

Abstract
Previous studies indicate that moderate-to-high ethanol (EtOH) concentrations enhance dopamine (DA) neurotransmission in the mesolimbic DA system from the ventral tegmental area (VTA) and projecting to the nucleus accumbens core (NAc). However, voltammetry studies demonstrate that moderate-to-high EtOH concentrations decrease evoked DA release at NAc terminals. The involvement of γ-aminobutyric acid (GABA) receptors (GABAARs), glycine (GLY) receptors (GLYRs) and cholinergic interneurons (CINs) in mediating EtOH inhibition of evoked NAc DA release were examined. Fast scan cyclic voltammetry, electrophysiology, optogenetics, and immunohistochemistry techniques were used to evaluate the effects of acute and chronic EtOH exposure on DA release and CIN activity in C57/BL6, CD-1, transgenic mice, and δ-subunit knock-out (KO) mice (δ−/−). Ethanol decreased DA release in mice with an IC50 of 80 mM ex vivo and 2.0 g/kg in vivo. GABA and GLY decreased evoked DA release at 1–10 mM. Typical GABAAR agonists inhibited DA release at high concentrations. Typical GABAAR antagonists had minimal effects on EtOH inhibition of evoked DA release. However, EtOH inhibition of DA release was blocked by the α4β3δ GABAAR antagonist Ro15–4513, the GLYR antagonist strychnine, and by the GABA ρ1 (Rho-1) antagonist TPMPA (10 μM), and reduced significantly in GABAAR δ−/− mice. Rho-1 expression was observed in CINs. Ethanol inhibited GABAergic synaptic input to CINs from the VTA and enhanced firing rate, both of which were blocked by TPMPA. Results herein suggest that EtOH inhibition of DA release in the NAc is modulated by GLYRs and atypical GABAARs on CINs containing δ- and Rho-subunits.
Keywords: Accumbens, alcohol, cholinergic, dopamine, GABA, nicotinic
Introduction
Dopamine (DA) release in the nucleus accumbens (NAc) has been implicated in the rewarding and reinforcing properties of drugs of abuse, including ethanol (EtOH;1). The mesolimbic DA system consists of projections from the ventral tegmental area (VTA) to limbic areas such as the NAc. Although it is clear that DA is involved in EtOH reinforcement, its role is complicated by results from neurochemical studies. For example, microdialysis studies have reported EtOH-induced increases in DA levels at moderate-to-high doses (0.5–2 g/kg;2,3), and others have reported no effect or DA reductions at high EtOH doses (2–5 g/kg;2–4). Voltammetry studies have reported increases in DA transient frequency after EtOH in freely-moving animals5,6. Striatal DA release can occur from firing of midbrain DA neurons, but can also be triggered by firing of local cholinergic interneurons (CINs) and subsequent depolarization of NAc DA terminals via nicotinic acetylcholine receptors (nAChRs;7). We have shown previously that EtOH enhances CIN-mediated NAc DA release at low concentrations (5–10 mM)8. In contrast to these excitatory effects, many in vivo and in vitro voltammetry studies report EtOH-induced decreases in DA release with moderate-to-high doses (>1g/kg or >40mM;9–15). The mechanisms underlying EtOH-mediated inhibition of DA release are complex and not fully understood.
Despite the many studies implicating the mesolimbic DA system in EtOH reward, consumption, and alcoholism (for recent review see16), there are relatively few studies elucidating the mechanism of action of EtOH effects on this system. The activity of the mesolimbic DA system is controlled or modulated by several neurotransmitters including γ-aminobutyric acid (GABA), glutamate (GLU), and glycine (GLY), metabolites including acetaldehyde17, enzymes including catalase18, receptors including the family of cysteine-loop gated ion channels including cholinergic nicotinic receptors19, GLYRs, GABAARs, and 5-HT3Rs, as well as the DA transporter (DAT) and D2 receptors. A series of studies from the Soderpalm lab have identified GLYRs in the NAc as a nexus for EtOH effects on the mesolimbic DA system20–24. However, the elucidation of the molecular mechanisms by which EtOH activates the system remains elusive.
Ethanol potentiates GABAergic inhibitory synaptic transmission throughout the brain25, and modulation of GABAergic synaptic transmission is regarded as one of the main factors underlying alcohol withdrawal-related phenomena26. GABAARs undergo allosteric modulation by EtOH, anesthetics, benzodiazepines and neurosteroids, and have been implicated in the acute and chronic effects of EtOH, including tolerance, dependence and withdrawal (for review see27). Typical GABAARs are pentameric ion channels consisting of two α-subunits (α1−α6), two β-subunits (β1−β3), and a fifth subunit that is comprised of either γ1−γ3, δ, ε, θ, or π subunits with the γ2-subunit being the most abundant28. GABAρ (or Rho) subunits are thought to form homopentameric GABAA Rho receptors (formerly classified as GABACRs;29). Although still under investigation, Rho-subunits may co-assemble with other GABAAR subunits, producing receptors with unique properties30–35.
VTA GABA neurons projects to the striatum onto CINs36 and may regulate DA release through an indirect CIN mediated mechanism on DA terminals. Furthermore, selective activation of VTA GABA neurons disrupts reward consumption37, and alters NAc CINs firing to enhance associative learning36. Together, these studies provide compelling evidence for the importance of VTA GABA neurons in regulating DA transmission via effects onto NAc CINs.
Given that we and others have shown that EtOH decreases evoked DA release in the NAc, we hypothesized that GABAergic projections from the VTA are co-activated with DA terminals and inhibit DA release, and that EtOH augments GABAergic inhibition to NAc CINs to reduce CIN-mediated DA release. Therefore, the effects of GABA and GLY agonists, antagonists, and modulators were examined on DA transmission and CIN activity in the context of EtOH.
Materials and Methods
Animal Subjects
The Institutional Animal Care and Use Committee (IACUC) of Brigham Young University approved the care and use of mice and experimental protocols in accordance with NIH guidelines. Two strains of adult mice (PND 60–90) were used in this study: CD-1 (white) and C57/BL6 (black). Five lines of mice were used based on these background strains: 1) GABAA δ-subunit knock-out (δ−/−; C57/BL6 background)38; 2) glutamate decarboxylase-67 (GAD67)-green fluorescent protein (GFP; GAD67-GFP; CD-1 background)39 were used in order to directly identify GABAergic neurons (i.e. in immunohistochemistry experiments); 3) Mice expressing channel rhodopsin (ChR2) and GFP on the vesicular GABA transporter (VGAT) promoter (VGAT-ChR2-GFP; JAX, B6.Cg-Tg(Slc32a1-COP4*H134R/EYFP)8Gfng/J); 4) VGAT-ires-Cre C57/BL6 mice (Jackson Labs, 028862) crossed with GAD67-GFP CD-1 mice to obtain a hybrid VGAT-Cre/GAD67-GFP mouse (Agouti), with GAD67-GFP mice used in order to facilitate identification of viral co-transfection in GAD67 expressing GABA neurons; and 5) choline acetyltransferase (ChAT) mice expressing ChR2 and eYFP reporter in CINs (ChAT-ChR2-eYFP; JAX, B6.Cg-Tg(Chat-COP4*H134R/EYFP,Slc18a3)6Gfng/J). Offspring of VGAT-Cre/GAD67-GFP were phenotyped at PND1–8 by visual inspection of cranial fluorescence with customized goggles containing filtered lenses of the corresponding wavelength with bright light illuminators of the appropriate activating wavelength. Mice were weaned at PND 21, housed in maximum groups of 5/cage and given ad libitum access to solid food and water and placed on a reverse light/dark cycle with lights ON from 2000 to 0800 hrs.
CIN Characterization
VTA GABA neuron inhibition of NAc CINs was the pathway focus for most of the electrophysiological recordings. In ChAT-ChR2-eYFP mice, CINs could be visualized with eYFP fluorescence. In VGAT-Cre/GAD67-GFP mice, CINs were visualized in the slice preparation as non-GFP neurons amongst predominantly GABA neurons. In mice without fluorescent identification, CINs were identifiable by their large size, morphology, tonic firing (~0.5–3Hz), high membrane capacitance (due to large size), and presence of an Ih current40. Fluorescent cells were imaged on Olympus BX5 and Nikon Eclipse FNI microscopes with 40x/0.80 n.a. objective lens. The filter cube for GFP detection and ChR2 stimulation was an Olympus GFP U-M6553 cube (Bandpass: 470–490 nm; Barrier: 510–560 nm; dichroic: 505 nm) or Nikon C-FL ENDOW GFP 96343 cube (Bandpass: 450–490 nm; Barrier: 500–550 nm; dichroic: 495 nm). Excitation was performed with a Thorlabs 470 nm LED. Cells were also imaged using infrared differential interference contrast imaging in order to facilitate physiological recordings.
Patch-clamp Recordings
For ex vivo whole-cell recordings, electrodes were pulled from borosilicate glass capillary tubes (1.5 mm o.d.; A-M Systems, Sequim, WA) filled with KCl pipette solutions (in mM: 128 KCl, 20 NaCl, 0.3 CaCl2, 1.2 MgCl2, 10 HEPES, 1 EGTA, 2 Mg-ATP, 0.25 Na-GTP and 5 QX-314; pH 7.3) for optically-evoked IPSC (oIPSC) studies. Pipettes having tip resistances of 2.5 – 5 MΩ, and series resistances typically ranging from 7 – 15 MΩ were used. Voltage clamp recordings were filtered (2 kHz) with an Axon Instruments Multiclamp 700B amplifier and digitized (5 kHz) using Axon 1440A digitizers. Axon Instruments pClamp ver10, Mini Analysis (Synaptsoft: Decatur, GA), and Igor Pro (Wavemetrics: Oswego, OR) software packages were used for data collection and analysis. The oIPSCs were recorded in the presence of kynurenic acid (2 mM) to block glutamate-mediated synaptic currents and isolate GABAAR-mediated synaptic currents. A holding potential of −70 mV was applied and ionic currents measured in response to drug application. The oIPSCs were evoked in VGAT-Cre/GAD67-GFP mice (injected in VTA with AAV-DIO-ChR2-mCherry) by 4 msec pulses of blue light (~470 nm).
