

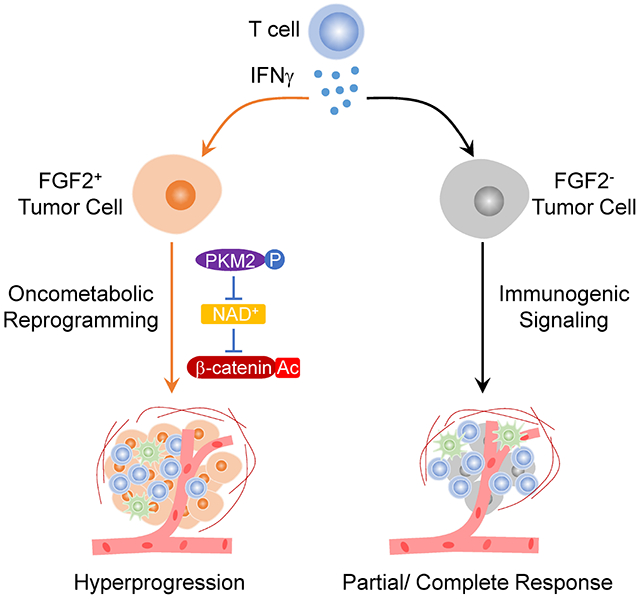
Summary
Immune checkpoint blockade (ICB) can produce durable responses against cancer. We and others have found that a subset of patients experience paradoxical rapid cancer progression during immunotherapy. It is poorly understood how tumors can accelerate their progression during ICB. In some preclinical models, ICB causes hyperprogressive disease (HPD). While immune exclusion drives resistance to ICB, counterintuitively, patients with HPD and complete response (CR) following ICB manifest comparable levels of tumor infiltrating CD8+ T cells and IFNγ–gene signature. Interestingly, patients with HPD but not CR exhibit elevated tumoral FGF2 and β-catenin signaling. In animal models, T cell-derived IFNγ promotes tumor FGF2 signaling, thereby suppressing PKM2 activity and decreasing NAD+, resulting in reduction of SIRT1-mediated β-catenin deacetylation and enhanced β-catenin acetylation, and consequently reprograming tumor stemness. Targeting the IFNγ-PKM2-β-catenin axis prevents HPD in preclinical models. Thus, the crosstalk of core immunogenic, metabolic, and oncogenic pathways via the IFNγ-PKM2-β-catenin cascade underlies ICB-associated HPD.
Keywords: Hyperprogressive disease, complete response, immune checkpoint blockade, glycolytic metabolism, oncogenesis, IFNγ, FGF2, β-catenin, PD-L1/PD-1 pathway, T-cell immunity
Graphical Abstract
eTOC Blurb
Li et al. uncover a crosstalk between core immunogenic, metabolic, and oncogenic pathways in cancer cells during immunotherapy, which enables hyperprogressive disease (HPD) in preclinical models and correlates with immunotherapy-associated HPD in cancer patients.
Introduction
The molecular determinants of tumor response to immunotherapy are incompletely defined. Immune checkpoint blockade (ICB) therapy unleashes T cell mediated anti-tumor immunity to promote partial or complete responses in a wide variety of cancers 1–4. However, ICB can also result in atypical patterns of response including pseudoprogression, in which an initial increase in tumor size is followed by subsequent clinical benefit from therapy 5. Unfortunately, most cancer patients develop stable or progressive disease on ICB 2. More recently, it has been suggested that initiation of ICB may promote hyperprogressive disease (HPD) 6, which manifests as an acceleration of cancer growth during ICB. Signaling induced by the ICB antibodies as well as amplifications of EGFR or MDM2/4 have been associated with HPD 7. Prior studies have identified T cell exclusion and immune signaling dysfunction as contributors to tumor progression on immunotherapy 1–4. However, the importance of tumoral immune composition to atypical but clinically relevant patterns of response to ICB remains poorly defined.
Interferon (IFN)γ signaling is a key immunogenic pathway and plays a decisive role in spontaneous and ICB-induced anti-tumor immunity 8. Host IFNγ signaling supports tumor antigen presentation, antigen presenting cell (APC) activation, effector T cell recruitment, and directly affects tumor cell proliferation and survival 1. Loss of IFNγ signaling in tumor cells results in immune evasion and resistance to immunotherapy 9–12. However, prolonged IFNγ exposure confers tumor resistance to ICB via multiple mechanisms, including PD-L1 induction and induction of cancer stemness 13–16. Hence, IFNγ signaling can play a dual role in cancer immune responses. However, whether IFNγ can directly promote tumor progression in the context of immunotherapy remains unknown.
An intertwined network of oncogenic and metabolic programs works in tandem to support cancer growth and viability 17–19. Oncogenes, including β-catenin, promote tumor stemness and invasiveness and increase metastatic potential 20. Additionally, β-catenin signaling enhances tumor cell survival by inducing MYC and other genes 21. Consequently, β-catenin signaling can promote resistance to immunotherapy 13,22. Oncogenic programs also upregulate aerobic glycolysis to support cancer progression 23. Furthermore, overexpression of tumor growth stimulatory signals, including epidermal growth factor (EGF) and fibroblast growth factor (FGF), enables unchecked growth 24,25. However, the importance and mechanism of how these oncogenic and metabolic drivers of cancer progression interact with immunogenic signaling induced by ICB therapy is poorly understood.
Here, we explored the interplay between key immunogenic (IFNγ), metabolic (glycolysis), and oncogenic (β-catenin) pathways in suspected ICB-associated HPD in tumor-bearing murine models and patients with cancer. We discover that core immunogenic, metabolic, and oncogenic pathway crosstalk provides a cellular and molecular basis for ICB-associated HPD. Our work suggests that targeting this crosstalk may prevent suspected iatrogenic cancer progression in patients receiving ICB.
Results
Rapid cancer progression occurs in a subset of patients during immunotherapy
Immunotherapy has altered the landscape of cancer treatment. It is not fully understood whether there are differences in clinical response to immunotherapy as compared to established cancer therapy. To explore this, we examined a cohort of metastatic melanoma patients treated with ICB or targeted therapy at the University of Michigan (Cohort 1, Table S1, 389 patients). ICB improved overall survival of patients when compared to targeted therapy (Figure 1A). Surprisingly, a subset of patients treated with immunotherapy progressed rapidly within three months as compared to those who received targeted therapy (Figure 1A, inset). Similarly, we noted that though ICB improved the progression-free survival of patients with metastatic melanoma (Figure 1B), a subset of patients rapidly progressed after receipt of ICB compared to targeted therapy (Figure 1B, inset). To extend our studies, we examined a cohort of non-small cell lung carcinoma (NSCLC) patients treated with ICB or chemotherapy at our institution (Cohort 2, Table S2, 375 patients). Receipt of ICB was also associated with inferior overall and progression-free survival at three months in NSCLC patients (Figures 1C and 1D). To substantiate this finding, we performed propensity-score matched multivariable modeling. After controlling for clinicopathologic variables, inferior overall survival, and progression-free survival (Figures S1A–S1D) remained in melanoma and NSCLC patients treated with ICB at three months as compared to other systemic therapies. To externally validate these observations, we conducted a pooled analysis of the prospective, randomized controlled trials which established the superiority of ICB over chemotherapy in metastatic NSCLC 26–28. Again, receipt of ICB was associated with inferior initial progression-free survival at three months (Figure 1E). This inferior progression-free survival was observed regardless of whether anti-PD-1 (pembrolizumab), anti-PD-L1 (atezolizumab), or anti-CTLA-4 and anti-PD-1 (ipilimumab and nivolumab) were utilized (Figure 1F). Progression within three months was associated with significantly inferior overall survival in melanoma and NSCLC patients receiving ICB (Figures 1G and 1H). Together, these data indicate that rapid cancer progression can occur in a subset of cancer patients during immunotherapy.
Figure 1: Rapid cancer progression occurs in a subset of patients during immunotherapy.
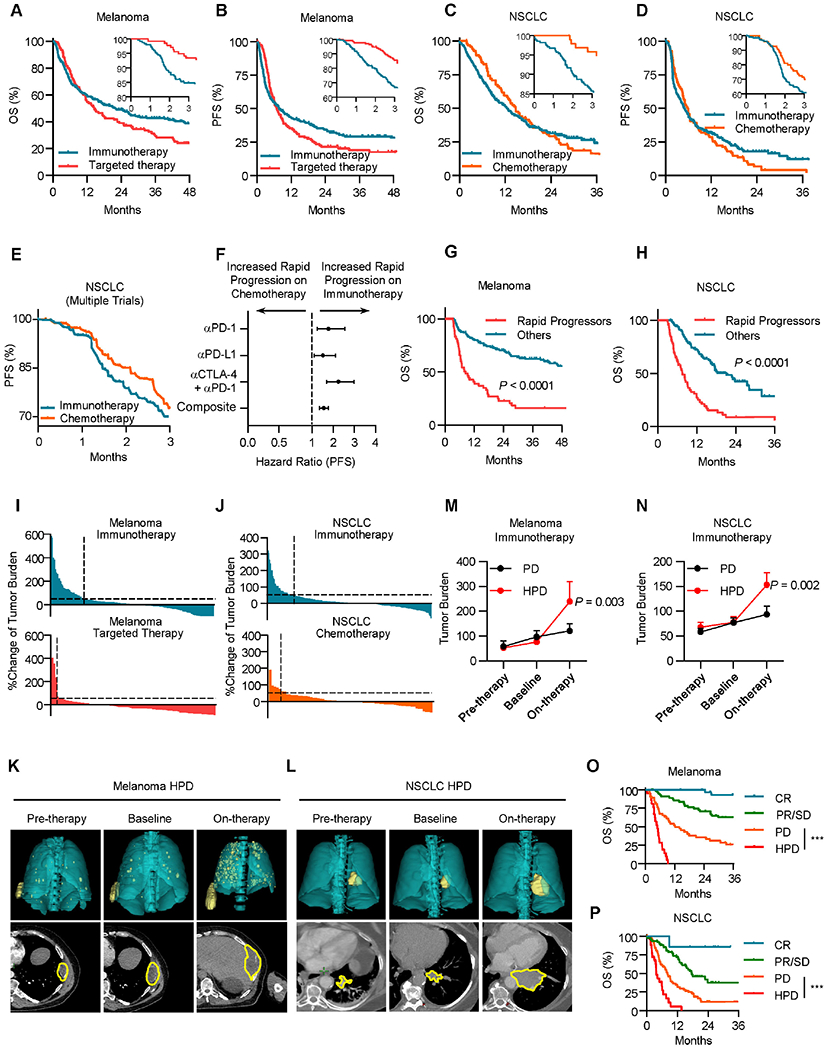
A, Overall survival (OS) of patients with metastatic melanoma (Cohort 1) stratified by therapy type; inset, 3-month OS; immunotherapy n = 251, targeted therapy n = 138, Restricted mean survival time (RMST) at 3 months, Hazard ratio (HR) = 0.95, P < 0.0001 by log-rank test.
B, Progression-free survival (PFS) of patients with metastatic melanoma stratified by therapy type. Inset, 3-month PFS. Progression-free RMST at 3 months HR = 0.89, immunotherapy n = 251, targeted therapy n = 138, P < 0.0001 by log-rank test.
C, OS of patients with metastatic NSCLC stratified by therapy type; Inset, 3-month OS; immunotherapy n = 279, chemotherapy n = 96, RMST at 3 months, HR = 0.94, P < 0.0001 by log-rank test.
D, PFS of patients with metastatic NSCLC stratified by therapy type. Inset, 3-month PFS; immunotherapy n = 279, chemotherapy n = 96, Progression-free RMST at 3 months, HR = 0.94, P < 0.0001 by log-rank test.
E, PFS of metastatic NSCLC patients treated with immunotherapy or chemotherapy, pooled analysis of Keynote-042, Poplar, and Checkmate 227 randomized control trials. Progression-free Log-rank HR at 3 months = 0.616, P = 0.0336.
F, Hazard ratios for 3-month PFS of metastatic NSCLC patients treated with immunotherapy or chemotherapy, pooled analysis of Keynote-042, Poplar, and Checkmate 227 randomized control trials. Two-sided t test, P = 0.0152.
G, OS of patients with metastatic melanoma treated with ICB (Cohort 1) stratified by timing of progression, other n = 146, rapid progression (PFS < 3 months) n = 53, Landmark analysis (3 months) hazard ratio (HR) = 0.291, P < 0.0001 by log-rank test.
H, OS of patients with metastatic NSCLC treated with ICB (Cohort 2) stratified by timing of progression, other n = 113, rapid progression (PFS < 3 months) n = 67, Landmark analysis (3 months) hazard ratio (HR) = 0.3251, P < 0.0001 by log-rank test.
I, Waterfall plot showing change of tumoral burden from initiation of therapy to first surveillance imaging in melanoma patients treated with indicated therapy, dotted line > 50% increase in tumor burden, immunotherapy n = 200, targeted therapy n = 96, Chi-square = 19.53, P < 0.0001. Data are shown as percentage change.
J, Waterfall plot showing change of tumoral burden from initiation of therapy to first surveillance imaging in NSCLC patients treated with indicated therapy, dotted line > 50% increase in tumor burden, immunotherapy n = 212, chemotherapy n = 68, Chi-square = 5.133, P = 0.0235. Data are shown as percentage change.
K-L, Representative cross-sectional (lower) and 3D reconstructed (upper) computed tomography (CT) images of a patient with metastatic melanoma (K) and a patient with NSCLC (L) with HPD preceding receipt of immunotherapy (left), at baseline preceding immunotherapy (middle), and at first reassessment following immunotherapy (right).
M-N, Longitudinal tumor burden assessment in melanoma (M) or NSCLC (N) patients who progressed while receiving ICB stratified by pattern of response. Baseline- cross sectional imaging immediately prior to ICB initiation. Pre-therapy- Imaging assessment prior to baseline evaluation. On therapy- next surveillance scan after baseline assessment. Melanoma patients with PD (progressive disease, per RECIST 1.1, n = 48) and HPD (hyperprogressive disease, per Champiat et al., n = 21); NSCLC patients with PD (n = 77) and HPD (n = 26), interrupted time series regression, Data are shown as mean ± s.d., P value indicated.
O. OS of metastatic melanoma patients (Cohort 1) stratified by best response, complete response (CR) n = 31, partial/stable disease (PR/SD) n = 58, progressive disease (PD) n = 48, and hyperprogressive disease (HPD) n = 21. Log-rank test, HPD vs PD HR = 0.3058, ***P < 0.001.
P. OS of metastatic NSCLC patients (Cohort 2) stratified by best response, CR n = 7, PR/SD n = 77, PD n = 77, and HPD n = 26. Log-rank test, HPD vs PD HR = 0.25, ***P < 0.001.
Next, we evaluated the patterns of early/initial radiographic response to cancer therapies in patients with metastatic melanoma and NSCLC (Cohorts 1 and 2). imRECIST criteria were utilized to exclude patients with pseudoprogression 29. Quantitative evaluation revealed that a higher proportion of metastatic melanoma had a substantial (> 50%) increase in tumor burden at the time of first surveillance imaging following receipt of ICB as compared to receipt of targeted therapy (Figure 1I). Similarly, in patients with metastatic NSCLC, a subset of patients had a substantial increase in tumor burden after receipt of ICB (Figure 1J). Substantial increases in tumor burden were associated with significantly inferior overall survival in melanoma and NSCLC patients receiving ICB (Figures S1E and S1F). These data collectively suggest that ICB is associated with a rapid and substantial increase in tumor burden in a subset of cancer patients.
Multiple groups have suggested that cancer hyperprogression may occur following receipt of immunotherapy 30. Tumor growth rate is a validated quantification of tumor kinetics over time 31. We observed that a proportion of patients had an acceleration of their tumor growth rate after ICB, targeted therapy, or chemotherapy in melanoma and NSCLC (Figures S1G and S1H). We next applied previously reported definitions for HPD whose criteria include rapid time to failure, an increase in tumor burden, and tumor growth acceleration, to our clinical cohorts (Table S3) 6,7,32–38. HPD occurred in ~11% patients with metastatic melanoma following receipt of ICB versus ~2% of patients following targeted therapy (Figure S1I). Likewise, we observed HPD in ~13% of patients with NSCLC following receipt of ICB and ~8% of patients following receipt of chemotherapy (Figure S1J). These proportions were similar regardless of which previously published definition of HPD was used 6,7,32–38. These data collectively suggest a small subset of cancer patients may experience rapid progression upon immunotherapy.
