

Abstract
Autologous chimeric antigen receptor (CAR) T cells targeting the CD19 antigen have demonstrated a high complete response rate in relapsed/refractory B cell malignancies. However, autologous CAR T cell therapy is not an option for all patients. Here we optimized conditions for clinical-grade manufacturing of allogeneic CD19-CAR T cells using CD45RA-depleted donor memory T cells for a planned clinical trial. Memory T cells were activated using the MACS GMP T Cell TransAct reagent and transduced in the presence of LentiBOOST with a clinical grade lentiviral vector that encodes a 2nd generation CD19-CAR with a 41BB.zeta endodomain. Transduced T cells were transferred to a G-Rex cell culture device for expansion and harvested on day 7 or 8 for cryopreservation. The resulting CD19-CAR(Mem) T cells expanded on average 34.2-fold, and mean CAR expression was 45.5%. The majority of T cells were CD4+ and had a central memory or effector memory phenotype, and retained viral specificity. CD19-CAR(Mem) T cells recognized and killed CD19-positive target cells in vitro and had potent antitumor activity in an ALL xenograft model. Thus we have successfully developed a cGMP-compliant process to manufacture donor-derived CD19-CAR(Mem) T cells. Our manufacturing process could be readily adapted for CAR(Mem) T cells targeting other antigens.
INTRODUCTION
Autologous chimeric antigen receptor (CAR) T cells targeting antigens expressed on tumor cells have proven to be efficacious in treating several types of hematological malignancies, especially relapsed/refractory CD19-positive acute lymphoblastic leukemia (ALL)(1–5). This has resulted in the FDA approval of several autologous CD19-CAR T cells(6, 7). However, autologous CD19-CAR T cell therapy is not an option for all patients. For some patients an autologous CAR product is unable to be generated, because of an inability to obtain enough T cells for CAR T cell production due to patient disease status. In addition, patients’ T cells might be dysfunctional due to previous therapies including high dose chemotherapy, leading to suboptimal CAR T cells (8). Accumulating evidence suggests that the quality of the autologous T cells used to produce CAR T cells is one of the major determinants of an efficacious cellular product (8, 9). However, several factors, including prior chemotherapy, host cancer environment, and high leukemia burden, can affect the quality of the autologous T cells, and thus the quality of the CAR T cell product made from those T cells.
To overcome these limitations, several approaches utilizing allogeneic T cells are currently being developed or evaluated clinically (10, 11). A major concern of allogeneic CAR T cells is their potential graft versus host disease (GVHD) activity, which can theoretically be overcome by several approaches. While the majority of efforts now focus on eliminating alloreactivity by disrupting genes such as TRAC and CD52 using genome editing technologies, a simpler approach might be the use of antigen-experienced memory T cells (Tm) that are depleted of alloreactive T cells, obtained from healthy donors to produce CAR T cells (10, 12–13). Alloreactive naïve T cells (Tn) express CD45RA and can be depleted using magnetic bead based technology (13–16). Several groups, including our own, have shown that infusion of CD45RA-depleted CD3+ T cells is safe, with resultant proliferation and persistence of infused Tm cells for up to 180 days without increasing severe GVHD (15, 17). The added benefit of using these cells was their anti-virus activity to control virus infection (18). The use of Tm cells from healthy donors may enable successful clinical scale production of high-quality CAR T cells.
In a preclinical study, CD19-CAR T cells generated from CD45RA-depleted T cells showed antigen-dependent tumor-killing activity both in vitro and in vivo in a xenograft NSG mouse model(19). Here we now report the development of a current good manufacturing practice (cGMP) compliant procedure to generate CD19-CAR T cells from memory T cells (CD19-CAR(Mem) T cells) by CD45RA- and CD14-depletion for a future clinical study evaluating the safety and efficacy of these cells in patients with relapsed/refractory leukemia. Our manufacturing process yielded sufficient cell numbers for the planned dose levels of the developed clinical study. Likewise, the generated CD19-CAR(Mem) T cells had the desired biologic characteristics, showing anti-ALL activity in vitro as well as in an ALL xenograft model.
MATERIALS AND METHODS
Lentiviral vector expressing CD19-CAR
The self-inactivating (SIN) 3rd generation lentivirus vector that encodes a CD19-CAR with a 41BB.zeta signaling domain under the control of a MND promoter has been described previously (19, 20). It was produced using an established producer cell line in the Children’s GMP, LLC and was purified by ion-exchange chromatography. The lentiviral vector is currently being used in an early phase clinical study (NCT03573700) that evaluates the safety and efficacy of autologous CD19-CAR T cells in pediatric patients with relapsed/refractory ALL. Vector copy numbers in transduced cells were measured by ddPCR as described in a previous study (21).
CD45RA+ and CD14+ cell depletion using CliniMACS
The CD45RA+ and CD14+ cell depletion process was modified from the previous report(15). Briefly, cryopreserved apheresis mononuclear cells from healthy donors (Cellero) were thawed in a 37°C water bath, washed with CliniMACS buffer (Miltenyi Biotec) supplemented with 0.5% Human serum albumin (HSA, Grifols, CA), and resuspended in 90 mL CliniMACS buffer containing 0.5% HSA. The cells were then incubated with 5 mL of IVIG (Grifols Therapeutics) at room temperature for 15 min, followed by 30 min labeling with 7.5 mL of human CD45RA MicroBeads (Miltenyi Biotec) and 7.5 mL of human CD14 MicroBeads (Miltenyi Biotec) at room temperature. The labeled cells were washed twice with CliniMACS buffer containing 0.5% HSA and resuspended in 120 mL CliniMACS buffer containing 0.5% HSA before being depleted using a CliniMACS Plus instrument. The Depletion 3.1 program was used with a CliniMACS depletion tubing set. After completion of the program, the CD45RA−CD14− cells from the collection bag were harvested for experiments.
Activation, transduction, and expansion of T(Mem) cells
On day 0, CD45RA−CD14− T cells (2×106 cells/mL) were cultured in X-VIVO15 media (Lonza) containing 5% Human AB serum (Valley Biomedicals), 10 ng/mL each of rhIL-7 (Miltenyi Biotec) and rhIL-15 (Miltenyi Biotec) and activated with 1:17.5 v/v of MACS GMP T Cell TransAct (Miltenyi Biotec) reagent for 18 to 24 hours in 6 well plates. On day 1, activated T cells were transduced with clinical-grade CD19-CAR vector in the presence of 4 μg/mL protamine sulfate for 18 to 24 hours. LentiBOOST (Sirion Biotech) is also added at this step in some groups. On day 2, cells were resuspended in X-VIVO15 medium containing 5% Human AB serum, 10 ng/mL each of rhIL-7 and rhIL-15 (complete media), and transferred to G-Rex-6M culture plates (Wilson Wolf) with a total volume of 100 mL for each well. On day 5, rhIL-7 (10 ng/mL) and rhIL-15 (10 ng/mL) were added again to the medium. On day 7 or 8, expanded T cells were harvested, washed with plasmaLyte A (Baxter) containing 4% HSA, and cryopreserved in plasmaLyte A containing 6% pentastarch (Preservation Solutions), 5.75% HSA, and 5% DMSO using a controlled-rate freezer.