For ex vivo cell-attached recordings, borosilicate electrodes (2.5–5 MΩ) filled with ACSF were used to form tight seals (>20 MΩ) on identified CINs. Spontaneous firing activity was then recorded in cell-attached, voltage-clamp mode with an Axon Instruments Multiclamp 700B amplifier, sampled at 10 kHz using NIDAQ (National Instruments, Austin, TX) data acquisition boards, and collected and analyzed using Axograph software. Firing rate recordings were performed in cell-attached mode in order to avoid dialyzing the contents of the cells and disrupting the cytoplasmic milieu (e.g., the Cl− ion gradient). A stable baseline recording of firing activity was obtained for 5–10 min before adding drugs. Neurons that did not achieve a stable baseline firing rate during this time were rejected from the study.
Carbon Fiber Electrodes and Fast Scan Cyclic Voltammetry
For in vivo and ex vivo voltammetry recordings a carbon fiber electrode (CFE; 7 μm diameter) was inserted into borosilicate capillary tubing (1.5 mm i.d.; A-M Systems, Sequim, WA) and pulled on a vertical pipette puller (Narishige, East Meadow, NY). The CFE was cut under microscopic control with 150–200 μm of bare fiber protruding from the sealed end of the glass micropipette and back-filled with 3 M KCl. Electrode calibration occurred post experiment as described previously9. For ex vivo voltammetry recordings, the CFE was positioned in the NAc. Dopamine release was evoked by a 4.0 msec, single-pulse electrical stimulation (monophasic, 350 μA; 2 min intervals) from a bipolar stimulating electrode (Plastics One, Roanoke, VA) placed 100–200 μm from the CFE. The CFE potential had a triangle command voltage applied (−0.4 to 1.3 V; 400V/s) and cyclic voltammograms were recorded (10 Hz; Ag/AgCl reference) by means of a ChemClamp voltage-clamp amplifier (Dagan Corporation, Minneapolis, MN). For in vivo voltametric recordings, mice were anesthetized with isoflurane and placed in a stereotaxic apparatus (David Kopf Instruments, Tejunga, CA). Bipolar, coated stainless steel electrodes were stereotaxically positioned into the medial forebrain bundle (MFB; −1.3 P, +1.0 L, 5.0–5.3 V), and the CFE in the NAc core (+1.3 A, +0.9 L, −3.8–4.5 V). CFE placement was verified as described9. The MFB was stimulated (60 biphasic pulses, 60 Hz, 350 μA, 4 ms pulse; 5 min intervals). Stimulation and recording electrodes were oriented by fine control to optimize DA release. Subsequently, the current was adjusted to that stimulus level which evoked DA release at 50% of the maximum DA current level to ensure that results were not skewed by overstimulation.
Drug Concentration/Dosage Justification
Drug concentration was typically selected based on maximal effects: APV (50 μM)41, picrotoxin (PTX; 50–100 μM)42,43, bicuculline (50 μM)44, pentobarbital (100 μM)45,46, SKF97541 (10 μM)47, TPMPA (10 μM)48. Chlordiazepoxide (30 μM) was selected based off previous studies showing an effective range within 3–300 μM49,50. Baclofen concentrations (0.001–2 mM) spanned and exceeded a known effective concentration range (1–100 μM)47. Muscimol concentrations (1–300 μM) were selected from and exceed the effective range previously used in our lab51. The lower concentration range selected for THIP (10 nM - 10 μM) was selected for its δ-subunit agonist selectivity43,52,53. For in vivo studies, Ro15–4513 dosage (1.5 mg/kg) was selected for effectiveness at reversing EtOH intoxicating effects54. EtOH concentration (5–160 mM) and dosage (0.5–4 gm/kg) was selected from numerous previous studies from our lab.
Statistical Analyses
For evoked DA release, results for control and drug treatment groups were derived from calculations performed on voltammetry current vs time plots. Peak amplitude was determined as previously described55. Briefly, three successive DA peak responses were averaged for control and drug conditions within each experiment. The means were then grand-averaged across animals. For neuronal firing rate, results are presented as percent of baseline firing rate ± standard error of the mean (SEM). Statistical significance required ≥ 95% level of confidence (p ≤ 0.05). Firing rate was determined from windowed and discriminated spike times and histogrammed into 10 sec bins. Averaged firing rate of 5 min was determined on ratemeters by rectangular integration in Igor Pro (Wavemetrics, Oswego, OR). The last minute of integrated firing rate before treatment (i.e., drugs) was compared against the last minute of drug effects 5 min after administration unless otherwise indicated. For whole cell recordings, IPSCs were recorded every 20 sec and averaged into 1 min bins in Excel (Microsoft). The last 5 min of 1 min averages before treatment (i.e., drugs or low frequency stimulation; LFS) were again averaged and all other values of the time course of effect were normalized to this average. Comparisons were made between 5 min of values before treatment and 5 min after drug treatment or 30–35 min after LFS. For comparison between groups, a mixed model ANOVA was used. A priori hypothesis testing was accomplished with Bonferroni or Tukey correction post-hoc tests. Analysis software included Demon Voltammetry55, Minianalysis, Clampfit, Axograph, Microsoft Excel, STATA (StataCorp, College Station, TX), and Igor Pro (Wavemetrics, Oswego, OR). Within-subjects comparisons were evaluated via one-way ANOVA and between-subject comparisons were evaluated via one-way ANOVA with Tukey post-hoc analysis where pertinent. For the comparison between δ−/− and WT mice, a two-way ANOVA was performed with genotype as the between-subject factor and EtOH concentration as the within-subject factor. Values were expressed as means ± SEM for cumulated data. Significance levels were indicated on graphs with asterisks *,**,***, corresponding to significance levels p<0.05, 0.01 and 0.001, respectively. Data will be shared on a per request basis.
Please see Supplemental for Methods on brain slice preparation, surgical procedures, drug administration, immunohistochemistry, and image analysis
Results
Effects of EtOH on Evoked Dopamine Release in CD-1 Mice: Ex Vivo vs In Vivo
The effects of EtOH on evoked DA release were evaluated using voltammetry (Fig. 1) ex vivo and in vivo in CD-1 mice, the background strain for some of the studies below. Superfusion of EtOH (5–80 mM) significantly reduced evoked DA release in the NAc slice preparation in CD-1 mice at the 80 and 160 mM levels (Fig. 1C; mean evoked DA release = 1.22 ± 0.16 μM; n=23; 80 mM: F1,23=19.8, p=0.0002; 160 mM: F1,11=14.5, p=0.003). In vivo, EtOH (0.5–4.0 g/kg; IP) reduced evoked DA release in the NAc of isoflurane-anesthetized CD-1 mice at the 1.0 – 4.0 g/kg levels (Fig. 1D; mean evoked DA release = 0.9 ± 0.13 μM; n=32; 1 g/kg: F1,9=6.5, p=0.03; 2.0 g/kg: F1,29=33.2, p<0.0001; 4.0 g/kg: F1,11=916, p<0.0001).
Figure 1. Effects of EtOH on evoked DA release ex vivo and in vivo in the NAc core of CD-1 mice.

(A) I vs V plot showing voltage and current responses associated with DA detection in voltammetry. (B) Color plot showing I vs t plots over 15 sec showing the DA signal obtained following local stimulation in the NAc slice. (C) Time course (top graph), I vs t plot (inset), and summary graph showing the representative effects of superfused EtOH and cumulated effects of EtOH (10 – 160 mM) on evoked DA release in the NAc slice. (D) Time course (top graph), I vs t plot (inset), and summary graph showing the representative effects of EtOH (IP 2 g/kg) and cumulated effects of EtOH (0.5–4 g/kg) on evoked DA release in the NAc in vivo. Asterisks *,**,*** indicate significance levels p<0.05, p<0.01, and p<0.001, respectively.
Glutamatergic NMDA Receptors do not Mediate Ethanol-Induced Inhibition of Dopamine Release
It was first tested if EtOH effects on NAc DA involved changes to glutamatergic NMDA receptor activity. Slices were pre-treated with APV (50 μM; NMDA receptor antagonist) for 1 hour prior to and during experimentation. There was no effect of APV on evoked DA release (Supplementary Fig. 1A; p>0.05). In APV, evoked DA release was inhibited by EtOH at 80 mM, the IC50 level of EtOH on evoked DA release9,10. Two-way repeated measures ANOVA of control vs EtOH revealed a main effect of EtOH (Supplementary Fig. 1B, F1,14=39.91, p<0.0001), but no significant effect of APV (F1,14=0.7, p=0.417) and no interactions between APV and EtOH (F1,14=0.88, p=0.36). Thus, EtOH is not inhibiting DA release through NMDA receptor mediated mechanisms.