HPD could represent unchecked intrinsic cancer growth in the face of ineffective therapy, or a paradoxical acceleration of cancer progression induced by therapy. To evaluate these possibilities, we examined serial radiographic cross-sectional images from the period preceding and following ICB initiation in patients with metastatic melanoma and NSCLC. Cross-sectional and reconstructed 3D imaging showed that a subset of patients had significant increases in tumor burden after receipt of ICB (Figures 1K, 1L, S1K and S1L). Longitudinal quantification of tumor burden demonstrated that a subset of melanoma and NSCLC patients had significant increases in tumor burden following but not preceding initiation of ICB as shown in individual patient growth curve (Figures S1M and S1N) as well as the composite values (Figures 1M and 1N). To further understand whether these patients with potential HPD differed from patients with progressive disease (PD) as defined by imRECIST, we stratified patients by their radiographic response and compared their disease burden longitudinally. We observed that unlike patients with PD, patients with HPD had dramatic increases in their tumor burden after receipt of ICB (Figures 1M and 1N). Interrupted time series analysis confirmed patients with HPD were on a distinct disease trajectory after initiation of ICB as compared to patients with PD. Metastatic melanoma and NSCLC patients with HPD had significantly diminished overall survival following receipt of ICB when compared to patients with PD (Figures 1O and 1P). In patients with metastatic melanoma, HPD following immunotherapy occurred regardless of the age, gender, performance status, lines of prior therapy, receipt of single or dual ICB, melanoma histologic subtype, or BRAF mutational status (Figure S1O). In patients with metastatic NSCLC, HPD following ICB occurred regardless of age, gender, performance status, smoking status, histology, and receipt of immunotherapy alone or in combination with chemotherapy (Figure S1P). Of note, ICB-associated HPD was not associated with increased tumor growth rates in the pretreatment period (Figures S1Q and S1R). These data suggest cancer may accelerate in a minority of patients following immunotherapy.
Immunogenic and oncogenic pathways correlate in patients with HPD
To investigate the molecular underpinnings of HPD in patients, we identified a cohort of patients who underwent comprehensive tumor and somatic sequencing, received ICB, and had evaluable cross-sectional imaging at our institution (Cohort 3, Table S4) 39. Through longitudinal radiographic quantitation of tumor burden, we identified patients who had a complete response (CR) as well as those patients who developed HPD in response to ICB (Figures S2A–S2D). Survival analysis confirmed that radiographic response to ICB impacted overall survival (Figure S2E). Limited IFNγ signaling and insufficient T cell infiltration are known to be associated with tumor progression and resistance to ICB 1,2,4. We hypothesized that limited T cell responses would characterize the tumor immune microenvironment in patients who subsequently developed HPD. Unexpectedly, patients with CR and patients with HPD demonstrated comparable levels of IFNγ and IRF1 as well as similar T cell clonal diversity, number of TCR clones, number of TCR reads, and CD8+ T cell infiltration (Figure 2A). Regulatory T cells, myeloid dendritic cells (DCs), and macrophages mediate immunosuppression in the tumor microenvironment via multiple mechanisms, such as the PD-L1-PD-1 pathway 40,41. Surprisingly, the levels of PDCD1 (PD-1), FOXP3, and CD68 were also comparable between patients who had CR and HPD in response to ICB (Figure 2A). To orthogonally confirm this observation, we evaluated CD8+ T cells in tumors in metastatic melanoma (Cohort 1) and NSCLC patients (Cohort 2) who had available tissues. Multiplex immunohistochemistry showed comparable levels of tumor CD8+ T cell infiltration between CR and HPD patients with melanoma (Figures 2B and 2C) and NSCLC (Figures 2D and 2E). These data indicate that patients with HPD are unexpectedly not immune excluded.
Figure 2: Immunogenic and oncogenic pathways correlate in patients with HPD.
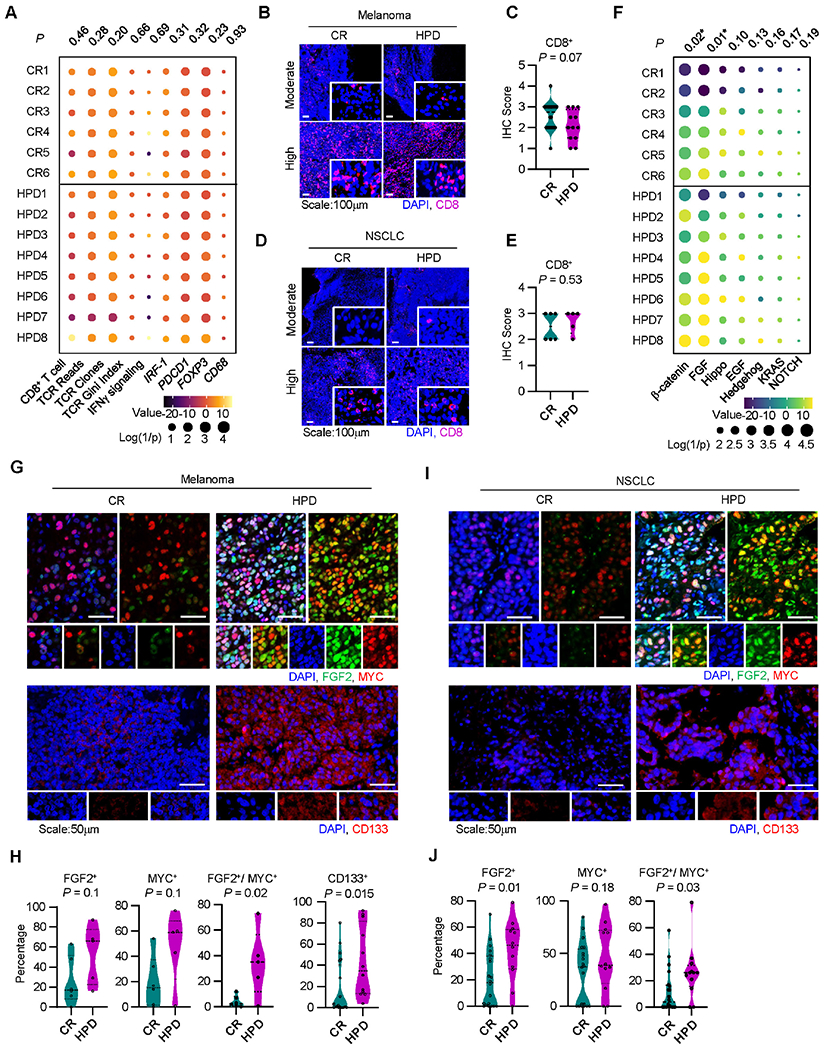
A. Immune gene signature analysis of patients receiving immunotherapy who developed a complete response (CR) or hyperprogressive disease (HPD) per Champiat et al. in Cohort 3, individual patients are shown; P-values were generated from multivariate mixed effect linear models controlling for biopsy site (fixed effect) and disease type (random effect).
B-C. Representative immunofluorescence staining (B) and quantitation (C) for baseline tumor infiltrating CD8+ T cells in melanoma patients with indicated response to therapy. Frequency of positive cells is shown; CR, n = 20, HPD, n =12. Two-sided t-test.
D-E. Representative immunofluorescence staining (D) and quantitation (E) for baseline tumor infiltrating CD8+ T cells in NSCLC patients with indicated response to therapy. Frequency of positive cells is shown; CR, n = 20, HPD, n =12. Two-sided t-test.
F. Oncogenic gene signature analysis of patients receiving immunotherapy who developed a complete response (CR) or hyperprogressive disease (HPD) in Cohort 3, individual patients are shown; P-values were generated from multivariate mixed effect linear models controlling for biopsy site (fixed effect) and disease type (random effect).
G-J. Multiplex immunofluorescence staining was conducted in tumor tissues from patients with melanoma (G, H) and NSCLC (I, J). Representative images showed FGF2, MYC, and CD133 expressing tumor cells in patients with HPD and CR (G, I). Percentages of single or double positive tumor cells are shown in patients with HPD and CR (H, J). Mean and interquartile range shown. Melanoma patients with CR (n = 20) and HPD (n =12); NSCLC patients with HPD (n = 5) and CR (n = 6). Two-sided t-test.
Given the lack of significant differences in the immune composition of patients who responded completely versus those who developed HPD following receipt of ICB, we then evaluated common oncogenic pathways 18. Gene signatures for several common oncogenic signaling pathways, including Sonic Hedgehog, Hippo, KRAS, NOTCH, and EGF were similarly expressed in patients with HPD and CR (Figure 2F). Interestingly, we found that FGF2 and Wnt-β-catenin gene signatures were highly expressed in patients who developed HPD as compared to patients who underwent a CR (Figures 2F and S2F). We detected comparable levels of expression of EGFR, MDM2, and MDM4, but higher levels of tumor stemness and invasiveness in HPD as compared to CR (Figure S2G). We found one case with BRAF mutation and no cases with MDM2, MDM4, and EGFR amplification in this cohort (Figure S2H). The results suggest that FGF2, Wnt-β-catenin, and stemness/invasiveness pathways may be activated in patients with HPD. To validate this finding, we examined FGF2 and MYC, surrogates for FGF signaling and β-catenin signaling, in tumors from patients with melanoma and NSCLC who had CR or HPD following receipt of ICB. Multiplex immunohistochemistry revealed nuclear FGF2 indicative of active FGF2 signal transduction 42 and MYC expression (Figure 2G, upper panel). Quantitation revealed higher levels of FGF2+MYC+ melanoma cells in patients with a HPD phenotype as compared to patients with a CR phenotype (Figure 2H). Similar results were obtained in patients with NSCLC (Figure 2I, upper panel and Figure 2J). Furthermore, we performed multiplex immunofluorescence staining for CD133, a marker for stemness in cancer tissues from patients with CR and HPD. There was higher frequency of CD133high tumor cells (Figures 2G, 2I, lower panels, and Figure 2H) and FGF2highCD133high tumor cells (Figures S2I and S2J) in HPD patients as compared to CR patients. The data suggest that immunotherapy-associated HPD is associated with activation of the FGF2 and β-catenin oncogenic pathways as well as increased tumor stemness.
CD8+ T cells drive cancer hyperprogression via IFNγ
Preclinical models mimicking ICB-triggered HPD have not been developed. To resolve if ICB may trigger HPD in melanoma, we sought to establish a melanoma murine model with HPD features by testing 4 cell lines: B16-F0, B16-F10, YUMM1.7, and YUMM5.2. We inoculated the 4 cell lines into C57BL/6 mice and treated them with anti-PD-L1 mAb (αPd-l1). αPD-L1 therapy showed minimal anti-tumor effect in mice bearing B16 melanoma 43,44 and YUMM5.2 tumor (Figure S3A). To our surprise, tumors progressed faster in YUMM1.7 melanoma-bearing mice under ICB (Figure 3A). In line with this, αCTLA-4 treatment also promoted YUMM1.7 tumor growth (Figure S3B). αPD-L1 resulted in increased tumor T cell infiltration in YUMM1.7 model (Figures 3B and 3C) as well as enhanced multiple tumor stemness- and invasiveness-associated genes, such as Myc and Cd44 proteins (Figures 3D) and transcripts (Figure 3E). In contrast, αPD-L1 therapy did not modulate these genes in YUMM5.2 model (Figure S3C). Notably, previous studies have revealed inconsistent efficacy of ICB in YUMM1.7 tumor bearing mice 45,46. To elucidate the necessity of immune activation as demonstrated in cancer patients (Figure 2) and recapitulate tumor stemness potential in vivo (Figures 1 and 2), we inoculated a limited number of tumor cells into animals and initiated treatment at an early time point.
Figure 3: CD8+ T cells drive cancer hyperprogression via IFNγ. See also Figure S3.
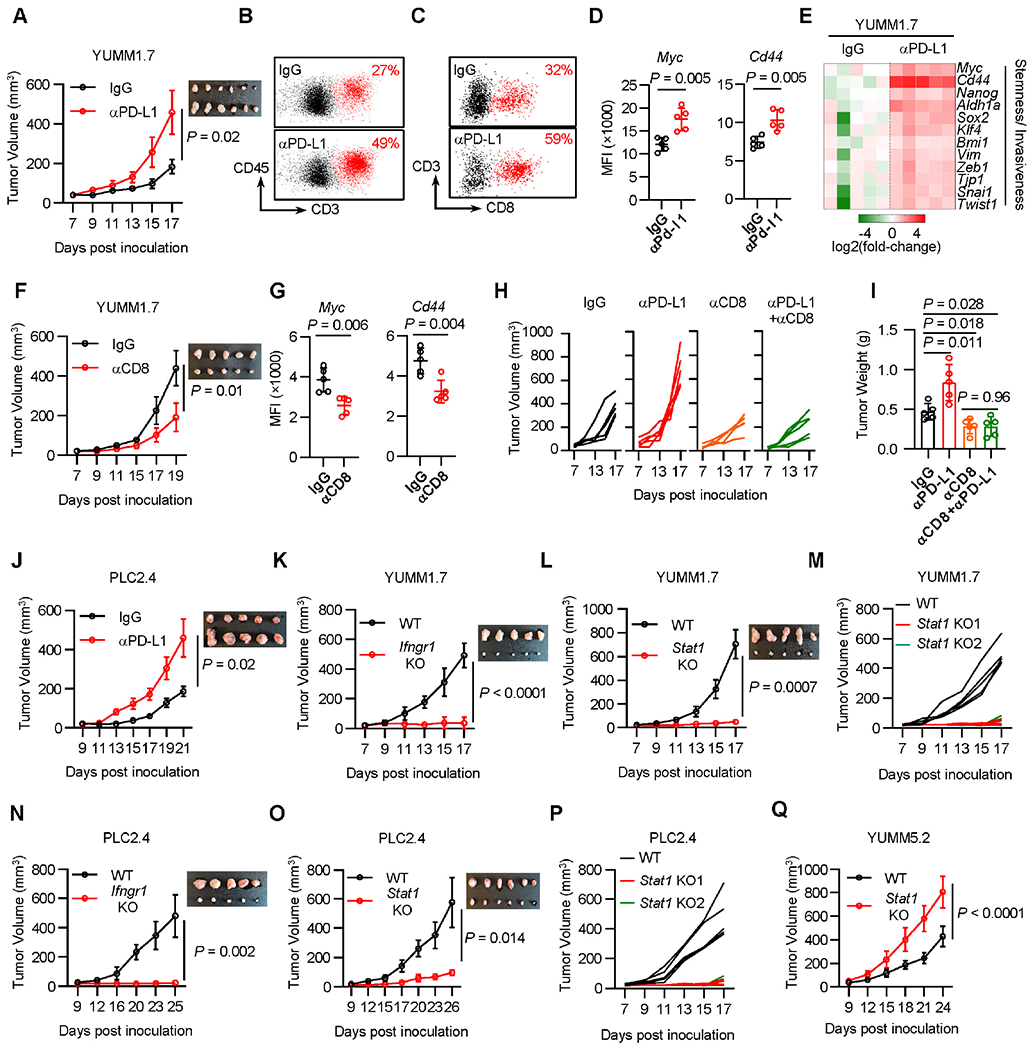
A-D. YUMM1.7 tumor bearing C57BL/6 mice were treated with control (IgG) or PD-L1 antibody. Tumor growth curves were plotted (A). FACS analysis showed tumor T cell infiltration (B, C) and tumor Myc and Cd44 expression (D). MFI, mean fluorescence intensity.
E. YUMM1.7 tumor bearing C57BL/6 mice were treated with control (IgG) or PD-L1 antibody. Day 14th after tumor inoculation, the indicated genes expression in tumors was determined by qPCR.
F-G. YUMM1.7 tumor bearing C57BL/6 mice were treated with control (IgG) or CD8 antibody. Tumor growth curves were plotted (F), and tumor Myc and Cd44 expression (MFI) were determined by FACS (G).
H-I. YUMM1.7 tumor bearing mice were treated with control (IgG), PD-L1 antibody, CD8 antibody, or the combination of PD-L1 and CD8 antibodies. Tumor growth curves were plotted (H) and end point tumor weight (I) were scaled.
J. PLC2.4 cells were inoculated in C57BL/6 wild type mice. Mice were treated with control (IgG) or PD-L1 antibody. Tumor growth curves were plotted.
K-L. Wild type and Ifngr1 KO (K) or Stat1 KO (L) YUMM1.7 cells were inoculated in C57BL/6 mice. Tumor growth curves were plotted.
