

Abstract
Cutaneous T-cell lymphoma is a form of non-Hodgkin lymphoma that manifests initially in the skin and disseminates systemically as the disease progresses. Mycosis fungoides and Sézary syndrome are the most common subtypes of cutaneous T-cell lymphoma. Advanced mycosis fungoides and Sézary syndrome are life threatening with few treatment options. We searched for new agents by high-throughput screening of selected targeted compounds and identified high-value targets, including phosphatidylinositol 3-kinase (PI3K) and cyclin-dependent kinases. To validate these hits from the screen, we developed patient-derived xenograft mouse models that recapitulated the cardinal features of mycosis fungoides and Sézary syndrome and maintained histologic and molecular characteristics of their clinical counterparts. Importantly, we established a blood-based biomarker assay using tumor cell-free DNA to measure systemic tumor burden longitudinally in living mice during drug therapy. A PI3K inhibitor, BKM120, was tested in our patient-derived xenograft model leading to disease attenuation and prolonged survival. Isoform-specific small interfering RNA knockdowns and isoform-selective PI3K inhibitors identified PI3K-δ as required for tumor proliferation. Additional studies showed a synergistic combination of PI3K-α/δ inhibitors with histone deacetylase inhibitors. The strong preclinical efficacy of this potent combination against multiple patient-derived xenograft models makes it an excellent candidate for further clinical development.
INTRODUCTION
Cutaneous T-cell lymphoma (CTCL) is a malignancy of skin-homing T cells that has various subtypes. The most common subtype of CTCL is mycosis fungoides (MF), followed by Sézary syndrome (SS). In general, early-stage MF is limited to the skin, is treated with skin-directed therapy, and has a favorable outcome. In contrast, SS and advanced-stage MF often require systemic therapy and have an aggressive clinical course with overall survival of 3.3–5.6 years, which has not improved over four decades (Agar et al., 2010; Kim et al., 2003;Talpur et al., 2012). This highlights the need for new treatments for advanced MF and/or SS.
The pathogenesis of CTCL remains elusive. However, our knowledge of genetic alterations in this disease has grown exponentially in recent years. Analysis of MF and/or SS samples from patients has revealed alterations in several signaling pathways (Choi et al., 2015; da Silva Almeida et al., 2015; Ungewickell et al., 2015;Wang et al., 2015), including TCR signaling and phosphatidylinositol 3-kinase (PI3K)/protein kinase B pathways. Importantly, many of these genetic alterations in signaling networks are shared among other cancers, which afforded an opportunity to examine whether targeted agents developed for other cancers can be translated to CTCL.
Discovery of the effective therapeutic targets in MF and/or SS has been hampered not only by a lack of candidate agents but also by a lack of translational drug-testing platforms. Few MF and/or SS cell lines are available, and xenograft models bearing CTCL cell lines yield subcutaneous masses that grow as solid tumors that do not reproduce the features of MF and/ or SS seen clinically (Krejsgaard et al., 2010; Thaler et al., 2004; Yano et al., 2007). In this study, we took a pathway-directed approach and identified signaling pathways important for cell growth. To validate potential new agents, we developed patient-derived xenograft (PDX) mouse models for MF and/or SS and demonstrated their utility as a drug-testing platform. Using this PDX platform, we validated that the PI3K pathway is of a high-value target and that inhibitions of PI3K and histone deacetylase (HDAC) are highly synergistic in growth inhibition (GI) and apoptosis induction in CTCL.
RESULTS
High-throughput screen
To dissect signaling pathways and identify new targeted agents in CTCL, we performed a high-throughput drug screening of 94 compounds representing Food and Drug Administration–approved and emerging targeted inhibitors of molecular pathways (Figure 1a and Supplementary Figure S1). We performed GI assays for each compound using four CTCL cell lines: HH, H9, Hut78, and MJ. We defined compounds with 50% growth inhibitory concentration (GI50) ≤ 1 μM as hits and organized all the hits on the basis of the molecular pathways that they target (Figure 1a and b). Pathways for which ≥50% of the tested compounds qualified as hits were deemed as important for cell proliferation. These pathways included PI3K, HDAC, proteasome, cyclin-dependent kinase (CDK), checkpoint 1 kinase, and HSP90. Of note, HDAC inhibitors and the proteasome inhibitor, bortezomib, have known clinical activity in CTCL (Heider et al., 2009; Zinzani et al., 2007). In our screen, the PI3K/mTOR pathway had the greatest number of compounds tested, and 10 of 14 tested compounds were qualified as hits, suggesting that the PI3K pathway is crucial for cell growth in CTCL. We chose to focus on BKM120, a pan-PI3K inhibitor, because it was most advanced in clinical development.
Figure 1. Pathway-directed high-throughput screen identified the PI3K pathway as important for the proliferation of CTCL cell lines.
