

Abstract
Developing radiation tumor biomarkers that can guide personalized radiotherapy clinical decision making is a critical goal in the effort towards precision cancer medicine. High-throughput molecular assays paired with modern computational techniques have the potential to identify individual tumor-specific signatures and create tools that can help understand heterogenous patient outcomes in response to radiotherapy, allowing clinicians to fully benefit from the technological advances in molecular profiling and computational biology including machine learning. However, the increasingly complex nature of the data generated from high-throughput and “omics” assays require careful selection of analytical strategies. Furthermore, the power of modern machine learning techniques to detect subtle data patterns comes with special considerations to ensure that the results are generalizable. Herein, we review the computational framework of tumor biomarker development and describe commonly used machine learning approaches and how they are applied for radiation biomarker development using molecular data, as well as challenges and emerging research trends.
INTRODUCTION
Radiation therapy is a core component of the treatment of cancer patients, with over 50% of patients receiving radiation therapy at some point during their treatment course, and approximately 60% of these patients are treated with curative intent1. Personalization of radiation therapy is a critical goal for radiation oncologists who aim to increase the chance of controlling disease while limiting the harmful side-effects and toxicities of treatment. Current strategies center around several frameworks – utilizing clinical characteristics to stratify patients, image guided radiation therapy that allows for higher doses to the tumor with improved sparing of normal tissue2, and biomarkers to guide treatment3–5. The first two are widely clinically adopted, but development of radiation tumor biomarkers has proven more difficult. Prognostic tests can help guide decisions about treatment intensification in high-risk patients or de-intensification in low-risk patients, but they provide limited insight into the most appropriate type of intervention. The ideal radiation tumor biomarker for the radiation oncologist would be treatment predictive, that is it would provide insight into whether a patient’s specific cancer is radiosensitive or radioresistant, or on the benefit of radiation. Such predictive biomarkers could then guide the decision on whether to treat with radiation and help determine the appropriate dose and fractionation schedule. Predictive biomarker discovery can be more difficult than identifying prognostic biomarkers, but advances in high-throughput molecular profiling technologies have provided an opportunity to create clinically useful radiation-based predictive biomarkers, and many biomarkers of radiation sensitivity and resistance have been developed using various machine learning techniques6 to harness the power of these assays. However, there are no tumor biomarkers specifically designed for radiation therapy that are clinically used today. One of the challenges with modern high-throughput assays is the complex nature of the data produced and the special analytical considerations required. In this review we set out to provide a broad overview of the computational machine learning methodologies used in of the development of high-throughput radiation biomarkers as well as review challenges and future possibilities.
High-throughput techniques and the “curse of dimensionality”
Modern high-throughput assays, or “omics” technologies, have revolutionized the study of biological systems. The assays provide an opportunity to comprehensively characterize the tumor DNA alterations (genomics), gene expression profiles (transcriptomics), protein abundance and modifications (proteomics), epigenetic modifications (epigenomics), metabolic profiles (metabolomics), etc. Herein, we will focus on the special analytical considerations needed for these molecular data. The common theme for all “omics” assays is that for every tumor sample (observation) there are hundreds to thousands to millions of variables measured (features, or dimensions, e.g. genes, proteins, etc.). The number of observations is usually lower than the number of features due to sample availability and cost. Given that 1) most features are typically not informative for the specific question asked, thus contributing only “noise”, and 2) there is a level of background random variability for each feature, there are usually features that can distinguish between groups by pure chance. Thus, adding hundreds or thousands of variables of which most are contributing noise does not necessarily make it easier to find the true biological signal in the data, a phenomenon that is sometimes called the “curse of dimensionality”. The key analytical challenge in biomarker development is to find the true signal in the noise.
This challenge is compounded by the power of modern machine learning techniques, which can be extremely efficient at finding subtle patterns in the data. Given the high-dimensional nature of “omics” data, it is easy for these methods to fit models on the noise in a specific dataset and the more flexible a model is, the easier it may be to pick up the noise in the data. By picking up noise instead of signal, the model weights are more sensitive to changes in the input data, and the model will have a high variance. Conversely, less flexible models usually have a bias towards the constraints of that model, making it more robust against noise, but also less flexible to the true signal. Ideally, a good model is flexible enough to find a close fit to the true signal (low bias) and have low variance. There is usually a trade-off between these properties, referred to as the bias-variance trade-off. In summary, these properties of high-dimensional data and the flexibility of models available in modern machine learning, requires a specific strategy and a set of steps for model development and validation where a model is trained, tuned, tested, and finally validated in independent data to ensure that the model has indeed found a true signal in the noise.
Sample handling and data pre-processing
Before the machine learning procedures can start, the data needs to be pre-processed for further analysis. The full details of these techniques are outside the scope of this review. Briefly, pre-processing typically includes normalization, batch-effect correction, and feature selection. Normalization is intended to reduce the technical variability between samples and can be done for a set of samples together (cohort) or on a single-sample basis. Single-sample techniques have the advantage of easier implementation in personalized medicine workflows as it can be performed continuously on each new sample that is processed and independently of other cohorts. Batch-effects refer to systematic technical variability in the data due to groups of samples having similar patterns due to experimental or technical factors, in contrast to biological differences. This can be due to different assay platforms being used, samples being processed at different centers or at different timepoints, differences in sample handling, storage, processing, etc. It is important to reduce these potential sources of variability, and to identify and account for batch-effects when present. Feature selection is intended to reduce the number of non-informative, or noise, features. It can be done based on unbiased data-driven methods (such as removing lowly expressed genes and selecting the most variable genes), by prior biological knowledge (e.g. selecting genes in specific biological pathways) or at later analytical stages by how the features contribute to model performance. The overall goal of the pre-processing is to yield a dataset with as little noise and as much true signal as possible.