M. Wild type or 2 single clones of Stat1 KO YUMM1.7 cells were inoculated in C57BL/6 mice. Tumor growth curves were plotted.
N-O. PLC2.4 wild type and Ifngr1 KO (N) or Stat1 KO (O) cells were inoculated in C57BL/6 wild type mice. Tumor growth curves were plotted, n = 5 (N), n = 6 (O).
P. Wild type or 2 single clones of Stat1 KO PLC2.4 cells were inoculated in C57BL/6 mice. Tumor growth curves were plotted.
Q. Wild type or Stat1 KO YUMM5.2 cells were inoculated in C57BL/6 mice. Tumor growth curves were plotted.
In all panels n=5 unless otherwise indicated. Data are shown as mean ± s.d., two-tailed t-test.
See also Figure S3.
As melanoma from HPD patients harbored activated CD8+ T cells (Figure 2), we wondered whether CD8+ T cells unexpectedly support tumor progression in the YUMM1.7 melanoma-bearing model. We treated YUMM1.7 melanoma-bearing mice with a CD8-depleting monoclonal antibody (αCD8) (Figure S3D). We observed that αCD8 slowed YUMM1.7 tumor progression (Figure 3F) and reduced tumoral Myc and Cd44 expression (Figures 3G and S3E). The data suggests that CD8+ T cells may activate oncogenic pathways in tumor cells. We further tested this possibility in the setting of ICB. We treated YUMM1.7 tumor-bearing mice with αPD-L1, αCD8, or the combination of αPD-L1 and αCD8. Again, αPD-L1 promoted tumor growth, but failed to do so when CD8+ T cells were depleted, as determined by tumor growth measurements (Figure 3H) and tumor weights (Figure 3I). These data suggest that ICB may enhance tumor growth in a CD8+ T cell-dependent manner.
To explore how CD8+ T cells activate oncogenic pathways, we cultured YUMM1.7 cells with activated CD8+ T cells or activated T cell media (TCM). T cells (Figure S3F) and TCM (Figure S3G) induced the expression of Myc in YUMM1.7 cells in a dose-dependent manner. Moreover, TCM promoted YUMM1.7 tumor sphere formation as compared to control media (Figure S3H). The data suggests that CD8+ T cell-derived factor(s) may stimulate tumorigenesis. IFNγ is a key effector cytokine released by CD8+ T cells which signals through the IFNγ receptor 1 (IFNGR1) and induces STAT1 phosphorylation 8. We hypothesized a potential role of IFNγ in inducing T cell-mediated tumorigenesis. To this end, we established Ifngr1 knock out (KO) YUMM1.7 cells and treated them with TCM. We found that TCM induced the expression of Myc and Cd44 in wild type (WT) YUMM1.7 cells, but not in Ifngr1 KO YUMM1.7 cells (Figures S3I and S3J). The data suggests that T cell-derived IFNγ may promote tumorigenesis. As a validation, we knocked out Stat1, the transcription factor responsible for IFNγ signaling, in YUMM1.7 cells. IFNγ strongly induced Cd44 expression and tumor sphere formation in YUMM1.7 WT cells, but not in Stat1 KO cells (Figures S3K and S3L). The data indicate that CD8+ T cell derived IFNγ signaling may promote tumor stemness, thereby enhancing tumorigenic potential. To solidify this finding, we examined the effect of recombinant IFNγ on YUMM1.7 and YUMM5.2 cancer cells in vitro. Treatment with IFNγ induced more spheres (Figures S3M and S3N), higher expression of stemness markers, including Cd44 and Cd133 (Figures S3O and S3P) on cancer cell membrane, and multiple stemness gene transcripts (Figure S3Q) in YUMM1.7 cells, but not in YUMM5.2 cells. Furthermore, we tested whether the effect of IFNγ on tumor stemness depended on tumor cell density. We cultured YUMM1.7 cells at different densities and observed that IFNγ strongly induced Myc and Cd44 expression in YUMM1.7 cells cultured at low density (10-30%), but not at high density (> 60%) (Figures S3R and S3S). Similar results were obtained in YUMM1.7 cell sphere formation at low density (Figures S3T and S3U). The data suggest that different melanoma cancer models, such as YUMM1.7 and YUMM5.2, can differentially respond to IFNγ and/or ICB.
To extend our observation to a lung cancer model, we inoculated Lewis lung carcinoma cells (LLC) into C57BL/6 mice and treated these mice with αPD-L1. In line with our previous results 44, LLC bearing mice were resistant to ICB. We isolated multiple tumor clones (PLC1.1-1.4 and PLC2.1-2.4) from mice bearing progressive LLCs (PLCs) and treated them with IFNγ. IFNγ stimulated Myc expression in all PLC clones (Figure S3V). We inoculated PLC2.4 cells into C57BL/6 mice and treated these mice with αPD-L1. Again, checkpoint blockade induced tumor progression (Figure 3J). Altogether, the results reveal an oncogenic role of IFN signaling in some mouse tumor models. We then stimulated multiple human melanoma and lung cancer cell lines with IFNγ. IFNγ was able to induce MYC expression in a minority of the cancer cell lines we examined (Figures S3W and S3X). To extend these findings, we inoculated A375 human melanoma cells into NSG mice and treated these mice with recombinant human IFNγ and observed that treatment with IFNγ supported tumor progression (Figure S3Y).
To determine a direct role of the IFNγ signaling pathway in YUMM1.7 tumor progression in vivo, we pooled 3 different Ifngr1 KO YUMM1.7 clones and 3 different Stat1 KO YUMM1.7 clones and inoculated these KO clones and wild type cells into wild type C57BL/6 mice. We observed that WT tumors progressed rapidly on treatment while Ifngr1 KO (Figure 3K) or Stat1 KO (Figure 3L) tumors grew slowly. Similar results were obtained when individual Stat1 KO YUMM1.7 cell clones were studied in vivo (Figure 3M). The data indicates that IFNγ signaling facilitates YUMM1.7 tumor progression in vivo. Moreover, pooled Ifngr1 KO (Figure 3N) or Stat1 KO (Figure 3O) PLC2.4 tumors progressed more slowly when compared to wild-type PLC2.4 tumors in vivo. Similar results were observed in individual Stat1 KO PLC2.4 clones (Figure 3P). Thus, IFNγ signaling promotes tumor growth in PLC2.4 model. As an additional control, we generated Stat1 KO YUMM5.2 cells and performed similar in vivo experiments. Contrary to the results from the Stat1 KO YUMM1.7 tumor model, the Stat1 KO YUMM5.2 tumors grew faster than their wild type counterparts (Figure 3Q). Thus, IFNγ signaling may promote tumor progression in a subset of preclinical models.
Altogether, the data suggest that in a small subset of cancer models, IFN signaling may activate oncogenic pathways and ICB may promote rapid tumor progression via the T cell-IFNγ signaling-activated oncogenic pathway.
IFNγ reduces NAD+ to activate β-catenin acetylation
We then explored the oncogenic pathway(s) activated by IFNγ and ICB. We analyzed the established oncogenic signaling genes in the lung cancer TCGA datasets 18. We found that IFNγ signaling positively and negatively correlated with the β-catenin and NOTCH signaling pathways, respectively. However, there was no correlation between IFNγ signaling pathway and KRAS, Hippo, and Hedgehog signaling pathways (Figure 4A). Furthermore, expression of IRF1 and MYC, target genes in the IFNγ and β-catenin signaling pathways, respectively, correlated positively across multiple cancer types in TCGA datasets (Figure 4B). This evidence, along with our observations on the relationship between CD8+ T cells/IFNγ and oncogenic gene expression in both mouse models and patients (Figure 2 and Figure 3), suggest a potential crosstalk between IFNγ and β-catenin signaling pathways in tumors. In line with this finding, real-time PCR (Figure S4A) and RNA-sequencing data (Figures 4C and 4D) demonstrated that IFNγ activated β-catenin signaling genes in A375 and A549 cells. We tested a role of IFNγ in modulating β-catenin signaling using a reporter. We treated human melanoma cell line A375 with IFNγ. IFNγ treatment induced the luciferase activity of β-catenin signaling reporter TOP-FLASH, but not the control reporter FOP-FLASH (Figure 4E). In line with this, IFNγ treatment resulted in nuclear translocation of β-catenin protein (Figure S4B) as well as induction of MYC, CCND1, VEGFA, and MMP14, classic target genes of β-catenin signaling, (Figures 4C, 4D and S4A). To determine if the increased gene expression was dependent on Wnt/ β-catenin signaling, we cultured A375 and YUMM1.7 cells with the Wnt-β-catenin signaling inhibitors DKK1 and Wnt-C59 in the presence of IFNγ. The two inhibitors diminished IFNγ-mediated expression of β-catenin signaling genes (Figure 4F), formation of tumor spheres (Figure 4G), and surface expression of the stemness markers Cd44 and Cd133 (Figures 4H and 4I). Furthermore, we established β-catenin KO A375 cells (Figure S4B). IFNγ activated the expression of MYC, CCND1, VEGFA, and MMP14 in WT cells, but not in CTNNB1 KO cells. (Figure 4J). Collectively, these data indicate that IFNγ activates β-catenin signaling in tumor cells.
Figure 4: IFNγ reduces NAD+ to activate β-catenin acetylation.
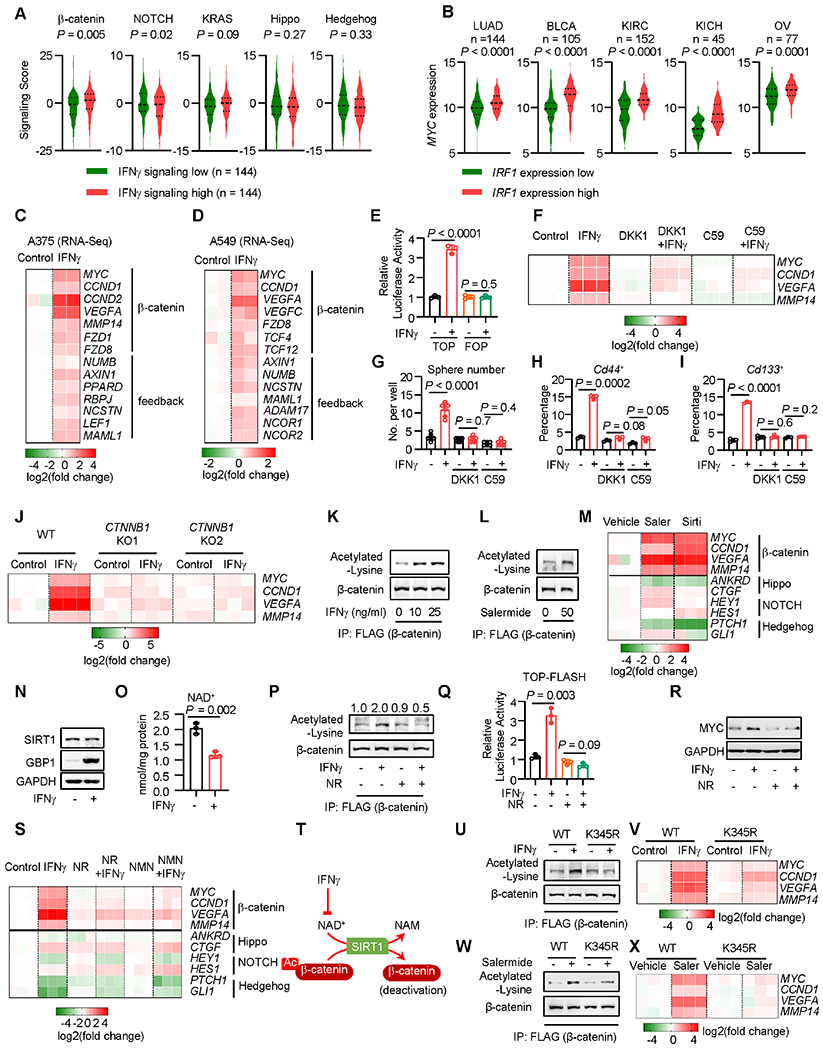
A. Correlation between the IFNγ and oncogenic signaling gene scores in lung adenocarcinoma in TCGA datasets. The expression of the indicated oncogenic gene signaling scores was plotted based on the top and bottom 25th percentiles of the IFNγ gene signaling scores. P value by two-tailed t-test.
B. Correlation between the IRF1 and MYC signaling gene scores in several cancer types in TCGA datasets. The expression of the MYC gene signaling scores was plotted based on the top and bottom 25th percentiles of the IRF1 gene signaling scores. P value by two-tailed t-test. LUAD, lung adenocarcinoma; BLCA, Bladder urothelial carcinoma; KIRC, Kidney renal clear cell carcinoma; OV, ovarian cancer.
C-D. Based on the RNA-seq datasets (GSE99299), the indicated gene expression was shown in A375 (C) or A549 (D) cells in the presence or absence of IFNγ. n = 2.
E. A375 cells were transfected with β-catenin signaling reporter TOP-FLASH or the mutant vector (FOP-FLASH). Cells were treated with IFNγfor 48 hours. Results are expressed as the relative luciferase activity. Data are shown as mean ± s.d., n = 3, P value by two-tailed t-test.
F. A375 cells were treated with IFNγ and recombinant DKK1 or Wnt-C59 (C59) for 48 hours. The indicated gene expression was determined by qPCR. n = 3.
G. YUMM1.7 cells were treated with IFNγ and DKK1 or Wnt-C59 (C59) for 48 hours, followed by 3D-sphere culture. Spheres were counted on day 7 after sphere culture. Data are shown as mean ± s.d., n = 6. Two-tailed t-test.
H-I. YUMM1.7 cells were treated with IFNγ and DKK1 or Wnt-C59 (C59) for 48 hours. Percentages of Cd44+ (H) or Cd133+ (I) cells were determined by FACS. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
J. Wild type or 2 clones of CTNNB1 KO A375 cells were treated with IFNγ for 48 hours. The indicated gene expression was determined by qPCR. n = 3.
K. Flag-tagged β-catenin expressing A375 cells were treated with IFNγ for 24 hours. Co-IP was performed with Flag antibody. Acetylated-lysine and β-catenin were detected in the IP products. 1 of 2 Western blots shown.
L. Flag-tagged β-catenin expressing A375 cells were treated with Salermide for 10 hours. Acetylated-lysine and β-catenin were determined in the Co-IP products with Flag antibody. 1 of 2 blots shown.
M. A375 cells were treated with Salermide (Saler) or Sirtinol (Sirti) for 24 hours. The indicated gene expression was determined by qRT-PCR. n = 3.
N. A375 cells were treated with IFNγ for 24 hours. SIRT1 and GBP1 (positive control) proteins were detected by Western blot. 1 of 2 Western blots shown.
O. A375 cells were treated with IFNγ for 24 hours. NAD+ levels were determined by quantitation kit. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
P. Flag-tagged β-catenin expressing A375 cells were treated with IFNγ, in the presence or absence of 0.7 mM nicotinamide riboside (NR), for 24 hours. Acetylated-lysine and β-catenin were detected in the Co-IP products with Flag antibody. 1 of 2 Western blots shown.
Q. A375 cells carrying TOP-FLASH were treated with IFNγ, in the presence or absence of 0.7 mM nicotinamide riboside (NR), for 24 hours. Luciferase activity of the β-catenin signaling reporter was determined. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
R. A375 cells were treated with IFNγ and nicotinamide riboside (NR) for 24 hours. MYC and GAPDH proteins were determined by Western blot. 1 of 3 Western blots shown.
S. A375 cells were treated with IFNγ and nicotinamide riboside (NR) or β-nicotinamide mononucleotide (NMN) for 48 hours. The indicated gene expression was determined by qPCR. n = 3.
T. Schematic diagram showing that IFNγ reduces NAD+ to suppress SIRT1-mediated β-catenin deacetylation, thereby activating β-catenin.
U-V. Wild type or CTNNB1 K345R mutant A375 cells were treated with IFNγ. 24 hours after treatment, acetylated β-catenin was determined by Western blotting following Flaĝ-catenin Co-IP (U). 48 hours after treatment, the indicated gene expression was determined by qPCR (V). 1 of 2 Western blots shown, n = 3 for qPCR.
W-X. Wild type or CTNNB1 K345R mutant A375 cells were treated with Salermide (Saler). 12 hours after treatment, acetylated β-catenin was determined by Western blotting following Flag-β- catenin Co-IP (W). 24 hours after treatment, the indicated gene expression was determined by qPCR (X). 1 of 2 Western blots shown, n = 3 for qPCR.