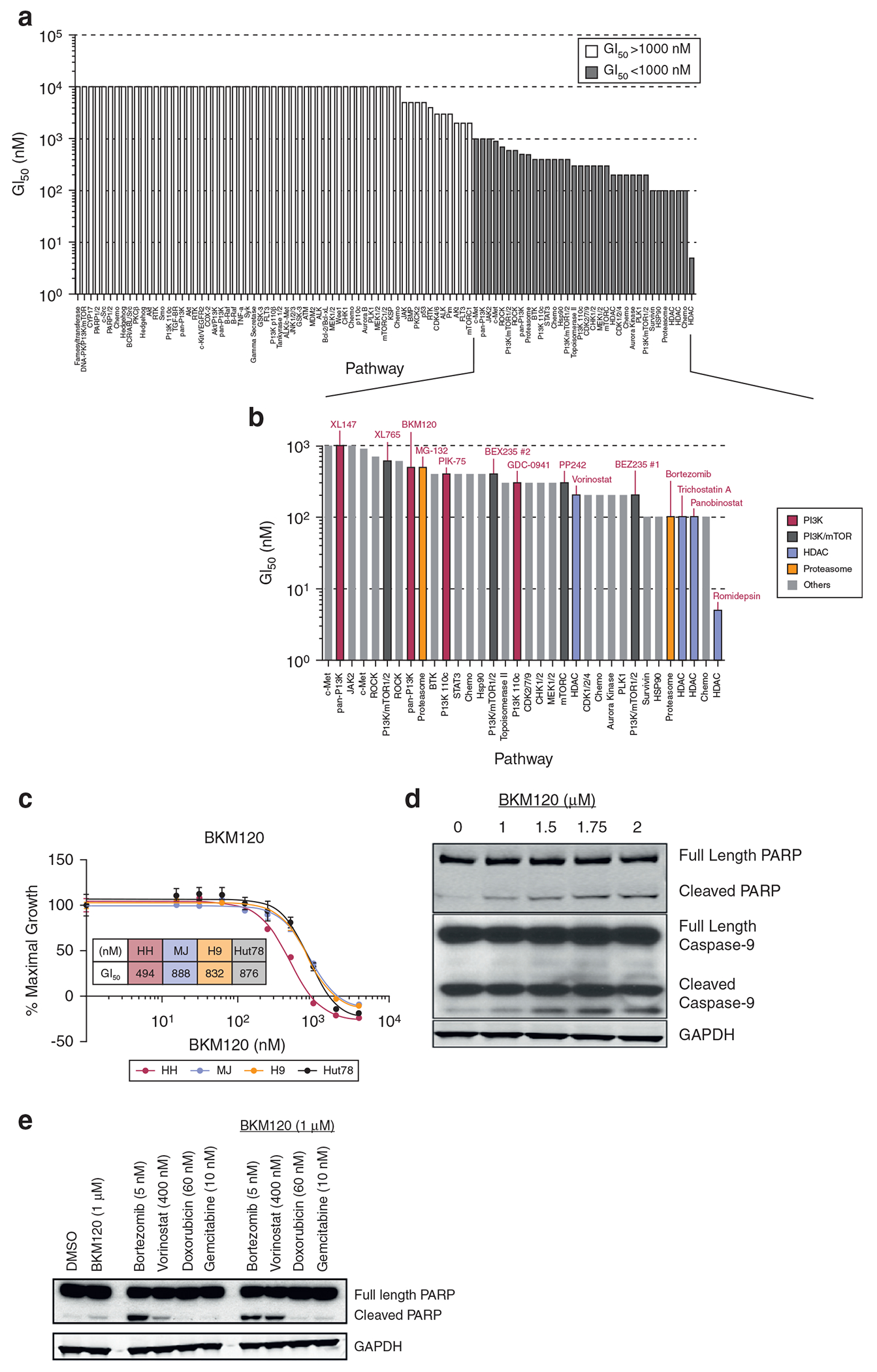
(a) The GI50 of the tested compounds in HH cells. White bars represent compounds with GI50 > 1 μM; gray bars represent compounds with GI50 < 1 μM. (b) The GI50 of the hits from the drug screen (y-axis) and corresponding molecular pathways that they target (x-axis). Red bars denote PI3K inhibitors, dark gray bars denote PI3K/mTOR inhibitors, blue bars denote HDAC inhibitors, green bars denote proteasome inhibitors, and gray bars denote inhibitors of other pathways. (c) Anti-proliferation activity of BKM120 in CTCL cell lines (n = 3). (d) BKM120 induces apoptosis of H9 cells in a dose-dependent fashion. (e) Apoptosis induction by the combination of BKM120 and other agents as hits in H9 cells. CTCL, cutaneous T-cell lymphoma; GI50, 50% growth inhibitory concentration; HDAC, histone deacetylase; PI3K, phosphatidylinositol 3-kinase.
To validate the results from the high-throughput screen, we treated CTCL cells with BKM120 and demonstrated that BKM120 indeed inhibited cell growth and induced apoptosis in CTCL cell lines (Figure 1c and d). Next, we examined whether BKM120 could potentiate apoptosis induction with four agents commonly used for treating CTCL in the clinic: bortezomib, vorinostat, doxorubicin, and gemcitabine. Vorinostat appeared to be the most synergistic partner with BKM120 (Figure 1e). In addition, we observed synergistic effects of BKM120 with other HDAC inhibitors, panobinostat and romidepsin, in both GI and apoptosis induction in Hut78 (Supplementary Figure S2). Both panobinostat and romidepsin have clinical activity in CTCL (Duvic et al., 2013; Piekarz et al., 2009).
Development and characterization of CTCL PDX models
To validate the candidate agents identified from the screen, we set out to establish CTCL PDX murine models. The characteristics of the patients whose tumors were used to establish the PDXs are described in Supplementary Table S1. To establish an MF PDX, we injected 6.6 million cells from Patient-1 into the flank of an NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse subcutaneously. After 19 days, the P0 mouse, PRS-1-P0, developed erythematous, scaly skin lesions and alopecia (Figure 2a). Necropsy revealed lymphoma infiltration in the skin, spleen, and liver. The affected skin showed atypical lymphocytes in the epidermis (Figure 2a). In addition, analysis of the affected skin and splenocytes from PRS-1-P0 demonstrated that malignant cells expressed human CD3 and CD4 without coexpression of CD7, which was an identical profile to that of the donor patient (Figure 2b and c).
Figure 2. Characterization of MF and SS PDX murine models.