GABA and GLY Inhibition of Evoked DA Release in the NAc Ex Vivo
Both GABA and GLY significantly inhibited DA release, but only from 1–10 mM (GABA: F7,45=23.89, p<0.0001; GLY: F7,44=7.998, p<0.001; Fig. 2A). The GABAAR/chloride ionophore blocker picrotoxin (PTX) significantly reduced both GABA (Two-way mixed ANOVA, GABA Control vs Picrotoxin, F1,40=11.93, p=0.0023; GABA concentration, F3,40=168.14, p<0.0001; Interaction, F3,40=11.39, p=0.002438) and GLY (Two-way mixed ANOVA, GLY Control vs Picrotoxin, F1,31=10.67, p=0.0061; GLY concentration, F3,31=21, p<0.0001; Interaction, F3,31=5.98, p=0.0024) inhibition of evoked DA release (Fig. 2B,C).
Figure 2. Effects of GABA and GLY on evoked DA release in the NAc core ex vivo.

(A) Concentration response curve of exogenous GABA and GLY (0.1 mM – 10 mM). Both GABA and GLY significantly reduced DA release at high concentrations (1–10 mM). (B,C) The chloride ionophore blocker picrotoxin reduces GABA and GLY inhibition of evoked DA release. Asterisks *,*** indicate significance levels p<0.05 and p<0.001, respectively.
Effects of GABAAR and GLYR Antagonists on EtOH Inhibition of Evoked NAc Dopamine Release Ex Vivo
The GLYR receptor and the α4β3δ subtype of the GABAAR may be working in tandem through co-localization, co-activation, and cross-talk to serve as a more potent regulatory system of DA control. GABA may narrow the time window for effective GLYergic inhibition56,57. Given the similarities in GABA, GLY, and EtOH inhibition of evoked DA release, we evaluated the effects of the GLYR antagonist strychnine (10 μM) and α4β3δ subtype GABAAR antagonist Ro15–4510 (10 μM) on EtOH inhibition of evoked DA release in NAC slices. Strychnine significantly decreased DA release (Supplementary Fig. 2C; 21.5 ± 7.7 %; t7=3.548, p=0.0094; n=8). Superfusion of Ro15–4513 slightly, but not significantly, increased evoked DA release (Supplementary Fig. 2A; t5=1.524, p=0.188; n=6). Superfusion of either strychnine (Fig. 3B,D) or Ro15–4513 (Fig. 3C,D) significantly reduced EtOH inhibition of DA release (Strychnine: F(1,15)=9.7, p=0.008; n=8; Ro15–4513: F(1,13)=15.5, p=0.002; n=7), compared to EtOH alone. Since strychnine had significant effects on its own, and also is known to block α7 nicotinic receptors (nAChRs) at the dose tested58, we evaluated the effects of the α7 nAChR antagonist methylylcaconitine (MLA; 100 nM), which did not significantly alter EtOH inhibition of DA release (data not shown; p>0.05; n=3), consistent with what we have reported previously9.
Figure 3. Effects of Strychnine and Ro15–4513 on EtOH inhibition of evoked DA release ex vivo.

(A) Representative example of the effects of superfusion of EtOH (80 mM) on evoked DA release in the NAc slice preparation. In this example, EtOH inhibited the DA signal approximately 50%. Insets show superimposed I vs t and I vs V cyclic voltammograms before and after EtOH. (B) Representative example of the effects of the GLYR antagonist strychnine on EtOH inhibition of evoked DA release. In this example, strychnine blocked EtOH’s typical inhibitory response. Insets show superimposed I vs t and I vs V cyclic voltammograms during strychnine (Strych) and strychnine+EtOH. (C) Representative example of the effects of the α4β3δ GABAAR antagonist Ro15–4513 (Ro15) on EtOH inhibition of evoked DA release. In this example, Ro15 reduced EtOH’s typical inhibitory response. Insets show superimposed I vs t and I vs V cyclic voltammograms during Ro15 and Ro15+EtOH. (D) Summary of strychnine and Ro15 on 80 mM EtOH inhibition of evoked DA release. Values in parentheses represent n values. Asterisks *,**,*** indicate significance levels p<0.05, p<0.01 and p<0.001, respectively.
Effects of GABAAR and GLYR Antagonists on EtOH Inhibition of Evoked NAc Dopamine Release In Vivo
As both strychnine and Ro15–4513 were effective in reducing EtOH inhibition of DA release ex vivo, we evaluated their effects on EtOH inhibition of DA release in vivo, compared to a saline injection control (Fig. 4). While 1.0 mg/kg strychnine and 1.5 mg/kg Ro15–4513 did not significantly affect evoked DA release in the NAc in vivo (Supplementary Fig. 2B,D; p>0.05), intraperitoneal administration strychnine (Fig. 4B,D) or Ro15–4513 (Fig. 4C,D) 15 min prior to EtOH significantly reduced the typical inhibition of DA release by EtOH (Strychnine: F(1,13)=21.5, p=0.0006; n=5; Ro15–4513: F(1,14)=7.7, p=0.016; n=6) produced by EtOH alone. Although Ro15–4513 (10 μM) did not significantly alter DA signals, it did significantly reduce EtOH inhibition of evoked DA release both ex vivo and in vivo, suggesting a role for δ-subunits. Subsequently, δ−/− mice were used to evaluate EtOH dose-response sensitivity at the same concentration levels (20 mM – 160 mM) as wild-type (WT) mice in brain slices. Interestingly, δ−/− mice demonstrated an increased evoked DA release with exposure to all concentrations of EtOH (Fig. 4E). Two-way ANOVA revealed significance of strain (F1,59=62.41, p<0.0001), concentration (F1,59=8.09, p=0.0006), and an interaction between strain and concentration variables (F1,59=4.11, p=0.0163). Thus, EtOH appears to interact with GABAARs containing δ subunits to influence DA release.
Figure 4. Effects of Strychnine and Ro15–4513 on EtOH inhibition of evoked DA release in vivo.

(A) Representative example of the effects of EtOH (2 g/kg; IP; Also shown in Fig. 1D) on evoked DA release in the NAc of an isoflurane-anesthetized mouse. In this example, EtOH inhibited the DA signal approximately 50%. Inset shows superimposed I vs t recordings before and after EtOH. (B) Representative example of the effects of strychnine (1 mg/kg) on 2.0 g/kg EtOH inhibition of evoked DA release. In this example, intraperitoneal administration of strychnine blocked the typical inhibition by EtOH. Inset shows superimposed I vs t recordings before and after strychnine and strychnine+EtOH. (C) Representative example of the effects of the α4β3δ GABAAR antagonist Ro15–4513 (Ro15) on 2.0 g/kg EtOH inhibition of evoked DA release in the NAc. Insets shows representative superimposed I vs t plots before and after Ro15 and Ro15+EtOH. (D) Summary of strychnine and Ro15 on 80 mM EtOH inhibition of evoked DA release. Values in parentheses represent n values. (E) Summary of EtOH effects in δ-KO vs WT mice. Knock-out mice were resistant to EtOH inhibition of evoked DA release. Asterisks *,*,*** indicate significance levels p<0.05, p<0.01, and p<0.001, respectively.
Typical GABAA Receptor Agonists and Antagonists Effects on Evoked NAc Dopamine Release
Since EtOH inhibition of DA release was sensitive to Ro15–4513 we hypothesized that co-activated GABA inhibition the NAc would be mediated by α4β3δs. As EtOH inhibition of DA release was sensitive to δ-receptor antagonists and reversed in δ-KO mice we tested the effects of the δ-subunit agonist GABAAR agonist THIP (10 nM – 10 μM), but it had no clear effect on evoked DA release on its own (Supplementary Fig. 3A, p>0.05). Thus, we tested the effects of the prototypic GABAAR agonist muscimol. Surprisingly, DA release was not sensitive to muscimol except at very high concentrations (Supplementary Fig. 3B, 100–350 μM; one-way ANOVA, F1,10=132.34, p<0.0001). Next, the effects of select GABA antagonists on evoked DA release and EtOH inhibition of evoked DA release were tested. The chloride channel GABA antagonist picrotoxin (100 μM) demonstrated some effectiveness in attenuating EtOH inhibition of DA release, however the results were not statistically significant (Supplementary Fig. 4; One-way ANOVA, F1,13=2.906, p=0.112). The GABAAR antagonist bicuculline (50 μM) had no effect on its own, and was ineffective at blocking EtOH inhibition of DA release (Supplementary Fig. 4; One-way ANOVA, F1,16=0.174, p=0.682). Benzodiazepines and barbiturates act as potent GABAA positive allosteric modulators, potentiating the effects of GABA at the GABAAR59 with distinct allosteric binding sites29. Benzodiazepine chlordiazepoxide (CDX; 30 μM) did not significantly affect evoked DA release or EtOH (80 mM) effects (Supplementary Fig. 4; One-way ANOVA, F1,13=1.269, p=0.280). The barbiturate pentobarbital (100 μM) had no effect on DA release and did not alter EtOH (80 mM) effects (Supplementary Fig. 4; One-way ANOVA, F1,13=0.043, p=0.838). Thus, GABA/muscimol effects on DA require high concentrations, suggestive of synaptic localization of receptors, and EtOH effects are not blocked by typical GABAA receptors, not modulated by typical GABAA allosteric modulators.
GABAB Agonist and Antagonist Effects on Evoked NAc Dopamine Release
Since typical GABAAR antagonists and modulators did not alter EtOH effects on evoked DA release, the effects of select GABAB receptor agonists were tested. The GABAB agonist baclofen (0.001 – 2 mM) did not significantly affect evoked DA release until 100 μM (Supplementary Fig. 3C; One-way ANOVA, F1,6=10.555, p=0.017). Since baclofen effects were limited, the effects of SKF97541 (GABAB agonist with ~10x baclofen potency) was tested47. SKF97541 (10 μM) did not reduce evoked NAc DA release (data not shown; One-way ANOVA, F1,11=0.460, p=0.512). Thus, GABAB effects on DA terminals are small and EtOH effects may involve GABAergic effects through atypical GABAA receptors.
