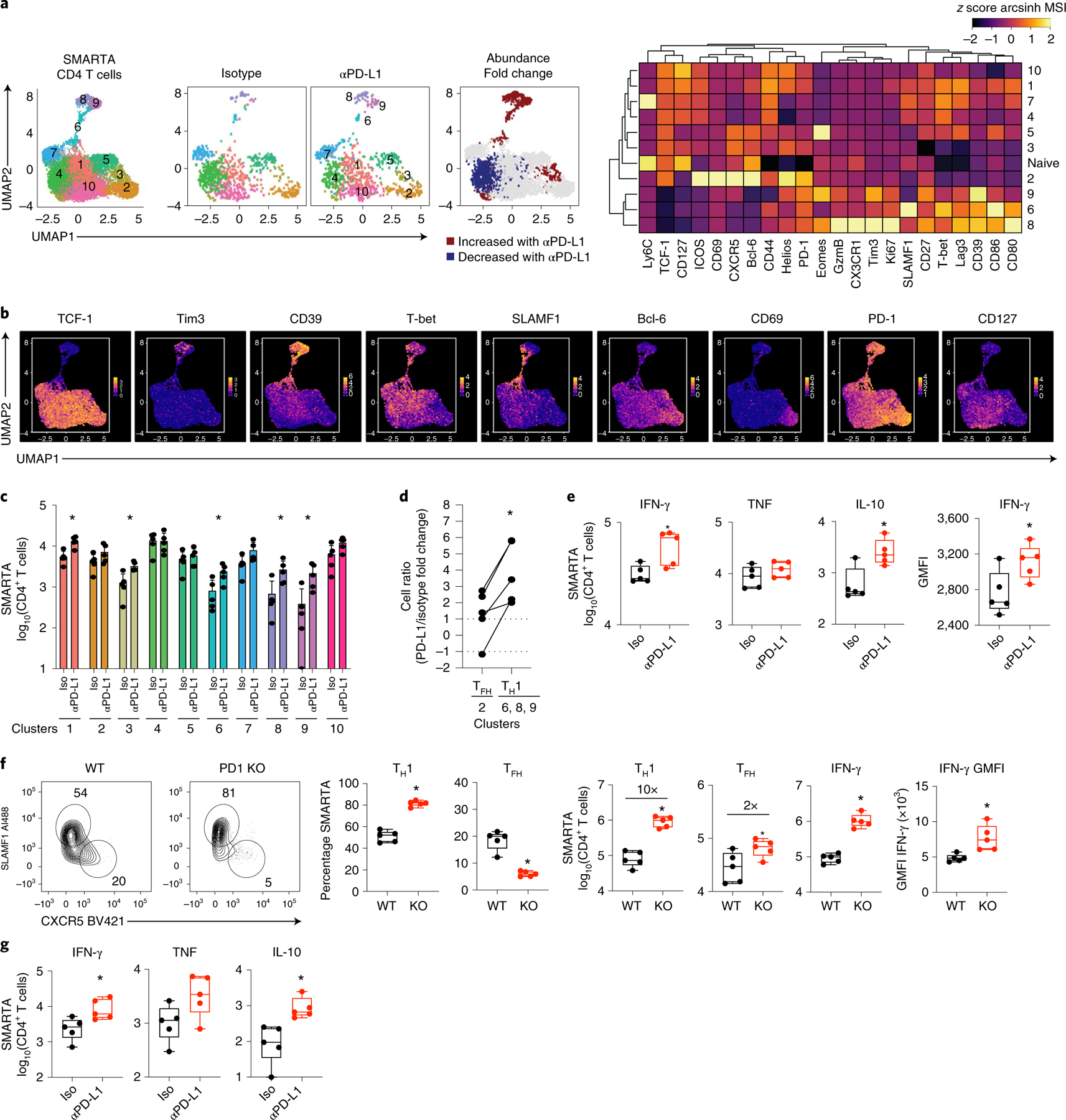
Of the TCF-1+ clusters, c2 exhibited the highest expression of Bcl-6, CXCR5, ICOS, Helios and was uniquely positive for CD69, suggesting the most differentiated TFH cells (Fig. 1a, b). Although all the CD4+ SMARTA cells were activated and expressed high levels of PD-1, the TFH c2 along with the TH1 clusters expressed the highest levels of PD-1, suggesting that these clusters were the most highly activated. The other TCF-1+ clusters c1, c3, c4, c5, c7 and c10 expressed IL-7Rα (CD127), and potentially signify less antigen-engaged, less terminally differentiated cells. Even among the less-differentiated/less-activated CD127+TCF-1+ clusters, substantial heterogeneity existed suggestive of hybrid TH1/TFH cells. For instance, c1, c4, c5 and c7 had high TCF-1 expression, but simultaneously expressed T-bet and some SLAMF1. Cluster 10, on the other hand, appeared to favor neither the TH1 or TFH lineages and had the highest expression of CD127, likely being the least differentiated of all the clusters. Thus, extensive heterogeneity exists amongst CD4+ T cells during chronic viral infection, and in addition to TH1 and TFH, multiple hybrid clusters emerge that have characteristics of both TH1 and TFH cells.
PD-L1 blockade restores virus-specific CD4+ TH1 cells.
PD-L1 blockade increased the proportions of the TH1 clusters (6, 8, 9), and drove a smaller increase in the proportion of c3, while decreasing the proportion of the TCF-1+ intermediate c4 (Fig. 1a and Extended Data Fig. 2b). The overall number of SMARTA T cells was elevated (p = 0.06) following the initial treatment (Extended Data Fig. 2c), primarily as a result of the increased TH1 clusters, and to a lesser extent from an increase in c3 and c1 (a transitional cluster associating next to the TH1 cells in UMAP space) (Fig. 1c). Gating on endogenous Foxp3−, PD-1+CD4+ T cells revealed that the TH1, but not TCF-1+ cells were also increased following anti-PD-L1 treatment, confirming that this was not an epitope, TCR or transgenic specific effect (Extended Data Fig. 2d). Unlike TH1 cells, the proportion and absolute number of the most differentiated TFH c2 was largely unaffected by PD-L1 blockade at this early timepoint, despite the high levels of PD-1 expression (Fig. 1a–c and Extended Data Fig. 2b). Direct comparison within the same mouse further revealed that the fold changes were significantly higher in the TH1 cells following PD-L1 blockade compared with the small increases observed in the TFH cluster (Fig. 1d). Functionally, anti-PD-L1 blockade increased IFN-γ-producing virus-specific CD4+ T cells, as well as the amount of IFN-γ produced per cell, while simultaneously increasing virus-specific CD4+ T cells producing IL-10 (Fig. 1e and Extended Data Fig. 2e), suggesting a negative feedback inhibition induced by the enhanced stimulation, but also consistent with observations that IL-10 production is specifically produced by TH1 cells during chronic viral infection23. On the other hand, there was no difference in the proportion or numbers of TNF-producing virus-specific CD4+ T cells (Fig. 1e and Extended Data Fig. 2e), indicating that not all cytokines were enhanced by blocking PD-L1. PD-1-deficient virus-specific CD4+ T cells also exhibited significantly increased TH1 differentiation by both proportion and number, as well as enhanced IFN-γ production, with only minimal effects on TFH numbers (Fig. 1f), thus demonstrating the CD4+ T cell-intrinsic nature of PD-1 signaling is to inhibit TH1 expansion and function throughout chronic viral infection.
After three anti-PD-L1 treatments (8 days after the initiation of treatment and 33 days after infection), there was a numerical increase in the majority of CD4+ SMARTA T cell clusters in the PD-L1 blocked mice, corresponding to the maintenance of these cells between these treatments (Extended Data Fig. 2f). Further, the number of IFN-γ and IL-10 expressing cells remained elevated following three anti-PD-L1 treatments (Fig. 1g). PD-L1 blockade also increased numbers of CD4+ TH1 SMARTA cells in the liver and lungs following three treatments, and although Bcl-6hi TFH populations were not present in these nonlymphoid organs, the population of Bcl-6lo TCF-1+ cells was less responsive to PD-L1 blockade (Extended Data Fig. 2g). Thus, anti-PD-L1 specifically increases the proportion and number of virus-specific CD4+ TH1 cells in both lymphoid and nonlymphoid organs.
Blocking PD-L1 targets precycling CD4+ TH1 populations.
To determine the origin of the increased TH1 cells following PD-L1 blockade, we examined the trajectory of virus-specific CD4+ T cells following anti-PD-L1 treatment. Overlaying the UMAP clusters onto a diffusion pseudotime map24 revealed three tips (T), which form the most distant endpoints of each branch and indicate the beginning or end of a differentiation state (Fig. 2a and Extended Data Fig. 3a). T1 was a TFH cell in c2; T2 was a cell from the most differentiated TH1 c8; and T3 was a cell in the immature/transitional SLAMF1+ TH1 c6. Pseudotime diffusion analysis identified two branches that mapped from the central SLAMF1+ TH1 cell T3 root, where it could either progress towards the TFH fate (as nor mally occurs during chronic viral infection as TH1 cells convert to TFH)2, or towards more differentiated TH1 clusters. To identify which of these directions was favored by PD-L1 blockade, we overlaid changes in cell number following anti-PD-L1 treatment onto the diffusion pseudotime map. Blocking PD-L1 rapidly increased both the proportion and numbers of the clusters on the branch towards the highly differentiated TH1 c8 in T2 (Fig. 2a), suggesting that anti-PD-L1 diverts CD4+ T cells from the normal TFH differentiation branch in chronic infection and can push less-committed TH1 cells towards the differentiated TH1 lineage.
Fig. 2 |. Pretherapy cycling CD4+ SMARTA T cells are expanded upon PD-L1 blockade.
a, Pseudotime diffusion maps of SMARTA T cells following the first anti-PD-L1 treatment. In the middle plot the clusters derived from PhenoGraph (left plot) were overlaid onto the diffusion pseudotime map. Tips (T1, T2, T3) designate the algorithm-derived cellular tips and the lines are the pathways derived from T3 as a starting point. The right plot shows the PhenoGraph clusters that are significantly increased in number (red) at 60 h following the first anti-PD-L1 treatment. b, The graphs show the proportion and total number of Ki67+ SMARTA cells 60 h following the first isotype or anti-PD-L1 treatment. c, UMAPs and the box plot depict the proportion of Ki67+ cells in each SMARTA PhenoGraph cluster 60 h following the first isotype or anti-PD-L1 treatment. d, Plots of CyTOF data show expression of the indicated protein combinations gated on Ki67+ SMARTA cells following the first and third isotype and anti-PD-L1 treatments. e, The heatmap depicts the arcsinh-transformed MSI of protein expression by Ki67+ and Ki67− SMARTA T cells following the first and third isotype and anti-PD-L1 treatments. Marker expression is normalized across columns by z-score. n = 5 mice per group and are representative of (a) n = 9 mice per group examined over two experiments and (b–e) a total of n = 15 isotype mice and n = 14 anti-PD-L1 mice over three experiments. Data are representative of two to three experiments with between four and six mice per group. *P < 0.05; (b,c) Student’s t-test (unpaired, two-tailed). Box plots indicate the median, upper and lower quartile, and the whiskers show the high and low value. n = x biologically independent samples/animals/cells/independent experiments/n = x cells examined over y independent experiments.
