

Abstract
Cancer-relevant mutations in the oligomerization domain (OD) of the p53 tumor suppressor protein, unlike those in the DNA binding domain, have not been well elucidated. Here we characterized the germline OD mutant p53(A347D), which occurs in cancer-prone Li-Fraumeni Syndrome (LFS) patients. Unlike wild-type p53, mutant p53(A347D) cannot form tetramers and exists as a hyper-stable dimeric protein. Further, p53(A347D) cannot bind or transactivate the majority of canonical p53 target genes. Isogenic cell lines harboring either p53(A347D) or no p53 yield comparable tumorigenic properties, yet p53(A347D) displays remarkable neomorphic activities. Cells bearing p53(A347D) possess a distinct transcriptional profile and undergo metabolic reprogramming. Further, p53(A347D) induces striking mitochondrial network aberration and associates with mitochondria to drive apoptotic cell death upon topoisomerase II inhibition in the absence of transcription. Thus, dimer-forming p53 demonstrates both loss of function (LOF) and gain of function (GOF) properties compared to the wild-type form of the protein.
Introduction
Tumor suppressor p53 sits at the regulatory nexus of intrinsic tumor suppressive responses including DNA repair, cell cycle arrest, regulation of metabolism, cell death and others that work in concert to preclude malignant transformation and progression (1,2). Consequently, p53 is mutated in more than 50% of all sporadic cancers, and germline mutations in p53, causative of Li-Fraumeni syndrome (LFS), result in a predisposition towards early-onset cancers, which frequently include breast carcinomas, acute leukemia, brain tumors, sarcomas, and adrenal cortical carcinomas (3–6). Unlike many other tumor suppressor genes, which are inactivated through frameshift or nonsense mutations, ~75–80% of TP53 mutations encode missense mutant proteins. This can result in a mutant p53 protein that has both lost canonical p53 transactivation ability and has gained not only dominant-repressive activity over wild-type p53 (WT p53) but also neomorphic properties that impart oncogenic ability (7,8). The spectrum of TP53 mutations, marked by highly frequent “hotspot” mutations at key residues, implicates the selective potential that certain mutations may confer to actively drive tumor development as the occurrence of hotspot mutations is a hallmark of a gain-of-function (GOF) oncogene. Such GOF activities in p53, however, vary across hotspot mutants with R175H and R273H but not R248W and D281G mutant p53 conferring the ability of SAOS-2 cells to grow in soft-agar, for instance (9). Thus, considering the mutation and context specificity of p53 mutations is essential in probing p53 GOF (10–12). Most work in understanding the oncogenic contributions of mutant p53 and wild-type p53 loss of function (LOF) has focused on hotspot mutations occurring in the DNA-binding domain (7). In contrast, mutations that occur within the oligomerization domain (OD) of p53, which generally compromise p53 oligomerization (13), have been poorly studied despite the high frequency of LFS mutations within the OD relative to its length (Supplementary Fig. S1A) (14). While the most common OD mutant form is the p.R337H variant, which is highly prevalent in Brazil presumably due to founder effect and may thereby be overrepresented in datasets of germline p53 variants (15–17), several other germ line p53 OD mutations have been documented in LFS patients (18). Among these, p.A347D is the most frequent and nearly only dimer-forming variant as identified from The TP53 Database (R20, 2019) (14). Elucidating the oncogenic gain-of-function and tumor suppressive loss-of-function effects of OD mutant p53 may reveal targetable vulnerabilities that will yield more effective therapies for a previously under-studied subset of patients harboring OD mutations.
p53 exists in monomeric, dimeric, and tetrameric states. Under unstressed conditions, p53 primarily exists as a dimer that is preferentially bound and degraded by its negative regulator MDM2 (19,20). Upon stress induction, p53 levels rise and the protein transitions into the tetrameric conformation to more efficiently bind DNA and transactivate canonical p53 targets (21,22). Over the years, much evidence has stressed the importance of the p53 OD as critical in transcriptional regulation, post-translational modification, degradation, and protein-protein interactions (23–26). Mutations within the OD affect oligomerization and p53 function in a mutation-specific and context-dependent manner. Kawaguchi et al. demonstrated that mutations impairing oligomerization vary in their impact on p53 transcriptional activity in yeast-based assays using several canonical targets (13). Furthermore, certain cancer-associated OD mutants have been shown to accumulate at abnormally high cellular levels, yet their impact on tumorigenicity is unknown (19,27). Previous studies have revealed that OD mutants may still stimulate transcription selectively, including the ability to activate cell cycle arrest but not pro-apoptotic genes (28). Moreover, limited evidence has shown that the monomer-forming p53-R337H mutant carriers may experience increased oxidative damage and elevated antioxidant status (29). Yet, the phenotype of OD mutants, especially in the context of GOF activity, remains largely uncharacterized, and most studies investigating the function of such mutants were performed under non-physiological conditions (24).
Here we focused on the cancer-associated p53(A347D) mutant, a dimer-forming (13), pathogenic p53 variant observed in ~15% of LFS patients with OD mutations and among a set of multimeric mutant p53 alleles that results in 80% disease penetrance (18). The prevailing, albeit limited, literature on OD mutant p53 focuses primarily on monomeric mutants while the pro-tumorigenic functions of p53(A347D), the third most frequent OD mutation and the most common dimeric mutant p53, remain unknown. We utilized three patient-derived primary fibroblast cell lines from a single family with one sibling expressing WT p53 and two expressing the germ-line p53(A347D) mutant. We generated stem cells (iPSCs) from these fibroblasts, which were then differentiated into mesenchymal stem cells (MSCs) and osteoblasts, the cell-of-origin for osteosarcoma, as a model for early LFS tumors. As osteosarcomas are one of the common tumors found in LFS patients, we engineered via CRISPR-Cas9 U2OS osteosarcoma cells that harbor wild-type p53 to express either no p53 or are heterozygous (p53+/AD) or homozygous (p53AD/AD) for the A347D mutation. These cell lines allowed us to demonstrate novel GOF and LOF activities of dimeric mutant p53.
Results
Oligomerization domain mutant p53(A347D) preferentially forms dimers
Single residue substitutions within the OD of TP53 range from completely abolishing p53 oligomerization to allowing tetramerization in a mutation-specific manner. It has been suggested that p53(A347D) specifically yields a dimeric protein unable to form the active, tetrameric structure and consequently lacking in canonical transcriptional activity (13). In concordance, we observed that both p53(A347D) and p53(A347T), when expressed via plasmid transfection, primarily forms dimers whereas the p53(L330A) mutant yields a monomeric protein (Supplementary Fig. S1B). We utilized CRISPR-Cas9 gene editing to generate single clones of U2OS either heterozygous (p53+/AD #1, p53+/AD #2, p53+/AD #3) or homozygous (p53AD/AD #1, p53+/AD #2) for the p53(A347D) mutation as well as U2OS cells that are p53 null (KO #1, KO #2, KO #3). Cells were pooled according to mutation status to account for clonal variation and are used where indicated. Furthermore, primary dermal fibroblast cells isolated from two LFS siblings (L53-M1, L53-M2) heterozygously expressing p53(A347D) were compared to primary dermal fibroblast cells isolated from a third sibling (L53-WT) with wild-type p53. We assessed the oligomeric status of p53 in U2OS cells (Fig. 1A) and LFS fibroblasts (Fig. 1B) following protein crosslinking with glutaraldehyde and observed that p53(A347D) preferentially forms dimers under basal conditions. Even after induction with etoposide, p53(A347D) expressed in U2OS p53AD/AD cells were completely unable to form tetramers, while the tetramer was the primary species observed in wild-type p53 cells (Fig. 1A). Interestingly, p53+/AD U2OS cells exhibited intermediate tetramer formation roughly half that of wild-type p53 cells after etoposide treatment, implying that hetero-tetramers of wild-type and mutant p53 may not form. Prior studies have suggested that p53 dimers form co-translationally whereas p53 tetramers form post-translationally, thereby resulting in 50% hetero-tetramer formation when wild-type and mutant p53 are co-expressed (30). However, we found that ectopically expressed wild-type and mutant p53 do not co-immunoprecipitate, further suggesting that hetero-oligomers of wild-type and dimeric mutant p53 do not form (Supplementary Fig. S1C).
Figure 1. Dimer-forming p53(A347D) is hyperstable and transcriptionally impaired.
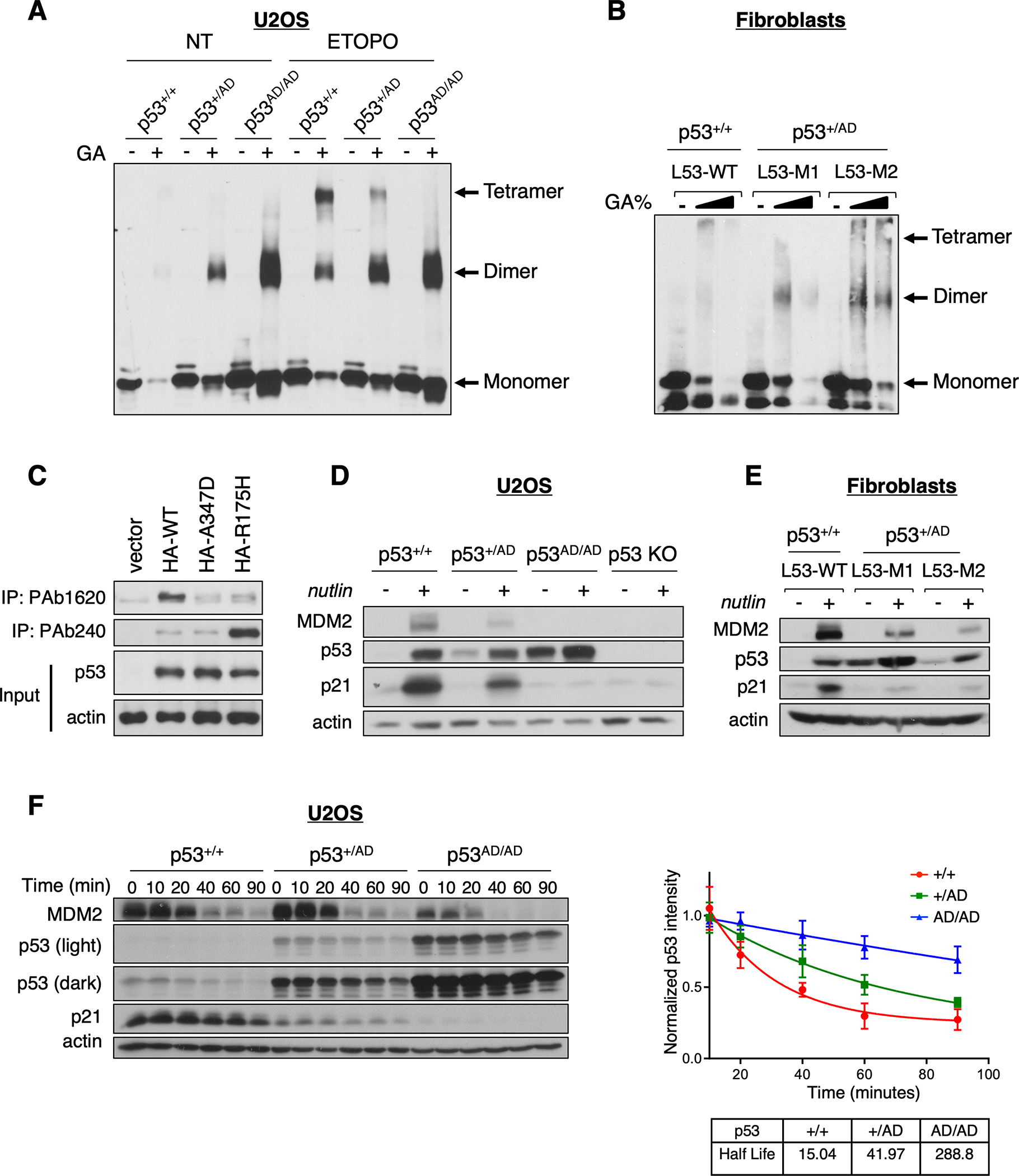
(A) Protein lysates from U2OS p53+/+, p53+/AD, and p53AD/AD cells treated with (ETOPO) or without (NT) 20 μM etoposide for 6 h were incubated in the presence or absence of 0.005% glutaraldehyde for 20 min at room temperature (RT) and subjected to immunoblot analysis with a monoclonal p53 antibody (DO1/1801) to detect p53 oligomeric species indicated at right.
(B) Protein lysates from primary dermal fibroblasts expressing WT p53 (L53-WT) or heterozygous p53(A347D) (L53-M1, L53-M2) were incubated with an increasing concentration of glutaraldehyde (0%, 0.01%, 0.05%) for 20 min at RT then subjected to immunoblot analysis as in A.
(C) U2OS p53 KO cells were transfected with plasmids expressing HA-WT-p53, HA-p53(A347D), HA-p53(R175H) or the empty vector pcDNA3. Protein lysates were subjected to immunoprecipitation with anti-p53 PAb240 or PAb1620 and immunoblot analysis with anti-p53 (DO-1).
(D) U2OS p53+/+, p53+/AD, p53AD/AD, and p53 KO cells were treated with 10 μM nutlin-3a for 24 h then lysed. Protein lysates were then subjected to immunoblot analysis with antibodies against indicated proteins.
(E) Protein lysates from primary dermal fibroblasts varying in p53 status were treated with 10 μM nutlin-3a for 24 h, and then processed for immunoblotting with antibodies against indicated proteins.
(F) (Left) Following the addition of cycloheximide (100 μg/mL), U2OS cells expressing p53+/+, p53+/AD, and p53AD/AD were harvested at the indicated times. Cell lysates were then subjected to immunoblotting. (Right) Densitometric analysis was performed using ImageJ to assess the half-life of p53. Each point represents the density of the p53 band at indicated time points relative to the initial time point. Data represent mean ± SEM for three biological replicates.
Previously published work has revealed that two antibodies can discriminate between wild-type and mutant forms of p53, namely PAb240, which can preferentially recognize the unfolded conformation of several common mutant forms of p53 by binding an epitope within the core domain that is normally hidden in wild-type p53 (31–33), and PAb1620, which appears to bind specifically to wild-type p53 (34). Surprisingly, while we confirmed increased immunoprecipitation of wild-type p53 by the 1620 antibody and superior immunoprecipitation of a common tumor-derived mutant form of p53(R175H) by PAb240, we observed that neither antibody preferentially immunoprecipitated p53(A347D), indicating that the conformation of p53(A347D) is neither fully wild-type or mutant and implying that p53(A347D) is only partially unfolded (Fig. 1C).
p53(A347D) cannot transactivate canonical wild-type p53 targets
Given the importance of p53 oligomerization for DNA binding and evidence highlighting the varied effects that p53 OD mutations exert on transcriptional activity, we sought to elucidate the transactivation potential of canonical p53 targets by p53(A347D) within an endogenous context. To assess the induction of downstream targets we treated cells varying in p53 status with nutlin-3a, a small molecule MDM2 antagonist that disrupts the p53-MDM2 complex and stabilizes p53 (35). We found that p53AD/AD U2OS cells were completely unable to increase expression of MDM2 and p21 proteins, while heterozygous p53+/AD U2OS cells (Fig. 1D and Supplementary Fig. S1D) and patient fibroblasts (Fig. 1E) revealed partially attenuated expression of these two proteins compared to wild-type p53-bearing cells as demonstrated by immunoblot analysis. Furthermore, p53(A347D) in p53AD/AD U2OS cells appeared to be hyper-stable, presumably as a result of the inability of p53(A347D) to activate its negative regulator MDM2. The observed hyper-stability of p53(A347D) was verified kinetically via a cycloheximide (CHX) chase assay (Fig. 1E and Supplementary Fig. S1E). Although we have previously reported that dimeric p53 is preferentially bound and degraded by MDM2, such enhanced degradation requires the presence of sufficient quantities of MDM2 (19). Quantitative RT-PCR similarly revealed the markedly attenuated expression of CDKN1A and MDM2 as well as additional canonical p53 target TIGAR mRNAs even upon nutlin-3a treatment in p53AD/AD U2OS cells, whereas p53+/AD U2OS cells (Supplementary Fig. S2A) and LFS fibroblasts (Supplementary Fig. S2B) exhibited intermediate transcriptional activity between p53+/+ and p53mt/mt cells. To further validate if p53(A347D) also loses its transactivation potential in LFS patients’ osteoblasts, the cell-of-origin of osteosarcoma, we generated WT and LFS-derived induced pluripotent stem cells (iPSCs) from L53-WT and L53-M1 fibroblasts, respectively (see Methods). We then differentiated these iPSCs to their corresponding mesenchymal stem cells (MSCs) and then to their osteoblast counterparts (see Methods). In concordance with p53+/AD U2OS cells, LFS mesenchymal stem cells also demonstrated partially attenuated transcription of MDM2 and CDNK1A (Supplementary Fig. S2C). Additionally, as p53 is known to bind DNA as a tetramer, we sought to assess whether the A347D mutation would disrupt its promoter binding ability. We performed quantitative chromatin immunoprecipitation (ChIP-qPCR) to assess the occupancy of p53(A347D) at select p53 targets, and, as expected, found that p53(A347D) is unable to bind promoter sequences of CDNK1A and PUMA even when treated with etoposide (Supplementary Fig. S2D).