Role of GABAA Rho-1 Receptors on Ethanol Inhibition of NAc Dopamine Release
None of the typical GABAA or GABAB modulators appeared to affect evoked DA release or EtOH inhibition of DA release. However, GABA δ-subtype receptors appeared to be involved in EtOH effects on evoked DA release. Therefore, we hypothesized that an atypical GABAAR was involved. Accordingly, the effects of the Rho-1 subunit antagonist TPMPA were examined on EtOH-mediated inhibition of DA release. TPMPA (10 μM) did not significantly alter evoked DA release in the NAc (Supplementary Fig. 5, p>0.05); however, it prevented EtOH-mediated inhibition of evoked DA release (Fig. 5A–B; F1,2=15.2, p=0.002; n=5). Thus, EtOH effects on NAc DA release involve atypical GABAA receptors.
Figure 5. Effect TPMPA on EtOH inhibition of evoked dopamine release ex vivo.
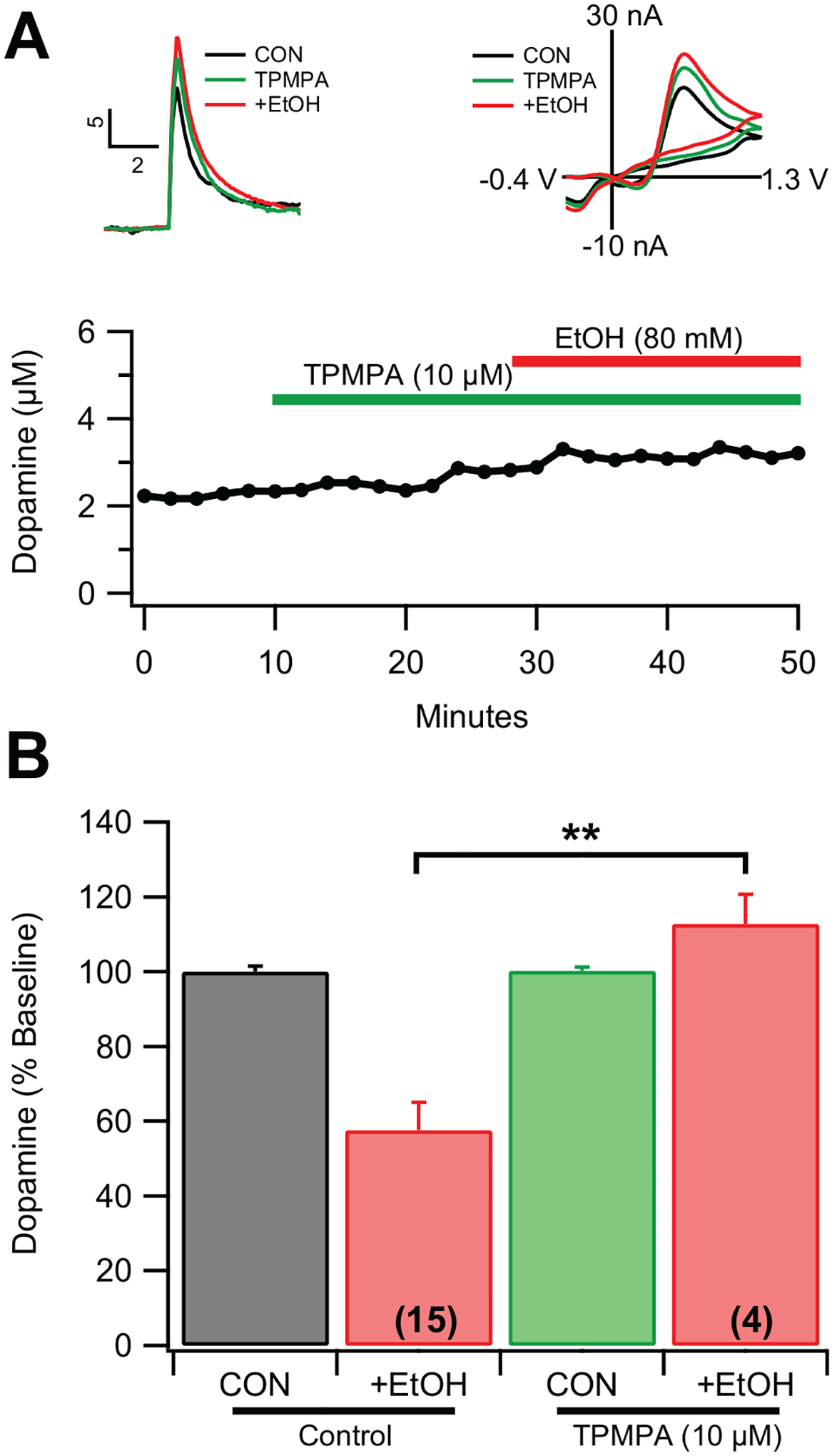
(A) Representative example of the effects of the of the Rho-1 subunit antagonist TPMPA (10 μM) on evoked DA release in the NAc ex vivo. Insets show representative superimposed I vs t and I vs V plots before and after TPMPA and EtOH. (B) Summary of TPMPA on 80 mM EtOH inhibition of evoked DA release. Values in parentheses represent n values. Values in parentheses represent n values. Asterisks *** indicate significance level p<0.001.
Immunohistochemistry of GABAA Rho-1 Subunit Receptors the Nucleus Accumbens and GABAergic Innervation of CINs by VTA GABA Neurons
We have shown in multiple reports that acute EtOH inhibits the firing rate of VTA GABA neurons in rats in vivo with an IC50 of 1.0 g/kg60–63, which is one order of magnitude more sensitive than EtOH effects on DA neurons64–66. In C57BL6 and CD-1 mice, VTA GABA neurons are even more sensitive to EtOH (IC50 of 0.25 g/kg)67. As VTA GABA neurons project to the NAc36, and GABA inhibition to the NAc appeared to be mediated by atypical GABAARs, we sought to determine the hodology of VTA GABA inputs to the NAc. Immunohistochemical experiments were performed to determine the expression of Rho-1 subunits and colocalization with ChAT expression in the NAc. Antibody labeling of Rho-1 subunits was observed in ChAT+ (solid arrows) and ChAT- (hollow arrows) neurons in the NAc core of C57/BL6 mice (Fig. 6A). GAD-GFP+ and GFP- neurons in the NAc core of GAD67-GFP mice also colabeled with Rho-1 subunits (Fig. 6B). GABA Rho-1 subunits typically homo-oligomerize to form GABAA Rho-1 receptors (formerly classified as GABACRs29), though Rho-1 subunits may also co-assemble with other GABAAR subunits, which would certainly affect their pharmacology32,33. Expression of Rho-1 receptors on CINs, and above pharmacology results, suggests that EtOH may be influencing GABA currents on CINs to affect DA release.
Figure 6. GABAAR Rho-1 subunit expression in NAc CINs.

(A) In C57/BL6 mice, GABAAR Rho-1 antibody-labeled (αRho-1) subunits (red) colocalized with immunostaining for choline acetyltransferase (αChAT) positive (green) cholinergic (ChAT+) neurons in the NAc core. Neuronal nuclear antigen labeling (αNeuN, blue) indicates general neuronal population in the NAc section. αRho-1 labeling was observed in some (solid arrow) but not all αChAT+ cells (putative αRho-1 negative ChAT+ neuron indicated by hollow arrow). αRho-1 colabeled with some αChAT- neurons (example indicated with asterisk). (B) In GAD67-GFP mice, Rho-1 subunits colabeled with GAD67-GFP negative (solid arrow) and positive (hollow arrow) neurons. Inset in B is a magnified image of the two neurons in the top center of the field. AC = anterior commissure. Calibration bar indicates 50 μm.
Effects of EtOH on GABAergic Inhibitory Synaptic Transmission to CINs
GABAergic synaptic transmission to CINs, and effects of EtOH were examined, including local inputs and distal GABAergic inputs from the VTA. Identification of CINs was reliable in in ChAT-ChR2-eYFP mice (Fig. 7A), and in non-labeled mice based on neuron morphology and presence of Ih in whole cell configuration (Fig. 7B, left), tonic firing and sensitivity to muscarine (10 μM; Fig. 8A). In VGAT-ChR2-eYFP mice, ChR2 stimulation (~470nm) reliably produced oIPSCs (Fig. 7B, right), likely due to direct depolarization of local GABA terminals onto CINs. Mean CIN oIPSC amplitude in VGAT-ChR2 mice was 740.9 ± 197.8 pA (n=10). EtOH (5–80 mM) significantly reduced (F5,45=4.5, p=0.002) oIPSCs in VGAT-ChR2 mice (Fig. 7B, right and bottom; 5 mM: reduced 19.9 ± 4.11 %; 5 mM: F1,19=21.1, p=0.0002; 10 mM: F1,19=31.4, p<0.0001; 20 mM: F1,19=25.4, p<0.0001; 40 mM: F1,19=30.6, p<0.0001; 80 mM: F1,15=24.2, p=0.0002).
Figure 7. EtOH reduces VTA GABAergic transmission to CINs.