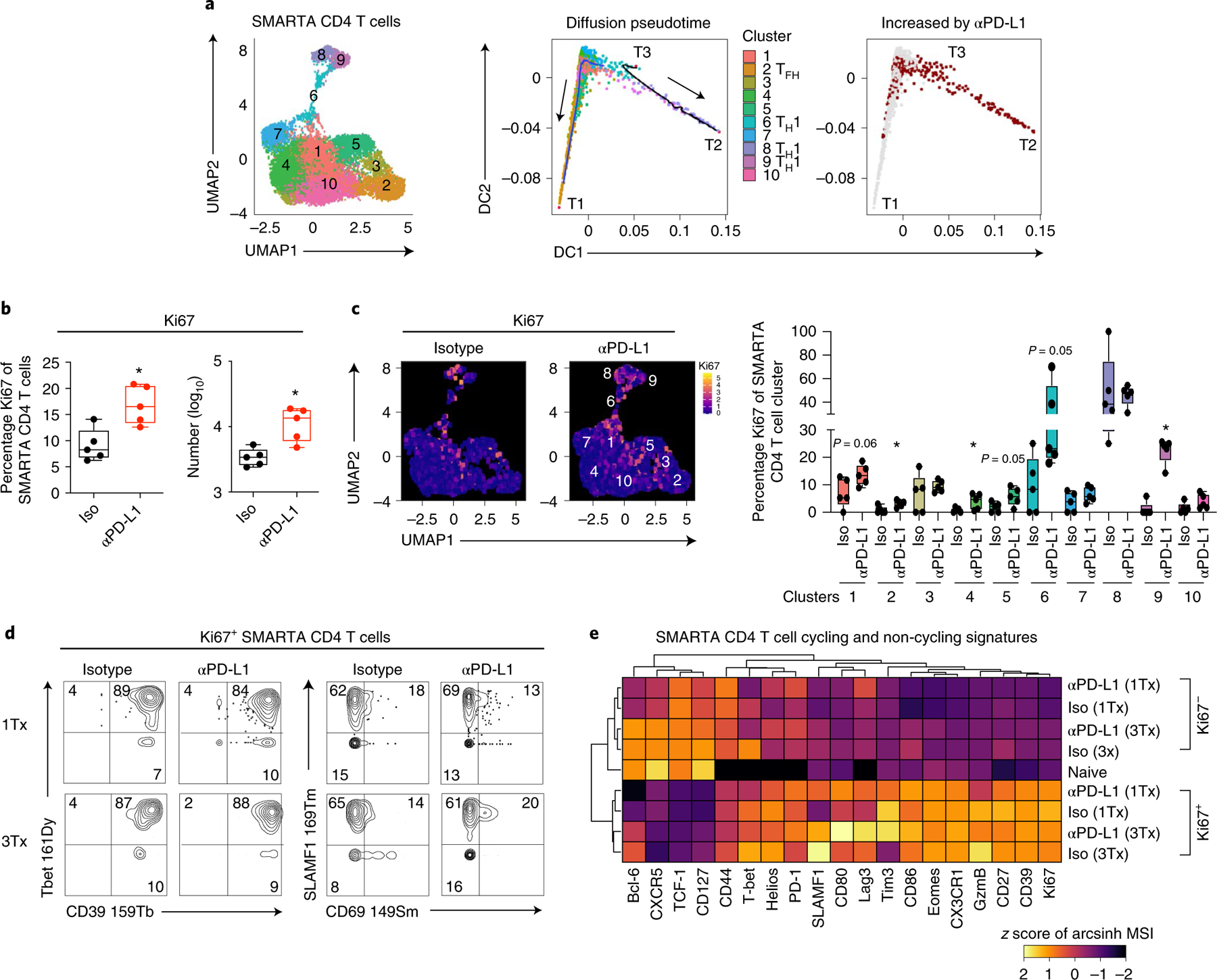
PD-L1 blockade rapidly increased the proportion and the number of Ki67+ (that is, cycling) virus-specific CD4+ T cells (Fig. 2b and Extended Data Fig. 3b). The vast majority of Ki67+ cells in the isotype-treated group (representing pretherapy) were in the TH1 c6 and c8, and the TH1-associating c1 (Fig. 2c). Critical for these analyses, the SMARTA cells were not clustered using Ki67 to prevent algorithmic clustering of cycling cells (Supplementary Table 2). Although the frequency of Ki67+ cells increased following PD-L1 blockade (Fig. 2b), the distribution of clusters that expressed Ki67 was generally unchanged (Fig. 2c). The exception to this was the TH1 c9, which exhibited low Ki67 expression in isotype-treated mice, but was robustly induced by anti-PD-L1 treatment (Fig. 2c). The increase in c9 is consistent with the pseudotime trajectory analysis indicating that activated cells in c6 progress through c9 toward the most differentiated population of TH1 cells in c8. Conversely, c8 started out with the highest frequency of Ki67+ cycling cells and this was then maintained following anti-PD-L1 therapy. Whereas the frequency of Ki67+ cells in TFH c2 and intermediates c4 and c5 were also enhanced upon PD-L1 blockade (Fig. 2c), the proportion of these cells remained small, and did not lead to an increase in the absolute numbers of those clusters, as was observed for the TH1 clusters (Fig. 1c).
The majority of Ki67+ cells in both isotype and anti-PD-L1 treated mice similarly expressed TH1 proteins CD39, T-bet and SLAMF1, and largely failed to express CD69 (Fig. 2d), a protein specifically on the c2 TFH. Further, the Ki67+ SMARTA cells in both isotype and anti-PD-L1 treated mice exhibited highly overlapping protein expression patterns with each other, and were more similar to each other than to their respective Ki67-negative cells (Fig. 2e and Extended Data Fig. 3c), further suggesting anti-PD-L1 blockade preferentially expands pretreatment cycling clusters. Even early in infection (day 10 after LCMV-Cl13 infection), Ki67 was predominantly confined on to the TH1 c4, c6, whereas the majority of TCF-1+ clusters, and particularly the most differentiated TFH subset c1, expressed less Ki67 (Extended Data Fig. 3d). Thus, PD-1 limits the expansion of the most proliferative CD4+ TH1 cell subset throughout chronic infection, and blocking PD-L1 overcomes these constraints.
PD-L1 blockade targets TH1-phenotype T regulatory cells.
Foxp3+ Treg cells comprise approximately 20% of total CD4+ T cells during chronic viral infection and can differentiate into different Th-like subsets to suppress antiviral responses25. Importantly, SMARTA cells in LCMV infection do not become Treg cells20, although in other infections virus-specific CD4+ T cells potentially could. The frequency of Treg cells rapidly increased after the first anti-PD-L1 treatment compared with isotype-treated mice and PhenoGraph clustering revealed eight distinct clusters that again broadly categorized into TCF-1+ (c1, c3, c4, c7, c8) and TCF-1-negative (c2, c5, c6) populations, with the TCF-1-negative Treg cells expressing T-bet to both higher levels and in a larger proportion of cells (Fig. 3a,b and Supplementary Table 3). PD-L1 blockade did not numerically affect the TCF-1+ clusters, but increased the TCF-1-negative, TH1-like c2, c5, c6 (Fig. 3b,c and Extended Data Fig. 4a,b). Although all Treg cells were gated based on Foxp3 positivity (Extended Data Fig. 1b), Foxp3 expression itself, along with TH1-associated inhibitory receptors CD39 and Tim3, as well as activation markers CD27, CD44 and SLAMF1 were all enriched in the TH1-phenotype clusters (Fig. 3b). These TH1 clusters also had the highest expression and contained the majority of ICOS+, CTLA4+ and PD-1+ cells, demonstrating heightened activation of the TH1-phenotype Treg cells during chronic infection.
Fig. 3 |. PD-L1 blockade specifically expands and activates TH1-like Treg cells.
a, Proportion of Foxp3+ cells (Treg cells) of total CD4+ T cells at 60 h following the first isotype or anti-PD-L1 treatment. b, UMAP plots show PhenoGraph clustering of CyTOF data of total Foxp3+CD4+ Treg cells (combined data from one and three isotype or anti-PD-L1 treatments). The heatmap depicts the arcsinh MSI of proteins in each cluster normalized across columns by z-score. The individual UMAPs depict single-cell expression of the indicated protein. c, Total number of TCF-1− T-bethi and TCF-1+ Treg cells 60 h following the first isotype or anti-PD-L1 treatment. UMAP clusters colored in red (TCF-1− clusters 2, 5 and 6) are significantly increased in number upon PD-L1 blockade. d, The heatmap colors designate the arcsinh ratio of the MSI change in Foxp3+ Treg cells for the indicated protein comparing anti-PD-L1 versus isotype treatment within each PhenoGraph cluster. Red indicates increased in anti-PD-L1 treated mice and blue indicates increased in isotype-treated mice. Values of p are calculated by the limma test in the diffcyt R package. *P < 0.05. e, Proportion of Ki67+ cells in virus-specific SMARTA T cells and Treg cells 60 h following the first isotype or anti-PD-L1 treatment. f, UMAPs depict Ki67 expression and box plot the proportion of Ki67+ Treg cells at 60 h following the first isotype or anti-PD-L1 treatment. g, Plots of CyTOF data show expression of the indicated protein combinations gated on Ki67+ Treg cells following the first isotype and anti-PD-L1 treatment. h, Total number of Treg cells in both isotype and anti-PD-L1 treated mice between one and three treatments. i, The heatmap depicts the arcsinh MSI of proteins expressed by Ki67+ and Ki67− Treg cells in isotype and anti-PD-L1 treated mice following one and three treatments. Marker expression is normalized across columns by z-score. n = 5 mice in the isotype group and n = 4 in the anti-PD-L1 group. The experiment is representative of n = 18 mice in the isotype group and n = 16 mice in anti-PD-L1 group examined over three experiments. *P < 0.05; unpaired, two-tailed Student’s t-test (a,c,e,f,h). Box plots indicate the median, upper and lower quartile, and the whiskers show the high and low value. Line graph shows the average value ± s.d.
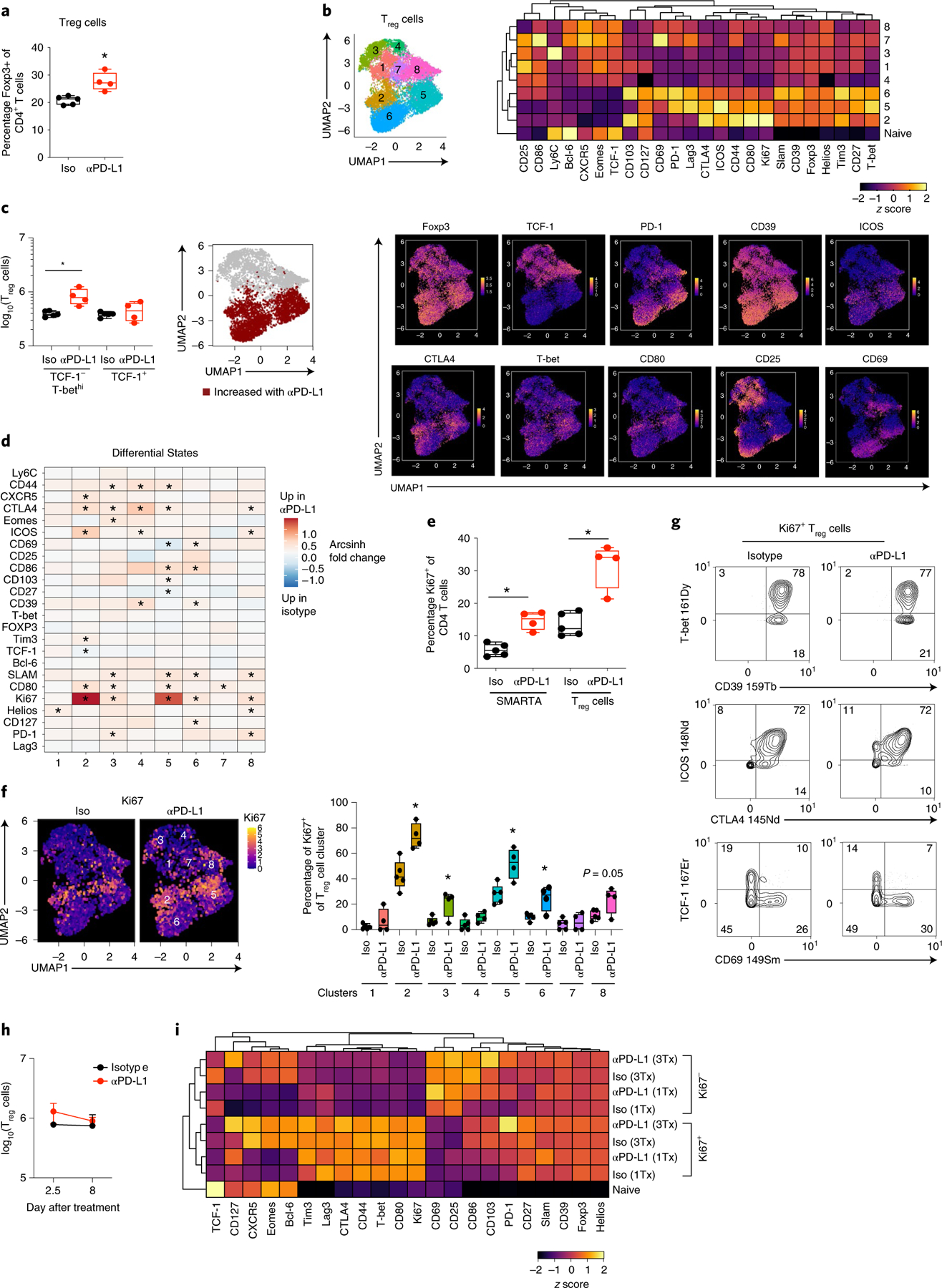
PD-L1 blockade also rapidly increased the expression of multiple activation/inhibitory proteins (including CTLA4, ICOS, CD80, CD86, SLAMF1) on the TH1-phenotype clusters compared with isotype treatment (Fig. 3d). Although anti-PD-L1 therapy did not bolster the number of TCF-1+ Treg cells, it did increase their expression of proteins such as PD-1, Helios, SLAMF1, ICOS and CTLA4 on multiple clusters (Fig. 3d), indicating that these populations were also being affected by the therapy. The inhibitory receptor CTLA4 was upregulated on multiple Treg cell clusters following anti-PD-L1 (c2–5, c8), but it had much lower/nondetectable expression on the virus-specific CD4+ T cells. Treg cell depletion led to increased numbers of CD4+ TH1 cells, but not TFH cells, with a particular restoration of GzmB+CD4+ TH1 cells (Extended Data Fig. 4c), indicating that consistent with their activated TH1-phentoype, Treg cells in the chronic infection preferentially suppress TH1 responses.
At baseline and following PD-L1 blockade, a higher proportion of Treg cells expressed Ki67 than did the virus-specific CD4+ T cells (Fig. 3e). PhenoGraph clustering was performed in the absence of Ki67 (Supplementary Table 3), and again, the Ki67+ cycling cell clusters at the onset of therapy were predominantly confined to the TH1-like Treg cells (c2, c5) (Fig. 3f). The cycling clusters that were present pretherapy (that is, in isotype-treated mice) were generally also the ones that were expanded by anti-PD-L1, and although an increase in the frequency of Ki67+ cells also occurred in Treg clusters c3, c6 and c8 (Fig. 3f), only the TH1-like c2, c5 and c6 were increased numerically (Fig. 3c and Extended Data Fig. 4a). Comparison of the Ki67+ cells in isotype and anti-PD-L1 conditions showed that cycling Treg cells exhibited similar protein expression patterns, with the vast majority of Ki67+ Treg cells expressing T-bet, ICOS and CTLA4, and high CD39, whereas the majority of the cycling cells in both conditions failed to express TCF-1 and CD69 (Fig. 3g). The similar overall protein expression patterns by cycling cells in isotype- and anti-PD-L1 treated mice suggested that specific pre-established populations of cycling virus-specific CD4+ TH1 cells and TH1-like Treg cells are specifically expanded by PD-L1 blockade.