In order to gain insight into the global transcriptional profiles of dimeric mutant-p53 bearing cells, we performed bulk RNA-sequencing (RNA-seq) of U2OS cells varying in p53 status treated with DMSO or etoposide as well as of LFS and WT osteoblasts (Supplementary Data S1–S3). Consistent with prior observations, RNA-seq analysis revealed significantly attenuated expression of high-confidence p53 target genes overall in p53AD/AD U2OS cells even after etoposide treatment, while p53+/AD U2OS cells retained partial ability to induce p53 targets upon activation (Fig. 2A) (28). Not surprisingly, p53 target gene expression was found to be higher overall in wild-type U2OS cells compared to p53AD/AD cells, both basally and following etoposide treatment (Supplementary Fig. S3A). Gene set enrichment analysis (GSEA) confirmed that the p53 signaling pathway is significantly enriched in U2OS p53+/+ and wild-type osteoblasts but not mutants (Supplementary Fig. S3B and S3C). Collectively, these results demonstrate the transcriptional impairment, a decidedly LOF phenotype, and the associated hyper-stability of p53(A347D).
Figure 2. Dimer-forming p53(A347D) is unable to transactivate canonical p53 target genes.
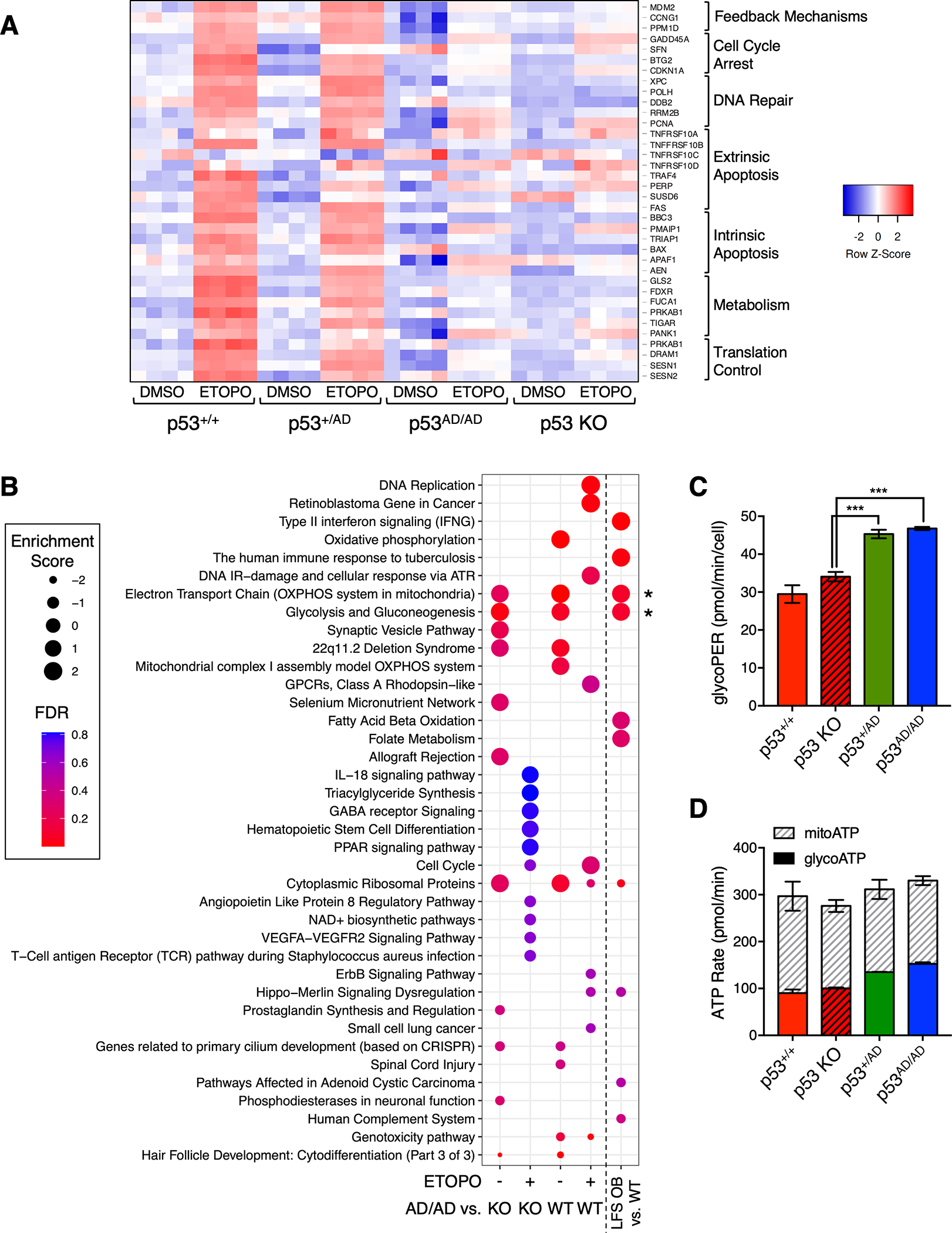
(A). Heatmap depicts differential gene expression of canonical p53 target genes in U2OS p53+/+, p53+/AD, p53AD/AD, or p53 KO cells that were subjected to RNA-sequencing (RNA-seq) following 24 h treatment with either DMSO or 20 μM etoposide (ETOPO). Columns represent individual technical replicates within each condition. Red indicates higher relative expression. Genes at right were assembled into different biological processes.
(B) Dot plot shows pathways positively or negatively enriched in U2OS p53AD/AD cells relative to wild-type p53 and p53 KO cells after DMSO or 20 μM etoposide (ETOPO) treatment and in LFS osteoblasts (OBs) relative to wild-type OBs. Relative dot size represents enrichment score, and color indicates false discovery rate according to legend.
(C) Basal glycolytic rate in p53+/+, p53 KO, p53+/AD, p53AD/AD U2OS cells seeded at equivalent densities was measured by the Seahorse XFp Analyzer. Glycolytic proton efflux rate (pmol/min) serves as a measure of glycolytic rate and was assessed in real-time. Data represent the mean ± SEM of three biologically independent experiments. (n=3).
(D) Glycolytic and mitochondrial ATP production rates (pmol/min) in p53+/+, p53 KO, p53+/AD, p53AD/AD U2OS cells were measured using the Seahorse XF Real-Time ATP Rate Assay kit. Data represent the mean ± SEM of two biologically independent experiments. (n=2).
Dimeric mutant p53 cells possess an altered transcriptional and metabolic profile
RNA-seq revealed not only that p53(A347D) has lost canonical transcriptional activity but also that the transcriptomes of mutant p53-bearing U2OS cells and LFS osteoblasts are decidedly altered, indicated by the presence of significantly upregulated and downregulated genes in such cells relative to their wild-type counterparts (Supplementary Fig. S4A–S4E). Pathway enrichment analysis via WebGestalt further demonstrated coordinate changes in biological processes within p53AD/AD U2OS cells under basal conditions and after etoposide treatment relative to both wild-type p53 and p53 KO U2OS cells (Fig. 2B). Pathway alterations were similarly observed in p53+/AD LFS osteoblasts compared to wild-type (Fig. 2B). Interestingly, fewer highly enriched pathways were observed in U2OS p53AD/AD cells relative to p53 KO cells after etoposide treatment than in the p53AD/AD vs. p53 KO etoposide-treated gene set, suggesting that transcriptional response to etoposide treatment may not be significantly different between mutant p53 and p53 KO cells (Fig. 2B). Amidst the myriad altered processes, glycolysis and electron transport chain pathways were strikingly found to be consistently enriched in both mutant p53 U2OS cells and osteoblasts under basal conditions. This concordance not only strongly implicates the potential GOF role of p53(A347D) in driving metabolic reprogramming, as the same enrichment was detected in two completely different cell types, but also suggests the validity of CRISPR-generated U2OS p53(A347D) cells as a model for what occurs in a patient-derived system.
Given that a significant proportion of genes involved in glycolysis were observed to be upregulated in p53AD/AD as compared to wild-type p53 cells (Supplementary Fig. S4F) and that gain-of-function activities of mutant p53 in driving increased glucose uptake and glycolysis have been demonstrated in the recent literature (although never with OD mutants) we sought to validate our findings (36,37). Metabolic flux analysis of U2OS cells varying in p53 status revealed that cellular glycolytic flux, as measured by proton efflux rate, was significantly elevated in both p53+/AD cells and p53AD/AD cells as compared to wild-type p53 and p53 KO cells (Fig. 2C). Additionally, ATP generated from glycolysis was highest in both p53+/AD cells and p53AD/AD cells whereas ATP generated in mitochondria remained similar across the cell lines evaluated (Fig. 2D). Overall, these data reveal a potential gain-of-function activity of p53(A347D) in eliciting a glycolytic switch.
p53(A347D) exhibits a unique chromatin association signature
In order to obtain a more global picture of mutant dimeric p53 occupancy across the genomic landscape and assess whether p53(A347D) may bind novel sequences, we performed chromatin immunoprecipitation of endogenously expressed p53 followed by next-generation sequencing (ChIP-seq) in both U2OS cells (Supplementary Data S4 and S5) and LFS osteoblasts (Supplementary Data S6). ChIP-seq analysis revealed significant genome-wide overlap in binding (389/520) between p53+/+ and p53+/AD cells. This suggested that p53(A347D) may not have dominant negative activity in preventing wild-type p53 from binding its transcriptional targets. Strikingly, p53(A347D) in p53AD/AD cells exhibited a starkly different binding landscape relative to wild-type p53 with only 9/571 peaks in common and 105/571 unique peaks, thereby indicating that p53(A347D) has not only lost wild-type DNA binding ability but also gained the capacity to associate with novel genomic loci (Fig. 3A). Corroborating this finding, p53 in LFS osteoblasts retained the ability to associate with regions bound by both wild-type p53 and dimeric mutant p53 in U2OS cells (Fig. 3B).
Figure 3. p53(A347D) exhibits a unique chromatin binding signature.
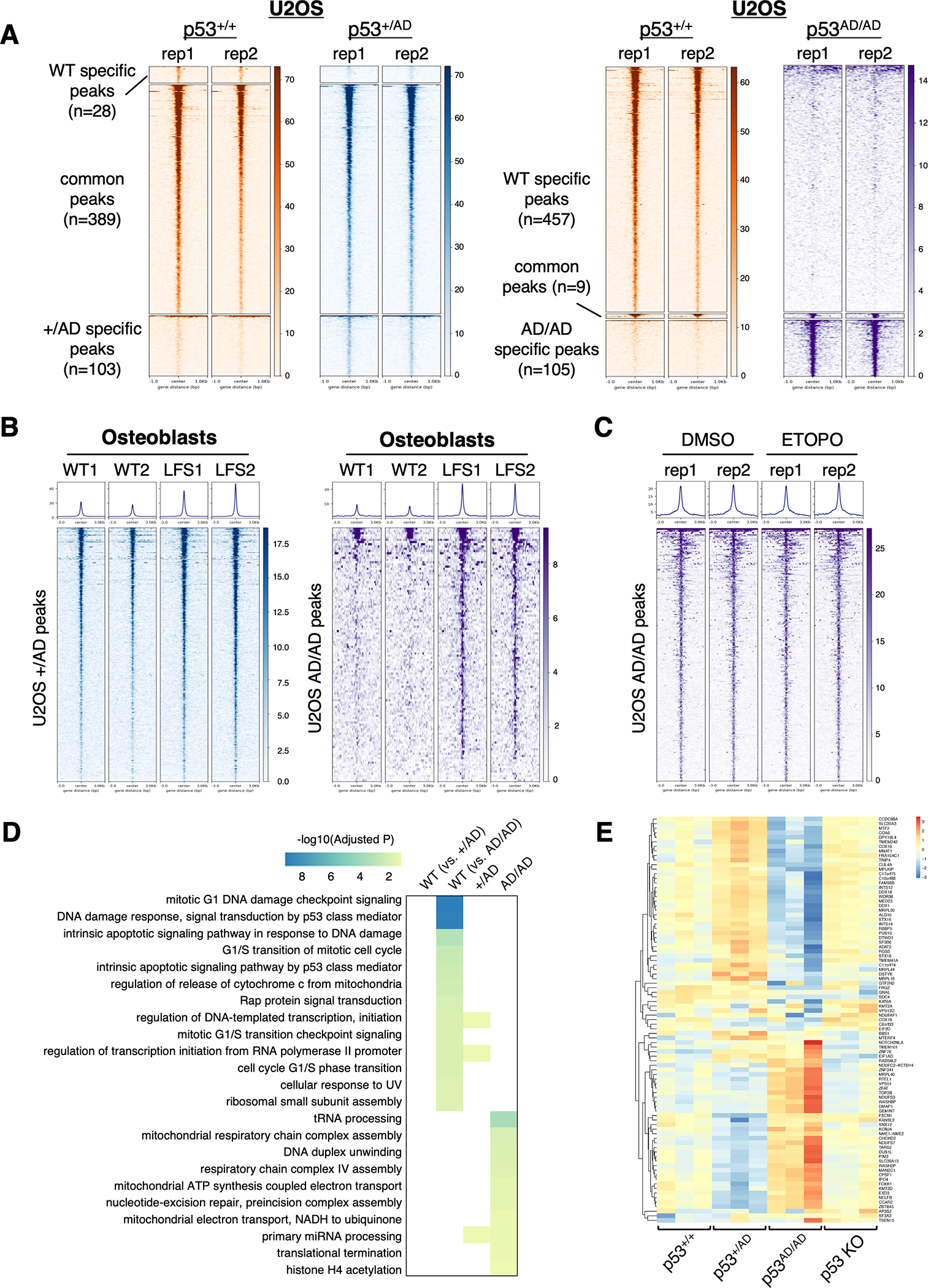
ChIP-Seq analysis was performed on either pooled clones of p53+/+, p53+/AD, and p53AD/AD U2OS cells as described in Methods with two biological replicates (rep1 and rep2) or two clones each of p53+/+ or p53+/AD osteoblasts (WT1 and WT2 or LFS1 and LFS2). U2OS p53 KO cells were used as a negative control in experiments with U2OS cells.