Flow cytometry analysis
The purity of CD45RA- and CD14-depleted cells were analyzed using the following monoclonal antibodies: CD3, CD4, CD8, CD14, CD19, CD45, CD45RA, CD45RO, CD56 (detailed antibody information is listed in the supplemental table 3) by flow cytometry. DAPI or 7-Aminoactinomycin D (7-AAD) was used as a dead cell marker. CD19-CAR expression was detected using a human CD19 Fc Chimera (Creative Biomart). CAR T cells were immunophenotyped using the following antibodies: CD3, CD4, CD8, CD45, CD27, PD1, TIM3, CCR7, and CD45RO. For FOXP3 staining, cryopreserved CD45RA−CD14− and CD45RA+CD14+ cells were thawed and stained with CD3 and CD4 antibodies follow by staining with a FOXP3 antibody using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). A CytoFlex flow cytometer (Beckman Coulter) was used to acquire immunofluorescence data. FlowJo software (BD Biosciences) and Prism software (GraphPad Software, San Diego, CA) were used for data analysis and generating the graph.
Cytotoxicity assay
Cytotoxicity of CD19-CAR T cells against BV173 (CD19+, German Collection of Microorganisms and Cell Cultures) or KG1a (CD19−, American Type Tissue Collection) target cells was determined in a flow cytometry-based cytotoxicity assay. Assays were performed with CD19-CAR T cells and non-transduced (NT) T cells of the same donors. Cells were thawed and rested for 2–3 days in complete media before performing the assay. Target cells were harvested and stained with CFSE (Invitrogen) in a final concentration of 5 μM. Afterward, they were washed and resuspended with RPMI-1640 media (GE Healthcare) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Avantor). Target cells and CD19-CAR T cells were combined in U-bottom shaped 96-well plates at an effector to target (E:T) ratios of 3:1 and 0.3:1 using 50,000 target cells per well in a total volume of 200 μl and incubated for 18–24 h at 37 °C with 5% CO2. Plates were centrifuged at 400 × g for 3 min and cells were resuspended in 200 μl PBS containing DAPI (BD Pharmingen) and 2 μL Count Bright™ Absolute Counting Beads (Invitrogen). 200 bead events were counted for each well using a BD FACS Lyrics flow cytometer or CytoFLEX flow cytometer. Live target cells were identified as CFSE+DAPI− and samples of target cells only were used as controls for spontaneous cell lysis. The percentage of cytotoxic activity was calculated using the following equation: Cytotoxicity % = (1- live target cell number in the assay well/live target cell number in the control well) *100%
Cytokine Assays
CAR T cells were thawed and rested for 2–3 days in complete media, and then co-cultured with BV173 or KG1a target cells at a 2:1 effector to target (E:T) ratio, using 500,000 target cells per well in a total volume of 2 ml in a 24-well plate. After 20–24 hours, culture supernatants were collected, and the Interferon-gamma (IFN-γ) and IL-2 levels were determined by ELISA using Quantikine® ELISA kit Human IFN-γ (R&D Systems) and Quantikine® ELISA kit Human IL-2 (R&D Systems) as per the manufacturer’s instructions.
Elispot assay
IFN-γ enzyme-linked immunospot (Elispot) assays were performed as previously described (22, 23). Briefly, cells were plated in triplicates at a seeding density of 1×105 – 2×105 cells/well, and antigen-specific T cell reactivity was measured after direct stimulation with peptide libraries (pepmixes; JPT Peptide Technology, Acton, MA) dissolved in DMSO consisting of 15 amino acid long peptides with an 11 amino acid overlap that covered the coding regions of the following proteins: Cytomegalovirus (CMV) pp65 (#PM-PP65–2) and IE-1 (#PM-IE-1); Adenovirus (AdV) serotype 5 penton protein (#PM-PM-HAdV5) and AdV serotype 3 hexon protein (#PM-HAdV3); Epstein Barr Virus (EBV) EBNA1 (#PM-EBV-EBNA1), LMP2 (#PM-EBV-LMP2), and BZLF1 (#PM-EBV-BZLF1); Human Herpes Virus 6 (HHV6) U54 (#PM-HHV6-U54) and U90 (#PM-HHV6-U90); BK Virus (BKV) VP1 (#PM-BKV-VP1) and Large T Antigen (#PM-BKV-LTA); Herpes Simplex Virus (HSV) type 2 E3 ICP0 (#PM-HSV2-E3), R1 (#PM-HSV2-R1), gD (#PM-HSV2-gD) and VP22 (#PM-HSV2-VP22); Varicella Zoster Virus (VZV) IE62 (#PM-VZV-IE62), IE63 (#PM-VZV-IE63), and gE (#PM-VZV-gE). Media (no peptide, DMSO only) served as a negative control, and Staphylococcus aureus Enterotoxin Type B (SEB) was used as a positive control. After 18–20 hours incubation, plates were developed, dried at room temperature, and sent to Zellnet Consulting (New York, NY) for quantification. The frequency of T cells specific to each antigen was noralized as spot-forming cells (SFC) per 5×105 cells.
Mixed Lymphocyte Reaction
De-identified peripheral blood (HemaCare, Memphis, TN) was fractionated over a Ficoll-hypaque gradient. The resulting PBMC fraction was depleted of CD19+ cells by magnetic isolation using CD19-MicroBeads. CAR T cell products were thawed, rested for 2 days in complete media. CAR T cells were washed, labeled with CellTrace CFSE and plated in triplicate on three replicate 96-well round bottom plates. CFSE labeled CAR T products (100,000 cells) were co-cultured with CD19-depleted PBMC (100,000 cells) at a 1:1 ratio for 3, 4, or 5 days. CAR T cells and CD19-depleted PBMC served as negative controls and CAR T cells stimulated with ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator served as positive controls. Plates were washed 2 times with PBS and resuspended in PBS with CountBright absolute counting beads and analyzed on a BD FACSLyric analyzer and analyzed with FlowJo software.