Following the third anti-PD-L1 treatment, the number of total and TH1-phenotype Treg cells had contracted to the same level as in isotype-treated mice (Fig. 3h and Extended Data Fig. 4d). Yet, PD-L1 blockade continued to drive higher levels of activation proteins, particularly in TH1-like c2, c5 and c6, whereas the TCF-1+ Treg cells appeared to be only minimally changed (Extended Data Fig. 4e). Despite the contraction, the TH1-like c2 and c5 still comprised the majority of the Ki67+ cells in both isotype and anti-PD-L1 groups, although the proportions of Ki67+ cells within these TH1 clusters were no longer significantly elevated in response to PD-L1 blockade (Extended Data Fig. 4f). Like the cycling virus-specific CD4+ T cell populations, the Ki67+ cells following the first and third anti-PD-L1 treatments had concordant protein expression patterns both between time points, and between isotype and anti-PD-L1 treatment (Fig. 3i and Extended Data Fig. 4g), indicating that like the virus-specific CD4+ T cells, the TH1-like Treg cells are targeted by PD-1/PD-L1 to limit their expansion and activation state.
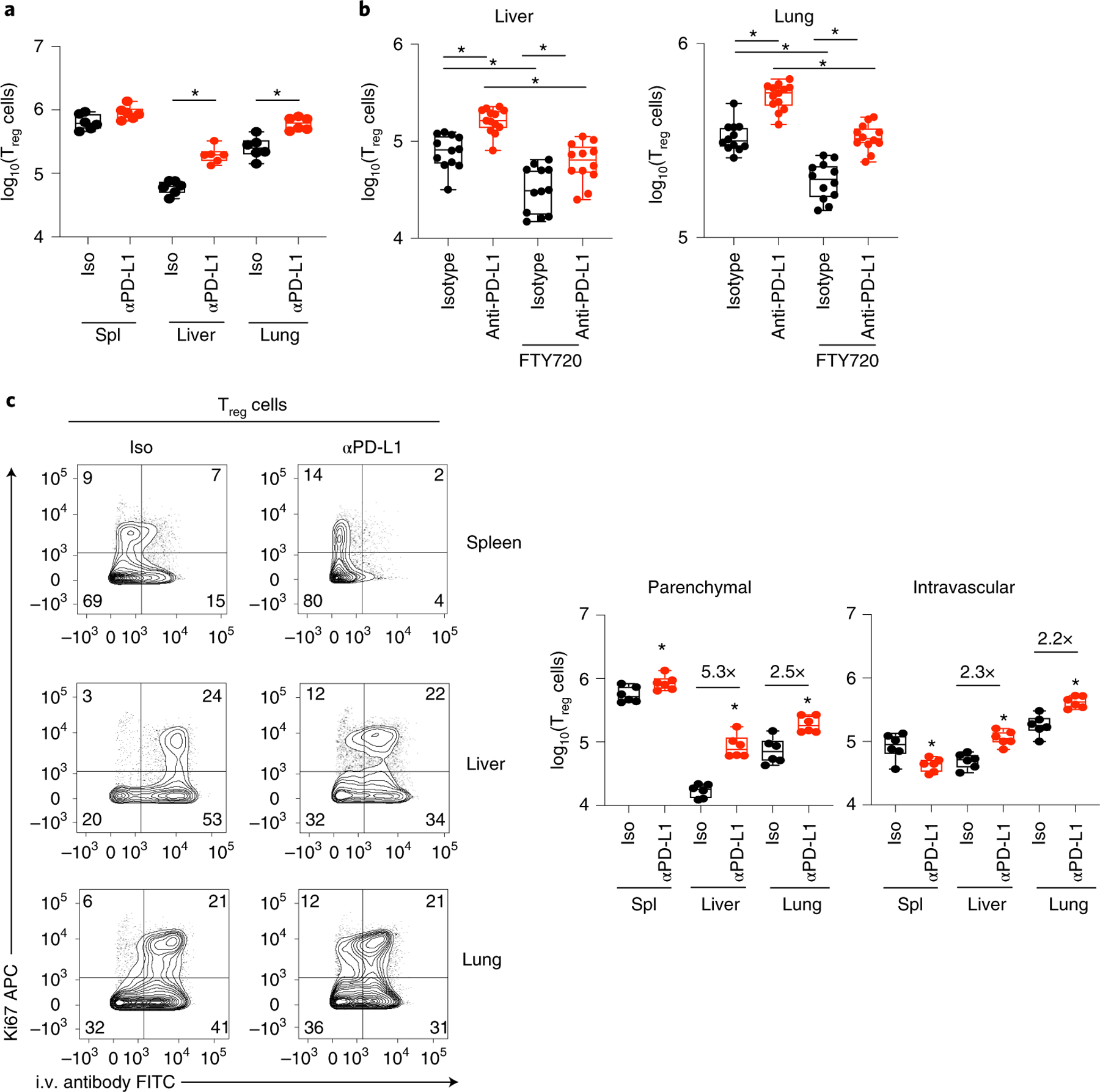
Treg cells in nonlymphoid organs (the liver and lung) increased following three anti-PD-L1 treatments, at a time when Treg cell amplification in the spleen had returned to baseline (Fig. 4a). To determine whether this increase was the result of local proliferation versus an influx of Treg cells from the lymphoid organs, we treated mice just before and then during anti-PD-L1 treatment with the drug FTY720 (fingolimod) to prevent cell emigration from lymphoid tissue. FTY720 treatment reduced Treg cells in the isotype conditions (Fig. 4b), suggesting that ongoing Treg cells in the lung and liver during chronic infection are replenished by homing from lymphoid organs. Yet, even with FTY720 treatment, PD-L1 blockade expanded the number of Treg cells in the liver and lung (Fig. 4b). Further, PD-L1 blockade increased Treg cell infiltration into the liver and lung tissue parenchyma, and within the spleen from the red pulp to the white pulp26 (Fig. 4c). Thus, PD-L1 blockade expands Treg cells directly in nonlymphoid organs, and functions to amplify Treg presence within the tissue parenchyma of these organs, where they are potentially positioned to interact and suppress effector T cells.
Fig. 4 |. PD-L1 blockade expands and induces tissue infiltration of Treg cells in nonlymphoid organs.
Naïve mice were infected with LCMV-Cl13 and 25 days later were treated with either isotype or anti-PD-L1 antibody and subsequently every 3 days for a total of three treatments. a, Total numbers of CD4+ Foxp3+ Treg cells in spleen, liver and lungs following anti-PD-L1 treatment. b, One day before isotype or anti-PD-L1 treatment (day 24 after LCMV-Cl13 infection) mice were injected with FTY720 or saline control and then treated daily until they were killed. Plots depict total numbers of Treg cells in liver and lung with and without FTY720 treatment during anti-PD-L1 therapy. c Mice were i.v. injected with anti-Thy1.2 just before being killed to distinguish intratissue parenchymal (Thy1.2-negative) versus intravascular (Thy1.2-positive) Treg cells. Flow plots depict expression of Ki67 versus i.v. antibody staining on Treg cells and box plots depict total SMARTA numbers in each organ. a,c, n = 6 mice per group and is representative of two experiments totaling 12 mice per group. b, Data is representative of two experiments totaling isotype (n = 12 mice), isotype+FTY720 (n = 12 mice), anti-PD-L1 (n = 13 mice), anti-PD-L1+ FTY720 (n = 12 mice). Data in (b) are a combination of two independent experiments with n = 12 isotype and n = 13 anti-PD-L1. Six or seven mice per group. Box plots indicate the median, upper and lower quartiles, and the whiskers show the high and low values. *P < 0.05. unpaired, two-tailed Student’s t-test (a–c).
Gene expression changes by virus-specific CD4+ T cells following PD-L1 blockade.
We next performed single-cell RNA-sequencing (scRNA-seq) on SMARTA T cells following isotype or anti-PD-L1 treatment. Analysis was performed after the third antibody treatment to allow for network redistributions to take hold, while viral titers were not yet decreased by PD-L1 blockade in this particular experiment (Extended Data Fig. 5a). Cell-cycle genes were regressed out before clustering (akin to clustering CyTOF data without Ki67). Of the eight clusters that emerged, only c2 and c3 increased following anti-PD-L1 blockade and these were characterized by expression of TH1-cell genes, including Ifngr, Ifng, Gzmb, Ccl5, Selplg (encoding PSGL1) and Nkg7 (Fig. 5a–c). c3 was a highly differentiated TH1 cluster (corresponding to c9 and some of c8 in the CyTOF analysis Fig. 1), and exclusively expressed Prdm1 (encoding Blimp1) and had the majority of Gzmb expression (Fig. 5c). Tcf7 (encoding TCF-1) was widely expressed by most clusters, but was largely absent from c3 and only present in the proportion of c2 that lacked Gzmb expression (Fig. 5c), suggesting c2 is a transitional population containing cells further differentiating to TH1, akin to c1 and c6 in the CyTOF clustering. All clusters were activated, although c0 was the least differentiated cluster, expressing Il7ra (CD127), Tcf7 and Ccr7, characteristic of more quiescent cells (Fig. 5b,c). c5 was comprised of bona fide TFH cells expressing Ascl2, Bcl-6 and Cxcr5, whereas c1 was composed of cells expressing TFH-associated genes, but not their lineage-defining transcription factors, thus we defined these as less-differentiated TFH. c7 expressed high amounts of IFN-stimulated genes (ISGs), and seemed to encompass both TH1 and TFH cells, indicating clustering was driven by the ISG signature and not a specific differentiation program. c6 had high expression of the genes Xcl1, Slamf7, Tnfrsf9 and Crtam, and appeared to be a hybrid between TH1 and TFH, expressing molecules such as Gzmk and Runx3, but also Tcf7, Sostdc1 and Bcl-6.
Fig. 5 |. PD-L1 blockade enhances TH1 gene programs and terminal differentiation of TH1 cells.
a, Virus-specific CD4+ SMARTA T cells were FACS-sorted and analyzed by single-cell RNA-seq. UMAP plots of isotype and anti-PD-L1 treated SMARTA cells clustered by Seurat. Bar graph depicts the proportion of each cluster with their respective treatments. b, Heatmap of top differentially expressed genes defining the clusters. c, UMAP plots depicting single-cell expression of the indicated gene. d, Average and percent expression of key TH1 and TFH defining genes in all SMARTA cell clusters (that is, total) following isotype or anti-PD-L1 treatment. UMAPs show single-cell expression of Sostdc1 and Il21. e, Volcano plot of differentially expressed genes in cluster 2 of CD4+ SMARTA T cells from PD-L1-blocked versus isotype-treated mice. Genes in red are increased or decreased twofold in expression and have an adjusted P < 0.05. Genes in blue have an adjusted P < 0.05. UMAPs show representative gene expression of Plac8 and Ctla2a, the two most increased genes in this cluster. f, Heatmap of top 30 upregulated and downregulated differentially expressed genes between TH1 c2 and c3. Violins depict expression of genes associated with terminal differentiation in clusters 2 and 3. Data are representative of one scRNA-seq experiment comprised of pooled cells from isotype (n = 4 mice pooled) and anti-PD-L1 (n = 4 mice pooled).
