

Abstract
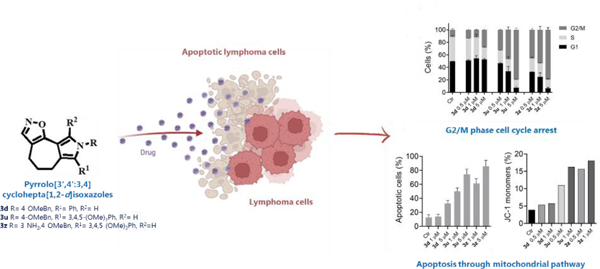
Unsatisfactory outcomes for relapsed/refractory lymphoma patients prompt continuing efforts to develop new therapeutic strategies. Our previous studies on pyrrole-based anti-lymphoma agents led us to synthesize a new series of twenty-six pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole derivatives and study their antiproliferative effects against a panel of four non-Hodgkin lymphoma cell lines. Several candidates showed significant anti-proliferative effects, with IC50’s reaching the sub-micromolar range in at least one cell line, with compound 3z demonstrating sub-micromolar growth inhibitory effects towards the entire panel. The VL51 cell line was the most sensitive, with an IC50 value of 0.10 μM for 3z.
Our earlier studies had shown that tubulin was a prominent target of many of our oxazole derivatives. We therefore examined their effects on tubulin assembly and colchicine binding. While 3u and 3z did not appear to target tubulin, good activity was observed with 3d and 3p.
Molecular docking and molecular dynamics simulations allowed us to rationalize the binding mode of the synthesized compounds toward tubulin. All ligands exhibited a better affinity for the colchicine site, confirming their specificity for this binding pocket. In particular, a better affinity and free energy of binding was observed for 3d and 3p. This result was confirmed by experimental data, indicating that, although both 3d and 3p significantly affected tubulin assembly, only 3d showed activity comparable to that of combretastatin A-4, while 3p was about 4-fold less active.
Cell cycle analysis showed that compounds 3u and especially 3z induced a block in G2/M, a strong decrease in S phase even at low compound concentrations and apoptosis through the mitochondrial pathway. Thus, the mechanism of action of 3u and 3z remains to be elucidated.
Very high selectivity toward cancer cells and low toxicity in human peripheral blood lymphocytes were observed, highlighting the good potential of these agents in cancer therapy and encouraging further exploration of this compound class to obtain new small molecules as effective lymphoma treatments.
Keywords: pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles; isoxazoles; antitumor agents; lymphoma; hematological malignancies
Graphical Abstract
1. Introduction
Lymphomas represent one of the most common hematological malignancies worldwide, affecting both children and adults. Lymphomas represent almost 5% of all cancers, with slow-growing forms not currently curable, and an annual incidence that has gradually increased in recent years [1,2]. Bone marrow/stem cell transplantation, radiation therapy and immunotherapy/targeted therapy, involving monoclonal antibodies, antibody drug conjugates or chimeric antigen receptor (CAR) T-cell therapy are among current treatment options. However, combination chemotherapy regimens, such as R-CHOP, ABVD or BEACOP, remain the cornerstone in the treatment of lymphomas [3,4], encouraging medicinal chemists to search for new agents with improved efficacy, tolerability, and specificity and that are not MDR substrates.
Pyrrole-based compounds have attracted much attention as bioactive molecules [5–12], and in this field, we have been involved in the synthesis of small molecules as anti-proliferative agents [13–17]. At the beginning of our studies, we identified a class of [1,2]oxazole isoindoles 1 (Chart 1) with potent activity in mesothelioma models. When these compounds were further explored, in terms of structure – activity relationships [18], we found that they had antitubulin activity and were active in refractory lymphoma models. In particular, micromolar – nanomolar IC50 values were obtained against four different lymphoma subtypes, and the compounds induced cell cycle arrest in the G2/M phase and caused apoptosis. These activities were confirmed by transcriptome analysis [18–20]. The insights gained regarding the chemical space led to the class of pyrrolo[2’,3’:3,4]cyclohepta[1,2-d][1,2]oxazoles 2 (Chart 1), which showed potent antitumor activity against the full NCI 60 cell line panel, with GI50 values at the nanomolar level and mean graph mid-points (MG_MID) of 0.08–0.41 μM. Moreover, they exhibited potent growth inhibitory activities against six lymphoma cell lines not included in the NCI panel, with IC50 values at the micromolar – submicromolar level. The most active compounds were able to induce cell cycle arrest in the G2/M phase, confirming the mechanism of the former class 1, along with apoptosis, mitochondrial depolarization, ROS generation and PARP cleavage activation [21]. The promising results obtained in refractory lymphoma models encouraged further exploration of the tricyclic pyrrole oxazole system, considering that so far only a few classes of small molecules have been reported as effective lymphoma treatments [2]. In this study, we examined the new class of pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles 3, positional isomers of class 2, to determine whether they would enhance the antitumor effects of [1,2]oxazoles in lymphomas (Chart 1). This structural manipulation combines the main structural features of compounds 1 and 2, based on the condensation of the pyrrole moiety in the tricyclic cyclohepta scaffold to yield a new chemical class of compounds 3 (Chart 1).
Chart 1.
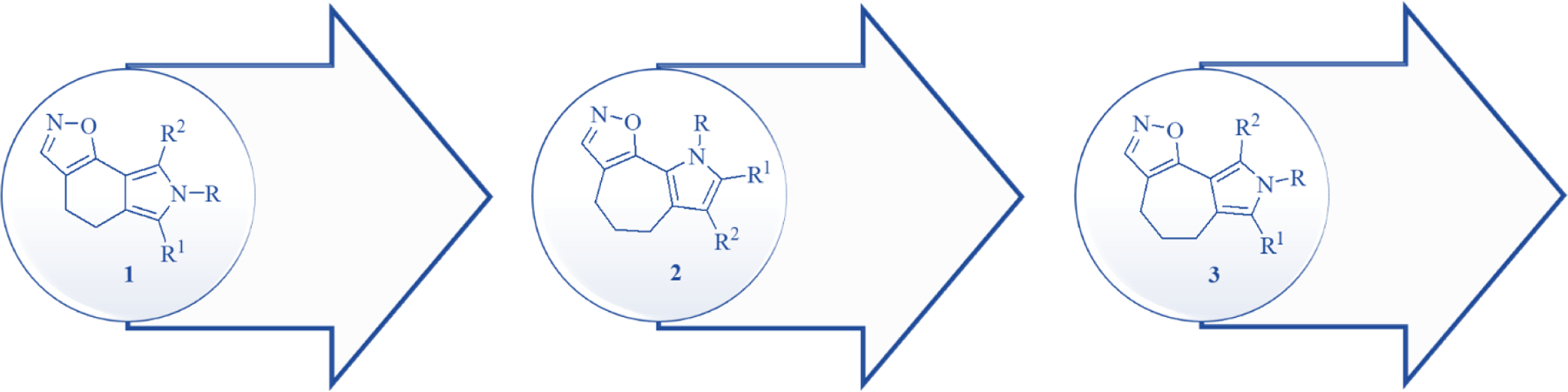
[1,2]Oxazolo[5,4-e]isoindoles (1), pyrrolo[2’,3’:3,4]cyclohepta[1,2-d][1,2]oxazoles (2), pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles (3).
2. Results and Discussion
2.1. Chemistry
Our synthetic approach to the pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole ring system began with the preparation of 5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-ones 8a-d as appropriate building blocks to achieve the tricyclic framework.
Based on our previous experience [19,22,23], we obtained ketones 8a,b by a multistep sequence described in Scheme 1. Commercially available cycloheptane-1,3-dione 4 was converted into the enamino derivative 5 in refluxing N,N-dimethylformamide dimethyl acetal (DMFDMA). The latter was reacted with phenylglycine or 3,4,5-trimethoxyphenylglycine, leading to intermediates 6a,b (80 – 82%), which were used in the following step without purification. Cyclization of compounds 6a,b in acetic anhydride and triethylamine yielded compounds 7a,b (53 – 73%) (Scheme 1), which were subjected to hydrolysis of acetyl groups in a (1:12) mixture of HCl (37%) and acetic acid (80%) at 60 °C, leading to compounds 8a,b (75 – 81%) (Scheme 1).
Scheme 1.
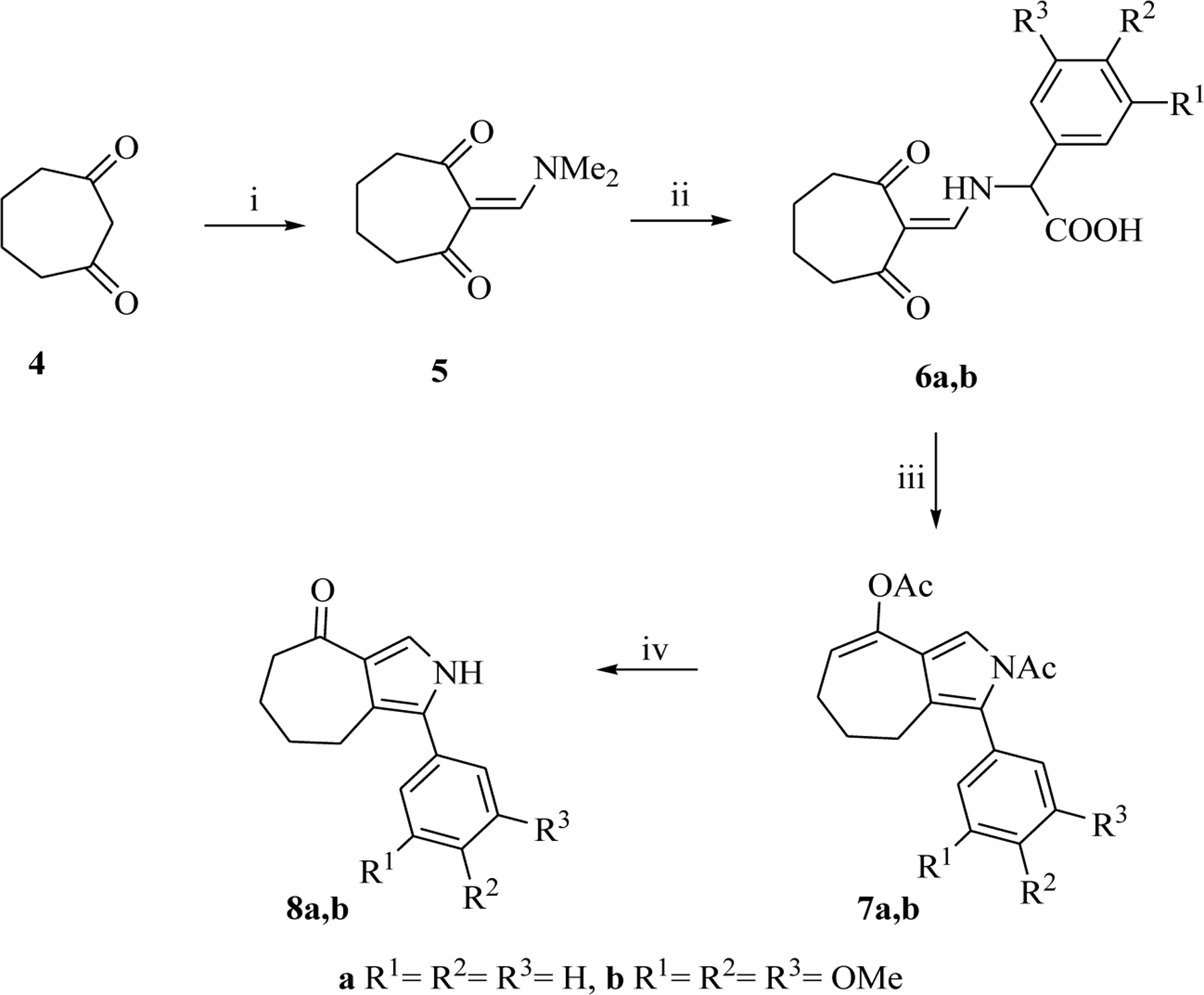
Synthesis of 1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8a) and 1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8b). Reagents and conditions: (i) DMFDMA, reflux, 1 h, 99%; (ii) phenylglycine or 3,4,5-trimethoxy phenylglycine, AcONa.3H2O, ethanol, reflux, 90 min, 80 – 82%; (iii) Et3N, Ac2O, 30 min, reflux, 30 min, 53 – 73%; (iv) 80% acetic acid/37% HCl (1:12), 15 min, 60 °C, 75 – 81%.
Ketones 8c,d were synthesized using a multistep sequence (Scheme 2). Ethyl 5-(4-methoxyphenyl)-1H-pyrrole-2-carboxylate 10 was obtained (74%) by reaction of 3-(4-methoxyphenyl)acrylaldehyde 9 with ethyl 2-azidoacetate. Friedel–Crafts acylation of compound 10 in the presence of glutaric anhydride and AlCl3, as acylating reagent and Lewis acid, respectively, led to compound 11 (60%) (Scheme 2). The subsequent reduction of the carbonyl group at the 2-position of the pyrrole ring and then cyclization by dehydration with an excess of trifluoroacetic anhydride gave the ethyl 3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate 8c (60%) (Scheme 2). Basic hydrolysis of the ethoxycarbonyl group and subsequent decarboxylation in the presence of 6 M HCl led to 8d in satisfactory yield (60%) (Scheme 2).
Scheme 2.
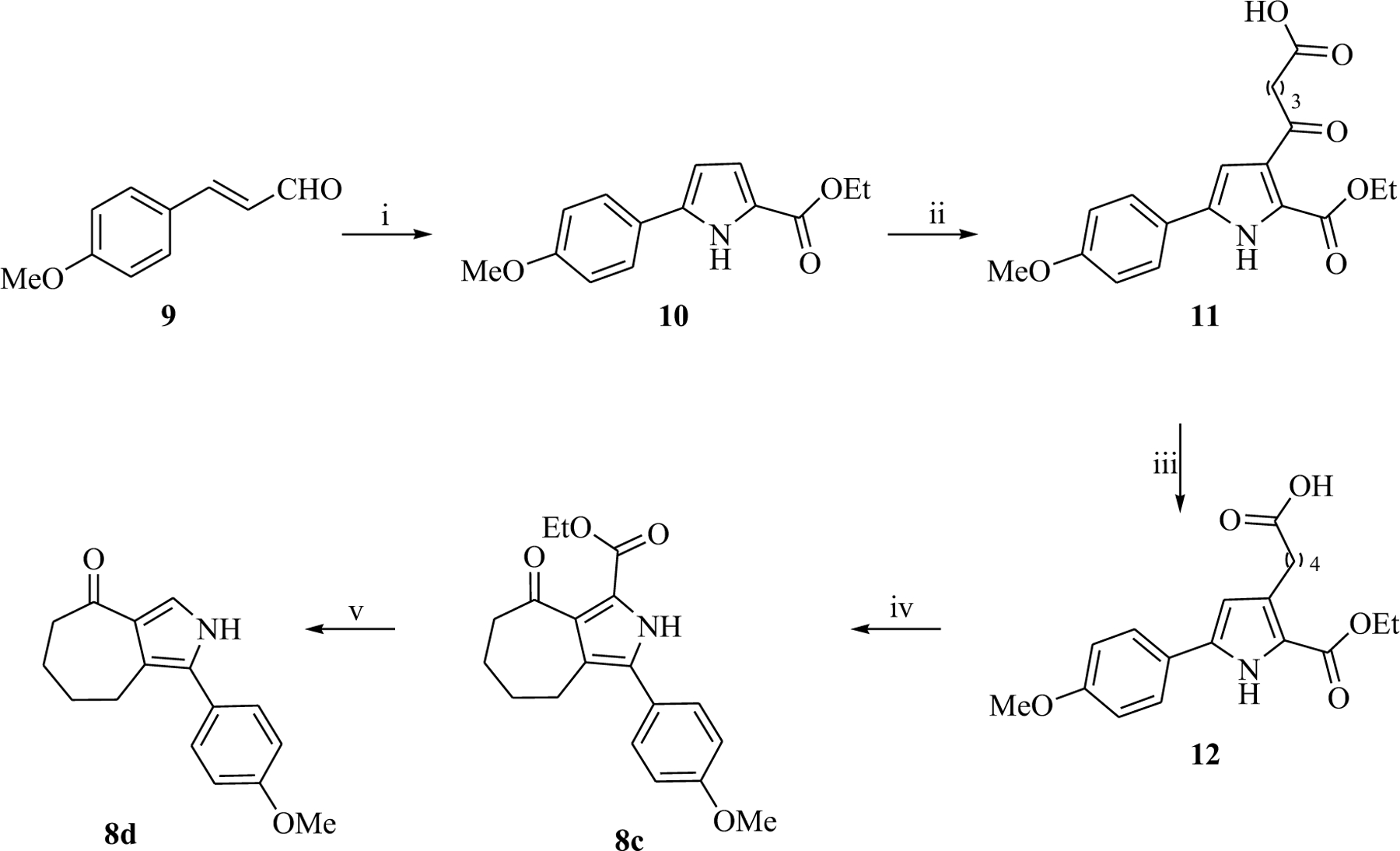
Synthesis of 3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate (8c) and 1-(4-methoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8d). Reagents and conditions: (i) a) ethyl azidoacetate, EtOK, ethanol, −20 °C, 4.5 h; b) toluene, reflux, 24 h, 74%; (ii) AlCl3, glutaric anhydride, DCM, rt, 1 h then 10, rt, 24 h, 60%; (iii) triethylsilane, trifluoroacetic acid, rt, 24 h, 61%; (iv) trifluoroacetic anhydride, rt, 1 h, 60%; (v) a) 50% KOH, ethanol, reflux, 3 h; b) HCl 6 M, ethanol, reflux, 1 h, 60%.
Ketones 8a-d were properly functionalized at the pyrrole nitrogen by reaction with benzyl halides and sodium hydride, as a base, to give the corresponding N-substituted derivatives 14a-h,j-y (60–98%) (Scheme 3). The 3-nitro,4-methoxybenzyl substituted derivatives 14h,y were subjected to catalytic reduction with ammonium formate and 10% Pd/C in ethyl acetate, furnishing the corresponding amino derivatives 14i,z (71 – 86%) (Scheme 3).
Scheme 3.
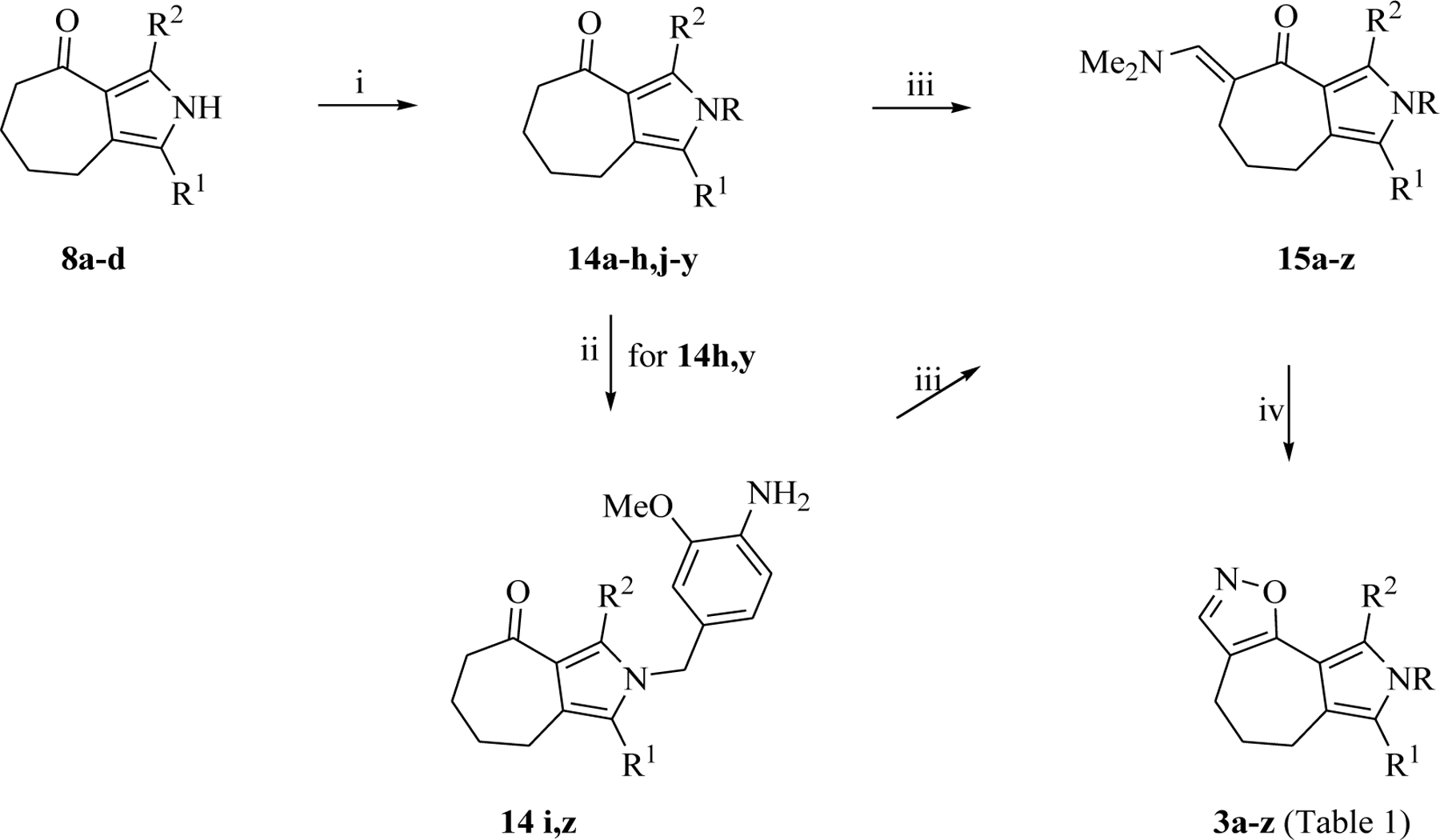
Synthesis of pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles 3a-z. Reagents and conditions: (i) NaH, DMF, 0 °C to rt, 1 h then benzyl halides at 0 °C to rt, 3–12 h, 60 – 98%; (ii) ammonium formate, 10% Pd/C, ethyl acetate, rt, 12 h, 71 – 86%; (iii) TBDMAM, toluene, reflux, 12 h; (iv) NH2OH·HCl, ethanol, reflux, 1 h, 60 – 93 %.