Xenograft model
In vivo experiments were performed under a protocol approved by St. Jude Children’s Research Hospital (St. Jude) Institutional Animal Care and Use Committee (IACUC). Animals were housed in specific pathogen-free rooms for the duration of the experiments. Female NSG mice (NOD-scid IL2Rgammanull, NOD-scid IL2Rgnull, NSG, NOD scid gamma) were obtained from the St. Jude breeding colony or purchased from the Jackson Laboratory and used at 8–10 weeks of age. Mice received by tail vein 3×106 BV173 tumor cells modified to express a GFP.ffluc fusion gene (BV173ffluc)(24). Seven days later, mice in the treatment groups were infused with effector T cells. Five control mice received non-transduced (NT) T cells. CD19-CAR(Mem) and CD19-CAR T cells, along with corresponding non-transduced (NT) mock control cells were thawed and allowed to recover in complete media with cytokines for 48 hours prior to administration. Mice were imaged at St. Jude’s Center for In Vivo Imaging and Therapeutics using the Xenogen IVIS®−200 imaging system (IVIS, Xenogen Corp., Alameda, CA) as previously described(25), and euthanized at predefined endpoints or when they met euthanasia criteria in accordance with SJCRH’s Animal Resource Center as previously described(25).
Statistical Analysis
For all experiments, the number of biological replicates and statistical analysis used are described in the figure legend. For comparison between two groups, a two-tailed t-test was used. For comparisons of three or more groups, values were log-transformed as needed and analyzed by ANOVA with Dunnett’s or Tukey’s post-hoc test. Survival was assessed by the log-rank test. Statistical analyses were conducted with Prism software (GraphPad Software, San Diego, CA).
RESULTS
Purification of memory T cells
Cryopreserved apheresis mononuclear cells from healthy donors were thawed and processed for CD45RA depletion. While CD45RA depletion removes naïve T cells along with most of B and NK cells, CD14+ mononuclear cells remain (14), which may lower the transduction efficiency(26). We therefore decided to combine CD45RA with CD14 depletion to obtain a purer memory T cell population as previously reported (27). We performed CD45RA and CD14 depletion from 4 healthy donors with an average yield of 4.9% of the total processed cells (Table 1). After the depletion, 94.5±1.6% of the nucleated cells were CD3+ (Fig. 1A, B). Within the CD3+ gate, 99.90±0.12% cells are CD45RO+CD45RA− memory T cells with 89.7±3.6% being CD4+ and 7.9±3.8% being CD8+ (Fig. 1C, D). CD45RA and CD14 depletion did not result in a selection of regulatory T cells as judged by intracellular staining for FOXP3 (Supplemental Fig. S1).
Table 1.
T-cell yield post CD45RA and CD14 depletion
| Donor | Starting volume of cryopreserved Apheresis MNC (mL) | Starting TNC count | Post-depletion CD45RA−/CD14− cell count (% of starting TNC count) |
|---|---|---|---|
| E19027 | 60 | 4.85×109 | 1.78×108 (3.7%) |
| E20002 | 60 | 9.36×109 | 6.26×108 (6.7%) |
| E19028 | 60 | 5.27×109 | 1.15×108 (2.2%) |
| E20003 | 102 | 6.85×109 | 4.87×108 (7.1%) |
MNC: mononuclear cell; TNC: total nucleated cell
Figure 1. Efficient depletion of CD45RA+ and CD14+ cells from cryopreserved apheresis MNCs.

Cryopreserved apheresis MNCs were thawed and labeled with anti-CD45RA and anti-CD14 MicroBeads for depletion using the CliniMACS Plus instrument. Pre-depletion and post-depletion samples were analyzed by flow cytometry for (A, B) CD45RA, CD14, CD3, CD19 and (C, D) CD45RO, CD4, CD8. (A, C) Representative data. (B, D) Summary data; n=4 donors, Two way ANOVA, **P<0.01, ****P<0.0001, NS: not significant.
Generation and phenotypic characterization of CAR T cells
CD19-CAR(Mem) T cell generation was adapted from our protocol to generate standard CD19-CAR T cells (Fig. 2A) with the only exception being the inclusion of LentiBOOST (LB) (28–30) to enhance lentiviral transduction. We first evaluated a range of LB concentrations (0.5 mg/mL, 1 mg/mL 2 mg/mL) in combination with different multiplicity of transduction (MOI) of our clinical-grade CD19-CAR vector (Supplemental Fig. S2A–G). At all three evaluated LB concentrations and MOIs (2.5, 5, and 10), LB increased the percentage of CD19-CAR-positive cells and VCN. Cell count on day 7, fold expansion, viability and the CD4 to CD8 T cell ratio was not affected by LB or MOI. Based on these results, we chose a concentration of 1 mg/mL LB and evaluated the benefit in three additional donors at MOIs of 5 and 10. LB did not affect expansion (Fig. 2B) but significantly increased CD19-CAR expression and VCN at an MOI of 5 or 10 (Fig. 2C, D). The percentage of CD4 and CD8, central memory, effector memory, and terminally differentiated subpopulations were not affected by LB (Fig. 2E, F). In all groups, the majority of the cells in the final product were CD4+ cells and were with an effector memory CD45RO+CCR7− phenotype (Fig. 2E, F). 1H-NMR analysis of the final drug product showed that the residual LB level is below the limit of detection of 0.94 μg/mL (data not shown).
Figure 2. Optimization of transduction of memory T cells with clinical-grade lentiviral CD19-CAR vector.