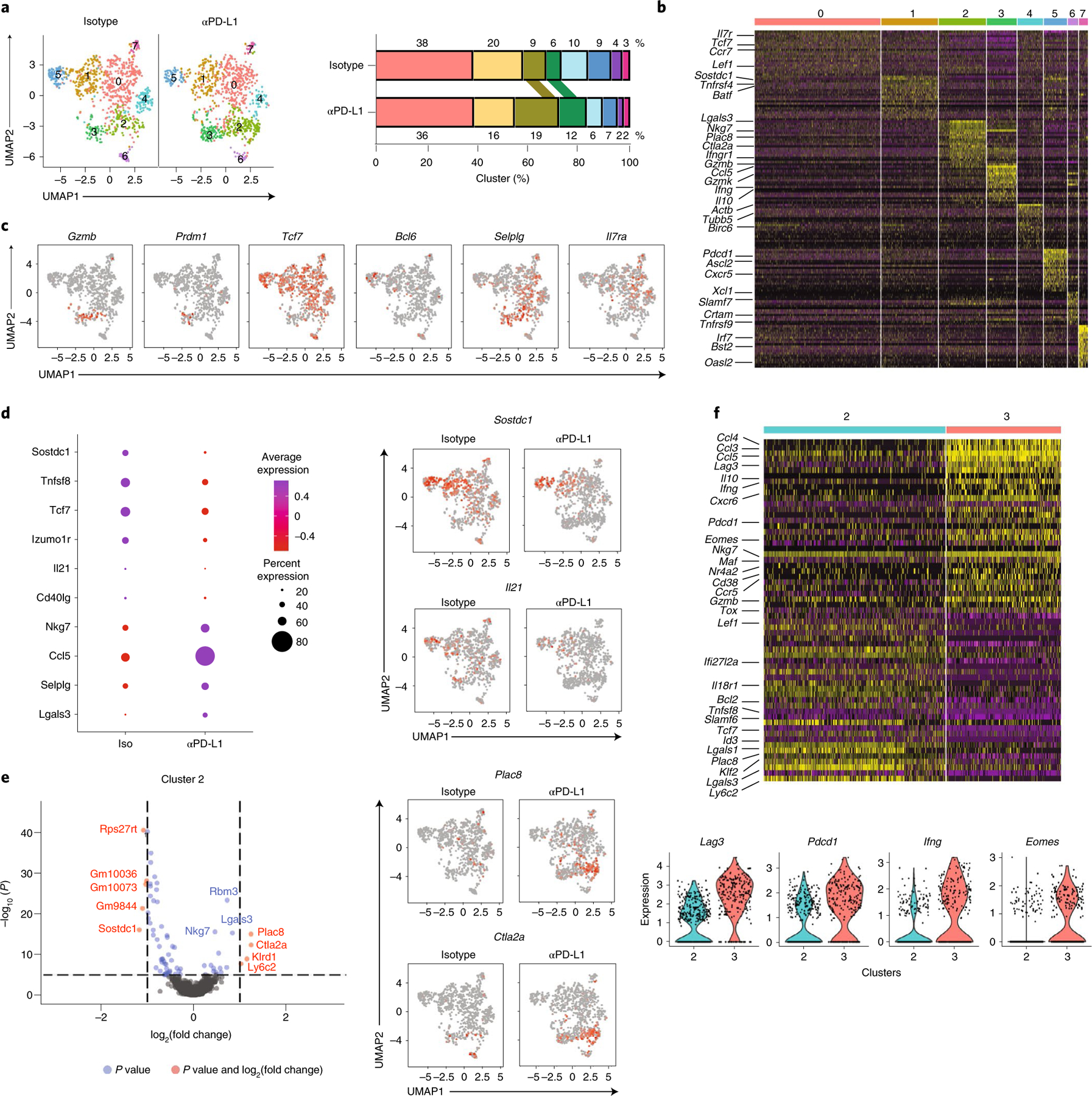
When all the clusters were combined, there were 461 significantly differentially expressed genes (DEG) following PD-L1 blockade (adjusted P < 0.05) (Supplementary Table 4). However, none increased more than twofold, and only Rps18-ps3, (ribosomal protein S18 pseudogene 3), and three predicted genes (Gm6133, Gm98433, Gm2000) reached twofold decreases upon blockade, indicating that at the population level the cells were transcriptionally similar following therapy. Of the DEG in the total SMARTA cell population, there was an overall increase in TH1 signature genes following PD-L1 blockade, including Nkg7, Ccl5, Selplg and Lgals3, which was accompanied by a global decrease in TFH signature genes including Sostdc1, Tnfsf8 (encoding CD30L), Tcf7, Izumo1r (encoding for JUNO), Il21 and CD40lg (Fig. 5d, Extended Data Fig. 5b). The decreased TFH signature gene expression also occurred within the TFH and TFH-like clusters, suggesting that despite a lack of expansion, the nature of these cells was also being fundamentally altered (Supplementary Table 4). Along with the diminished TFH gene expression, PD-L1 blockade decreased Il21 production (a factor critical for control of chronic viral infection19,27,28), which was produced by both TH1 and TFH in isotype conditions and now had only minimal residual expression in TFH phenotype cells (Fig. 5d). Although the decreased TFH gene signature was evident across all SMARTA cell clusters following PD-L1 blockade, the increase in TH1 driving genes was largest in c2 (the transitional TH1 cluster) and in some cases the less-differentiated, centrally located c0. c2 itself expressed 64 DEG in anti-PD-L1 versus isotype treatment, with more than twofold increased expression of TH1-associated genes, including Plac8, (placenta specific 8) Ctla2a (cytotoxic T lymphocyte-associated protein 2 alpha), Klrd1 (killer cell lectin like receptor D1) and Ly6c2, as well as Nkg7 and Lgals3 (encoding galectin3) (Fig. 5e and Extended Data Fig. 5b). c0 and c1 expressed 140 and 68 DEG respectively, whereas clusters 3–7 expressed fewer than 40 DEG each (Supplementary Table 4). Many of the DEG in c0 and c1 were ribosomal proteins that were downregulated upon PD-L1 blockade. TH1 genes were also upregulated in these clusters upon PD-L1 blockade (particularly c0) with genes such as Nkg7, Ccl5 increased in c0 and c1, and genes such as Sostdc1, Tnfsf8 (encoding CD30L), Tnfrsf4 (encoding OX40) and Il21 decreased in c0 (Supplementary Table 4). In addition, anti-PD-L1 therapy increased Il7ra and Bcl2 expression in c0, suggesting enhanced cell survival of this less-differentiated population that likely were precursors to TH1 cells. Thus, not only were TH1 clusters enhanced by proportion and number by PD-L1 blockade, but there was a system-wide recalibration towards the TH1 differentiation state.
To analyze the transcriptional programs pushing the transition of the TH1 cells, we compared the genes differentially regulated from the transitional TH1 c2 with the terminally differentiated TH1 c3. Compared with c2, c3 exhibited increased expression of multiple TH1 defining and modulating genes, including increased expression of Ifng and multiple inhibitory receptors, including Lag3, Pdcd1 and Entpd1 (encoding CD39) (Fig. 5f and Extended Data Fig. 5c). Further, c3 was the only cluster that produced Il10 (Extended Data Fig. 5c), indicating feedback inhibition by the most terminally differentiated TH1 cells. PD-L1 blockade itself did not directly enhance these genes in c3 compared with isotype, indicating instead that PD-L1 directed the less-differentiated TH1 cells down a terminal differentiation pathway leading up to c3. The expression of transcription factor Tox (which drives CD8 T cell exhaustion29–31), Eomes (which associates with exhausted CD8 T cells32) and Maf which can drive Il10 expression33 were increased in c3 compared with c2 (Fig. 5f), indicating that the transcriptional transformations associated with CD8 T cell exhaustion underlay the transition to CD4+ TH1 terminal differentiation during chronic infection.
Network profiling reveals global transcriptional changes.
We next measured putative upstream regulators of DEGs in total cells, and focused on the TH1 c2 and c3, as well as the TFH c5, to understand how blocking PD-L1 modified the downstream TH1 and TFH cell transcriptional programs. Upstream regulator analysis predicted PD-L1 blockade-induced activation of central TH1 differentiation networks (for example, Tbx21, Id2, Tet2, IRF4), and inhibition of TFH driving cytokine networks (IL6, IL27, IL21, STAT3) (Fig. 6a). These occurred in conjunction with the predicted downregulation of TFH-associated regulators CD40lg and CD28 in c5, and the predicted activation of multiple upstream regulators for networks that control cellular proliferation (for example, Myc, Mycn, Itk, Ccnd1), particularly in c2. Chronic IFN-I signaling suppresses antiviral CD4+ T cell proliferation and function in chronic viral infection34,35, and multiple IFN-I related upstream regulators (IFN-α/β, IFN-α1/ IFN-α13, IFN-α2, IFN-αR1, JAK1/2) were predicted to be inhibited upon PD-L1 blockade in both TH1 and TFH clusters, whereas regulation through the SOCS1 network, an inhibitor of IFN-I signaling, was predicted to be elevated in the transitional TH1 population c2, as well as the TFH population c5. In line with diminished IFN-I mediated regulation, PD-L1 blockade decreased expression of numerous ISGs, such as IRF7 and OAS1a, while reciprocally elevating TH1-driving ISGs, such as Ccl5 and Ifngr (Fig. 6b and Extended Data Fig. 6a). Unexpected with the increased cycling and activation states of virus-specific CD4+ TH1 cells following PD-L1 blockade, TCR signaling (TCR, CD3, NR4A1 (encoding Nur77)) and multiple key immune-activating signaling pathways induced by TCR signaling (NFATC, RICTOR, NFKB) were predicted to be inhibited by PD-L1 blockade. Deeper examination revealed downregulation of many TCR-stimulated transcription factors by PD-L1 blockade (Fig. 6c), corresponding to a global diminution of TCR signaling strength when PD-L1 is inhibited. Only a few TCR-driven transcription factors were increased following PD-L1 blockade, particularly in the transitional TH1 c2, and these were specifically associated with TH1-fate determination and included Prdm1 and Klf2, as well as Tsc22d3 which is stimulated by IL-10 (Fig. 6c). Further, Nur77 protein expression, which is directly associated with the strength of TCR signaling36, was decreased rapidly (and before decrease in viral titers) in virus-specific CD4+ T cells from anti-PD-L1 treated compared with isotype-treated mice (Fig. 6d and Extended Data Fig. 6b). Both heightened and chronic TCR signaling promote conversion of TH1 to TFH cells2,37, suggesting that blocking PD-L1 reduces TCR signaling networks to re-establish TH1 programming.
Fig. 6 |. PD-L1 blockade reorients intracellular interferon and TCR signaling.
a, Bar graphs depict activation z-scores of IPA-predicted upstream regulator molecules in total (that is, all clusters combined) SMARTA CD4+ T cells, and individually in clusters 2, 3 and 5. b,c, Changes in gene expression that define inhibition of the (b) interferon alpha/beta receptor (IFNAR) complex or (c) TCR signaling as an upstream regulator. Data is across total SMARTA cells, c2 and c3 cells comparing anti-PD-L1 with isotype treatment. Green and red coloring of genes depicts downregulation or upregulation of gene expression, respectively. Red and blue lines indicate a decrease would lead to observed gene activation or inhibition, respectively. Orange lines signify that predicted gene expression change is inconsistent with the state of the downstream molecule, and a gray line signifies that the effect cannot be predicted. d, Flow cytometric analysis of Nur77 protein expression in SMARTA cells following the third isotype or anti-PD-L1 treatment. e, Rows in the heatmap illustrate the z-score of the RAS average (representing the activity of the regulons associated with the indicated transcription factor) and the number of downstream target genes (g) coexpressed with each transcription factor. P values (Wilcoxon test) comparing RAS means in anti-PD-L1 versus isotype-treated cells for each regulon and Seurat cluster corrected for the false discovery rate (FDR) for multiple hypothesis testing. ***FDR < 0.001, **FDR < 0.01, *FDR < 0.05 indicate a significant difference in the indicated regulon between anti-PD-L1 and isotype treatment. f, Gene set enrichment analysis of differentially expressed pathways in TH1 clusters 2 and 3 in CD4+ SMARTA T cells from anti-PD-L1 treated (red) or isotype-treated (black) mice. Pathways are ranked by normalized enrichment score (NES) and have P values <0.05. a–c,e,f, Data are representative of a single scRNA-seq experiment comprised of pooled cells from isotype (n = 4 mice pooled) and anti-PD-L1 (n = 4 mice pooled). d, Box plots show n = 5 isotype and n = 5 anti-PD-L1 and the experiment is representative of n = 9 isotype and n = 10 anti-PD-L1 mice examined over two experiments. *P < 0.05; unpaired, two-tailed Student’s t-test (d). Box plots indicate the median, upper and lower quartiles, and the whiskers show the high and low values.
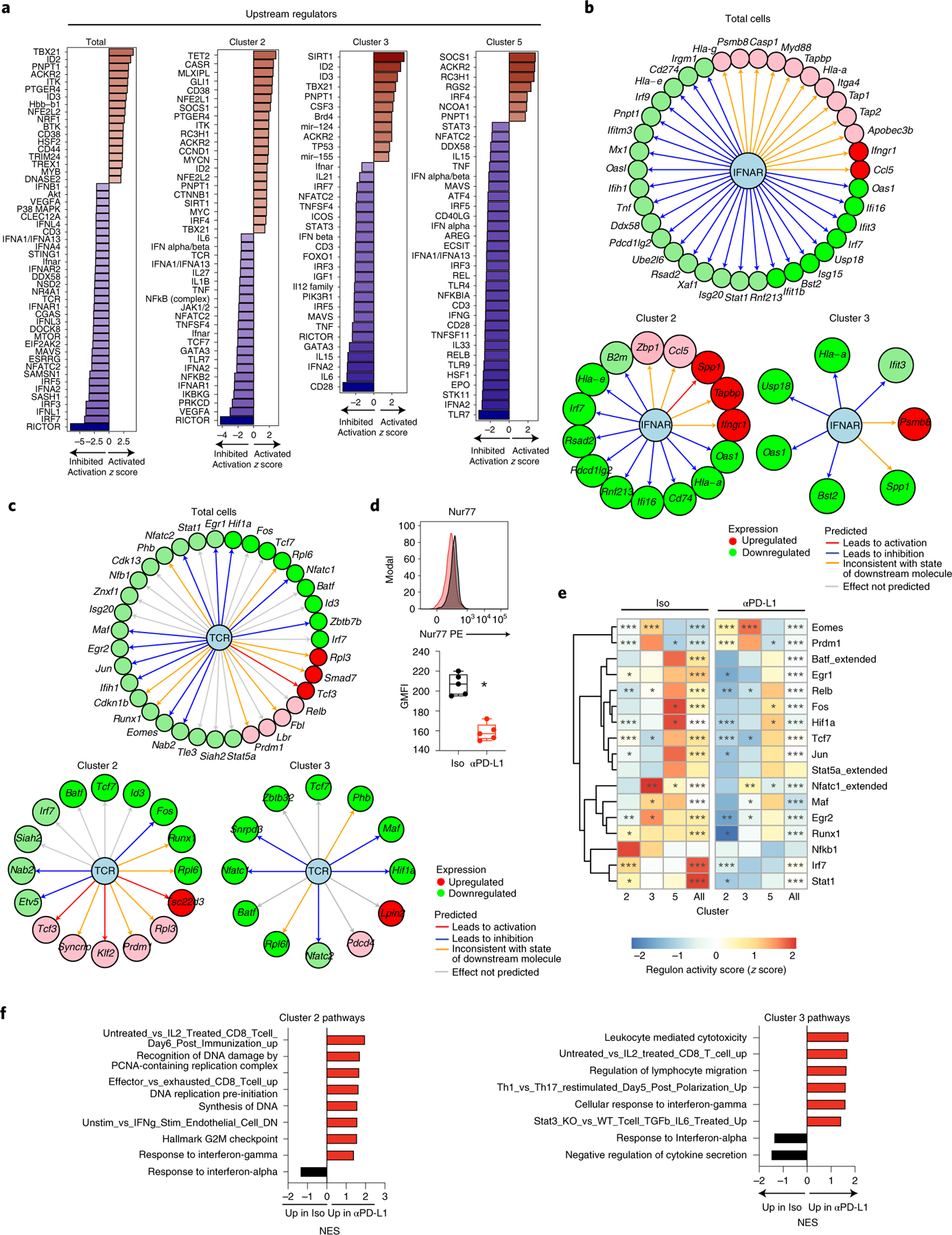
To further probe anti-PD-L1 mediated changes in transcription factor activity and their downstream target gene networks, we used the algorithm SCENIC (single-cell regulatory network interference and clustering)38. SCENIC identifies each transcription factor and its direct binding targets in the same cell (termed regulons) based on coexpression profiles and cis-regulatory motif enrichment. Quantification of the fold change in average regulon activity indicated that the decreased expression by scRNA-seq of multiple transcription factors involved in TCR signaling following PD-L1 blockade corresponded to a similar decrease in their target RNAs (regulons), including regulons driven by Fos, Jun, Nfatc1, Maf and Relb (Fig. 6e). The TFH c5 had a generally higher activity score for many of these regulons than the TH1 clusters during chronic infection, consistent with strong TCR signaling driving the TFH phenotype. Yet, even in this TFH c5, target regulons driven by Fos, Hif1a and Nfatc1 were significantly decreased upon anti-PD-L1 therapy. Although Eomes RNA was decreased following PD-L1 blockade, its target regulons remained elevated in c2 and c3, suggesting a delay in the transcriptional decrease in its expression and the functional activity of the protein. Conversely, the increased expression of Prdm1 following PD-L1 blockade was mirrored in increased expression of its target regulons, further corroborating the re-enforcement of TH1-like genes programing.