STRATEGIES FOR MACHINE LEARNING ANALYSIS
Definition Overview
The terms artificial intelligence (AI) and machine learning (ML) are used colloquially and often interchangeably, but they do hold separate meanings. AI refers to any technique utilized on a computer that seems to replicate a form of intelligence. ML represents a subset of AI where computers can learn directly from labeled (i.e. patient outcomes or radiosensitivity status of a sample is known) or unlabeled data. The goal of all ML methods is to take data as input and create a more structured and simplified output. There are two primary methodologies to perform this task – unsupervised learning and supervised learning. The primary difference between these two methods is that supervised models train on labeled data for a specific outcome – a continuous variable (regression) such as the survival fraction of cells after receiving 2 Gy of radiation (SF2) or a discrete variable (classification) such as whether a tumor is known to be radiation sensitive or resistant – while unsupervised models are used to identify the intrinsic structure in a dataset without utilizing any labels (e.g. clustering).
Data Spending
Since the number of observations usually is limited, how the limited data gets used is one of the most important aspects of ML to ensure generalizability to future samples. Data spending refers to the task of splitting up data at steps throughout the workflow so that the model reproducibly finds signal instead of noise. The data used to build a model is called training data, while we refer to the data that the final and locked model is run on to validate the performance as independent validation data. The training data can be further split for model tuning, which we refer to as a hold-out test set. A true validation data set is meant to be entirely separate from the model training workflow and only used as a final independent confirmation of model performance. Various strategies have been utilized to train radiation signatures, such as Eschrich et al. utilizing the NCI-60 cell lines7, Zhao et al. utilizing orthotopic glioblastoma patient-derived xenografts8, and Sjöström et al. utilizing breast cancer patient samples9. All three studies had independent clinical validation datasets that were used to confirm the clinical validity and generalizability of the models. A critical requirement for radiation biomarkers is a validation dataset which is independent from training7,8,10–15, such that no validation data is used to adjust the model. If the validation data is used for training, or informing the training (i.e. information leakage), the model may pick up noise that is present in both the training and validation data, instead of true signal, and will not generalize to future datasets. Validation data may come from a different source than the training data. In that case, differences in platforms or experimental protocols may need to be accounted for via normalization and/or batch correction strategies described above. However, this would not generally be considered a major component of information leakage, since the validation data is not being directly trained on the outcome of interest.
Hyperparameter Fitting and Model Selection
As discussed above, ML models can be so flexible and effective in finding patterns that they find noise that does not represent true biological signal and is only specific to that data set, which is referred to as overfitting16. Thus, a model that is over-fit to a particular training dataset usually performs poorly on validation. A bias term making the model less flexible can be added so that the model’s fit is reduced on training data, increasing the generalizability and ideally with improved performance on validation (regularization, described in detail later) (Figure 1A). The bias term represents a hyperparameter, which is a feature that can only be optimized through evaluation of a model’s performance, unlike a traditional parameter in a linear regression model which has a best-fit solution. Both the selection of an appropriate model with the optimal level of flexibility and multiple hyperparameters need to be tested to identify the optimal model and settings. However, selecting the best model and settings in the training data commonly leads to over-fitting, while selection based on the validation data would be information leakage. Thus, one strategy is to further split the training data into a model building set and a hold-out test set from which optimal model and bias hyperparameters are selected (Figure 1B), so that all of this occurs only within the training data, keeping the validation data separate.
Figure 1 – Machine Learning Toolkit.
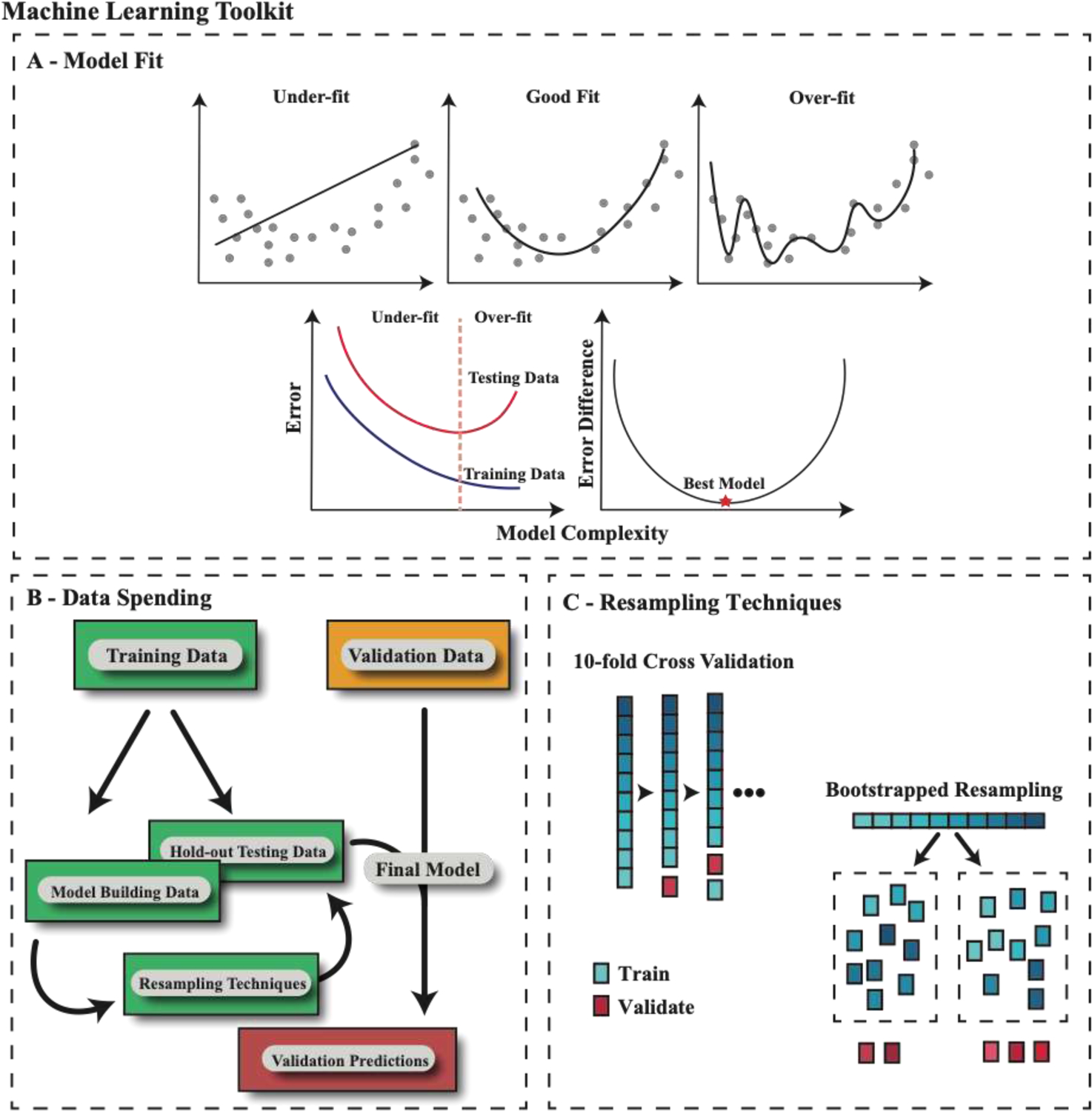
A – A model that tries to overlearn features in the training data is at risk of over-fitting that data and performing poorly on the testing (or validation) set. A bias term can be added to the model training procedure so that it performs worse on training but better on testing (or validation), finding a minimum point in the error difference. B – Depiction of an appropriate data spending schema where all modeling is performed on the training data and an independent validation dataset is utilized only to confirm the prediction of the model. The training data is further split into model building and a hold-out testing set, where resampling is performed only on the model building data. C – Depiction of the two most common resampling techniques, 10-fold cross validation (CV) and bootstrapped resampling. Note, CV will train on all but one partition of the data at a time, cycling through all combinations, while bootstrapped resampling will create N resampled groups with repeat samples so that every bootstrapped group matches the size of the original dataset.