The N-substituted derivatives 14 were converted into the α-enaminoketones 15a-z using tert-butoxybis(dimethylamino)methane (TBDMAM), which upon reaction with hydroxylamine hydrochloride as dinucleophile yielded pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles 3a-z in good to excellent yields (60 – 93%) (Scheme 3, Table 1).
Table 1.
Pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles 3a-z.
| [1,2-oxazole] | Starting ketone | R | R1 | R2 | Yieldsa (%) |
|---|---|---|---|---|---|
| 3a | 14a | Bn | Ph | H | 73 |
| 3b | 14b | 2-OMeBn | Ph | H | 78 |
| 3c | 14c | 3-OMeBn | Ph | H | 68 |
| 3d | 14d | 4-OMeBn | Ph | H | 83 |
| 3e | 14e | 2,5-(OMe)2Bn | Ph | H | 80 |
| 3f | 14f | 3,4-(OMe)2Bn | Ph | H | 80 |
| 3g | 14g | 3,4,5-(OMe)3Bn | Ph | H | 69 |
| 3h | 14h | 3-NO2,4-OMeBn | Ph | H | 60 |
| 3i | 14i | 3-NH2,4-OMeBn | Ph | H | 63 |
| 3j | 14j | Bn | OMe-Ph | COOEt | 93 |
| 3k | 14k | 2-OMeBn | OMe-Ph | COOEt | 83 |
| 3l | 14l | 3-OMeBn | OMe-Ph | COOEt | 75 |
| 3m | 14m | 4-OMeBn | OMe-Ph | COOEt | 66 |
| 3n | 14n | 2-OMeBn | OMe-Ph | H | 60 |
| 3o | 14o | 3-OMeBn | OMe-Ph | H | 64 |
| 3p | 14p | 4-OMeBn | OMe-Ph | H | 70 |
| 3q | 14q | 3,4,5-(OMe)3Bn | OMe-Ph | H | 64 |
| 3r | 14r | Bn | 3,4,5-(OMe)3Ph | H | 66 |
| 3s | 14s | 2-OMeBn | 3,4,5-(OMe)3Ph | H | 60 |
| 3t | 14t | 3-OMeBn | 3,4,5-(OMe)3Ph | H | 68 |
| 3u | 14u | 4-OMeBn | 3,4,5-(OMe)3Ph | H | 71 |
| 3v | 14v | 2,5-(OMe)2Bn | 3,4,5-(OMe)3Ph | H | 77 |
| 3w | 14w | 3,4-(OMe)2Bn | 3,4,5-(OMe)3Ph | H | 71 |
| 3x | 14x | 3,4,5-(OMe)3Bn | 3,4,5-(OMe)3Ph | H | 71 |
| 3y | 14y | 3-NO2,4-OMeBn | 3,4,5-(OMe)3Ph | H | 67 |
| 3z | 14z | 3-NH2,4-OMeBn | 3,4,5-(OMe)3Ph | H | 68 |
Obtained at the final reaction step.
2.2. Antiproliferative activity
Pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles 3a-z were submitted to the NCI and initially tested at a 10−5 M dose in the full panel of 60 human cell lines derived from nine human cancer cell types (leukemia, non-small-cell lung, colon, central nervous system, melanoma, ovarian, renal, prostate and breast) (Tables S1–S3). Two compounds 3u and 3z were then selected for further screening over a five-dose concentration range (10−4–10−8 M) in each of the 60 tumor cell lines, defining their antiproliferative activity in terms of GI50 values. Both compounds showed growth inhibitory effects against all tested human tumor cell lines, with GI50 values in the low micromolar to submicromolar range (Table 2).
Table 2.
Overview of the NCI in vitro human tumor cell line screening for derivatives 3u,3z.
GI50 = concentration that inhibits 50% net cell growth (μM)
Number of cell lines investigated.
Number of cell lines giving positive GI50 values.
MG_MID = mean graph midpoint (μM); the arithmetic mean value for all tested cancer cell lines. If the indicated effect was not attainable under the concentration range used, the highest tested concentration was used for the calculation.
The 3,4,5-trimethoxyphenyl substituted derivative 3z, bearing a 3-amino, 4-methoxybenzyl group at the pyrrole nitrogen, emerged as the most potent candidate, with a mean graph_midpoint (MG_MID) of 0.69 μM on the full NCI panel. The analysis of the GI50 values listed in Table 3 showed that leukemia and prostate cell lines were particularly responsive to treatment with 3z, with GI50 values of 0.30 – 0.65 μM and 0.43 – 0.84 μM, respectively, maintaining submicromolar activity against all the tested cell lines. Comparable potency was also exerted against the renal (GI50 0.36 – 0.84 μM) and colon (GI50 0.38 – 0.56 μM) cancer subpanels, with the exception of the TK-10 (GI50 96.6 μM) and HCC-2998 (GI50 1.66 μM) cell lines, respectively. The best antiproliferative effect in specific subpanels was observed for the melanoma MDA-MB-435 cell line (GI50 0.24 μM), the breast cancer line BT-549 (GI50 0.25 μM), and the non-small cell lung line NCI-H522 (GI50 0.26 μM).
Table 3.
In vitro GI50 (μM) values for compounds 3u and 3z in the full NCI panel.
| Cell lines | 3u | 3z | Cell lines | 3u | 3z |
|---|---|---|---|---|---|
| Leukemia | M14 | 0.39 | 0.48 | ||
| CCRF-CEM | 0.41 | 0.44 | MDA-MB-435 | 0.23 | 0.24 |
| HL-60(TB) | 0.35 | 0.30 | SK-MEL-2 | 1.61 | - |
| K-562 | 0.42 | 0.39 | SK-MEL-28 | - | 2.93 |
| MOLT-4 | 0.80 | 0.65 | SK-MEL-5 | 2.00 | 1.13 |
| RPMI-8226 | 1.56 | 0.61 | UACC-257 | - | 0.58 |
| SR | 0.36 | 0.36 | UACC-62 | 0.91 | 0.43 |
| Non-Small Cell Lung Cancer | Ovarian Cancer | ||||
| A549/ATCC | 2.34 | 0.95 | IGROV1 | 0.56 | 1.56 |
| EKVX | 3.00 | 2.67 | OVCAR-4 | 4.97 | - |
| HOP-62 | 2.22 | 0.69 | OVCAR-5 | 3.72 | 1.44 |
| HOP-92 | 1.27 | 0.52 | OVCAR-8 | 4.30 | 1.57 |
| NCI-H226 | 1.99 | 2.54 | NCI/ADR-RES | 0.54 | 0.60 |
| NCI-H23 | 2.62 | 1.34 | SK-OV-3 | 4.18 | 0.68 |
| NCI-H322M | 3.28 | 0.84 | Renal Cancer | ||
| NCI-H460 | 0.50 | 0.40 | 786–0 | 0.67 | 0.52 |
| NCI-H522 | 0.98 | 0.26 | A498 | 0.69 | 0.36 |
| Colon Cancer | ACHN | 0.90 | 0.69 | ||
| COLO 205 | 1.45 | 0.38 | CAKI-1 | 0.49 | 0.58 |
| HCC-2998 | 1.61 | 1.66 | RXF 393 | 0.66 | 0.45 |
| HCT-116 | 0.39 | 0.40 | SN12C | 4.60 | 0.84 |
| HCT-15 | 0.48 | 0.53 | TK-10 | - | 96.6 |
| HT29 | 1.13 | 0.42 | UO-31 | 0.92 | 0.69 |
| KM12 | 0.69 | 0.56 | Prostate Cancer | ||
| SW-620 | 0.47 | 0.43 | PC-3 | 0.85 | 0.43 |
| CNS cancer | DU-145 | 4.11 | 0.84 | ||
| SF-268 | 4.72 | 1.45 | Breast Cancer | ||
| SF-295 | 0.51 | 0.50 | MCF7 | 0.44 | 0.42 |
| SF-539 | 0.70 | 0.40 | MDA-MB-231/ATCC | 2.54 | 1.15 |
| SNB-19 | 1.53 | 0.81 | HS 578T | 1.54 | 0.40 |
| SNB-75 | 0.87 | 0.38 | BT-549 | 0.48 | 0.25 |
| U251 | 4.47 | 0.51 | T-47D | 1.85 | 0.39 |
| Melanoma | MDA-MB-468 | 0.56 | 0.65 | ||
| LOX IMVI | 0.99 | 0.70 |
Compound 3u, a 3,4,5-trimethoxyphenyl derivative with a 4-methoxybenzyl group at the pyrrole nitrogen, was second in overall potency with high selectivity against the leukemia and colon cancer subpanels (GI50 values of 0.35 – 1.56 μM and 0.39 – 1.61 μM, respectively). The calculated MG_MID for these two subpanels were 0.65 and 0.88 μM, respectively, much lower than the overall cell line MG_MID value of 1.41 μM. For compound 3u, the most sensitive cell lines were MDA-MB-435, with a GI50 of 0.23 μM, and the two leukemic cell lines HL-60(TB) and SR, with GI50 values of 0.35 and 0.36 μM, respectively.
2.3. Screening results in lymphoma models
Because of our interest in lymphoma models, the pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles 3a-z were evaluated against non-Hodgkin lymphoma (NHL) cells. Cell viability was assessed in cultures of HBL1 (activated B cell-like diffuse large B cell lymphoma, ABC-DLBCL), SU-DHL-10 (germinal center B cell-like diffuse large B cell lymphoma, GCB-DLBCL), MINO (mantle cell lymphoma, MCL) and VL51 (marginal zone lymphoma, MZL) cells by means of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays. In order to select compounds with the highest activity, a concentration of 1 μM was chosen for the initial assay. After a 72 h incubation, compounds 3u and 3z showed a reduction in the percentage of cellular proliferation higher than 50% towards each cell line, and they were therefore tested in a wider range of concentrations (0.15 – 10 μM) to establish IC50 values. These are shown in Table 4, with IC50 values all in the low micromolar – submicromolar range. Among the four NHL cell lines, VL51 was the most sensitive to the two compounds, with IC50 values < 500 nM. In all cell lines, 3z was more active than 3u, with IC50 values ranging from 0.1 to 0.5 μM.
Table 4.
IC50 values (μM) of 3u and 3z against NHL cell lines. Cell lines were exposed to the compounds at 0.15 – 10 μM for 72 h.
| CPD | VL51 (MZL) | MINO (MCL) | HBL1 (ABC DLBCL) | SU-DHL-10 (GCB DLBCL) |
|---|---|---|---|---|
| 3u | 0.4±0.07 | 0.9±0.4 | 1±0.3 | 1.8±0.7 |
| 3z | 0.1±0.06 | 0.4±0.02 | 0.5±0.7 | 0.5±0.05 |
Marginal zone lymphoma (MZL); mantle cell lymphoma (MCL); activated B cell-like diffuse large B cell lymphoma (ABC-DLBCL); germinal center B cell-like diffuse large B cell lymphoma (GCB-DLBCL)
From a structure-activity relationship point of view, the presence of a 3,4,5-trimethoxyphenyl ring and a 4-methoxybenzyl group at the pyrrole nitrogen were structural requirements for the higher antiproliferative activity of this new class of [1,2]oxazoles. Furthermore, the amino group at position 3 of the benzene ring seemed crucial to obtain the best growth inhibitory effect. The presence of an ethoxycarbonyl group in position 9 was not essential and generally reduced activity compared to the corresponding parent compound.
Compared to the previously reported classes of compounds 1 and 2, the derivatives of group 3 displayed interesting antiproliferative activity, which was, however, slightly reduced with respect to the class 1 compound bearing a cyclohexyl central ring, indicating that the enlargement of the central ring results in reduction of activity.
2.4. Effects of compounds in human peripheral blood lymphocytes (PBLs)
With the aim of obtaining a preliminary indication of the cytotoxic potential of these derivatives in non-tumoral human cells, three representative compounds (3d, 3u and 3z) were evaluated in vitro against PBLs from healthy donors. As shown in Table 5, all three compounds showed IC50 values greater than 10 μM both in resting lymphocytes and in lymphocytes in an active phase of proliferation induced by phytohematoagglutinin (PHA), a mitogenic stimulus. These results suggest that these compounds have very high selectivity toward cancer cells and low toxicity in normal cells.
Table 5.
Cytotoxicity of compounds 3d, 3u and 3z in human PBLs.
Compound concentration required to inhibit cell growth by 50%
PBL not stimulated with PHA
PBL stimulated with PHA
2.5. Tubulin binding assay
Since [1,2]oxazolo[5,4-e]isoindoles 1, pyrrolo[2’,3’:3,4]cyclohepta[1,2-d][1,2]oxazoles 2 and other derivatives structurally related to this new class of compounds were previously shown to affect tubulin polymerization [18,21,24], we investigated their antitubulin activity in comparison with reference compound combretastatinA-4 (CA-4), which potently inhibits tubulin assembly by interacting with the colchicine site on tubulin. The inhibition of tubulin assembly was assessed for all compounds in a reaction mixture containing 10 μM (1.0 mg/mL) tubulin. Those compounds having IC50 values < 5 μM in the assembly assay were further evaluated for their ability to compete with the colchicine-tubulin interaction. The colchicine assay was performed with 0.5 μM tubulin, 5.0 μM [3H]colchicine and 5.0 μM inhibitor (Table 6). We found that only 3d and 3p significantly affected tubulin assembly, with 3d having activity similar to that of CA-4, while 3p was about 4-fold less active. Neither compound had significant activity inhibiting colchicine binding at 5 μM, but weak inhibition was observed when the concentration of 3d was increased to 25 μM. Neither 3u nor 3z caused significant inhibition of tubulin assembly at low concentrations, thus excluding tubulin as their intracellular target, unless they undergo intracellular conversion to a more active agent.
Table 6.
Inhibition of tubulin assembly and colchicine binding by compounds 3d,3p,3u,3z.
| CPD | Inhibition of tubulin assembly | Inhibition of colchicine binding | |
|---|---|---|---|
|
| |||
| IC50 (μM) ± SD | % Inhibition ± SD 5 μM inhibitor | % Inhibition ± SD 25 μM inhibitor | |
| CA-4 | 0.75 ± 0.06 | 98 ± 2 | - |
|
| |||
| 3d | 1.1 ± 0.09 | 1.8 ± 4 | 19 ± 3 |
| 3p | 4.6 ± 0.2 | 7.7 ± 5 | - |
| 3u | > 20 | - | - |
| 3z | 18 ± 3 | - | |
2.6. Molecular modeling
Computational studies were focused on compounds 3d, 3p, 3u, and 3z to elucidate their potential interactions with tubulin [25]. Thus, we performed docking studies directed toward the colchicine (4O2B PDB code) [26] and vinblastine (1Z2B PDB code) [27] sites by selecting the pose with the best G-Score (kcal/mol) for each compound. The ligands showed insufficient affinity for being accommodated at the vinblastine site, as demonstrated by their G-scores ranging between −4.16 and −5.07 Kcal/mol, due to the formation of numerous poor contacts and steric hindrances. Conversely, all ligands exhibited a better affinity for the colchicine site (Table S4), confirming their specificity for this binding pocket.
The experimental data indicate that none of the compounds have a better affinity than colchicine for tubulin, since inhibition of colchicine binding was relatively weak with 3d and 3p, while 3u and 3z were poor inhibitors of tubulin assembly. The best interactions with tubulin were thus observed with 3d and 3p. The docking studies were also performed on the 3N2G PDB model [28] to investigate the binding mode of our ligands towards two additional neighboring pockets (zones 2 and 3) in the tubulin colchicine domain. Thus, we obtained results only for 3d, while no pose was generated for the other ligands (Table S4). Although zone 1 is still preferred, 3d showed a better affinity than colchicine for zones 2 and 3. As shown in Figure S1, 3d established a hydrogen bond (H-bond) acceptor between its isoxazole ring and the backbone of βA250. Moreover, we observed good contact with an additional hydrophobic pocket of the β subunit, formed by residues L255, A316, A317, A354, C241, and K352. For comparison, in the best docking pose of 3d with tubulin structure 4O2B [26], containing zones 1 and 2 of the colchicine site, we observed strong hydrophobic interactions with β-tubulin residues V181, L248, A250, L255, A354, and A316 (Figure 1A). Most importantly, 3d displayed a binding geometry characterized by the isoxazole moiety facing the C241 and the benzene ring being accommodated in zone 1 and interacting with N258, V181, and A316. The other tubulin-active compound 3p shared with 3d the same binding geometry and the same hydrophobic contribution within the colchicine site (Figure 1B). In contrast, the best poses for 3u and 3z placed the tricyclic system more planarly than the 3d and 3p poses, with the isoxazole ring facing L255 and the pyrrole ring interacting with the side chain of K352 through a π-cation bond (Figure 1C–D). In addition, 3z established two H-bonds between its isoxazole ring and aniline group with N258 and Q247, respectively. Despite the ability of 3u and 3z to establish π-cation interactions and hydrogen bonds with tubulin, their lower affinity for the binding site could be explained by several steric hindrances in their docking poses. Specifically, we observed a significant clash between the cycloheptane ring of the 3u tricyclic system and βA354, while 3z showed a poor contact that involved the side chain of βS178 and its methylene group group linking the pyrrole ring to the 2-methoxy-5-aniline moiety. Furthermore, we observed that the trimethoxyphenyl group was not well located in zone 1, leading to the loss of hydrophobic interactions with β-tubulin residues V181, L248, A250, L255, A354, and A316.
Figure 1.