See also Figure S4.
To dissect how IFNγ activates β-catenin, we investigated the expression and posttranslational modification of β-catenin. IFNγ treatment did not alter the protein levels of total and phosphorylated β-catenin (Figures S4C and S4D) but increased β-catenin acetylation in A375 cells (Figure 4K). Acetylation of β-catenin increases its activity to stimulate gene transcription. P300 and Sirtuins are the main enzymes to catalyze acetylation and deacetylation of β-catenin, respectively 47–49. We treated A375 cells with L002, an inhibitor of P300 50, in the presence of IFNγ. As expected, L002 treatment reduced MYC expression. However, IFNγ was still able to increase MYC expression in L002 pretreated A375 cells, suggesting that P300 may not be involved in IFNγ-induced β-catenin signaling (Figure S4E). Then, we treated A375 cells with Salermide, a Sirtuin inhibitor 51. We observed that Salermide treatment induced β-catenin acetylation (Figure 4L) and target gene expression (Figure 4M). In addition, IFNγ failed to increase the expression of β-catenin target genes in cells co-treated with Salermide (Figure S4F–S4I). In line with this, Sirtinol, another Sirtuin inhibitor, also induced β-catenin signaling gene expression (Figure 4M). Notably, Salermide and Sirtinol failed to trigger the signaling gene expression of Hippo, NOTCH, and Hedgehog (Figure 4M). The Wnt-β-catenin signaling inhibitors, DKK1 and Wnt-C59, abolished the effect of Salermide on the expression of MYC, CCND1, VEGFA, and MMP14 (Figure S4J). The data suggest that IFNγ may induce the β-catenin signaling via reducing Sirtuin-mediated β-catenin deacetylation. Sirtuins are a class of NAD+ dependent deacetylases 52. IFNγ stimulated guanylate binding protein (GBP)-1 expression (positive control) 53, had no effect on SIRT1 expression (Figure 4N), but reduced the intracellular level of NAD+ (Figure 4O). To restore NAD+ and Sirtuin activity, we treated A375 cells with nicotinamide riboside (NR), an NAD+ precursor. Interestingly, NR treatment diminished IFNγ altered β-catenin acetylation (Figure 4P), TOP-FLASH reporter activity (Figure 4Q), and β-catenin target gene expression (Figure 4R and 4S), as compared with vehicle controls. Similarly, treatment with nicotinamide mononucleotide (NMN), also diminished the effect of IFNγ on the expression of β-catenin signaling genes, while expression of other oncogenic pathways, such as Hippo, NOTCH, and Hedgehog signaling, were not affected by NR or NMN (Figure 4S). Altogether, these data suggest that IFNγ enhances β-catenin acetylation via reducing NAD+ levels (Figure 4T). β-catenin can be acetylated at K49 and K345. We found that IFNγ did not affect the expression of K49-acetylated β-catenin (Figure S4K). Sirtuin may catalyze the deacetylation of β-catenin at K345 47. We established stable cells carrying K345R β-catenin mutation. Upon IFNγ (Figures 4U and 4V) and Salermide (Figures 4W and 4X) treatment, β-catenin acetylation (Figures 4U and 4W) and downstream gene activation (Figures 4V and 4X) were largely abrogated in K345R mutant cells. The data suggest that IFNγ may affect β-catenin acetylation at K345 via reducing Sirtuin activity. As an additional control, we treated non-HPD-prone mouse YUMM5.2 cells with IFNγ. IFNγ had no effect on the β-catenin signaling activity (Figure S4L) and intracellular NAD+ levels (Figure S4M). Thus, that IFNγ selectively alters the NAD+-β-catenin signaling activities in HPD-prone tumor models.
IFNγ regulates PKM2 phosphorylation to alter NAD+ and β-catenin signaling
Glycolysis ferments glucose into lactate, functioning as a metabolic regulator of NAD*/ NADH in cancer cells 54. We investigated whether IFNγ regulates tumor glycolysis, thereby altering NAD+ and the β-catenin signaling pathway. Seahorse experiments revealed that IFNγ treatment resulted in lower glycolytic rate in A375 cells as shown by extracellular acidification rate (ECAR). This effect was abolished by 2-DG, a glycolysis inhibitor (Figures 5A, S5A and S5B). To determine if IFNγ-regulated glycolysis is related to tumor cell proliferation, we treated A375 cells with IFNγ, in the presence or absence of different concentrations of Palbociclib, a CDK4/6 inhibitor. IFNγ reduced tumor lactate production regardless of Palbociclib treatment (Figure S5C). Additionally, IFNγ diminished lactate production in wild type A375 cells but not in STAT1 KO A375 cells (Figure S5D). Thus, IFNγ signaling abrogates tumor glycolysis.
Figure 5: IFNγ regulates PKM2 phosphorylation to alter NAD+/ β-catenin signaling.
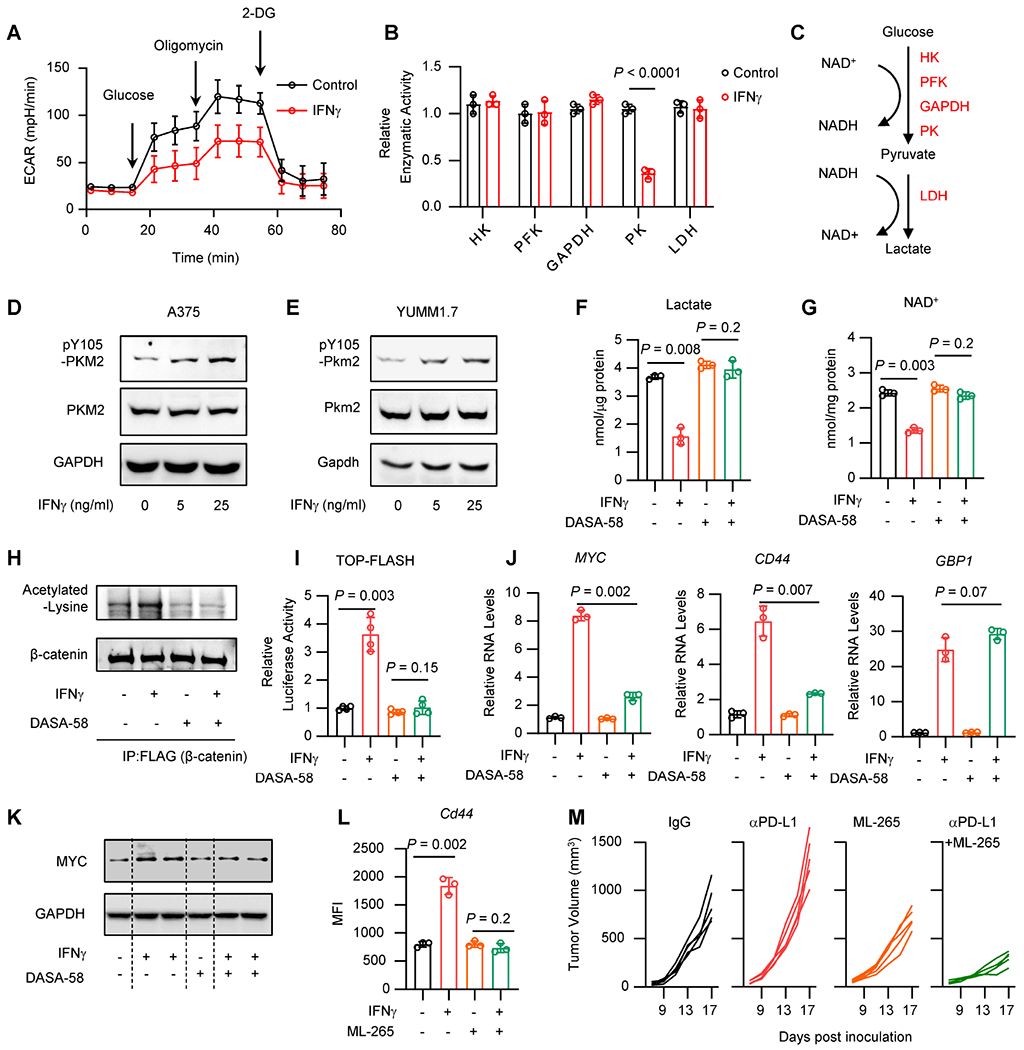
A. A375 cells were treated with IFNγ for 24 hours. Seahorse analysis showed the extracellular acidification rate (ECAR) in control cells and IFNγ-treated cells in the presence of glucose, oligomycin or 2-DG. Data are shown as mean ± s.d., n = 3.
B. A375 cells were treated with IFNγ for 24 hours. Catalytic activities of glycolysis rate-limiting enzymes were determined by quantitation kits. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
C. Schematic diagram showing the glycolysis pathway and the NAD+/ NADH balance.
D-E. A375 (D) or YUMM1.7 (E) cells were treated with IFNγ for 24 hours. Phosphorylated or total protein levels of PKM2 were detected by Western blot. 1 of 2 Western blots shown.
F-G. A375 cells were treated with IFNγ, in the presence or absence of DASA-58, for 48 hours. Lactate production (F) or intracellular levels of NAD+ (G) were determined by quantitation kit. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
H. A375 cells carrying Flag-tagged β-catenin were treated with IFNγ, in the presence or absence of DASA-58. Acetylated-lysine and β-catenin were detected in the Co-IP products with Flag antibody. 1 of 2 Western blots shown.
I. A375 cells carrying TOP-FLASH were treated with IFNγ, in the presence or absence of DASA-58, for 24 hours. Relative luciferase activity was determined. Data are shown as mean ± s.d., n = 4. Two-tailed t-test.
J. A375 cells were treated with IFNγ, in the presence or absence of DASA-58, for 24 hours, β- catenin signaling genes and IFNγ signaling gene (GBP1) (positive control) were determined by qRT-PCR. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
K. A375 cells were treated with IFNγ, in the presence or absence of DASA-58, for 48 hours. MYC and GAPDFI proteins were determined by Western blot. 1 of 2 Western blots shown.
L. YUMM1.7 cells were treated with IFNγ, in the presence or absence of ML-265, for 48 hours. Surface expression of Cd44 was determined by FACS. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
M. YUMM1.7 tumor bearing C57BL/6 mice were treated with anti-PD-L1, ML-265, or the combination of anti-PD-L1 and ML-265. Tumor growth curves were plotted, n = 5 / group.
See also Figure S5.
To validate this observation in the context of T cells, we co-cultured A375 cells with TCM. Consistent with our prior findings, TCM reduced lactate production in wild type A375 cells but not IFNGR1 KO A375 cells (Figure S5E). The data suggest that T cells regulate tumor glycolysis via IFNγ. To understand how IFNγ affects glycolysis, we measured the catalytic activities of the rate-limiting enzymes in the glycolysis pathway. Interestingly, IFNγ impaired the activity of pyruvate kinase (PK), but not that of hexokinase (FHK), phosphofructokinase (PFK), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), or lactate dehydrogenase (LDH) (Figure 5B). Inhibition of PK diminishes pyruvate production, in turn decreasing the reaction by LDH, in which pyruvate and NADH will be converted into lactate and NAD+, respectively (Figure 5C). Indeed, we detected reduced levels of pyruvate in A375 cells treated with IFNγ as compared to control (Figure S5F). PKM2 is the predominant isotype of PK in tumor cells 55. IFNγ failed to regulate lactate production in shPKM2 A375 cells (Figure S5G). Thus, IFNγ regulates tumor glycolysis in a PK dependent manner.
We observed that IFNγ induced PKM2 phosphorylation without altering total PKM2 expression in A375 and YUMM1.7 cells (Figures 5D and 5E). Notably, IFNγ induced the phosphorylation of PKM2 at Y105 (Figures 5D and 5E), but not at S37 56 (Figure S5F–H). Phosphorylation of PKM2 at Y105 inhibits the formation of active, tetrameric PKM2 by disrupting binding of the PKM2 cofactor fructose-1,6-bisphosphate, thereby reducing its catalytic activity 57. To link PKM2 with NAD+-β-catenin signaling, we knocked down Pkm2 with shRNA in YUMM1.7 cells and PLC2.4 cells. We detected a decrease in the levels of pyruvate, lactate, and NAD+ (Figures S5I–S5K) and an increase in the levels of Myc (Figure S5L) in both shPkm2 YUMM1.7 cells and shPkm2 PLC2.4 cells as compared to controls. Moreover, knocking down Pkm2 enhanced Cd44 expression and nicotinamide riboside (NR) blocked this effect (Figure S5M). The data suggest that knocking down PKM2 can activate β-catenin signaling and promote tumorigenesis. To validate this observation in an in vivo system, we inoculated shPkm2 YUMM1.7 and shPkm2 PLC2.4 cells into C57BL/6 mice. As expected, shPkm2 tumors progressed faster than control tumors (Figures S5N and S5O). Thus, restriction of PKM2 activity could promote β-catenin signaling and accelerate tumor progression.
To rescue the effect of IFNγ on PKM2, we applied DASA-58, a selective activator of PKM2 58. We treated A375 cells with DASA-58 in the presence of IFNγ. DASA-58 treatment reversed the effect of IFNγ on lactate production (Figure 5F), NAD+ (Figure 5G), β-catenin acetylation (Figure 5H), TOP-FLASH reporter activity (Figure 5I), and β-catenin signaling gene expression (Figures 5J and 5K). Notably, DASA-58 failed to alter the impact of IFNγ on GBP1 expression (Figure 5J). The data suggest that DASA-58 blocks β-catenin activation by IFNγ, rather than the blockade of global IFNγ signaling. Similarly, ML-265, another PKM2 activator 58, diminished the effect of IFNγ on Cd44 expression in YUMM1.7 cells (Figure 5L). Collectively, PKM2 activation blocks IFNγ-mediated β-catenin signaling activation. To extend this observation in vivo, we treated YUMM1.7 tumor bearing mice with αPD-L1, ML-265, and the combination of αPD-L1 and ML-265. Although αPD-L1 alone promoted tumor progression, ML-265 alone inhibited tumor growth, and the combination therapy completely reversed αPD-L1-mediated HPD, thereby resulting in a potent tumor inhibition (Figure 5M). As an experimental control, we observed that IFNγ had no effect on lactate production (Figure S5P) and PKM2 phosphorylation (Figure S5Q) in non-HPD prone YUMM5.2 cells. Altogether, targeting PKM2 blocks β-catenin activation and subverts ICB-triggered HPD.
IFNγ targets FGF2 to control PKM2-NAD+-β-catenin signaling
Next, we explored the upstream signaling genes involved in the regulation of the PKM2-NAD+-β-catenin axis by IFNγ. Several kinases, such as EGFR and FGFR, can induce PKM2 phosphorylation 56,57. We treated A375 cells with Gefitinib, an EGFR inhibitor, and Dovitinib, a FGFR inhibitor. We found that inhibition of FGFR, but not EGFR, abolished the stimulatory effect of IFNγ on MYC expression (Figure 6A). In addition to MYC, Dovitinib diminished the effect of IFNγ on the expression of CCND1 but had no impact on GBP1 expression (Figure 6B). RNA-seq datasets demonstrated that IFNγ induced expression of FGF2, but not the other FGF/FGFR family members, in A375 melanoma cells, while IFNγ failed to induce FGF2 in non-HPD prone B16F10 melanoma cells (Table S5). In support of this data, we observed that IFNγ stimulated FGF2 mRNA and protein expression in A375, YUMM1.7, and PLC2.4 cells (Figure 6C and 6D). IRF1 was enriched in the promoter of FGF2 in the ChIP-seq datasets on K562 cells (Figure S6A). The IRF1 motif was conserved across human and mouse species (Figure S6A). We inserted the FGF2 promoter and exon 1 sequence upstream of luciferase coding sequence in PGL3-basic plasmid and generated a FGF2 promoter reporter. We found that IFNγ induced the luciferase activity of FGF2 promoter and failed to do so when the IRF1 binding site was deleted (Figure S6B). Thus, IFNγ activates FGF2 transcription via IRF1.
Figure 6: IFNγ induces FGF2 to control PKM2/ NAD+/ β-catenin signaling.