Pathway analysis of the individual cell clusters indicated enrichment of pathways involved in positive regulation of translation, RNA splicing, cell cycling and cellular metabolic processes when PD-L1 was inhibited (Extended Data Fig. 6c), indicating metabolic reprogramming of virus-specific CD4+ T cells. Corresponding to the overall virus-specific CD4+ T cell survival observed following PD-L1 blockade, stress response pathways such as “response to cold”, and pathways of negative regulation of apoptosis were increased across most of the clusters. Specifically, within the less-differentiated CD4+ TH1 c2, but not the terminally differentiated TH1 c3, PD-L1 blockade amplified multiple pathways involved in DNA replication and cell-cycle progression, further indicating cell-intrinsic modulation of cell-cycle machinery fueling expansion of the TH1 c2 population and c3 arising from these cells. Conversely, following PD-L1 blockade, c3 was enriched for pathways of TH1 functional activity, regulation of lymphocyte migration, leukocyte-mediated cytotoxicity and decreased STAT3 signaling (STAT3 is known to inhibit TH1 differentiation39) (Fig. 6f). Like the widespread inhibition of upstream regulation by IFN-I, the pathway “response to interferon-alpha” was one of the few pathways downregulated in c2 and c3 of the anti-PD-L1 treated group (Fig. 6f). Conversely, both c2 and c3 had increased signatures of IFN-γ activation and signaling, indicating a switch from chronic IFN-I toward IFN-γ signaling in response to PD-L1 blockade. Both c2 and c3 had gene signatures associated with CD8 T cell effector function following anti-PD-L1 therapy (c2 up in “effector vs exhausted CD8 T cells”; c3 up in “leukocyte-mediated cytotoxicity”) (Fig. 6f), suggesting the molecular acquisition of a gene signature pushed toward CD8 T cell effector functions.
PD-L1 blockade drives a CTL gene signature and resurrects CD4+ killing capacity.
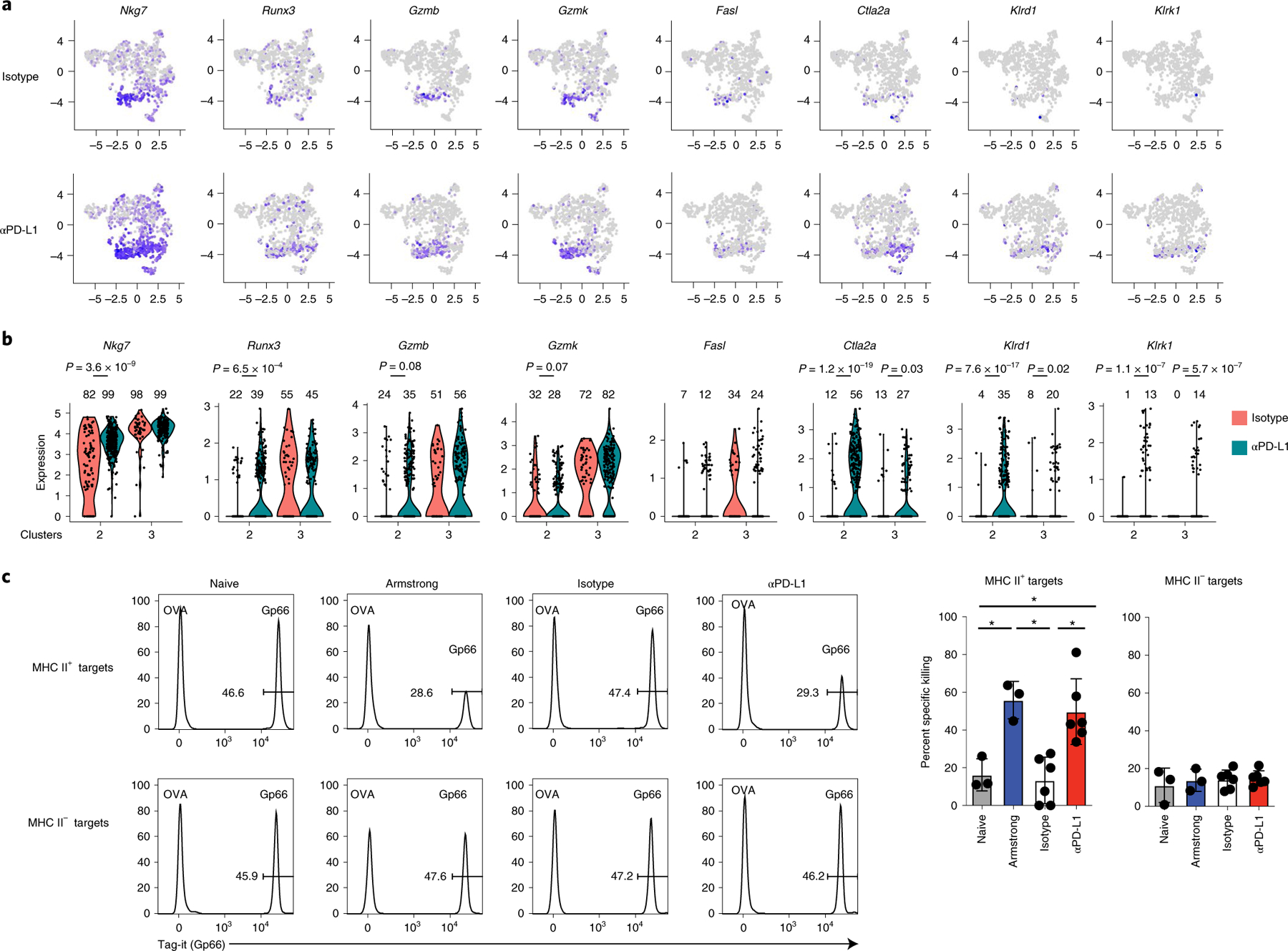
The increased signatures of CD8 T cell effector functions and IFN-γ signaling suggested that the TH1 may acquire cytolytic function when PD-L1 signals are inhibited. Indeed, multiple cytotoxic mediators including Nkg7, Runx3 (the transcription factor that drives cytotoxic function in CD8 T cells), Gzmb, Gzmk, Fasl, Ctla2a, Klrd1 and Klrk1 (encoding NKG2D) were preferentially expressed in TH1 clusters 2 and 3 (Fig. 7a and Extended Data Fig. 7), and were increased in expression and/or by proportion upon PD-L1 blockade. (Fig. 7b). By contrast, the non-TH1 clusters lacked or had minimal expression of these cytotoxic mediators and failed to upregulate them (Fig. 7a and Extended Data Fig. 7), suggesting the cytotoxic signature was localized to the TH1 cells. To directly test whether PD-L1 blockade-induced changes in virus-specific CD4+ T cell cytolytic function, we performed an in vivo CD4+ cytotoxic T lymphocyte (CTL) assay. To ensure killing was mediated by CD4+ T cells and was CD8+ T cell independent, we used β2-microglobulin (β2M) deficient splenocytes (that is, lacking MHC class I expression) as target cells. The splenocytes were labeled with LCMV-GP61–80 peptide (the SMARTA epitope) or an irrelevant peptide (OVA323–339) and then coinjected into LCMV-Cl13 infected mice after the third anti-PD-L1 treatment. Naïve mice demonstrated no specific killing of MHC II+ target cells compared with OVA323–339 labeled cells (Fig. 7c). Mice acutely infected with LCMV-Armstrong exhibited approximately 55% specific killing of transferred LCMV-GP61–80 labeled MHC II+ target cells, whereas this killing was completely abolished in chronically infected isotype-treated mice. Interestingly, PD-L1 blockade completely restored CD4+ T cell mediated killing to levels observed in the acutely infected mice. The CD4+-mediated killing was MHC class II dependent, as target cells that lacked MHC class II failed to be killed within the same mice (Fig. 7c). Thus, anti-PD-L1 enhances CD4+ cytotoxic gene networks and restores CD4+ T cell mediated killing during chronic infection, identifying a new mechanism of action of PD-L1 blockade therapy to restore immune function.
Fig. 7 |. PD-L1 blockade restores virus-specific CD4+ CTL function.
a,b, Virus-specific CD4+ SMARTA T cells from mice treated with three isotype or anti-PD-L1 treatments were analyzed by scRNA-seq as in Fig. 5. a, UMAPs depict expression of cytotoxic genes across the clusters in isotype and anti-PD-L1 treated virus-specific CD4+ SMARTA T cells. b, Violin plots depict expression of genes associated with cytotoxic function in the TH1 c2 and c3 following isotype and anti-PD-L1 treatment. Numbers above each violin indicate the percentage of cells positive for the indicated RNA in each cluster and the P value (Welch’s t-test) is indicated. c, In vivo CD4+ CTL assay. β2M−/− splenic target cells were labeled with Gp66–80 (LCMV) or OVA323–339 (nonspecific) peptide. Plots depict ratios of LCMV versus OVA peptide-labeled MHC II+ and MHC II− target cells in the spleen after 18 h in: (i) naïve mice (n = 3); (ii) mice infected with acute LCMV-Armstrong for 7 days (n = 3); or LCMV-Cl13 infected mice following (iii) the third isotype (n = 6); or (iv) anti-PD-L1 (n = 6) treatment. Graphs depict the average ± s.d. of specific killing of MHC II+ and MHC II− target cells in spleen. Data are representative of two experiments with a total of 6 naïve mice, 6 mice infected with LCMV-Armstrong, 12 isotype-treated LCMV-Cl13 infected mice, and 12 anti-PD-L1 treated LCMV-Cl13 infected mice. *P < 0.05. one-way ANOVA, Tukey’s multiple comparisons test.
Discussion
To date, the role of PD-L1 blockade to restore exhausted antigen-specific CD4+ T cells has been unclear. Our data now demonstrate that anti-PD-L1 therapy in chronic viral infection specifically and cell-intrinsically enhances TH1 subsets. Indeed, as chronic viral infection persists, TH1 cells are gradually lost, exasperating CD8 T cell dysfunction2,6. In addition to conventional virus-specific CD4+ T cells, we identify that TH1-phenotype Treg cells are highly responsive to PD-L1 blockade, through expansion, upregulation of suppressive factors and migration into nonlymphoid tissue. TH1-phenotype Treg cells specifically limit TH1 responses40, and consistent with this, we observed that Treg cell depletion during chronic infection preferentially increased TH1 cells, with minimal effect on TFH cells. The rapid increase in Treg cells by blocking PD-L1 likely reflects feedback inhibition to prevent excessive immunopathology, but likely also explains how they impede CD8 T cell restoration following PD-L1 blockade in chronic infection41. Thus, while restoring virus-specific CD4+ TH1 cells, PD-L1 blockade simultaneously releases counter-measures to cull back this response. PD-L1 blockade did not indiscriminately target CD4+ T cells, but instead the pre-established cycling capacity of specific populations was a major determinant of responding cells, with only limited incorporation of noncycling ones into the therapeutic response. In this way, PD-L1 specifically acts to limit cellular proliferation and makes the highly proliferative TH1 subsets the prime target in chronic virus infection. Thus, in cases when CD4+ T cells fail to respond to PD-1/L1 blockade therapies, pre-existing cycling antigen-specific subsets may not be present. Yet, it is important to point out that in other disease states where different CD4+ TH subsets are highly proliferative, these too may be targeted.