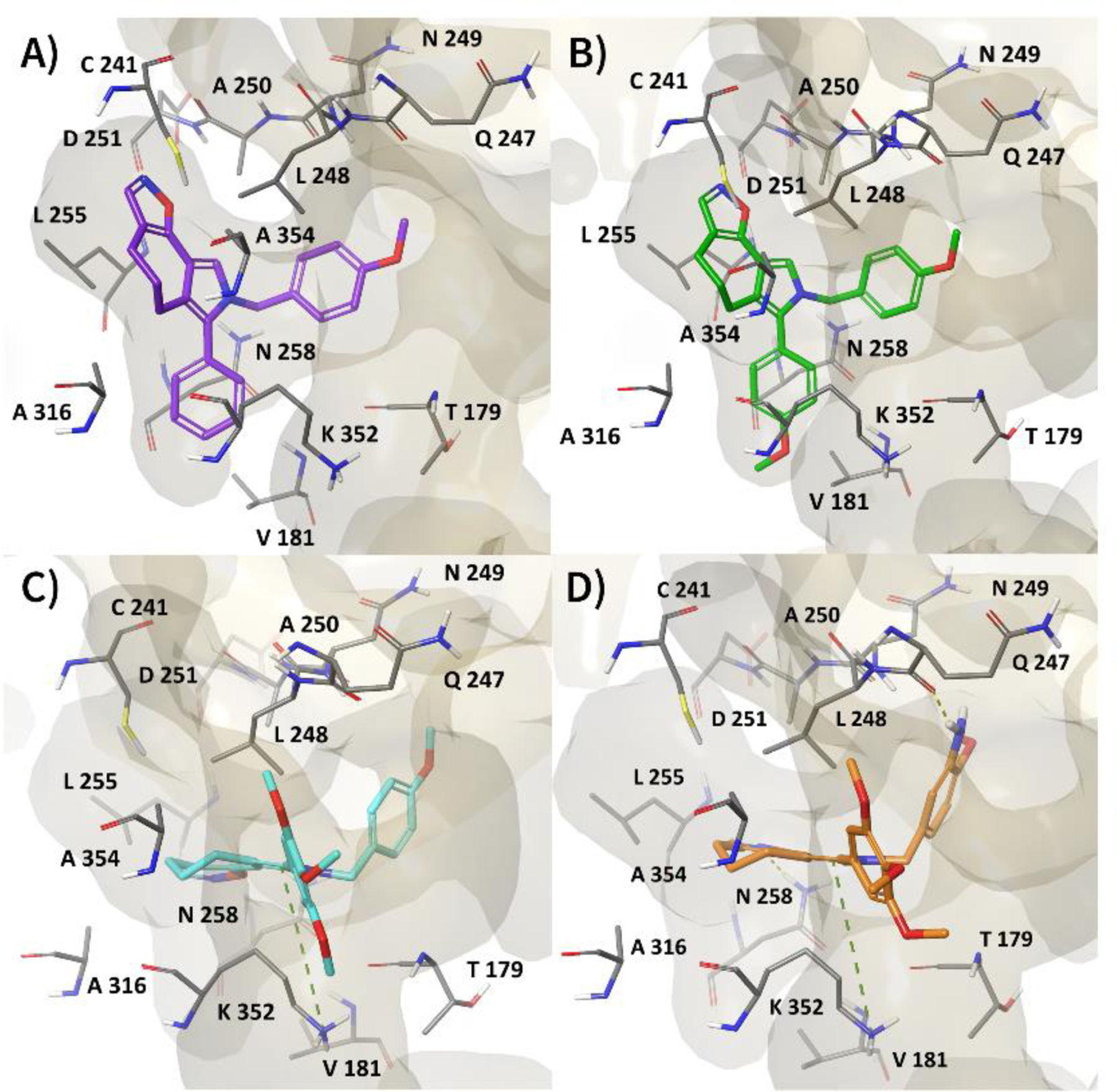
Best docked pose of A) 3d, B) 3p, C) 3u, and D) 3z with the 4O2B crystal structure of tubulin, depicting zones 1 and 2 of the colchicine site. Tubulin is shown in a faded yellow surface, while ligand and residues, involved in the most important interactions, are shown as sticks. H-bond and π-cation interactions are indicated as dashed yellow and green lines, respectively.
MDs in water as solvent were performed on the best docking poses of 3d, 3p, 3u, and 3z against the 4O2B model to better assess their binding stability and to evaluate the presence of induced-fit phenomena in the tubulin recognition process of these ligands. The 3N2G model complexed with 3p was also investigated through MDs, and colchicine was used as a reference compound. Thus, we analyzed the geometric behavior of all MDs and we computed the related binding free energy and the global number of contacts for the MDs most representative structures (Table S5).
Interestingly, the most active compounds 3d and 3p during MDs improved their interactions with the colchicine site with respect to their docking pose (Figure 1A–B), thanks to their ability to engage in further electrostatic contacts, such as H-bond and π-cation interactions (Figure 2A–B). Specifically, 3d established a π-cation interaction between its phenolic ring and the side chain of βK352 (Figure 2A), while 3p exhibited an H-bond between its pyrrole moiety and the backbone of βN249 (Figure 2B). Conversely, for 3u (Figure 2C), the loss of the π-cation interaction, previously reported in the docking pose (Figure 1C), along with the reduction of hydrophobic contacts, could explain its lower activity in the tubulin experiments. Finally, in the most representative MDs structure of 3z (Figure 2D), we observed two different H-bonds compared to its docking pose (Figure 1D). Specifically, the trimethoxyphenyl group and the isoxazole ring formed two H-bonds with the backbone of T179 and of D251, respectively. Despite these favorable interactions, its reduced affinity for the colchicine site may be explained by a higher solvation energy penalty and the lower number of good contacts, as reported in the Supplementary Material.
Figure 2.
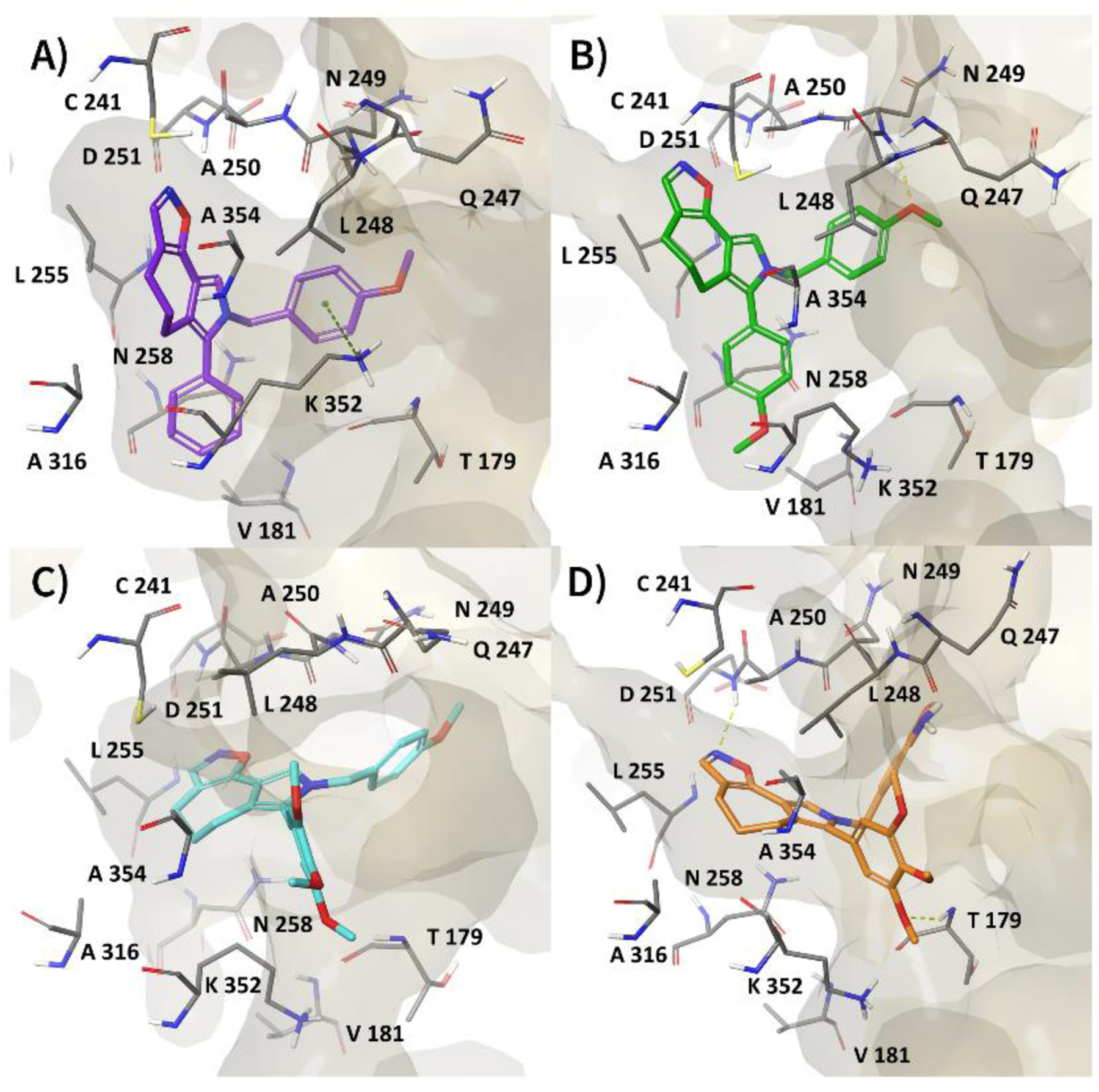
Most representative MDs structure of tubulin (PDB code 4O2B) complexed with A) 3d, B) 3p, C) 3u, and D) 3z. Tubulin is depicted as a pale yellow surface, while ligand and residues, involved in the most important interactions, are shown as sticks. H-bond and π-cation interactions are indicated as dashed yellow and green lines, respectively.
Finally, the MDs confirmed the binding mode predicted by the docking calculation for 3d (Figure S1A) towards the 3N2G model, with the sole exception being the residue engaged in the H-bond with the isoxazole ring. Indeed, in the most representative MDs structure, an induced-fit process allowed the interaction with the backbone of βN249 (Figure S1B). This finding highlights the possibility that 3d interacts with tubulin through two distinct, stable binding modes.
Regarding the in-silico ADME (absorption, distribution, metabolism, excretion) assessment [29], the most active new derivatives (3d, 3p, 3u and 3z) exhibited a pharmacokinetics profile similar to those of colchicine, with the only exception of the logP (QPlogPo/w). As shown in Table S7, all new compounds showed a logP value > 5, violating Lipinski’s rule (RO5) but achieving better oral bioavailability.
At the end, we performed a target prediction using the Molinspiration virtual screening engine tool (Table S8) [30], with the aim of exploring possible off-target effects for our best compounds. Based on the drug-likeness score, the bioactivity of the ligand molecules can be divided into three categories, such as active (>0.0), moderate (from −5.0 to 0.0), and inactive (<−5.0). All the most effective compounds exhibited active drug-likeness scores as nuclear receptor and GPCR ligands, especially 3d. They also showed good scores as kinase and enzyme inhibitors. Conversely, they did not exhibit a significant potential active profile as either an ion channel modulator or a protease inhibitor.
2.7. Cell cycle analysis
To investigate the mechanism of action of the new derivatives, we evaluated their influence on the cell cycle in three cell lines, A549, CCRF-CEM and VL51. As shown in Figure 3 (Panel A-C), compound 3d, although endowed with significant activity as an inhibitor of tubulin polymerization (Table 6), induced in the three cell lines only a modest increase in G2/M phase, which was observed only at the maximum concentration used (5 μM for A549 and VL51 and 10 μM for CCRF-CEM). Conversely compounds 3u, and even more markedly 3z, both of which did not show significant activity in the tubulin assay, induced a block in G2/M accompanied by a strong decrease in S phase cells even at low concentrations (1.0 μM).
Figure 3.
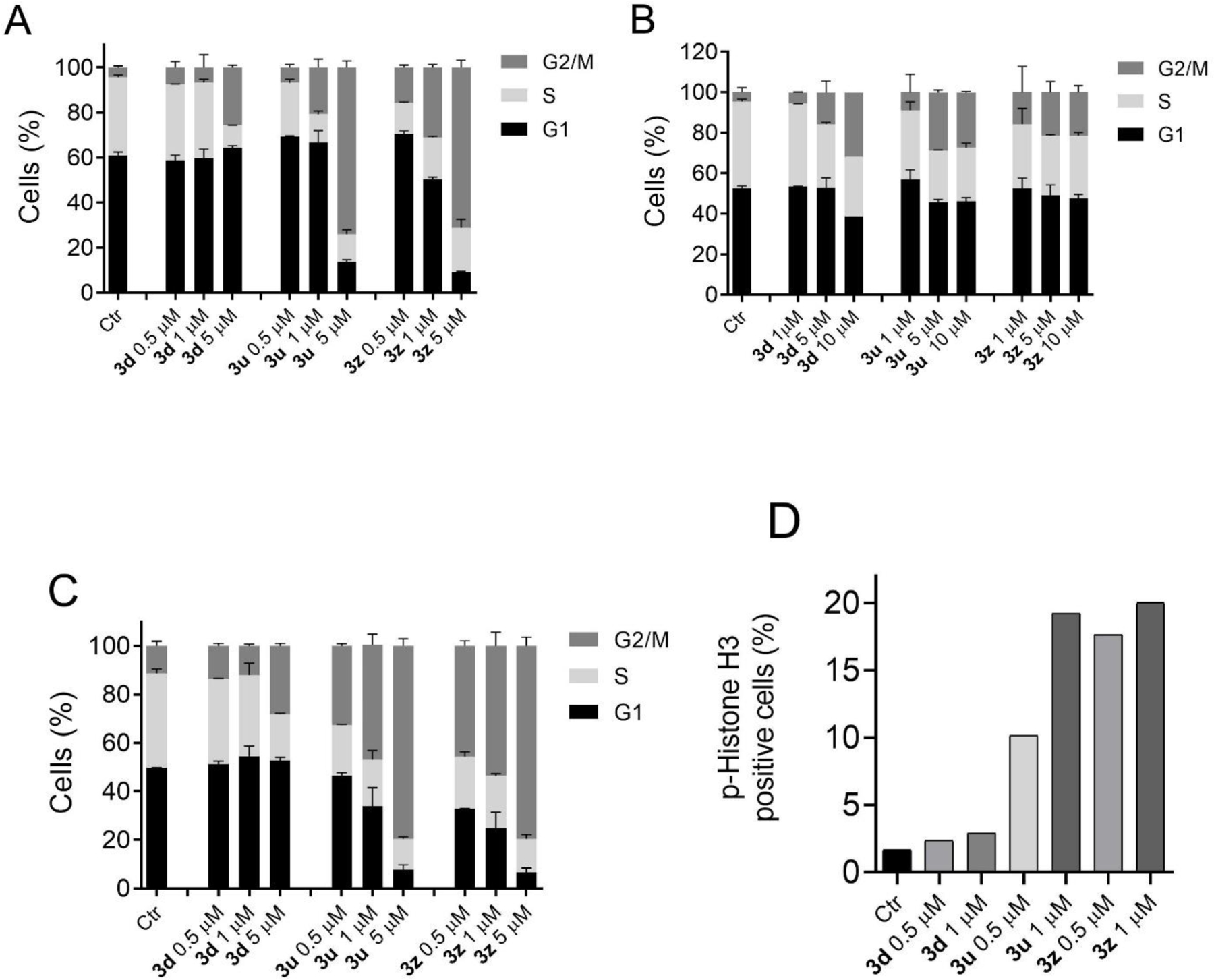
Cell cycle analysis of A549 (A), CCRF-CEM (B) and VL51 (C) cells treated for 24 h at the indicated concentrations with 3d, 3u and 3z. Cells were fixed and labeled with PI and analyzed by flow cytometry as described in the Experimental section. Data are presented as mean of two independent experiments ± SEM. (D) Percentage of p-histone H3 positive cells (mitotic cells) obtained from flow cytometric analysis of VL51 cells immunofluorescently labeled with an antibody to p-histone H3, following treatment with the indicated concentrations of compounds for 24 h.
In order to determine whether these compounds were able to block cells at the mitotic phase (M), cells were stained with an immunofluorescent antibody to phospho-histone H3 [31], a well-known mitotic marker, as well as propidium iodide (PI), and analyzed by flow cytometry. As shown in Figure 3 (Panel D), VL51 cells arrested in M phase, represented by p-histone H3 positive cells, which increased in a concentration dependent manner only for compounds 3u and 3z. In particular, the percentage of mitotic cells increased from about 1.5% observed in untreated cells to about 18% at the concentration of 1 μM for both compounds. In good agreement with the cell cycle analysis, we also did not observe a significant increase of mitotic cells with compound 3d.
2.8. The new derivatives induce apoptosis through the mitochondrial pathway
With the goal of studying the mode of cell death induced by the new derivatives, we evaluated the induction of apoptosis using double labeling of treated cells with annexin-V conjugated with FITC and with PI. Annexin-V binds to the phosphatidylserine exposed on the outer surface of the cytoplasmic membrane during the process of apoptosis, while PI binds to DNA, indicating cells undergoing necrosis.
In excellent agreement with the cytotoxicity data, the results shown in Figure 4 (Panels A-C) demonstrate that the more active 3u and 3z induced, after a 48 h incubation, massive apoptosis in a dose-dependent manner, while 3d induced apoptosis to a lesser extent . It should be emphasized that apoptosis occurred in all three cell lines examined, but to a greater extent in the VL51 cells, suggesting a particular tropism of these compounds towards lymphomas.
Figure 4.
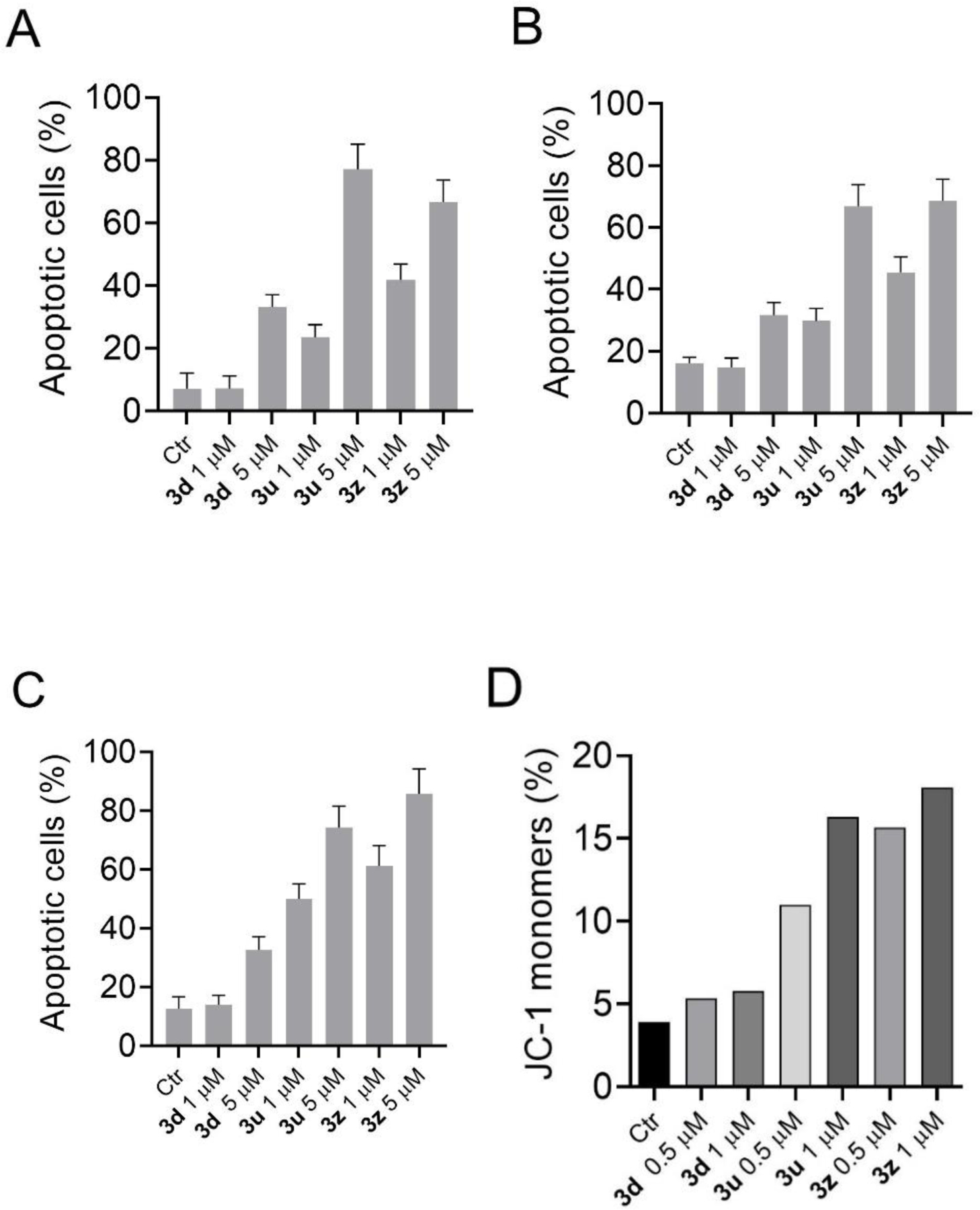
Compounds 3d, 3u and 3z induced apoptosis in A549 (A), CCRF-CEM (B) and VL51 (C) cells. Cells were treated with the compounds for 48 h at the indicated compound concentrations. The cells were then harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are presented as mean ± S.E.M. for three independent experiments. The percentage of apoptotic cells refers to the sum of annexin-V positive and Annexin-V and PI double positive cells. (D) Assessment of mitochondrial membrane potential by flow cytometry with the fluorescent probe JC-1 after treatment for 24 h of VL51 cells with the indicated compounds at 0.5 and 1.0 μM.
One of the early events that precede apoptosis is the decrease of the membrane mitochondrial potential [32]. To determine if this happened with our compounds, we used the JC-1 fluorescent dye and analyzed the VL51 cells after treatment for 24 h with compounds 3d, 3u and 3z. The results shown in Figure 4D demonstrate that 3d caused a slight depolarization of the mitochondrial membrane, while depolarization was extensive following treatment of the cells with 3u or 3z, in excellent agreement with their relative abilities to induce apoptosis. This suggests that apoptosis follows the mitochondrial pathway.