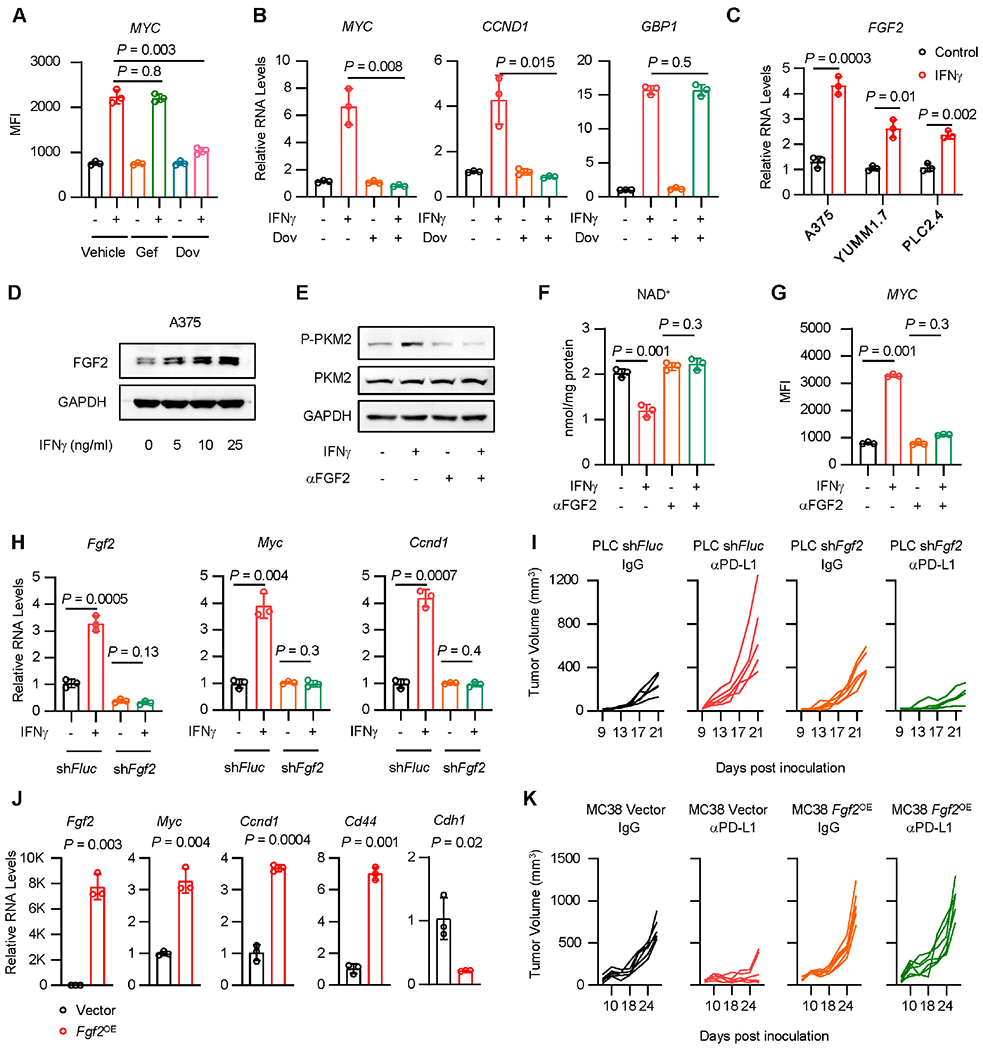
A. A375 cells were treated with IFNγ in the presence of Gefitinib (Gef) or Dovitinib (Dov), for 36 hours. MYC protein was determined by FACS. Data are shown as mean ± s.d., n = 3. Two-tailed t- test.
B. A375 cells were treated with IFNγ in the presence of Dovitinib (Dov), for 48 hours, β-catenin signaling genes (MYC, CCND1) and IFNγ signaling gene (GBP1) were determined by qRT-PCR. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
C. FGF2 transcripts were quantified by qRT-PCR in IFNγ-treated A375, YUMM1.7 and PLC2.4 cells. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
D. FGF2 protein was determined by Western blot in A375 cells treated with IFNγ. 1 of 2 Western blots shown.
E-G. A375 cells were treated with IFNγ, in the presence or absence of FGF2 neutralizing antibody (αFGF2). Phosphorylated (Y105) and total protein levels of PKM2 were determined at 24 hours by Western blot (E). Cellular NAD+ levels were quantified at 24 hours by kit (F). MYC expression was determined at 48 hours by FACS (G). 1 of 2 Western blots shown (E). Data are shown as mean ± s.d., n = 3. Two-tailed t-test (F, G).
H. Fgf2, Myc or Ccnd1 transcripts were detected by qRT-PCR in PLC2.4 shFlue or shFgf2 cells. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
I. shFluc or shFgf2 PLC2.4 tumor bearing C57BL/6 mice were treated with anti-PD-L1 and isotype IgG. Tumor growth curves were plotted, n = 5 animals.
J. MC38 cells were forced expression of Fgf2 (Fgf2OE). RNA levels of Fgf2, β-catenin signaling genes (Myc, Ccnd1, Cd44), and epithelial marker gene (Cdh1) were determined by qRT-PCR. Data are shown as mean ± s.d., n = 3. Two-tailed t-test.
K. Fgf2OE MC38 tumor bearing C57BL/6 mice were treated with anti-PD-L1 and isotype IgG. Tumor growth curves were plotted, n = 6 animals.
To determine the involvement of FGF2 on the effect of IFNγ on β-catenin, we treated A375 cells with IFNγ, in the presence or absence of FGF2 neutralizing antibody (αFGF2). αFGF2 abrogated the effect of IFNγ on PKM2 phosphorylation, NAD+, and β-catenin signaling gene expression (Figures 6E–6G) and had no effect on the levels of total and phosphorylated STAT1 (Figure S6C). In line with this, treatment with recombinant FGF2 protein induced the expression of phosphorylated-PKM2, MYC, and CD44, and reduced the levels of NAD+ (Figures S6D–S6F). Moreover, knockdown of FGF2 resulted in reduced expression of Myc and Cd44 induced by IFNγ in PLC2.4 and YUMM1.7 cells (Figures 6H, S6G and S6H). In addition, we treated multiple mouse and human cancer cell lines with IFNγ. Consistent with MYC activation (Figures 3D, S3C, S3S, S3U, and S3V), IFNγ stimulated FGF2 protein in a minority of cancer cell lines (Figures S6I–S6K). Together, IFNγ promotes FGF2 signaling to control PKM2-NAD+-β-catenin signaling in tumor cells.
To determine whether FGF2 expression is involved in ICB-triggered HPD, we inoculated shFGF2 PLC2.4 cells into C57BL/6 mice and treated these mice with αPD-L1. As expected, αPD-L1 accelerated tumor progression in WT PLC2.4 tumors, but inhibited shFgf2 PLC2.4 tumor progression (Figure 6I). The data suggest that FGF2 signaling contributes to ICB-triggered HPD. To solidify this finding, we overexpressed Fgf2 (Fgf2OE) in MC38 cells. We found that Fgf2OE MC38 cells expressed higher levels of Myc, Ccnd1, and Cd44, and lower levels of Cdh1 as compared to vector carrying MC38 cells (Figure 6J). We inoculated Fgf2OE MC38 tumors into C57BL/6 mice and treated these mice with αPD-L1. αPD-L1 slowed down control MC38 tumor progression, however, failed to control Fgf2OE MC38 tumors (Figure 6K). Collectively, these data suggest that tumor FGF2 activates oncogenic signaling and shapes immunotherapy outcome.
Oncometabolic reprogramming drives cancer hyperprogression during immunotherapy
Finally, we evaluated whether oncogenic and immunogenic signaling pathways converged in patients with progressive disease on ICB 59–62. While radiographic quantification required for HPD ascertainment is not available in public datasets, we hypothesized that a subset of patients with PD would have HPD. We confirmed that IFNγ signaling strongly correlated with CD8+ T cell infiltration in these cohorts (Figure S7A). Our data suggests that FGF may regulate Wnt signaling in preclinical models of cancer HPD (Figures 6A–6K). Interestingly, we observed a positive correlation between FGF and Wnt-β-catenin signaling signatures in patients receiving immunotherapy (Figure S7B). Our experimental work also suggests that IFNγ signaling promotes FGF and β-catenin signaling in HPD. Interestingly, in patients who receive ICB and develop PD, we found that FGF and Wnt-β-catenin signaling scores were increased in the subset of patients with elevated IFNγ signaling (Figure S7C). In contrast, there was no elevation of FGF or Wnt-β-catenin in patients who developed a CR (Figure S7D). We confirmed that within PD patients, a triple-high (IFNγ/ FGF/ β-catenin) gene signature score was associated with diminished overall survival as compared to patients with triple-low signatures (Figure S7E). In line with this, we analyzed a single cell RNA-seq dataset from patients with cutaneous malignancies treated with ICB 63. Tumors from non-responders manifested higher levels of IFNγhighFGFhighβ-cateninhigh (triple-high) gene signatures as compared with responders (Figure S7F). Interestingly, there existed 2 distinct tumor cell subtypes within the same tumor: one population that was sensitive to ICB and another one that was resistant to ICB (Figure S7G). The triple-high gene signature was enriched in the ICB-resistant subtype (Figure S7H). The data suggest that immunotherapy fails to eradicate tumor cells expressing triple-high gene signature. Thus, CD8+/IFNγ immunogenic signaling and FGF2/β-catenin oncogenic signaling is enriched in patients who derive limited benefit from ICB (Figure 7).
Figure 7: Oncometabolic reprogramming drives cancer hyperprogression during immunotherapy.
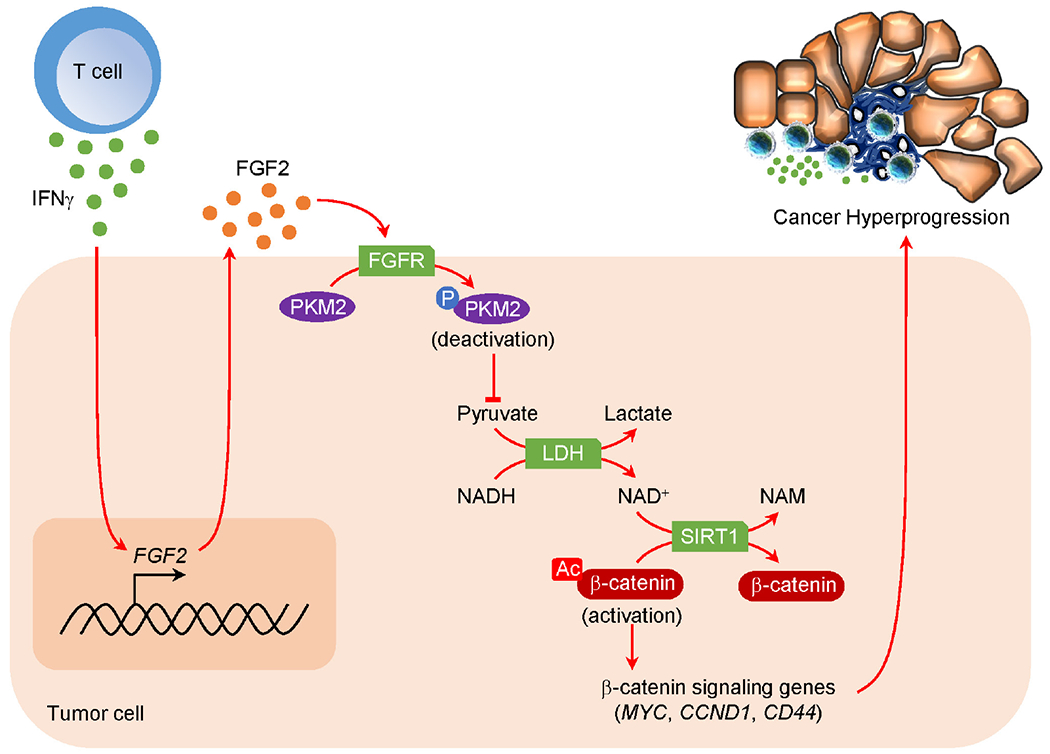
Mechanistic scheme of HPD development. IFNγ produced by ICB-activated T cells targets tumor FGF2 signaling, inducing PKM2 phosphorylation at Y105 and decreasing NAD+ levels, thereby diminishing SIRT1 activity and lessening β-catenin deacetylation. Consequently, β-catenin signaling pathway is activated resulting in enhanced oncogenic potential and HPD.
Discussion
In this work, we have conducted comprehensive sequencing, immunological, clinical, and radiographic evaluations in multiple cohorts of patients receiving ICB. These analyses demonstrate that HPD likely occurs in a small subset of patients. In addition, we have established the syngeneic immunocompetent animal models of HPD, enabling the dissection of the cellular, molecular, and immunological mechanisms of accelerated cancer progression due to ICB. We report that the interplay between core immunogenic, metabolic, and oncogenic mechanisms enables cancer hyperprogression via the IFNγ-PKM2-β-catenin signaling cascade in preclinical models and patients.
Heterogeneity of response to cancer therapeutics is frequently observed. While ICB improves progression-free and overall survival at a population level, we found that in both prospective 26–28 and institutional cohorts, a minority of patients develop hyperprogression after receipt of ICB. This is similar to previous reports demonstrating an association between HPD and ICB 6,7,32. It remains to be prospectively validated whether HPD is enriched in patients receiving ICB. Although medically challenging, in the future the generation of a humanized mouse with tumors and matched autologous T cells from patients experiencing HPD could enable additional mechanistic insights. Our data suggest that akin to ICB-triggered autoimmune reactions, HPD may be an immune-related adverse event.
Given the prognostic impact of HPD, it is extremely important to define molecular mechanisms and identify molecular biomarkers for this clinical outcome. By integrating information from comprehensive sequencing with clinical and radiographic platforms at the University of Michigan Rogel Cancer Center 39,64,65, we have immunologically and genetically characterized HPD. Previous clinical reports suggest amplification of oncogenic genes, including EGFR, MDM2, and MDM4 7, and enriched Foxp3+ regulatory T (Treg) cells 66 in HPD tumors. Myeloid cells and the PD-L1-PD-1 pathway is also known to mediate immunosuppression in the human tumor microenvironment 40,41,67. However, we have detected comparable levels of EGFR, MDM2, MDM4, FOXP3, CD68, and PDCD1 expression in tumors from patients with CR and HPD. Moreover, it has been speculated that the Fc region of ICB antibodies may be involved in HPD development 68. However, HPD has been observed in patients treated with anti-PD-L1, anti-PD-1, and anti-CTLA4 monoclonal antibodies in different isotypes, including lgG1, lgG2, and lgG4 (demonstrated in this work) 33,69–79. Thus, prior to our study, the cellular and molecular basis of HPD remained elusive.
Our experiments demonstrate that HPD is driven by the interplay among core immunogenic, metabolic, and oncogenic pathways via the IFNγ-PKM2-β-catenin molecular cascade. Immune exclusion and mutations in the IFNγ and MHC genes are known resistance mechanisms to immunotherapy, promoting disease progression. It is commonly assumed that HPD tumors are “cold” with poor immune infiltration. To our surprise, we discovered that intratumoral CD8+ T cells and active IFNγ signaling are required for HPD in preclinical models. This mirrors the unexpected finding that patients with HPD and CR had similar tumoral T cell infiltration and IFNγ signaling. This extends previous reports which highlight the duality of interferon signaling in both promoting antitumor immunity and enabling tumor immune evasion 80. Hence, CD8+ T cells and IFNγ signaling may unexpectedly contribute to HPD in both mouse models and cancer patients.
We observed CD8+ T cell-derived IFNγ targets FGF2 to selectively inhibit PKM2, a dominant rate-limiting enzyme in glycolysis, resulting in reduced NAD+ production. Consequently, β-catenin activity is increased in tumor cells, thereby promoting cancer stemness and tumorigenic potential. Corroborating this mechanism, patients with HPD had increases in MYC and FGF2 pathway activities, accompanied by high levels of invasiveness and stemness gene signatures and expression of cancer stem-like marker CD133. In line with this, it has been reported that tumor cells with stem-like properties manifest high metastatic potential 81,82 and are resistant to T cell-mediated cytotoxicity 83,84.