(A) Heatmaps depict p53 binding to 1-kb genomic loci surrounding identified ChIP-seq peaks in p53+/+ vs. p53+/AD U2OS cells (left) and p53+/+ vs. p53AD/AD cells (right) and are grouped by WT p53-specific peaks, common peaks, and either p53+/AD or p53AD/AD specific peaks.
(B) Heatmaps depict p53 binding to 3-kb genomic loci surrounding peaks associated with either p53+/AD (left) or p53AD/AD (right) U2OS cells in either WT or LFS osteoblasts.
(C) Heatmaps demonstrate dimeric mutant p53 binding to 3-kb genomic loci surrounding peaks identified p53AD/AD U2OS cells upon treatment with DMSO or 20 μM etoposide (ETOPO) for 24 h.
(D) Heatmap depicts GO biological processes highly enriched in p53+/+ cells relative to both p53+/AD and p53AD/AD U2OS cells, p53+/AD cells, and p53AD/AD cells.
(E) Heatmap illustrates expression of genes in p53 whose loci are identified to be bound by dimeric mutant p53 in p53+/+. p53 KO, p53+/AD, and p53AD/AD U2OS cells clustered by upregulated and downregulated genes.
The genomic distribution of p53 binding sites in p53AD/AD cells was found to be proportionally distinct from both p53+/+ and p53+/AD cells with over three-quarters (90/117) detected at promoters, defined as regions within ±1000 bp of transcription start sites, as compared to roughly six percent (30/468) in cells bearing only wild-type p53 (Supplementary Fig. S5A). De novo motif enrichment analysis revealed that, as expected, p53 motifs were the top enriched in p53+/+ and p53+/AD cells, suggesting that p53 binding at canonical elements is permitted even in the presence of mutant p53 (Supplementary Fig. S5B). However, only cells expressing p53(A347D) exhibited enrichment of ETS transcription factor family motifs, albeit at much lower confidence than p53 motifs in wild-type p53 cells, which may support a potential chromatin-binding GOF by p53(A347D) (Supplementary Fig. S5B). While these data are intriguing and seemingly in concordance with prior studies demonstrating that certain cancer-related mutant p53 proteins associate with ETS sites and ETS family members(11,38,39), such canonical p53 mutant proteins harbor mutations within the DNA binding domain, whereas the p53(A347D) mutant has a wild-type DNA binding domain.
In fact, when ectopically expressed in H1299 cells, only p53(R175H) and not p53(A347D) could be co-immunoprecipitated with ETS1 (Supplementary Fig. S5C) or ETS2 (Supplementary Fig. S5D), suggesting that any association of p53(A347D) with ETS motifs may be through direct binding or at the very least independent of ETS proteins. A related study by Gencel-Augusto et al. has identified cooperative binding of p53(A347D) and PPARα at PPAR response elements to promote a PPAR transcriptional program in mice harboring the same OD mutation (40). Although we were unable to detect PPAR motif enrichment in U2OS cells bearing mutant p53, we found that p53(A347D) could be co-immunoprecipitated with PPARα to a greater degree than could wild-type p53 (Supplementary Fig. S5E). Furthermore, in a similar assay, p53(A347D) was found to associate with PPARγ although this binding interaction necessitated the presence of PPARα (Supplementary Fig. S5F).
We next asked whether etoposide treatment might either increase or alter the distribution of genomic binding of dimeric mutant p53. While, as expected, wild-type p53 chromatin association significantly increased following etoposide treatment (Supplementary Fig. S5G), binding to mutant-specific peaks did not increase after etoposide in U2OS p53AD/AD cells (Fig. 3C). We then performed functional enrichment analysis on genomic regions occupied by wild-type and/or mutant p53 (Fig. 3D). Those ontologies that were highly enriched in p53+/+ vs. p53AD/AD cells were not enriched in p53+/AD cells. This indicated that p53 in the heterozygous mutants may retain wild-type DNA binding ability. However, several gene ontologies, including those related to mitochondrial respiration, were identified to be highly enriched in the p53AD/AD cells exclusively, which was in concordance with pathway analysis of differentially expressed genes in U2OS p53AD/AD cells (Fig. 3D). On the other hand, glycolysis was not seen to be enriched in the p53AD/AD ChIP-seq gene set, which suggests that the glycolytic switch may not be directly transcriptionally induced by dimeric mutant p53. Furthermore, by intersecting RNA-seq expression data with ChIP-seq peaks, we were able to identify significant differential regulation of genes bound uniquely by p53(A347D) relative to cells with either wild-type p53 or no p53 (Fig. 3E). Gene set enrichment analysis of osteoblast expression data also revealed that ChIP targets identified in p53+/AD and p53AD/AD U2OS cells were enriched in LFS osteoblasts compared to wild-type osteoblasts, ultimately indicating that patient-derived cells may express transcriptional targets of p53(A347D) (Supplementary Fig. S5H). Consistency between two cellular contexts not only strengthens the case for novel chromatin binding by p53(A347D) but also suggests the relevance of studying LFS-derived mutant p53 with either CRISPR-generated or patient-derived cells. While there are far fewer genes potentially regulated by p53(A347D) than wild-type p53, these results implicate a neomorphic role of dimeric mutant p53 in transcriptional regulation that may result in mitochondrial alterations.
p53(A347D) promotes mitochondrial network aberration
Since the results from both RNA-seq and ChIP-seq analyses revealed that the expression of mitochondrial electron transport proteins may be altered in cells expressing p53(A347D), we used confocal microscopy to assess mitochondrial morphology in the U2OS series of cells by staining with MitoTracker Red. This revealed dramatic mitochondrial network aberration in p53AD/AD U2OS cells characterized by greater mitochondrial area and greater percentage of cells with altered mitochondria when compared to p53+/+, p53 KO, or p53+/AD cells (Fig. 4A and 4B and Supplementary Fig. S6A and S6B). Knockdown of mutant p53 significantly reduced the aberrant morphology of mitochondria. The morphological change we observed was similar to that seen in cells with wild-type p53 that were treated with etoposide (Fig. 4C and Supplementary Fig. S6A). Since p53+/AD cells did not exhibit altered mitochondrial morphology, this suggests that wild-type p53 may be haplosufficient in maintaining normal mitochondrial networks. These results implicate an extra-nuclear function of p53(A347D) in potentially inducing mitochondrial stress. While p53 knockdown was able to revert the stressed mitochondrial phenotype basally in p53AD/AD cells, etoposide-mediated changes in mitochondrial morphology were unable to be so rescued (Supplementary Fig. S6A). It remained possible that the altered mitochondrial morphology seen in the untreated p53AD/AD cells evolved during the generation of these CRISPR cells as part of the selection process. Thus, we sought to confirm this key observation using an orthogonal method: introducing mutant p53 into U2OS KO cells by retroviral expression. Importantly, the ectopically expressed mutant p53(A347D) phenocopied the aberrant mitochondrial morphology observed in U2OS p53AD/AD cells (Fig. 4D).
Figure 4. Dimeric mutant p53 promotes mitochondrial network aberration.
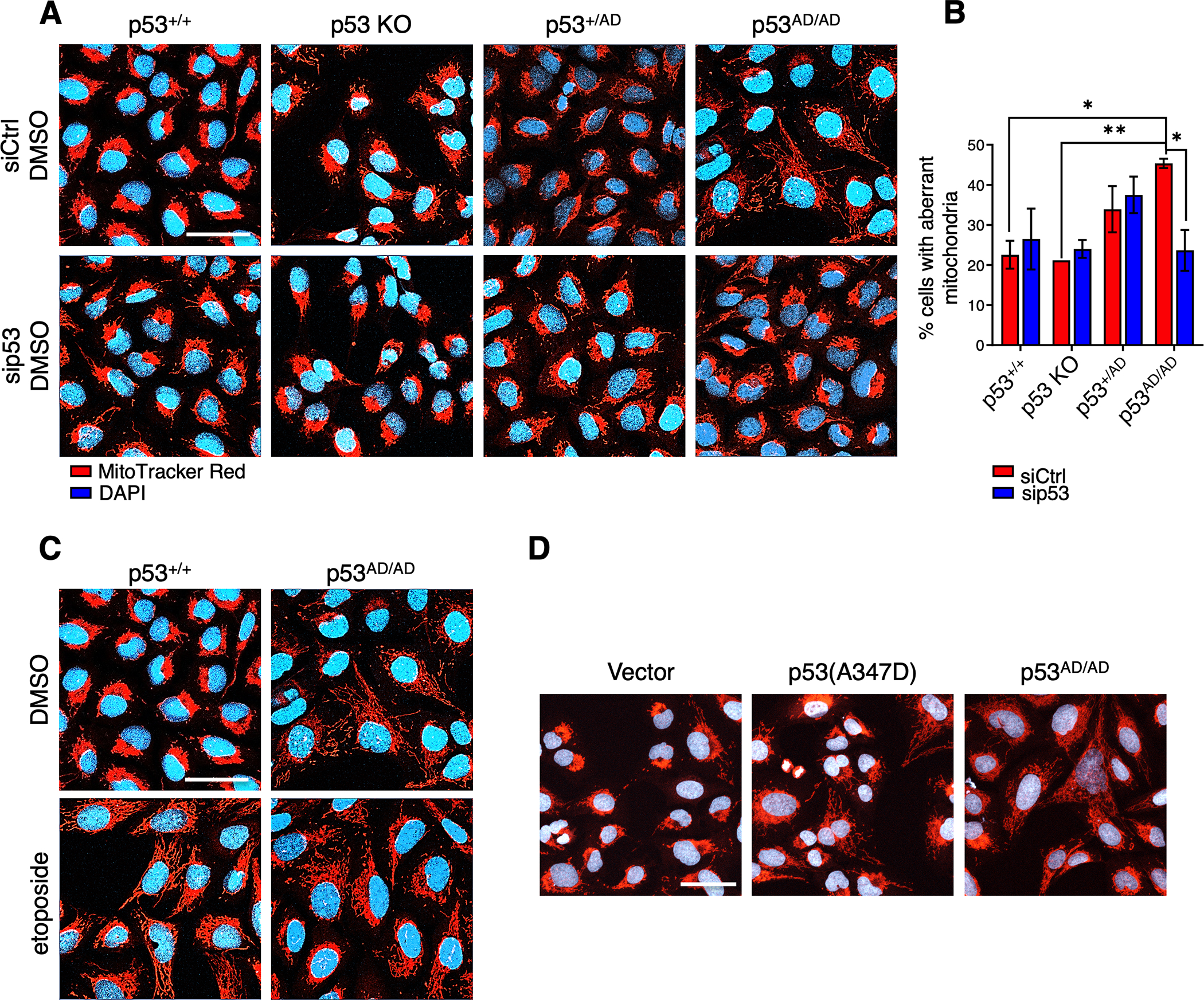
(A) Representative images of fixed p53+/+, p53 KO, p53+/AD, and p53AD/AD U2OS cells transfected with 25 nM of either a non-targeting siRNA pool (siCtrl) or siRNA pool against p53 (sip53) for 48 h and treated with DMSO for 24h. Mitochondria were visualized by MitoTracker Red staining (red). Nuclei were stained with DAPI (blue). (n=3). Scale bar, 50 μm.
(B) Relative mitochondrial aberrance was determined by a trained observer counting the number of cells with aberrant mitochondria and total number of cells within each field. Images were blinded and randomized prior to counting. Bars represent the mean ± SEM of three biologically independent experiments. (n=3, 7–12 images captured per group).
Statistical significance was assessed by two-tailed t-test. *p<0.05, **p<0.01
(C) Representative images of p53+/+ and p53AD/AD U2OS cells treated with etoposide for 24 h after transfection with non-targeting siRNA for 24 h then stained with MitoTracker Red (red) and DAPI (blue). (n=3). Scale bar, 50 μm.
(D) Representative images of U2OS p53 KO cells stably expressing either vector or p53(A347D) and U2OS p53AD/AD cells stained with MitoTracker Red (red) and DAPI (blue). Scale bar, 50 μm.
Mutant p53 cells undergo transcription-independent apoptosis under genotoxic stress
Mitochondrial fragmentation reflects a shift in the dynamic mitochondrial equilibrium towards mitochondrial fission and is ultimately associated with alterations in metabolism and mitochondrial dysfunction (41–43). Although a causal link between mitochondrial fragmentation and apoptosis is not yet clear, evidence suggests that a shift towards mitochondrial fission sensitizes cells to apoptosis and vice versa (44). Accordingly, we sought to assess whether dimeric mutant p53-dependent mitochondrial network changes may drive sensitization to apoptosis both basally and under stress. In addition to its well-characterized role as an activator of transcription, p53 possesses transactivation-independent, cytosolic effects, including the inhibition of autophagy and induction of apoptosis (45). Cytoplasmic p53 rapidly localizes to mitochondria under cell death-inducing conditions to drive mitochondrial outer membrane permeabilization and subsequent cytochrome c release independent of its transcriptional activity, which is supported by the observation that transactivation-deficient mutant p53 retains the ability to induce apoptosis (46–49). Along with our findings on aberrant mitochondrial morphology, we hypothesized that mutant p53(A347D)-bearing cells may similarly retain pro-apoptotic function(s) and thereby be more vulnerable to genotoxic stress. Indeed, p53AD/AD U2OS cells displayed markedly attenuated viability following treatment with etoposide as compared to p53+/+, p53 KO, and p53+/AD cells (Fig. 5A and Supplementary Fig. S7A). Decreased cell viability upon etoposide treatment was rescuable by inhibiting caspase 3 with zVAD-FMK confirming that p53AD/AD cells preferentially undergo apoptosis. Further, the viability of cells with ectopically expressed p53(A347D) was also significantly decreased upon etoposide treatment (Fig. 5B) indicating that the increased death seen upon treatment with this chemotherapeutic agent was not an indirect outcome of selecting the mutant p53 expressing cells. Interestingly as well, expression of other dimer-forming mutants including E343K, L344A, A347S (13) rendered cells somewhat more sensitive to etoposide treatment, while the monomeric A347P mutation (13) did not elicit such sensitivity (Fig. 5B). Enhanced PARP and caspase 3 cleavage were detected in p53AD/AD cells upon etoposide treatment, corroborating the finding that etoposide induces apoptosis (Fig. 5C). Additionally, MSCs derived from LFS patient iPSCs expressing heterozygous p53(A347D) mutant protein exhibited increased PARP and caspase-3 cleavage following etoposide treatment (Fig. 5D). While p53+/AD U2OS cells were similarly sensitive to etoposide as both wild-type p53 and p53 KO cells, cell cycle profiling revealed that both p53+/AD and p53AD/AD U2OS cells arrested to a greater degree after etoposide treatment (Supplementary Fig. S7B). Notably, our results indicating severe transcriptional impairment of mutant p53(A347D) suggested that cell death caused by etoposide was independent of transcription. This was confirmed when we treated cells with actinomycin D and found that the mutant p53(A347D) cells still underwent cell death upon etoposide treatment (Fig. 5E). Verifying that etoposide sensitivity was not due to an off-target effect, we observed propensity for apoptotic cell death in p53AD/AD cells after treatment with daunorubicin, another inhibitor of topoisomerase II (Supplementary Fig. S7C).
Figure 5. Mutant p53 cells preferentially undergo apoptosis under genotoxic stress.