TCF-1+ memory-like CD8 T cells rapidly proliferate and have been shown to be the primary target of PD-L1 blockade7–13. On the other hand, the TCF-1+ CD4+ T cells were only minimally affected by therapy during chronic infection, suggesting that TCF-1+CD4+ and CD8+ T cells may function at different ends of the spectrum in response to PD-L1 blockade. CD4+ TFH can respond to immunotherapy in some cancers, particularly breast cancer42, although these therapies often incorporate anti-CTLA4 which can expand CD4+ T cells16, enhance TFH cells43 and indirectly alter CD4+ T cell responses through modulation of Treg cells44,45. Anti-PD-1-mediated tuberculosis reactivation in cancer patients exhibits increased Mtb-specific CD4+ TH1, but not TH17 cells, suggesting that TH1 cells can indeed respond to anti-PD-1 therapy in contexts other than viral infection46. Thus, based on these dichotomies, it will be important to further uncover the basis for disease-specific CD4+ TH restoration by PD-L1 blockade and how they contribute to disease control (or exasperation).
Considering that PD-1 functions to attenuate TCR signaling47, it was unexpected that blocking PD-L1 would decrease TCR signaling and downregulate expression of multiple transcription factors and their target genes. However, increased TCR signaling can potentiate CD8 T cell exhaustion1 and pushes virus-specific CD4+ T cell differentiation toward TFH2. Not all transcription factors were decreased following PD-L1 blockade, with specific increases in transcription factors associated with TH1 programming and differentiation. Thus, blocking PD-L1 and decreasing TCR signaling likely enables the cells to reroute toward TH1 amplification and functions; and paradoxically, the decrease in chronic TCR signaling favors restoration of CD4+ TH1 cells.
Combined with diminishing CD4+ TH1 responses, we identify the loss of CTL capacity as a CD4+ T cell dysfunction during chronic infection and demonstrate that CD4+ CTL transcriptional networks and killing capacity are completely restored by blocking PD-L1. In this way, not only can anti-PD-L1 enhance CD4+ TH1 cell help for CD8 T cells, but may also facilitate killing of infected MHC II+ cells. CD4+ T cells with CTL function have recently been identified in various cancers that can target MHC II+ tumors, including melanoma and bladder cancer48,49. Thus, it is interesting to speculate that anti-PD-1/L1 restoration of cytolytic CD4+ T cell function may be a mechanism of action of these therapies towards MHC II+ tumors, and may at least partially drive the high response rates to anti-PD-1 therapy in Hodgkin’s lymphoma (a B cell tumor) where the tumors are often largely MHC I negative and response rates are correlated with MHC class II expression14,15. Although the ability of PD-L1 blockade to restore CD8 CTL killing during chronic viral infection undoubtedly accounts for much of the therapy’s efficacy in lowering viral titers, we postulate that CD4+ CTL killing complements and likely further enhances the cytotoxicity potential of anti-PD-L1 therapy, particularly through targeting of virus-infected MHC II+ antigen presenting cell (APC) populations. Thus, these data high-light that the suppression of CD4+ CTL killing is a CD4+ T cell dysfunction during chronic infection regulated by PD-L1, and that blocking PD-L1 reprograms and functionally restores CD4+ CTL killer cells. Ultimately, these mechanisms have important implications for multiple therapeutic contexts of anti-PD-L1 therapy.
Methods
Mice.
All mice used for experiments were between 6 and 10 weeks old at the initiation of the experiment. Female C57BL/6 mice (CD45.2+) were purchased from the Princess Margaret Cancer Center (PMCC) or The Jackson Laboratory. LCMV-GP61-80-specific CD4+ TCR transgenic (SMARTA; CD45.1+) mice have been described previously18 and were bred at PMCC. Pdcd1−/− mice were purchased from The Jackson Laboratory and subsequently crossed to SMARTA mice to generate PD-1−/− SMARTA mice. These mice were then bred at PMCC. β2M−/− mice and Foxp3DTR mice were purchased from The Jackson Laboratory. All mice were housed under specific pathogen-free conditions. Mice were sex and age matched for experiments. Mouse handling conformed to the experimental protocols approved by the OCI Animal Care Committee at PMCC/University Health Network. No statistical tests were used to predetermine sample sizes. Because the models are well established, sample sizes were chosen based on previous studies of our own and by others in the field50,51. Group numbers in each experiment are described in the figure legends.
LCMV infection and T cell adoptive transfer.
Mice were infected i.v. via the retro-orbital sinus with 2 × 106 plaque-forming units (p.f.u.) of LCMV-Cl13. 2 × 105 p.f.u. of LCMV-Armstrong was injected intraperitoneally (i.p.) into mice as a control for the in vivo CTL assay. Virus stocks were prepared and viral titers were quantified as described previously18. LCMV-specific CD4+ SMARTA T cells were isolated from the spleens of transgenic mice by negative selection (StemCell Technologies). Then 3,000 CD45.1+ SMARTA cells (donors) were transferred i.v. into the retro-orbital sinus of naïve CD45.2+ C57BL/6 mice (recipients) that were then infected with LCMV-Cl13 one day later.
In vivo antibody administration.
For in vivo PD-L1 blockade experiments, 250 μg of anti-PD-L1 (10 F.9G2) or isotype control (LTF-2) antibody (BioXcell) was administered i.p. beginning 25 days after cell transfer, and then every 3 days thereafter, as described in the figure legends. Allocation of infected mice to treatment groups was random. Mice were killed 60 h following the first antibody treatment or one day after the third antibody treatment. In some experiments mice were injected i.v. with 3 µg of Thy1.2 fluorescein isothiocyanate antibody and killed 5 min later to examine intravascular versus extravascular localization of cells in the various organs.
FTY720 treatment and Treg cell depletion.
FTY720 was reconstituted in saline and i.p. injected into mice at a dose of 1 mg kg−1 daily starting 1 day before the first anti-PD-L1 treatment (that is, day 24 after infection) until the time of death. For Treg depletion mice were injected with 50 µg kg−1 of diphtheria toxin at days 23 and 24 after LCMV-Cl13 infection and then every 3 days (day 27 and day 30) until mice were killed on day 33.
Time-of-flight mass cytometry.
The CyTOF antibody panel is listed in Supplementary Table 1. Purified unconjugated antibodies were labeled with metal tags at the SickKids-UHN Flow and Mass Cytometry Facility using the MaxPar Antibody Labeling Kit from Fluidigm. Directly conjugated antibodies were purchased from Fluidigm. All working antibody concentrations were determined by titration.
For staining, single-cell suspensions from individual samples were first stained for 15 min at room temperature (20 °C) with antibodies that did not perform well after fixation. The samples were then washed with PBS and pulsed with 12.5 μM cisplatin (BioVision) in PBS for 1 min at room temperature (20 °C) before quenching with CyTOF staining media (Mg+/Ca+ HBSS containing 2% fetal bovine serum (FBS) (Multicell), 10 mM HEPES (pH 7.3; Corning), and FBS underlay. Cells were then fixed for 12 min at rom temperature (20 °C) with transcription factor fixative (eBiosciences) and permeabilized, and individual samples were barcoded before being combined using the 20-Plex Pd Barcoding Kit according to the manufacturer’s instructions (Fluidigm). Combined samples were resuspended in staining media containing metal-tagged surface antibodies (Supplementary Table 1) and Fc block for 30 min at 4 °C. Cells were then permeabilized and stained with metal-tagged intracellular antibodies using the Foxp3 Transcription Factor Staining Kit (eBiosciences) according to the manufacturer’s instructions. Cells were then incubated overnight in PBS (Multicell) containing 0.3% (w/v) saponin, 1.6% (v/v) paraformaldehyde (Polysciences Inc.) and 1 nM iridium (Fluidigm). Cells were then washed and kept in PBS with 1.6% paraformaldehyde in 4 °C for approximately 1 week until acquisition. Cells were analyzed on a Helios or Helios2 mass cytometer (Fluidigm) at Sick Kids-UHN Flow and Mass Cytometry Facility. EQ Four Element Calibration Beads (Fluidigm) were used to normalize signal intensity over time on CyTOF software version 6.7. FCS files were manually de-barcoded and analyzed using Cytobank 6.2 (Cytobank, Inc).
Heatmaps were plotted in R using the viridis color package and the gplots package. Arcsinh-transformed median of spectral indices (MSI) values were used to generate the heatmaps. Naïve cell data in heatmaps were obtained from clustering total CD4+ T cells from the same experiments as the virus-specific T cells and graphing MSIs of the naïve cluster defined as PD-1-neg, CD44lo, TCF-1hi and CD127hi. The R implementation of UMAP (k = 100) and the PhenoGraph algorithm52 were used for cluster analysis of arcsinh-transformed CyTOF data. Arcsinh cofactors were manually determined by staining intensity. Marker channels used to cluster each of the cell populations can be found in Supplementary Tables 2 and 3. For clustering, equal sampling of cells was performed on each of the time points based on the lowest common denominator of all groups before clustering. Owing to lower overall numbers of SMARTAs, all cells were used for clustering and the numbers displayed in UMAPs were balanced to accurately display the data. Differential states and differential abundance of clusters were calculated using the limma and edgeR tests respectively, through the “diffcyt” R package and plotted using ggplot2 (ref. 53). The R package “destiny”24 was used to make diffusion maps and do the pseudotime analyses of SMARTA cells.
Flow cytometry and intracellular cytokine staining.
Single-cell suspensions were prepared from organs and were stained ex vivo using antibodies to CD4 (GK1.5), CD45.1 (A20), SLAMF1 (TC15-12F12.2), CXCR5 (2G8) and B220 (RA3-6B2), all purchased from BioLegend. Staining for Ki67 (35/Ki-67 RUO) (BD Biosciences) and Nur77 (12.14) (eBiosciences) was performed as directed using the Foxp3 Transcription Factor Staining kit (eBiosciences). Samples were run on a FACS Verse or a FACS Lyric (BD Biosciences) and data analyzed using Flow Jo software (v.9 or v.10; Treestar).
For cytokine quantification, splenocytes were restimulated for 5 h at 37 °C with 5 μg ml−1 of MHC class II-restricted LCMV peptide GP61-80 in the presence of 50 U ml−1 recombinant murine IL-2 and 1 mg ml−1 brefeldin A (Sigma). Following the 5 h in vitro restimulation, cells were stained with the fixable viability stain zombie aqua (BioLegend), extracellularly stained as above with CD4+, CD45.1+, and then fixed, permeabilized (BioLegend cytokine staining kit) and stained with IFN-γ (XMG1.2), TNF-α (MP6-XT22) and IL-10 (JES5-16E3) (BioLegend).
Single-cell RNA-sequencing.
SMARTA T cells were transferred into naïve mice that were infected with LCMV-Cl13 a day later. Anti-PD-L1-blocking antibody or isotype control antibody was administered i.p. beginning 25 days after infection and subsequently on day 28 and day 31 for a total of three treatments. Mice were killed following the third treatment (day 33 post infection) and splenocytes from four anti-PD-L1 treated mice or four isotype-treated mice were separately pooled and B cell depleted with anti-CD19 beads (Miltenyi). Single-cell suspensions were then stained for virus-specific SMARTA T cells using CD4 and CD45.1 and FACSorted on a Moflo Astrios (Beckman Coulter) or a BD FACSAria Fusion cell sorter (Schematic for Sort is shown in Extended Data Fig. 8). Sorted anti-PD-L1 treated and isotype-treated SMARTA cells were then washed twice with PBS + 0.04% BSA and mixed with 10× Genomics Chromium single-cell RNA master mix and loaded onto a 10× chromium chip according to the manufacturer’s protocol to obtain single-cell complementary DNA (cDNA). Library generation was performed following the Chromium Single Cell 3ʹ Reagents Kits v.2 User Guide: CG00052 Rev B. Each library was sequenced on the Illumina HiSeq 2500 platform to achieve an average of 50,000 reads per cell. Sequencing was done at the Princess Margaret Genomics Center. Plasma LCMV titers were measured to confirm that virus was not decreased at the time of death.
For the anti-PD-L1 and isotype-treated SMARTA samples raw sequencing data was processed using Cell Ranger (v.1.3.1, 10× Genomics) to generate expression matrix of RNAs (gene-level counts) for each cell in each sample. Cell Ranger identified 965 cells for the isotype sample and 1,743 cells for the anti-PD-L1 sample. All subsequent downstream analysis was performed in the R statistical programming language using the Seurat (v.3.1.0) package54. For various quality control steps involving cell filtering, normalization and removal of technical variation, we merged both samples into a single Seurat object. In total, 179 low-quality cells were filtered out using selected threshold for nUMI ≥ 500, nGene ≥ 200 and <2,500, log10(GenesPerUMI) > 0.80, mitochondrial ratio < 5 and genes with zero counts. The final dataset used for analysis consisted of 2,529 cells (965 cells for isotype and 1,618 cells for anti-PD-L1) and 10,575 genes. The SCTransform function was used to normalize data and regress out factors related to mitochondrial and cell-cycle genes. To identify clusters, we performed an integrated analysis of the cells from isotype and anti-PD-L1 samples using 3,000 highly variable genes and the first 30 principal components. Eight clusters were identified at resolution 0.4. Differential expression analyses for the eight clusters were performed using the MAST function in the Seurat package. Cluster 2 volcano plot was made using the R package Enhancedvolcano.