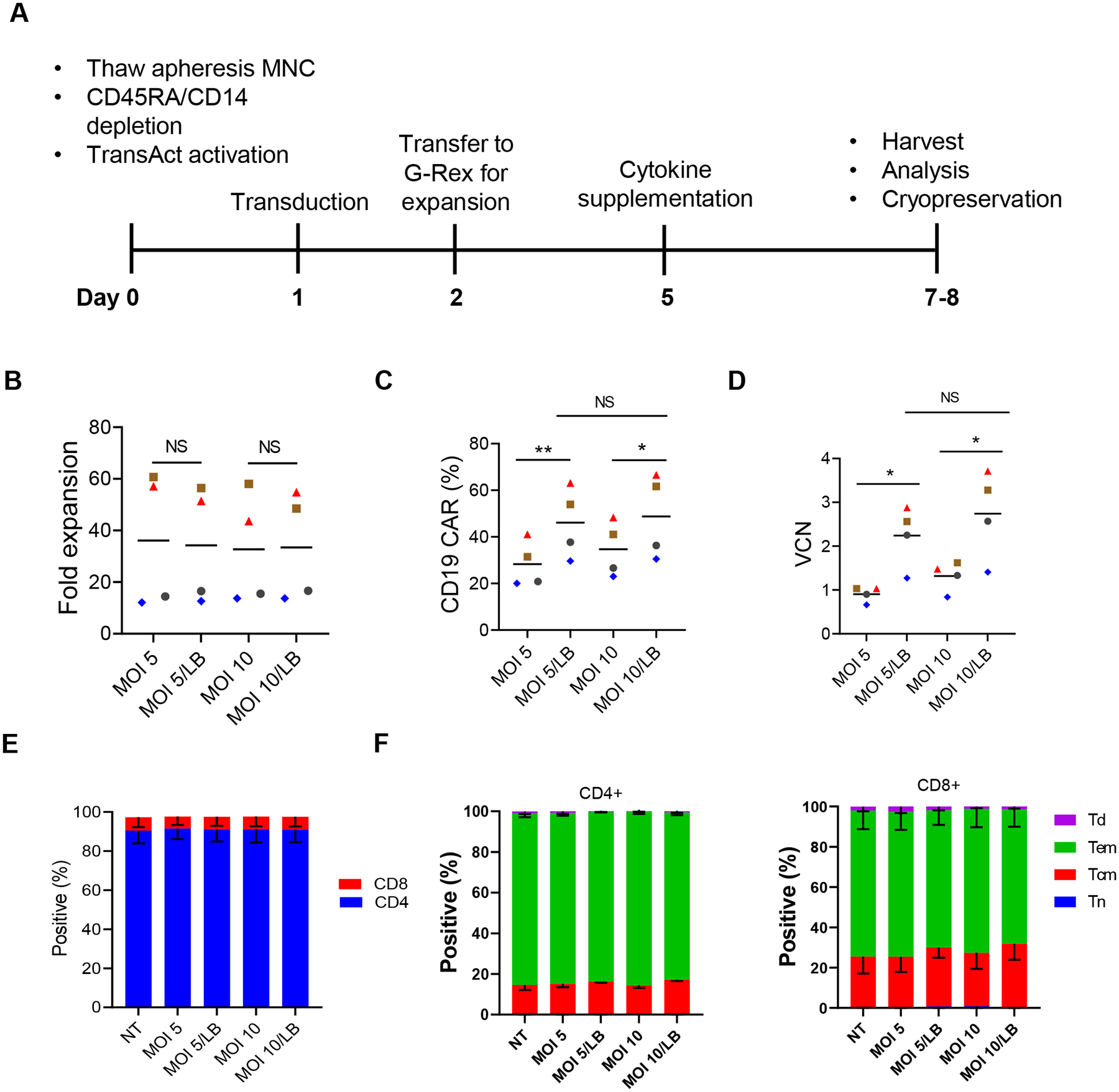
(A) Schema of CAR T cell manufacturing. (B) Fold expansion (n=4 donors; Mean values depicted as bars). (C) CAR expression based on flow cytometry (n=4 donors; Mean values depicted as bars). (D) Vector copy number (VCN; n=4 donors; Mean values depicted as bars). (E) CD4 and CD8 expression (n=4 donors; Mean values depicted as bars). (F) T cell subpopulations based on CCR7 and CD45RO expression (n=3 donors). Tn: Naïve-like, CCR7+CD45RO; Tcm: central memory, CCR7+CD45RO+; Tem: effector memory, CCR7−CD45RO+; Td: terminally differentiated, CCR7−CD45RO−; n=4 donors, Paired t-test, **P<0.01, *P<0.05, NS: not significant. Data for one donor is also shown in part in Supplemental Figure S1.
Optimization of cell yield of CD19-CAR(Mem) T cells
For manufacturing standard CD19-CAR T cell clinical-grade products, typically 2×106 transduced cells per 10 cm2 are transferred to a G-Rex100M-CS resulting in an expansion of >50-fold within 7 days (data not shown). In contrast, the expansion of CD19-CAR(Mem) T cells was only in the range of ~10 – 60 fold (Fig. 2B, n=4). In order to ensure sufficient number of CD19-CAR(Mem) T cells for the prescribed dose levels for our approved clinical study without extending expansion time or using multiple G-Rex devices, we evaluated a range of seeding cell numbers, ranging from 1.5×106 to 1×107 cells per 10 cm2, in G-Rex culture devices for three donors to evaluate if we can achieve the target cell number by increasing the seeding cell number (Fig. 3). On day 7, the number of harvested cells was directly proportional to the initial seeding number (Fig. 3A). However, fold expansion (Fig. 3B), cell viability (Fig. 3C), and CD19-CAR expression (Fig. 3D) were not affected by seeding density. We therefore selected a starting cell range of 5×106 – 1×107 cells per 10 cm2 for our clinical-grade CD19-CAR(Mem) T cell manufacturing protocol. We have transferred the manufacturing process to the Children’s GMP at St Jude and two tech-transfer runs and two engineering runs confirmed the robustness of the developed manufacturing process (Supplemental Tables 1 and 2).
Figure 3. Optimizing cell seeding number in G-Rex device for cell expansion.