Resampling Methods
Selecting a hold-out test dataset within the training data can result in very different results depending on how the data are split. Resampling by repeatedly generating the hold-out test set multiple times is one way to minimize this variability. The two most common resampling techniques are k-fold cross validation (CV) and bootstrapped resampling (Figure 1C). CV involves randomly splitting the data into k groups, with a model trained on k-1 groups and validated on that final hold out group (repeated k times). Leave-one-out CV is a special case where k equals the number of observations. Bootstrapping is where observations are randomly sampled from the data (allowing for repeats). The model is then trained on that sampled group and tested on the remaining observations. This can be performed n times and averaged out. To use this technique for hyperparameter optimization, one approach is performing resampling within the training data. Once the best hyperparameters are selected, the model can be locked and validated on that hold out test set. This is called double-loop resampling, with Sjöström et al.9 using this approach to develop a breast cancer radiation sensitivity signature. The training data they collected was split 50/50 into a model building and hold-out test set, where both gene selection and model hyperparameter optimization were performed using cross validation within the model building set. The model was first validated against their own hold-out test set and then on independent, publicly available datasets, demonstrating the clinical utility of the signature as prognostic for ipsilateral breast tumor recurrence and predictive of radiotherapy benefit in estrogen receptor positive patients. In summary, strict data spending schema and proper training using resampling minimizes information leakage, while maximizing the performance and generalizability of machine learning models.
MACHINE LEARNING APPROACHES
Unsupervised Learning
Unsupervised learning searches for intrinsic structure in a dataset while blinded to any labels. Clustering describes an unsupervised learning approach to grouping based on similarity. The two most common clustering algorithms used in biomarker development are k-means clustering and hierarchical clustering17. K-means clustering algorithm partitions data into k clusters with an algorithm that first randomly selects k observations (centroids), then calculates the distance from all other observations to the centroids (e.g. Euclidian distance), designates observations to a cluster based on the minimum distance to a centroid, and finally recalculates new centroids based on the mean values of all the observations in a cluster. This process is repeated until observations stop changing clusters, producing groups that minimize within-cluster variances. Hierarchical clustering tries to define clusters through either a bottom-up approach, where each observation starts in its own cluster, and are iteratively paired as the hierarchy ascends, or a top-down approach, where all observations start in one cluster and splits are made recursively as the hierarchy descends. Hierarchical approaches are advantageous as they produce readily interpretable structure among clusters. However, they are limited by the increased computational complexity with larger datasets. Conversely, k-means clustering is more computationally efficient but often produces more homogenous clusters with less readily interpretable structure18. Both methods have been used in radiation biomarker development, with Piening et al. using hierarchical clustering of differentially expressed genes (cell lines compared before and after radiation exposure) to identify prognostic breast cancer patient clusters19 and Weichselbaum et al. utilizing k-means clustering of interferon-related DNA damage resistance genes in breast cancer patients to identify patient clusters, creating clusters that had utility in predicting recurrence after radiation therapy20.
Dimensionality reduction aims to reduce the number of features in a dataset (e.g. genes) into a lower dimensional space. Each feature in a dataset can be thought of as an additional dimension. There are three primary reasons for reducing the dimensions in a feature set – 1) too many features can be uninterpretable, 2) many features may represent experimental noise unrelated to the structure of the data, and 3) many features may be redundant, such as two genes that are correlated in all samples. While low dimensional datasets can be understood more intuitively, such as plotting each sample’s expression of three genes in a 3-D scatter plot, it’s difficult to visualize data in higher dimensions. Thus, dimensionality reduction is a key strategy for visualizing data in a lower dimensional space that can be more interpretable.