3. Conclusions
In the current study, twenty-six derivatives were evaluated for their antiproliferative activity in the NCI-60 cell panel and in four different NHL histotypes (ABC-DLBCL, GCB-DLBCL, MCL and MZL). All tested compounds showed antiproliferative activity in the low micromolar – submicromolar range, with the greatest growth inhibition activity observed with 3u and 3z. Overall, the new structural modification confirmed a promising antiproliferative effect, although reduced compared to the [1,2]oxazolo[5,4-e]isoindoles 1. Contrary to our hypothesis, based on the results obtained from our previous series of compounds, the best candidates 3u and 3z probably have a different primary target, as they were found to have modest activity as inhibitors of tubulin polymerization.
Nevertheless, 3u and 3z block the cell cycle in metaphase as demonstrated by the increase of p-histone H3 positive cells. In this context, further experiments are needed to verify whether the action of these compounds is linked to an inhibition of proteins which regulate the cell cycle, in particular for those proteins involved in the regulation of spindle associated events. In addition, these compounds induce apoptosis by a mechanism that follows the mitochondrial pathway. We should also note that we cannot exclude metabolic conversion of 3u and/or 3z to a tubulin-active compound. Overall our results provide new perspectives for pyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles.
4. Experimental Section
4.1. Chemistry. Synthesis and characterization
All melting points were taken on a Büchi melting point M-560 apparatus. IR spectra were determined in bromoform with a Shimadzu FT/IR 8400S spectrophotometer. 1H and 13C NMR spectra were measured at 200 and 50.0 MHz, respectively, in DMSO-d6 or CDCl3 solution using a Bruker Avance II series 200 MHz spectrometer. Column chromatography was performed with Merck silica gel (230–400 mesh ASTM) or a Büchi Sepacor chromatography module (prepacked cartridge system). Elemental analyses (C, H, N) were within ±0.4% of theoretical values and were performed with a VARIO EL III elemental analyzer. The purity of all the tested compounds was >95%, determined by HPLC (Agilent 1100 series).
4.1.1. Procedure for the preparation of 2-((dimethylamino)methylene)cycloheptane-1,3-dione (5).
A solution of cycloheptane-1,3-dione 4 (1 g, 8 mmol) in anhydrous DMFDMA (2.6 mL) was heated under reflux for 1 h. After cooling, the solvent was evaporated at reduced pressure, and the oily residue was triturated with diethyl ether with the solvent removed by filtration. Brown solid; yield: 99%; mp: 102 – 103 °C; IR (cm−1): 1660 (CO) 1585 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.84 – 1.88 (m, 4H, 2 x CH2), 2.59 (t, 4H, J= 6.2 Hz, 2 x CH2), 2.80 (s, 3H, CH3), 3.30 (s, 3H, CH3), 7.72 (s, 1H, CH); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.2 (2 x C), 40.5 (2 x C), 43.2, 47.9, 112.8, 159.6 (2 x C), 200.2. Anal Calcd. for C10H15NO2: C, 66.27; H, 8.34; N, 7.73. Found: C, 65.98; H, 8.12; N, 7.89.
4.1.2. General procedure for the preparation of 2-{[(2,7-dioxocycloheptylidene)methyl]amino}-arylacetic acid (6a,b).
To a solution of 5 (16 mmol) in ethanol (30 mL), a solution of the appropriate phenylglycine (19 mmol) and sodium acetate trihydrate (0.26 g) in ethanol was added, and the reaction mixture was heated under reflux until the reaction was complete (TLC). After cooling, the reaction mixture was filtered, and the filtrate was dried under reduced pressure. To the residue, ice and water were added, and the resulting solution was acidified with 6 M HCl. The solid obtained was filtered and dried.
4.1.2.1. 2-{[(2,7-Dioxocycloheptylidene)methyl]amino}-2-phenylacetic acid (6a).
This compound was obtained from reaction of 4 with phenylglycine after 1–1/2 h. Brown oil; yield: 80%; IR (cm−1): 3422 (NH), 3287 (OH), 1703 (CO), 1658 (CO), 1621 (CO); 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.70 (s, 4H, 2 x CH2), 2.55 – 2.60 (m, 4H, 2 x CH2), 3.50 (s, 1H, OH), 5.63 (d, 1H, J= 7.2 Hz, CH), 7.33 – 7.46 (m, 5H, Ar), 7.92 (d, 1H, J= 14.0 Hz, CH), 11.44 – 11.51 (m, 1H, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 21.5, 21.6, 30.1 (2 x C), 63.8, 111.8, 127.8 (2 x C), 129.1, 129.7 (2 x C), 137.7, 158.4, 171.3, 198.9, 201.1. Anal Calcd. for C16H17NO4: C, 66.89; H, 5.96; N, 4.88. Found: C, 66.74; H, 5.81; N, 4.99.
4.1.2.2. 2-{[(2,7-Dioxocycloheptylidene)methyl]amino}-2-(3,4,5-trimethoxyphenyl)acetic acid (6b).
This compound was obtained from reaction of 4 with 3,4,5-trimethoxyphenylglycine after 1–1/2 h. Brown solid; yield: 82%; mp: 102–103 °C; IR (cm−1): 3401 (NH), 3299 (OH), 1698 (CO), 1652 (CO), 1633 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.82 (s, 4H, 2 x CH2), 2.66 (s, 4H, 2 x CH2), 3.53 (s, 1H, OH), 3.83 (s, 3H, CH3), 3.84 (m, 6H, 2 x CH3), 5.08 (d, 1H, J= 6.8 Hz, CH), 6.59 (s, 2H, Ar), 8.03 (d, 1H, J= 14.1 Hz, Ar), 11.57 – 11.64 (m, 1H, NH); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.4, 21.5, 40.1, 40.6, 56.2 (2 x C), 60.8, 65.2, 104.5 (2 x C), 112.2, 130.7, 138.6, 153.8 (2 x C), 159.1, 170.7, 201.7, 202.1. Anal Calcd. for C19H23NO7: C, 60.47; H, 6.14; N, 3.71. Found: C, 60.19; H, 6.38; N, 3.56.
4.1.3. General procedure for the synthesis of 2-acetyl-1-substituted-2,6,7,8-tetrahydrocyclohepta[c]pyrrol-4-yl acetate (7a,b).
To a solution of 6a,b (8 mmol) in acetic anhydride (25 mL), triethylamine was added (5.7 mmol, 8 mL). The reaction mixture was heated under reflux until the reaction was complete (TLC). After cooling, the reaction mixture was poured into water and ice and formed a rubbery solid. The liquid phase was decanted, and the remaining solid was stirred with a saturated solution of Na2CO3 (50 mL). The solid obtained was filtered and dried. The solid was dissolved in dichloromethane and purified using a chromatography column (dichloromethane).
4.1.3.1. 2-Acetyl-1-phenyl-2,6,7,8-tetrahydrocyclohepta[c]pyrrol-4-yl acetate (7a).
This compound was obtained from reaction of 6a after 30 min. Brown solid; yield: 73%; mp: 120–121 °C; IR (cm−1): 1769 (CO) 1711 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.75 – 1.86 (m, 2H, CH2), 2.08 (s, 3H, CH3), 2.28 (s, 3H, CH3), 2.41 – 2.53 (m, 4H, 2 x CH2), 5.49 (t, 1H, J= 5.1 Hz, CH), 7.22 – 7.28 (m, 3H, Ar and H-3), 7.35 – 7.47 (m, 3H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.1, 24.7, 25.1, 26.6, 28.5, 117.2, 119.6, 121.8, 125.9, 128.2, 128.3 (2 x C), 130.3, 130.6 (2 x C), 133.0, 141.0, 168.8, 170.1. Anal Calcd. for C19H19NO3: C, 73.82; H, 7.12; N, 4.30. Found: C, 74.03; H, 7.33; N, 3.99.
4.1.3.2. 2-Acetyl-1-(3,4,5-trimethoxyphenyl)-2,6,7,8-tetrahydrocyclohepta[c]pyrrol-4-yl acetate (7b).
This compound was obtained from reaction of 6b after 30 min. Brown oil; yield: 53% IR (cm−1): 1753 (CO) 1722 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.87 (m, 2H, CH2), 2.12 (s, 3H, CH3), 2.28 (s, 3H, CH3), 2.44 –2.48 (m, 2H, CH2), 2.52 – 2.56 (m, 2H, CH2), 3.85 (s, 6H, 2 x CH3), 3.91 (s, 3H, CH3), 5.50 (t, 1H, J= 5.0 Hz, CH), 6.45 – 6.49 (m, 3H, Ar and H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.0, 24.6, 24.8, 26.7, 28.3, 56.0 (2 x C), 61.0, 108.0 (2 x C), 117.1, 119.5, 121.7, 125.9, 128.3, 129.7, 130.1, 141.1, 153.2 (2 x C), 168.6, 169.9. Anal Calcd. for C22H25NO6: C, 66.15; H, 6.31; N, 3.51. Found: C, 66.27; H, 6.45; N, 3.28.
4.1.4. General procedure for the preparation of 1-substituted-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8a,b).
To a solution of 7a,b (3.3 mmol) in AcOH (80%, 20 mL), HCl (37%, 1.7 mL) was added dropwise. The reaction mixture was heated at 60 °C until the reaction was complete (TLC). After cooling, the reaction mixture was poured into water and ice. The solid obtained was filtered and dried. The solid was purified using column chromatography (dichloromethane : ethyl acetate 95 : 5).
4.1.4.1. 1-Phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8a).
This compound was obtained from 7a after 15 min. Brown solid; yield: 75%; mp: 103 – 104 °C; IR (cm−1): 3248 (NH), 1651 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.84 – 1.94 (m, 4H, 2 x CH2), 2.73 (t, 2 H, J= 5.9 Hz, CH2), 2.91 (t, 2H, J= 5.9 Hz, CH2), 7.29 – 7.47 (m, 6H, Ar and H-3), 8.80 (s, 1H, NH); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.3, 23.9, 26.3, 41.6, 121.3, 122.2, 127.1, 127.2, 127.6 (2 x C), 128.9 (2 x C), 129.5, 132.4, 200.0. Anal Calcd. for C15H15NO: C, 79.97; H, 6.71; N, 6.22. Found: C, 80.12; H, 6.47; N, 6.39.
4.1.4.2. 1-(3,4,5-Trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8b).
This compound was obtained from 7b after 15 min. Brown solid; yield 81%; IR (cm−1): 3258 (NH), 1657 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.86 – 1.95 (s, 4H, 2 x CH2), 2.77 (t, 2H, J= 6.4 Hz, CH2), 2.92 (t, 2H, J= 6.4 Hz, CH2), 3.89 (s, 3H, CH3), 3.90 (s, 6H, 2 x CH3), 6.61 (s, 2H, H-2” and H-6”), 7.52 (s, 1H, H-3), 9.26 (s, 1H, NH); 13C NMR (CDCl3) (ppm): 22.4, 24.1, 26.3, 41.6, 56.2 (2 x C), 61.0, 105.2 (2 x C), 121.0, 128.5, 129.5, 129.6, 129.7, 137.5, 153.5 (2 x C), 199.9. Anal Calcd. for C18H21NO4: C, 68.55; H, 6.71; N, 4.44. Found: C, 68.67; H, 6.46; N, 4.18.
4.1.5. Procedure for the preparation of ethyl 5-(4-methoxyphenyl)-1H-pyrrole-2-carboxylate (10).
To a solution of ethyl azidoacetate (7g, 54 mmol) in anhydrous ethanol (10 mL), a solution of 9 (1.62 g, 10 mmol) in anhydrous ethanol (30 mL) was added at −20 °C, followed by the dropwise addition of a solution of potassium ethoxide (52 mmol) in ethanol (50 mL). The reaction was stirred for 4–1/2 h at −20 °C. The reaction mixture was then allowed to reach room temperature, and the solvent was evaporated under reduced pressure. The residue was dissolved in water and was extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was dissolved in toluene, and the reaction mixture was heated under reflux for 24 h. The solvent was evaporated under reduced pressure, and the crude product was purified using a chromatography column (dichloromethane). Yellow solid; yield 74%; IR (cm−1): 3421 (NH), 1679 (CO); 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.30 (t, 3H, J= 7.1 Hz, CH3), 3.78 (s, 3H, CH3), 4.26 (q, 2H, J= 7.1 Hz, CH2), 6.53 (d, 1H, J= 3.5 Hz, Ar), 6.84 (d, 1H, J= 3.5 Hz, Ar), 6.96 (d, 2H, J= 8.8 Hz, H-3’ and H-5’), 7.80 (d, 2H, J= 8.8 Hz, H-2’ and H-6’), 11.97 (s, 1H, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 14.4, 55.1, 59.4, 106.7, 114.0 (2 x C), 116.6, 122.5, 124.1, 126.5 (2 x C), 137.2, 158.6, 160.3. Anal Calcd. for C14H15NO3: C, 68.56; H, 6.16; N, 5.71. Found: C, 68.39; H, 5.82; N, 6.01.
4.1.6. Procedure for the preparation of 5-(2-(ethoxycarbonyl)-5-(4-methoxyphenyl)-1H-pyrrol-3-yl)-5 oxopentanoic acid (11).
A suspension of AlCl3 (6.56 g, 49mmol) and glutaric anhydride (1.86 g, 16 mmol) in anhydrous dichloromethane (30 mL) was stirred at room temperature. After 1 h, a solution of 10 (2 g, 8.2 mmol) in anhydrous dichloromethane was added dropwise at 0 °C, and the reaction mixture was stirred for 24 h. The reaction mixture was poured into water and ice and formed a rubbery solid, which was extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. Brown oil; Yield 60%; IR (cm−1): 3449 (NH), 3344 (OH), 1702 (CO), 1682 (CO), 1644 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.30 (t, 3H, J= 7.1 Hz, CH3), 1.63 – 1.80 (m, 2H, CH2), 2.25(t, 2H, J= 7.2 Hz, CH2), 2.77 (t, 2H, J= 7.2 Hz, CH2), 3.81 (s, 3H, CH3), 4.27 (q, 2H, J= 7.1 Hz, CH2), 6.97 (d, 2H, J=8.8 Hz, H-3’ and H-5’), 7.33 (s, 1H, Ar), 7.50 (d, 2H, J=8.8 Hz, H-2’ and H-6’), 12.12 (s, 1H, OH), 12.46 (s, 1H, NH); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.3, 19.9, 32.7, 49.4, 55.2, 59.9, 113.0 (2 x C), 117.6, 121.2, 121.7, 123.2, 131.2 (2 x C), 139.9, 159.5, 160.0, 174.1, 195.1. Anal Calcd. for C19H21NO6: C, 63.50; H, 5.89; N, 3.90. Found: C, 63.31; H, 6.08; N 4.02.
4.1.7. Procedure for the preparation of 5-(2-(ethoxycarbonyl)-5-(4-methoxyphenyl)-1H-pyrrol-3-yl)pentanoic acid (12).
To a solution of 11 (4.15 g, 12 mmol) in trifluoroacetic anhydride (28 mL), triethylsilane (6.6 mL) was added at 0 °C. The reaction mixture was stirred at room temperature for 24 h. The solvent was evaporated under reduced pressure, and the residue was added to water and ice. The solid that formed was filtered, dried and purified using column chromatography (dichloromethane : ethyl acetate 84 : 16). Brown oil; yield 61%; IR (cm−1): 3434 (NH), 3355 (OH), 1700 (CO), 1675 (CO); 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.28 (t, 3H, J= 7.1 Hz, CH3), 1.47 – 1.57 (m, 4H, 2 x CH2), 2.19 (t, 2H, J= 6.5 Hz, CH2), 2.46 – 2.52 (m, 2H, CH2), 3.79 (s, 3H, CH3), 4.22 (q, 2H, J= 7.1 Hz, CH2), 6.71 (s, 1H, Ar), 6.99 (d, 2H, J= 8.7 Hz, H-2’ and H-6’), 7.43 (d, 2H, J=8.7 Hz, H-3’ and H-5’), 11.68 (s, 1H, OH), 12.00 (s, 1H, NH); 13 C NMR (DMSO-d6, 200 MHz, ppm): δ 14.4, 24.3, 25.5, 29.7, 33.4, 55.1, 59.3, 113.8 (2 x C), 116.2, 120.7, 121.4, 124.4, 129.2 (2 x C), 133.9, 158.4, 160.3, 174.5. Anal Calcd. for C19H23NO5: C, 66.07; H, 6.71; N, 4.06. Found: C, 66.23; H, 6.54; N, 4.18.
4.1.8. Procedure for the preparation of ethyl 3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta [c]pyrrole-1-carboxylate (8c).
To a solution of 12 (4 g, 12 mmol) in anhydrous dichloromethane, trifluoroacetic anhydride was added (10 mL) at 0 °C. The reaction mixture was stirred at room temperature for 1 h. The solvent was evaporated under reduced pressure, and the residue was purified using column chromatography (petroleum ether : ethyl acetate 9 : 1). Brown solid; yield 61%; mp: 108 – 109 °C; IR (cm−1): 3438 (NH), 1681 (CO), 1667 (CO); 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.24 (t, 3H, J= 7.1, CH3), 1.69 – 1.84 (m, 4H, 2 x CH2), 2.60 – 2.73 (m, 4H, 2 x CH2), 3.80 (s, 3H, CH3), 4.20 (q, 2H, J= 7.1, CH2), 7.02 (d, 2H, J=8.7 Hz, H-3’ and H-5’), 7.38 (d, 2H, J=8.7 Hz, H-2’ and H-6’), 12.14 (s, 1H, NH); 13C NMR (DMSO-d6, 200 MHz, ppm): δ 14.0, 23.3, 23.6, 26.1, 42.2, 55.1, 59.9, 113.8 (2 x C), 120.1, 121.0, 123.3, 130.0 (2 x C), 130.1, 131.8, 158.8, 160.2, 199.3. Anal Calcd. for C19H21NO4: C, 69.71; H, 6.47; N, 4.28. Found: C, 69.56; H, 6.59; N, 4.39.
4.1.9. Procedure for the preparation of 1-(4-methoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (8d).
To a solution of 8c (0.73 g, 2.2 mmol) in ethanol (31 mL), 50% aqueous KOH (1.74 mL) was added. The reaction mixture was heated under reflux for 3 h. After cooling, the solvent was evaporated under reduced pressure. The residue was poured into water and ice and acidified with 6 M HCl. The formed solid was filtered and dried. A solution of this solid (0.47 g, 1.6 mmol) in ethanol (22 mL) was heated almost to boiling and 6 M HCl (10 mL) was then added. The reaction mixture was heated under reflux for 1 h. The solvent was evaporated under reduced pressure, and the residue was poured into water and ice. The solution was extracted with ethyl acetate and dried on Na2SO4, and the solvent evaporated at reduced pressure. Brown solid; mp: 135 – 136 °C; yield: 60%; IR (cm−1): 3425 (NH), 1668 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.81 – 1.91 (m, 4H, 2 x CH2), 2.70 (t, 2H, J= 5.8 Hz, CH2), 2.85 (t, 2H, J= 5.8 Hz, CH2), 3.83 (s, 3H, CH2), 6.95 (d, 2H, J=8.6 Hz, H-3’ and H-5’), 7.33 (d, 3H, J=8.6 Hz, H-2’ and H-6’), 7.40 (s, 1H, H-3), 9.17 (s, 1H, NH); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 23.9, 26.3, 41.6, 55.4, 114.2 (2 x C), 120.5, 121.9, 125.1, 126.9, 129.0 (2 x C), 129.4, 158.7, 200.3. Anal Calcd. for C16H17NO2: C, 75.27; H, 6.71; N, 5.49. Found: C, 74.48; H, 6.97; N, 5.63.
4.1.10. General procedure for the preparation of (4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate (14a-h,j-y).
To a solution of 8a-d (10 mmol) in anhydrous DMF (15 mL), NaH (0.24 g, 10 mmol) was added at 0 °C, and the reaction mixture was stirred for 1 h at room temperature. The appropriate benzyl halide (20 mmol) was added at 0 °C, and the reaction mixture was stirred at room temperature until the reaction was complete (TLC). The reaction mixture was poured into ice and brine, then the aqueous solution was extracted with dichloromethane (3 x 50 mL). The organic phase was dried over Na2SO4, and the solvent evaporated at reduced pressure. The crude product was purified using chromatography column (petroleum ether: ethyl acetate 9 : 1).
4.1.10.1. 2-benzyl-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14a).
This compound was obtained from reaction of compound 8a with benzyl bromide after 3 h. Yellow oil; yield 90%; IR (cm−1): 1661 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.81 – 1.93 (m, 4H, 2 x CH2), 2.66 – 2.74 (m, 4H, 2 x CH2), 4.96 (s, 2H, CH2), 6.98 (t, 2H, J= 7.0 Hz, Ar), 7.20 – 7.28 (m, 4H, Ar), 7.36 – 7.40 (m, 5H, Ar and H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.3, 26.3, 41.9, 51.4, 122.8, 125.1, 125.3, 127.2, 127.7, 128.0 (2 x C), 128.4 (2 x C), 128.7 (2 x C), 130.9 (2 x C), 131.5, 131.7, 137.1, 199.4. Anal. Calcd. for C22H21NO: C, 83.78; H, 6.71; N, 4.44. Found: C, 84.02; H, 6.49; N, 4.72.