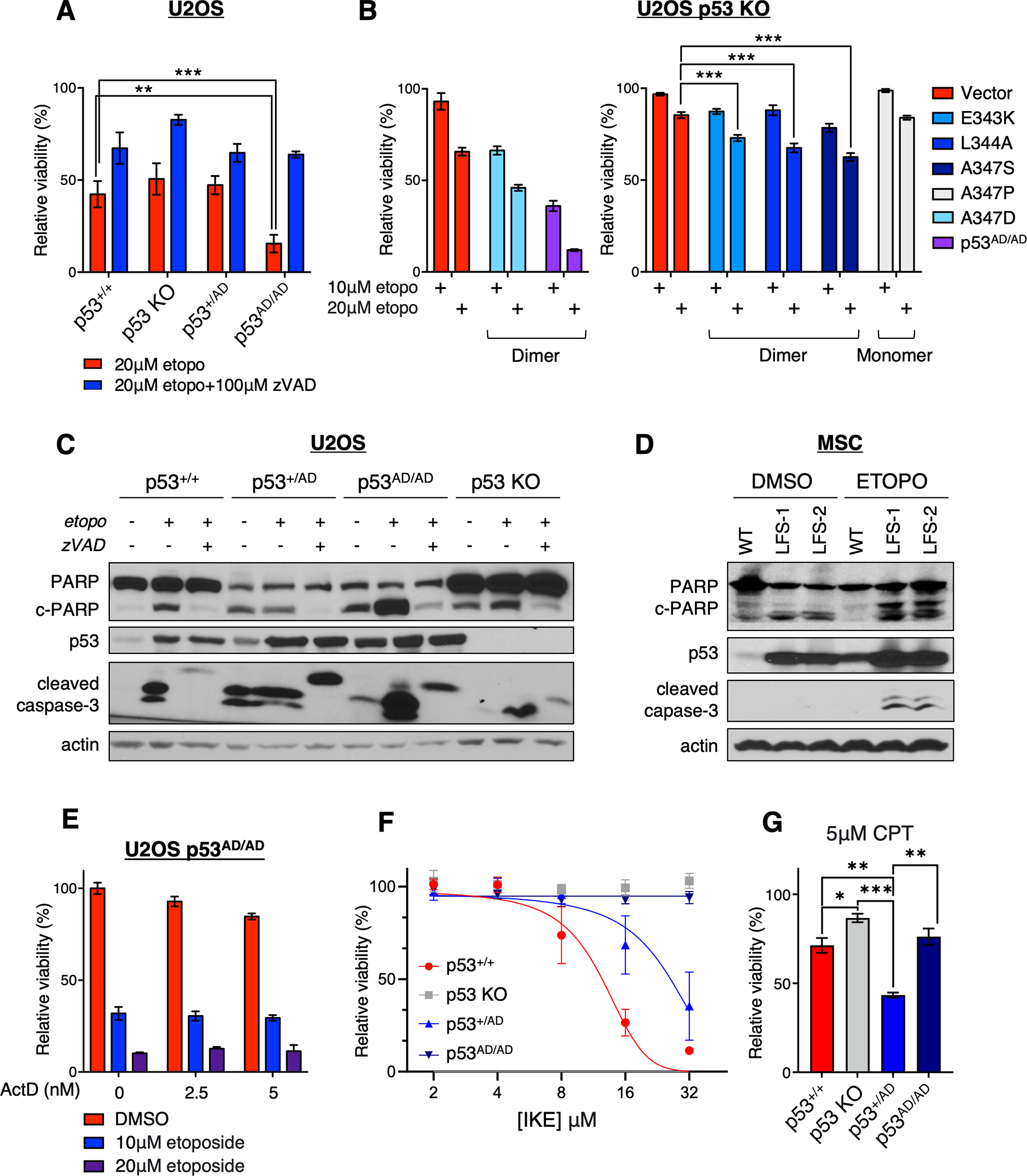
(A) Viability of indicated U2OS cells treated with 20 μM etoposide (etopo) or 20 μM etoposide + 100 μM zVAD-FMK (zVAD) as assessed by neutral red uptake and normalized to the DMSO control. Data represent mean ± SEM of four biologically independent experiments each with four technical replicates.
(B) Viability of U2OS p53 KO cells stably expressing either vector or the following p53 mutations (E343K, L344A, A347S, A347D) and U2OS p53AD/AD cells treated with 10 μM or 20 μM etoposide (etopo) as assessed by neutral red uptake and normalized to the DMSO control. Viability experiment with vector, transduced p53(A347D), and p53AD/AD cells (left) performed separately. Data represent mean ± SEM of three biologically independent experiments each with two or three technical replicates.
(C) U2OS cells varying in p53 status (p53+/+. p53 KO, p53+/AD, and p53AD/AD) were treated with DMSO, 20 μM etoposide, or 20 μM etoposide + 100 μM zVAD-FMK (zVAD) for 48 h and subjected to immunoblotting with the indicated antibodies.
(D) Mesenchymal stem cells (MSC) derived from primary dermal fibroblasts through the protocol described in the Methods were treated with 20 μM etoposide for 24 h, harvested, and subjected to immunoblot analysis with the indicated antibodies.
(E) Viability of U2OS p53AD/AD cells treated with etoposide and/or actinomycin D (ActD) at indicated concentrations for 48 h as assessed by neutral red uptake and normalized to the DMSO control. Two biologically independent experiments were performed in triplicate, and representative data are shown as mean ± SD.
(F) Viability of U2OS cells varying in p53 status treated with increasing concentrations of IKE for 48 h as assessed by neutral red uptake and normalized to DMSO-treated cells. Data represent mean ± SEM of three biologically independent experiments each with at least three technical replicates.
(G) Viability of U2OS cells varying in p53 status treated with 5 μM camptothecin (CPT) for 48 h as assessed by neutral red uptake and normalized to the DMSO control. Data represent mean ± SEM of three biologically independent experiments each with three technical replicates.
Statistical significance was assessed by two-tailed t-test. *p<0.05, **p<0.01, ***p<0.001
We went on to assess whether the mutant p53(A347D) cells were also sensitive to other known agents that produce wild-type p53-dependent cell death. When we tested the impact of treatment with the ferroptosis inducer IKE (50), only cells with wild-type p53 (p53+/+ and p53+/AD) underwent ferroptosis under these conditions, which was confirmed via rescue with the ferroptosis inhibitor ferrostatin-1 (fer-1) (51) (Fig. 5F and Supplementary Fig. S7D). Further, treatment with the topoisomerase I inhibitor camptothecin did not kill mutant p53(A347D) cells (Fig. 5G)
Given the well-defined apoptogenic role cytoplasmic p53 plays at mitochondria and the hyperstability of p53(A347D), we posited that p53(A347D) drives basal levels of stress, which leads to enhanced apoptotic cell death after topoisomerase-II inhibition (46). In order to test this hypothesis, we assessed the association of p53 protein from the different U2OS allelic series with mitochondria. Cell fractionation analysis revealed that p53 protein from U2OS p53+/+ and p53+/AD cells was associated with the mitochondrial fraction to a much lesser extent than p53 protein from p53AD/AD cells, with p53+/AD cells exhibiting intermediate mitochondrial p53 levels (Fig. 6A). The elevated levels of dimeric mutant p53 at mitochondria may be due to the greater amounts of this hyper-stable protein or may reflect preferential localization or binding to mitochondria. The amount of the p53AD/AD variant associated with mitochondria was increased to an even greater extent when cells were treated with etoposide, suggesting that protein levels alone may not explain increased levels of mitochondrial mutant p53 (Fig. 6A and 6B).
Figure 6. p53(A347D) preferentially associates with mitochondria to drive increased cell death.
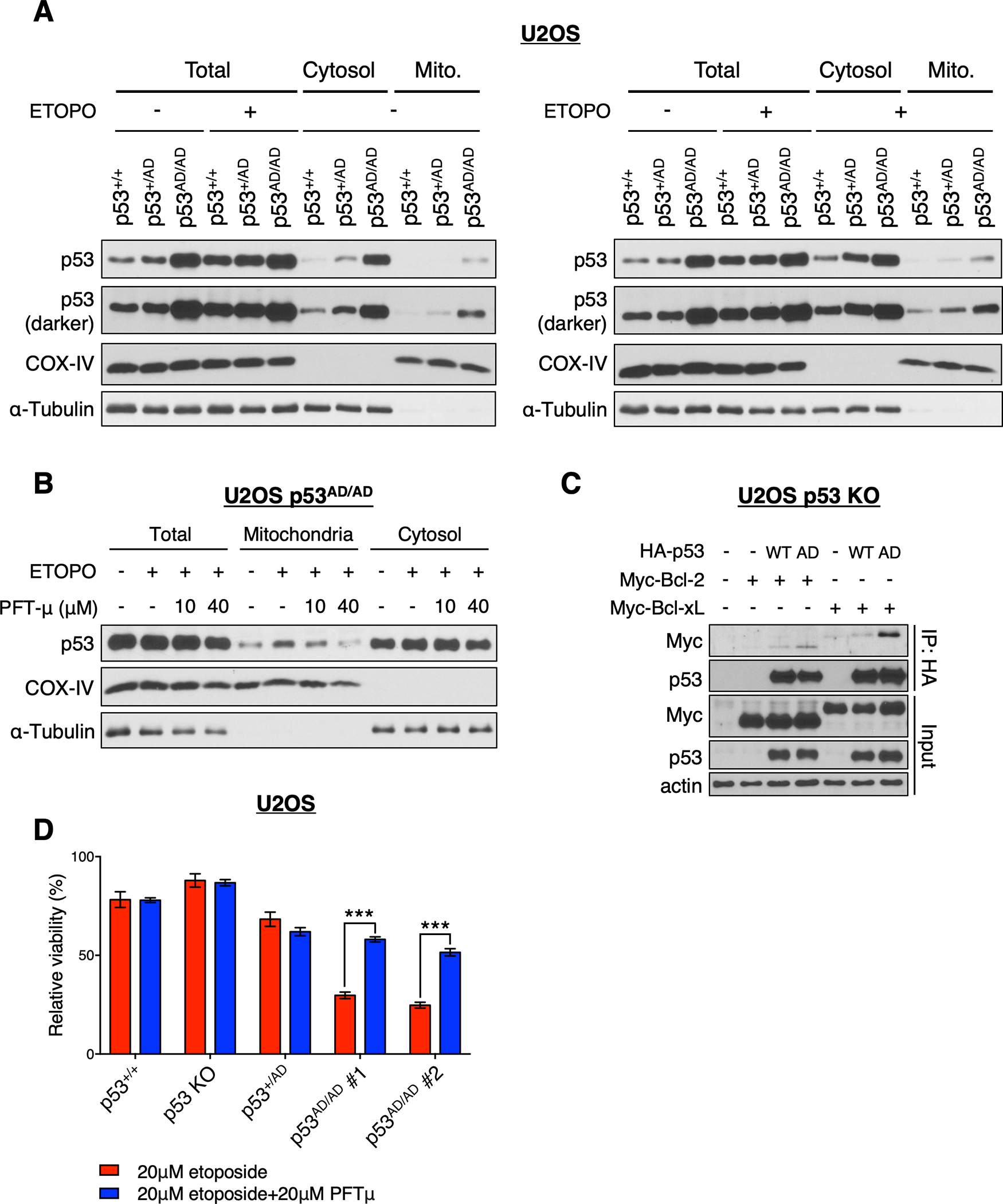
(A) Mitochondrial and cytosolic fractions were isolated from U2OS p53+/+, p53+/AD, and p53AD/AD cells treated with or without 10 μM etoposide for 24 h. Isolated fractions and total cell lysate were subjected to immunoblot analysis with indicated antibodies.
(B) Mitochondrial and cytosolic fractions were isolated from U2OS p53AD/AD cells treated with or without 10 μM etoposide in the presence or absence of pifithrin-μ (10 μM or 40 μM) for 20 h. Isolated fractions and total cell lysate were subjected to immunoblot analysis with indicated antibodies.
(C) U2OS p53 KO cells were co-transfected with the empty vector pcDNA3, HA-WT p53, or HA-p53(A347D) and myc-DDK-Bcl-2 or myc-DDK-Bcl-xL at indicated combinations and treated with 10 μM etoposide for 6 h. Protein lysates were immunoprecipitated with anti-HA agarose beads and subjected to immunoblot analysis with indicated antibodies.
(D) Viability of indicated U2OS cells treated with 20 μM etoposide or 20 μM etoposide + 20 μM pifithrin-μ (PFTμ) as assessed by neutral red uptake and normalized to the DMSO control. Data represent mean ± SEM of three biologically independent experiments each with four technical replicates.
We utilized pifithrin-μ (PFTμ), a small molecule inhibitor reported to disrupt p53 binding to mitochondrial anti-apoptotic proteins and prevent p53-mediated apoptosis (52). Remarkably, treatment with this compound dramatically reduced the level of mutant p53 within the mitochondrial fraction (Fig. 6B and Supplementary Fig. S8A and S8B). In accordance, we found that p53(A347D) could be found associated with the mitochondrial anti-apoptotic proteins, Bcl-2 and Bcl-xL to a significantly greater extent than does wild-type p53 (Fig. 6C). Relevantly, p53 association with Bcl-2 and Bcl-xL inhibits their ability to block the pro-apoptotic activities of BH3-only proteins (53), implicating the mechanism by which p53(A347D) promotes apoptosis. These observations prompted us to determine whether the interaction of mutant p53 with mitochondria might be involved in its ability to render cells hypersensitive to etoposide treatment. At an etoposide dose that did not affect significantly the viability of p53+/+ or p53 KO cells, co-treatment of PFTμ and etoposide dramatically rescued the decreased cell viability in p53AD/AD cells observed upon etoposide treatment alone (Fig. 6D and Supplementary Fig. S8C). Finally, etoposide treatment attenuated reduced (GSH) to oxidized (GSSG) glutathione ratios in p53(A347D)-bearing cells relative to both untreated cells and p53+/+ and p53 KO cells (Supplementary Fig. S8D). This suggests that etoposide induces oxidative stress to a greater extent in mutant p53-bearing cells, thereby corroborating the direct apoptogenic role of p53(A347D) at mitochondria.
Taken together, our findings indicate that dimeric mutant p53 may inhibit mitochondrial anti-apoptotic proteins to induce cell death to a greater extent than wild-type p53 under induction of specific apoptotic stimuli. Ultimately, we discovered that though mutant dimeric p53 has lost the ability to induce the expression of pro-apoptotic genes, it retains the license to kill through transactivation-independent effects at mitochondria, thereby driving elevated sensitivity to certain DNA-damaging agents.
Dimeric p53 mutants demonstrate enhanced tumorigenic capacity
We next examined functional consequences of the LFS-associated A347D mutation that might be more directly associated with certain aspects of tumorigenesis using different assays. First, we examined growth characteristics of the U2OS cell line series and found that p53AD/AD and p53+/AD U2OS cells exhibited higher in vitro growth rate in monolayer cell culture compared to wild-type p53 and p53 KO cells (Fig. 7A). We also assessed the ability of the U2OS allelic series of cells to engraft and proliferate in vivo using xenograft tumor assays and observed that while U2OS cells harboring wild-type p53 (p53+/+ or p53+/AD) did not form tumors over the time course of the experiments, both U2OS KO or p53AD/AD cells formed tumors at equivalent rates (Fig. 7B).
Figure 7. p53(A347D) mutants demonstrate enhanced tumorigenic capacity.