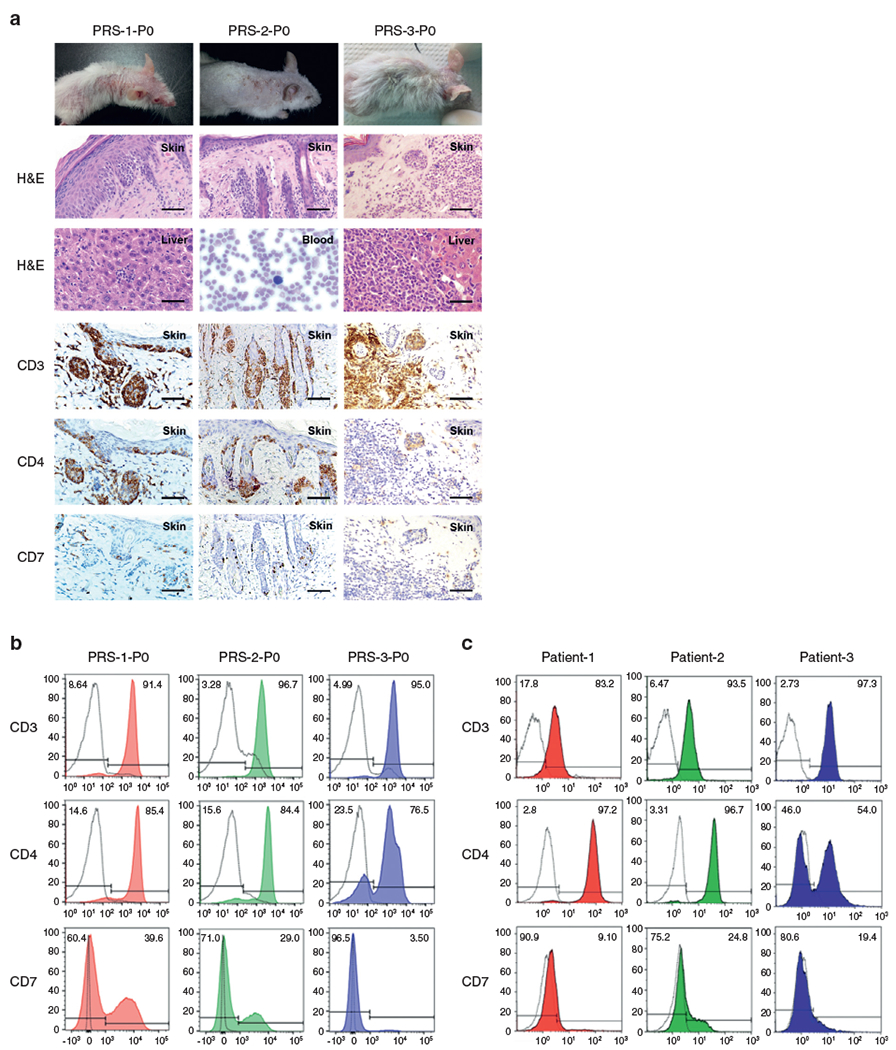
(a) H&E and IHC of the skin and other organs involved with lymphoma in P0 PDX mice. Bars = 25 μm. Peripheral blood is shown at a magnification of ×40. (b) Flow cytometry analysis of lymphocytes from the spleens harvested from P0 PDX mice. (c) Flow cytometry analysis of donor patients′ specimens: lymph node FNA biopsy from Patient-1 and peripheral blood from Patient-2 and Patient-3. Black dotted lines represent samples stained with isotype-control antibodies; color-filled lines represent samples stained with the indicated antibodies. FNA, fine-needle aspiration; IHC, immunohistochemistry; MF, mycosis fungoides; PDX, patient-derived xenograft; SS, Sézary syndrome.
To establish SS PDXs from Patient-2 and Patient-3, PBMCs from these patients were injected intravenously through a tail vein into NSG mice named as PRS-2-P0 and PRS-3-P0, respectively. Both P0 animals developed erythematous, scaly skin lesions and disseminated disease involving the spleen and viscera organs (Figure 2a). We also observed Sézary cells in the blood from the PRS-2-P0 mouse, a clinical and diagnostic hallmark of SS. The affected skin and splenocytes from PRS-2-P0 mouse demonstrated that malignant cells expressed human CD4 without coexpression of CD7 (Figure 2a and b). In the PRS-3-P0 mouse, we identified two malignant cell populations in the splenocytes, one CD4+ and the other CD4−, identical to that of Patient-3 (Figure 2b and c).
Our CTCL PDXs had markedly enlarged spleens compared with normal controls (Figure 3a). To propagate PDXs, we harvested splenocytes from P0 mice and injected them into naive NSG mice. PDX splenocytes were also cryopreserved for future experiments. All three lines of PDXs were successfully passaged to P3. The spleen sizes and survival times of each passage from P0 to P2 are shown in Figure 3b and c.
Figure 3. Stable passage of MF and/or SS PDXs and establishment of tumor cfDNA for measuring tumor burden in PDXs.
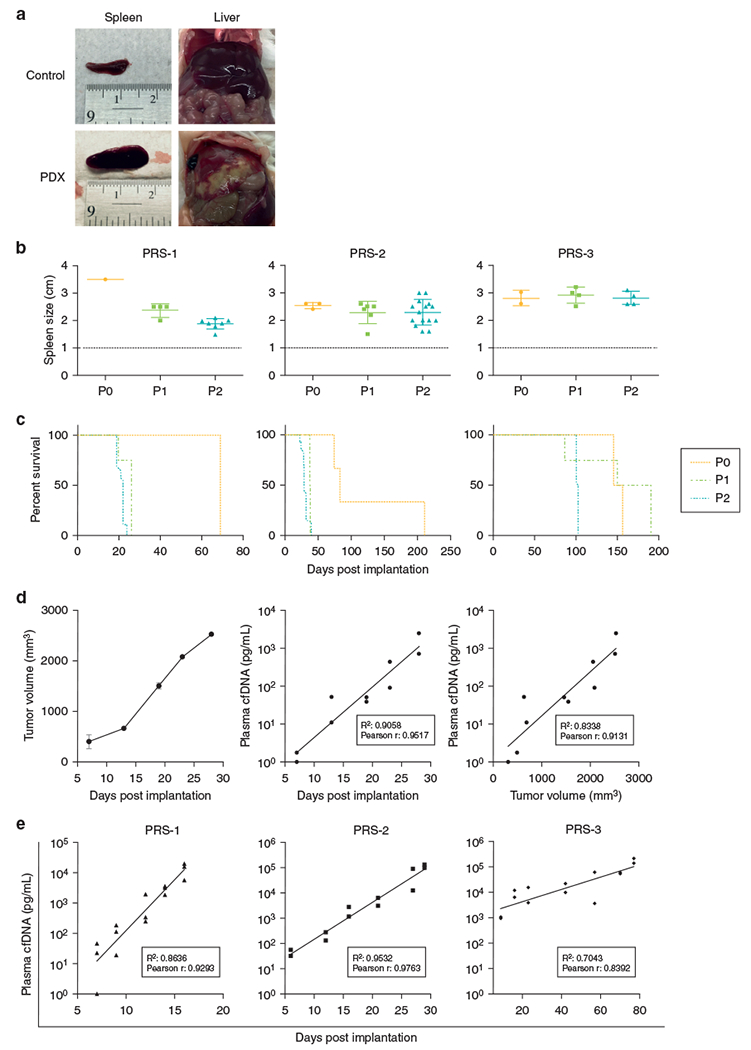
(a) Tumor-infiltrated liver and spleen from a PDX mouse. (b) Spleen size and (c) life span of each passage of PDXs. (d) Validation of tumor cfDNA as a surrogate for tumor burden. Postimplantation tumor volume measured by the calipers (left panel) and the concentration of tumor cfDNA in the plasma (middle panel). The concentration of tumor cfDNA in plasma (right panel) correlated with tumor size measured by the calipers method in the xenografts-bearing HH cells. (e) Tumor cfDNA is correlated with tumor progression in PDXs. The data were analyzed by Pearson correlation. cfDNA, cell-free DNA; MF, mycosis fungoides; PDX, patient-derived xenograft; SS, Sézary syndrome.