4.1.10.2. 2-(2-methoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14b).
This compound was obtained from reaction of compound 8a with 2-methoxybenzyl chloride after 8 h. Yellow oil; yield 60%; IR (cm−1): 1656 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.66 – 1.85 (m, 4H, 2 x CH2), 2.33 – 2.48 (m, 4H, 2 x CH2), 3.81 (s, 3H, CH3), 5.11 (s, 2H, CH2), 6.81 – 6.91 (m, 2H, Ar), 7.09 – 7.17 (m, 2H, Ar), 7.31 – 7.48 (m, 3H, Ar), 7.57 – 7.67 (m, 3H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.6, 24.3, 26.4, 41.7, 46.3, 55.3, 110.2, 120.6, 122.4, 123.8, 124.7, 125.2, 128.1 (2 x C), 128.5, 128.7, 129.0, 129.2 (2 x C), 131.2, 156.7, 159.4, 199.5. Anal. Calcd. for C23H23NO2: C, 79.97; H, 6.71; N, 4.05. Found: C, 80.12; H, 6.92; N, 3.87.
4.1.10.3. 2-(3-methoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14c).
This compound was obtained from reaction of compound 8a with 3-methoxybenzyl chloride after 5 h. Yellow oil; yield 78%; IR (cm−1): 1657 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.78 – 1.92 (m, 4H, 2 x CH2), 2.62 – 2.73 (m, 4H, CH2), 3.73 (s, 3H, CH3), 4.92 (s, 2H, CH2), 6.47 – 6.58 (m, 2H, Ar), 6.75 – 6.80 (m, 2H, Ar), 7.14 – 7.24 (m, 3H, Ar), 7.32 – 7.42 (m, 3H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 41.9, 51.3, 55.2, 112.9, 113.0, 119.5, 122.8 (s), 125.1 (s), 125.3, 128.0, 128.5 (2 x C), 129.8, 130.9 (2 x C), 131.3, 131.7, 138.6, 159.8, 199.5. Anal. Calcd. for C23H23NO2: C, 79.97; H, 6.71; N, 4.05. Found: C, 80.18; H, 7.02; N, 3.88.
4.1.10.4. 2-(4-methoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14d).
This compound was obtained from reaction of compound 8a with 4-methoxybenzyl chloride after 6 h. Yellow oil; yield 67%; IR (cm−1): 1658 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.77 – 1.91 (m, 4H, 2 x CH2), 2.61 – 2.72 (m, 4H, 2 x CH2), 3.77 (s, 3H, CH3), 4.87 (s, 2H, CH2), 6.70 – 6.92 (m, 4H, Ar), 7.15 – 7.41 (m, 6H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 41.9, 50.9, 55.3, 114.1 (2 x C), 122.8, 124.9, 125.1, 128.0, 128.4 (2 x C), 128.7 (2 x C), 128.9, 130.9 (2 x C), 131.4, 131.6, 159.0, 199.5. Anal. Calcd. for C23H23NO2: C, 79.97; H, 6.71; N, 4.05. Found: C, 79.71; H, 6.98; N, 3.87.
4.1.10.5. 2-(2,5-Dimethoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14e).
This compound was obtained from reaction of compound 8a with 2,5-dimethoxybenzyl chloride after 4–1/2 h. Yield 82%; oil; IR (cm−1): 1655 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.92 (m, 4H, 2 x CH2), 2.66 – 2.73 (m, 4H, 2 x CH2), 3.68 (s, 3H, CH3), 3.71 (s, 3H, CH3), 4.94 (s, 2H, CH2), 6.30 (s, 1H, Ar), 6.75 (s, 2H, Ar), 7.29 – 7.42 (m, 6H, Ar and H-3); 13C NMR (CDCl3, 200 MHz, ppm): δ 22.4, 24.2, 26.3, 41.9, 46.3, 55.6, 55.8, 111.3, 113.2, 115.2, 122.6, 124.9, 125.6, 126.5, 127.8, 128.4 (2 x C), 130.9 (2 x C), 131.6, 132.7, 151.0, 153.6, 199.3. Anal. Calcd. for C24H25NO3: C, 76.77; H, 6.71; N, 3.73. Found: C, 77.03; H, 6.47; N, 3.95.
4.1.10.6. 2-(3,4-Dimethoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14f).
This compound was obtained from reaction of compound 8a with 3,4-dimethoxybenzyl chloride after 4 h. Yellow oil; yield 73%; IR (cm−1): 1660 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.89 (m, 4H, 2 x CH2), 2.66 – 2.70 (m, 4H, 2 x CH2), 3.74 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.89 (s, 2H, CH2), 6.40 (s, 1H, Ar), 6.55 (d, 1H, J= 8.0 Hz, Ar), 6.75 (d, 1H, J= 8.0 Hz, Ar), 7.23 (d, 2H, J= 6.6 Hz, Ar), 7.37 – 7.44 (m, 4H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 41.8, 51.3, 55.8, 55.9, 110.4, 110.6, 110.9, 111.0, 111.2, 119.3, 119.9, 120.5, 125.2, 127.9, 128.5 (2 x C), 129.3, 130.9 (2 x C), 131.5, 199.5. Anal. Calcd. for C24H25NO3: C, 76.77; H, 6.71; N, 3.73. Found: C, 76.65; H, 6.49; N, 4.01.
4.1.10.7. 2-(3,4,5-Trimethoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14g).
This compound was obtained from reaction of compound 8a with 3,4,5-trimethoxybenzyl chloride after 6 h. Yield 98%; oil; IR (cm−1): 1662 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.79 – 1.92 (m, 4H, 2 x CH2), 2.64 – 2.73 (m, 4H, 2 x CH2), 3.73 (s, 6H, 2 x CH3), 3.81 (s, 3H, CH3), 4.89 (s, 2H, CH2), 6.13 (s, 2H, H-2” and H-6”), 6.62 (s, 1H, Ar), 7.36 – 7.45 (m, 5H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.3, 24.1, 26.2, 41.8, 51.8, 56.0 (2 x C), 60.8 (q), 104.5 (2 x C), 123.1, 125.0, 125.2, 128.0, 128.5 (2 x C), 131.0 (2 x C), 131.4, 131.5, 132.3, 132.4, 153.3 (2 x C), 199.5. Anal. Calcd. for C25H27NO4: C, 74.05; H, 6.71; N, 3.45. Found: C, 74.23; H, 6.48; N, 3.61.
4.1.10.8. 2-(4-Methoxy-3-nitrobenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14h).
This compound was obtained from reaction of compound 8a with 3-nitro,4-methoxybenzyl chloride after 6 h. Brown solid; yield 90%; mp: 194 – 195 °C; IR (cm−1): 1656 (CO), 1535 (NO2); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.68 – 175 (m, 4H, 2 x CH2), 2.51 – 2.58 (m, 4H, 2 x CH2), 3.85 (s, 3H, CH3), 5.10 (s, 2H, CH2), 7.12 (d, 1H, J = 8.5 Hz, Ar), 7.23 (d, 3H, J= 8.4 Hz, Ar), 7.34 – 7.45 (m, 4H, Ar), 7.59 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.1, 23.9, 26.1, 41.7, 49.7, 57.1, 115.0, 122.9, 124.3, 124.9, 126.0, 128.4, 129.0 (2 x C), 130.3, 131.0 (2 x C), 131.1, 131.3, 133.8, 139.1, 151.8, 197.8. Anal. Calcd. for C23H22N2O4: C, 70.75; H, 5.68; N, 7.18. Found: C, 70.51; H, 5.89; N, 7.36.
4.1.10.9. Ethyl 2-benzyl-3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate (14j).
This compound was obtained from reaction of 8c with benzyl bromide after 3 h. Yellow oil; yield 63%; IR (cm−1): 1688 (CO), 1661 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.19 (t, 3H, J = 7.1 Hz, CH3), 1.76 – 1.84 (m, 2H, CH2), 1.90 – 1.99 (m, 2H, CH2), 2.56 (t, 2H, J = 6.0 Hz, CH2), 2.78 (t, 2H, J = 6.0 Hz, CH2), 3.84 (s, 3H, CH3), 4.19 (q, 2H, J = 7.1 Hz, CH2), 5.28 (s, 2H, CH2), 6.83 – 6.86 (m, 2H, Ar), 6.90 (d, 2H, J = 8.7 Hz, Ar), 7.09 (d, 2H, J = 8.7 Hz, Ar), 7.21 – 7.28 (m, 3H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 13.7, 23.9, 24.5, 26.7, 42.9, 49.1, 55.3, 61.0, 114.0 (2 x C), 122.0, 122.7, 122.9, 126.2 (2 x C), 127.1, 128.4 (2 x C), 129.8, 132.2 (2 x C), 134.9, 138.1, 159.8, 162.1, 200.6. Anal. Calcd. for C26H27NO4: C, 74.80; H, 6.52; N, 3.35. Found: C, 74.68; H, 6.74; N, 3.48.
4.1.10.10. Ethyl 2-(2-methoxybenzyl)-3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate (14k).
This compound was obtained from reaction of 8c with 2-methoxybenzyl chloride after 5 h. Yellow oil; yield 71%; IR (cm−1): 1691 (CO), 1665 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.14 (t, 3H, J = 7.1 Hz, CH3), 1.72 – 1.85 (m, 2H, CH2), 1.88 – 1.97 (m, 2H, CH2), 2.55 (t, 2H, J = 5.9 Hz, CH2), 2.78 (t, 2H, J = 5.9 Hz, CH2), 3.70 (s, 3H, CH3), 3.81 (s, 3H, CH3), 4.16 (q, 2H, J = 7.1 Hz, CH2), 5.23 (s, 2H, CH2), 6.47 (d, 1H, J = 7.5 Hz, CH), 6.73 – 6.88 (4H, m, Ar), 7.03 – 7.21 (3H, m, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 13.6, 23.9, 24.5, 26.8, 42.9, 44.9, 55.1, 55.3, 60.9, 109.6, 113.8 (2 x C), 120.5, 121.9, 122.8, 123.3, 126.5, 126.8, 128.0, 129.6, 132.0 (2 x C), 135.0, 155.8, 159.6, 161.8, 200.8. Anal. Calcd. for C27H29NO5: C, 72.46; H, 6.53; N, 3.13. Found: C, 72.61; H, 6.67; N, 2.95.
4.1.10.11. Ethyl 2-(3-methoxybenzyl)-3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate (14l).
This compound was obtained from reaction of 8c with 3-methoxybenzyl chloride after 6 h. Yellow oil; yield 76%; IR (cm−1): 1692 (CO), 1667 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.19 (t, 3H, J = 7.2 Hz, CH3), 1.70 – 1.83 (m, 2H, CH2), 1.87 – 1.99 (m, 2H, CH2), 2.54 (t, 2H, J = 5.9 Hz, CH2), 2.76 (t, 2H, J = 5.9 Hz, CH2), 3.71 (s, 3H, CH3), 3.82 (s, 3H, CH3), 4.19 (q, 2H, J = 7.2 Hz, CH2), 5.24 (s, 2H, CH2), 6.37 – 6.44 (m, 2H, Ar), 6.69 – 6.75 (m, 1H, Ar), 6.90 (d, 2H, J = 8.7 Hz, Ar), 7.06 – 7.17 (m, 3H, Ar); 13C NMR (CDCl3, 200 MHz, ppm): δ 13.8, 23.9, 24.5, 26.8, 42.9, 49.0, 55.1, 55.3, 61.1, 111.9, 112.6, 114.0 (2 x C), 118.6, 122.0, 122.7, 122.9, 129.5, 129.9, 132.2 (2 x C), 134.9, 139.8, 159.6, 159.8, 162.0, 200.7. Anal. Calcd. for C27H29NO5: C, 72.46; H, 6.53; N, 3.13. Found: C, 72.32; H, 6.41; N, 3.29.
4.1.10.12. Ethyl 2-(4-methoxybenzyl)-3-(4-methoxyphenyl)-8-oxo-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrrole-1-carboxylate (14m).
This compound was obtained from reaction of 8c with 4-methoxybenzyl chloride after 4 h. Yellow oil; yield 75%; IR (cm−1): 1690 (CO), 1662 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.21 (t, 3H, J = 7.1 Hz, CH3), 1.74 – 1.95 (m, 4H, 2 x CH2), 2.53 (t, 2H, J = 5.8 Hz, CH2), 2.75 (t, 2H, J = 5.8 Hz, CH2), 3.74 (s, 3H, CH3), 3.83 (s, 3H, CH3), 4.20 (q, 2H, J = 7.1 Hz, CH2), 5.19 (s, 2H, CH2), 6.70 – 6.79 (m, 4H, Ar), 6.90 (d, 2H, J = 8.8 Hz, Ar), 7.27 (d, 2H, J = 8.8 Hz, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 13.8, 23.8, 24.4, 26.7, 42.9, 48.5, 55.2, 55.3, 61.1, 113.8 (2 x C), 113.9 (2 x C), 122.1, 122.8, 127.7 (2 x C), 128.7, 129.7, 130.2, 132.2 (2 x C), 134.7, 158.7, 159.7, 162.2, 200.6. Anal. Calcd. for C27H29NO5: C, 72.46; H, 6.53; N, 3.13. Found: C, 72.29; H, 6.82; N, 3.02.
4.1.10.13. 2-(2-Methoxybenzyl)-1-(4-methoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14n).
This compound was obtained from reaction of compound 8d with 2-methoxybenzyl chloride after 7 h. Yellow oil; yield 70%; IR (cm−1): 1658 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.77 – 1.94 (m, 4H, 2 x CH2), 2.60 – 2.72 (m, 4H, 2 x CH2), 3.75 (s, 3H, CH3), 3.83 (s, 3H, CH3), 4.92 (s, 2H, CH2), 6.68 – 6.95 (m, 5H, Ar), 7.12 – 7.27 (m, 3H, Ar), 7.33 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.5, 24.3, 26.3, 41.9, 46.3, 55.3 (2 x C), 110.2, 113.8 (2 x C), 120.6, 122.4, 123.8, 124.7, 125.2, 125.45, 128.7, 129.0, 131.5, 132.1 (2 x C), 156.6, 159.2, 199.5. Anal. Calcd. for C24H25NO3: C, 76.77; H, 6.71; N, 3.73. Found: C, 77.02; H, 7.03; N, 3.58.
4.1.10.14. 2-(3-Methoxybenzyl)-1-(4-methoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14o).
This compound was obtained from reaction of compound 8d with 3-methoxybenzyl chloride after 6 h. Yellow oil; yield 62%; IR (cm−1): 1657 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.77 – 2.00 (m, 4H, 2 x CH2), 2.60 – 2.77 (m, 4H, 2 x CH2), 3.73 (s, 3H, CH3), 3.83 (s, 3H, CH3), 4.88 (s, 2H, CH2), 6.43 – 6.62 (m, 2H, Ar), 6.74 – 6.98 (m, 3H, Ar), 7.09 – 7.26 (m, 3H, Ar), 7.35 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.3, 26.3, 41.9, 51.2, 55.2, 55.3, 101.0, 112.9, 113.0, 113.9 (2 x C), 119.4, 122.6, 123.5, 125.0, 129.8, 131.4, 132.2 (2 x C), 138.8, 159.4, 159.8, 199.5. Anal. Calcd. for C24H25NO3: C, 76.77; H, 6.71; N, 3.73. Found: C, 76.49; H, 6.98; N, 3.85.
4.1.10.15. 2-(4-Methoxybenzyl)-1-(4-methoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14p).
This compound was obtained from reaction of compound 8d with 4-methoxybenzyl chloride after 12 h. Yellow oil; yield 78% IR (cm−1): 1659 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.71 – 1.91 (m, 4H, 2 x CH2), 2.59 – 2.71 (m, 4H, 2 x CH2), 3.77 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.84 (s, 2H, CH2), 6.78 (d, 2H, J = 8.8 Hz, Ar), 6.88 – 6.95 (m, 4H, Ar), 7.12 (d, 2H, J = 8.8 Hz, Ar), 7.33 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.3, 26.2, 42.0, 50.89, 55.3 (2 x C), 113.8 (2 x C), 114.1 (2 x C), 122.7, 123.6, 124.7, 124.8, 128.7 (2 x C), 129.1, 131.3, 132.2 (2 x C), 159.2, 159.3, 199.5. Anal. Calcd. for C24H25NO3: C, 76.77; H, 6.71; N, 3.73. Found: C, 76.89; H, 6.64; N, 3.98.
4.1.10.16. 1-(4-Methoxyphenyl)-2-(3,4,5-trimethoxybenzyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14q).
This compound was obtained from reaction of compound 8d with 3,4,5-trimethoxybenzyl chloride after 12 h. Yellow oil; yield 63%; IR (cm−1): 1655 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.73 – 1.83 (m, 4H, 2 x CH2), 2.47 – 2.52 (m, 4H, 2 x CH2), 3.80 (s, 6H, 2 x CH3), 3.81 (s, 3H, CH3), 3.82 (s, 3H, CH3), 5.18 (s, 2H, CH2), 6.51 (m, 2H, H-2” and H-6”), 7.04 (d, 2H, J = 7.1 Hz, H-3’ and H-5’), 7.11 (s, 1H, H-3), 7.53 (d, 2H, J = 7.1 Hz, H-2’ and H-6’); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 41.6, 49.2, 56.0 (2 x C), 56.1, 60.8, 104.8 (2 x C), 105.2, 107.8, 113.8 (2 x C), 122.8, 125.2, 130.8, 131.8 (2 x C), 136.0, 136.2 153.1 (2 x C), 159.4, 200.0. Anal. Calcd. for C26H29NO5: C, 71.70; H, 6.71; N, 3.22. Found: C, 71.46; H, 6.51; N, 3.49.
4.1.10.17. 2-Benzyl-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14r).
This compound was obtained from reaction of compound 8b with benzyl bromide after 3 h. Yellow oil; yield 68%; IR (cm−1): 1659 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.97 (m, 4H, 2 x CH2), 2.68 – 2.75 (m, 4H, 2 x CH2), 3.69 (s, 6H, 2 x CH3), 3.89 (s, 3H, CH3), 4.98 (s, 2H, CH2), 6.33 (s, 2H, H-2” and H-6”), 6.99 – 7.02 (m, 2H, Ar), 7.25 – 7.32 (m, 3H, Ar), 7.43 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.3, 24.1, 26.2, 30.1, 41.8, 51.4 (2 x C), 56.0, 107.8 (2 x C), 122.7, 125.0, 125.4, 126.5, 126.8 (2 x C), 127.7, 128.7 (2 x C), 131.6, 137.5, 137.9, 152.9 (2 x C), 199.2. Anal. Calcd. for C25H27NO4: C, 74.05; H, 6.71; N, 3.45. Found: C, 74.27; H, 6.56; N, 3.32.
4.1.10.18. 2-(2-Methoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14s).
This compound was obtained from reaction of compound 8b with 2-methoxybenzyl chloride after 7 h. Yellow oil; yield 68%; IR (cm−1): 1659 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.68 – 1.84 (m, 4H, 2 x CH2), 2.33 – 2.56 (m, 4H, CH2), 3.78 (s, 6H, 2 x CH3), 3.80 (s, 3H, CH3), 3.82 (s, 3H, CH3), 5.20 (s, 2H, CH2), 6.81 – 6.92 (m, 2H, Ar), 7.01 (1H, s, Ar), 7.10 (s, 2H, Ar), 7.14 – 7.21 (m, 2H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.9, 24.6, 26.7, 41.0, 46.3, 55.3, 56.8 (3 x C), 60.7, 106.2 (2 x C), 110.8, 120.2, 122.6, 123.9, 124.5, 125.8, 125.6, 128.9, 129.1, 131.6, 153.9 (2 x C), 156.9, 159.1, 199.5. Anal. Calcd. for C26H29NO5: C, 71.70; H, 6.71; N, 3.22. Found: C, 71.55; H, 6.37; N, 3.48.
4.1.10.19. 2-(3-Methoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14t).