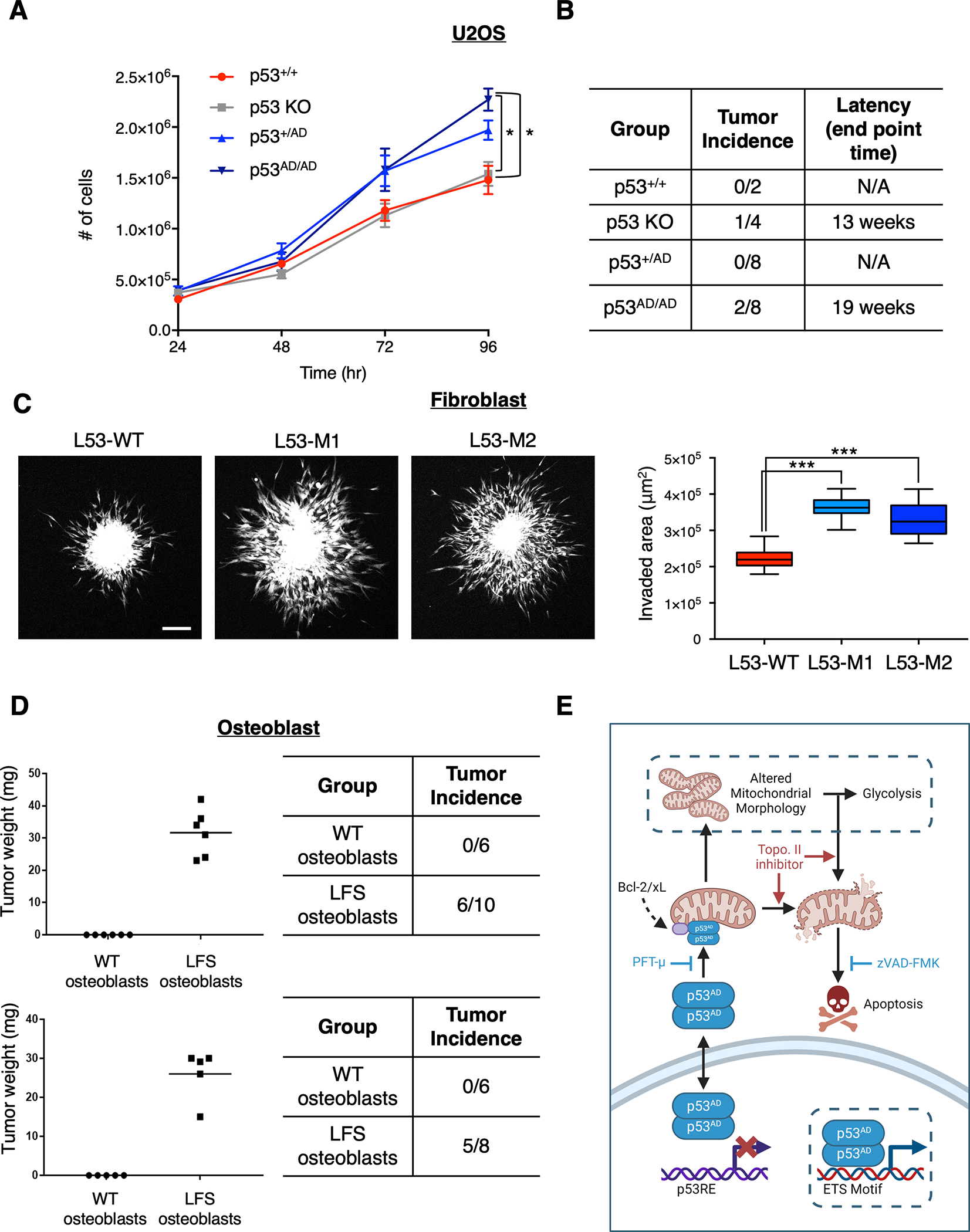
(A) The allelic series of U2OS cells were seeded at equivalent densities and counted at indicated timepoints. Data show relative cell number of indicated cell lines normalized to U2OS parental and represent mean ± SEM of three biologically independent experiments each with three technical replicates per condition.
(B) p53+/+, p53 KO, p53+/AD, and p53AD/AD U2OS cells were injected subcutaneously into the right and left dorsal flanks of NU/NU mice. Tumors were extracted at indicated end points post injection and weighed.
(C) Spheroids of primary dermal fibroblasts harboring either p53+/+ (L53-WT) or p53+/AD (L53-M1, L53-M2) were formed and implanted into a collagen matrix as described in Methods. Fluorescent microscopy images were taken 1 hour and 24 hours after implantation from which a representative confocal microscopy image (maximum projection) is shown (left) and invaded area was calculated by subtracting the area of initial spheroid from the ellipse covering the invaded area at endpoint of the experiments for each individual spheroid (right). The data is presented in a box plot depicting the median and second and third quartiles, with whiskers representing the data from 5% to 95%. Squares indicate mean values. Data was pooled from three biologically independent experiments and contains 24 spheroids for condition L53-WT, 28 spheroids for condition L53-M1, and 26 spheroids for condition L53-M2. Scale bar, 200 μm.
(D) WT and LFS iPSC-derived osteoblasts were injected subcutaneously into the right and left dorsal flanks of NU/NU mice. Tumors were extracted 1 month post injection and weighed (left). Table (right) demonstrates tumor incidence from two biologically independent experiments and indicates the number of injections per condition (n=2, 6–10 injections per group).
Statistical significance was assessed by two-tailed t-test. ***p<0.001, **p<0.01, *p<0.05
(E) Model depicting novel activities of mutant p53(A347D). Dimer-forming p53(A347D) has lost the ability to bind and transactivate canonical p53 target genes yet gains the ability to bind select genes with ETS motifs, which may lead to their activation or repression. p53(A347D) can translocate to mitochondria and interact with anti-apoptotic proteins Bcl-2 and Bcl-xL, leading to apoptosis following topoisomerase ii inhibition. Although p53(A347D) induces mitochondrial network aberrations, it is yet unclear whether altered mitochondrial morphology is a result of either direct mitochondrial interactions or the novel transcriptional activity of dimeric mutant p53, which are denoted with dotted borders. Altered mitochondrial morphology and function may cause a compensatory increase in glycolysis. Model diagram was created using Biorender.com
To examine possible early stages of the metastatic process, we used an invasion assay described previously by Guzman et al. (54), which involves generating cell spheroids under low adhesions conditions, then embedding them within a 3D biopolymer matrix, followed by monitoring the spontaneous invasion of cells into the matrix. Strikingly, LFS patient-derived p53+/AD fibroblasts exhibited significantly greater invasion into the surrounding matrix compared to wild-type p53 fibroblasts (Fig. 7C). As it had been predetermined that U2OS cells are not amenable to this assay, we did not pursue the invasion potential of the A347D mutation in our U2OS allelic series of cells.
Finally, we examined the ability of LFS iPSC-derived osteoblasts to engraft and grow in nude mice. We found that 6/10 and 5/8 LFS (p53+/AD) osteoblasts successfully engrafted and formed tumors whereas none of the WT p53 (p53+/+) osteoblasts produced palpable tumors in two independent experiments (Fig. 7D). Although monolayer growth suggests p53(A347D) GOF in driving enhanced proliferation, these results overall indicate that increased tumorigenicity (in a xenograft model) may be a loss of function phenotype. It is also possible that while p53(A347D) may promote greater proliferation than both wild-type p53 and p53 KO cells, the ability to induce mitochondrial stress and promote cell death imparts a selective disadvantage. Although the precise mechanism by which p53(A347D) may promote tumorigenesis through either GOF or LOF effects is not yet fully elucidated, it is clear from these data and the observation that individuals bearing the p53(A347D) in the germline suffer from cancers that cells bearing p53(A347D) demonstrate greater oncogenic potential and malignant properties over cells with wild-type p53. Ultimately, we describe p53(A347D) as a hyper-stable dimeric protein that has lost canonical transcriptional activity, which facilitates tumorigenesis, while gaining the ability to induce mitochondrial changes and apoptosis, thereby retaining certain tumor suppressive characteristics (Fig. 7E).
Discussion
We have characterized key activities of the LFS mutant p53(A347D) that preferentially forms dimers and not tetramers and is unable to form hetero-oligomers with wild-type p53 when co-expressed ectopically. Although we identified p53(A347D) to have significantly lost canonical transcriptional activity and wild-type DNA binding ability, selective p53-dependent transactivation may still occur, given our finding of a set of upregulated and downregulated genes whose loci are occupied by dimeric mutant p53. We also observed the enrichment of glycolysis and mitochondrial ETC-related transcripts in both U2OS p53AD/AD cells and LFS osteoblasts, which translated to elevated glycolysis in mutant p53-bearing U2OS cells. Furthermore, dimeric mutant p53 drives mitochondrial network aberrations, which implicates glycolytic reprogramming as a compensatory response. Surprisingly, we found that despite an impaired potential to activate many pro-apoptotic targets, p53(A347D) retains the ability to preferentially stimulate apoptosis through the direct mitochondrial cell death pathway in a transactivation-independent manner. Finally, we discovered that cells bearing dimeric mutant p53 are more proliferative, invasive, and tumorigenic than wild-type p53 cells.
A growing body of work has examined myriad aspects of the impact of mutant p53 gain-of-function activities on key, pro-tumorigenic processes such as metabolic reprogramming, invasion, and stemness as well as the mechanistic underpinnings and clinical implications of mutant p53 GOF (11,12,55). However, these insights have been near-exclusively associated with DNA-binding domain mutants, and little is known about the functions of tumor-associated p53 OD variants. To our knowledge, this is the first study to perform a deep dive into the molecular functions of oligomerization-defective mutant p53 that may ultimately possess therapeutic implications. Here, we studied the p53(A347D) mutation expressed in a diverse set of endogenous contexts (U2OS cells, patient-acquired primary fibroblasts from three siblings, one with wild-type p53, and patient-derived iPSC-p53-A347D osteoblasts) and utilized orthogonal experimental approaches to characterize the gain-of-function and loss-of-function activities of this dimeric p53 variant, which are summarized in Supplementary Tables S1 and S2. Given striking similarities between derivatives from LFS patient-derived cells and CRISPR-generated mutants, we posit the relevance of both systems to study other LFS-derived p53 variants.
Tetramerization of p53 has been well demonstrated to be necessary for efficient DNA binding, and certain mutations within the p53 OD were shown to abolish both tetramer formation and transcriptional activity to varying degrees (13,23). While prior findings have demonstrated that p53(A347D) may have lost selective transcriptional activity, our observations demonstrate more globally in a physiologically relevant context that endogenously expressed p53(A347D) has significantly impaired ability to activate a canonical p53 transcriptional program. This loss in transactivation potential most likely leads to the accumulation and hyperstability of p53(A347D) due to the inability of this p53 variant to induce its negative regulator, MDM2. Although we have demonstrated previously that dimeric p53, in both wild-type and tumor-derived variants, is preferentially bound and degraded by MDM2, such an enhanced degradation may only occur in contexts where MDM2 is present in adequate quantities (19). Ultimately, these results implicate impaired canonical transcriptional activity as a clear loss-of-function phenotype of p53(A347D) that may promote tumorigenesis. Yet, p53(A347D) gains a novel chromatin binding signature characterized by the enrichment of ETS-binding motifs at predicted mutant p53 binding sites. While this finding implies cooperativity with ETS family members, especially ETS1 and ETS2 which have previously been demonstrated to interact with p53 DNA binding domain mutants to drive transcription(38,39,56) the lack of such an interaction by p53(A347D) suggests that this dimer-forming variant may independently bind DNA, albeit less efficiently than wild-type p53. This is a distinct possibility given that p53(A347D) possesses a conformation that is not fully denatured, implicated by inaccessibility to PAb240, yet not completely well-folded. We cannot rule out the possibility that p53(A347D) either interacts with other ETS family members to regulate transcription or coordinates with PPAR proteins, although we did not observe evidence for the upregulation of a PPAR-dependent transcriptional program.
Transcriptomics revealed both glycolysis and mitochondrial respiration-related genes to be enriched in dimeric mutant p53-bearing cells yet only mitochondrial electron transport genes were predicted to be bound by p53(A347D). This seeming discrepancy suggests that mitochondrial changes may be regulated, in part, transcriptionally by mutant p53 while glycolytic reprogramming may be an indirect compensatory outcome of mitochondrial network alterations that occur. We additionally report that dimeric mutant p53 is found associated with mitochondria at higher levels both basally and after etoposide treatment, which may also contribute significantly to mitochondrial network changes. Although difficult to delineate whether these changes are mediated through gain of transcriptional or transactivation-independent function, it is clear that p53(A347D) drives mitochondrial aberrations that are characteristic of stress.
The regulation of mitochondrial fission and fusion dynamics are critical in sustaining mitochondrial health and function under metabolically or environmentally stressful conditions (43,57). Excessive mitochondrial fission leads to fragmentation and is both a sensitizer to and requisite step in apoptosis (57–59). Furthermore, mitochondrial fragmentation has been linked to glycolytic reprogramming in cancer cells and cancer-associated myofibroblasts (60,61). Wild-type p53 has been shown to regulate fission-fusion dynamics by transcriptionally inducing mitofusins (Mfn1 and Mfn2), which are essential for mitochondrial fusion, and by promoting the mitochondrial translocation of dynamin-related protein 1 (Drp1), which induces fission (46,62). Though we cannot definitively label mitochondrial aberrations driven by p53(A347D) as either mitochondrial fission or fusion, when placed into the context of apoptotic sensitivity and increased glycolysis, unopposed fission may be the more likely explanation for this phenotype. Further investigation into the precise mechanism by which p53(A347D) may tilt the mitochondrial dynamic balance either transcriptionally or through direct protein-protein interactions is warranted especially given that (a) we found elevated levels of mutant dimeric p53 associated with mitochondria, (b) wild-type p53 already functions in this capacity, and (c) dysregulated mitochondrial dynamics may be a targetable vulnerability of cancers (46,63). However, of note is that though p53AD/AD cells may be primed to die, they do not undergo apoptosis under basal conditions, implying that mitochondrial network alteration may be necessary, but not sufficient, for cell death.
Mitochondrial fission may additionally be tumor-supportive in certain contexts. Higher levels of mitochondrial fission have been associated with increased proliferative capacity and invasion in some cancer cell lines whereas mitochondrial fusion opposes growth (64). Mitochondrial fission may thereby serve as a potential mechanism of tumorigenesis in cells bearing p53(A347D). Furthermore, mitochondrial fragmentation has been linked to glycolytic reprogramming in cancer cells and cancer-associated myofibroblasts (60,61). Elevated glycolysis is a well demonstrated and frequent pro-tumorigenic alteration that supports the increased anabolic demands of rapidly proliferating cancers and helps maintain cellular redox potential (65). Therefore, we can hypothesize that mitochondrial fission and associated metabolic reprogramming may be mechanisms by which p53(A347D) mediates the observed enhanced proliferative, invasive, and tumorigenic capacities of cells bearing this mutation.
In addition to its pro-apoptotic functions as a transcription factor, p53 possesses transactivation-independent apoptogenic roles. Wild-type p53 rapidly translocates to mitochondria in response to stress signals where it can interact with and induce the oligomerization of both pro-apoptotic BH3-only proteins Bax and Bak and VDAC, leading to pore formation in the outer mitochondrial membrane and subsequent release of apoptogenic factors from mitochondria (66,67). Additionally, p53 physically interacts with and inhibits anti-apoptotic proteins Bcl-2, Bcl-xL, and Mcl-1, thereby releasing pro-apoptotic tBid, Bax, and Bak from the Bcl-2/Bcl-xL inhibitory complex and leading to the induction of apoptosis (46,53). The DNA-binding domain (DBD) of p53 was determined to be the binding interface for Bcl-2, and mutations within the DBD abolish this binding interaction (68,69). Consequently, several tumor-derived DBD missense mutants were found to be unable to promote cytochrome c release while a C-terminally deleted p53 protein preserved the ability to induce apoptosis (70,71). Yet, some recent evidence suggests that p53(R181E) preserves the ability to induce apoptotic cell death, suggesting mutation specificity in terms of apoptogenic potential (72). We demonstrate that p53(A347D), which has a wild-type DNA-binding domain, binds both Bcl-2 and Bcl-xL to an even greater extent than wild-type p53 does, suggesting that higher levels of the mutant protein basally and after induction alone do not account for mutant p53 cells’ propensity to apoptose. The direct mitochondrial apoptogenic role for p53(A347D) is supported by our finding that PFT-μ, which has been shown to attenuate p53 association with mitochondria and preclude p53 binding to Bcl-2 and Bcl-xL, both decreases the amount of p53 found in the mitochondrial fraction and rescues p53AD/AD cells from etoposide-mediated cell death (52,73). Not only p53(A347D) but other dimeric p53 mutants may similarly possess transactivation-independent pro-apoptotic activity, given that ectopically expressed dimer but not monomer mutants were able to sensitize cells to etoposide. This pro-apoptotic ability may not be a universal feature of dimeric p53, however, as the dimer-forming K351N mutant demonstrates defective mitochondrial trafficking in ovarian cancer cells and attenuated p53-dependent mitochondrial fragmentation in yeast (74). Furthermore, our observation that not all inducers of genotoxic stress can promote heightened cell death in mutant p53 cells indicates specificity in terms of what can induce p53(A347D) to kill. Ultimately, these data describe p53(A347D) as a mutant tumor suppressor that retains the license to kill but only under specific orders.
