


Abstract
Transcription-associated cyclin-dependent kinases (CDKs) regulate the transcription cycle through sequential phosphorylation of RNA polymerase II (RNAPII). Herein, we report that dual inhibition of the highly homologous CDK12 and CDK13 impairs splicing of a subset of promoter-proximal introns characterized by weak 3′ splice sites located at larger distance from the branchpoint. Nascent transcript analysis indicated that these introns are selectively retained upon pharmacological inhibition of CDK12/13 with respect to downstream introns of the same pre-mRNAs. Retention of these introns was also triggered by pladienolide B (PdB), an inhibitor of the U2 small nucelar ribonucleoprotein (snRNP) factor SF3B1 that recognizes the branchpoint. CDK12/13 activity promotes the interaction of SF3B1 with RNAPII phosphorylated on Ser2, and disruption of this interaction by treatment with the CDK12/13 inhibitor THZ531 impairs the association of SF3B1 with chromatin and its recruitment to the 3′ splice site of these introns. Furthermore, by using suboptimal doses of THZ531 and PdB, we describe a synergic effect of these inhibitors on intron retention, cell cycle progression and cancer cell survival. These findings uncover a mechanism by which CDK12/13 couple RNA transcription and processing, and suggest that combined inhibition of these kinases and the spliceosome represents an exploitable anticancer approach.
Graphical Abstract
Graphical Abstract.
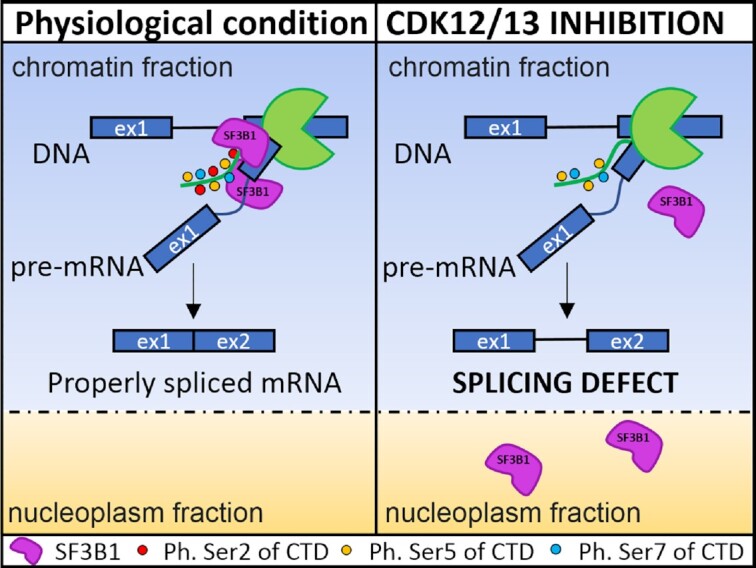
CDK12/13 promote splicing of proximal introns by enhancing the interaction between RNA polymerase II and the splicing factor SF3B1.
INTRODUCTION
Transcription of protein-coding RNAs and most non-coding RNAs is regulated by RNA polymerase II (RNAPII). The transcription cycle consists of initiation, promoter-proximal pausing, elongation and termination (1). All these steps need to be tightly controlled to ensure proper production of transcripts from each gene unit. A key node for the control of this transcription cycle is represented by the C-terminal domain (CTD) of RNAPII (2,3). In humans, the CTD is composed of 52 repeats of the YSPTSPS heptapeptide, which acts as a platform for the recruitment of factors that contribute to the regulation of RNAPII function and to co-transcriptional processing of the nascent transcripts, such as splicing of introns and cleavage and polyadenylation of the 3′ end (3,4). The flexible and modular function of the CTD is mainly achieved through phosphorylation of different residues of the heptapeptide by transcription-associated cyclin-dependent kinases (CDKs). These CDKs operate in a sequential manner during the transcription cycle to determine the transition from one phase to the next (1,4). Initial phosphorylation of Ser5 of the CTD by CDK7 allows RNAPII to escape from the gene promoter and to pause in the promoter-proximal region (5), thus favouring quality control and capping of the 5′ end of the transcript (1). Phosphorylation of Ser2 by CDK9 is then required to release RNAPII from the pausing zone and to start the elongation phase (1). Interestingly, Ser2 phosphorylation of the CTD steadily increases along the transcription unit and reaches a peak near the cleavage and polyadenylation (pA) site (2–4). Ser2 phosphorylation within the gene body is largely mediated by the homologous CDK12 and CDK13, which maintain a fast elongation rate of RNAPII and ensure transcription through the end the gene (6–9).