This compound was obtained from reaction of compound 8b with 3-methoxybenzyl chloride after 6 h. Yellow oil; yield 82%; IR (cm−1): 1656 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.83 – 1.94 (m, 4H, 2 x CH2), 2.68 – 2.74 (m, 4H, 2 x CH2), 3.71 (s, 6H, 2 x CH3), 3.75 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.94 (s, 2H, CH2), 6.36 (s, 2H, H-2” and H-6”), 6.53 (s, 1H, Ar), 6.60 (1H, d, J = 7.5 Hz, Ar), 6.79 (1H, dd, J = 8.1, 2.1 Hz, Ar), 7.22 (t, 1H, J= 7.9 Hz, Ar), 7.42 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.4, 26.3, 41.9, 51.5, 55.2, 55.9 (2 x C), 60.9, 108.1 (2 x C), 112.7, 112.8, 119.1, 122.7, 125.0, 125.4, 129.8, 131.6, 135.4, 137.8, 139.1, 153.0 (2 x C), 160.0, 199.4. Anal. Calcd. for C26H29NO5: C, 71.70; H, 6.71; N, 3.22. Found: C, 72.01; H, 6.39; N, 3.45.
4.1.10.20. 2-(4-Methoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14u).
This compound was obtained from reaction of compound 8b with 4-methoxybenzyl chloride after 6 h. Yield 66%; yellow oil; IR (cm−1): 1658 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.81 – 1.93 (m, 4H, 2 x CH2), 2.66 – 2.73 (m, 4H, 2 x CH2), 3.74 (s, 6H, 2 x CH3), 3.78 (s, 3H, CH3), 3.90 (s, 3H, CH3), 4.90 (s, 2H, CH2), 6.36 (s, 2H, H-2” and H-6”), 6.81 (d, 2H, J = 8.7 Hz, Ar), 6.93 (d, 2H, J = 8.7 Hz, Ar), 7.39 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.4, 26.3, 41.9, 51.1, 55.3, 56.0 (2 x C), 60.9, 108.1 (2 x C), 114.1 (2 x C), 122.7, 124.8, 125.2, 126.7, 128.4 (2 x C), 129.3, 131.5, 137.8, 153.0 (2 x C), 159.2, 199.3. Anal. Calcd. for C26H29NO5: C, 71.70; H, 6.71; N, 3.22. Found: C, 71.47; H, 6.56; N, 3.39.
4.1.10.21. 2-(2,5-Dimethoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14v).
This compound was obtained from reaction of compound 8b with 2,5-dimethoxybenzyl chloride after 8 h. Yellow oil; yield 61%; IR (cm−1): 1659 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.81 – 1.93 (m, 4H, 2 x CH2), 2.68 – 2.74 (m, 4H, 2 x CH2), 3.68 (s, 3H, CH3), 3.72 (s, 9H, 3 x CH3), 3.88 (s, 3H, CH3), 4.94 (s, 2H, CH2), 6.32 (s, 1H, Ar), 6.40 (s, 2H, H-2” and H-6”), 6.75 (s, 2H, Ar), 7.41 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.7, 23.6, 25.5, 41.0, 45.9, 54.9, 55.0, 55.2 (2 x C), 60.1, 107.2 (2 x C), 110.4, 112.0, 114.4, 121.9, 123.8, 125.2, 126.0, 130.9, 149.9, 150.0, 152.2 (2 x C), 152.9, 156.7, 199.0. Anal. Calcd. for C27H31NO6: C, 69.66; H, 6.71; N, 3.01. Found: C, 69.51; H, 6.88; N, 3.29.
4.1.10.22. 2-(3,4-Dimethoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14w).
This compound was obtained from reaction of compound 8b with 3,4-dimethoxybenzyl chloride after 6 h. Yellow oil; yield 61%; IR (cm−1): 1656 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.68 – 1.85 (m, 4H, 2 x CH2), 2.34 – 2.65 (m, 4H, 2 x CH2), 3.77 (s, 6H, 2 x CH3), 3.79 (s, 9H, 3 x CH3), 5.15 (s, 2H, CH2), 6.77 – 7.01 (m, 5H, Ar), 7.20 (s, 1H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 41.8, 51.3, 55.4 (2 x C), 55.8, 55.9, 60.3, 106.2 (2 x C), 110.1, 110.4, 111.1, 111.7, 112.2, 119.1, 119.7, 120.7, 124.8, 129.9, 131.7, 133.2, 153.0 (2 x C), 199.4. Anal. Calcd. for C27H31NO6: C, 69.66; H, 6.71; N, 3.01. Found: C, 69.47; H, 6.92; N, 3.54.
4.1.10.23. 2-(3,4,5-Trimethoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14x).
This compound was obtained from reaction of compound 8b with 3,4,5-trimethoxybenzyl chloride after 12 h. Yellow oil; yield 64%; IR (cm−1): 1661 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.91 (m, 4H, 2 x CH2), 2.66– 2.72 (m, 4H, 2 x CH2), 3.71 (s, 6H, 2 x CH3), 3.72 (s, 6H, 2 x CH3), 3.88 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.81 (s, 2H, CH2), 6.15 (s, 2H, Ar), 6.34 (s, 2H, Ar), 7.23 (s, 1H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.3, 24.2, 26.3, 41.8, 56.0 (2 x C), 56.1 (2 x C), 60.7, 60.8, 60.9, 104.8 (2 x C), 107.8 (2 x C), 122.9, 124.2, 124.9, 125.0, 126.5, 131.1, 136.1, 142.0, 153.1 (2 x C), 153.2 (2 x C), 199.3. Anal. Calcd. for C28H33NO7: C, 67.86; H, 6.71; N, 2.83. Found: C, 68.03; H, 6.99; N, 2.61.
4.1.10.24. 2-(4-Methoxy-3-nitrobenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14y).
This compound was obtained from reaction of compound 8b with 3-nitro,4-methoxybenzyl chloride after 3 h. Yellow solid; yield 65%; mp: 198 – 199 °C; IR (cm−1): 1660 (CO), 1531 (NO2); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.77 – 1.87 (m, 4H, 2 x CH2), 2.62 – 2.70 (m, 4H, 2 x CH2), 3.75 (s, 6H, 2 x CH3), 3.86 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.94 (s, 2H, CH2), 6.34 (s, 2H, H-2” and H-6”), 6.97 (d, 1H, J= 8.7 Hz, Ar), 7.14 (dd, 1H, J= 8.7, 2.0 Hz, Ar), 7.34 (s, 2H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.3, 24.3, 26.2, 41.9, 50.2, 56.1 (2 x C), 56.6, 60.9, 108.0 (2 x C), 113.8, 123.2, 124.4, 124.8, 125.3, 126.4, 129.7, 131.4, 132.8, 138.1, 139.5, 152.3, 153.2 (2 x C), 199.3. Anal. Calcd. for C26H28N2O7: C, 64.99; H, 5.87; N, 5.83. Found: C, 64.74; H, 5.99; N, 5.67.
4.1.11. General procedure for the preparation of 2-(3-amino-4-methoxybenzyl)-1-substituted-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14i,z).
To a solution of 14h,y (1 mmol) in ethyl acetate (12 mL), ammonium formate (1 mmol) and Pd/C were added. The reaction mixture was stirred at room temperature for 12 h. Pd/C was removed by filtration through celite using ethyl acetate as eluent. The solvent was evaporated under reduced pressure, giving the desired compound.
4.1.11.1. 2-(3-Amino-4-methoxybenzyl)-1-phenyl-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14i).
This compound was obtained from reaction of compound 14h. Yellow oil; yield 86%; IR (cm−1): 3461–3389 (NH2), 1651 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.92 (m, 4H, 2 x CH2), 2.66 – 2.73 (m, 4H, 2 x CH2), 3.83 (s, 3H, CH3), 4.78 (s, 2H, NH2), 4.81 (s, 2H, CH2), 6.36 – 6.39 (m, 2H, Ar), 6.61 – 6.70 (m, 1H, Ar), 7.23 – 7.43 (m, 6H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 41.5, 51.1, 55.5, 110.2, 113.9, 117.5, 122.7, 125.2, 127.3, 128.1, 128.4 (2 x C), 129.4, 130.9 (2 x C), 130.9, 130.1, 131.4, 136.4, 199.5. Anal. Calcd. for C23H24N2O2: C, 76.64; H, 6.71; N, 7.77. Found: C, 76.33; H, 7.04; N, 7.98.
4.1.11.2. 2-(3-Amino-4-methoxybenzyl)-1-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (14z).
This compound was obtained from reaction of compound 14y. Yellow oil; yield 71%; IR (cm−1): 3444–3361 (NH2), 1655 (CO); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.91 (m, 4H, 2 x CH2), 2.66 – 2.72 (m, 4H, 2 x CH2), 3.73 (s, 6H, 2 x CH3), 3.81 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.76 (s, 2H, NH2), 4.81 (s, 2H, CH2), 6.33 – 6.37 (m, 4H, Ar), 6.66 (d, 1H, J= 8.0 Hz, Ar), 7.38 (s, 1H, Ar); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.6, 23.6, 25.5, 41.1, 50.5, 54.8, 55.2 (2 x C), 60.1, 107.4 (2 x C), 109.5, 112.6, 116.2, 116.9, 121.8, 123.9, 124.6, 125.9, 129.1, 130.8, 135.8, 146.0, 152.1 (2 x C), 198.7. Anal.Calcd. for C26H30N2O5: C, 69.31; H, 6.71; N, 6.22. Found: C, 69.54; H, 6.43; N, 6.39.
General procedure for the preparation of 5-((dimethylamino)methylene)-1-substituted-5,6,7,8-tetrahydrocyclohepta[c]pyrrol-4(2H)-one (15a-z).
To a solution of ketone 14a-z (1 mmol) in anhydrous toluene (2.5 mL), TBDMAM (3 mmol) was added, and the reaction mixture was heated under reflux for 12 h. After cooling, the solvent was removed under reduced pressure. The residue was used in the following step without further purification.
4.1.12. General procedure for the preparation of 4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazoles (3a-z).
To a solution of 15a-z (5 mmol) in ethanol (15 mL) and acetic acid (3 mL), hydroxylamine hydrochloride was added (7.5 mmol). The reaction mixture was heated under reflux for 1 h. After cooling, the solvent was removed under reduced pressure. The crude product was poured into water and ice. The solid formed was obtained by filtration, dried and purified using a chromatography column (dichloromethane).
4.1.12.1. 8-Benzyl-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3a).
This compound was obtained from reaction of compound 15a. White solid; yield 73%; mp: 107 – 108 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 2.02 (s, 2H, CH2), 2.86 (s, 4H, 2 x CH2), 5.11 (s, 2H, CH2), 7.10 – 7.49 (m, 11H, Ar), 8.12 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.5, 24.7, 27.3, 51.2, 111.2, 112.1, 119.3, 120.0, 127.0 (2 x C), 127.7, 127.9, 128.5 (2 x C), 128.8 (2 x C), 130.9 (2 x C), 131.5, 131.8, 137.9, 151.9, 162.1. Anal. Calcd. for C23H20N2O: C, 81.15; H, 5.92; N, 8.23. Found: C, 80.89; H, 5.67; N, 8.39.
4.1.12.2. 8-(2-methoxybenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3b).
This compound was obtained from reaction of compound 15b. White solid; yield 78%; mp: 114 – 115 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.85 – 1.96 (m, 2H, CH2), 2.69 – 2.78 (m, 4H, 2 x CH2), 3.76 (s, 3H, CH3), 4.99 (s, 2H, CH2), 6.69 – 6.87 (m, 3H, Ar), 7.19 – 7.39 (m, 7H, Ar), 7.98 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.3, 46.2, 55.3, 110.1, 111.0, 111.7, 119.5, 119.6, 120.6, 126.2, 127.6, 128.2, 128.3 (2 x C), 128.8, 130.8 (2 x C), 131.6, 131.7, 151.7, 156.5, 162.3. Anal. Calcd. for C24H22N2O2: C, 77.81; H, 5.99; N, 7.56. Found: C, 77.56; H, 6.08; N, 7.78.
4.1.12.3. 8-(3-methoxybenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3c).
This compound was obtained from reaction of compound 15c. White solid; yield 68%; mp: 125 – 126 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.86 – 1.97 (m, 2H, CH2), 2.70 – 2.78 (m, 4H, 2 x CH2), 3.73 (s, 3H, CH3), 4.96 (s, 2H, CH2), 6.50 – 6.61 (m, 2H, Ar), 6.77 (dd, 1H, J = 8.2, 2.4 Hz, Ar), 7.15 – 7.26 (m, 4H, Ar), 7.34 – 7.44 (m, 3H, Ar), 8.00 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.2, 51.0, 55.2, 111.2, 112.1, 112.7, 112.8, 119.2, 119.3, 119.9, 127.8, 128.4 (2 x C), 129.8, 130.8 (2 x C), 131.4, 131.7, 139.4, 151.8, 159.8, 162.1. Anal. Calcd. for C24H22N2O2: C, 77.81; H, 5.99; N, 7.56. Found: C, 78.02; H, 6.08; N, 7.34.
4.1.12.4. 8-(4-Methoxybenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3d).
This compound was obtained from reaction of compound 15d. White solid; yield 83%; mp: 194 – 195 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.85 – 1.96 (m, 2H, CH2), 2.70 – 2.77 (m, 4H, 2 x CH2), 3.77 (s, 3H, CH3), 4.91 (s, 2H, CH2), 6.79 (d, 2H, J = 8.7, H-3’ and H-5’), 6.92 (d, 2H, J = 8.7, H-2’ and H-6’), 7.16 – 7.26 (m, 3H, Ar), 7.35 – 7.41 (m, 3H, Ar), 7.99 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.2, 50.6, 55.3, 111.1, 111.9, 114.0 (2 x C), 119.0, 119.9, 127.8, 128.4 (4 x C), 129.7, 130.8 (2 x C), 131.5, 131.6, 151.8, 159.0, 162.1. Anal. Calcd. for C24H22N2O2: C, 77.81; H, 5.99; N, 7.56. Found: C, 78.11; H, 5.78; N, 7.87.
4.1.12.5. 8-(2,5-Dimethoxybenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3e).
This compound was obtained from reaction of compound 15e. White solid; yield 80%; mp 174 – 175 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 2.03 (2H, s, CH2), 2.86 (4H, s, 2 x CH2), 3.79 (3H, s, CH3), 3.85 (3H, s, CH3), 5.10 (2H, s, CH2), 6.34 – 6.42 (1H, m, Ar), 6.74 – 6.92 (2H, m, Ar), 7.25 – 7.55 (6H, m, Ar and H-9), 8.12 (1H, s, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.5, 24.7, 27.3, 46.2, 55.7, 55.9, 111.2, 112.8, 114.3, 114.9, 119.6, 119.8, 127.4, 127.7, 128.4 (2 x C), 130.8 (2 x C), 131.7, 150.8, 150.9, 151.8, 153.6, 153.7, 162.3. Anal. Calcd. for C25H24N2O3: C, 74.98; H, 6.04; N, 7.00. Found: C, 75.21; H, 5.77; N, 7.15.
4.1.12.6. 8-(3,4-Dimethoxybenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3f).
This compound was obtained from reaction of compound 15f. White solid; yield 80%; mp 114 – 115 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.80 – 1.93 (2H, m, CH2), 2.65 – 2.76 (4H, m, 2 x CH2), 3.76 (3H, s, CH3), 3.86 (3H, s, CH3), 4.90 (2H, s, CH2), 6.41 (1H, s, Ar), 6.56 (1H, d, J= 7.8 Hz, Ar), 6.76 (1H, d, J= 7.8 Hz, Ar), 7.25 – 7.43 (7H, m, Ar, H-9 and H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.4, 24.2, 26.2, 51.4, 55.8, 55.9, 110.3, 111.2, 119.9 (2 x C), 123.0, 124.9 125.2 (2 x C), 127.9 (2 x C), 128.4 (2 x C), 129.3, 131.0 (2 x C), 131.4, 131.5, 148.6, 149.0. Anal. Calcd. for C25H24N2O3: C, 74.98; H, 6.04; N, 7.00. Found: C, 74.79; H, 6.22; N, 7.12.
4.1.12.7. 8-(3,4,5-trimethoxybenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3g).
This compound was obtained from reaction of compound 15g. White solid; yield 69%; mp 155 – 156 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.91 – 1.96 (2H, m, CH2), 2.73 – 2.78 (4H, m, 2 x CH2), 3.75 (6H, s, 2 x CH3), 3.88 (3H, s, CH3), 4.93 (2H, s, CH2), 6.18 (2H, s, Ar), 7.25 – 7.28 (3H, m, Ar), 7.38 – 7.45 (3H, m, Ar), 8.02 (1H, s, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.2, 51.5, 56.0 (2 x C), 60.9, 104.2 (2 x C), 111.2, 112.0, 119.2, 120.2, 127.8, 128.4 (2 x C), 130.8 (2 x C), 131.5, 131.6, 133.2, 137.3, 151.8 (d), 153.3 (2 x C), 162.0. Anal. Calcd. for C26H26N2O4: C, 72.54; H, 6.09; N, 6.51. Found: C, 72.35; H, 6.27; N, 6.80.
4.1.12.8. 8-(4-Methoxy-3-nitrobenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3h).
This compound was obtained from reaction of compound 15h. White solid; yield 60%; mp: 173 – 174 °C IR (cm−1): 1533 (NO2); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.89 – 1.96 (m, 2H, CH2), 2.73 – 2.77 (m, 4H, 2 x CH2), 3.94 (s, 3H, CH3), 4.99 (s, 2H, CH2), 6.99 (d, 1H, J= 8.7 Hz, Ar), 7.13 (dd, 1H, J = 8.7, 1.8, Ar), 7.19 – 7.22 (m, 3H, Ar), 7.37 – 7.42 (m, 4H, Ar), 8.03 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.2, 24.6, 27.1, 49.9, 56.6, 111.5, 112.5, 113.8, 118.9, 120.5, 124.3, 128.1, 128.4, 128.6 (2 x C), 130.1, 130.7 (2 x C), 131.1, 131.6, 132.6, 151.8, 152.3, 161.7. Anal. Calcd. for C24H21N3O4: C, 69.39; H, 5.10; N, 10.11. Found: C, 69.12; H, 5.26; N, 10.27.
4.1.12.9. 8-(4-Methoxy-3-aminobenzyl)-7-phenyl-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3i).
This compound was obtained from reaction of compound 15i. Yellow oil; yield 63%; IR (cm−1): 3441–3354 (NH2); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.89 – 1.96 (m, 2H, CH2), 2.72 – 2.79 (m, 4H, 2 x CH2), 3.83 (s, 3H, CH3), 4.85 (s, 2H, CH2), 5.23 (s, 2H, NH2), 6.38 – 6.40 (m, 2H, Ar), 6.68 (d, 1H, J= 8.7 Hz, Ar), 7.19 (s, 1H, Ar), 7.25 – 7.28 (m, 2H, Ar), 7.37 – 7.45 (m, 3H, Ar), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.2, 50.8, 55.5, 110.3, 111.0, 111.7, 113.7, 117.3, 119.2, 119.7, 127.7, 128.3 (2 x C), 130.3, 130.8 (2 x C), 131.5, 131.6, 136.2, 146.8, 151.7, 162.2. Anal. Calcd. for C24H23N3O2: C, 74.78; H, 6.01; N, 10.90. Found: C, 74.65; H, 6.33; N, 10.54.
4.1.12.10. Ethyl 8-benzyl-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole-9-carboxylate (3j).
This compound was obtained from reaction of compound 15j. Yellow oil; Yield 93%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.25 (t, 3H, J = 7.1 Hz, CH3), 1.95 – 2.01 (m, 2H, CH2), 2.58 – 2.61 (m, 2H, CH2), 2.80 (t, 2H, J = 6.4 Hz, CH2), 3.84 (s, 3H, CH3), 4.29 (q, 2H, J = 7.1 Hz, CH2), 5.34 (s, 2H, CH2), 6.87 (d, 2H, J = 6.7 Hz, Ar), 6.92 (d, 2H, J = 8.8 Hz, Ar), 7.12 (d, 2H, J = 8.8 Hz, Ar), 7.17 – 7.26 (m, 3H, Ar), 8.08 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.0, 24.3, 25.1, 25.7, 49.1, 55.3, 61.2, 114.0 (2 x C), 114.2, 115.0, 120.8, 122.5, 122.7, 126.1 (2 x C), 127.0, 128.4 (2 x C), 132.2 (2 x C), 135.4, 138.5, 151.8, 159.8, 160.3, 162.4. Anal. Calcd. for C27H26N2O4: C, 73.28; H, 5.92; N, 6.33. Found: C, 73.59; H, 5.71; N, 6.45.