(A) IR-DIC image of a patched CIN in the NAc slice preparation from a ChAT-ChR2-eYFP mouse. (B) CINs could be identified by presence of an Ih current (top; −60 to 120 mV step). Blue light stimulation (470nm) induced oIPSCs in NAc CINs of VGAT-ChR2 mice. Ethanol (5–80 mM) significantly decreases oIPSCs in CINs in VGAT-ChR2 mice. (C) In VGAT-Cre/GAD67-GFP mice injected with AAV-flex-mRuby2 into the VTA punctate mRuby labeling was evident on both CINs and putative GABAergic MSNs. Color wheel shows relative number of ChAT+ and GAD67+ neurons that demonstrated adjacent mRuby labeling. (D) Kernel density estimation (KDE) values were used to separate cells expressing AAV-flex-mRuby2 from cells that did not. Values to the right of the cut-off (blue dotted line) indicate cells expressing AAV-flex-mRuby2. Note the bimodal distribution. (E) Representative traces showing that optical stimulation in VGAT-Cre/GAD67-GFP mice injected with AAV-DIO-ChR2 into the VTA, with oIPSCs reduced by 5 and 10 mM EtOH. (F) Summary of VTA-NAc oIPSCs in CINs and their response to 5 mM EtOH. Asterisks *,** indicate significance levels p<0.05 and p<0.01, respectively.
Figure 8: EtOH markedly enhances CIN firing rate with block by TPMPA.

(A) CINs visualized in ChAT mice are characterized by tonic firing and auto-receptor inhibition by the M2 agonist muscarine (10 μM). Insets are 5 sec representative spike recordings before (a), during (b), after (c) muscarine (wash). Times for (a, b, c) are indicated on the representative rate meter below. (B) In identified CINs, firing rate is enhanced by increasing concentrations of EtOH. (C) Block of EtOH enhancement of CIN firing rate by TPMPA (10 μM) in a representative neuron. (D) Summary of TPMPA effects on EtOH enhancement of CIN firing rate by TPMPA. Asterisks **,*** indicate significance levels p<0.01 and p<0.001, respectively.
Since VTA GABA neurons also project to the NAc36, the effects of EtOH on these inputs was tested. VGAT-Cre/GAD67-GFP mice were injected bilaterally with AAV-flex-mRuby2 into the VTA and mRuby puncta were observed near ChAT+ and GAD67+ neurons (Fig. 7C,D). In order to determine ROIs that were positive for virus staining, the distribution of mean fluorescent intensity (MFI) was visualized in a histogram (Fig. 7D). A clear bimodal distribution was observed. Kernel density estimation (KDE) was used to approximate a cutoff point for virus positive and virus negative ROIs. The minimum point between the two peaks of the bimodal distribution was used as an approximate cutoff. This usage is conservative, and likely underestimates the number of virus positive neurons. This imaging data provide supporting evidence for VTA GABA innervation of NAc CINs.
In VGAT-Cre/GAD67-GFP mice injected in the VTA with AAV-DIO-ChR2, GABAergic oIPSCs were observed in CINs (Fig. 7E), with mean oIPSC amplitude at 207.0 ± 46.9 pA (n=9). The VTA-NAc stimulated oIPSCs were inhibited by 5 mM EtOH, with a 25.5 ± 7.7% decrease in oIPSC amplitude (F(1,15)=10.9, p=0.005; Fig. 7E,F). This was surprising, given that EtOH typically enhances GABAergic synaptic transmission (e.g., IPSCs) in most systems25, including our studies with VTA GABA neurons68. Thus, VTA GABA neurons project onto CINs, and GABAergic input to CINs is reduced by EtOH administration.
Role of GABAA Rho-1 Subunit Receptors on Ethanol Enhancement of CIN Firing Fate
Since CIN oIPSCs are inhibited by EtOH, CIN firing may also be affected. Thus, the effects of EtOH on CIN firing rate were examined (Fig. 8). CINs are inhibited by muscarinic receptor agonists due to autoreceptor inhibition, which is a pharmacological way to distinguish them from other neurons in the striatum (Fig. 8A). Baseline firing rate of CINs was 0.47 ± 0.08 Hz (n=10). Consistent with a role for Rho-1 GABAARs in modulating CINs, TPMPA (10 μM) significantly increased CIN firing rate (139.4 ± 12.5%; F(1,13) = 7.99, p<0.01). CIN firing rate was markedly enhanced by increasing concentrations of EtOH (5–60 mM) in VGAT-Cre mice (Fig. 8B; F(5,90) = 26.3, p<0.0001). Interestingly, TPMPA pretreatment blocked EtOH-induced enhancement of CIN firing (Fig. 8C–D; 10 mM: F(1,14) = 17.2, p<0.001; 20 mM: F(1,15) = 13.1, p<0.003; 40 mM: F(1,13) = 14.2, p<0.003; 60 mM: F(1,12) = 33.3, p<0.0001). Thus, EtOH enhances NAc CIN firing rate through decreased activity in TPMPA sensitive GABAA receptors on CINs, supporting a role for atypical Rho-1 GABA receptors in EtOH effects.
Discussion
Ethanol reduced evoked DA release in the NAc of CD-1 mice both in vivo and ex vivo, similar to our previous findings in rats and C57/BL6 mice9–11. The higher doses necessary for DA inhibition (IC50 ex vivo ~80 mM and in vivo ~2g/kg or ~40mM) are likely involved in EtOH-mediated impairment of learning associated processes. However, ethanol impairment of learning processes is extremely complex and likely involves many different mechanisms (for review see69). We hypothesized that EtOH inhibition of DA release was mediated by co-activation of GABAergic inhibitory projections from the VTA. We first evaluated GABA and GLY on evoked DA release. Both inhibited DA release from 1–10 mM, which is a very high concentration, consistent with their low potency as agonists at GABA and GLY receptors, but consonant with their known concentrations in the synapse as neurotransmitters. GABA and GLY inhibition of DA release was blocked by PTX, suggesting that GABA and GLY receptor coupling to the chloride ionophore was involved. Surprising, none of the typical GABAAR or GABABR modulators significantly affected DA release. However, EtOH inhibition of DA release was blocked by the δ-subunit GABAAR antagonist Ro15–4513 and by the GLYR antagonist strychnine. Strychnine and Ro15–4513 produced nearly the same results in blocking EtOH’s effects on terminal DA release in the NAc. The similarities between GABA and GLY receptors and evidence for co-activation or cross-talk between these two receptors is substantial. At the molecular level, the two inhibitory receptor sites GABA and GLY are shown to function almost identically by forming a pseudo-ring that binds the amino acids ARG and GLU. This binding enacts a conformational change that puts both receptors in an optimum position over the chloride ion channel and attracting chloride ions70. GABA and GLY receptors are members of the cys-loop receptor family and share in their role of inhibition. In many synapses, GABA and GLY are co-localized and co-released. Co-release of GABA and GLY were demonstrated to occur from the same vesicle in spinal cord slices71 and GABA can act as an endogenous ligand for synaptic GLYRs.57,72. Strong simultaneous activation of GABA and GLY receptors could cause an increase of local chloride concentration in the cell reducing further chloride influx73,74. The implication of cross-talk between GABA and GLY receptors in spinal cord slices, motorneurons, olfactory, and auditory synapses represent a pattern of co-activation characteristic of these two inhibitory receptors. Additionally, the chance of post-synaptic GABA and GLY receptor clustering is greatly increased due to the ability of gephyrin to cluster both of these receptors75,76. Further work needs to be done in order to identify the α4β3δ subtype of the GABAAR as one that is co-activated with GLYR. Previous work has shown the efficacy of Ro15–4513 to antagonize low to moderate doses of EtOH enhancement of δ-subunit-containing GABAARs, as well as implicating them as the most rapidly regulated in response to high-dose EtOH or chronically exposed EtOH in rats77. It has been shown that reduction in the expression of the δ GABAAR subunit in the NAc decreases alcohol intake78.
GABA receptor sensitivity is highly dependent upon subunit composition and where those receptors are located. For instance, δ-subunit containing receptors are typically found extrasynpatically and typically have a high sensitivity to GABA79–81. Indeed, extrasynaptic (i.e. δ containing) GABAA receptors are typically highly sensitive to GABA, with an EC50 below 1 μM (α6β3δ are ~300nM). In contrast, synaptic (i.e. non-δ containing) GABA receptors are relatively insensitive to GABA and GABA like ligands (such as muscimol), which is thought to contribute to the rapid dissociation required fast synaptic transmission80. The GABA EC50 values for synaptic receptors are in the 10–100 μM range80–82. This is really surprising in the context of the present results where δ antagonism/knockout results in a preclusion of ethanol’s effects, but GABA agonists have a low potency. Combined with data showing a similar blocking effect with the rho antagonist TPMPA, this suggests that the current composition of GABA receptors doesn’t follow typical GABA pharmacology.
As VTA GABA neurons project to the NAc, we sought to further evaluate the hodology and pharmacology of GABAergic projections and their role in EtOH inhibition and found that Rho-1 receptors were also involved in EtOH effects, suggesting an atypical GABAAR in the NAc. Ethanol induced reductions in GABA currents in CINs are associated with increases in CIN firing that are prevented by the Rho-1 GABAAR antagonist TPMPA. From previous results, blocking α6 subunit containing (α6*) nAChRs attenuates both the high-dose inhibitory effects of EtOH9,11 and low-dose (5–10 mM) enhancement of spontaneous DA release by EtOH8, suggesting overlap in the mechanisms involved in these opposing effects and the GABAergic effects observed herein. Striatal CINs are powerful heterosynaptic regulators of DA terminal function. CIN activation results in DA release due to nAChR-mediated depolarization of DA terminals7,8,83. CINs receive both local and distal GABAergic input, and the present results highlight the importance of GABA regulation of CIN activity in EtOH’s indirect actions on DA terminals.