Germline mutations in the p53 OD have been found to have variable impacts on clinical outcome dependent on the quaternary structure the specific p53 mutant may form (18). While clinical data stratifying survival by monomeric and dimeric p53 status is limited, the association with cancer development is clear. Limited in vitro evidence has demonstrated increased proliferation and colony formation in both monomeric and dimeric p53 mutants expressed ectopically in H1299 cells relative to both WT p53 and tetrameric p53 variants (18). We expand on this finding to observe that endogenously expressed mutant p53 may facilitate augmented proliferation in 2D culture, invasion into an experimentally-derived extracellular environment in a 3D setting and tumor engraftment and growth in ectopic xenograft models. Despite the tumorigenic characteristics imparted by p53(A347D), this mutant still preserves apoptogenic ability, thereby suggesting that chemotherapies may retain efficacy in cancers bearing such a mutant. Although we suspect that the GOF activities of p53(A347D) may be applicable to other transcriptionally inert, dimeric p53 mutants that accumulate to intracellular high levels, whether other OD mutants demonstrate this phenotype remains to be shown. Ultimately, our results serve to delineate the LOF and GOF abilities of the dimer-forming p53(A347D) mutant, which not only hint at the complexities inherent in different p53 mutants but also implicates the efficacy of specific pro-apoptotic therapeutic strategies.
Methods
Chemicals and Antibodies
Dimethyl sulfoxide (Sigma), nutlin-3a (Sigma), etoposide (Sigma), zVAD-FMK (AdooQ Biosciences), daunorubicin (Sigma), camptothecin (Sigma), imidazole ketone erastin (Selleck Chemicals), ferrostatin-1 (Selleck Chem), actinomycin D (Calbiochem), and pifithrin-μ (Calbiochem) were utilized at indicated concentrations and times.
The following commercial primary antibodies, supplemented with 0.02% sodium azide, were used: anti-MDM2 (1:1000; Cell Signaling Technology, D1V2Z), anti-p53 (1:1000; Bio-Rad, VMA00019), anti-p53 (1:500; Santa Cruz, sc-126), anti-p53 PAb1620 (1:1000; EMD Millipore, OP33), anti-p53 PAb240 (1:500; Santa Cruz, sc-99), anti-β-actin (1:5000; Sigma, A2228), anti-p21 (1:2000; Cell Signaling Technology, 12D1), anti-Myc-tag (1:1000; Cell Signaling Technology, 9B11), anti-α-Tubulin (1:1000; Cell Signaling Technology, 2144S), anti-PARP (1:1000; Cell Signaling Technology, 9542S), anti-caspase 3 (1:1000; Cell Signaling Technology, 9662S), anti-COX-IV (1:1000; Cell Signaling Technology, 4844S), CD24 (BD Biosciences, 555427), CD73 (BD Biosciences, 560847), CD105 (eBioscience, 12–1057), OCT4 (Santa Cruz, sc-9081), and SSEA4 (R&D Systems, FAB1435P). In-house mAb anti-p53 (1:10; mixture of DO1/1801 hybridoma supernatant) was also used. Goat anti-mouse and goat anti-rabbit horseradish peroxidase conjugated secondary antibodies (1:5000; Sigma) were utilized. Mouse TruBlot ULTRA: anti-mouse Ig HRP (ROCKLAND, eB144) secondary antibody was also used.
Plasmids
The HA-p53 and FLAG-p53 plasmids were described previously (19). The following p53 mutations (L330A, E343K, L344A, A347S, A347P, A347D) were introduced into the HA-p53 construct as previously reported using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) (19). Myc-DDK-tagged BCL2 (RC204498), Myc-DDK-tagged BCL2L1 (RC201314), Myc-DDK-tagged PPARα (RC216237) and Myc-DDK-tagged PPARγ (RC201538) were purchased from OriGene Technologies. Myc-ETS1 and myc-ETS2 were kindly gifted by Dr. Luis A. Martinez.
Study Participants
This study was approved by the National Heart, Lung, and Blood Institute (NHLBI)–NIH Internal Review Board (ClinicalTrials.gov identifier NCT00406445 and NCT01143454) and the University of Utah Institutional Review Board (IRB #41211, Cancer Genetics Study). The study was conducted in accordance with the U.S. Common Rule and after the subjects provided written informed consent for their participation.
Cell Lines
U2OS osteosarcoma cell lines varying in p53 status (U2OS parental; U2OS CRISPR p53 KO, p53+/AD, p53AD/AD), primary dermal fibroblasts with either wild-type p53 (L53-WT) or p53+/AD (L53-M1, L53-M2), the p53-null lung carcinoma H1299 cell line, and HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C in a humidified 5% CO2 environment. Dermal fibroblasts were obtained from a LFS family in which two siblings harbored the germline p53 A347D mutation (L53-M1 and L53-M2) and a third sibling harbored wild-type p53 (L53-WT). Human fibroblasts (L53-M1) from one of the LFS siblings were prepared at the NHLBI–NIH, and the fibroblasts (L53-WT and L53-M2) from the two additional siblings were prepared at the University of Utah using standard protocols. Either wild-type p53 dermal fibroblasts or L53-M1 fibroblasts were utilized to generate iPSCs, which were then differentiated into MSCs and osteoblasts according to the methods provided below. Single clones were isolated, and patient fibroblast derivatives with mutant p53 are termed LFS-1, LFS-2, LFS-3, LFS-4, and LFS-5. Wild-type p53 derivatives are termed WT-1, WT-2, and WT-3. Cell lines were routinely assessed for Mycoplasma infection using the Lookout Mycoplasma PCR Detection Kit (Sigma).
Gene Editing by CRISPR-Cas9 in U2OS cells
U2OS p53(A347D) mutant cells were generated using a modification of CRISPR-Cas9 genome-editing technology (75). A 100-base-pair single-stranded oligodeoxynucleotide (ssODN) flanking the cut site and containing the mutation of interest was used as a donor template. U2OS cells (2 × 106) were electroporated with 200 pmol of ssODN (IDT), 100 pmol of EnGen Cas9 NLS (New England Biolabs), and 120 pmol of single-guide RNA (Synthego) using an Amaxa Nucleofector II device. Single-cell clones were selected via limiting dilution, and mutant clones were confirmed by genomic DNA and cDNA sequencing. Three single cell clones of p53+/AD cells, two single cell clones of p53AD/AD cells, and two single cell clones of p53 KO cells were pooled after confirming similar growth rates and are used for all experiments with U2OS cells unless otherwise indicated. U2OS p53 KO cells were generated as described previously (19).
Generating U2OS cells stably expressing OD variants by retroviral transduction
Mutant p53 constructs (E343K, L344A, A347S, A347P, A347D) were cloned into the pMXs-IRES-Puro retroviral expression vector (Cell Biolabs) and co-transfected with packaging plasmids into HEK293T cells to produce viruses. Each virus with either empty vector or mutant p53 constructs was infected into U2OS p53 KO cells and selected by 1 μg/mL of puromycin for at least one week.
Somatic Cell Reprogramming of Dermal Fibroblasts
Somatic cell reprogramming with Sendai viruses (SeVs) was described previously (76–79). Wild-type and LFS (L53-M1) fibroblasts were reprogrammed by transducing SeVs expressing Yamanaka four factors (OCT4, SOX2, KLF4, and MYC) using the CytoTune-iPS 2.0 reprogramming kit (Invitrogen). The reprogrammed iPSCs were maintained in hESC media (DMEM/F12 (Corning) containing 20% (vol/vol) KnockOut Serum Replacement (Invitrogen), β-mercaptoethanol, non-essential amino acids, L-glutamine, Penicillin-Streptomycin antibiotics, and 10 ng/ml bFGF) for four weeks. The iPSC clones with hESC morphology and positive expression of the hESC surface marker SSEA4 were picked and expanded in StemMACS iPSC-Brew XF medium (MiltenyBiotec). Expression of hESC pluripotency factor OCT4, hESC surface marker SSEA4, and alkaline phosphatase was confirmed by immunofluorescent staining (Supplementary Fig. S9A).
In Vitro Differentiation of LFS iPSCs to MSCs and then to Osteoblasts
In vitro differentiation of iPSCs to MSCs was performed by an SB-431542/7.5% CO2-based MSC differentiation method described previously (78,80). iPSCs were cultured in StemMACS iPSC-Brew XF medium supplemented with 10 μM TGFβ inhibitor SB-431542 and 7.5% CO2. After culturing for one month, the majority of cells acquired an MSC-like spindle-shaped morphology. The differentiated cells were then maintained and expanded in the modified MSC differentiation medium (KnockOut DMEM (Gibco) supplemented with 10% KnockOut serum replacement, β-mercaptoethanol, non-essential amino acids, penicillin-streptomycin, glutamine, 20 ng/ml EGF, and 20 ng/ml bFGF) under regular culture conditions. The expression of MSC markers CD73 and CD105 but not pluripotent stem cell marker CD24 was verified by immunofluorescent staining (Supplementary Fig. S9B). For osteogenic differentiation, 2 × 104 CD73+/CD105+/CD24− MSCs were plated and cultured in osteogenic differentiation medium (α-MEM supplemented with 10% FBS, 10 mM β-glycerol phosphate, 200 μM ascorbic acid, and 0.1 μM dexamethasone) for 15 days (76–79).
Immunoblot
Cells were lysed with TEB Lysis Buffer (10 mM Tris-HCl pH 7.5, 137 mM NaCl, 10% glycerol, and 1% Nonidet P-40 with 50 nM PMSF and inhibitor cocktail containing 100 μM Benzamidine, 300 μg/μL Leupeptin, 100 mg/mL Bacitracin, and 1 mg/mL a2-macroglobulin). Lysates were cleared by 10,000-rpm centrifugation at 4°C, and protein concentration measured using the Bradford assay (Bio-Rad). Equivalent lysates were separated by SDS-PAGE and electrotransferred onto nitrocellulose membranes (Bio-Rad) and blocked with PBS containing 0.1% Tween 20 (Sigma) and 5% non-fat dry milk for 30 min. The above antibodies were applied. Pierce ECL Western Blotting Substrate (Thermo-Fisher) or Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore) were used for protein visualization.
Immunoprecipitation
PAb240/PAb1620 IP and PPARα/PPARγ co-IP
U2OS p53 KO cells were transfected with HA-p53, HA-p53(A347D), HA-p53(R175H), or empty vector pcDNA3 using Lipofectamine 2000 (Thermo-Fisher) according to indicated combinations. Cells were lysed with IP buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% NP40, 10% glycerol, protease inhibitor). Protein lysates and Protein G Sepharose 4 Fast Flow beads (Cytiva) were incubated with anti-p53 PAb1620, PAb240, or DO-1 (Santa-Cruz) overnight at 4°C. Samples were washed with IP buffer three times and subjected to immunoblotting with anti-p53 (DO-1, Santa-Cruz), anti-Myc-tag, or anti-β-actin antibodies.
ETS1/ETS2 co-IP
H1299 cells were co-transfected with HA-p53, HA-p53(A347D), HA-p53(R175H) or the empty vector pcDNA3 and/or myc-ETS1 and myc-ETS2 using Lipofectamine 2000 (Thermo-Fisher) according to indicated combinations. Cells were lysed with IP buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.0 or 7.4), 1 mM EDTA, 1% NP40, 10% glycerol, protease inhibitor). Protein lysates were incubated with Pierce Anti-HA Agarose (Fisher Scientific) overnight at 4°C. Samples were washed with IP buffer three times and subjected to immunoblotting with anti-Myc-tag, anti-p53 (DO-1), or anti-β-actin antibodies.
Bcl-2/Bcl-xL co-IP
U2OS p53 KO cells were co-transfected with indicated plasmids and combinations using Lipofectamine 2000 (Thermo-Fisher) and treated with 10 μM for 6 h. Cells were lysed with IP buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1% NP40, 10% glycerol, protease inhibitor) and protein lysates were incubated with Pierce Anti-HA Agarose (Fisher Scientific) overnight at 4°C. Samples were washed with IP buffer three times and subjected to immunoblotting with anti-Myc-tag, anti-p53 (DO-1), or anti-β-actin antibodies.
Crosslinking Analysis
Following harvesting cells by scraping with PBS, cells were resuspended in Phosphate Buffer (PBS +10% glycerol +10 mM EDTA +0.5% NP-40, 0.1M KCl, protease inhibitor mixture), incubated on a rolling shaker at RT for 20 min in the presence or absence of glutaraldehyde at indicated concentrations (Sigma). Samples were then boiled with protein sample buffer containing SDS and subjected to immunoblot analysis as described previously.
Cycloheximide Chase
Cells were treated with 100 μg/mL cycloheximide (Sigma) and harvested at the indicated time points. Cell lysates were subjected to immunoblotting with the indicated antibodies. Signal intensity of p53 and actin was measured using ImageJ software (version 1.53K). p53 signal was normalized to the actin signal.
Quantitative Real-Time PCR
RNA extraction was performed using RNeasy Mini Kit (Qiagen) according to manufacturer protocol, and concentration measured using the NanoDrop 2000 (Thermo-Fisher). QuantiTect Reverse Transcription Kit (Qiagen) was used to generate cDNA. Power SYBR Green Master Mix (Thermo-Fisher) was mixed with cDNA and following real-time PCR primers. The RPL32 gene is used as a housekeeping control. Quantitative PCR was performed with the StepOnePlus Real-Time PCR System (Applied Biosystems) and quantified by double delta Ct analysis.
Quantitative Real-Time PCR
The following qRT-PCR primers were used: RPL32_F 5’-TTCCTGGTCCACAACGTCAAG-3’, RPL32_R 5’-TGTGAGCGATCTCGGCAC-3’, MDM2_F 5’-TTGGCGTGCCAAGCTTCTCT-3’, MDM2_R 5’-TACCTGAGTCCGATGATTCC-3’, CDKN1A_F 5’-GGCGGCAGACCAGCATGACA-3’, CDKN1A_R 5’-GCAGGGGGCGGCCAGGGTAT-3’, TIGAR_F 5’-CCAGGCTCGCAGCTTCA-3’, TIGAR_R 5’-GGTTTCGACTCCAGGTGCAA-3’.
RNA-Sequencing
Conditions and RNA Library Preparation
Bulk RNA was extracted from cells grown in 15 cm plates using Trizol (Invitrogen) and the RNeasy Mini Kit (Qiagen). Cells grown on separate 15 cm plates were considered biological replicates. RNA-sequencing was performed on RNA libraries prepared from two biologically independent experiments in U2OS cells. The first prep contained U2OS parental and U2OS CRISPR p53 KO, p53+/AD, and p53AD/AD cells treated with either DMSO or 20 μM etoposide for 24 h with four biological replicates per condition. The second prep contained U2OS parental and U2OS CRISPR p53 KO, and p53AD/AD cells treated with either DMSO or 20 μM etoposide for 24 h with two biological replicates per condition. Additionally, bulk RNA was prepared from 3 clones of LFS osteoblasts and 2 clones of wild-type osteoblasts grown in 10 cm plates. U2OS RNA samples from the first prep were sent in Trizol to BGI Genomics for further sample preparation, QC, and sequencing. U2OS RNA samples from the second prep and osteoblast RNA samples were sent to the CPRIT UTHealth Cancer Genomics Center for further library preparation, QC and sequencing.