It has previously been observed that FGF and β-catenin signaling pathways promote T cell exclusion and resistance to immunotherapy 85. In contrast, we have uncovered a previously unappreciated mode of action of IFNγ controlling an intertwined metabolic and oncogenic signaling pathway: IFNγ targets PKM2 to diminish Sirtuin-mediated β-catenin deacetylation via NAD+ reduction, thereby causing β-catenin acetylation and activation. These data suggest that similar to oncogene addiction, there is a state of interferon addiction where tumor cells can be supported by and dependent on immune signaling axes. While tumoral regulation of T cell metabolism to promote immunosuppression is well established, our work suggests T cells may also regulate tumoral metabolism. Further, this work suggests that tumor glycolysis is not only a downstream effect of oncogenic signaling, but also an upstream regulator of oncogenesis. In line with this, FGF2 and β-catenin oncogenic signatures were enriched in patients with HPD. Hence, we suggest a causal link between immune activation and accelerated tumor progression during immunotherapy. Collectively, our data suggest that HPD is a form of immunopathology.
In addition to its scientific importance, our work may be translationally meaningful. We identify a triple-high (IFNγ/ FGF/ β-catenin) gene signature score associated with HPD in patients. This may serve as a surrogate marker for HPD, enabling prediction of HPD. In addition, our study may inform the development of therapeutic strategies to limit the devastating consequences of cancer hyperprogression. Mechanistically, IFNγ-FGF2-PKM2-β-catenin signaling promotes HPD. FGF2R inhibitors are approved as anti-oncologic agents in cholangiocarcinoma and urothelial carcinoma 86. PKM2 activators are currently being clinically pursued (NCT04328740). Our studies highlight that these agents may be repurposed to limit HPD. Thus, our molecular studies provide insight and justification for future clinical trials to further our understanding of ICB-associated HPD in order to search for biomarker(s) and improve clinical management of HPD patients.
In summary, our work demonstrates that ICB is likely associated with HPD in a small subset of patients. The interaction between the core immunogenic, metabolic, and oncogenic pathways via the IFNγ-FGF2-β-catenin axis is a plausible mechanism and may serve as potential biomarkers for HPD. This work suggests that targeting this axis may prevent the development of HPD in patients receiving immunotherapy and provides a rationale for investigating this in prospective clinical studies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for materials should be directed to the lead contact: Weiping Zou (wzou@umich.edu)
Materials availability
This study did not generate new unique reagents
Data and code availability
The expression sequencing from the University of Michigan cohort was previously deposited (National Center for Biotechnology Information database of Genotypes and Phenotypes (dbGaP) under accession number phs000673.v2. p1.) 39,64,65. The expression sequencing data and corresponding clinical annotation for patients receiving ICB was previously deposited (Gene Expression Omnibus (GEO) under accession number GSE91061; National Center for Biotechnology Information database of Genotypes and Phenotypes (dbGaP) under accession number phs001919. v1.p1. 59–62. The expression profile of cell lines was previously deposited (Gene Expression Omnibus (GEO) under accession number GSE99299) 11. The single cell RNA sequencing data of cancer patients upon immunotherapy was previously deposited (Gene Expression Omnibus (GEO) under accession number GSE123814) 63. All raw data supporting the findings of this study are available from the corresponding authors upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Studies
Patients were recruited through the University of Michigan Hospital, Ann Arbor, MI, USA. All clinical records in this study were obtained with the approval of Institutional Review Boards and patients’ consents was waived following Institutional Review Board protocol review (HUM00146400, HUM00139259, HUM00163915, HUM00161860, and HUM00046018). Cohort 1 represents metastatic melanoma patients who received treatment at the University of Michigan from 2013-2020. Cohort 2 represents metastatic NSCLC patients who received treatment at the University of Michigan from 2013-2019. Cohort 3 represents patients at the University of Michigan who have undergone comprehensive tumor and somatic bulk RNA sequencing as previously described who received immunotherapy and had evaluable cross-sectional imaging 39. Tumor radiographic response and tumor growth rate were evaluated at the first imaging assessment preceding and following therapy initiation in the subset of patients with evaluable longitudinal cross-sectional imaging. Radiologists blinded to the hypothesis conducted evaluation of clinical imaging to define tumor burden and all measurements were validated by a board-certified radiation oncologist. Patients with pseudoprogression were identified by using imRECIST criteria29 and excluded from all cohorts. RECIST1.187 was utilized for response assessment. Hyperprogression was defined as previously published (Table S3); analysis using Champiat et al. criteria are displayed. Tumor burden was defined as the sum of the longest cross-sectional diameters of lesions noted on radiographic studies. Progression-free survival and overall survival were calculated from initiation of therapy. 3D reconstructions were constructed in Eclipse within the ARIA Oncology Information System (Varian Oncology, Version 15).
Cell lines
Human melanoma cell lines included A375 (CRL-1619), MeWo (HTB-65), A2058 (CRL-11147), CHL-1(CRL-9446), Malme-3M (HTB-64), G361 (CRL-1424), SKMEL-1 (HTB-67), SKMEL-2 (HTB- 68), SKMEL-5 (HTB-70), and SKMEL-28 (HTB-72). Human lung cancer cell lines included A549 (CCL-185), H1299 (CRL-5803), H292 (CRL-1848), H23 (CRL-5800), H69 (HTB-119), H460 (HTB-177), H661 (HTB-183), H1437 (CRL-5872), and H1975 (CRL-5908). Mouse melanoma cell lines were YUMM1.7 (CRL-3362), YUMM5.2 (CRL-3367), and B16-F0 (CRL-6322). Mouse lung cancer cell lines were Lewis lung carcinoma (LLC, CRL-1642) and its subclones. Human embryonic kidney cell 293T (CRL-3216) was used in the study. These cell lines were from the American Type Culture Collection (ATCC). Mouse colon cancer cell line MC38 from previously studies was used 44,88. To generate LLC subclones, LLC tumor bearing mice were treated with anti-PD-L1. Tumor cells were dissected from these mice bearing ICB-resistant tumors and serially diluted and seeded into 96 well plates. Ten days after seeding, single cell clones (PLC1.1-PLC2.4) were collected by trypsinization. Using CRISPR Cas9, IFNGR1 KO and STAT1 KO A375 cells, Ifngr1 KO and Stat1 KO YUMM1.7 cells and PLC2.4 cells, and Stat1 KO YUMM5.2 cell lines were generated in this study. YUMM1.7 cells were maintained at low confluence (less than 70%) to avoid clumping. All cell lines were tested for mycoplasma contamination routinely and confirmed negative for mycoplasma. Cells were cultured in pyruvate-free medium supplemented with 10% FBS. A375, MeWo, A2058, CHL-1, Malme-3M, SKMEL-1, SKMEL-2, SKMEL-5, SKMEL-28, A549, 293T, LLC, and PLC2.4 cells were cultured in DMEM (Gibco #11965), H1299, H292, H23, H69, H460, H661, H1437, H1975 YUMM1.7, YUMM5.2, B16, and MC38 cells were cultured in RPMI (Gibco #11875). G361 cells were cultured in McCoy’s 5a medium (Gibco #16600). All cells were maintained in 37°C and 5% CO2.
Tumor models
Six- to eight-week-old female C57BL/6 (C57BL/6J, Stock# 000664) and NSG (#005557) mice were obtained from the Jackson Laboratory. All mice were maintained under pathogen-free conditions. The animal room is a controlled environment: temperature (18-23°C), humidity (40-60%), and a 12-hour light/dark cycle. YUMM1.7 (105), YUMM5.2 (105), PLC2.4 (105), and MC38 (2.5 × 106) cells were subcutaneously injected into the right flank of C57BL/6 mice. A375 (5 × 105) cells were subcutaneously injected into the right flank of NSG mice. Tumor diameters were measured using calipers. Tumor volume was calculated by Length × Width × Width/2. Tumor weight was measured with an analytical balance. Animal studies were conducted under the approval of the Institutional Animal Care and Use Committee at the University of Michigan (PRO00008278). The study is compliant with all relevant ethical regulations regarding animal research. In none of the experiments did xenograft tumor size surpass 2 cm in any dimensions, and no animal had severe abdominal distension (≥ 10% original body weight increase). Sample size was chosen based on preliminary data. After tumor inoculation, mice were randomized and assigned to different groups for treatment.
METHOD DETAILS
Multiplex immunofluorescence staining and analysis
Multiplex immunofluorescence staining was performed on pretreatment formalin fixed paraffin embedded (FFPE) human melanoma or non-small cell lung carcinoma sections with OPAL 4-Color IHC kits (Akoya Biosciences, NEL810001KT). Antibodies against human CD8 (C8/144B, Abcam, 1:100), FGF2 (EP1735, Abcam, 1:500), MYC (Y69, Abcam, 1:200), and CD133 (AC133, Miltenyi Biotec, 1:50) were used in the study. Imaging was completed using the Mantra Quantitative Pathology Workstation. Tissue core images were captured at x4 and x40 magnifications. All cube filters were used for each image capture (DAPI, CY3, CY5, Texas Red). The incorporated saturation protection feature was set at an exposure time of 250 ms. Images were analyzed using in Form Cell Analysis software (Perkin Elmer). Images were batch analyzed using a subset of randomly chosen tissue core images. Using the inform software, both tissue and cell compartments were identified and segmented. Tissue was segmented into stroma and epithelial cancer compartments, while cells were segmented into nucleus compartments. DAPI counterstain was used to determine the size and shape of each nucleus. After cell segmentation, based on single staining, CD8+ T cells and tumor cells were determined and quantified using the inform software after selected cells were manually assigned.
Animal experiments
Six- to eight-week-old female C57BL/6 (C57BL/6J, Stock# 000664) and NSG (#005557) mice were obtained from the Jackson Laboratory. All mice were maintained under pathogen-free conditions. The animal room is a controlled environment: temperature (18-23°C), humidity (40-60%), and a 12-hour light/dark cycle. YUMM1.7 (105), YUMM5.2 (105), PLC2.4 (105), and MC38 (2.5 × 106) cells were subcutaneously injected into the right flank of C57BL/6 mice. A375 (5 × 105) cells were subcutaneously injected into the right flank of NSG mice. For anti-PD-L1 treatment in MC38 model, 5 mg/kg anti-PD-L1 (InVivoMAb, 10F.9G2) and control antibody (InVivoMAb, LTF-2) were intraperitoneally administered on day 6, 9, and 12 post tumor inoculation. For anti-PD-L1 treatment in YUMM1.7 and PLC2.4 model, 5 mg/kg anti-PD-L1 (InVivoMAb, 10F.9G2) and control antibody (InVivoMAb, LTF-2) were intraperitoneally administered on day 0, 3, 6, 9, 12, and 15 post tumor inoculation. For anti-CTLA4 treatment in YUMM1.7 model, 5 mg/kg anti-CTLA4 (InVivoMAb, 9H10) and control antibody (InVivoMAb, LTF-2) were intraperitoneally administered on day 0, 3, 6, 9, 12, and 15 post tumor inoculation. For anti-CD8 treatment in YUMM1.7 model, 5 mg/kg anti-CD8α (InVivoMAb, YTS169.4) and control antibody (InVivoMAb, LTF-2) were intraperitoneally administered on day −1, 2, 5, and 8 post tumor inoculation. For IFNγ treatment in A375 model, 0.5 μg IFNγ (R&D, 285-IF) was intraperitoneally administered every 3 days post tumor inoculation. For ML-265 treatment in YUMM1.7 model, 50 mg/kg ML-265 was intraperitoneally administered every other day post tumor inoculation. Tumor diameters were measured using calipers. Tumor volume was calculated by Length × Width × Width/2. Tumor weight was measured with an analytical balance. Animal studies were conducted under the approval of the Institutional Animal Care and Use Committee at the University of Michigan (PRO00008278). The study is compliant with all relevant ethical regulations regarding animal research. In none of the experiments did xenograft tumor size surpass 2 cm in any dimensions, and no animal had severe abdominal distension (≥ 10% original body weight increase). Sample size was chosen based on preliminary data. After tumor inoculation, mice were randomized and assigned to different groups for treatment.
Cell Culture
To generate knock down cell lines, lentiviral particles were produced by transfection of PLKO.1 shRNA plasmid with psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) (4:3:1) into 293T cells, and subsequently transduced into tumor cells with polybrene (Sigma-Aldrich, 8 μg/ml) overnight. 48 hours after transfection, cells were selected with puromycin (1-2 μg/ml) for an additional 2 weeks. To establish knock out cell lines, PX459-sgRNA plasmids were transfected into tumor cells for 2 days and selected by puromycin (1-2 μg/ml) for an additional 2 days. The cells were then serially diluted and seeded into 96 well plates. After 2-3 weeks, single cell colonies were dissociated and re-plated into 6 well plates. Upon cell confluency, half of the cells were harvested and validated for knock out (KO) efficiency via Western blotting, β-catenin K345R mutant plasmid was expressed in CTNNB1 KO cells to generate β-catenin K345R mutant cells. All transfections were conducted with lipofectamine 2000 (Thermo Fisher) at a ratio of 1 μg plasmid: 2 μl transfection regent. The transfection dosage was determined by titration.
Plasmids
To generate FGF2 promoter luciferase reporter, DNA sequences corresponding to Fgf2 promoter and exon1 were synthesized (Origene) and inserted into PGL3-basic plasmid (Promega). TOP-FLASH (#12456), FOP-FLASH (#12457), and FLA-β-catenin (#16828) were obtained from Addgene. Site directed mutagenesis was conducted to generate β-catenin K345R mutant plasmid. To force mouse Fgf2 expression, the coding sequence of Fgf2 was PCR amplified from cDNA generated from IFNγ-treated YUMM1.7 cells and subsequently inserted into PCI-Flag plasmid. PCI-Flag plasmid was prepared by inserting the Kozak sequence plus Flag tag plus 5 × Glycine sequence into the PCI-neo plasmid (Promega) between NheI and XhoI. To knock down Pkm2 and Fgf2, shRNAs were designed and inserted into PLKO.1 plasmid (Addgene #10879). The shRNA targeting firefly luciferase (shFluc) served as a negative control. To knock out IFNGR1 and STAT1, sgRNA was designed and inserted into PX459 plasmid (Addgene #48139). The target sequences are listed in Table S6. The primer sequences are listed in Table S7.
Luciferase activity assay
A375 cells were transfected with TOP-FLASH or FOP-FLASH, along with PRL-SV40P (Addgene #27163) for 24 hours, then treated with IFNγ, NR, and DASA-58 for additional 24 hours. Luciferase activity for firefly luciferase (TOP-FLASH) and renilla luciferase (PRL-SV40P) was measured with Dual-Luciferase Reporter Assay System (Promega). Relative firefly luciferase activity was normalized with renilla luciferase activity.
Flow cytometry analysis (FACS)
Cells were trypsinized and washed with MACS buffer (PBS, 2%FBS, 1 mM EDTA). Surface staining was performed by adding the following antibodies to the cell suspension in 50 μl MACS buffer: anti-CD45 (30-F11, BD Biosciences), anti-CD44 (IM7, BD Biosciences), and anti-CD133 (315-2C11, BioLegend). For intracellular staining, cell suspension was incubated with anti-MYC (D3N8F, Cell Signaling Technology), followed by washing and incubating with secondary antibody (#A-11011, Invitrogen). After 30 minutes incubation, cells were washed with MACS buffer and analyzed on BD Fortessa flow cytometer.
Extracellular acidification (ECAR) and oxygen consumption rate (OCR)
ECAR and OCR were measured in control cells or IFNγ-treated cells with a Seahorse XF96 Analyzer (Seahorse Bioscience, Billerica, MA, USA). In brief, 3 × 105 A375 cells were seeded in RPMI-1640 with 10% FBS and incubated for 1 to 2 hours to allow cell adhesion. The media were removed and replaced with Seahorse assay media with 2 mM glutamine without glucose. The plates containing cells were incubated for 1 hour at 37°C without CO2. Extracellular flux analysis was performed at 37°C without CO2 in the XF96 analyzer (Seahorse Bioscience) following the manufacturer’s instructions. Port additions and times are indicated in the figures. Glucose (10 mM), Oligomycin (1.25 μM), and 2-DG (5 mM), were injected where relevant, and ECAR (mpH/min) or OCR (pmol O2/min) were measured in real time.
Quantification of enzymatic activity
A375 cells were treated with 10 ng/ml IFNγ for 36 hours. Cells were collected and the catalytic activity of glycolysis rate-limiting enzymes were quantitated by Hexokinase Colorimetric Assay Kit (MAK091, Sigma-Aldrich), Phosphofructokinase (PFK) Activity Colorimetric Assay Kit (MAK093, Sigma-Aldrich), GAPDH Activity Assay Kit (MAK277, Sigma-Aldrich), Pyruvate Kinase Activity Assay Kit (MAK072, Sigma-Aldrich), and Lactate Dehydrogenase Activity Assay Kit (MAK066, Sigma-Aldrich), respectively, according to the manufacturer’s instructions.