Cholinergic activity and underlying modulation of DA release is highly regulated and increases in acetylcholine release are associated with both increases and decreases in DA release that appear to be concentration dependent. Low concentrations of acetylcholinesterase inhibitors and positive allosteric modulation of nAChRs results in increased DA release84. In contrast, high concentrations of acetylcholinesterase inhibitors results in decreased evoked DA release84,85. Further, high concentrations of acetylcholine (1mM, via localized puff application) results in decreased evoked DA release, an effect mimicked by desensitizing concentrations of nicotine9,84,85. We have previously reported low concentration positive allosteric effects of EtOH at nAChRs (in heterologous expression systems), which was associated with increases in spontaneous dopamine release at lower concentrations of ethanol (5–10mM), an effect that disappeared at higher EtOH concentrations (>20mM)8. This suggests that the range for enhanced DA release from increased CIN firing is limited to lower EtOH concentrations, likely due in part to increased acetylcholine levels and subsequent nAChR desensitization at higher EtOH concentrations. Indeed, blocking nAChRs attenuates EtOH’s excitatory8 and inhibitory effects on DA release9,11. Since DA signals are smaller in the presence of nAChR blockers, it is important to note that the magnitude of the EtOH effect is not related to DA signal amplitude. This was tested previously
Ethanol’s effects are diverse, targeting many excitatory and inhibitory ion channels, including GLU, GABA, and nAChRs to name a few86. A gamut of GABAergic pharmacological agents was examined in order to determine other possible mechanisms for inhibition of DA release (for review of GABA receptor pharmacology, see29). GABA (1–10 mM) and muscimol (100–300 μM) reduced evoked DA release, albeit at higher concentrations, suggesting that NAc GABA effects are complex, and likely involve interactions with competing circuitry possibly distinguished by GABA subunit composition. GABAA modulating sedative-hypnotics CDX (30 μM) and pentobarbital (100 μM) did not reduce DA release nor attenuate the effects of EtOH on DA release. The typical GABAAR antagonist bicuculline and chloride channel blocker picrotoxin no effect on evoked DA release, and EtOH’s effects on DA release were not blocked by these antagonists. GABAB agonists had minimal effect on DA release, and thus antagonist interactions with EtOH were not examined. Others have shown that α4β3δ GABAARs have distinct pharmacology and are insensitive to modulation by classical benzodiazepine ligands like diazepam52, which is consistent with the lack of effect observed with CDX and pentobarbital. The competitive and atypical α4β3δ GABAAR selective antagonist Ro15–4513 effectively blocked EtOH inhibition of DA release in the NAc both ex vivo and in vivo. Ro15–4513 has reported effectiveness on EtOH intoxication87 and consumption88. The direct effects of Ro15–4513 on alcohol behavioral antagonism are somewhat controversial89, however, the compound’s potential interactions with EtOH include specific alcohol antagonist actions on δ-subunit containing GABAARs (for review see90). In δ−/− mice, EtOH superfusion increased evoked DA release, further implicating GABAA δ-subunits in EtOH’s inhibitory effects on evoked NAc DA transmission.
Atypical GABAA Rho receptors are expressed in many brain regions including the striatum. Furthermore, GABAA Rho-subunits may co-assemble with GABAA subunits, “which would mask the typical GABA Rho pharmacology”32. The high sequence conservation between Rho- and β-subunits has been hypothesized to “account for some GABA receptors in the CNS that exhibit atypical characteristics”33. The high correlation of our findings related to these characteristics of Rho-subunits and recent publications, indicate the possibility of Rho-subunits incorporating with other GABAAR subunits91. Additionally, the Allen Brain Atlas, which demonstrates in situ hybridization of protein expression, notes a significant distribution of GABRR1 (Rho-1 subunit) in the ventral striatum92, indicating that somata in this region express Rho-1 subunit mRNA and could therefore express such subunits. The immunohistochemistry studies herein indicate GABAAR Rho-1 subunits on cells that co-express choline acetyltransferase, supporting a model for GABAAR Rho-1 containing receptors postsynaptic to GABAergic terminals on CINs.
The selective, competitive Rho-1 antagonist TPMPA demonstrated a mild, but not significant, increase in evoked DA release on its own but completely blocked EtOH inhibition of DA release. This antagonist effect on EtOH-mediated inhibition of DA was similar to observed effects of strychnine and Ro15–4513, supporting a common mechanism of action. Similar to δ-subunit pharmacology, Rho-1 subunit pharmacology is hampered by the lack of specific agonist availability. Indeed, the Rho-1 agonist TACA was tested, but did not produce any effect on either evoked DA release or on EtOH inhibition of DA release, which may be due to atypical subunit composition. Thus, although CINs appear to express Rho-1 containing GABAARs, current pharmacological tools due not support a known subunit composition.
Recently, CINs have been implicated in EtOH-mediated effects on DA levels in the NAc, as measured by microdialysis, with cholinergic antagonists preventing EtOH-induced DA increases93. Ethanol has also been shown to inhibit CINs in the dorsal striatum94. In contrast, EtOH enhanced NAc CIN firing, suggesting that regional effects may exist that could be due to local GABA subunit composition on CINs amongst other factors. Interestingly, the increased CIN firing observed was associated with decreased GABA input. Specifically, GABAergic oIPSCs were reduced by EtOH. Reductions were observed in VGAT-ChR2 mice, and in VGAT-Cre/GAD67-GFP mice with AAV-DIO-ChR2 injected into the VTA. In the former experiments, GABA terminals were activated in every GABA terminal to CINs, while the latter experiments selectively activated GABA terminals from VTA GABA neurons. Since EtOH reduced NAc oIPSCs in CINs in both VGAT-ChR2 and VGAT-Cre/GAD67-GFP mice and concomitantly enhanced CIN firing rate, EtOH appears to reduce general GABAergic transmission to CINs to elevate CIN firing.
The present results indicate an interesting and developing story regarding the effects of EtOH on evoked DA release from terminals in the NAc involving an atypical Rho-1 subunit containing GABAARs. This conclusion is supported by both pharmacological and immunohistochemical data, but the presence of these receptors has also been suggested by others32,91,95,96. GABA receptors composed of the Rho-1 subunit possess a markedly different pharmacological profile and are characterized by unique inhibitory responses to EtOH97. In fact, Mihic et al. demonstrated a dose response curve and IC50 of EtOH on current recordings closely identical to our findings demonstrating reduction of GABAergic transmission to CINs, with its accompanying enhancement of CIN firing rate, and ultimately modulation of DA release97. However, CIN modulation of DA release is complex because of desensitization of nAChRs at high acetylcholine release levels. Regardless, Rho-1 GABAARs are likely involved in CIN modulation of DA release as superfusion of the Rho-1 antagonist TPMPA prevented EtOH mediated inhibition of NAc CIN firing and EtOH effects on evoked DA release.
Given the previous published data expressing the high sequence similarity of Rho- and β-subunits33, the pharmacological results herein suggest the presence of an atypical GABAAR in the NAc that modulates DA release. While the full subunit composition of this atypical GABAAR will need to be determined, the pharmacological and immunohistohemical evidence herein support a composition of GABAARs containing Rho-1 and δ-subunits (ρ1δ*), likely also containing two α4-subuints and possibly a β-subunit (α4βρ1δ). A similar subunit composition has been observed in medium spiny neurons98, albeit Rho-1 subunits were not observed in CINs in this previous study. However, the proposed atypical GABAAR is strongly supported by our pharmacological, molecular, physiological, neurochemical, and immunohistochemistry data, in addition to multiple recent publications that suggest the possibility of atypical GABAARs of this type in the nervous system32,91,95–97. Atypical GABAARs appear to underlie EtOH’s inhibitory effects on DA transmission in the NAc, at least at high doses. Future work will need to be performed to confirm the exact identity of this atypical receptor, but the evidence of the techniques demonstrated herein strongly confirm an atypical GABAAR with Rho-1 and δ subunits.
Supplementary Material
Acknowledgments:
Funding for this study was provided by NIH NIAAA grants AA020439 to JTY and AA020919 and DA035958 to SCS. The authors have no other conflicts of interest to declare.