Principal Component Analysis (PCA) is the most common and easily interpretable form of dimensionality reduction21. PCA takes the original features of a dataset and creates new principal components (PC) that are linear combinations of those original features. For example, if a dataset had 10 genes, PCA would instead produce 10 PCs that are linear combinations of each gene. Importantly, these new PCs are explicitly de-correlated (i.e., no variable is redundant) and the PCs are ordered based on how much variability in the data each explains (e.g., PC1 will explain the most variance). By plotting PCs in a 2D or 3D space, this reduced representation of the data can readily be visualized to help understand the structure of the samples. Importantly, because PCA relies on linear combinations, any non-linear effects (gene A increases exponentially with respect to gene B) will not be accurately captured. PCA is often used for radiation signature development, with Kim et al. utilizing PCA for dimensionality reduction to plot the expression of their radiation signature, showing that radiosensitive and radioresistant cell lines were well separated in a 3D space22. Starmans et al. were able to use PCA to identify a batch effect between cell lines in their hypoxic radiosensitivity signature, warranting downstream analysis that accounted for this confounding variable23.
A methodology that tries to improve upon some of the limitations with PCA is the t-distributed stochastic neighbor embedding (t-SNE) method24, in which distances between points are scaled onto a t-distributed curve and those scaled distances are then used to map samples to a lower dimensional space, preserving some of the higher dimensional clustering structure that can be lost with PCA. Uniform Manifold Approximation and Projection (UMAP)25 is a more recent and very similar algorithm that has allows for increased computational efficiency. These techniques are still dimensionality reduction techniques and do not explicitly map samples to a specified cluster, however, samples can be color coded in a 2D t-SNE or UMAP projection based on the groupings from a k-means or hierarchical clustering, a technique that is commonly used to identify cell types from single cell RNA (scRNA) expression data26. Gao et al. utilized scRNA data from a breast cancer cell line treated with and without radiation and identified clusters on a t-SNE plot that identified heterogeneity of response to radiation27.
Supervised Learning
In contrast to unsupervised techniques which are aimed at finding unknown structures within the data, supervised learning methods utilize models that are trained using known labeling of the data, such as radiosensitive vs. radioresistant tumors. Supervised learning can be structured based on the goal of the learning task – either developing inferential models or predictive models. An inferential model is used to identify the significance of a specific association, where the priority is producing a highly interpretable model. Conversely, the goal of a predictive model is to optimize prediction performance, often sacrificing interpretability. The most common inferential models are those utilized in clinical studies, such as a univariate or multivariate Cox regression or logistic regression. Another example commonly used in biomarker discovery is differential gene expression analysis of RNA-seq28, where a generalized linear model provides a log-fold change and significance test for the comparison between two experimental groups across every gene. Differential expression analysis is commonly used for radiosensitivity biomarker identification, such as in comparisons of gene expression levels in irradiated and non-irradiated cell lines to identify induced and repressed genes19, performing cell line survival experiments and utilizing integral survival29 or SF230 to define radioresistant and sensitive cell lines, and evaluating varying hypoxic conditions in cell lines23,31 and patients32 to identify hypoxia related genes relevant for radiation response. While inference and prediction can be two separate goals, the reality is that there is a spectrum – ideally inferential models are predictive and vice versa. Below, we review several commonly used supervised learning predictive models for radiation biomarkers, again focusing on studies with independent clinical validation. Many other ML algorithms exist beyond these (support vector machines, Bayesian networks, etc.), but will not be covered below.
Generalized Linear Models
Simple univariate and multivariate generalized linear models (GLMs; including linear, logistic, Poisson, negative binomial, Cox regressions, etc.) all represent supervised ML methods, as they utilize labeled training data to build models that predict a specific outcome. The benefit of GLMs is that they are highly interpretable and have a single best-fit solution, allowing a model to be built that utilizes all data without necessarily needing resampling-based hyperparameter optimization. Nevertheless, there are many other decisions that need to be made – picking a generalized linear model distribution, transforming non-linear variables, or adding interaction terms. A major advantage of GLMs is their interpretability. The importance of each variable is directly proportional to its weight in the model.
A common strategy in radiation biomarker development is to use GLMs for feature selection, such as evaluating the correlation between gene expression and SF212,22,33. Yard et al. performed the largest such analysis to date, where they radiated 533 cell lines from the Cancer Cell Line Encyclopedia (CCLE) and evaluated the correlation coefficients between gene expression and cell viability (in addition to evaluating DNA variant and copy number changes)34. Eschrich et al. used an approach whereby many multivariate linear models were trained to identify the genes for their radiation sensitivity index (RSI)7, representing one model for each expressed gene. Included in each model was the value of the expressed gene, p53 and RAS mutation status, and tissue of origin which were trained on cell lines to predict SF2. The 500 gene-models with the lowest error in this procedure was used to identify 10 genes based on gene hub identity, which was then used to train a multivariate linear model that predicts radiosensitivity.
Regularized Models
As introduced above, regularization is a procedure where a bias term is added to a GLM to prevent over-fitting and reduce the model’s reliance on less important variables. Regularization is used in both Ridge and the least absolute shrinkage and selection operator (LASSO) regression methods35. In a linear model, each feature is multiplied by a weight, which are then added to a y-axis intercept to produce a prediction. With regularization, a bias (penalization) term is added to this linear model (weight squared for Ridge and the absolute value of the weight for LASSO) to make the model less flexible and thus less prone to overfitting. These penalization values are then scaled by the regularization hyperparameter λ (ranging from 0 to 1) and added to the linear model. The benefit of LASSO is that it can penalize the weights of features to zero, removing less important variables from the model, representing a type of within-model feature selection. Elastic Net regression36 is a hybrid approach that adds a second hyperparameter scaling the amount of each type of regularization bias (Ridge or LASSO).