Quantification of lactate, NAD+/NADH, and pyruvate
To analyze the metabolites, cells were washed with warm fresh medium and incubated in warm fresh medium for 1 hour to balance the metabolites. Then, extracellular lactate was measured with a Lactate Assay Kit (MAK064, Sigma-Aldrich). Intracellular NAD+/ NADH and pyruvate were measured with a NAD+/NADH quantification kit (MAK037, Sigma-Aldrich) and Pyruvate assay kit (MAK071, Sigma-Aldrich), respectively, according to the manufacturer’s instructions.
Quantitative PCR (qPCR)
Total RNA was isolated from cells by column purification (Direct-zol RNA Miniprep Kit, Zymo Research) with DNase treatment. cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) with random hexamer primers. Quantitative PCR (qPCR) was performed on cDNA using Fast SYBR Green Master Mix (Thermo Fisher Scientific) on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). Gene expression was quantified using the primers listed in Table S8. Fold changes in mRNA expression were calculated by the ΔΔCt method using ACTB as an endogenous control. Results are expressed as fold change by normalizing to the controls.
Western blotting
Cells were washed in cold PBS and lysed in 1 × RIPA lysis buffer (Pierce) with 1 × protease inhibitor (Pierce). Lysates were incubated on ice for 10 min and cleared by centrifugation at 15,000g for 15 minutes. Protein concentration was quantified using a BCA protein assay kit (Thermo Fisher). Thirty microgram protein was mixed with sample buffer (Thermo Fisher) with β-ME and denatured at 95°C for 5 minutes. Sample was separated by SDS–PAGE and transferred to a Nitrocellulose Membrane (Bio-Rad). Membranes were blocked with 5% w/v non-fat dry milk and incubated with primary antibodies overnight at 4 °C and HRP-conjugated secondary antibodies (CST) for 1 hour at room temperature. Signal was detected using Clarity and Clarity Max Western ECL Blotting Substrates (Bio-Rad) and captured using ChemiDoc Imaging System (Bio-Rad). Antibodies were as follows: anti-FGF2 (Abcam, #EP1735, 1:1000), anti-phosphorylated PKM2 (Y105) (CST, #3827, 1:1000), anti-phosphorylated PKM2 (S37) (Signalway, #11456, 1:1000), anti-PKM2 (Proteintech, #15822-1-AP, 1:1000), anti-GBP1 (Proteintech, #15303, 1: 1,000), anti-GBP2 (Proteintech, #11854, 1:1000), anti-SIRT1 (CST, #2028, 1: 1,000), anti-phospho-STAT1 (CST, #9167, 1:1000), anti-STAT1 (CST, #9172, 1: 1,000), anti-MYC (CST, #13987, 1:1,000), anti-acetylated-lysine (CST, #9441, 1:1000), anti-acetyl-β-catenin (K49) (CST, #9030, 1:1000), anti-non-phospho (Active) β-catenin (CST, #8814, 1:1000), anti-CD44 (CST, #37259, 1:1000), anti-Histone H3 (CST, #4499, 1:1000), anti-Tubulin (CST, #2148, 1:1000), and anti-GAPDH (Proteintech, #60004, 1: 5,000). Subcellular fractionation was performed with NEPER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, #78833).
Co-Immunoprecipitation (Co-IP)
Cells were collected with IP lysis buffer (Pierce, 87787) plus protease inhibitor. Protein concentration was determined with BCA protein assay kit. 200-500 μg protein samples were added with 20 μl EZview Red ANTI-FLAG M2 Affinity Gel (Sigma Aldrich), then incubated with gentle rocking at 4°C overnight. Samples were then centrifuged at 7500 ×g for 30 seconds at 4°C. Cell pellets were washed 4 times with IP lysis buffer, resuspended with 40 μl 2 × sample buffer with β-ME, and heated for 5 minutes at 95°C. The denatured protein samples were analyzed by Western blot.
Intratumoral immune cell profiling
To analyze intratumoral T cells, single-cell suspensions were prepared from fresh tumor tissues by physically passing through 100 μm cell strainers. Immune cells were enriched by density gradient centrifugation. 2-3 μl of Anti-CD45 (30-F11, BD Biosciences), anti-CD90 (53-2.1, BD Biosciences), anti-CD3 (145-2C11, BD Biosciences), anti-CD4 (GK1.5, BD Biosciences), and anti-CD8 (53-6.7, BD Biosciences) antibodies were added for 20 minutes for surface staining. The cells were then washed and resuspended in 1 ml of freshly prepared Fix/Perm solution (BD Biosciences) at 4 °C overnight, followed by washing with Perm/Wash buffer (BD Biosciences). All samples were read on an LSR Fortessa cytometer and analyzed with FACS DIVA software v. 8.0 (BD Biosciences).
Signature score computation
We used normalized expression of genes (Z-score) to define the following signatures: CD8+ T cell infiltration (CD8A, CD8B, PRF1, and GZMB), IFNγ signaling (IFNG, STAT1, IRF1, GBP1, CXCL9, IFIT1, IFITM1, and IFI35), FGF signaling (FGF2, FGFR1, FRS2, GRB2, SOS1, FOS, MET, RUNX2, SHC1, PTK2B, RPS6KA1, and SSH1), β-catenin signaling (CTNNB1, MYC, CCND2, LEF1, TCF7, ADAM17, AXIN1, AXIN2, CUL1, DKK1, DKK4, DVL2, FZD1, FZD8, MAML1, NCOR2, NCSTN, NKD1, NUMB, PPARD, PSEN2, RBPJ, and SKP2), Shh signaling (GLI1, PTCH1, TLE1, SHH, SCG2, RTN1, SLIT1, OPHN1, and NRCAM), Hippo signaling (YAP1, WTIP, AMOT, WWC1, WWTR1, LATS2, SCHIP1, MARK3, WWC2, LATS1, and PJA1), KRAS signaling (ABCB1, AKAP12, APOD, ARG1, BMP2, BTBD3, CBL, EPHB2, and MAP7), NOTCH signaling (NOTCH1, HEY1, HEY2, JAG1, JAG2, HES1, SKP1, KAT2A, MAML2), EGF signaling (EGF, EGFR, ETS2, IER3, EGR1, IER2, NEDD9, EGR3, EGR4, ID1, ID3, ARC, NAB2, and NAP1L1), and stemness/ invasiveness gene signature (MYC, CD44, NANOG, SOX2, KLF4, BMI1, VIM, ZEB1, FN1, TJP1, SNAI1, SNAI2, and TWIST1).
Statistical analysis
For cell-based experiments, biological triplicates were performed in each single experiment, unless otherwise stated. For animal studies, no less than 5 replicates per group were employed. Animals were randomized into different groups after tumor cell inoculation. The investigators were not blinded to allocation during experiments and outcome assessment. Data are shown as mean values with standard derivation. Statistical analysis was performed using GraphPad Prism8 software (GraphPad Software, Inc.) or in R. Two-tailed t-test was used to compare treatment groups with control groups; Survival function was estimated by Kaplan–Meier methods and log-rank test was used to calculate statistical differences. In cases with non-proportional hazards, the restricted mean survival time was utilized. Inverse-probability weighted (IPW) estimator was used for multivariable modeling. The weights were estimated using the covariate balancing propensity score method with consideration for age, gender, histology, performance status, number of lines of prior therapy, BRAF mutational status (Cohort 1 only), and smoking status (Cohort 2 only). For all tests, P < 0.05 was considered significant. Sample size was not predetermined. Unless noted, samples were independent biological replicates.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| anti-FGF2 | Abeam | Cat # ab92337, RRID: AB_2049652 |
| anti-phospho-PKM2 (Y105) | Cell Signaling Technology | Cat# 3827, RRID: AB_1950369 |
| anti-phospho-PKM2 (S37) | Signalway | Cat# 11456 |
| anti-PKM2 | Proteintech | Cat# 15822, RRID: AB_1851537 |
| anti-GBP1 | Proteintech | Cat# 15303, RRID: AB_2247448 |
| anti-GBP2 | Proteintech | Cat# 11854, RRID: AB_2109336 |
| anti-SIRT1 | Cell Signaling Technology | Cat # 2028, RRID: AB_1196631 |
| anti-phospho-STAT1 | Cell Signaling Technology | Cat #9167, RRID: AB_561284 |
| anti-STAT1 | Cell Signaling Technology | Cat #9172, RRID: AB_2198300 |
| anti-MYC | Cell Signaling Technology | Cat# 13987, RRID: AB_2631168 |
| anti-acetylated-lysine | Cell Signaling Technology | Cat #9441, RRID: AB_331805 |
| anti-acetyl-β-catenin (K49) | Cell Signaling Technology | Cat # 9030, RRID: AB_2797689 |
| anti-non-phospho-β-catenin | Cell Signaling Technology | Cat #8814, RRID: AB_11127203 |
| anti-CD44 | Cell Signaling Technology | Cat # 37259, RRID: AB_2750879 |
| anti-Histone H3 | Cell Signaling Technology | Cat #4499, RRID: AB_10544537 |
| anti-Tubulin | Cell Signaling Technology | Cat # 2148, RRID: AB_2288042 |
| anti-GAPDH | Proteintech | Cat # 60004, RRID: AB_2107436 |
| V500 Rat Anti-Mouse CD45 | BD Biosciences | Cat #561487, RRID: AB_10697046 |
| PE Rat Anti-Mouse CD44 | BD Biosciences | Cat # 561860, RRID: AB_10895375 |
| APC anti-mouse CD133 | Biolegend | Cat # 141207, RRID: AB 10898121 |
| FITC Rat Anti-Mouse CD90.2 | BD Biosciences | Cat # 553004, RRID: AB_394543 |
| PerCP-Cy5.5 Anti-Mouse CD3e | BD Biosciences | Cat #551163, RRID: AB_394082 |
| APC-Cy7 Anti-Mouse CD4 | BD Biosciences | Cat #552051, RRID: AB_394331 |
| Alexa Fluor 700 Rat Anti-Mouse CD8a | BD Biosciences | Cat # 557959; RRID: AB_396959 |
| anti-mouse PD-L1 | Bio X Cell | Cat # BE0101, RRID: AB_10949073 |
| rat lgG2b isotype control | Bio X Cell | Cat# BE0090, RRID: AB_1107780 |
| anti-mouse CTLA-4 | Bio X Cell | Cat # BE0131, RRID: AB 10950184 |
| anti-mouse CD8α | Bio X Cell | Cat # BE0117, RRID: AB_10950145 |
| anti-Human CD8α | Abcam | Cat # ab17147, RRID: AB 443686 |
| anti-Human FGF2 | Abcam | Cat # ab92337, RRID: AB_2049652 |
| anti-Human MYC | Abcam | Cat # ab32072, RRID: AB_731658 |
| anti-Human CD133 | Miltenyi Biotec | Cat# 130-090-422, RRID: AB_244339 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DASA-58 | Cayman Chemical | Cat # 13941 |
| ML-265 | Cayman Chemical | Cat # 13942 |
| L002 | Cayman Chemical | Cat # 17778 |
| Wnt-C59 | Cayman Chemical | Cat # 16644 |
| Nicotinamide riboside | Cayman Chemical | Cat # 23132 |
| β-Nicotinamide Mononucleotide | Cayman Chemical | Cat # 16411 |
| Sirtinol | Cayman Chemical | Cat# 10523 |
| Salermide | Cayman Chemical | Cat # 13178 |
| Palbociclib | Cayman Chemical | Cat # 16273 |
| human IFNγ | R&D Systems | Cat # 285-IF |
| mouse IFNγ | R&D Systems | Cat # 485-MI |
| human DKK1 | R&D Systems | Cat # 5439-DK |
| mouse DKK1 | R&D Systems | Cat # 5897-DK |
| human FGF2 | R&D Systems | Cat # 233-FB |
|
| ||
| Critical commercial assays | ||
|
| ||
| Lactate Assay Kit | Sigma-Aldrich | Cat # MAK064 |
| NAD/NADH Quantitation Kit | Sigma-Aldrich | Cat # MAK037 |
| Pyruvate Assay Kit | Sigma-Aldrich | Cat # MAK071 |
| Hexokinase Colorimetric Assay Kit | Sigma-Aldrich | Cat # MAK091 |
| Phosphofructokinase (PFK) Activity Colorimetric Assay Kit | Sigma-Aldrich | Cat # MAK093 |
| GAPDH Activity Assay Kit | Sigma-Aldrich | Cat # MAK277 |
| Pyruvate Kinase Activity Assay Kit | Sigma-Aldrich | Cat # MAK072 |
| Lactate Dehydrogenase Activity Assay Kit | Sigma-Aldrich | Cat # MAK066 |
|
| ||
| Deposited data | ||
|
| ||
| Gene expression profile of patient samples | 39,64,65 | dbGaP: phs000673.v2. p1 |
|
| ||
| Gene expression profile of patient samples | 60 | GEO datasets: GSE91061 |
|
| ||
| Gene expression profile of patient samples | 62 | dbGaP: phs001919. v1.p1 |
|
| ||
| Gene expression profile of cell samples | 11 | GEO datasets: GSE99299 |
|
| ||
| Gene expression profile of single cell patient samples | 63 | GEO datasets: GSE123814 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human Cell line: A375 | ATCC | Cat # CRL-1619 |
| Human Cell line: MeWo | ATCC | Cat # HTB-65 |
| Human Cell line: A2058 | ATCC | Cat # CRL-11147 |
| Human Cell line: CHL-1 | ATCC | Cat # CRL-9446 |
| Human Cell line: Malme-3M | ATCC | Cat # HTB-64 |
| Human Cell line: G361 | ATCC | Cat # CRL-1424 |
| Human Cell line: SKMEL-1 | ATCC | Cat # HTB-67 |
| Human Cell line: SKMEL-2 | ATCC | Cat # HTB-68 |
| Human Cell line: SKMEL-5 | ATCC | Cat # HTB-70 |
| Human Cell line: SKMEL-28 | ATCC | Cat # HTB-72 |
| Human Cell line: A549 | ATCC | Cat # CCL-185 |
| Human Cell line: H1299 | ATCC | Cat # CRL-5803 |
| Human Cell line: H292 | ATCC | Cat # CRL-1848 |
| Human Cell line: H23 | ATCC | Cat # CRL-5800 |
| Human Cell line: H69 | ATCC | Cat# HTB-119 |
| Human Cell line: H460 | ATCC | Cat # HTB-177 |
| Human Cell line: H661 | ATCC | Cat# HTB-183 |
| Human Cell line: H1437 | ATCC | Cat # CRL-5872 |
| Human Cell line: H1975 | ATCC | Cat # CRL-5908 |
| Mouse Cell line: YUMM1.7 | ATCC | Cat # CRL-3362 |
| Mouse Cell line: YUMM5.2 | ATCC | Cat # CRL-3367 |
| Mouse Cell line: B16-F0 | ATCC | Cat # CRL-6322 |
| Mouse Cell line: LLC | ATCC | Cat # CRL-1642 |
| Human Cell line: 293T | ATCC | Cat # CRL-3216 |
| Mouse Cell line: MC38 | 44 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laborat Cat # JAX: 000664 | |
| Mouse: NSG | The Jackson Laborat Cat # JAX: 005557 | |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers | Integrated DNA Technologies | See Tables S6–S8 |
|
| ||
| Recombinant DNA | ||
|
| ||
| PGL-3 Basic | Promega | Cat # E1751 |
| PCI-neo | Promega | Cat # E1841 |
| TOP-FLASH | Addgene | Cat # 12456 |
| FOP-FLASH | Addgene | Cat # 12457 |
| FLAG-β-catenin | Addgene | Cat # 16828 |
| PLKO.1 | Addgene | Cat # 10879 |
| PX459 | Addgene | Cat # 48139 |
| Fgf2 promoter and exon1 | Origene | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| BD FACSDiva Software | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software#Overview |
| Graphpad Prism 8.0 softwan | GraphPad Software | http://www.graphpad.com/scientificsoftware/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Highlights.
Hyperprogressive disease (HPD) occurs during immunotherapy
HPD is associated with high levels of IFNγ, FGF2, and β-catenin signaling
CD8+ T cell derived IFNγ promotes HPD via rewiring cancer oncometabolic pathways
High IFNγ-FGF2-β-catenin signature is a potential biomarker and target for HPD
Acknowledgments
We thank our laboratory members for intellectual input. This work was supported in part by the research grants from the U.S. NIH/NCI R01 grants (CA217648, CA123088, CA099985, CA193136, and CA152470), VA (VA150CU000182, I01BX005267), foundations (MRA689853, L863220), and the NIH/NCI through the University of Michigan Rogel Cancer Center Grant (CA46592).