Abbreviations
- CFE
carbon fiber electrode
- CIN
cholinergic interneuron
- DA
dopamine
- EtOH
ethanol
- GABA
gamma aminobutyric acid
- GLU
glutamate
- MFB
medial forebrain bundle
- NAc
nucleus accumbens
- nAChR
nicotinic acetylcholine receptor
- oIPSC
optogenetic inhibitory postsynaptic potential
- VTA
ventral tegmental area
- WT
wild type
References
- 1.Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5(6):483–494. [DOI] [PubMed] [Google Scholar]
- 2.Blomqvist O, Ericson M, Engel JA, Soderpalm B. Accumbal dopamine overflow after ethanol: localization of the antagonizing effect of mecamylamine. Eur J Pharmacol. 1997;334(2–3):149–156. [DOI] [PubMed] [Google Scholar]
- 3.Imperato A, Di Chiara G. Preferential stimulation of dopamine release in the nucleus accumbens of freely moving rats by ethanol. J Pharmacol Exp Ther. 1986;239(1):219–228. [PubMed] [Google Scholar]
- 4.Blanchard BA, Steindorf S, Wang S, Glick SD. Sex differences in ethanol-induced dopamine release in nucleus accumbens and in ethanol consumption in rats. Alcohol Clin Exp Res. 1993;17(5):968–973. [DOI] [PubMed] [Google Scholar]
- 5.Cheer JF, Wassum KM, Sombers LA, et al. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci. 2007;27(4):791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson DL, Howard EC, McConnell S, Gonzales RA, Wightman RM. Disparity between tonic and phasic ethanol-induced dopamine increases in the nucleus accumbens of rats. Alcohol Clin Exp Res. 2009;33(7):1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yorgason JT, Zeppenfeld DM, Williams JT. Cholinergic Interneurons Underlie Spontaneous Dopamine Release in Nucleus Accumbens. J Neurosci. 2017;37(8):2086–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao F, Chen D, Ma X, et al. Alpha6-containing nicotinic acetylcholine receptor is a highly sensitive target of alcohol. Neuropharmacology. 2019;149:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schilaty ND, Hedges DM, Jang EY, et al. Acute ethanol inhibits dopamine release in the nucleus accumbens via alpha6 nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 2014;349(3):559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yorgason JT, Ferris MJ, Steffensen SC, Jones SR. Frequency-dependent effects of ethanol on dopamine release in the nucleus accumbens. Alcohol Clin Exp Res. 2014;38(2):438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yorgason JT, Rose JH, McIntosh JM, Ferris MJ, Jones SR. Greater ethanol inhibition of presynaptic dopamine release in C57BL/6J than DBA/2J mice: Role of nicotinic acetylcholine receptors. Neuroscience. 2015;284:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Budygin EA, Mathews TA, Lapa GB, Jones SR. Local effects of acute ethanol on dopamine neurotransmission in the ventral striatum in C57BL/6 mice. Eur J Pharmacol. 2005;523(1–3):40–45. [DOI] [PubMed] [Google Scholar]
- 13.Budygin EA, Phillips PE, Robinson DL, Kennedy AP, Gainetdinov RR, Wightman RM. Effect of acute ethanol on striatal dopamine neurotransmission in ambulatory rats. J Pharmacol Exp Ther. 2001;297(1):27–34. [PubMed] [Google Scholar]
- 14.Budygin EA, Phillips PE, Wightman RM, Jones SR. Terminal effects of ethanol on dopamine dynamics in rat nucleus accumbens: an in vitro voltammetric study. Synapse. 2001;42(2):77–79. [DOI] [PubMed] [Google Scholar]
- 15.Hedges DM, Yorgason JT, Brundage JN, et al. Corticotropin releasing factor, but not alcohol, modulates norepinephrine release in the rat central nucleus of the amygdala. Neuropharmacology. 2020;179:108293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soderpalm B, Ericson M. Neurocircuitry Involved in the Development of Alcohol Addiction: The Dopamine System and its Access Points. Curr Top Behav Neurosci. [DOI] [PubMed] [Google Scholar]
- 17.Melis M, Diana M, Enrico P, Marinelli M, Brodie MS. Ethanol and acetaldehyde action on central dopamine systems: mechanisms, modulation, and relationship to stress. Alcohol. 2009;43(7):531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ, Deitrich RA. Enzymatic mechanisms of ethanol oxidation in the brain. Alcohol Clin Exp Res. 2006;30(9):1500–1505. [DOI] [PubMed] [Google Scholar]
- 19.Soderpalm B, Ericson M, Olausson P, Blomqvist O, Engel JA. Nicotinic mechanisms involved in the dopamine activating and reinforcing properties of ethanol. Behav Brain Res. 2000;113(1–2):85–96. [DOI] [PubMed] [Google Scholar]
- 20.Adermark L, Clarke RB, Olsson T, Hansson E, Soderpalm B, Ericson M. Implications for glycine receptors and astrocytes in ethanol-induced elevation of dopamine levels in the nucleus accumbens. Addiction biology. 2011;16(1):43–54. [DOI] [PubMed] [Google Scholar]
- 21.Clarke RB, Soderpalm B, Lotfi A, Ericson M, Adermark L. Involvement of Inhibitory Receptors in Modulating Dopamine Signaling and Synaptic Activity Following Acute Ethanol Exposure in Striatal Subregions. Alcohol Clin Exp Res. 2015;39(12):2364–2374. [DOI] [PubMed] [Google Scholar]
- 22.Jonsson S, Adermark L, Ericson M, Soderpalm B. The involvement of accumbal glycine receptors in the dopamine-elevating effects of addictive drugs. Neuropharmacology. 2014;82:69–75. [DOI] [PubMed] [Google Scholar]
- 23.Molander A, Soderpalm B. Accumbal strychnine-sensitive glycine receptors: an access point for ethanol to the brain reward system. Alcohol Clin Exp Res. 2005;29(1):27–37. [DOI] [PubMed] [Google Scholar]
- 24.Soderpalm B, Lof E, Ericson M. Mechanistic studies of ethanol’s interaction with the mesolimbic dopamine reward system. Pharmacopsychiatry. 2009;42 Suppl 1:S87–94. [DOI] [PubMed] [Google Scholar]
- 25.Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther. 2006;111(3):533–554. [DOI] [PubMed] [Google Scholar]
- 26.Liang J, Olsen RW. Alcohol use disorders and current pharmacological therapies: the role of GABA(A) receptors. Acta pharmacologica Sinica. 2014;35(8):981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enoch MA. The role of GABA(A) receptors in the development of alcoholism. Pharmacol Biochem Behav. 2008;90(1):95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60(3):243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bormann J. The ‘ABC’ of GABA receptors. Trends in pharmacological sciences. 2000;21(1):16–19. [DOI] [PubMed] [Google Scholar]
- 30.Enz R, Cutting GR. GABAC receptor rho subunits are heterogeneously expressed in the human CNS and form homo- and heterooligomers with distinct physical properties. Eur J Neurosci. 1999;11(1):41–50. [DOI] [PubMed] [Google Scholar]
- 31.Hartmann K, Stief F, Draguhn A, Frahm C. Ionotropic GABA receptors with mixed pharmacological properties of GABAA and GABAC receptors. Eur J Pharmacol. 2004;497(2):139–146. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Delgado G, Estrada-Mondragon A, Miledi R, Martinez-Torres A. An Update on GABA rho Receptors. Curr Neuropharmacol. 2010;8(4):422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milligan CJ, Buckley NJ, Garret M, Deuchars J, Deuchars SA. Evidence for inhibition mediated by coassembly of GABA(A) and GABA(C) receptor subunits in native central neurons. Journal of Neuroscience. 2004;24(33):7241–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan Y, Qian H. Interactions between rho and gamma2 subunits of the GABA receptor. J Neurochem. 2005;94(2):482–490. [DOI] [PubMed] [Google Scholar]
- 35.Pan ZH, Zhang D, Zhang X, Lipton SA. Evidence for coassembly of mutant GABAC rho1 with GABAA gamma2S, glycine alpha1 and glycine alpha2 receptor subunits in vitro. Eur J Neurosci. 2000;12(9):3137–3145. [DOI] [PubMed] [Google Scholar]
- 36.Brown MT, Tan KR, O’Connor EC, Nikonenko I, Muller D, Luscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492(7429):452–456. [DOI] [PubMed] [Google Scholar]
- 37.van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012;73(6):1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mihalek RM, Bowers BJ, Wehner JM, et al. GABA(A)-receptor delta subunit knockout mice have multiple defects in behavioral responses to ethanol. Alcohol Clin Exp Res. 2001;25(12):1708–1718. [PubMed] [Google Scholar]
- 39.Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. The Journal of comparative neurology. 2003;467(1):60–79. [DOI] [PubMed] [Google Scholar]
- 40.Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20(22):8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies J, Francis AA, Jones AW, Watkins JC. 2-Amino-5-phosphonovalerate (2APV), a potent and selective antagonist of amino acid-induced and synaptic excitation. Neurosci Lett. 1981;21(1):77–81. [DOI] [PubMed] [Google Scholar]
- 42.Eccles JC, Schmidt R, Willis WD. Pharmacological Studies on Presynaptic Inhibition. J Physiol. 1963;168:500–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wafford KA, Thompson SA, Thomas D, Sikela J, Wilcox AS, Whiting PJ. Functional characterization of human gamma-aminobutyric acidA receptors containing the alpha 4 subunit. Mol Pharmacol. 1996;50(3):670–678. [PubMed] [Google Scholar]
- 44.Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J Neurosci. 1997;17(2):625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathers DA, Barker JL. (−)Pentobarbital opens ion channels of long duration in cultured mouse spinal neurons. Science. 1980;209(4455):507–509. [DOI] [PubMed] [Google Scholar]
- 46.Mathers DA, Wan X, Puil E. Barbiturate activation and modulation of GABA(A) receptors in neocortex. Neuropharmacology. 2007;52(4):1160–1168. [DOI] [PubMed] [Google Scholar]
- 47.Bon C, Galvan M. Electrophysiological actions of GABAB agonists and antagonists in rat dorso-lateral septal neurones in vitro. British journal of pharmacology. 1996;118(4):961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ragozzino D, Woodward RM, Murata Y, Eusebi F, Overman LE, Miledi R. Design and in vitro pharmacology of a selective gamma-aminobutyric acidC receptor antagonist. Mol Pharmacol. 1996;50(4):1024–1030. [PubMed] [Google Scholar]