4.1.12.11. Ethyl 8-(2-methoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole-9-carboxylate (3k).
This compound was obtained from reaction of compound 15k. Yellow oil; yield 83%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.21 (t, 3H, J = 7.1 Hz, CH3), 1.94 – 2.09 (m, 2H, CH2), 2.54 – 2.61 (m, 2H, CH2), 2.79 (t, 2H, J = 6.3 Hz, CH2), 3.72 (s, 3H, CH3), 3.80 (s, 3H, CH3), 4.25 (q, 2H, J = 7.1 Hz, CH2), 5.29 (2H, s, CH2), 6.49 (d, 1H, J = 7.4 Hz, Ar), 6.74 – 6.88 (4H, m, Ar), 7.05 – 7.20 (3H, m, Ar), 8.07 (1H, s, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 13.9, 24.3, 25.2, 25.7, 44.9, 55.2, 55.3, 61.1, 109.7, 113.8 (2 x C), 114.1, 118.4, 120.6, 121.0, 122.4, 122.8, 126.2, 127.2, 127.9, 132.0 (2 x C), 135.4, 151.9, 155.8, 159.6, 160.5, 162.2. Anal. Calcd. for C28H28N2O5: C, 71.17; H, 5.97; N, 5.93. Found: C, 70.89; H, 6.11; N, 6.18.
4.1.12.12. Ethyl 8-(3-methoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole-9-carboxylate (3l).
This compound was obtained from reaction of compound 15l. Yellow solid; yield 75%; mp 106–107 °C; 1H (CDCl3, 200 MHz, ppm): δ 1.25 (t, 3H, J = 7.1 Hz, CH3), 1.91 – 2.01 (m, 2H, CH2), 2.54 – 2.59 (m, 2H, CH2), 2.78 (t, 2H, J = 6.3 Hz, CH2), 3.71 (s, 3H, CH3), 3.82 (s, 3H, CH3), 4.29 (q, 2H, J = 7.1 Hz, CH2), 5.29 (s, 2H, CH2), 6.43 (d, 2H, J = 7.6 Hz, Ar), 6.72 (dd, 1H, J = 8.0, 2.4 Hz, Ar), 6.87 – 6.94 (m, 2H, Ar), 7.08 – 7.17 (m, 3H, Ar), 8.06 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.1, 24.3, 25.1, 25.7, 49.1, 55.1, 55.3, 61.2, 111.8, 112.5, 114.0 (2 x C), 114.2, 115.0, 118.4, 120.7, 122.6, 122.7, 129.5, 132.2 (2 x C), 135.4, 140.2, 151.9, 159.7, 159.8, 160.3, 162.4. Anal. Calcd. for C28H28N2O5: C, 71.17; H, 5.97; N, 5.93. Found: C, 71.31; H, 6.08; N, 5.72.
4.1.12.13. Ethyl 8-(4-methoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole-9-carboxylate (3m).
This compound was obtained from reaction of compound 15m. White solid; yield 60%; mp 131–132 °C; 1H NMR(CDCl3, 200 MHz, ppm): δ 1.26 (t, 3H, J = 7.1 Hz, CH3), 1.89 – 2.01 (m, 2H, CH2), 2.54 – 2.59 (m, 2H, CH2), 2.77 (t, 2H, J = 6.3 Hz, CH2), 3.74 (s, 3H, CH3), 3.83 (s, 3H, CH3), 4.30 (q, 2H, J = 7.1 Hz, CH2), 5.24 (s, 2H, CH2), 6.71 – 6.81 (m, 4H, Ar), 6.88 – 6.94 (m, 2H, Ar), 7.09 – 7.13 (m, 2H, Ar), 8.06 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.1, 24.3, 25.1, 25.7, 48.5, 55.2, 55.3, 65.1, 113.8 (2 x C), 113.9 (2 x C), 114.1, 114.9, 120.7, 122.5, 122.8, 127.5 (2 x C), 130.6, 132.2 (2 x C), 135.3, 151.9, 158.6, 159.7, 160.3, 162.6. Anal. Calcd. for C28H28N2O5: C, 71.17; H, 5.97; N, 5.93. Found: C, 70.96; H, 6.18; N, 5.81.
4.1.12.14. 8-(2-Methoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3n).
This compound was obtained from reaction of compound 15n. Yellow oil; yield 60%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.90 – 1.97 (m, 2H, CH2), 2.73 – 2.77 (m, 4H, 2 x CH2), 3.80 (s, 3H, CH3), 3.85 (s, 3H, CH3), 4.98 (s, 2H, CH2), 6.72 (d, 1H, J = 7.3 Hz, Ar), 6.83 – 6.89 (m, 2H, Ar), 6.93 (d, 2H, J = 8.6 Hz, Ar), 7.17 – 7.28 (m, 4H, Ar), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.4, 23.5, 27.5, 38.1, 55.4, 55.7, 110.1, 110.8, 113.7, 114.2 (2 x C), 118.3, 119.1, 120.6, 121.8, 127.4, 128.3, 128.8, 129.1, 129.3 (2 x C), 132.0, 150.6, 160.0, 160.8. Anal. Calcd. for C25H24N2O3: C, 74.98; H, 6.04; N, 7.00. Found: C, 75.11; H, 5.89; N, 6.69.
4.1.12.15. 8-(3-Methoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3o).
This compound was obtained from reaction of compound 15o. Yellow oil; yield 64%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.89 – 1.96 (m, 2H, CH2), 2.73 – 2.76 (m, 4H, 2 x CH2), 3.76 (s, 3H, CH3), 3.85 (s, 3H, CH3), 4.95 (s, 2H, CH2), 6.53 (s, 1H, Ar), 6.61 (d, 1H, J = 7.6 Hz, Ar), 6.79 (dd, 1H, J = 8.2, 2.3 Hz, Ar), 6.93 (d, 2H, J = 8.7 Hz, Ar), 7.14 – 7.23 (m, 4H, Ar), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.1, 23.5, 27.6, 43.2, 55.2, 55.3, 92.23, 105.8, 112.8, 113.1, 114.0 (2 x C), 114.5, 121.5, 127.4, 127.6, 129.2, 129.7, 132.0, 139.5, 150.9, 151.7, 159.4, 159.9, 161.9. Anal. Calcd. for C25H24N2O3: C, 74.98; H, 6.04; N, 7.00. Found: C, 74.73; H, 6.26; N, 7.39.
4.1.12.16. 8-(4-Methoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3p).
This compound was obtained from reaction of compound 15p. Yellow oil; yield 70%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.84 – 1.95 (m, 2H, CH2), 2.72 (t, 4H, J = 5.5 Hz, 2 x CH2), 3.78 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.88 (s, 2H, CH2), 6.80 (d, 2H, J = 8.7 Hz, Ar), 6.92 (d, 4H, J = 8.5 Hz, Ar), 7.12 – 7.16 (m, 3H, Ar), 7.98 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.2, 50.5, 55.3 (2 x C), 111.0, 111.7, 113.8 (2 x C), 114.0 (2 x C), 118.6, 119.6, 123.8, 128.4 (2 x C), 129.8, 131.3, 132.1 (2 x C), 151.7, 159.0, 159.2, 162.2. Anal. Calcd. for C25H24N2O3: C, 74.98; H, 6.04; N, 7.00. Found: C, 74.77; H, 5.83; N, 6.71.
4.1.12.17. 8-(3,4,5-trimethoxybenzyl)-7-(4-methoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3q).
This compound was obtained from reaction of compound 15q. Yellow oil; yield 64%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.89 (s, 2H, CH2), 2.72 (s, 2H, CH2), 2.84 (s, 2H, CH2), 3.69 (s, 6H, 2 x CH3), 3.79 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.82 (s, 2H, CH2), 6.13 (s, 2H, Ar), 6.86 (d, 2H, J = 8.3 Hz, Ar), 7.02 – 7.07 (m, 3H, Ar and H-9), 8.00 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.5, 27.2, 31.4, 48.9, 55.2, 55.9 (2 x C), 60.8, 104.8 (2 x C), 107.5, 113.8 (2 x C), 118.7, 119.8, 123.3, 123.8, 131.4, 131.7 (2 x C), 131.9, 136.1, 152.3, 152.4, 153.1 (2 x C), 159.2. Anal. Calcd. for C27H28N2O5: C, 70.42; H, 6.13; N, 6.08. Found: C, 70.56; H, 5.88; N, 6.34.
4.1.12.18. 8-Benzyl-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3r).
This compound was obtained from reaction of compound 15r. Yield 66%; white solid; mp 122 – 123 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.93 – 1.99 (2H, m, CH2), 2.75 – 2.82 (4H, m, 2 x CH2), 3.69 (6H, s, 2 x CH3), 3.89 (3H, s, CH3), 5.02 (2H, s, CH2), 6.36 (2H, s, H-2” and H-6”), 7.03 – 7.05 (2H, m, Ar), 7.25 – 7.33 (4H, m, Ar and H-9), 8.02 (1H, s, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.3, 51.2, 55.9 (2 x C), 60.9, 107.9 (2 x C), 111.2, 111.9, 119.3, 119.8, 126.5 (2 x C), 126.7, 127.5, 128.7 (2 x C), 131.6, 137.7, 138.3, 151.7, 152.9 (2 x C), 162.0. Anal. Calcd. for C26H26N2O4: C, 72.54; H, 6.09; N, 6.51. Found: C, 72.27; H, 5.87; N, 6.79.
4.1.12.19. 8-(2-Methoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3s).
This compound was obtained from reaction of compound 15s. Yellow oil; yield 60%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.93 – 2.00 (m, 2H, CH2), 2.74 – 2.84 (m, 4H, 2 x CH2), 3.69 (s, 6H, 2 x CH3), 3.80 (s, 3H, CH3), 3.89 (s, 3H, CH3), 5.01 (s, 2H, CH2), 6.41 (s, 2H, H-2” and H-6”), 6.77 (d, 1H, J = 7.5 Hz, Ar), 6.76 – 6.92 (m, 2H, Ar), 7.22 – 7.27 (m, 2H, Ar), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.4, 46.5, 55.3, 55.9 (2 x C), 60.9, 107.8 (2 x C), 110.1, 111.0, 111.7, 119.5, 119.6, 120.6, 126.6, 126.9, 127.9, 128.7, 131.6, 137.5, 151.7, 152.9 (2 x C), 156.3, 162.2. Anal. Calcd. for C27H28N2O5: C, 70.42; H, 6.13; N, 6.08. Found: C, 70.63; H, 6.29; N, 5.72.
4.1.12.20. 8-(3-Methoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3t).
This compound was obtained from reaction of compound 15t. Yellow oil; yield 68%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.92 – 1.98 (2H, m, CH2), 2.74 – 2.81 (4H, m, 2 x CH2), 3.70 (6H, s, 2 x CH3), 3.76 (3H, s, CH3), 3.89 (3H, s, CH3), 4.98 (2H, s, CH2), 6.38 (2H, s, H-2” and H-6”), 6.57 (1H, d, J = 1.8 Hz, Ar), 6.64 (1H, dd, J = 7.6, 0.6 Hz, Ar), 6.79 (1H, dd, J = 8.0, 2.4 Hz, Ar), 7.20 – 7.24 (2H, m, Ar), 8.01 (1H, s, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.3, 51.2, 55.2, 55.9 (2 x C), 60.9, 107.9 (2 x C), 111.9, 112.5, 112.7, 118.9, 119.4, 119.7, 126.7, 129.8, 129.9, 131.6, 137.7, 139.9, 151.7, 152.9 (2 x C), 160.0, 162.0. Anal. Calcd. for C27H28N2O5: C, 70.42; H, 6.13; N, 6.08. Found: C, 70.33; H, 6.38; N, 5.83.
4.1.12.21. 8-(4-Methoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3u).
This compound was obtained from reaction of compound 15u. Yellow oil; yield 51%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.92 – 1.98 (2H, m, CH2), 2.74 – 2.80 (4H, m, 2 x CH2), 3.74 (6H, s, 2 x CH3), 3.79 (3H, s, CH3), 3.90 (3H, s, CH3), 4.94 (2H, s, CH2), 6.39 (2H, s, H-2” and H-6”), 6.83 (2H, d, J = 8.8 Hz, H-3’ and H-5’), 6.96 (2H, d, J = 8.8 Hz, H-2’ and H-6’), 7.21 (1H, s, H-9), 8.01 (1H, s, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.3, 50.8, 55.3, 56.0 (2 x C), 60.9, 108.0 (2 x C), 111.1, 111.8, 114.1 (2 x C), 119.1, 119.7, 126.9, 128.1, 130.1 (2 x C), 131.5, 137.7, 151.7, 152.9 (2 x C), 159.1, 162.0. Anal. Calcd. for C27H28N2O5: C, 70.42; H, 6.13; N, 6.08. Found: C, 70.23; H, 6.41; N, 6.27.
4.1.12.22. 8-(2,5-Dimethoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3v).
This compound was obtained from reaction of compound 15v. Yellow oil; yield 77%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.92 – 1.99 (m, 2H, CH2), 2.74 – 2.83 (m, 4H, 2 x CH2), 3.68 (s, 3H, CH3), 3.72 (s, 6H, 2 x CH3), 3.75 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.98 (s, 2H, CH2), 6.37 (s, 1H, Ar), 6.43 (s, 2H, H-2” and H-6”), 6.75 – 6.76 (m, 2H, Ar), 7.23 (s, 1H, H-9), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.4, 46.4, 55.7, 55.8, 55.9 (2 x C), 60.9, 107.8 (2 x C), 111.0, 111.7, 112.4, 114.8, 119.5, 119.6, 126.9, 127.7, 127.8 (s), 131.5, 137.5, 150.6, 151.7, 152.9 (2 x C), 153.7, 162.2. Anal. Calcd. for C28H30N2O6: C, 68.56; H, 6.16; N, 5.71. Found: C, 68.31; H, 6.42; N, 5.55.
4.1.12.23. 8-(3,4-Dimethoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3w).
This compound was obtained from reaction of compound 15w. Yellow oil; yield 71%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.92 – 1.99 (m, 2H, CH2), 2.74 – 2.83 (m, 4H, 2 x CH2), 3.69 (s, 3H, CH3), 3.72 (s, 6H, 2 x CH3), 3.76 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.98 (s, 2H, CH2), 6.37 (s, 1H, Ar), 6.43 (s, 2H, H-2” and H-6”), 6.76 (s, 2H, Ar), 7.24 (s, 1H, H-9), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.4, 46.4, 55.7, 55.8, 55.9 (2 x C), 60.9, 107.8 (2 x C), 111.0 (2 x C), 111.7, 112.4, 114.8, 119.6, 119.7, 126.9, 127.7, 131.5, 137.5, 150.6, 151.7, 152.9, 153.7 (2 x C), 162.2. Anal. Calcd. for C28H30N2O6: C, 68.56; H, 6.16; N, 5.71. Found: C, 68.32; H, 5.88; N, 5.92.
4.1.12.24. 8-(3,4,5-Trimethoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3x).
This compound was obtained from reaction of compound 15x. Yellow oil; yield 71%; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.90 – 1.95 (m, 2H, CH2), 2.73 – 2.78 (m, 4H, 2 x CH2), 3.70 (s, 6H, 2 x CH3), 3.71 (s, 6H, 2 x CH3), 3.75 (s, 3H, CH3), 3.79 (s, 3H, CH3), 4.86 (s, 2H, CH2), 6.18 (s, 2H, Ar), 6.36 (s, 2H, Ar), 7.07 (s, 1H, H-9), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.3, 49.1 (q), 56.0 (2 x C), 56.1 (2 x C), 60.8, 60.9, 104.9 (2 x C), 107.6, 107.7 (2 x C), 111.2, 111.9, 119.0, 119.9, 124.0, 126.6, 131.9, 136.2, 151.8, 153.0 (2 x C), 153.2 (2 x C), 161.9. Anal. Calcd. for C29H32N2O7: C, 66.91; H, 6.20; N, 5.38. Found: C, 67.09; H, 5.99; N, 5.57.
4.1.12.25. 8-(4-Methoxy-3-nitrobenzyl)-7-(3,4,5-trimethoxyphenyl)-4,5,6,8-tetrahydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d][1,2]oxazole (3y).
This compound was obtained from reaction of compound 15y. Yellow oil; yield 67%; IR (cm−1): 1532 (NO2); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.90 – 1.98 (m, 2H, CH2), 2.73 – 2.78 (m, 4H, 2 x CH2), 3.79 (s, 6H, 2 x CH3), 3.90 (s, 3H, CH3), 3.93 (s, 3H, CH3), 5.00 (s, 2H, CH2), 6.40 (s, 2H, H-2” and H-6”), 7.01 (d, 1H, J = 8.7 Hz, Ar), 7.18 – 7.22 (m, 2H, Ar), 7.41 (d, 1H, J = 2.1 Hz, Ar), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.3, 24.5, 27.2, 50.0, 56.1 (2 x C), 56.7, 60.9, 107.91 (2 x C), 111.5, 112.5, 113.8, 118.7, 120.4, 124.2, 126.5, 130.4, 131.5, 132.5, 138.0, 139.6, 151.8, 152.3, 153.2 (2 x C), 161.7. Anal. Calcd. for C27H27N3O7: C, 64.15; H, 5.38; N, 8.31. Found: C, 64.31; H, 5.54; N, 8.11.
4.1.12.26. 2-Methoxy-5-((7-(3,4,5-trimethoxyphenyl)-5,6-dihydropyrrolo[3’,4’:3,4]cyclohepta[1,2-d]isoxazol-8(4H)-yl)methyl)aniline (3z).
This compound was obtained from reaction of compound 15z. Yellow oil; yield 60%; IR (cm−1): 3463–3381 (NH2); 1H NMR (CDCl3, 200 MHz, ppm): δ 1.90 – 1.97 (m, 2H, CH2), 2.71 – 2.81 (m, 4H, 2 x CH2), 3.78 (s, 6H, 2 x CH3), 3.86 (s, 3H, CH3), 3.89 (s, 3H, CH3), 4.94 (s, 2H, CH2), 6.48 (s, 2H, Ar), 6.80 (s, 2H, NH2), 7.12 (s, 1H, Ar), 7.89 (s, 1H, Ar), 8.00 (d, 1H, J = 6.5 Hz, Ar), 8.17 (s, 1H, Ar), 8.41 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz, ppm): δ 24.4, 24.6, 27.3, 50.8, 55.9, 56.1 (2 x C), 60.9, 108.0 (2 x C), 110.1, 111.1, 111.9, 118.9, 119.2, 119.6, 122.7, 126.8, 127.0, 130.6, 131.7, 147.2, 151.7, 153.0 (2 x C), 158.8, 162.1. Anal. Calcd. for C27H29N3O5: C, 68.19; H, 6.15; N, 8.84. Found: C, 68.48; H, 5.87; N, 8.63.
4.2. Biology
4.2.1. Cell lines
All the cell lines used in this paper were of human origin and purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Non-small cell lung carcinoma (A549) and T-acute lymphoblastic leukemia (CCRF-CEM) cells were grown in DMEM or RPMI (A549) medium (Gibco, Milano, Italy). Both media were supplemented with 115 units/mL of penicillin G (Gibco, Milano, Italy), 115 μg/mL of streptomycin (Invitrogen, Milano, Italy) and 10% fetal bovine serum (Invitrogen, Milano, Italy).
Lymphoma cell lines were cultured as recommended, using RPMI-1640 medium, supplemented with 20% (v/v) fetal bovine serum, Penicillin-Streptomycin-Neomycin (~5,000 units penicillin, 5 mg streptomycin and 10 mg neomycin/mL, Sigma) and L-glutamine (1%). Cell line identities were validated by CellCheck test (IDEXX, BioResearch) or with the Promega GenePrint 10 System kit, and all experiments were performed within one month after the cells were thawed. Cells were periodically tested to confirm mycoplasma negativity using the MycoAlert Mycoplasma Detection Kit (Lonza). Cells were incubated at 37 °C with 5% CO2 and were subcultured every three days.
4.4.2. Preparation of compounds for in vitro screening
All compounds (solids or oils) were dissolved in dimethyl sulfoxide (DMSO) to obtain a stock concentration of 10 mM and were stored frozen at 4 °C. For each experiment, fresh dilutions of compounds were made from the stock solutions to obtain the indicated concentrations. The DMSO concentration did not exceed 0.1% in any experiment.
4.2.3. Cell proliferation analysis
For each screening experiment, cells were seeded in 96-well plates (non-tissue culture treated) at a density ranging from 5,000 to 10,000 cells/well, depending on the doubling time of the specific cell line. For distributing cells into wells of the plates either a VIAFLO 96 hand-held electronic channel pipette (Integra Biosciences) or manual 12-channel pipet was used. Cells were initially treated with a single dose of 1 μM compound for 72 h. Selected compounds, which reached proliferation inhibition of about 60%, were further tested in the appropriate tissue culture medium with increasing compound doses ranging from 0 to 10 μM, using 1:2 dilution in series to obtain IC50 values. These assays were performed in triplicate. To 100 μL of cells suspended in medium, 100 μL of drug suspension was added, for a total seeding volume of 200 μL/well. After preparation of the microplates, they were incubated at 37 °C in a 5% CO2, 95% air atmosphere with 100% relative humidity for 72 h. Wells containing medium only were included on each plate and used as blanks for absorbance readings.