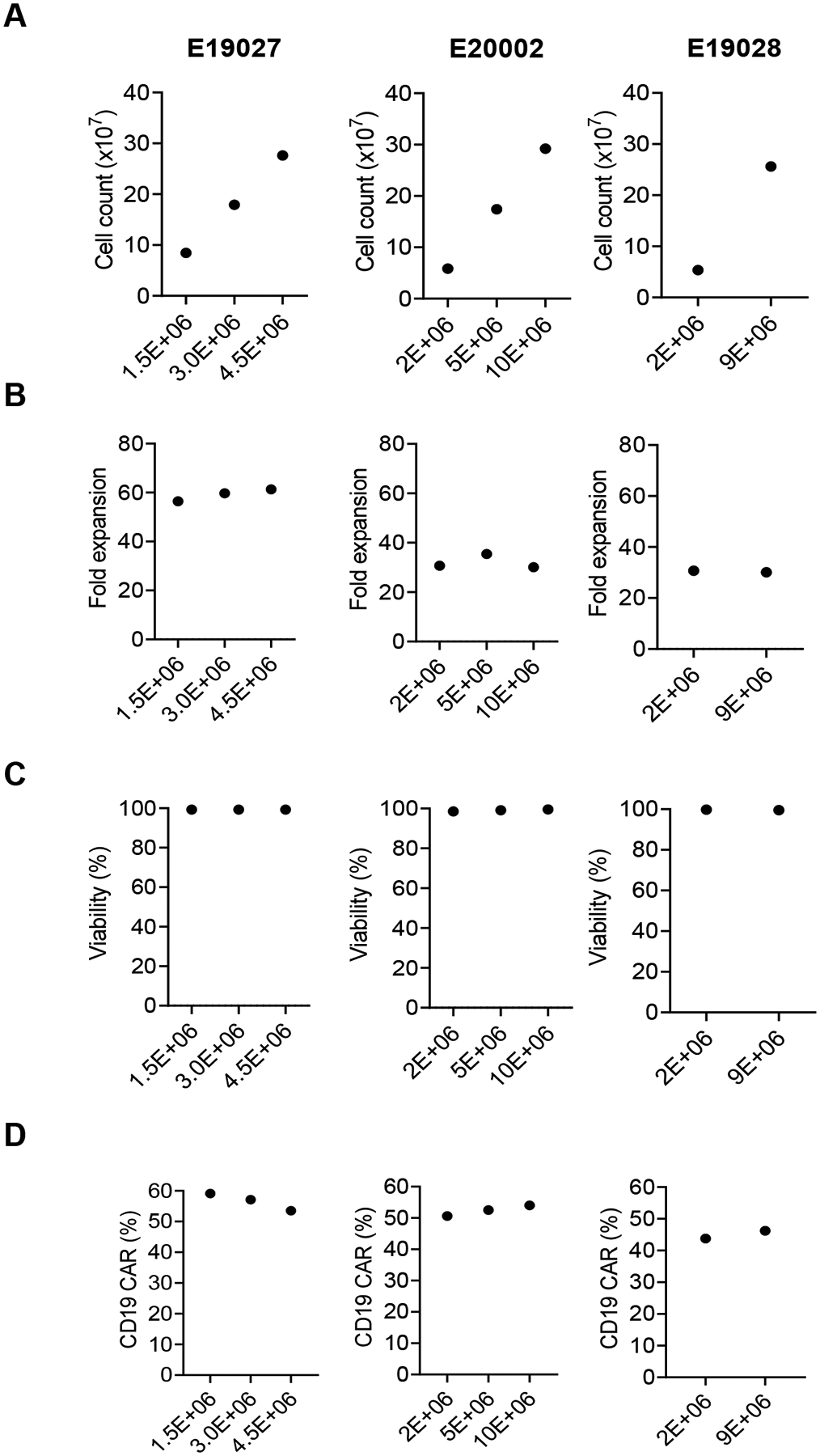
After transduction on day 2, various number of cells were transferred into the G-Rex device for expansion. Cells were harvested on day 7. (A) Total cell count. (B) Fold expansion. (C) Cell viability, as determined by AOPI exclusion. (D) Percentage of CD19-CAR expression as determined by flow cytometry. Donor identifications are shown at the top of each column. Each column represents one donor. Numbers on the X-axis represent the number of cells seeded per cm2 in G-Rex®6M (10 cm2). The data from the 2.0×106 groups are also included in the data shown in Figure 2B, D).
CD19-CAR(Mem) T cells maintain viral-specificity post-expansion and have reduced alloreactivity
To assess if during the ex vivo expansion the virus-specificity of memory T cells is retained, we performed IFN-γ Elispot assay on day 0 and on day 7 on NT and CD19-CAR(Mem) T cells that were transduced at an MOI 5 +/− LB. T cell populations were stimulated with pepmixes specific for CMV, AdV, EBV, HHV6, BKV, HSV2, and VZV with DMSO and SEB serving as controls. While there was an increase in IFN-γ secreting cells in response to DMSO (baseline) and SEB (positive control), virus-specific T cell responses persisted (Fig. 4A–D), with no significant differences in frequency when normalized to baseline (Fig. 4E). To demonstrate that CD19-CAR(Mem) T cells have reduced alloreactivity we performed a standard MLR with ‘third party’ PBMCs in which CD19-positive B cells were depleted. Standard CD19-CAR T cells proliferated briskly as judged by CSFE dilution; in contrast the proliferation of CD19-CAR(Mem) T cells was significantly lower, indicative of reduced alloreactivity (Supplemetal Fig. 3A–C).
Figure 4. CD19-CAR(Mem) T cells maintain virus specificity post-expansion.

IFN-γ Elispot assay using indicated pepmixes as stimulators; DMSO: negative control; SEB: positive control; n= 4 donors. (A) Day 0, (B) Day 7: NT, (C) Day 7: MOI 5, (D) Day 7: MOI 5 + LB. (E) To compare data presented in A-B, normalized values were plotted (value minus negative control); n=4 per group, Two-way ANOVA, ****P<0.0001; NS: not significant.
CD19-CAR(Mem) T cells recognize and kill CD19+ target cells
We next evaluated the cytotoxicity of CD19-CAR(Mem) T cells against CD19+ BV173 and CD19− KG1a cell lines (Fig. 5A). CD19-CAR(Mem) T cells, generated with or without LB at an MOI of 5 or 10, significantly killed BV173 cells but not KG1a cells at an E:T ratio of 3:1 or 0.3:1, demonstrating antigen-specific cytotoxic activity. This was confirmed in coculture assays, in which CD19-CAR(Mem) T cells only produced significant amounts of IFN-γ and IL-2 in the presence of BV173 (Fig. 5B).
Figure 5. In vitro functional characterization of CD19-CAR(Mem) T cells.