Predicted upstream regulator analysis of differentially expressed genes was performed by Ingenuity Pathway Analysis software (Qiagen). Gene Set Enrichment Analysis was performed55,56 on the Enrichment Map gene set “Mouse_GOBP_AllPathways_no_GO_iea_August_01_2017_symbol.gmt”57 and ImmuneSigDB55,58 using DE genes preranked by P value between anti-PD-L1 and isotype treatments on total cells and in each individual cluster. ImmuneSigDB gene lists were converted from human genes to orthologous mouse genes using Ensembl BioMart59.
The SCENIC R package (v.1.2.4)38 was used to assess regulon activity on a cellular level. A regulon is formed of a transcription factor (TF) and its putative direct targets. Briefly, SCENIC defines coexpressed modules based on TF and gene coexpression using the GENIE3 R package (v.1.12.0). Regulons are then defined by excluding indirect targets from the coexpressed modules based on enrichment of DNA TF-binding motifs in targets using the RcisTarget R package (v.1.10.0).
Finally, a regulon activity score (RAS) is calculated for each regulon using the AUCell R package (v.1.13.1) based on the enrichment of a regulon for genes located at the top of a list of all genes ranked by their expression in decreasing order. SCENIC was run on 7,669 genes having at least 75 (3 UMI × 25 (1%) cells) counts per cell, expressed in at least 1% of isotype and anti-PD-L1 treated cells, and available in the mm10 mouse RcisTarget database. SCENIC identified 158 regulons in 2,494 cells out of 3,514 coexpressed modules, of which 107 regulons were active in more than 1% of isotype and anti-PD-L1 treated cells. Regulon RAS average was calculated for cells in each Seurat cluster in isotype and anti-PD-L1 treated groups. The z-scores are then calculated by regulon (row normalized). Heatmaps were generated using the pheatmap R package (v.1.0.12).
Bulk RNA-sequencing of CD4+ TH1 and TFH cells during acute LCMV-Armstrong infection.
SMARTA T cells were transferred into naïve mice that were infected i.p. with 2 × 105 p.f.u. of LCMV-Armstrong a day later. Seven days after infection, mice were killed and splenocytes from ten mice each pooled into three groups for separate replicates and B cell depleted with anti-CD19 beads (Miltenyi). Single-cell suspensions were then stained for virus-specific CD4+ TH1 and TFH SMARTA cells using CD4+, CD45.1+, SLAMF1 and CXCR5, and FACSorted on a Moflo Astrios (Beckman Coulter) directly into RLT buffer (Qiagen). RNA was isolated using a single-cell RNA purification kit (Norgen Biotech) according to manufacturer’s instructions. SMART-Seq v.4 Ultra Low Input RNA Kit for Sequencing (Clontech) was used per manufacturer’s instructions for amplification of RNA and subsequent cDNA synthesis. All samples proceeded through NexteraXT DNA Library Preparation (Illumina) using NexteraXT Index Kit V1 or V2 Set A (Illumina) following manufacturer’s instructions. A portion of this library pool was sent for sequencing on an Illumina NextSeq HighOutput, single read at the Princess Margaret Genomics Centre. An average of 400 × 106 reads were obtained per pool, with an average of 40M reads/sample across the entire data set.
Illumina reads were aligned to the Mus musculus GRCm38 genome build 88 (ref. 60) using HISAT2 (ref. 61). Alignments were compressed and sorted using SAMtools62. The alignments were quantified using HTSeq63–65 to obtain gene counts. Differential analysis was conducted using edgeR64,65 with modified code from the rnaseq.wiki protocol66. Low-count genes were excluded from the analysis if at least three samples did not have at least one count per million reads mapped (CPM) for that gene. Gene counts were normalized using Trimmed Mean of M values normalization55,58,67. Figure was plotted in R using the ggplot2 package68.
In vivo CTL assay.
Splenocytes from naïve β2M knockout (β2M−/−) mice were isolated for target cells and pulsed with either 5 μg ml−1 of LCMV-specific GP61-80 peptide or a nonspecific peptide (OVA323-339) and then respectively labeled with either Tag-it violet proliferation dye (BioLegend) or carboxyfluorescein succinimidyl ester (Sigma). GP61-80 and OVA323-33 labeled β2M−/− splenocytes were mixed at a ratio of 1:1 and 2 million cells were injected into either naïve mice, mice infected for 9 days with LCMV-Armstrong, or mice infected with LCMV-Cl13 that had been administered either their third anti-PD-L1 or isotype antibody treatment a day earlier (treatment schedule described above). Eighteen hours after target cell transfer, mice were killed and the ratio of specific (Tag-it) to nonspecific (carboxyfluorescein succinimidyl ester) killing of B220+ target cells (MHC II positive) or CD4+ target cells (MHC II negative) was subsequently analyzed by flow cytometry in the spleen. The percentage of specific killing was determined by the following equation: [1 − (ratio of Gp66 to OVA)] ×100.
Data exclusion and blinding.
No data was excluded from this study. On some occasions, SMARTA cells were not detected in recipient mice at the experimental time point, and these were excluded from the analysis (that is, if there are no SMARTA cells, we cannot phenotype or analyze them). Note, this occurred in both isotype- and anti-PD-L1 treated mice, indicating it is not a result of a specific treatment. Data collection and analysis were not performed blind to the conditions of the experiments.
Statistical analysis.
All statistical parameters are described in the figure legends. Student’s t-tests (two-tailed, unpaired) or one-way ANOVA (two-tailed, unpaired) were performed using GraphPad Prism v.6 (GraphPad Software). Data distribution was assumed to be normal but this was not formally tested. As a result, individual data distribution (individual data points) are shown. In the line and bar graphs the error bars indicate standard deviation (s.d.). In the box and whisker plots the box represents the median and upper and lower quartiles and the whiskers the minimum and maximum values.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. CyTOF gating scheme.
Plots show the successive gating scheme for (a) SMARTA CD4 + T cells and (b) Treg cells.
Extended Data Fig. 2 |. Enhancement of virus-specific CD4+ TH1 cells by PD-L1 blockade occurs prior to decreased virus titers.
Virus-specific CD4+ SMARTA T cells were transferred into naïve mice which were subsequently infected with LCMV-Cl13 a day later. Twenty-five days after infection mice were treated with either isotype or anti-PD-L1 antibody and subsequently every 3 days for a total of 3 treatments. Mice were sacrificed 60 h following the first treatment or two days after the third treatment and splenic virus-specific CD4+ SMARTA T cells were analyzed. (a) Longitudinal plasma virus titers pre-treatment and then following one (n = 5) or three (n = 5) isotype or anti-PD-L1 treatments. (b) Log fold change of abundance of CD4+ SMARTA T cell PhenoGraph clusters at 60 h after the first antibody treatment. P-values of cluster abundances are calculated by the edge R test in the diffcyt R package. *, p < 0.05. (c) Total virus-specific CD4+ SMARTA T cell numbers at 60 h after the first isotype (n = 5) or anti-PD-L1 (n = 5) treatment. (d) Proportions and total number of endogenous (that is, non-SMARTA) TH1 cells (CD39 hi) vs TCF-1+ cells after the first isotype (n = 5) or anti-PD-L1 (n = 5) treatment. Foxp3+ Treg cells and SMARTA cells were gated out and plots are gated on the remaining CD4+ PD-1+ (activated) cells. (e) Proportion of virus-specific CD4+ T cells producing IFN-γ, TNF, and IL-10 following ex vivo LCMV-GP61–80 peptide stimulation. Data are from 60 h after the first isotype (n = 5) or anti-PD-L1 (n = 5) treatment. (f) Kinetics of CD4+ SMARTA T cells in each PhenoGraph cluster between 1 and 3 isotype or anti-PD-L1 treatments. (g) Total number of CD4+ SMARTA TH1 (TCF-1-negative) and TCF-1+ virus-specific T cells in liver and lung following 3 anti-PD-L1 treatments. (a–c) n = 5 mice per group examined over four experiments totalling n = 19 isotype and n = 20 anti-PDL1 mice. (d–f) n = 5 mice per group examined over 2 experiments totaling 10 mice per group. Data in (G) is cumulative of 2 experiments with n = 11 for isotype mice and n = 12 for anti-PDL1 treatment groups. *: p < 0.05. unpaired, two-tailed Student’s t-test (a, c–g). Box plots indicate the median, upper and lower quartile, and the whiskers show the high and low value. Line graphs show the average and error bars indicate the standard deviation (SD).
Extended Data Fig. 3 |. Pre-therapy cycling CD4+ SMARTA T cell populations are targets of anti-PD-L1 blockade.
(a) Diffusion pseudotime map of CD4+ SMARTA T cells colored by pseudotime starting at Tip (T)3. T1 – T3 designate the algorithm-derived cellular tips and the lines and arrows the pathways derived from T3 as a starting point. (b) Representative flow plots of Ki67+ SMARTA cells following 1 treatment in spleen. (c) UMAPs depict Ki67 expression in CD4+ SMARTA T cells and the graph the proportion of Ki67+ of SMARTA cells in each cluster following the third isotype or anti-PD-L1 treatment. (d) CD4+ SMARTA T cells in the spleen 10 days after LCMV-Cl13 infection mice. (Top) UMAP and heatmap of CyTOF data of CD4 + SMARTA T cells clustered with PhenoGraph. (Bottom) UMAPs illustrating the single cell expression of the indicated protein. The graph shows proportion of SMARTA cells that are Ki67+ in each PhenoGraph cluster. (a) n = 5 mice per group and are representative of total n = 9 mice examined over two experiments. (b, c) n = 5 mice per group and are representative of total n = 10 mice per group over 2 experiments. (d) n = 4 mice per group and representative of n = 9 mice total over 2 experiments. *: p < 0.05. unpaired, two-tailed Student’s t-test between isoytpe and aPD-L1 of each cluster (c), or between clusters 1 (Tfh) and Th1 clusters 4 and 6 (d). Box plots indicate the median, upper and lower quartile, and the whiskers show the high and low value.
Extended Data Fig. 4 |. PD-L1 blockade targets cycling, TH1-phenotype Treg cells.
(a) Total number of Foxp3+ Treg cells in each cluster at 60 h after the first isotype or anti-PD-L1 treatment. (b) Representative 2D CyTOF plots of T-bet, Bcl-6 and TCF-1 expression in total Treg cells. (c) To deplete Treg cells, LCMV-Cl13 infected Foxp3DTR mice were treated with DT or as a control PBS on days 23, day 24, day 27 and day 30 after infection. Mice were sacrificed at day 33 and splenocytes isolated for flow cytometric analysis. Representative FACS plots depict SLAM vs GzmB expression in splenic Foxp3-negative effector CD4+ T cells. Box plots depict total numbers of TH1, TFH and GzmB+ Foxp3-negative CD4+ T cells. (d) Total number of Foxp3+ Treg cells in each cluster following 3 treatments. (e) The colors in the heatmap designate the arcsinh ratio of the MSI change for the indicated protein within each PhenoGraph cluster of Treg cells. The graph is comparing protein expression in Foxp3+ Treg cells following the third anti-PD-L1 versus isotype treatment, with (red) increased with anti-PD-L1 treated mice or (blue) increased in isotype-treated mice. P-values are calculated by the limma test in the diffcyt R package. *, p-value<0.05. (f) UMAPs and bar graph depict the expression and proportion of Ki67+ cells in each Treg PhenoGraph cluster following 3 anti-PD-L1 or isotype treatments. (g) Representative protein expression plots gated on Ki67+ Treg cells in spleens of mice following 3 anti-PD-L1 or isotype treatments. (a) n = 5 mice in the isotype group and n = 4 in the anti-PDL1 group. The experiment is representative of n = 18 isotype and n = 16 anti-PDL1 mice examined over three experiments. (c) n = 5 mice in the PBS group and n = 7 mice in the DT group. The experiment is representative of n = 8 mice in the isotype and n = 11 mice in the DT group examined over two experiments. (d–g) n = 5 mice per group.The experiment is representative of n = 15 mice per group examined over three experiments. *: p < 0.05; (unpaired, two-tailed Student’s t-test (a, c, d, f). Box plots indicate the median, upper and lower quartile, and the whiskers show the high and low value.
Extended Data Fig. 5 |. Single cell transcriptomic analyses of virus-specific CD4+ T cells following PD-L1 blockade.
(a) Serum Viral Titers in mice prior and post 3 anti-PD-L1 treatments. Line graph indicates average +/−SD of four mice per group. Data are representative of 1 single cell experiment with a total of n = 4 mice per group. Unpaired t-test was used to compare isotype pre and post therapy, and between PDL1 pre and post therapy. (b, c) Data are from one scRNA-seq experiment comprised of pooled cells from four individual mice per group. SMARTA cells were FACS-sorted following the third antibody treatment and analyzed by scRNA-seq. (b) Bar graph depicts differential expression of genes in sorted SLAMF1+ TH1 versus CXCR5+ TFH SMARTA cells from bulk RNA-seq at day 7 after LCMV-Armstrong infection. (c) UMAPs show IFNγ and IL10 gene expression.
Extended Data Fig. 6 |. Pathway analysis of CD4+ SMARTA T cells following PD-L1 blockade.