Zhao et al. utilized the strategy of regularization in the development of the Post-Operative Radiation Therapy Outcomes Score (PORTOS)13. PORTOS performed feature selection through utilization of previously defined gene sets that were then filtered based on the genes that had interaction effects with radiation in Cox models for predicting metastasis in post-operative prostate cancer patients. This filtered down gene list was then used to build two separate ridge penalized Cox models (radiation and no radiation), trained to predict metastasis, with the final PORTOS score representing the difference between the radiation and no radiation model predictions.
Decision Based Models
A limitation of the GLM framework is that these models do not incorporate non-linear effects or interaction terms, and while such features can be directly added to the model, that requires creation of a new variable (feature engineering - such as squaring the expression of one gene or multiplying the expression of two genes together). Decision-based models are desirable as they can account for these effects and thus no added feature engineering steps are required. The most used decision-based models in radiation biomarker development are tree-based models and top scoring pair (TSP) classifiers.
Central to the idea of tree-based methods is recursive partitioning37, where the best possible split of the data is identified. For example, if trying to find how to best separate radiation sensitive and resistant cells based on the expression of 5 genes, a recursive partitioning strategy would evaluate each gene and find the split that has the best classification performance. The same would hold for a regression problem where the split creates groups of samples where the mean of those new groups produces the lowest error. The tree-based model can then repeat these splits, creating multiple “branches”, until you have a tree that has separated samples at a level sufficient to produce good predictions. Unfortunately, while individual tree models are advantageous for their highly interpretable structure, overfitting is a perennial problem.
Tree-based methods can have considerable improvement in predictive performance with a method called ensemble learning, where the combined predictions of many tree models are aggregated into a “forest”. This can be done by producing N trees through the bootstrapped resampling technique, and getting the prediction based on the call with the highest number of votes (classification) or the average for continuous predictions (regression). Unfortunately, another issue can arise, where there may be such a strong signal from one gene that almost every tree has the same first split, resulting in correlated trees. The random forest algorithm38 solves this problem by allowing for random sampling of the features that get used in each tree, thus decorrelating the trees.
Another strategy that has been very successful is to use an ensemble of “weak” models. With respect to tree-based methods, such a strategy will create trees with fewer splits, creating a weak model that explains just part of the data. That model is used to get a first round of predictions and a resulting error for those predictions. Rather than creating a completely independent tree for the next step, like was done with the previously described models, the next tree is created to reduce that error as much as possible through a gradient descent algorithm (a gradient boosted tree). A popular iteration of this is extreme gradient boosting (XGBoost)39. A non-tree, decision-based algorithm that also functions by aggregating “weak” models is the k-Top Scoring Pairs (TSP) classifier. TSP methods are based on pairwise comparisons of feature values40, with a commonly used iteration being k-TSP that creates ensembles of top scoring, non-overlapping feature pairs to create model decision rules (e.g. expression of gene A is higher than gene B), a method that has shown similar efficacy to many other machine learning based methods in evaluating gene expression profiles41. The k-TSP method is similar to the ensemble methods used in gradient boosting decision trees where each new tree works to reduce the error. The difference is that each rule involves a comparison of two features that were not present in any prior model. An advantage of TSP methods is that they are based on relative values of features within one observation, and not absolute values, making them better suited to generalize across different assay platforms. A trade-off of the more complex forest-based approaches is the reduction in interpretability of the model.
A range of decision-based methods have been used in the development of radiation biomarkers. Speers et al. utilized a random forest model trained on breast cancer cell lines to predict SF212. Lewis et al. utilized the XGBoost algorithm to integrate multiple data types into a model to predict radiation response, which combined genomic, transcriptomic, kinetic, and thermodynamic parameters into a flux balance analysis (FBA) model42. Luxton et al. also used XGBoost, trained on telomere length from blood samples, before and after receiving 4 Gy, and used that to predict telomere length post radiation43. Weichselbaum et al. utilized a k-TSP model trained on cell lines to predict radioresistant and radiosensitive clusters20, while Sjöström et al. utilized k-TSP trained on breast cancer samples to predict in breast tumor recurrence after lumpectomy with/without radiation therapy9.
Beyond Classical ML Techniques: Deep Learning
Deep learning (DL) represents a subset of ML methodologies that rely on multi-layered neural networks. Neural networks have existed for many decades, but recent advances in GPU-based processing have made these computationally expensive models feasible. The advantage of DL compared to other ML methods is its ability to model non-linear and interaction effects, perform higher order feature engineering, and produce excellent performance, especially on image analysis tasks. An example of this higher order feature engineering is in convolutional neural networks trained on large image databases, where an inner layer of the neural network may represent a square or circle feature. Prior to deep learning techniques, feature engineering had to be performed to include such features in a model, such as the use of radiomics in medical imaging prediction44, where higher-order features are extracted from images with mathematical formulae and then used in machine learning models. This type of feature engineering is similar to an unsupervised learning approach, as these intermediate radiomic features are created with no knowledge of the outcome of interest. DL allows for bypassing this step through creation of higher order features within the network that are optimized to the outcome of interest.
Deep learning is most advantageous in settings with very large training datasets, and thus has not been as popular for radiation signature development. Transfer learning is a strategy in deep learning that allows smaller datasets to build a model utilizing features from a previously trained model, which has been beneficial in radiology based deep learning models45, where models trained on large databases of non-radiologic images can be utilized to improve model prediction on radiologic images, and could be one way of overcoming limited sample sizes for radiation signatures. Neural networks can also be utilized in unsupervised learning tasks, as was used in the radiosensitivity signature developed by Chen et al., which utilized a neural network autoencoder for feature selection46. Finally, digital pathology-based DL represents an emerging technique for biomarker development47, that may be applicable for radiation biomarker development.