Declaration of Interests
W.Z. has served as a scientific advisor or consultant for NGM, CrownBio, Cstone, ProteoVant, Hengenix, NextCure, and Intergalactic. L.A.F receives clinical trial support from Array, Kartos, BMS, EMD Serono, Pfizer and is a consultant for Elsevier. C.D.L. receives clinical trial support from BMS, Merck, and Novartis. A.Q. has research funding from Merck and Clovis. A.A. serves as a consultant for Merck, AstraZeneca, Bristol-Myers Squibb and Pfizer/EMD Serono. A.A. receives research funding through the University of Michigan from Merck, Genentech, Prometheus Laboratories, Mirati Therapeutics, Roche, Bayer, Progenics, Astellas Pharma, Arcus Biosciences, AstraZeneca, Bristol-Myers Squibb and Clovis Oncology. All other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
References:
- 1.Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Marabelle A, Herrscher H, Caramella C, Rouby P, Fizazi K, and Besse B (2020). Immunotherapy discontinuation - how, and when? Data from melanoma as a paradigm. Nat Rev Clin Oncol 17, 707–715. 10.1038/s41571-020-0399-6. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, and Pardoll DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou W, Wolchok JD, and Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324. 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiou VL, and Burotto M (2015). Pseudoprogression and Immune-Related Response in Solid Tumors. J Clin Oncol 33, 3541–3543. 10.1200/JCO.2015.61.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria JC, and Ferte C (2017). Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin Cancer Res 23, 1920–1928. 10.1158/1078-0432.CCR-16-1741. [DOI] [PubMed] [Google Scholar]
- 7.Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, and Kurzrock R (2017). Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res 23, 4242–4250. 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castro F, Cardoso AP, Goncalves RM, Serre K, and Oliveira MJ (2018). Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol 9, 847. 10.3389/fimmu.2018.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer discovery 7, 188–201. 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404 e399. 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du W, Hua F, Li X, Zhang J, Li S, Wang W, Zhou J, Wang W, Liao P, Yan Y, et al. (2021). Loss of optineurin drives cancer immune evasion via palmitoylation-dependent IFNGR1 lysosomal sorting and degradation. Cancer Discov. 10.1158/2159-8290.CD-20-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spranger S, and Gajewski TF (2018). Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 18, 139–147. 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. (2019). Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 178, 933–948 e914. 10.1016/j.cell.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554 e1512. 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh S, Kumar S, Srivastava RK, Nandi A, Thacker G, Murali H, Kim S, Baldeon M, Tobias J, Blanco MA, et al. (2020). Loss of ELF5-FBXW7 stabilizes IFNGR1 to promote the growth and metastasis of triple-negative breast cancer through interferon-gamma signalling. Nat Cell Biol 22, 591–602. 10.1038/s41556-020-0495-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e310. 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 20.Clevers H, and Nusse R (2012). Wnt/beta-catenin signaling and disease. Cell 149, 1192–1205. 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 21.MacDonald BT, Tamai K, and He X (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17, 9–26. 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 23.Gatenby RA, and Gillies RJ (2004). Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4, 891–899. 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 24.Turner N, and Grose R (2010). Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10, 116–129. 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 25.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, and Salomon DS (2006). Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16. 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, Park K, Alexandru A, Lupinacci L, de la Mora Jimenez E, et al. (2019). Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med 381, 2020–2031. 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 27.Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, Castro G Jr., Srimuninnimit V, Laktionov KK, Bondarenko I, et al. (2019). Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393, 1819–1830. 10.1016/S0140-6736(18)32409-7. [DOI] [PubMed] [Google Scholar]
- 28.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, et al. (2016). Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 387, 1837–1846. 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 29.Hodi FS, Ballinger M, Lyons B, Soria JC, Nishino M, Tabernero J, Powles T, Smith D, Hoos A , McKenna C, et al. (2018). Immune-Modified Response Evaluation Criteria In Solid Tumors (imRECIST): Refining Guidelines to Assess the Clinical Benefit of Cancer Immunotherapy. J Clin Oncol 36, 850–858. 10.1200/JCO.2017.75.1644. [DOI] [PubMed] [Google Scholar]
- 30.Ferte C, Fernandez M, Hollebecque A, Koscielny S, Levy A, Massard C, Balheda R, Bot B, Gomez-Roca C, Dromain C, et al. (2014). Tumor growth rate is an early indicator of antitumor drug activity in phase I clinical trials. Clin Cancer Res 20, 246–252. 10.1158/1078-0432.CCR-13-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park HJ, Kim KW, Won SE, Yoon S, Chae YK, Tirumani SH, and Ramaiya NH (2021). Definition, Incidence, and Challenges for Assessment of Hyperprogressive Disease During Cancer Treatment With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Netw Open 4, e211136. 10.1001/jamanetworkopen.2021.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matos I, Martin-Liberal J, Garcia-Ruiz A, Hierro C, Ochoa de Olza M, Viaplana C, Azaro A, Vieito M, Brana I, Mur G, et al. (2020). Capturing Hyperprogressive Disease with Immune-Checkpoint Inhibitors Using RECIST 1.1 Criteria. Clin Cancer Res 26, 1846–1855. 10.1158/1078-0432.CCR-19-2226. [DOI] [PubMed] [Google Scholar]
- 33.Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, Mazieres J, Zalcman G, Brosseau S, Le Moulec S, et al. (2018). Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol 4, 1543–1552. 10.1001/jamaoncol.2018.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim CG, Kim KH, Pyo KH, Xin CF, Hong MH, Ahn BC, Kim Y, Choi SJ, Yoon HI, Lee JG, et al. (2019). Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann Oncol 30, 1104–1113. 10.1093/annonc/mdz123. [DOI] [PubMed] [Google Scholar]
- 35.Saada-Bouzid E, Defaucheux C, Karabajakian A, Coloma VP, Servois V, Paoletti X, Even C, Fayette J, Guigay J, Loirat D, et al. (2017). Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol 28, 1605–1611. 10.1093/annonc/mdx178. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y, Kim CH, Lee HY, Lee SH, Kim HS, Lee S, Cha H, Hong S, Kim K, Seo SW, et al. (2019). Comprehensive Clinical and Genetic Characterization of Hyperprogression Based on Volumetry in Advanced Non-Small Cell Lung Cancer Treated With Immune Checkpoint Inhibitor. J Thorac Oncol 14, 1608–1618. 10.1016/j.jtho.2019.05.033. [DOI] [PubMed] [Google Scholar]
- 37.Tunali I, Gray JE, Qi J, Abdalah M, Jeong DK, Guvenis A, Gillies RJ, and Schabath MB (2019). Novel clinical and radiomic predictors of rapid disease progression phenotypes among lung cancer patients treated with immunotherapy: An early report. Lung Cancer 129, 75–79. 10.1016/j.lungcan.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrioli R, Mazzei MA, Giorgi S, Cesqui E, Gentili F, Francini G, Volterrani L, and Francini E (2020). Hyperprogressive disease in advanced cancer patients treated with nivolumab: a case series study. Anticancer Drugs 31, 190–195. 10.1097/CAD.0000000000000864. [DOI] [PubMed] [Google Scholar]
- 39.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. (2017). Integrative clinical genomics of metastatic cancer. Nature 548, 297–303. 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou W (2005). Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5, 263–274. 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 41.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. (2003). Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med 9, 562–567. 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 42.Maher PA (1996). Nuclear Translocation of fibroblast growth factor (FGF) receptors in response to FGF-2. J Cell Biol 134, 529–536. 10.1083/jcb.134.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128, 1708. 10.1172/JCI120803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nikbakht N, Tiago M, Erkes DA, Chervoneva I, and Aplin AE (2019). BET Inhibition Modifies Melanoma Infiltrating T Cells and Enhances Response to PD-L1 Blockade. J Invest Dermatol 139, 1612–1615. 10.1016/j.jid.2018.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, et al. (2016). Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol Res 4, 845–857. 10.1158/2326-6066.CIR-16-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chocarro-Calvo A, Garcia-Martinez JM, Ardila-Gonzalez S, De la Vieja A, and Garcia-Jimenez C (2013). Glucose-induced beta-catenin acetylation enhances Wnt signaling in cancer. Mol Cell 49, 474–486. 10.1016/j.molcel.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 48.Wolf D, Rodova M, Miska EA, Calvet JP, and Kouzarides T (2002). Acetylation of beta-catenin by CREB-binding protein (CBP). J Biol Chem 277, 25562–25567. 10.1074/jbc.M201196200. [DOI] [PubMed] [Google Scholar]
- 49.Simic P, Zainabadi K, Bell E, Sykes DB, Saez B, Lotinun S, Baron R, Scadden D, Schipani E, and Guarente L (2013). SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol Med 5, 430–440. 10.1002/emmm.201201606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, Jahn SC, Saldanha SA, Chase P, Planck J, et al. (2013). Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol Cancer Ther 12, 610–620. 10.1158/1535-7163.MCT-12-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, Varela-Rey M, Rotili D, Nebbioso A, Ropero S, et al. (2009). Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 28, 781–791. 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 52.Imai S, and Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol 24, 464–471. 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li G, Kryczek I, Nam J, Li X, Li S, Li J, Wei S, Grove S, Vatan L, Zhou J, et al. (2021). LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nat Cell Biol 23, 526–537. 10.1038/s41556-021-00672-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liberti MV, and Locasale JW (2016). The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218. 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, et al. (2011). Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283. 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, and Lu Z (2011). Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480, 118–122. 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et al. (2009). Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2, ra73. 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. (2012). Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8, 839–847. 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nathanson T, Ahuja A, Rubinsteyn A, Aksoy BA, Hellmann MD, Miao D, Van Allen E, Merghoub T, Wolchok JD, Snyder A, and Hammerbacher J (2017). Somatic Mutations and Neoepitope Homology in Melanomas Treated with CTLA-4 Blockade. Cancer Immunol Res 5, 84–91. 10.1158/2326-6066.CIR-16-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e916. 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abril-Rodriguez G, Torrejon DY, Liu W, Zaretsky JM, Nowicki TS, Tsoi J, Puig-Saus C, Baselga-Carretero I, Medina E, Quist MJ, et al. (2020). PAK4 inhibition improves PD-1 blockade immunotherapy. Nat Cancer 1, 46-+. 10.1038/s43018-019-0003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, Rizvi SM, Qin A, Waninger JJ, Lang X, et al. (2021). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med 27, 152–164. 10.1038/s41591-020-1131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tay C, Qian Y, and Sakaguchi S (2020). Hyper-Progressive Disease: The Potential Role and Consequences of T-Regulatory Cells Foiling Anti-PD-1 Cancer Immunotherapy. Cancers (Basel) 13. 10.3390/cancers13010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, Thaker YR, Zhang Q, McArdel SL, Juneja VR, et al. (2021). PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med 218. 10.1084/jem.20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, Ferro S, Ganzinelli M, Gasparini P, Huber V, et al. (2019). Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin Cancer Res 25, 989–999. 10.1158/1078-0432.CCR-18-1390. [DOI] [PubMed] [Google Scholar]
- 69.Bosch-Barrera J, Oliva E, Sais E, Vasquez CA, Rosello A, and Menendez JA (2019). Hyperprogression after first dose of immunotherapy in a patient with radioresistant metastasis from nonsmall cell lung cancer. Anticancer Drugs 30, 1067–1070. 10.1097/CAD.0000000000000837. [DOI] [PubMed] [Google Scholar]
- 70.Fricke J, Mambetsariev I, Pharaon R, Subbiah S, Rajurkar S, and Salgia R (2020). Hyperprogression on immunotherapy with complete response to chemotherapy in a NSCLC patient with high PD-L1 and STK11: A case report. Medicine (Baltimore) 99, e22323. 10.1097/MD.0000000000022323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng Y, Zhang L, Zeng T, Liu L, Liu X, Yang Y, Zhang H, and Ruan Z (2020). Characterization of Hyperprogression After Immunotherapy in a Lung Adenocarcinoma Patient With Strong Expression of Programmed Death Ligand 1. J Thorac Oncol 15, e4–e8. 10.1016/j.jtho.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 72.Bernal Vaca L, Mendoza SD, Vergel JC, Rueda X, and Bruges R (2019). Hyperprogression in Pediatric Melanoma Metastatic to the Breast Treated with a Checkpoint Inhibitor. Cureus 11, e3859. 10.7759/cureus.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schuiveling M, Tonk EHJ, Verheijden RJ, and Suijkerbuijk KPM (2020). Hyperprogressive disease rarely occurs during checkpoint inhibitor treatment for advanced melanoma. Cancer Immunol Immunother. 10.1007/s00262-020-02716-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yilmaz M, and Akovali B (2020). Hyperprogression after nivolumab for melanoma: A case report. J Oncol Pharm Pract 26, 244–251. 10.1177/1078155219845436. [DOI] [PubMed] [Google Scholar]
- 75.Forschner A, Hilke FJ, Bonzheim I, Gschwind A, Demidov G, Amaral T, Ossowski S, Riess O, Schroeder C, Martus P, et al. (2020). MDM2, MDM4 and EGFR Amplifications and Hyperprogression in Metastatic Acral and Mucosal Melanoma. Cancers (Basel) 12. 10.3390/cancers12030540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J, Wang X, Yang X, Zhao H, and Huo L (2020). FDG PET Findings of Hyperprogression During Immunotherapy in a Patient With Hepatocellular Carcinoma. Clin Nucl Med 45, 92–93. 10.1097/RLU.0000000000002849. [DOI] [PubMed] [Google Scholar]
- 77.Hoshal SG, Wickwire PC, Gandour-Edwards RF, Rajappa P, and Cates DJ (2020). Metastatic Renal Cell Carcinoma Presenting as a Rapidly Enlarging Endotracheal Mass Due to Hyperprogression on Anti-PD1 Immunotherapy. Ear Nose Throat J, 145561320931215. 10.1177/0145561320931215. [DOI] [PubMed] [Google Scholar]
- 78.Chan AS, Ng VY, Snider J, Kallen ME, and Miller KD (2020). Hyperprogression of Liver Metastasis With Neoadjuvant Immunotherapy for Soft Tissue Sarcoma. Cureus 12, e8575. 10.7759/cureus.8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ji Z, Peng Z, Gong J, Zhang X, Li J, Lu M, Lu Z, and Shen L (2019). Hyperprogression after immunotherapy in patients with malignant tumors of digestive system. BMC Cancer 19, 705. 10.1186/s12885-019-5921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forys JT, Kuzmicki CE, Saporita AJ, Winkeler CL, Maggi LB Jr., and Weber JD (2014). ARF and p53 coordinate tumor suppression of an oncogenic IFN-beta-STAT1-ISG15 signaling axis. Cell Rep 7, 514–526. 10.1016/j.celrep.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, and Parada LF (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oshimori N, Oristian D, and Fuchs E (2015). TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160, 963–976. 10.1016/j.cell.2015.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T, Falcone M, et al. (2019). Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 572, 254–259. 10.1038/s41586-019-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ, et al. (2010). Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 466, 133–137. 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, and Gajewski TF (2016). Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res 4, 563–568. 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, Fleming M, Rezazadeh A, Mellado B, Varlamov S, et al. (2019). Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med 381, 338–348. 10.1056/NEJMoa1817323. [DOI] [PubMed] [Google Scholar]
- 87.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. (2009). New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45, 228–247. 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 88.Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, Green MD, Wang W, Lin H, Li W, et al. (2020). Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. 10.1172/JCI134402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The expression sequencing from the University of Michigan cohort was previously deposited (National Center for Biotechnology Information database of Genotypes and Phenotypes (dbGaP) under accession number phs000673.v2. p1.) 39,64,65. The expression sequencing data and corresponding clinical annotation for patients receiving ICB was previously deposited (Gene Expression Omnibus (GEO) under accession number GSE91061; National Center for Biotechnology Information database of Genotypes and Phenotypes (dbGaP) under accession number phs001919. v1.p1. 59–62. The expression profile of cell lines was previously deposited (Gene Expression Omnibus (GEO) under accession number GSE99299) 11. The single cell RNA sequencing data of cancer patients upon immunotherapy was previously deposited (Gene Expression Omnibus (GEO) under accession number GSE123814) 63. All raw data supporting the findings of this study are available from the corresponding authors upon request.