(a) Histograms depict Log10 transformed RNA expression of the ISGs Irf7 and Oas1a, and TH1-related genes Ifngr1 and Ccl5 in total virus-specific SMARTA CD4+ T cells, in the TH1 c2, c3, and in the TFH c5 from the scRNA-seq data from Fig. 5. Numbers in each histogram represent the percent of cells expressing the RNA. Blue histograms are anti-PD-L1 treated and those outlined in black are isotype treated. (b) Box plots indicate the GMFI of Nur77 protein expression in SMARTA cells 60 h after the first isotype (n = 5) or anti-PD-L1 (n = 6) antibody treatment. Data are representative of two experiments totaling 11 isotype and 12 anti-PD-L1 treated mice. *: p < 0.05, unpaired, two-tailed Student’s t-test. (c) The graph shows gene set enrichment analysis (GSEA) of differentially expressed pathways upregulated in CD4+ SMARTA T cells in anti-PD-L1 treatment vs isotype treatment. Pathways are colored by normalized enrichment score (NES). Data are representative of one scRNA-seq experiment comprised of pooled cells from four individual mice per group (Isotype or anti-PD-L1).
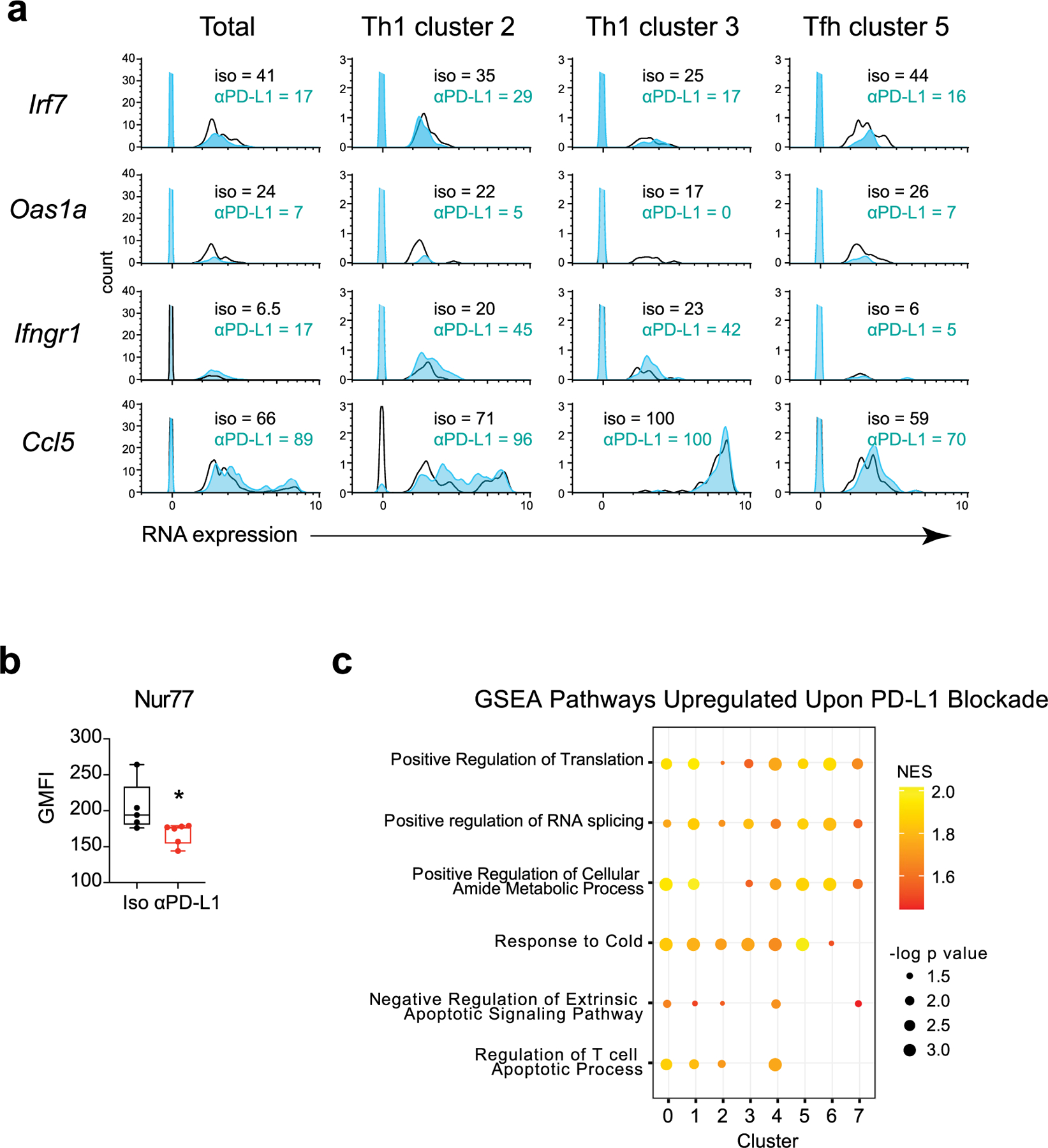
Extended Data Fig. 7 |. Cytotoxic gene expression in all SMARTA clusters from single cell analysis.
Violin plots depict expression of genes associated with cytotoxic function in all clusters following isotype and anti-PD-L1 treatment. Data are representative of one scRNA-seq experiment comprised of pooled cells from four individual mice per group.
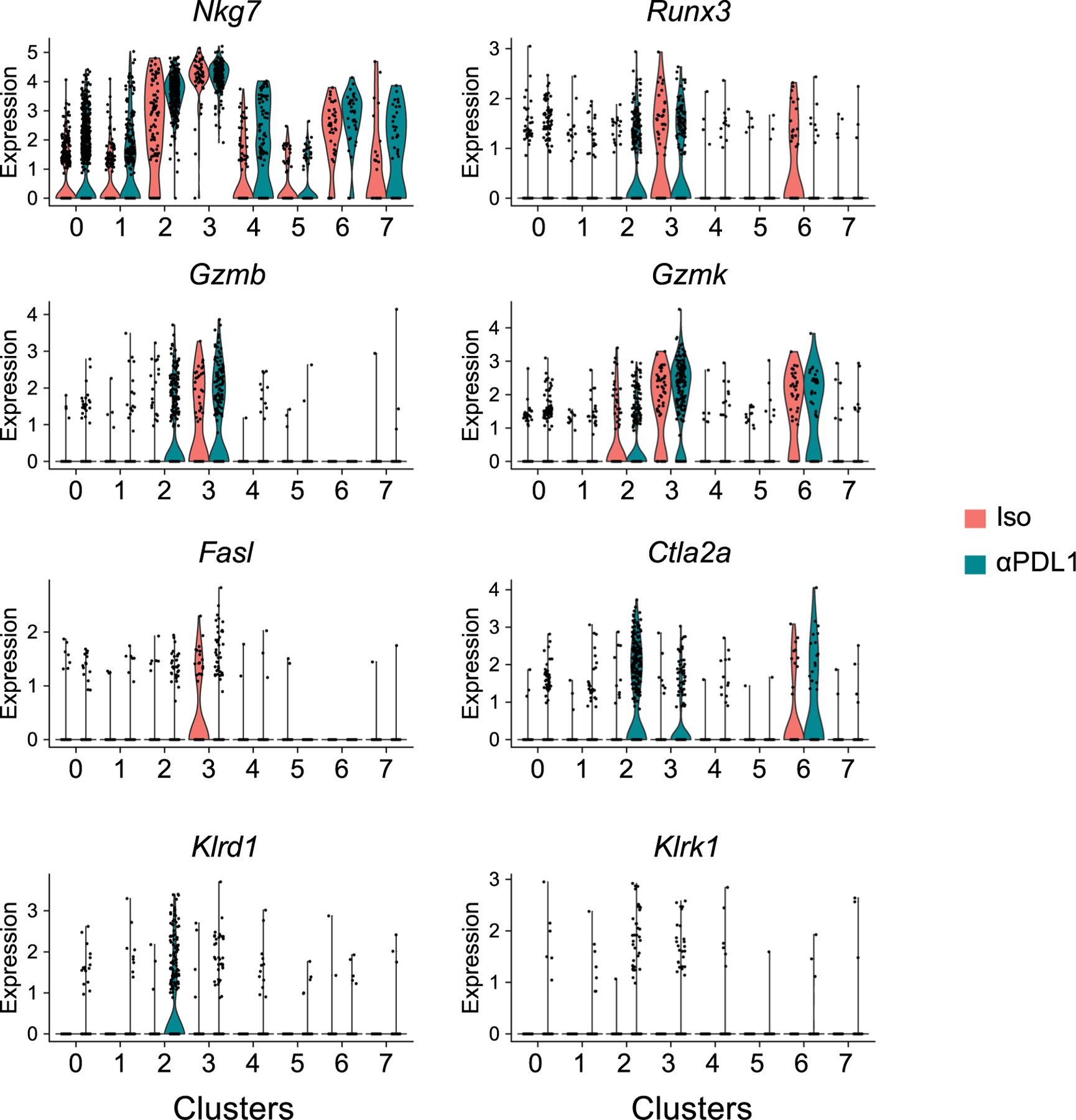
Extended Data Fig. 8 |. Flow gating scheme for sorting.
Plots show the successive gating scheme for the sorting of SMARTA CD4 + T cells.
Supplementary Material
Acknowledgements
We thank past and present members of the Brooks laboratory for technical help and discussion. This work was supported by the Canadian Institutes of Health Research (CIHR) Foundation Grant FDN148386 (D.G.B.), the National Institutes of Health (NIH) grant AI085043 (D.G.B.), the Scotiabank Research Chair to D.G.B, and a training grant from the Fonds de la Recherche en Santé du Québec (L.M.S.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-021-01060-7.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41590-021-01060-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41590-021-01060-7.
Peer review information Nature Immunology thanks Robert Thimme, Santosha Vardhana and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. L. A. Dempsey was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
The RNA-seq data generated in this paper have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE163345. Source data are provided with this paper.
References
- 1.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Fahey LM et al. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J. Exp. Med 208, 987–999 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrovas C et al. CD4 T follicular helper cell dynamics during SIV infection. J. Clin. Invest 122, 3281–3294 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindqvist M et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Invest 122, 3271–3280 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng J et al. Patients with chronic hepatitis C express a high percentage of CD4(+)CXCR5(+) T follicular helper cells. J. Gastroenterol 47, 1048–1056 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Snell LM et al. Overcoming CD4 Th1 cell fate restrictions to sustain antiviral CD8 T cells and control persistent virus infection. Cell Rep 16, 3286–3296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He R et al. Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature 537, 421–428 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Utzschneider DT et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siddiqui I et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Kurtulus S et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(−)CD8(+) tumor-infiltrating T cells. Immunity 50, 181–194.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller BC et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roemer MGM et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J. Clin. Oncol 36, 942–950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cader FZ et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med 26, 1468–1479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei SC et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang AC et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks DG, McGavern DB & Oldstone MB Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J. Clin. Invest 116, 1675–1685 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elsaesser H, Sauer K & Brooks DG IL-21 is required to control chronic viral infection. Science 324, 1569–1572 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks DG, Teyton L, Oldstone MB & McGavern DB Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol 79, 10514–10527 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu L et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat. Immunol 16, 991–999 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Choi YS et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat. Immunol 16, 980–990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parish IA et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J. Clin. Invest 124, 3455–3468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angerer P et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Macleod BL et al. A network of immune and microbial modifications underlies viral persistence in the gastrointestinal tract. J. Exp. Med 217, e20191473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson KG et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frohlich A et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 324, 1576–1580 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Yi JS, Du M & Zajac AJ A vital role for interleukin-21 in the control of a chronic viral infection. Science 324, 1572–1576 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan O et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabrysova L et al. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4(+) T cells. Nat. Immunol 19, 497–507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson EB et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teijaro JR et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Au-Yeung BB et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc. Natl Acad. Sci. USA 111, E3679–E3688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fazilleau N, McHeyzer-Williams LJ, Rosen H & McHeyzer-Williams MG The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol 10, 375–384 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray JP et al. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity 40, 367–377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine AG et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Penaloza-MacMaster P et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med 211, 1905–1918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollern DP et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell 179, 1191–1206.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang CJ et al. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc. Natl Acad. Sci. USA 112, 524–529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wing JB, Ise W, Kurosaki T & Sakaguchi S Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 41, 1013–1025 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Sage PT, Paterson AM, Lovitch SB & Sharpe AH The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 41, 1026–1039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barber DL et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci. Transl. Med 11, eaat2702 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokosuka T et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med 209, 1201–1217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sledzinska A et al. Regulatory T cells restrain interleukin-2- and Blimp-1-dependent acquisition of cytotoxic function by CD4(+) T cells. Immunity 52, 151–166 e156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oh DY et al. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 181, 1612–1625. e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snell LM et al. CD8+ T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity. Immunity 49, 678–694.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barber DL et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Levine JH et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber LM, Nowicka M, Soneson C & Robinson MD diffcyt: differential discovery in high-dimensional cytometry via high-resolution clustering. Commun. Biol 2, 183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Merico D, Isserlin R, Stueker O, Emili A & Bader GD Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Godec J et al. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity 44, 194–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinsella RJ et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011, bar030 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yates A et al. Ensembl 2016. Nucleic Acids Res 44, D710–D716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, Pyl PT & Huber W HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCarthy DJ, Chen Y & Smyth GK Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Griffith M, Walker JR, Spies NC, Ainscough BJ & Griffith OL Informatics for RNA sequencing: a web resource for analysis on the cloud. PLoS Comput. Biol 11, e1004393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wickham H ggplot2: Elegant Graphics for Data Analysis (Springer, 2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data generated in this paper have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE163345. Source data are provided with this paper.