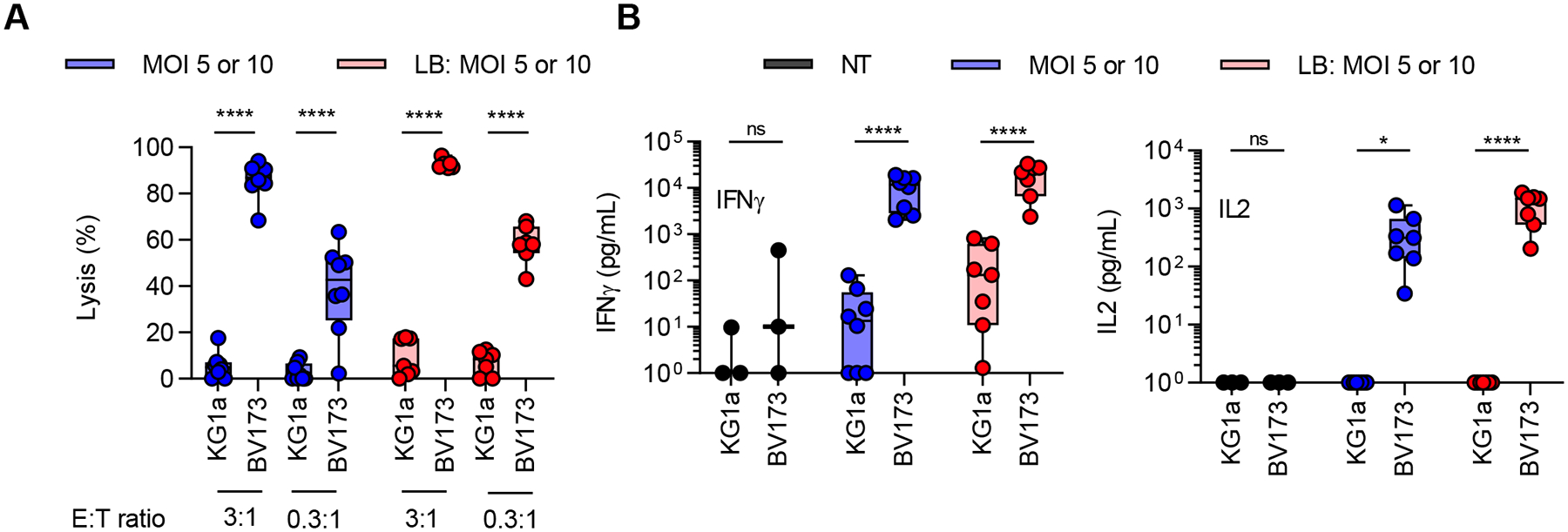
(A) Flow cytometry-based cytotoxicity against CD19+ BV173 and CD19− KG1a leukemia cell line. (B) IFN-γ and IL-2 production after coculturing CAR T cells with tumor cells at an effector to target (E:T) ratio of 2:1. MOI 5 and 10, and MOI 5 and 10 + LB are plotted together. LB: LentiBoost; MOI: multiplicity of Infection; n=4 per group, Two-way ANOVA, *P<0.05, ****P<0.0001; for IL-2 data analysis one outlier value (IL-2: −13.7 pg/mL) was excluded; negative data values were changed to 1 for graphing purposes.
CD19-CAR(Mem) and standard CD19-CAR T cells have comparable anti-tumor activity in vivo
Finally, we compared the in vivo anti-tumor activity of CD19-CAR(Mem) and standard CD19-CAR T cells generated from two donors, E20003 (Fig. 6A–D) and E19028 (Fig. 6E–H) using our standard BV173 NSG murine xenograft model(24). BV173.ffluc-bearing mice received a single i.v. dose of CD19-CAR(Mem), standard CD19-CAR, or NT T cells (Supplemental Fig. S4A–C). We used low CAR T cell doses to observe potential differences between T cell populations. Each mouse received 2×106 CD19-CAR(Mem)+ or 1.3×106 standard CD19-CAR+ T cells for donor E20003, and 2×106 CD19-CAR(Mem)+ or standard CD19-CAR+ T cells for donor E19028. Tumor burden was followed weekly by bioluminescence imaging. For both donors, CD19-CAR(Mem)+ or standard CD19-CAR+ T cells decreased tumor growth (Fig. 6B, C, F, G), resulting in significant survival advantages in comparison to mice that received NT T cells (Fig. 6D, H). No significant difference in overall survival between groups of mice that received CD19-CAR(Mem)+ or standard CD19-CAR+ T cells was observed.
Figure 6. CD19-CAR(Mem) and standard CD19-CAR T cells have comparable antitumor activity in vivo.
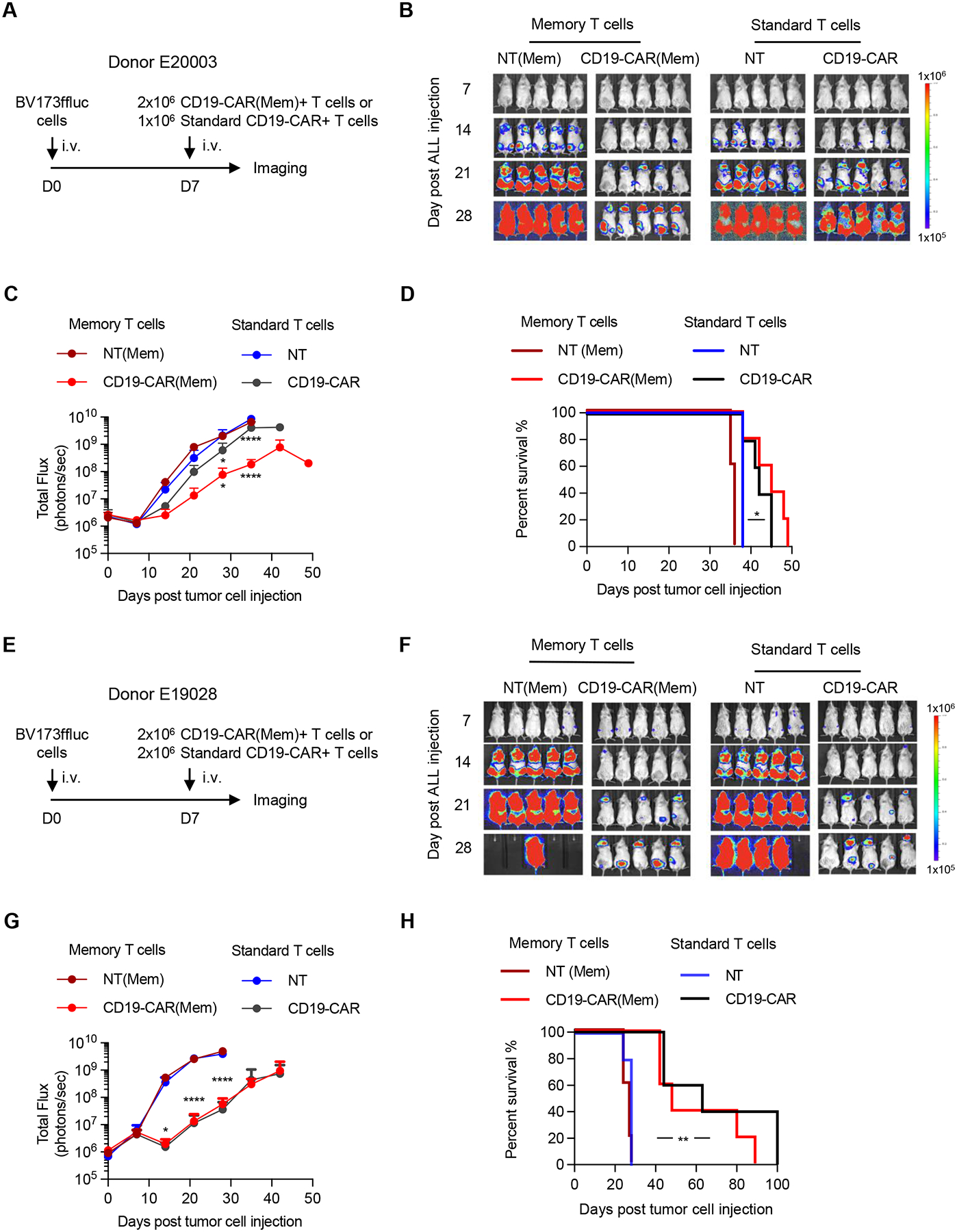
CD19-CAR(Mem) and standard CD19-CAR T cells were generated from two donors (A-D) E20003 and (E-H) E19028 and evaluated in BV173.ffluc xenograft NSG mouse model (n=5 per group). Non-transduced (NT) memory or standard T cells served as control. (B, F) Bioluminescence images. (C, G) Quantification of bioluminescence signal; n=5 per group, Two-way ANOVA, *P<0.05, ***P<0.001, ****P<0.0001. (D, H) Kaplan-Meier survival analysis, Log-rank test, *P<0.05, **P<0.01.