SUMMARY, CURRENT CHALLENGES, AND EMERGING TRENDS
The machine learning methodologies described herein provide a broad range of strategies for developing radiation tumor biomarkers, but there are some consistently used approaches across the literature. Most studies start with feature selection to determine what features to use in the biomarker, which can be done by utilizing previously curated lists of known biology, e.g. genes involved in pathways that are known to be involved in radiation response20,48, differential expression analysis29,30,32, correlative analysis8,22,34,49, or through feature selection as part of a modeling approach7,9,42,46,50. Next, a method is used to stratify patients, either through an unsupervised clustering approach19,22, a supervised model-based approach7–9,12,13,33, or with a combination of both20. Finally, independent clinical validation is the most critical step in the development of radiation biomarkers. Good validation datasets should include clinical samples with clearly defined outcomes, be independent (i.e. not used, and preferably from a different dataset altogether to asses generalizability) from the training set, and ideally should include patients that did and didn’t receive radiation or patients that had different doses. This last feature is critical as it distinguishes a treatment-predictive biomarker, such as identifying a benefit of radiation in those classified as radiosensitive and no benefit in those classified as radioresistant, from prognostic biomarkers. In Table 1 we provide examples of tumor biomarkers that were developed with an explicit goal of modeling radiation sensitivity or benefit (in contrast to many of the currently on the market genomic biomarkers), focusing on those that have independent clinical validation. We also identify markers that have been statistically assessed as predictive biomarkers, defined via an interaction term between radiation treatment/dose and the biomarker, and adjustment for treatment selection bias.
Table 1 –
Examples of Radiation Biomarkers with Independent Validation.
| Signature | Feature selection | ML method | Training | Validation | Validation outcome | Prognostic | Predictive (pint reported) | Treatment selection bias corrected (if pint) |
|---|---|---|---|---|---|---|---|---|
| Weichselbaum 2008 - IRDS20 | Correlation | Clustering, k-TSP | Breast Cancer Patients | Breast Cancer | LRC | X | ||
| Piening 200919 | Differential Expression | Hierarchical Clustering | Cell Lines | Breast Cancer Patient Survival | OS | X | ||
| Eschrich 2012 - RSI10 | Linear Regression | Linear Regression | Cell Lines | Breast Cancer Recurrence Free Survival | RFS | X | X | None |
| Speers 2015 - RSS12 | Correlation | Random Forest | Cell Lines | Breast Cancer Patient LRR and Survival | LRR and OS | X | ||
| Zhao 2016 - PORTOS13 | Interaction Terms | Ridge Regression | Post-operative Prostate Cancer Patients | Post-operative Prostate Cancer Patients | DM | X | X | Matching |
| Sjöström2018 - SSP9 | Random Forest | k-TSP | Breast Cancer Patients | Breast Cancer | IBTR | X | X | None |
| Cui 2018 - RSS and IMS51 | Cox Regression | Ridge and LASSO Regression | Breast Cancer Patients | Breast Cancer | DSS | X | X | Matching |
| Sjöström2019 - ARTIC50 | Cox Regression | Ridge Regression | Breast Cancer Patients | Breast Cancer | LRR | X | X | RCT |
| Zhao 2019 - RT-GS and ChemoRT-GS8 | Correlation | Average | Glioblastoma PDXs | Glioblastoma | OS | X | X | MVA |
| Chen 202046 | Neural Network Autoencoder | LASSO Regression | Breast Cancer Patients | Breast Cancer | OS | X | ||
| Speers 202052 | Correlation | Elastic Net Regression | Breast Cancer Patients | Breast Cancer | Early vs Late Recurrence | X | ||
| Scott 2021 - GARD15 | See RSI | Scaled RSI | Cell Lines | Pan-Cancer | Time to first recurrence and OS | X | X | None |
Abbreviations: ML – Machine Learning, IRDS – Interferon-related DNA damage resistance signature, RSI – Radiosensitivity index, RSS – Radiation Sensitivity signature, PORTOS – Post-operative radiation therapy outcome score, SSP – Single sample predictor, IMS – Immune signature, ARTIC – Adjuvant Radiotherapy Intensification Classifier, RT-GS – Radiotherapy Gene Signature, GARD – Genome-based model for adjusting radiotherapy dose, MVA – Multivariate analysis, TSP – Top scoring pairs, LASSO - Least absolute shrinkage and selection operator, PDX – Patient-derived xenograft, LRC – Locoregional control, OS – Overall survival, RFS – Recurrence free survival, LRR – Locoregional recurrence, DM – Distant metastasis, IBTR – Ipsilateral breast tumor recurrence, DSS – Disease specific survival, pint – interaction p-value between receipt of radiotherapy and the signature, RCT – Randomized Control Trial.
While many radiation tumor biomarkers have been developed, none are currently used in the clinic. Surprisingly, there is little overlap in many of the genes used in these signatures6, despite many being trained on the same data and/or outcomes. A major challenge is the availability of high-quality datasets. A common strategy has been to train models on cell lines7,12,19, but these models inherently lack features from the tumor microenvironment, host immune system, and vasculature. Patient-derived xenografts may provide an opportunity to better replicate the host environment, a strategy used by Zhao et al. in development of their glioblastoma radiation and chemo-radiation gene signatures8, but xenograft development and radiation experimentation is more resource intensive than cell line based experimentation. Collaboration with veterinary colleagues using companion species with de novo tumors treated with radiation is another potential model system, though these genomes are not identical to humans. Clinical datasets are preferred, but typically have other limitations, such as the lack of robust annotation of radiation-related clinical endpoints such as locoregional recurrence, or a limited number of patients. Non-randomized data should account for clinicopathologic variables potentially leading to treatment selection bias (e.g. with matching, propensity adjustment, multi-variate analysis, etc.). Large, randomized trials represent the ideal training and validation datasets, but availability of molecular data is a challenge. Improvements in data sharing and availability, and integration of biomarker development into clinical trials are critical in advancing the development of radiation tumor biomarkers that will be able to provide clinical utility in radiation oncology decision making.