To confirm that engrafted T cells in PDX mice indeed represented malignant T cells from respective donor patients, we compared TCR clonality in patient biopsies with that in corresponding PDX mice using high-throughput deep sequencing of TCR (Table 1). The PRS-1-P0 and PRS-2-P0 mice each had a dominant TCR that accounted for >99% of all TCRs in the splenocytes. More importantly, the dominant TCR clones in the PRS-1-P0 and PRS-2-P0 mice shared the same variable, diversity, and joining (VDJ) gene usage as that in the biopsy specimens from the corresponding donors, Patient-1 and Patient-2, respectively. Furthermore, in both PRS-1 and PRS-2 mice, the TCR clonality was maintained through passages from P0 to P2. In PRS-3-P0, we identified two dominant TCR clones: clone 1 constituted 76% of all TCRs and clone 2 made up 16% of all the TCRs. These two dominant clones were identical to those found in the biopsy specimens of Patient-3 as demonstrated by VDJ gene usage. Next, we characterized the genomic alterations in PDXs by exome sequencing and found recurrent somatic mutations that recapitulated those in humans reported previously (Choi et al., 2015; da Silva Almeida et al., 2015; Wang et al., 2015). For instance, mutations in PLCG1 and NRAS were identified in PRS-1, and mutations in RARA were identified in 2 of 3 PDXs (Supplementary Table S2). Taken together, the PDX models recapitulated the clinical syndrome of MF and/or SS and maintained the morphological, immunophenotypical, and molecular characteristics of their clinical counterparts.
Table 1.
Comparison of VDJ Gene Usage of TCRs in Biopsies from Patients and in Splenocytes from their Corresponding PDXs
| Patients and PDXs | V Gene |
D Gene |
J Gene |
Malignant Clone (%) |
|---|---|---|---|---|
| TCRBV | TCRBD | TCRBJ | ||
| Patient-1 (MF) | 29-01 | 01-01 | 01-05 | 79.52 |
|
| ||||
| PRS-1-P0 | 29-01 | 01-01 | 01-05 | 99.99 |
|
| ||||
| PRS-1-P1 | 29-01 | 01-01 | 01-05 | 99.99 |
|
| ||||
| PRS-1-P2 | 29-01 | 01-01 | 01-05 | 99.93 |
|
| ||||
| Patient-2 (SS) | 07-02 | unknown | 02-05 | 94.91 |
|
| ||||
| PRS-2-P0 | 07-02 | unknown | 02-05 | 99.85 |
|
| ||||
| PRS-2-P1 | 07-02 | unknown | 02-05 | 99.93 |
|
| ||||
| PRS-2-P2 | 07-02 | unknown | 02-05 | 99.93 |
|
| ||||
| Patient-3 (SS)_clone 1 | 02-01 | 01-01 | 01-04 | 85.99 |
|
| ||||
| PRS-3-P0 | 02-01 | 01-01 | 01-04 | 76.44 |
|
| ||||
| PRS-3-P1 | 02-01 | 01-01 | 01-04 | 74.04 |
|
| ||||
| PRS-3-P2 | 02-01 | 01-01 | 01-04 | 62.41 |
|
| ||||
| Patient-3 (SS)_clone 2 | 04-03 | 01-01 | 01-02 | 0.06 |
|
| ||||
| PRS-3-P0 | 04-03 | 01-01 | 01-02 | 16.35 |
|
| ||||
| PRS-3-P1 | 04-03 | 01-01 | 01-02 | 25.34 |
|
| ||||
| PRS-3-P2 | 04-03 | 01-01 | 01-02 | 37.57 |
Abbreviations: MF, mycosis fungoides; PDX, patient-derived xenograft; SS, Sézary syndrome; VDJ, variable, diversity, joining.
PRS-1, PRS-2, and PRS-3 refer to PDX derived from Patient-1, Patient-2, and Patient-3, respectively. P0 denotes the original mouse receiving cells directly from each patient. P1 and P2 denote the first and second subsequent mice receiving cells passaged from the P0 mice.
To use these PDXs to test potential therapeutic agents, which have not been identified previously, we sought to develop a blood-based quantitative assay for tumor burden. We investigated whether human β-actin cell-free DNA (cfDNA) in the plasma of the PDX mice could serve as a surrogate of tumor burden in these animals with systemically disseminated lymphoma. We first examined whether tumor cfDNA concentration, represented by human β-actin cfDNA, was in proportion to the tumor volume. Cells from the HH cell line were injected into the flank of an NSG mouse, resulting in a subcutaneous tumor mass, which was measured by the calipers method (Figure 3d, left panel). Concurrently, plasma was collected at multiple time points to measure human β-actin cfDNA concentration. As shown in Figure 3d, the concentration of tumor cfDNA in the plasma after inoculation increased in a linear fashion (middle panel) and was correlated with the tumor size with an R2 = 0.8338 (right panel). Finally, we demonstrated that in the PDX mice, it was feasible to obtain serial measurements of plasma tumor cfDNA and that these cfDNA concentrations increased with tumor growth after implantation (Figure 3e).
Preclinical screening of prioritized compounds
We first investigated the in vivo activity of BKM120 in HH xenografts. HH was chosen because it is most sensitive to BMK120 (Figure 1c). In comparison with the control mice, BKM120 dramatically inhibited tumor growth, leading to prolonged survival (Figure 4a and b).
Figure 4. Antitumor activity of BKM120 in cell line and PDX xenografts.