Sequencing and Downstream Analysis
Resulting .fastq files were aligned to human genome reference hg38 using the kallisto pseudoalignment and quantification algorithm, as described by Bray et al. (81). Quantification of RNA read counts was performed at the gene level for downstream analysis. No outliers were observed with respect to total sequencing depth or gene coverage. In order to perform integrated analysis of expression data from two separate library preps performed in U2OS cells, ComBat-Seq was utilized to correct for batch effect (82). Using the DESeq2 analysis package (83), the combined read-counts matrix of all samples annotated by treatment and genotype was converted to a variance-stabilized gene expression matrix, and projected into two-dimensional visualization by Uniform MAnifold Projection (UMAP). On this projection, replicates of the same genotype and treatment condition can be observed to co-cluster, without concerning outliers. The major axis of separation between data points was found to be etoposide treatment, though within each treatment arm co-segregation of gene expression patterns is seen corresponding to genotype.
Within each treatment arm, pairwise differential gene expression was computed by DESeq2 negative binomial test for p53AD/AD vs. p53+/+ genotype, p53 KO vs. p53+/+ genotype, and p53AD/AD vs. p53 KO genotype, with adjustment of P-values for multiple hypothesis testing by Benjamini-Hochberg correction. Differential gene expression for LFS OB vs WT OB was also computed similarly. The full data tables ranked by log-fold-change are provided in the Supplementary Data files.
In order to highlight biological pathways in addition to individual genes of interest, pathway enrichment was performed by gene set enrichment analysis for each gene set described above, on the WebGestalt online platform (84) using the entire set of biological pathways defined in the Human Wikipathways database (85). Enriched pathways were ranked by enrichment score and weighted set cover was used to reduce redundancies. Pathways that were found to positively or negatively enriched in indicated datasets were plotted as a dot plot with relative dot size indicative of enrichment score and relative color representing the false-discovery rate. All analyses were performed in the R version 4.1.2 statistical computing environment. Additionally, gene set enrichment analyses for the set of differentially expressed genes in LFS osteoblasts vs. WT osteoblasts were performed using the GSEA software (86) using the p53 signaling pathway defined in the KEGG Pathway Database (87).
Quantitative Chromatin Immunoprecipitation
Chromatin immunoprecipitation experiments were performed as previously described (88). U2OS cells with either p53+/+ or p53AD/AD grown under indicated conditions were harvested and lysed with RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris–HCl pH 8.0, 5 mM EDTA, and protease inhibitors) and incubated on ice for 20 minutes. Lysates were sonicated to generate DNA fragments with an average length of ~500–1000 bp. Following sonication, samples were incubated overnight with indicated antibodies and protein A/G beads pre-blocked with 1 mg/mL BSA and 0.3 mg/mL salmon sperm DNA. The following antibodies were utilized for ChIP: anti-p53 (DO-1, Santa Cruz Biotechnology, sc-126) and rabbit IgG (Diagenode, #C15410206). DNA was purified using the QIAquick PCR Purification Kit (Qiagen). Quantitative ChIP was performed was performed on a StepOnePlus Real-Time PCR system (Thermo-Fisher) using Power SYBR Green Master Mix (Thermo-Fisher) versus genomic standard DNA and input DNA. ChIP primers designed with Primer Express (Applied Biosystems) were derived from the UCSC Human Genome Browser hg19 assembly and sequence specificity was confirmed with the UCSC Human Genome Browser in silico PCR tool. Primer sequences are provided below.
ChIP-qPCR Primer Sequences
The following ChIP-qPCR primers were utilized: CDKN1A_F 5’-GTGGCTCTGATTGGCTTTCTG-3’, CDKN1A_R 5’-CTGAAAACAGGCAGCCCAAG-3’, PUMA_F 5’-GAACGCCCGTCGGTCGGTCTGT-3’, PUMA_R 5’-CAAGTCAGGACTTGCAGGCGC-3’.
ChIP-Sequencing
Experimental Conditions
Two biologically independent ChIP-seq experiments were performed in U2OS cells. The first contained two biological replicates of U2OS p53+/+, p53+/AD, and p53AD/AD cells and one replicate of U2OS p53 KO cells as a control. The second experiment contained two biological replicates of U2OS p53+/+ and p53AD/AD cells treated with either DMSO or 20 μM etoposide for 24 h and one replicate of U2OS p53 KO cells treated under the same conditions as a control. Additionally, ChIP-seq was performed on two clones of wild-type osteoblasts (WT-1 and WT-2) and two clones of LFS osteoblasts (LFS-1 and LFS-2).
ChIP-seq Library Preparation
p53 ChIP-seq was performed using modified previous methods (78). Cells were fixed in 1% formaldehyde at room temperature for 10 min and quenched by 125 mM glycine. Collected cells were sonicated in ChIP lysis buffer (50mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.1% Na-Deoxycholate, 0.1% SDS, 1% Triton X-100, protease inhibitor) on ice. Anti-p53 antibody (Santa Cruz, sc-126) was added to lysate supernatants for overnight incubation at 4°C. The cell lysates and antibody mixtures were diluted by the same amount of ChIP dilution buffer (50mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100) and incubated with magnetic protein G Dynabeads (Thermo-Fisher Scientific, 10004D) for 3 h at 4°C. Immunoprecipitates on beads were washed with ChIP high salt buffer (20mM Tris-HCl pH 8.0, 500mM NaCl, 2mM EDTA, 0.1% SDS, 1% Triton X-100) and eluted in TE buffer. DNA isolated from the immunoprecipitates was used for library preparation and sequencing at the CPRIT UTHealth Cancer Genomics Center.
ChIP-sequencing data analyses were conducted at Galaxy (usegalaxy.org) following the tutorials provided by Galaxy Training!. In brief, .fastq files were trimmed by Trim Galore! and mapped to human reference genome (hg19) by Bowtie2. PCR duplicates were removed by SAMtools RmDup before MACS2 was used for calling peaks. Unique peaks were identified by bedtools intersect. Heatmaps were generated using deepTools (bamCoverage, computeMatrix and plotHeatmap). Peak annotation and motif prediction were performed using HOMER software (89). Identification of enriched biological processes (Fig. 3D) was performed using EnrichR. GSEA for Supplementary Fig. S3B and C was performed using default KEGG pathway gene sets with the metric for ranking genes as “Signal2Noise” and the enrichment statistic as “classic”. GSEA for Supplementary Fig. S5H was performed using default U2OS p53+/+, p53+/AD, and p53AD/AD ChIP targets as gene sets with the metric for ranking genes as “Diff_of_Classes” and the enrichment statistic as “classic”. The analyzed results were considered significant when the false discovery rate (FDR) q value was less than 0.25 and nominal (NOM) p value was less than 0.05.
Mitochondrial Network Imaging and Quantification
3×105 U2OS cells varying in p53 status were seeded in 6-well plates, allowed to adhere overnight, and transfected with SMARTpool sip53 (Horizon Discovery) and DharmaFECT1 Transfection Reagent for 48 h. Cells were then split 1:2 into 6-well plates containing coverslips, let adhere overnight, and treated with either DMSO or 10 μM etoposide for 24 h. Treated cells were washed twice with 1x PBS and MitoTracker® Red CMXRos was added in final concentration of 100nM. Cells were incubated at 37°C for 30 min, and later fixed with 4% paraformaldehyde (Thermo-Fisher) for 1 h, washed twice with 1x PBS, and permeabilized with PBS/0.5% Triton X-100 (Sigma) for 15 min followed by blocking with 2% goat serum (Sigma) for 30 min at room temperature. Immunofluorescence staining was performed by adding 100 μl of diluted (1:1000) primary antibodies (mouse anti-p53 (DO-1) solution to coverslips. After 1 h incubation at room temperature, coverslips were washed with 1X PBS and then incubated for 1 h with 100 μl of diluted (1:100) secondary antibody (Alexa Fluor 488 goat anti-rabbit or Alexa Fluor 594 goat anti-mouse antibodies; Life Technologies). Coverslips were washed three times with 1 X PBS after each of the above steps, mounted on a microscope slide with Vectashield (Mounting Medium with DAPI; Vector Laboratories), and images were analyzed by confocal laser scanning microscopy (Zeiss LSM 800). Quantification of the percentage of cells with aberrant mitochondria was performed by a trained observer using blinded and randomized files. Quantification of mitochondrial area/cell was performed in ImageJ. Mitochondrial network visualization was also performed for U2OS p53 KO cells stably expressing either vector or p53(A347D) with U2OS p53AD/AD cells in the same experiment as a comparison according to above protocol.
Mitochondrial Fractionation
Mitochondrial and cytosolic fractions were isolated with the Pierce Mitochondria Isolation Kit for Culture Cells (Thermo-Fisher) according to manufacturer’s protocol. Signal intensity of p53 and COX-IV was measured using ImageJ software (version 1.53K). p53 signal was normalized to the COX-IV signal.
Metabolic Assays
U2OS parental and U2OS CRISPR cells (p53 KO, p53+/AD, p53AD/AD) were plated at a density of 10,000 cells/well in Seahorse XFp cartridges the day prior to analysis. Seahorse XFp Glycolytic Rate Assay and ATP Production Assay were performed according to manufacturer’s protocol. To assess GSH:GSSG ratios, cells varying in p53 status were plated in triplicate at 1×104 cells/well in 96-well plates. After cells adhered to the plate overnight, media containing 0.1% FBS and either 20 μM etoposide or no treatment was added for 24 h. Reduced and oxidized glutathione levels were assessed using the GSH/GSSG-Glo Assay (Promega) according to manufacturer’s protocol. Lysates were transferred to a white-walled 96-well plate prior to luminescence detection.
Cell Viability Assay
Cells (5×103) were plated in a 96-well plate (Corning) in appropriate growth medium and allowed to adhere overnight. Cells were then treated with indicated drugs for 48 h. Neutral Red Cell Viability Assay (BioVision) was utilized to measure cell viability according to manufacturer’s protocol.
Cell Cycle Profiling
U2OS parental and U2OS CRISPR cells (p53 KO, p53+/AD, p53AD/AD) were seeded in 6-well plates in triplicate at an appropriate density to yield sub-confluent plates at harvest and let adhere to plate overnight. Cells were treated with either DMSO or 20 μM etoposide for 24 h, collected with trypsin, and counted. ~5×105 cells were resuspended in 200 μL cold PBS after which 200 μL cold 100% ethanol was added to fix and permeabilize cells. Cells were incubated for 30 min on ice, washed once with cold PBS, and resuspended in 400 μL propidium iodide staining solution (50 μg/mL ribonuclease A and 62.5 μg/mL propidium iodide (Sigma) in PBS). Cells were transferred to FACS tubes and incubated at 37°C for 15 min protected from light. Samples were immediately analyzed with the FACSCelesta flow cytometer (Becton Dickinson) using a 488 nm laser and collecting fluorescence emission with a 610/20 bandpass filter. FlowJo software (Becton Dickinson) was used to determine the percentage of cells at each stage of the cell cycle.
Growth Curve
U2OS parental and U2OS CRISPR cells (p53 KO, p53+/AD, p53AD/AD) were seeded at an initial density of 2.5×105 cells/well in a 6-well plate with three technical replicates/cell line. Cells were trypsinized and counted with the Countess 2 FL Automated Cell Counter.
3D Invasion Assay
Reagents
Accutase was purchased from MP Biomedicals. Pepsin-treated (PT) bovine type I collagen was obtained from Advanced BioMatrix as a 5.9–6.1 mg/ml solution. Growth factor-reduced, Phenol Red-free BME/Matrigel was obtained as an 8.9–10 mg/ml solution from BD Biosciences. DMEM solution (10×), NaOH (1 N) and sodium bicarbonate solution (7.5%) were purchased from Sigma Aldrich and sterile filtered before use. Gibco 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (1 M) was obtained from Invitrogen and sterile filtered before use. CellMask membrane dye was obtained from Molecular Probes by Thermo Fisher Scientific.
Generation of multicellular tumor spheroids and patient-derived organoids
Spheroids were formed from 2000 cells per spheroid in appropriate growth medium using a centrifugation method described previously(90) employing ultralow attachment 96-well U-bottom Nunclon Sphera plates (Thermo Fisher Scientific). Spheroids and organoids were allowed to form for 24 h at 37°C under 5% carbon dioxide.
Preparation of collagen solutions
Collagen solution (1 mg/ml) was prepared by diluting the high-concentration PT collagen monomer stock solution. Appropriate amounts of collagen stock solution were prepared with 10% (v/v) 10× DMEM, 2.5% (v/v) HEPES buffer, 2.5% (v/v) sodium bicarbonate and distilled deionized H2O. To prevent self-assembly of collagen monomers, all solutions were held and mixed at 4°C. NaOH was added to adjust the pH to 7.4.
Preparation of cell-embedded gels
To prepare gels loaded with a single spheroid, 1 mg/ml collagen solution was prepared as described above at 4°C to prevent gelation. Neutralized collagen (200 μl) was added to a chamber consisting of a 5 mm glass cylinder glued to a coverslip-bottom cell culture dish. A nylon mesh was placed on the inner circumference of the cylinder to anchor the collagen gel. The spheroid was added to the liquid collagen in 5 μl growth medium. The chamber was then transferred immediately to a 37°C incubator. The collagen gels were overlaid with 50 μl growth medium after 1 h and surrounded by additional liquid to prevent drying during extended incubation periods. To prepare collagen gels loaded with dispersed cells, the 1 mg/ml collagen solution was prepared omitting 50% of the water and neutralized at 4°C. The water volume was substituted with ice-cold growth medium containing the desired number of cells (20000–40000 cells/ml). Subsequently 100 μl of the cell-loaded collagen or collagen/BME was added to the chamber and gelled at 37°C as described above for spheroid-loaded collagen gels. After 1 h, gels were overlaid with 30 μl growth medium. In all cell experiments, time points are described considering time zero (t=0 min) to be the time at which the cells or spheroids were placed into the collagen or collagen/BME solution.
Imaging and image analysis
For invasion analysis, spheroids were generated from cells fluorescently labelled with CellTracker Orange (7 μM dye for 40 minutes at 37°C) and imaged at 10× magnification using transmitted light microscopy at 1 h and fluorescent confocal imaging at 24 h after implantation (z-stacks from 80–120 μm total with step size of 10 μm). We used these magnified fluorescence images to determine the invaded area, defined as the difference between the area of the spheroid at t=1h and an ellipse that circumscribes 90% of the invasive cells at t=24 h. Maximum intensity projections were thresholded and used to generate masks from which the non-invading spherical core of the spheroid, which is well discernable under these matrix conditions, was removed. The remaining structures, termed invasive structures, were analyzed regarding their area which reflects how far the cells have invaded. The total area of invasive structures per spheroid was used to assess invasion efficiency.
Tumor Xenograft Assay
All mouse experiments were reviewed and approved by the UTHealth Houston Animal Welfare Committee following IACUC guidelines. Tumor xenografts were performed as described previously (77,78,91). 1 × 107 wild-type and LFS iPSC-derived osteoblasts were injected subcutaneously into the right and left flanks of 7–8 weeks old NU/NU mice. After 3-month inoculation, mice were sacrificed, and tumors were extracted and weighed. For U2OS cells, 1 × 107 parental and U2OS CRISPR pools (p53 KO, p53+/AD, p53AD/AD) were injected subcutaneously into the right and left flanks of 7–8 weeks old NU/NU mice. Mice were sacrificed and tumors were extracted and weighed at indicated time.