FUNDING
S.G. Zhao is funded by the DOD (W81XWH2010799, W81XWH2110205, W81XWH2210830), NIH (1DP2CA271832-01, UH2CA260389), and the University of Wisconsin OVCRGE Promoting Industry Collaboration Initiative. M. Sjöström is a Prostate Cancer Foundation Young Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
S.G. Zhao has patent applications with Veracyte on molecular signatures in prostate cancer unrelated to this work, and a family member employed by Artera, and with stock in Exact Sciences. M. Bootsma has a family member employed by Luminex – A DiaSorin Company. K. Helzer has a family member employed by Epic Systems.
References
- 1.Barnett GC et al. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer 9, 134–142 (2009). 10.1038/nrc25872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho B Intensity-modulated radiation therapy: a review with a physics perspective. Radiat Oncol J 36, 1–10 (2018). 10.3857/roj.2018.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cagney DN et al. Heterogeneity in high-risk prostate cancer treated with high-dose radiation therapy and androgen deprivation therapy. BMC Urol 17, 60 (2017). 10.1186/s12894-017-0250-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li A et al. Characterizing advanced breast cancer heterogeneity and treatment resistance through serial biopsies and comprehensive analytics. NPJ Precis Oncol 5, 28 (2021). 10.1038/s41698-021-00165-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsoutsou PG et al. Emerging Opportunities of Radiotherapy Combined With Immunotherapy in the Era of Breast Cancer Heterogeneity. Front Oncol 8, 609 (2018). 10.3389/fonc.2018.00609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manem VS & Dhawan A RadiationGeneSigDB: a database of oxic and hypoxic radiation response gene signatures and their utility in pre-clinical research. Br J Radiol 92, 20190198 (2019). 10.1259/bjr.20190198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eschrich SA et al. A gene expression model of intrinsic tumor radiosensitivity: prediction of response and prognosis after chemoradiation. Int J Radiat Oncol Biol Phys 75, 489–496 (2009). 10.1016/j.ijrobp.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao SG et al. Xenograft-based, platform-independent gene signatures to predict response to alkylating chemotherapy, radiation, and combination therapy for glioblastoma. Neuro Oncol 21, 1141–1149 (2019). 10.1093/neuonc/noz090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sjöström M et al. Identification and validation of single-sample breast cancer radiosensitivity gene expression predictors. Breast Cancer Res 20, 64 (2018). 10.1186/s13058-018-0978-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eschrich SA et al. Validation of a radiosensitivity molecular signature in breast cancer. Clin Cancer Res 18, 5134–5143 (2012). 10.1158/1078-0432.CCR-12-0891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres-Roca JF et al. Integration of a Radiosensitivity Molecular Signature Into the Assessment of Local Recurrence Risk in Breast Cancer. Int J Radiat Oncol Biol Phys 93, 631–638 (2015). 10.1016/j.ijrobp.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speers C et al. Development and Validation of a Novel Radiosensitivity Signature in Human Breast Cancer. Clin Cancer Res 21, 3667–3677 (2015). 10.1158/1078-0432.CCR-14-2898 [DOI] [PubMed] [Google Scholar]
- 13.Zhao SG et al. Development and validation of a 24-gene predictor of response to postoperative radiotherapy in prostate cancer: a matched, retrospective analysis. Lancet Oncol 17, 1612–1620 (2016). 10.1016/S1470-2045(16)30491-0 [DOI] [PubMed] [Google Scholar]
- 14.Scott JG et al. A genome-based model for adjusting radiotherapy dose (GARD): a retrospective, cohort-based study. Lancet Oncol 18, 202–211 (2017). 10.1016/S1470-2045(16)30648-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott JG et al. Pan-cancer prediction of radiotherapy benefit using genomic-adjusted radiation dose (GARD): a cohort-based pooled analysis. Lancet Oncol 22, 1221–1229 (2021). 10.1016/S1470-2045(21)00347-8 [DOI] [PubMed] [Google Scholar]
- 16.Servant N et al. Search for a gene expression signature of breast cancer local recurrence in young women. Clin Cancer Res 18, 1704–1715 (2012). 10.1158/1078-0432.CCR-11-1954 [DOI] [PubMed] [Google Scholar]
- 17.Andreopoulos B, An A, Wang X & Schroeder M A roadmap of clustering algorithms: finding a match for a biomedical application. Brief Bioinform 10, 297–314 (2009). 10.1093/bib/bbn058 [DOI] [PubMed] [Google Scholar]
- 18.Peterson AD, Ghosh AP & Maitra R Merging K-means with hierarchical clustering for identifying general-shaped groups. Stat (Int Stat Inst) 7 (2018). 10.1002/sta4.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piening BD, Wang P, Subramanian A & Paulovich AG A radiation-derived gene expression signature predicts clinical outcome for breast cancer patients. Radiat Res 171, 141–154 (2009). 10.1667/RR1223.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weichselbaum RR et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A 105, 18490–18495 (2008). 10.1073/pnas.0809242105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jolliffe IT & Cadima J Principal component analysis: a review and recent developments. Philos Trans A Math Phys Eng Sci 374, 20150202 (2016). 10.1098/rsta.2015.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HS et al. Identification of a radiosensitivity signature using integrative metaanalysis of published microarray data for NCI-60 cancer cells. BMC Genomics 13, 348 (2012). 10.1186/1471-2164-13-348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starmans MH et al. The prognostic value of temporal in vitro and in vivo derived hypoxia gene-expression signatures in breast cancer. Radiother Oncol 102, 436–443 (2012). 10.1016/j.radonc.2012.02.002 [DOI] [PubMed] [Google Scholar]