MTT (Sigma, Buchs) was prepared as a 5 mg/mL stock in phosphate-buffered saline and filter-sterilized, as we previously performed also for solid cell lines [33]. At the end of the incubation period, 20 μL of MTT solution was added to each well, and microplates were incubated at 37 °C for 4 h. Cells were then lysed with 50 μL per well of 25% sodium dodecyl sulfate lysis buffer, and absorbance was read at 570 nm using a Beckman Coulter-AD340 plate reader. The % of proliferation by the cells was obtained by quantifying the linear relationship between live cells and the A570 signal produced.
4.2.4. Antiproliferative activity in PBLs
Additional experiments were carried out with PBLs from healthy donors obtained as described previously [34]. For cytotoxicity evaluations in proliferating PBL cultures, non-adherent cells were resuspended at 5 x 105 cells/mL in a growth medium containing 2.5 g/mL PHA (Irvine Scientific). The same cellular density was used also for resting PBL cultures but without the addition of PHA. Scalar concentrations of the test compounds were added, and viability was determined after a 72 h incubation by the MTT test.
4.2.5. Analysis of cell cycle by flow cytometry
For these experiments, the A549, CCRF-CEM and VL51 cell lines were used. They were seeded in 6-well plates at a density of 5 x 104, 2.5 x 105 and 2.0 x 105, respectively, in a final volume of 2 mL culture medium. The cells were then treated with the test compounds for 24 h at the indicated concentrations. After this incubation period, the cells were detached with trypsin-EDTA (A549) and harvested by centrifugation. The pellet thus obtained was fixed in 70% ice cold ethanol. Then the cells were processed and analyzed as described previously [35], except that for the acquisition of data with labeled cells, a Cytomics FC500 flow cytometer (Beckman Coulter) was used.
Further experiments were performed to distinguish cells in G2 from those in metaphase by combining cell cycle analysis with phosphohistone H3 (p-H3, Cell Signalling) staining. This assay specifically identifies cells in metaphase.
4.2.6. Apoptosis assay
The quantification of apoptosis induced by the test compounds was carried out by flow cytometric analysis using the Annexin-V Fluos kit (Roche Diagnostics) following the manufacturer’s instructions. The cells treated with different concentrations of the test compounds after a 48 h incubation were labeled with annexin V/FITC and PI and analyzed with a Coulter Cytomics FC500 instrument (Beckman Coulter) in the FL1 and FL3 channels, respectively
4.2.7. Analysis of mitochondrial potential
The analysis of the mitochondrial potential was carried out by flow cytometric analysis. Briefly, cells treated with the test compound were labeled with the JC-1 dye as previously described [35]. The labeled cells were analyzed using the Coulter Cytomics FC500 instrument (Beckman Coulter) in the FL1 and FL2 channels, respectively.
4.2.8. Molecular modeling
All molecular modeling simulations were carried out using the Schrödinger Suite version 2018 [36]. For compounds 3d, 3p, 3u and 3z, we performed docking, molecular dynamics simulations, and thermodynamics evaluations by following the previously described protocol [21].
QikProp software [37] was applied for calculating the drug-like properties of the active derivatives (3d, 3p, 3u and 3z) and for evaluating their pharmacokinetic properties, by considering their absorption, distribution, metabolism and excretion (ADME) profile [38].
Finally, we used the Molinspiration virtual screening engine v2018.08 to explore potential off-target effects, by predicting the biological activity of the given ligands quickly and efficiently towards other targets [39]. In particular, this tool provides a druglikeness score of our ligands towards GPCR ligands, ion channel modulators, kinase inhibitors, nuclear receptor ligands, protease inhibitors and other enzyme targets based on Molinspiration technology [40,41].
Supplementary Material
HIGHLIGHTS.
3u and 3z showed IC50 values at submicromolar level against different lymphoma cells
3u and 3z displayed very high selectivity toward cancer cells and low toxicity in PBLs
3u and 3z induced cell cycle arrest in G2/M
3u and 3z induced apoptosis through the mitochondrial pathway
Acknowledgements
This work was financially supported by Ministero dell’Università e della Ricerca (MUR). This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations used
- NHL
non-Hodgkin’s lymphoma
- DLBCL
activated B-cell like diffuse large B cell lymphoma
- ABC
activated B-cell
- GCB
germinal center B-cell
- FL
follicular lymphoma
- MCL
mantle cell lymphoma
- MZL
marginal zone lymphoma
- NCI
National Cancer Institute
- MG_MID
mean graph mid-points
- DMFDMA
N,N-dimethylformamide dimethyl acetal
- TFAA
trifluoroacetic anhydride
- DCM
dichlomethane
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- TBDMAM
tert-butoxy bis(dimethylamino)methane
- CA-4
combretastatin A-4
- PBLs
peripheral blood lymphocytes
- DMSO
dimethyl sulfoxide
- CDCl3
deuterated chloroform
- s
singlet
- d
doublet
- t
triplet
- q
quartet
- m
multiplet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Francesco Bertoni reports financial support was provided by ADC Therapeutics, Bayer AG, Cellestia, Helsinn, HTG Molecular Diagnostics, ImmunoGen, Menarini Ricerche, NEOMED Therapeutics 1, Nordic Nanovector ASA, Helsinn, Menarini. Eugenio Gaudio reports a relationship with Helsinn Healthcare SA that includes: employment.
Declaration of competing interest
Eugenio Gaudio: currently, employee of Helsinn Healthcare SA, Lugano, Switzerland. Francesco Bertoni: institutional research funds from ADC Therapeutics, Bayer AG, Cellestia, Helsinn, HTG Molecular Diagnostics, ImmunoGen, Menarini Ricerche, NEOMED Therapeutics 1, Nordic Nanovector ASA; consultancy fee from Helsinn, Menarini; travel grants from Amgen, Astra Zeneca. The other Authors have nothing to disclose.
References
- [1].Barreca M, Spanò V, Raimondi MV, Bivacqua R, Giuffrida S, Montalbano A, Cavalli A, Bertoni F, Barraja P, GPCR Inhibition in Treating Lymphoma, ACS Med. Chem. Lett 13 (2022) 358–364. doi: 10.1021/acsmedchemlett.1c00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barreca M, Stathis A, Barraja P, Bertoni F, An overview on anti-tubulin agents for the treatment of lymphoma patients, Pharmacol. Ther 211 (2020) 107552. doi: 10.1016/j.pharmthera.2020.107552. [DOI] [PubMed] [Google Scholar]
- [3].PDQ Adult Treatment Editorial Board. Adult Non-Hodgkin Lymphoma Treatment (PDQ®): Health Professional Version. 2022. Jul 6. In: PDQ Cancer Information Summaries [Internet] Bethesda (MD): National Cancer Institute (US); 2002, 2002. [Google Scholar]
- [4].PDQ Adult Treatment Editorial Board. Adult Hodgkin Lymphoma Treatment (PDQ®): Health Professional Version. 2022. Jul 15. In: PDQ Cancer Information Summaries [Internet] Bethesda (MD): National Cancer Institute (US); 2002, 2002. [Google Scholar]
- [5].Yang J, Liu Y, Lan S, Yu S, Luo D, Shan H, Zhong X, Yan G, Li R, Discovery of 2‑Methyl-2-(4-(2-methyl-8-(1H‑pyrrolo[2,3‑b]pyridin-6-yl)‑1H‑naphtho[1,2‑d]imidazol-1-yl)phenyl)propanenitrile as a Novel PI3K/mTOR Inhibitor with Enhanced Antitumor Efficacy In Vitro and In Vivo, J. Med. Chem 65 (2022) 12781–12801. doi: 10.1021/acs.jmedchem.2c00572. [DOI] [PubMed] [Google Scholar]
- [6].Narva S, Chitti S, Rao B, Alvala M, Jain N, Gowri V, Sekhar C, Synthesis and biological evaluation of pyrrolo [2,3-b]pyridine analogues as antiproliferative agents and their interaction with calf thymus DNA, Eur. J. Med. Chem 114 (2016) 220–231. doi: 10.1016/j.ejmech.2016.02.059. [DOI] [PubMed] [Google Scholar]
- [7].Spanò V, Barreca M, Cilibrasi V, Genovese M, Renda M, Montalbano A, Galietta LJV, Barraja P, Evaluation of fused pyrrolothiazole systems as correctors of mutant CFTR protein, Molecules 26 (2021) 1–25. doi: 10.3390/molecules26051275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barreca M, Spanò V, Montalbano A, Cueto M, Díaz Marrero AR, Deniz I, Erdoğan A, Bilela LL, Moulin C, Taffin-De-Givenchy E, Spriano F, Perale G, Mehiri M, Rotter A, Thomas OP, Barraja P, Gaudêncio SP, Bertoni F, Marine anticancer agents: An overview with a particular focus on their chemical classes, Mar. Drugs 18 (2020) 619. doi: 10.3390/md18120619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang XX, Xiao Y, Yan YY, Wang YM, Jiang H, Wu L, Shi J, Liu XH, Discovery of the Novel 1H‑Pyrrolo[2,3‑b]pyridine Derivative as a Potent Type II CDK8 Inhibitor against Colorectal Cancer, J. Med. Chem 65 (2022) 12095–12123. doi: 10.1021/acs.jmedchem.2c00820. [DOI] [PubMed] [Google Scholar]
- [10].Spanò V, Venturini A, Genovese M, Barreca M, Raimondi MV, Montalbano A, Galietta LJV, Barraja P, Current development of CFTR potentiators in the last decade, Eur. J. Med. Chem 204 (2020) 1–15. doi: 10.1016/j.ejmech.2020.112631. [DOI] [PubMed] [Google Scholar]
- [11].Kilic-kurt Z, Bakar-ates F, Aka Y, Kutuk O, Design , synthesis and in vitro apoptotic mechanism of novel pyrrolopyrimidine derivatives, Bioorg. Chem 83 (2019) 511–519. doi: 10.1016/j.bioorg.2018.10.060. [DOI] [PubMed] [Google Scholar]
- [12].Barreca M, Spanò V, Raimondi MV, Tarantelli C, Spriano F, Bertoni F, Barraja P, Montalbano A, Recurrence of the oxazole motif in tubulin colchicine site inhibitors with anti-tumor activity, Eur. J. Med. Chem. Reports 1 (2021) 100004. doi: 10.1016/j.ejmcr.2021.100004. [DOI] [Google Scholar]
- [13].Grillone K, Riillo C, Rocca R, Ascrizzi S, Spanò V, Scionti F, Polerà N, Maruca A, Barreca M, Juli G, Arbitrio M, Di Martino MT, Caracciolo D, Tagliaferri P, Alcaro S, Montalbano A, Barraja P, Tassone P, The new microtubule-targeting agent SIX2G induces immunogenic cell death in multiple myeloma, Int. J. Mol. Sci 23 (2022) 10222. doi: 10.3390/ijms231810222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barreca M, Ingarra AM, Raimondi MV, Spanò V, De Franco M, Menilli L, Gandin V, Miolo G, Barraja P, Montalbano A, Insight on pyrimido[5,4-g]indolizine and pyrimido[4,5-c]pyrrolo[1,2-a]azepine systems as promising photosensitizers on malignant cells, Eur. J. Med. Chem 237 (2022) 114399. doi: 10.1016/j.ejmech.2022.114399. [DOI] [PubMed] [Google Scholar]
- [15].Labbozzetta M, Barreca M, Spanò V, Raimondi MV, Poma P, Notarbartolo M, Barraja P, Montalbano A, Novel insights on [1,2]oxazolo[5,4-e]isoindoles on multidrug resistant acute myeloid leukemia cell line, Drug Dev. Res (2022) 1–11. doi: 10.1002/ddr.21962. [DOI] [PMC free article] [PubMed]
- [16].Barreca M, Ingarra AM, Raimondi MV, Spanò V, Piccionello AP, De Franco M, Menilli L, Gandin V, Miolo G, Barraja P, Montalbano A, New tricyclic systems as photosensitizers towards triple negative breast cancer cells, Arch. Pharm. Res 45 (2022) 806–821. doi: 10.1007/s12272-022-01414-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Montalbano A, Parrino B, Diana P, Barraja P, Carbone A, Spanò V, Cirrincione G, Synthesis of the new oligopeptide pyrrole derivative isonetropsin and its one pyrrole unit analogue, Tetrahedron 69 (2013) 2550–2554. doi: 10.1016/j.tet.2013.01.076. [DOI] [Google Scholar]
- [18].Barreca M, Spanò V, Rocca R, Bivacqua R, Abel AC, Maruca A, Montalbano A, Raimondi MV, Tarantelli C, Gaudio E, Cascione L, Rinaldi A, Bai R, Steinmetz MO, Prota AE, Alcaro S, Hamel E, Bertoni F, Barraja P, Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: Further chemical modifications and potential therapeutic effects against lymphomas, Eur. J. Med. Chem 243 (2022) 114744. doi: 10.1016/j.ejmech.2022.114744. [DOI] [PubMed] [Google Scholar]
- [19].Spanò V, Pennati M, Parrino B, Carbone A, Montalbano A, Cilibrasi V, Zuco V, Lopergolo A, Cominetti D, Diana P, Cirrincione G, Barraja P, Zaffaroni N, Preclinical activity of new [1,2]oxazolo[5,4-e]isoindole derivatives in diffuse malignant peritoneal mesothelioma, J. Med. Chem 59 (2016) 7223–7238. doi: 10.1021/acs.jmedchem.6b00777. [DOI] [PubMed] [Google Scholar]
- [20].Spanò V, Pennati M, Parrino B, Carbone A, Montalbano A, Lopergolo A, Zuco V, Cominetti D, Diana P, Cirrincione G, Zaffaroni N, Barraja P, [1,2]Oxazolo[5,4-e]isoindoles as promising tubulin polymerization inhibitors, Eur. J. Med. Chem 124 (2016) 840–851. doi: 10.1016/j.ejmech.2016.09.013. [DOI] [PubMed] [Google Scholar]
- [21].Spanò V, Rocca R, Barreca M, Giallombardo D, Montalbano A, Carbone A, Raimondi MV, Gaudio E, Bortolozzi R, Bai R, Tassone P, Alcaro S, Hamel E, Viola G, Bertoni F, Barraja P, Pyrrolo[2’,3’:3,4]cyclohepta[1,2-d][1,2]oxazoles, a new class of antimitotic agents active against multiple malignant cell types, J. Med. Chem 63 (2020) 12023–12042. doi: 10.1021/acs.jmedchem.0c01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barraja P, Spanò V, Diana P, Carbone A, Cirrincione G, Synthesis of the new ring system 6,8-dihydro-5H-pyrrolo[3,4-h]quinazoline, Tetrahedron Lett 50 (2009) 5389–5391. doi: 10.1016/j.tetlet.2009.07.045. [DOI] [Google Scholar]
- [23].Spanò V, Montalbano A, Carbone A, Parrino B, Diana P, Cirrincione G, Castagliuolo I, Brun P, Issinger OG, Tisi S, Primac I, Vedaldi D, Salvador A, Barraja P, Synthesis of a new class of pyrrolo[3,4-h]quinazolines with antimitotic activity, Eur. J. Med. Chem 74 (2014) 340–357. doi: 10.1016/j.ejmech.2013.10.014. [DOI] [PubMed] [Google Scholar]
- [24].Spanò V, Barreca M, Rocca R, Bortolozzi R, Bai R, Carbone A, Raimondi MV, Piccionello AP, Montalbano A, Alcaro S, Hamel E, Viola G, Barraja P, Insight on [1,3]thiazolo[4,5-e]isoindoles as tubulin polymerization inhibitors, Eur. J. Med. Chem 212 (2021) 113122. doi: 10.1016/j.ejmech.2020.113122. [DOI] [PubMed] [Google Scholar]
- [25].Maruca A, Ambrosio FA, Lupia A, Romeo I, Rocca R, Moraca F, Talarico C, Bagetta D, Catalano R, Costa G, Artese A, Alcaro S, Computer-based techniques for lead identification and optimization i: Basics, Phys. Sci. Rev 4 (2019) 20180113. doi: 10.1515/psr-2018-0113. [DOI] [Google Scholar]
- [26].Prota AE, Danel F, Bachmann F, Bargsten K, Buey RM, Pohlmann J, Reinelt S, Lane H, Steinmetz MO, The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization, J. Mol. Biol 426 (2014) 1848–1860. doi: 10.1016/j.jmb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- [27].Gigant B, Wang C, Ravelli RBG, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M, Structural basis for the regulation of tubulin by vinblastine, Nature 435 (2005) 519–522. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- [28].Barbier P, Dorléans A, Devred F, Sanz L, Allegro D, Alfonso C, Knossow M, Peyrot V, Andreu JM, Stathmin and interfacial microtubule inhibitors recognize a naturally curved conformation of tubulin dimers, J. Biol. Chem 285 (2010) 31672–31681. doi: 10.1074/jbc.M110.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rocca R, Costa G, Artese A, Parrotta L, Ortuso F, Maccioni E, Pinato O, Greco ML, Sissi C, Alcaro S, Distinto S, Moraca F, Hit Identification of a Novel Dual Binder for h-telo/c-myc G-Quadruplex by a Combination of Pharmacophore Structure-Based Virtual Screening and Docking Refinement, ChemMedChem 11 (2016) 1721–1733. doi: 10.1002/cmdc.201600053. [DOI] [PubMed] [Google Scholar]
- [30].Molinspiration Cheminformatics free web services https://www.molinspiration.com, Slovensky Grob, Slovakia, 2021 (accessed 15 Oct 2021), (2022) 2022.
- [31].Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M, Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation, J. Biol. Chem 274 (1999) 25543–25549. doi: 10.1074/jbc.274.36.25543. [DOI] [PubMed] [Google Scholar]
- [32].Gottlieb E, Armour SM, Harris MH, Thompson CB, Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis, Cell Death Differ 10 (2003) 709–717. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- [33].Celano M, Schenone S, Cosco D, Navarra M, Puxeddu E, Racanicchi L, Brullo C, Varano E, Alcaro S, Ferretti E, Botta G, Filetti S, Fresta M, Botta M, Russo D, Cytotoxic effects of a novel pyrazolopyrimidine derivative entrapped in liposomes in anaplastic thyroid cancer cells in vitro and in xenograft tumors in vivo, Endocr. Relat. Cancer 15 (2008) 499–510. doi: 10.1677/ERC-07-0243. [DOI] [PubMed] [Google Scholar]
- [34].Romagnoli R, Baraldi PG, Salvador MK, Prencipe F, Lopez-Cara C, Schiaffino Ortega S, Brancale A, Hamel E, Castagliuolo I, Mitola S, Ronca R, Bortolozzi R, Porcù E, Basso G, Viola G, Design, synthesis, in vitro, and in vivo anticancer and antiangiogenic activity of novel 3-arylaminobenzofuran derivatives targeting the colchicine site on tubulin, J. Med. Chem 58 (2015) 3209–3222. doi: 10.1021/acs.jmedchem.5b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Viola G, Vedaldi D, Dall’Acqua F, Fortunato E, Basso G, Bianchi N, Zuccato C, Borgatti M, Lampronti I, Gambari R, Induction of γ-globin mRNA, erythroid differentiation and apoptosis in UVA-irradiated human erythroid cells in the presence of furocumarin derivatives, Biochem. Pharmacol 75 (2008) 810–825. doi: 10.1016/j.bcp.2007.10.007. [DOI] [PubMed] [Google Scholar]
- [36].Schrödinger LLC, New York (USA), (2018). [Google Scholar]
- [37].QikProp Version 3.5, Schrödinger LLC, New York, NY (USA), 2012, (2012). [Google Scholar]
- [38].Khanna V, Ranganathan S, Physiochemical property space distribution among human metabolites, drugs and toxins, BMC Bioinformatics 10 (2009) 1–18. doi: 10.1186/1471-2105-10-S15-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Molinspiration – cheminformatics [last accessed: Dicember 2022] www.molinspiration.com, (2022).
- [40].Khan T, Dixit S, Ahmad R, Raza S, Azad I, Joshi S, Khan AR, Molecular docking PASS analysis, bioactivity score prediction, synthesis, characterization and biological activity evaluation of a functionalized 2-butanone thiosemicarbazone ligand and its complexes, J. Chem. Biol 10 (2017) 91–104. doi: 10.1007/s12154-017-0167-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cilibrasi V, Spanò V, Bortolozzi R, Barreca M, Raimondi MV, Rocca R, Maruca A, Montalbano A, Alcaro S, Ronca R, Viola G, Barraja P, Synthesis of 2H-Imidazo[2′,1’:2,3] [1,3]thiazolo[4,5-e]isoindol-8-yl-phenylureas with promising therapeutic features for the treatment of acute myeloid leukemia (AML) with FLT3/ITD mutations, Eur. J. Med. Chem 235 (2022) 114292. doi: 10.1016/j.ejmech.2022.114292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.