DISCUSSION
Here we report the development of a cGMP compliant process to manufacture CD19-CAR(Mem) T cells from leukapheresis products for a future clinical study, which will evaluate the safety and efficacy of allogeneic CD19-CAR(Mem) T cells in pediatric patients with relapsed/refractory ALL. The manufacturing protocol consists of 4 simple steps: CD45RA/CD14-depletion, activation, transduction with lentiviral vectors in the presence of LentiBOOST, and expansion in the presence of IL-7 and IL-15. Resulting CD19-CAR(Mem) T cell products consisted predominately of CD4+ T cells with an effector memory phenotype. Functional characterization revealed that they retained their virus-specificity and recognized CD19+ ALL cells in vitro and in vivo.
Several approaches are currently being pursued to reduce the alloreactivity of allogeneic CAR T cells (10, 12). We focused here on depleting CD45RA+ (naïve) T cells, based on our and other investigators’ experience that infusion of CD45RA-depleted T cells is associated with a significantly lower risk of GVHD(15, 17). We combined CD45RA with CD14-depletion to reduce monocytes prior to T cell activation. Other investigators have previously generated genetically-modified CD8+ T cells out of the central memory pool by CD45RA+, CD4+, and CD14+ depletion followed by CD62L selection and transduction with i) lentiviral vectors encoding either a CD19-CAR with a zeta- or CD28.zeta-signaling domain(13, 31), or ii) a retroviral vector encoding a αβTCR(32). Infusion of autologous CD8+ memory CD19-CAR T cells was safe and associated with clinical benefit. We decided not to include a CD4+ depletion step since CAR-CD4 T cells have shown promising anti-tumor activity in preclinical models(33, 34). While studies have demonstrated that T cells derived from the central memory compartment (CD45RA−/CD62L+) have improved persistence in vivo(35), we decided not to include a CD62L selection step to enrich for central memory T cells. First, others have reported that including a CD62L selection step results in an average cell yield of 0.4% of input cells, which is 10-fold lower than the average yield (4.9%) obtained on our study without selection. In addition, virus-specific T cells expressing CD19-CAR that were generated without CD62L selection have demonstrated promising activity in one early phase clinical study(36).
CD19-CAR(Mem) T cell yield was lower within 7–8 days post activation in comparison to standard CD19-CAR T cells manufactured in the Children’s GMP, LLC facility at St. Jude. Since longer production time may affect cell quality(37), we optimized the seeding density post-transduction in G-Rex culture devices. In addition, we explored the use of LentiBOOST to enhance the percentage of CAR+ T cells in order to minimize the number of untransduced allogeneic T cells in the infused product. LentiBOOST consistently improved T cell transduction by our clinical-grade VSV-G pseudotyped lentiviral vector as judged by CD19-CAR expression and VCN with no effect on phenotype, expansion, or in vitro functionality. LentiBOOST has been used to enhance lentiviral transduction efficiencies of human hematopoietic stem and progenitor cells for clinical gene therapy studies. In addition, preclinical studies have demonstrated its effectiveness in improving transduction efficiencies in primary murine T cells(30). Our study extends these findings and highlights its usefulness for the manufacturing of clinical-grade CAR T cell products.
We evaluated the frequency of virus-specific T cells in CD19-CAR(Mem) T cell products prior to and 7–8 days post activation/expansion. There was an increase in activation of CD19-CAR(Mem) T cells in the presence of DMSO that had been exposed to LentiBOOST during transduction, and further studies are needed to investigate this finding. Nevertheless, we could unequivocally demonstrate that the frequency of virus-specific T cells was preserved or even increased after activation and expansion in IL-7 and IL-15. Unspecific T cell expansion of non-CD45RA-depleted T cells has been reported to decrease the frequency of virus-specific T cells, most likely due to the proliferative advantage of naïve T cells(38), and our study suggests that this problem can be overcome by CD45RA-depletion prior to activation. Since our donors were not HLA typed, we could not perform tetramer or pentamer analysis of memory T cell populations before and after expansion, however, we are planning to conduct these types of studies in the future.
CD19-CAR(Mem) T cells recognized and killed CD19-positive target cells in vitro, and had significant antitumor activity in our BV173 xenograft model. Importantly, CD19-CAR(Mem) T cells had comparable antitumor activity to CD19-CAR T cells from the same donor that were generated by standard CD4/CD8 selection, confirming results by others that CAR T cells that are predominately CD4+ can have potent antitumor activity(33, 34).
In summary, we have developed a process for manufacturing clinical grade CD19- CAR(Mem) T cells by simple CD45RA and CD14 depletion. The resulting cell products were predominately CD4+, retained virus-specificity, and had significant anti-leukemia activity. We will evaluate the safety and efficacy of these cells in a clinical study entitled ‘A Phase I study evaluating allogeneic memory T cells engineered to express chimeric antigen receptors for CD19 for the treatment of pediatric and young adult patients ≤ 21 years of age with relapsed or refractory CD19-positive leukemia (MEMCAR19)’, which recently received Investigational New Drug (IND) approval by the FDA. If allogeneic CD19-CAR(Mem) T cells have the desired properties in humans, allogeneic CAR(Mem) T cells targeting other tumor-associated antigens could be readily evaluated in early phase clinical studies.