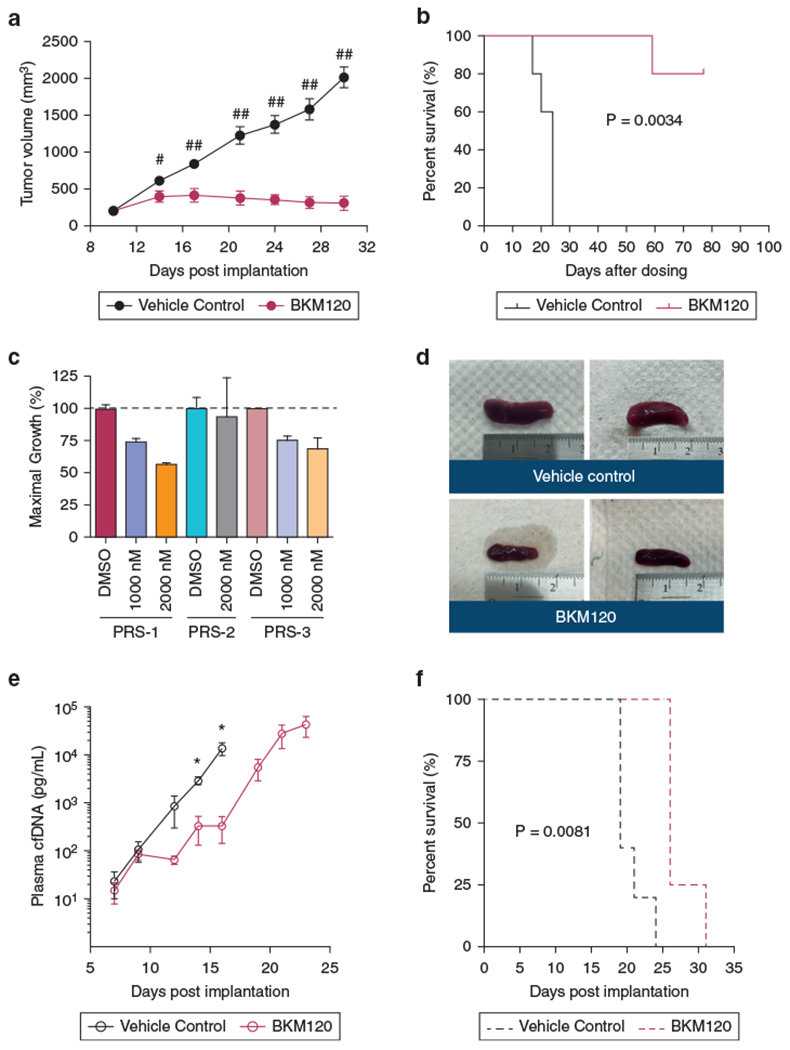
(a, b) Effect of BKM120 on tumor growth and survival in HH xenografts (n = 5 per group). *P < 0.01, **P < 0.001 by unpaired t-test with one-tailed P-values. (c) GI of BKM120 in primary tumor cells from PDXs. (d–f) Comparison of spleen sizes and tumor burden measured by tumor cfDNA and survival in PRS-1 treated with BKM120 versus vehicle control (n = 5 per group). Tumor volume and cfDNA are presented as mean ± SEM and analyzed by unpaired t-test with one-tailed P-value (*P < 0.05). The survival curves were generated by the Kaplan–Meier method and analyzed by the Gehan–Breslow–Wilcoxon test. cfDNA, cell-free DNA; GI, growth inhibition; PDX, patient-derived xenograft.
We next investigated the antitumor activity of BM120 in PDX mice. We first demonstrated that BKM120 inhibited cell growth of human malignant T cells from the PRS-1 and PRS-3 mice but not from PRS-2 (Figure 4c); thus, PRS-1 was chosen for in vivo experiments. Compared with the control mice, BKM120-treated mice had a lower tumor burden represented by smaller spleens, reduced tumor growth measured by tumor cfDNA, and prolonged survival (Figure 4d–f).
Identification of PI3K isoforms that mediate the BKM120 antitumor activity in CTCL
To better characterize the molecular targets that mediate the observed antitumor activity of the pan-PI3K inhibitor, BKM120, we investigated the effect of isoform-specific small interfering RNA (siRNA) knockdown on proliferation of CTCL cell lines. First, we verified that the α, β, δ, and γ isoform-specific siRNAs and their doublet combinations were able to reduce the respective targeted mRNA expression level by 50–85% compared with the scrambled siRNA (Supplementary Figure S3). Next, we demonstrated that in CTCL cell lines, the δ isoform knockdown had the biggest impact on cell proliferation. In addition, among the six doublets of siRNA knockdowns (αβ, αδ, αγ, βδ, βγ, δγ), those containing δ-isoform siRNA were more potent on GI than the other combinations (Figure 5a).
Figure 5. Copanlisib potentiates the effect of panobinostat on GI and apoptosis induction in primary CTCL tumor cells.
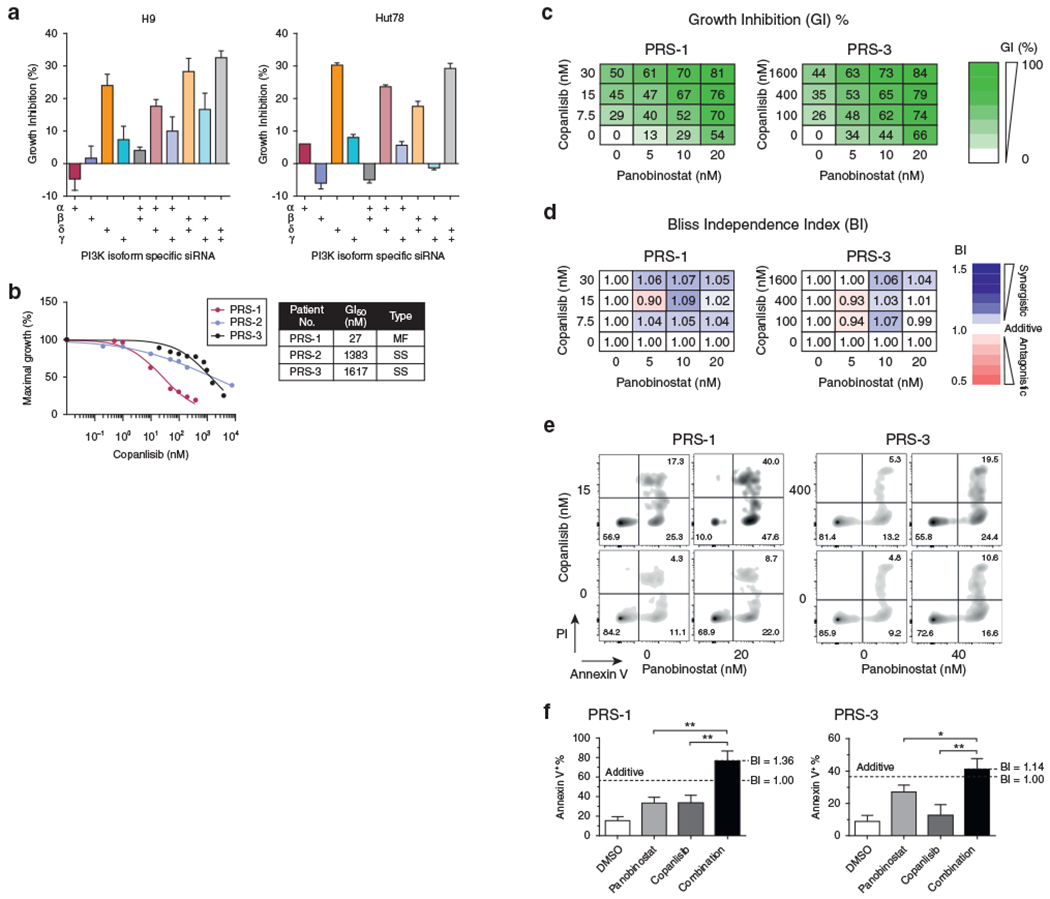
(a) The effect of isoform-specific siRNA knockdowns, alone or in doublets, on GI in cell lines. Data are presented as mean ± SD from three independent experiments. (b) Antiproliferation activity of copanlisib in primary tumor cells from PDXs. (c, d) Antiproliferation activities of copanlisib in combination with panobinostat in primary tumor cells from PDXs and are analyzed by the BI method. (e, f) The effect of the combination of copanlisib and panobinostat on apoptosis induction in primary tumor cells from PDXs. Data are presented as mean ± SD from three independent experiments and analyzed by unpaired t-test with one-tailed P-values (*P < 0.05, **P < 0.01). BI, Bliss independence index; CTCL, cutaneous T-cell lymphoma; GI, growth inhibition; GI50, 50% growth inhibitory concentration; PDX, patient-derived xenograft; PI, propidium iodide; PI3K, phosphatidylinositol 3-kinase; siRNA, small interfering RNA.