- 49.Sur C, Wafford KA, Reynolds DS, et al. Loss of the major GABA(A) receptor subtype in the brain is not lethal in mice. J Neurosci. 2001;21(10):3409–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mierlak D, Farb DH. Modulation of neurotransmitter receptor desensitization: chlordiazepoxide stimulates fading of the GABA response. J Neurosci. 1988;8(3):814–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson AC, Williams SB, Pistorius SS, et al. Ventral Tegmental Area GABA Neurons Are Resistant to GABA(A) Receptor-Mediated Inhibition During Ethanol Withdrawal. Front Neurosci. 2018;12:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. British journal of pharmacology. 2002;136(7):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marowsky A, Vogt KE. Delta-subunit-containing GABAA-receptors mediate tonic inhibition in paracapsular cells of the mouse amygdala. Front Neural Circuits. 2014;8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzdak PD, Paul SM, Crawley JN. Effects of Ro15–4513 and other benzodiazepine receptor inverse agonists on alcohol-induced intoxication in the rat. J Pharmacol Exp Ther. 1988;245(3):880–886. [PubMed] [Google Scholar]
- 55.Yorgason JT, Espana RA, Jones SR. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods. 2011;202(2):158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fucile S, de Saint Jan D, David-Watine B, Korn H, Bregestovski P. Comparison of glycine and GABA actions on the zebrafish homomeric glycine receptor. J Physiol. 1999;517 (Pt 2):369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu T, Rubio ME, Trussell LO. Glycinergic transmission shaped by the corelease of GABA in a mammalian auditory synapse. Neuron. 2008;57(4):524–535. [DOI] [PubMed] [Google Scholar]
- 58.Matsubayashi H, Alkondon M, Pereira EF, Swanson KL, Albuquerque EX. Strychnine: a potent competitive antagonist of alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 1998;284(3):904–913. [PubMed] [Google Scholar]
- 59.Carter CR, Kozuska JL, Dunn SM. Insights into the structure and pharmacology of GABA(A) receptors. Future medicinal chemistry. 2010;2(5):859–875. [DOI] [PubMed] [Google Scholar]
- 60.Gallegos RA, Criado JR, Lee RS, Henriksen SJ, Steffensen SC. Adaptive responses of GABAergic neurons in the ventral tegmental area to chronic ethanol. J Pharmacol Exp Ther. 1999;291:1045–1053. [PubMed] [Google Scholar]
- 61.Steffensen SC, Walton CH, Hansen DM, Yorgason JT, Gallegos RA, Criado JR. Contingent and non-contingent effects of low-dose ethanol on GABA neuron activity in the ventral tegmental area. Pharmacology, biochemistry, and behavior. 2009;92(1):68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stobbs SH, Ohran AJ, Lassen MB, Allison DW, Brown JE, Steffensen SC. Ethanol suppression of ventral tegmental area GABA neuron electrical transmission involves NMDA receptors. J Pharmacol Exp Ther. 2004;311(1):282–289. [DOI] [PubMed] [Google Scholar]
- 63.Ludlow KH, Bradley KD, Allison DW, et al. Acute and chronic ethanol modulate dopamine D2-subtype receptor responses in ventral tegmental area GABA neurons. Alcohol Clin Exp Res. 2009;33(5):804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mereu GK -W. Y, Gessa G, Naes L, Westfall T. Preferential stimulation of ventral tegmental area dopaminergic neurons by nicotine. Eur J Pharmacol. 1987;141:395–399. [DOI] [PubMed] [Google Scholar]
- 65.Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Research. 1983;277:119–127. [DOI] [PubMed] [Google Scholar]
- 66.Brodie MS, Appel SB. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin Exp Res. 1998;22(1):236–244. [PubMed] [Google Scholar]
- 67.Steffensen SC, Bradley KD, Hansen DM, et al. The role of connexin-36 gap junctions in alcohol intoxication and consumption. Synapse. 2010;65(8):695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steffensen SC, Shin SI, Nelson AC, et al. alpha6 subunit-containing nicotinic receptors mediate low-dose ethanol effects on ventral tegmental area neurons and ethanol reward. Addict Biol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Skike CE, Goodlett C, Matthews DB. Acute alcohol and cognition: Remembering what it causes us to forget. Alcohol. 2019;79:105–125. [DOI] [PubMed] [Google Scholar]
- 70.Aprison MH, Galvez-Ruano E, Lipkowitz KB. Comparison of binding mechanisms at cholinergic, serotonergic, glycinergic and GABAergic receptors. Journal of neuroscience research. 1996;43(2):127–136. [DOI] [PubMed] [Google Scholar]
- 71.Jonas P, Bischofberger J, Sandkuhler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281(5375):419–424. [DOI] [PubMed] [Google Scholar]
- 72.Singer JH. GABA is an endogenous ligand for synaptic glycine receptors. Neuron. 2008;57(4):475–477. [DOI] [PubMed] [Google Scholar]
- 73.Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors [see comments]. Science. 1995;269(5226):977–981. [DOI] [PubMed] [Google Scholar]
- 74.Segal M, Barker JL. Rat hippocampal neurons in culture: voltage-clamp analysis of inhibitory synaptic connections. Journal of Neurophysiology. 1984;52(3):469–487. [DOI] [PubMed] [Google Scholar]
- 75.Kirsch J, Betz H. The postsynaptic localization of the glycine receptor-associated protein gephyrin is regulated by the cytoskeleton. J Neurosci. 1995;15(6):4148–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nature neuroscience. 1998;1(7):563–571. [DOI] [PubMed] [Google Scholar]
- 77.Olsen RW. Extrasynaptic GABAA receptors in the nucleus accumbens are necessary for alcohol drinking. Proc Natl Acad Sci U S A. 2011;108(12):4699–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nie H, Rewal M, Gill TM, Ron D, Janak PH. Extrasynaptic delta-containing GABAA receptors in the nucleus accumbens dorsomedial shell contribute to alcohol intake. Proc Natl Acad Sci U S A. 2011;108(11):4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18(5):1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6(3):215–229. [DOI] [PubMed] [Google Scholar]
- 81.Meera P, Wallner M, Otis TS. Molecular basis for the high THIP/gaboxadol sensitivity of extrasynaptic GABA(A) receptors. Journal of neurophysiology. 2011;106(4):2057–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benkherouf AY, Taina KR, Meera P, et al. Extrasynaptic delta-GABAA receptors are high-affinity muscimol receptors. J Neurochem. 2019;149(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brimblecombe KR, Threlfell S, Dautan D, Kosillo P, Mena-Segovia J, Cragg SJ. Targeted Activation of Cholinergic Interneurons Accounts for the Modulation of Dopamine by Striatal Nicotinic Receptors. eNeuro. 2018;5(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang L, Zhou FM, Dani JA. Cholinergic drugs for Alzheimer’s disease enhance in vitro dopamine release. Mol Pharmacol. 2004;66(3):538–544. [DOI] [PubMed] [Google Scholar]
- 85.Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4(12):1224–1229. [DOI] [PubMed] [Google Scholar]
- 86.Dopico AM, Lovinger DM. Acute alcohol action and desensitization of ligand-gated ion channels. Pharmacol Rev. 2009;61(1):98–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suzdak PD, Glowa JR, Crawley JN, Schwartz RD, Skolnick P, Paul SM. A selective imidazobenzodiazepine antagonist of ethanol in the rat. Science. 1986;234(4781):1243–1247. [DOI] [PubMed] [Google Scholar]
- 88.Melon LC, Boehm SL 2nd. GABAA receptors in the posterior, but not anterior, ventral tegmental area mediate Ro15–4513-induced attenuation of binge-like ethanol consumption in C57BL/6J female mice. Behavioural brain research. 2011;220(1):230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Britton KT, Ehlers CL, Koob GF. Is ethanol antagonist Ro15–4513 selective for ethanol? Science. 1988;239(4840):648–650. [DOI] [PubMed] [Google Scholar]
- 90.Wallner M, Hanchar HJ, Olsen RW. Alcohol selectivity of beta3-containing GABAA receptors: evidence for a unique extracellular alcohol/imidazobenzodiazepine Ro15–4513 binding site at the alpha+beta- subunit interface in alphabeta3delta GABAA receptors. Neurochem Res. 2014;39(6):1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blednov YA, Benavidez JM, Black M, et al. GABAA receptors containing rho1 subunits contribute to in vivo effects of ethanol in mice. PLoS One. 2014;9(1):e85525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–176. [DOI] [PubMed] [Google Scholar]
- 93.Loften A, Adermark L, Ericson M, Soderpalm B. An acetylcholine-dopamine interaction in the nucleus accumbens and its involvement in ethanol’s dopamine-releasing effect. Addict Biol. 2020:e12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blomeley CP, Cains S, Smith R, Bracci E. Ethanol affects striatal interneurons directly and projection neurons through a reduction in cholinergic tone. Neuropsychopharmacology. 2011;36(5):1033–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frazao R, Nogueira MI, Wassle H. Colocalization of synaptic GABA(C)-receptors with GABA (A)-receptors and glycine-receptors in the rodent central nervous system. Cell Tissue Res. 2007;330(1):1–15. [DOI] [PubMed] [Google Scholar]
- 96.Rosas-Arellano A, Machuca-Parra AI, Reyes-Haro D, Miledi R, Martinez-Torres A. Expression of GABArho receptors in the neostriatum: localization in aspiny, medium spiny neurons and GFAP-positive cells. J Neurochem. 2012;122(5):900–910. [DOI] [PubMed] [Google Scholar]
- 97.Mihic SJ, Harris RA. Inhibition of rho1 receptor GABAergic currents by alcohols and volatile anesthetics. J Pharmacol Exp Ther. 1996;277(1):411–416. [PubMed] [Google Scholar]
- 98.Maguire EP, Macpherson T, Swinny JD, et al. Tonic inhibition of accumbal spiny neurons by extrasynaptic alpha4betadelta GABAA receptors modulates the actions of psychostimulants. J Neurosci. 2014;34(3):823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.