Statistics
Statistical analyses were conducted in Excel and GraphPad Prism 7. Results, unless indicated otherwise, are expressed as mean ± SEM. A P-value of <0.05 determined via two-tailed Student’s t-test of unknown variance was considered statistically significant for hypothesis testing. The Benjamini-Hochberg procedure was utilized to correct for multiple hypothesis where appropriate. The following format was used to denote significance based on P-value: P<0.05 (*), P<0.01 (**), P<0.001 (***).
Supplementary Material
Statement of Significance.
We have characterized dimer-forming mutant p53 (A347D) associated with increased cancer susceptibility in LFS patients. We found that this variant wields a double-edged sword, driving tumorigenesis through LOF while gaining enhanced apoptogenic activity as a new GOF, which yields a therapeutic vulnerability potentially targetable by select approaches.
Acknowledgements:
We are grateful to David R. Tong for his invaluable generation of the U2OS CRISPR cell lines used in this study. David passed away in 2022 and is deeply missed for his contributions to this study and to the Prives laboratory in general. We are grateful to the members of the Prives and Lee laboratories for helpful discussions. Ella Freulich, Ruoyu Wang, and Ping Xu are thanked for expert technical assistance. Ping-Yuan Wang is thanked for help with key protocols. J.D.S. is the Helen Clise Presidential Endowed Chair in Li-Fraumeni Syndrome Research at Huntsman Cancer Institute at the University of Utah. This work was supported by NIH/NCI grant R35CA220526 to C.P., NIH/NCI grant R01CA246130 to D.-F.L. and R.Z., and NIH/NCI grant R15CA213426 to Y.Z. A.X. was a CPRIT Postdoctoral Fellow in the Biomedical Informatics, Genomics and Translational Cancer Research Training Program (BIG-TCR, CPRIT grant RP210045). The UTHealth Cancer Genomics Core is supported by the Cancer Prevention and Research Institute of Texas (CPRIT RP180734). Research reported in this publication utilized the Genetic Counseling and Biorepository and Molecular Pathology Shared Resources at Huntsman Cancer Institute at the University of Utah and was supported by NIH/NCI grant P30CA042014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest Statement:
The authors declare no conflicts with the research conducted in this study.
Data Availability
The SuperSeries containing all four sequencing experiments as well as each individual sequencing experiment have been deposited to the Gene Expression Omnibus (GEO. The GEO accession number for the SuperSeries is as follows: GSE226407.
References
- 1.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014;14(5):359–70 doi 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009;137(3):413–31 doi 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 3.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991;253(5015):49–53. [DOI] [PubMed] [Google Scholar]
- 4.Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016;122(23):3673–81 doi 10.1002/cncr.30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleihues P, Schäuble B, zur Hausen A, Estève J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 1997;150(1):1–13. [PMC free article] [PubMed] [Google Scholar]
- 6.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell 2017;170(6):1062–78 doi 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev 2012;26(12):1268–86 doi 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007;28(6):622–9 doi 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 9.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet 1993;4(1):42–6 doi 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 10.Hassin O, Nataraj NB, Shreberk-Shaked M, Aylon Y, Yaeger R, Fontemaggi G, et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nature Communications 2022;13(1):2800 doi 10.1038/s41467-022-30481-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ 2018;25(1):161–8 doi 10.1038/cdd.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol 2018;15(1):13–30 doi 10.1038/nrclinonc.2017.151. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi T, Kato S, Otsuka K, Watanabe G, Kumabe T, Tominaga T, et al. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 2005;24(46):6976–81 doi 10.1038/sj.onc.1208839. [DOI] [PubMed] [Google Scholar]
- 14.de Andrade KC, Lee EE, Tookmanian EM, Kesserwan CA, Manfredi JJ, Hatton JN, et al. The TP53 Database: transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death & Differentiation 2022;29(5):1071–3 doi 10.1038/s41418-022-00976-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achatz MI, Zambetti GP. The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harb Perspect Med 2016;6(12) doi 10.1101/cshperspect.a026195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garritano S, Gemignani F, Palmero EI, Olivier M, Martel-Planche G, Le Calvez-Kelm F, et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a founder effect. Hum Mutat 2010;31(2):143–50 doi 10.1002/humu.21151. [DOI] [PubMed] [Google Scholar]
- 17.Pinto EM, Billerbeck AE, Villares MC, Domenice S, Mendonça BB, Latronico AC. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol 2004;48(5):647–50 doi 10.1590/s0004-27302004000500009. [DOI] [PubMed] [Google Scholar]
- 18.Fischer NW, Prodeus A, Tran J, Malkin D, Gariépy J. Association Between the Oligomeric Status of p53 and Clinical Outcomes in Li-Fraumeni Syndrome. JNCI: Journal of the National Cancer Institute 2018;110(12):1418–21 doi 10.1093/jnci/djy114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz C, Low-Calle AM, Choe JH, Laptenko O, Tong D, Joseph-Chowdhury JN, et al. Wild-type and cancer-related p53 proteins are preferentially degraded by MDM2 as dimers rather than tetramers. Genes Dev 2018;32(5–6):430–47 doi 10.1101/gad.304071.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaglia G, Guan Y, Shah JV, Lahav G. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci U S A 2013;110(38):15497–501 doi 10.1073/pnas.1311126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 1999;18(6):1660–72 doi 10.1093/emboj/18.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J Mol Biol 2004;341(5):1145–59 doi 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- 23.Chene P The role of tetramerization in p53 function. Oncogene 2001;20(21):2611–7 doi 10.1038/sj.onc.1204373. [DOI] [PubMed] [Google Scholar]
- 24.Gencel-Augusto J, Lozano G. p53 tetramerization: at the center of the dominant-negative effect of mutant p53. Genes Dev 2020;34(17–18):1128–46 doi 10.1101/gad.340976.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warnock LJ, Knox A, Mee TR, Raines SA, Milner J. Influence of tetramerisation on site-specific post-translational modifications of p53: comparison of human and murine p53 tumor suppressor protein. Cancer Biol Ther 2008;7(9):1481–9. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Zhou X, Wang J, Li Z, Kong X, Qian J, et al. RhoGAPs attenuate cell proliferation by direct interaction with p53 tetramerization domain. Cell Rep 2013;3(5):1526–38 doi 10.1016/j.celrep.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 27.Park JH, Li J, Starost MF, Liu C, Zhuang J, Chen J, et al. Mouse Homolog of the Human TP53 R337H Mutation Reveals Its Role in Tumorigenesis. Cancer research 2018;78(18):5375–83 doi 10.1158/0008-5472.Can-18-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer NW, Prodeus A, Malkin D, Gariepy J. p53 oligomerization status modulates cell fate decisions between growth, arrest and apoptosis. Cell cycle (Georgetown, Tex) 2016;15(23):3210–9 doi 10.1080/15384101.2016.1241917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macedo GS, Lisboa da Motta L, Giacomazzi J, Netto CB, Manfredini V, Vanzin CS, et al. Increased oxidative damage in carriers of the germline TP53 p.R337H mutation. PloS one 2012;7(10):e47010 doi 10.1371/journal.pone.0047010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicholls CD, McLure KG, Shields MA, Lee PW. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J Biol Chem 2002;277(15):12937–45 doi 10.1074/jbc.M108815200. [DOI] [PubMed] [Google Scholar]
- 31.Gannon JV, Greaves R, Iggo R, Lane DP. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J 1990;9(5):1595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994;265(5170):346–55 doi 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 33.Stephen CW, Lane DP. Mutant conformation of p53. Precise epitope mapping using a filamentous phage epitope library. J Mol Biol 1992;225(3):577–83 doi 10.1016/0022-2836(92)90386-x. [DOI] [PubMed] [Google Scholar]
- 34.Cook A, Milner J. Evidence for allosteric variants of wild-type p53, a tumour suppressor protein. Br J Cancer 1990;61(4):548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004;303(5659):844–8 doi 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 36.Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, Tamura RE. p53 and metabolism: from mechanism to therapeutics. Oncotarget 2018;9(34):23780–823 doi 10.18632/oncotarget.25267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nature Communications 2013;4(1):2935 doi 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev 2012;26(8):830–45 doi 10.1101/gad.181685.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015;525(7568):206–11 doi 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gencel-Augusto J, Su X, Qi Y, Whitley EM, Pant V, Xiong S, et al. Dimeric p53 mutant elicts unique tumor suppressive activities through an altered metabolic program. Cancer Discovery 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 2005;6(8):657–63 doi 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 42.Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab 2016;27(2):105–17 doi 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012;337(6098):1062–5 doi 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otera H, Mihara K. Mitochondrial Dynamics: Functional Link with Apoptosis. International Journal of Cell Biology 2012;2012:821676 doi 10.1155/2012/821676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009;458(7242):1127–30 doi 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchenko ND, Moll UM. Mitochondrial death functions of p53. Mol Cell Oncol 2014;1(2):e955995 doi 10.1080/23723548.2014.955995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolff S, Erster S, Palacios G, Moll UM. p53’s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Research 2008;18(7):733–44 doi 10.1038/cr.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murphy ME, Leu JIJ, George DL. p53 Moves to Mitochondria: A Turn on the Path to Apoptosis. Cell cycle (Georgetown, Tex) 2004;3(7):834–7 doi 10.4161/cc.3.7.956. [DOI] [PubMed] [Google Scholar]
- 49.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004;6(5):443–50 doi 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 50.Larraufie MH, Yang WS, Jiang E, Thomas AG, Slusher BS, Stockwell BR. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg Med Chem Lett 2015;25(21):4787–92 doi 10.1016/j.bmcl.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149(5):1060–72 doi 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2006;2(9):474–9 doi 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- 53.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003;11(3):577–90 doi 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 54.Guzman A, Sánchez Alemany V, Nguyen Y, Zhang CR, Kaufman LJ. A novel 3D in vitro metastasis model elucidates differential invasive strategies during and after breaching basement membrane. Biomaterials 2017;115:19–29 doi 10.1016/j.biomaterials.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 55.Stein Y, Aloni-Grinstein R, Rotter V. Mutant p53 oncogenicity: dominant-negative or gain-of-function? Carcinogenesis 2020;41(12):1635–47 doi 10.1093/carcin/bgaa117. [DOI] [PubMed] [Google Scholar]
- 56.Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, et al. Mutant p53 Cooperates with ETS and Selectively Up-regulates Human MDR1 Not MRP1 *. Journal of Biological Chemistry 2001;276(42):39359–67 doi 10.1074/jbc.M103429200. [DOI] [PubMed] [Google Scholar]
- 57.Moulder DE, Hatoum D, Tay E, Lin Y, McGowan EM. The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers (Basel) 2018;10(6) doi 10.3390/cancers10060189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev 2008;22(12):1577–90 doi 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 2010;10(6):640–8 doi 10.1016/j.mito.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 60.Dai W, Jiang L. Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front Endocrinol (Lausanne) 2019;10:570 doi 10.3389/fendo.2019.00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012;3(8):798–810 doi 10.18632/oncotarget.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang DB, Kinoshita C, Kinoshita Y, Morrison RS. p53 and mitochondrial function in neurons. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2014;1842(8):1186–97 doi 10.1016/j.bbadis.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson GR, Wardell SE, Cakir M, Yip C, Ahn Y-r, Ali M, et al. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nature Communications 2018;9(1):1677 doi 10.1038/s41467-018-04033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen H, Chan DC. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell metabolism 2017;26(1):39–48 doi 10.1016/j.cmet.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell metabolism 2016;23(1):27–47 doi 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta 2009;1787(5):414–20 doi 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death & Differentiation 2018;25(1):104–13 doi 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem 2006;281(13):8600–6 doi 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 69.Wei H, Qu L, Dai S, Li Y, Wang H, Feng Y, et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nature Communications 2021;12(1):2280 doi 10.1038/s41467-021-22655-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giorgi C, Bonora M, Missiroli S, Morganti C, Morciano G, Wieckowski MR, et al. Alterations in Mitochondrial and Endoplasmic Reticulum Signaling by p53 Mutants. Frontiers in Oncology 2016;6(42) doi 10.3389/fonc.2016.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev 1995;9(17):2170–83 doi 10.1101/gad.9.17.2170. [DOI] [PubMed] [Google Scholar]
- 72.Timofeev O, Klimovich B, Schneikert J, Wanzel M, Pavlakis E, Noll J, et al. Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. The EMBO journal 2019;38(20):e102096 doi 10.15252/embj.2019102096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hagn F, Klein C, Demmer O, Marchenko N, Vaseva A, Moll UM, et al. BclxL Changes Conformation upon Binding to Wild-type but Not Mutant p53 DNA Binding Domain *. Journal of Biological Chemistry 2010;285(5):3439–50 doi 10.1074/jbc.M109.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muscolini M, Montagni E, Palermo V, Di Agostino S, Gu W, Abdelmoula-Souissi S, et al. The cancer-associated K351N mutation affects the ubiquitination and the translocation to mitochondria of p53 protein. J Biol Chem 2011;286(46):39693–702 doi 10.1074/jbc.M111.279539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014;3:e04766 doi 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim H, Yoo S, Zhou R, Xu A, Bernitz JM, Yuan Y, et al. Oncogenic role of SFRP2 in p53-mutant osteosarcoma development via autocrine and paracrine mechanism. Proc Natl Acad Sci U S A 2018;115(47):E11128–e37 doi 10.1073/pnas.1814044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee DF, Su J, Kim HS, Chang B, Papatsenko D, Zhao R, et al. Modeling familial cancer with induced pluripotent stem cells. Cell 2015;161(2):240–54 doi 10.1016/j.cell.2015.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tu J, Huo Z, Yu Y, Zhu D, Xu A, Huang MF, et al. Hereditary retinoblastoma iPSC model reveals aberrant spliceosome function driving bone malignancies. Proc Natl Acad Sci U S A 2022;119(16):e2117857119 doi 10.1073/pnas.2117857119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jewell BE, Xu A, Zhu D, Huang MF, Lu L, Liu M, et al. Patient-derived iPSCs link elevated mitochondrial respiratory complex I function to osteosarcoma in Rothmund-Thomson syndrome. PLoS Genet 2021;17(12):e1009971 doi 10.1371/journal.pgen.1009971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao Q, Gregory CA, Lee RH, Reger RL, Qin L, Hai B, et al. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc Natl Acad Sci U S A 2015;112(2):530–5 doi 10.1073/pnas.1423008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 2016;34(5):525–7 doi 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Parmigiani G, Johnson WE. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genomics and Bioinformatics 2020;2(3) doi 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Research 2019;47(W1):W199–W205 doi 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martens M, Ammar A, Riutta A, Waagmeester A, Slenter Denise N, Hanspers K, et al. WikiPathways: connecting communities. Nucleic Acids Research 2021;49(D1):D613–D21 doi 10.1093/nar/gkaa1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 2005;102(43):15545–50 doi doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 2015;44(D1):D457–D62 doi 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JPt, et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019;176(3):564–80.e19 doi 10.1016/j.cell.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38(4):576–89 doi 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ivascu A, Kubbies M. Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J Biomol Screen 2006;11(8):922–32 doi 10.1177/1087057106292763. [DOI] [PubMed] [Google Scholar]
- 91.Zhou R, Xu A, Tu J, Liu M, Gingold JA, Zhao R, et al. Modeling Osteosarcoma Using Li-Fraumeni Syndrome Patient-derived Induced Pluripotent Stem Cells. J Vis Exp 2018(136):57664 doi 10.3791/57664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SuperSeries containing all four sequencing experiments as well as each individual sequencing experiment have been deposited to the Gene Expression Omnibus (GEO. The GEO accession number for the SuperSeries is as follows: GSE226407.