Antitumor activity of PI3K isoform-specific inhibitors in primary CTCL tumor cells
To corroborate the result from the siRNA experiment, we examined the antiproliferative activity of the isoform-specific PI3K inhibitors, duvelisib and copanlisib, which are δγ-specific and αδ-specific inhibitors, respectively. Both agents have been approved by the Food and Drug Administration for B-cell lymphomas. Copanlisib appeared to be significantly more potent in the three CTCL cell lines tested, with a GI50 10—20 times lower than that of duvelisib (Supplementary Figure S4a). In addition, copanlisib appeared to be a more synergistic partner than duvelisib when combining with panobinostat (Supplementary Figure S4b). Recently, duvelisib has shown clinical activity in T-cell lymphoma (Horwitz et al., 2018), which led us to hypothesize that copanlisib would be active in T-cell lymphoma as well. Indeed, using PDX tumor cells, we demonstrated that copanlisib exerted dose-dependent GI with a GI50 ranging from 27 to 1,617 nM (Figure 5b).
Having demonstrated the activity of copanlisib in primary tumor cells, we explored whether copanlisib, identified as a potential new CTCL agent, could potentiate the antitumor activity of another drug that is already used for treating CTCL, such as HDAC inhibitors. To choose HDAC inhibitors to pair with copanlisib, we tested three Food and Drug Administration—approved HDAC inhibitors in a primary PDX culture (PRS-3) and showed that romidepsin and panobinostat were markedly more potent than vorinostat with GI50 of 3 nM, 14 nM, and 347 nM, respectively (data not shown). Romidepsin is administered as a 4-hour intravenous infusion, and panobinostat is an oral HDAC inhibitor, which is advantageous when combined with an intravenous formulated copanlisib. Next, we performed combination assays of copanlisib and panobinostat choosing three concentrations for both agents, yielding nine pairwise combinations, and using the Bliss independence index model to examine the synergistic interactions (Zhao et al., 2014) (Figure 5c and d). We found that copanlisib potentiated the antiproliferation and apoptosis induction activities of panobinostat across a broad range of concentrations in both PRS-1 and PRS-3 lines despite that PRS-3 was only modestly sensitive to copanlisib as a single agent (Figure 5c–f). Thus, the combination of copanlisib and panobinostat represents a favorable drug combination for CTCL that merits further development.
DISCUSSION
Recent advancement in understanding the genetic landscape of MF and/or SS signifies a new era of utilizing targeted agents against these diseases. To identify targetable pathways in CTCL, we took an approach of clustering the hits from a high-throughput drug screen on the basis of the pathways that they inhibited. Using this approach, we identified PI3K and CDK pathways among others to be of importance for CTCL cell proliferation. Our results are supported by published data. For example, recurrent genetic alterations in both PI3K and CDK have been reported (Choi et al., 2015; da Silva Almeida et al., 2015; Ungewickell et al., 2015; Wang et al., 2015), and recent trials demonstrated the efficacy of PI3K inhibitors in CTCL (Horwitz et al., 2018; Oki et al., 2018). As for the CDK pathway, investigators have reported genetic changes and inactivation of tumor suppressor genes, p15 and p16, upstream of CDK in skin biopsies of patients with CTCL, suggesting that cell cycle dysregulation plays a role in the pathogenesis of CTCL (Gallardo et al., 2004; Navas et al., 2002; Scarisbrick et al., 2002). Finally, recent studies using T-cell lymphoma cell lines showed that copanlisib and duvelisib are synergistic with several inhibitors of key signaling pathways, including panobinostat, CDK inhibitor, venetoclax, romidepsin, and mTOR (Faia et al., 2018; Tarantelli et al., 2020).
Testing novel agents requires a predictive preclinical model that accurately reflects the potential antitumor activity in patients. For this purpose, we developed MF and/or SS PDX murine models. Our PDX models have several unique features that make them an excellent and reliable preclinical platform to translate drug discovery. First, the models captured the quintessential clinical features of the disease and maintained the genetic and molecular heterogeneity of the original tumor. Second, we established a highly sensitive biomarker assay using tumor cfDNA to assess systemic tumor burden longitudinally in living animals, enabling using response rate and disease progression rather than survival alone as primary endpoints for drug therapy. Because the implanted tumors are the only human cells in these mice, in principle, we could design human-specific primers for any human gene. We chose β-actin because it is a reliable genomic target that is rarely mutated or deleted in human cancers and because robust primers and PCR conditions were established in our hands. We tested BKM120 using the PDX platform and validated the potential of our PDX as a much-needed reliable preclinical model for translating drug discovery in CTCL.