- 24.van der Maaten L, H. G Visualizing Data using t-SNE. Journal of Machine Learning Research 9, 2579–2605 (2008). [Google Scholar]
- 25.McInnes L, H. J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv e-prints 1802.03426 (2018). [Google Scholar]
- 26.Wu Y & Zhang K Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nat Rev Nephrol 16, 408–421 (2020). 10.1038/s41581-020-0262-0 [DOI] [PubMed] [Google Scholar]
- 27.Gao Y, Duan Q, Wu N & Xu B A heterogeneous cellular response to ionizing radiation revealed by single cell transcriptome sequencing. Am J Cancer Res 11, 513–529 (2021). [PMC free article] [PubMed] [Google Scholar]
- 28.Costa-Silva J, Domingues D & Lopes FM RNA-Seq differential expression analysis: An extended review and a software tool. PLoS One 12, e0190152 (2017). 10.1371/journal.pone.0190152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jong MC et al. Pretreatment microRNA Expression Impacting on Epithelial-toMesenchymal Transition Predicts Intrinsic Radiosensitivity in Head and Neck Cancer Cell Lines and Patients. Clin Cancer Res 21, 5630–5638 (2015). 10.1158/1078-0432.CCR-15-0454 [DOI] [PubMed] [Google Scholar]
- 30.Amundson SA et al. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res 68, 415–424 (2008). 10.1158/0008-5472.CAN-07-2120 [DOI] [PubMed] [Google Scholar]
- 31.van Malenstein H et al. A seven-gene set associated with chronic hypoxia of prognostic importance in hepatocellular carcinoma. Clin Cancer Res 16, 4278–4288 (2010). 10.1158/1078-0432.CCR-09-3274 [DOI] [PubMed] [Google Scholar]
- 32.Toustrup K et al. Gene expression classifier predicts for hypoxic modification of radiotherapy with nimorazole in squamous cell carcinomas of the head and neck. Radiother Oncol 102, 122–129 (2012). 10.1016/j.radonc.2011.09.010 [DOI] [PubMed] [Google Scholar]
- 33.Torres-Roca JF et al. Prediction of radiation sensitivity using a gene expression classifier. Cancer Res 65, 7169–7176 (2005). 10.1158/0008-5472.CAN-05-0656 [DOI] [PubMed] [Google Scholar]
- 34.Yard BD et al. A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nat Commun 7, 11428 (2016). 10.1038/ncomms11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melkumova LE, S. SY Comparing Ridge and LASSO estimators for data analysis. Procedia Engineering 201, 746–755 (2017). 10.1016/j.proeng.2017.09.615 [DOI] [Google Scholar]
- 36.Zou H, H. T Regularization and variable selection via the elastic net. J. R. Statist. Soc. B 67, 301–320 (2004). [Google Scholar]
- 37.Strobl C, Malley J & Tutz G An introduction to recursive partitioning: rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol Methods 14, 323–348 (2009). 10.1037/a0016973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biau G Analysis of a Random Forests Model. Journal of Machine Learning Research 13, 1063–1095 (2012). 10.1177/1536867X20909688 [DOI] [Google Scholar]
- 39.Tianqi Chen CG XGBoost: A Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining KDD ‘16, 785–794 (2016). https://doi.org:http://doi.acm.org/10.1145/2939672.2939785 [Google Scholar]
- 40.Geman D, d’Avignon C, Naiman DQ & Winslow RL Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol Biol 3, Article19 (2004). 10.2202/1544-6115.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan AC, Naiman DQ, Xu L, Winslow RL & Geman D Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics 21, 3896–3904 (2005). 10.1093/bioinformatics/bti631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis JE & Kemp ML Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance. Nat Commun 12, 2700 (2021). 10.1038/s41467-021-22989-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luxton JJ et al. Telomere Length Dynamics and Chromosomal Instability for Predicting Individual Radiosensitivity and Risk via Machine Learning. J Pers Med 11 (2021). 10.3390/jpm11030188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayerhoefer ME et al. Introduction to Radiomics. J Nucl Med 61, 488–495 (2020). 10.2967/jnumed.118.222893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HE et al. Transfer learning for medical image classification: a literature review. BMC Med Imaging 22, 69 (2022). 10.1186/s12880-022-00793-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Zheng J, Zhuo ML, Zhang A & You Z A six-gene-based signature for breast cancer radiotherapy sensitivity estimation. Biosci Rep 40 (2020). 10.1042/BSR20202376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Esteva A et al. Prostate cancer therapy personalization via multi-modal deep learning on randomized phase III clinical trials. NPJ Digit Med 5, 71 (2022). 10.1038/s41746-022-00613-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winter SC et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res 67, 3441–3449 (2007). 10.1158/0008-5472.CAN-06-3322 [DOI] [PubMed] [Google Scholar]
- 49.Abazeed ME et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res 73, 6289–6298 (2013). 10.1158/0008-5472.CAN-13-1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjöström M et al. Clinicogenomic Radiotherapy Classifier Predicting the Need for Intensified Locoregional Treatment After Breast-Conserving Surgery for Early-Stage Breast Cancer. J Clin Oncol 37, 3340–3349 (2019). 10.1200/JCO.19.00761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui Y, Li B, Pollom EL, Horst KC & Li R Integrating Radiosensitivity and Immune Gene Signatures for Predicting Benefit of Radiotherapy in Breast Cancer. Clin Cancer Res 24, 4754–4762 (2018). 10.1158/1078-0432.CCR-18-0825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Speers C et al. A Signature That May Be Predictive of Early Versus Late Recurrence After Radiation Treatment for Breast Cancer That May Inform the Biology of Early, Aggressive Recurrences. Int J Radiat Oncol Biol Phys 108, 686–696 (2020). 10.1016/j.ijrobp.2020.05.015 [DOI] [PubMed] [Google Scholar]