Supplementary Material
Acknowledgments
We thank Suzette Whittaker, Rebecca Banks-Spivey, Jeeba Bellot, Amanda Burton, and Madhuri Kalathur for assistance with engineering runs. We thank Sarah Schell and MaCal Tuggle-Brown for performing Elispot assays. The work was supported by Cookies for Kids’ Cancer and the American Lebanese Syrian Associated Charites. AT received additional support from the American Society of Transplantation and Cellular Therapy (ASTCT) New Investigator Award. Animal imaging was performed by the Center for In Vivo Imaging and Therapeutics, which is supported in part by NIH grants P01CA096832 and R50CA211481. Cellular images were acquired at St. Jude Cell & Tissue Imaging Center which is supported by St. Jude and NCI P30 CA021765.
Conflict of Interest
SG has a research collaboration with TESSA Therapeutics, is a DSMB member of Immatics, and on the scientific advisory board of Tidal. BMT had travel support from Miltenyi Biotec.
REFERENCES
- 1.Cappell KM, Sherry RM, Yang JC, Goff SL, Vanasse DA, McIntyre L, et al. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J Clin Oncol. 2020;38:3805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey NV, Gill S, Hexner EO, Schuster S, Nasta S, Loren A, et al. Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J Clin Oncol. 2020;38:2862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134:2361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol. 2019;19:73–4. [DOI] [PubMed] [Google Scholar]
- 5.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullard A FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov. 2021;20:166. [DOI] [PubMed] [Google Scholar]
- 8.Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. 2019;129:2123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–99. [DOI] [PubMed] [Google Scholar]
- 11.Caldwell KJ, Gottschalk S, Talleur AC. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front Immunol. 2020;11:618427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9(374). [DOI] [PubMed] [Google Scholar]
- 13.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bleakley M, Heimfeld S, Jones LA, Turtle C, Krause D, Riddell SR, et al. Engineering human peripheral blood stem cell grafts that are depleted of naive T cells and retain functional pathogen-specific memory T cells. Biol Blood Marrow Transplant. 2014;20:705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Triplett BM, Shook DR, Eldridge P, Li Y, Kang G, Dallas M, et al. Rapid memory T-cell reconstitution recapitulating CD45RA-depleted haploidentical transplant graft content in patients with hematologic malignancies. Bone Marrow Transplant. 2015;50:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez L, Fernandez A, Mirones I, Escudero A, Cardoso L, Vela M, et al. GMP-Compliant Manufacturing of NKG2D CAR Memory T Cells Using CliniMACS Prodigy. Front Immunol. 2019;10:2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. 2015;125:2677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamcarz E, Madden R, Qudeimat A, Srinivasan A, Talleur A, Sharma A, et al. Improved survival rate in T-cell depleted haploidentical hematopoietic cell transplantation over the last 15 years at a single institution. Bone Marrow Transplant. 2019;55:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan WK, Suwannasaen D, Throm RE, Li Y, Eldridge PW, Houston J, et al. Chimeric antigen receptor-redirected CD45RA-negative T cells have potent antileukemia and pathogen memory response without graft-versus-host activity. Leukemia. 2015;29:387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–84. [DOI] [PubMed] [Google Scholar]
- 21.Bauler M, Roberts JK, Wu CC, Fan B, Ferrara F, Yip BH, et al. Production of Lentiviral Vectors Using Suspension Cells Grown in Serum-free Media. Mol Ther Methods Clin Dev. 2020;17:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–6. [DOI] [PubMed] [Google Scholar]
- 23.Louis CU, Straathof K, Bollard CM, Gerken C, Huls MH, Gresik MV, et al. Enhancing the in vivo expansion of adoptively transferred EBV-specific CTL with lymphodepleting CD45 monoclonal antibodies in NPC patients. Blood. 2009;113:2442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Velasquez MP, Torres D, Iwahori K, Kakarla S, Arber C, Rodriguez-Cruz T, et al. T cells expressing CD19-specific Engager Molecules for the Immunotherapy of CD19-positive Malignancies. Sci Rep. 2016;6:27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riberdy JM, Zhou S, Zheng F, Kim YI, Moore J, Vaidya A, et al. The Art and Science of Selecting a CD123-Specific Chimeric Antigen Receptor for Clinical Testing. Mol Ther Methods Clin Dev. 2020;18:571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noaks E, Peticone C, Kotsopoulou E, Bracewell DG. Enriching leukapheresis improves T cell activation and transduction efficiency during CAR T processing. Mol Ther Methods Clin Dev. 2021;20:675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012;35:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hauber I, Beschorner N, Schrodel S, Chemnitz J, Kroger N, Hauber J, et al. Improving Lentiviral Transduction of CD34(+) Hematopoietic Stem and Progenitor Cells. Hum Gene Ther Methods. 2018;29:104–13. [DOI] [PubMed] [Google Scholar]
- 29.Jang Y, Kim YS, Wielgosz MM, Ferrara F, Ma Z, Condori J, et al. Optimizing lentiviral vector transduction of hematopoietic stem cells for gene therapy. Gene therapy. 2020;27:545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delville M, Soheili T, Bellier F, Durand A, Denis A, Lagresle-Peyrou C, et al. A Nontoxic Transduction Enhancer Enables Highly Efficient Lentiviral Transduction of Primary Murine T Cells and Hematopoietic Stem Cells. Mol Ther Methods Clin Dev. 2018;10:341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casati A, Varghaei-Nahvi A, Feldman SA, Assenmacher M, Rosenberg SA, Dudley ME, et al. Clinical-scale selection and viral transduction of human naive and central memory CD8+ T cells for adoptive cell therapy of cancer patients. Cancer Immunol Immunother. 2013;62:1563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agarwal S, Hanauer JDS, Frank AM, Riechert V, Thalheimer FB, Buchholz CJ. In Vivo Generation of CAR T Cells Selectively in Human CD4(+) Lymphocytes. Mol Ther. 2020;28:1783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. 2018;3:e99048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lapteva N, Gilbert M, Diaconu I, Rollins LA, Al-Sabbagh M, Naik S, et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor-Expressing T Cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019;25:7340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol Res. 2018;6:1100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sili U, Huls MH, Davis AR, Gottschalk S, Brenner MK, Heslop HE, et al. Large-scale expansion of dendritic cell-primed polyclonal human cytotoxic T-lymphocyte lines using lymphoblastoid cell lines for adoptive immunotherapy. J Immunother. 2003;26:241–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.