Combination therapies are important to enhance the clinical efficacy of targeted agents. However, for MF and SS, it is extremely difficult to obtain an adequate number of primary tumor cells to test novel drug combinations in vitro. Using primary cells harvested from PDXs, we demonstrated the feasibility of rapid screening of multiple drug combinations in dose-dependent matrices. Our result showed that inhibitions of PI3K α and δisoforms and HDAC are synergistic in GI and apoptosis induction in CTCL (Figure 5). The mechanism of synergy between HDAC and PI3K inhibitions remains to be elucidated. Recently, Horwitz et al. (2018) analyzed duvelisib-sensitive versus resistance cell lines by phosphoproteomics, which suggested that epigenetic modification could mitigate the effects of PI3K inhibition in resistant lines. Subsequently, they demonstrated that romidepsin and duvelisib have synergistic activities in systemic T-cell lymphoma cell lines (Horwitz et al., 2018).
In summary, our results indicate that the δ-isoform of PI3K is a suitable therapeutic target in CTCL, and inhibition of PI3K potentiates the antitumor activity of HDAC inhibitors. Furthermore, the combination of copanlisib and panobinostat is especially potent against multiple lines of PDX and represents an excellent candidate for clinical development.
MATERIALS AND METHODS
Patient specimens and animals
Primary lymphoma cells were obtained from patients at the University of California, San Francisco. All patients signed informed consent, and specimen collection was performed according to a protocol approved by the University of California, San Francisco, Human Research Protection Program Institutional Review Board. The 6–8-week-old NSG mice (The Jackson Laboratory, Bar Harbor, ME) were used to generate PDXs. Mice were fed rodent chow diet and water ad libitum and housed under specific pathogen-free conditions. All animal procedures were reviewed and approved by the University of California, San Francisco, Institutional Animal Care and Use Committee.
Establishment of PDX models
To establish MF and/or SS PDXs, lymphoma cells were obtained from either the involved lymph nodes by fine-needle aspiration biopsy or from peripheral blood. Erythrocytes in the specimen were either lysed with ammonium-chloride-potassium lysis buffer (Thermo Fisher Scientific, Waltham, MA) or separated by a density gradient using Histopaque-1077 (Sigma-Aldrich, Saint Louis, MO). Isolated leukocytes were washed with PBS, and 5 × 106–10 × 106 cells were suspended in RPMI-1640 with matrigel basement membrane matrix (Corning, Corning, NY) at a 1:1 ratio before getting injected subcutaneously into the flank of an NSG mouse. For intravenous injection, cells were suspended in PBS and injected through the tail vein of an NSG mouse. Once tumor cells engrafted and the tumor-bearing mice (referred to as P0) met humane or experimental protocol endpoint criteria, they were euthanized. Cells harvested from the spleen of P0 mice were passaged serially into naive NSG mice to generate P1 and P2 PDX mice.
Quantification of tumor cfDNA
Serial blood samples (250 μl) were collected by retro-orbital bleeding from PDX mice in microtainer tubes with dipotassium EDTA (BD Biosciences, San Jose, CA). cfDNA was purified from 100 μl of the plasma samples using the NucleoSpin Plasma XS kit (Macherey-Nagel, Bethlehem, PA) according to the manufacturer′s instructions. Plasma cfDNA concentrations were subsequently determined by TaqMan qPCR assay (Applied Biosystems, Waltham, MA). The following human β-actin primer pair and probe set were used: forward primer 5′-ATCCTAAAAGCCACCCCACT-3′; reverse primer 5′-CTCAAGTTGGGGGACAAAAA-3′; and probe 5′-FAM-CACAGGGGAGGTGATAGCAT-TAMURA-3′. Serial dilutions of genomic DNA extracted from the Hut78 and H9 CTCL cell lines were used to calibrate for cfDNA quantification. cfDNA concentrations were extrapolated from the standard curve using Prism software, version 6.0 (GraphPad Software, La Jolla, CA).
High-throughput drug screen
GI activity of 94 compounds was examined in four MF and/or SS cell lines as indicated. All drugs underwent 10-fold serial dilutions, ranging from 0.01–10 μg/ml, and transferred were to a 384-well plate with four replicates per dilution. The 2 × 104 cells were incubated with drugs at 37 °C for 72 hours, and GI was measured by the alamarBlue assay (Thermo Fisher Scientific). The results were analyzed using MATLAB software (MathWorks, Natick, MA) and a computational algorithm developed by Dr Sourav Bandyopadhyay (University of California) to plot dose–response curves and GI50 for each drug (defined as the drug concentration that inhibits cell growth by 50% compared with controls).
In vivo activity of BKM120 in CTCL cell line xenograft and PDX models
To establish cell line xenografts, ten 8-week-old female NSG mice were each subcutaneously injected with 10 × 106 cells in the flank. Drug treatment was started 10 days after implantation when the tumor volume reached ~200 mm3. The mice were assigned to vehicle control and BKM120 groups, with five mice per group. BKM120 was prepared in 0.5% methylcellulose (Sigma-Aldrich), 0.2% Tween-80 (Sigma-Aldrich), and 6% DMSO. The vehicle control contained 0.5% methylcellulose, 0.2% Tween-80, and 6% DMSO. The mice in vehicle control and BKM120 (30 mg/kg body weight) groups were treated by oral gavage at 5 days on and 2 days off until disease reached the point of euthanasia per institutional guidelines. Tumor growth was assessed twice weekly by calipers measurement, and animal weight was monitored twice weekly.
For the therapeutic trial with BKM120 in MF and/or SS PDXs, ten 9-week-old female NSG mice were implanted with 8 × 106 PRS-1 P1 cells in the flank subcutaneously (day 0). On day 7, mice were assigned to the vehicle control or BKM120 group. The preparation of the BKM120 and vehicle control as well as the treatment schema were as described earlier. Plasma was collected from the mice twice weekly beginning on day 7 for measurements of tumor cfDNA levels.
Isoform-specific knockdown of PI3K
Cells were transfected with Silencer Select siRNA human PIK3CA (s10520), PIK3CB (s10524), PIK3CD (s10530), and PIK3CG (s10532) (Thermo Fisher Scientific) using the nucleofector technology. The Nucleofector 2b Device (Lonza, Cologne, Germany) and Amaxa Cell Line Nucleofector Kits (Lonza) were used to perform electroporation according to the manufacturer′s instructions. Briefly, cells were subcultured 2–3 days before nucleofection. The 1 × 106–2 × 106 cells were harvested and suspended in Nucleofector Solution with 2 μM of siRNA. Cell and/or siRNA suspension was transferred into a cuvette for electroporation using an appropriate nucleofector program. After electroporation, 500 μl culture medium was added to the cuvette, and the sample was transferred into a 6-well plate with 2 ml of culture medium in each well. Cells were then cultured in recommended media for 48 hours; the cell viabilities were determined by the RealTime-Glo MT cell viability assay (Promega, Madison, WI).
Statistical analysis
The correlation between plasma tumor cfDNA concentration and tumor volume per day after implantation was determined by parametric Pearson′s correlation calculations. The results of tumor volume and plasma tumor cfDNA for each treatment group were expressed as mean ± SEM. The differences between tumor volumes were analyzed by unpaired, one-tailed t-test. For animal studies, survival curves were generated by the Kaplan–Meier method and compared using the Gehan–Breslow–Wilcoxon test. The combination synergy between PI3K inhibitors and HDAC inhibitors was evaluated using the Bliss independence index model (Zhao et al., 2014). The Bliss independence index > 1 indicates synergism, Bliss independence index = 1 indicates addition, and Bliss independence index < 1 indicates antagonism. Differences in apoptosis between the single agents and the combinations were analyzed by unpaired, one-tailed t-test. Statistical analysis was performed by using Prism software, version 6.0. P < 0.05 was considered statistically significant.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, WZA, upon reasonable request.
Supplementary Material
ACKNOWLEDGMENTS
We thank Matthew L. Springer, Karin M. Gaensler, Marc Shuman, Sung Eun Kim, and Andrew Wolfe for their critical review of the manuscript. This study is generously supported by the Martin and Dorothy Spatz Foundation, the Summit Bank Foundation, and the Donna L. and Edward E. Martins Foundation.
Abbreviations:
- CDK
cyclin-dependent kinase
- cfDNA
cell-free DNA
- CTCL
cutaneous T-cell lymphoma
- GI
growth inhibition
- GI50
50% growth inhibitory concentration
- HDAC
histone deacetylase
- MF
mycosis fungoides
- PDX
patient-derived xenograft
- PI3K
phosphatidylinositol 3-kinase
- siRNA
small interfering RNA
- SS
Sézary syndrome
Footnotes
CONFLICT OF INTEREST
FM is a cofounder and a consultant for BridgeBio Pharma and a consultant with Leidos Biomedical Research in his role as Scientific Director for the National Cancer Institute’s RAS Initiative. The remaining authors state no conflicts of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2020.05.110.
REFERENCES
- Agar NS, Wedgeworth E, Crichton S, Mitchell TJ, Cox M, Ferreira S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol 2010;28:4730–9. [DOI] [PubMed] [Google Scholar]
- Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet 2015;47:1011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat Genet 2015;47:1465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M, et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 2013;49:386–94. [DOI] [PubMed] [Google Scholar]
- Faia K, White K, Murphy E, Proctor J, Pink M, Kosmider N, et al. The phosphoinositide-3 kinase (PI3K)-γ,δ inhibitor, duvelisib shows preclinical synergy with multiple targeted therapies in hematologic malignancies. PLoS One 2018;13:e0200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo F, Esteller M, Pujol RM, Costa C, Estrach T, Servitje O. Methylation status of the p15, p16 and MGMT promoter genes in primary cutaneous T-cell lymphomas. Haematologica 2004;89:1401–3. [PubMed] [Google Scholar]
- Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I, et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol 2009;82:440–9. [DOI] [PubMed] [Google Scholar]
- Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, et al. Activity of the PI3K-γ,δ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018;131:888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol 2003;139:857–66. [DOI] [PubMed] [Google Scholar]
- Krejsgaard T, Kopp K, Ralfkiaer E, Willumsgaard AE, Eriksen KW, Labuda T, et al. A novel xenograft model of cutaneous T-cell lymphoma. Exp Dermatol 2010;19:1096–102. [DOI] [PubMed] [Google Scholar]
- Navas IC, Algara P, Mateo M, Martínez P, García C, Rodriguez JL, et al. p16(INK4a) is selectively silenced in the tumoral progression of mycosis fungoides. Lab Invest 2002;82:123–32. [DOI] [PubMed] [Google Scholar]
- Oki Y, Haverkos B, Zain JM, Lechowicz MJ, Devata S, Korman NJ, et al. Tenalisib, a dual PI3K δ/γ inhibitor: safety and efficacy results from an ongoing phase I/Ib study in relapsed/refractory T-cell lymphoma. J Clin Oncol 2018;36(Suppl. 15):7510. [Google Scholar]
- Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 2009;27:5410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarisbrick JJ, Woolford AJ, Calonje E, Photiou A, Ferreira S, Orchard G, et al. Frequent abnormalities of the p15 and p16 genes in mycosis fungoides and sezary syndrome. J Invest Dermatol 2002;118:493–9. [DOI] [PubMed] [Google Scholar]
- Talpur R, Singh L, Daulat S, Liu P, Seyfer S, Trynosky T, et al. Long-term outcomes of 1,263 patients with mycosis fungoides and Sezary syndrome from 1982 to 2009. Clin Cancer Res 2012;18:5051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantelli C, Lange M, Gaudio E, Cascione L, Spriano F, Kwee I, et al. Copanlisib synergizes with conventional and targeted agents including venetoclax in B- and T-cell lymphoma models. Blood Adv 2020;4: 819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler S, Burger AM, Schulz T, Brill B, Bittner A, Oberholzer PA, et al. Establishment of a mouse xenograft model for mycosis fungoides. Exp Dermatol 2004;13:406–12. [DOI] [PubMed] [Google Scholar]
- Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic analysis of mycosis fungoides and Sézary syndrome identifies recurrent alterations in TNFR2. Nat Genet 2015;47:1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of Sézary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet 2015;47:1426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Ishida T, Inagaki A, Ishii T, Ding J, Kusumoto S, et al. Defucosylated anti CC chemokine receptor 4 monoclonal antibody combined with immunomodulatory cytokines: a novel immunotherapy for aggressive/refractory mycosis fungoides and Sezary syndrome. Clin Cancer Res 2007;13:6494–500. [DOI] [PubMed] [Google Scholar]
- Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A new Bliss independence model to analyze drug combination data. J Biomol Screen 2014;19:817–21. [DOI] [PubMed] [Google Scholar]
- Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol 2007;25:4293–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, WZA, upon reasonable request.