Recent evidence indicates that, in addition to regulating the transcription cycle, transcription-associated CDKs also modulate RNA processing (4,10). For instance, CDK12 and CDK13 contain an N-terminal arginine–serine (RS)-rich domain found in many splicing factors, which mediates their protein–protein interactions and promotes recruitment of the spliceosome to the splice sites (11). Moreover, CDK12/13 are localized in nuclear speckles where many splicing factors are also stored (12). These observations suggested that CDK12/13 could couple transcription with RNA processing events. In line with this hypothesis, genetic or pharmacological inhibition of CDK12 or CDK13 caused widespread defects in splicing of introns and led to selection of alternative last exons (ALEs) (7,8,13). Although the mechanism(s) involved in splicing regulation was not elucidated, these studies suggested that the reduced RNAPII elongation rate within long introns exposes the transcript to premature termination at cryptic intronic pA (IPA) sites (6–8). A role in splicing regulation was also recently proposed for CDK7, which phosphorylates several splicing factors, including the spliceosome-associated factors SF3B1 and U2AF65 (14). Accordingly, CDK7 inhibition caused defects in splicing (14). However, since CDK7 also phosphorylates CDK9, CDK12 and CDK13, it is unclear whether the effect was direct or mediated by other transcription-associated CDKs.
In this study, by using bioinformatics analyses and functional experiments, we show that pharmacologic inhibition of CDK12/13 selectively impairs splicing of a class of promoter-proximal introns characterized by weak 3′ splice sites that are distant from the branchpoint. Mechanistically, we demonstrate that CDK12/13 activity promotes the interaction of the U2 small nuclear ribonucleoprotein (snRNP) factor SF3B1 with RNAPII phosphorylated on Ser2 of the CTD. Inhibition of CDK12/13 disrupts this interaction, and impairs association of SF3B1 with chromatin and its recruitment to the 3′ splice site of target introns. Furthermore, we describe a synergic effect of the CDK12/13 inhibitor THZ531 and the SF3B1 inhibitor pladienolide B (PdB), which leads to enhanced intron retention (IR), cell cycle arrest and apoptosis. Thus, our studies uncover a CDK12/13-dependent mechanism of coupling between transcription and splicing and suggest a new approach to increase the efficacy of splicing inhibitors that are currently being tested in clinical trials as chemotherapeutic agents.
MATERIALS AND METHODS
Cell culture and treatments
MiaPaCa-2 and HEK293T cells were cultured in D5796 Dulbecco’s modified Eagle’s medium (DMEM; Sigma Aldrich), and MDA-MB-231 and OVCAR-3 cells in RPMI 1640 medium (Euroclone), all supplemented with 10% fetal bovine serum (FBS; Gibco), 100× non-essential amino acids (Thermo Fischer), 1% penicillin and 200× streptomycin and gentamicin (Thermo Fischer). Cells were grown in a 37°C humidified atmosphere of 5% CO2. Cells were treated with THZ1 (HY-80013), THZ531 (HY-103618), SY-1365 (HY-128587) and PdB (sc-391691) at the indicated doses, or dimethylsulphoxide (DMSO) as a control.
Production and transduction of lentiviral particles
The pLKO.1 plasmids containing short hairpin RNA (shRNA) sequences targeting CDK12 (5′ TTCAGAGTTATAGAGCCGAGC 3′), CDK13 (5′ TAAATCAGCAAGAAGACATCG 3′), CDK7 (5′ GCTGTAGAAGTGAGTTTGTAA 3′) and non-target control sequence were transfected with pCMV-dR8.2-dvpr and pCMV-VSV-G helper plasmids into HEK293T cells using Lipofectamine 2000 (ThermoFisher Scientific, Invitrogen). After 48 h, the supernatant containing lentiviral particles was collected and centrifuged at 3000 rpm for 5 min. MiaPaCa-2 cells were transduced with the supernatant in the presence of polybrene (8 μg/ml) for 24 h before replacement of medium supplemented with puromycin (1 μg/ml). Cells were collected 96 h post-transduction and RNA was isolated as described below.
Bioinformatics analysis
The GSE121273 and GSE222493 datasets were analysed for gene expression and splicing. Repartition of quality reads, inner distance size estimation, gene body coverage and strand specificity of the library were performed using FastQC v0.11.2, Picard-Tools v1.119, Samtools v1.0 and RSeQC v2.3.9. Reads were mapped using STAR v2.4.0f1 (15) on the human hg19 genome assembly, and read count was performed using feature Count from SubRead v1.5.0 and normalized with DESeq2 (16) using ERCC spike-in as the control gene. Gene expression estimation was performed as described previously (17) using Human FAST DB v2018_1 annotations. Genes were considered as expressed if their fragments per kilobase of transcript per million mapped reads (FPKM) value was greater than the FPKM of 98% of the intergenic regions (background), and only genes expressed in at least one of the two conditions were evaluated. For differential expression, we used as thresholds fold change (FC) ≥1.5 and P-value ≤0.05.
Splicing analyses were performed taking into account only exon reads and flanking exon–exon junction reads (‘EXON’ analysis) to potentially detect new alternative events that are differentially regulated (i.e. without limiting them to known alternative events). The analysis was also performed by taking into account known patterns (‘PATTERN’ analysis) using the FAST DB splicing patterns annotation. ‘EXON’ and ‘PATTERN’ analyses are based on the splicing-index calculation as described (19,20). Results were considered statistically significant for P-values ≤0.05 and FCs ≥1.5 for ‘PATTERN’ analysis, and P-values ≤0.01 and FCs ≥2.0 for ‘EXON’ analysis. Significant results from ‘EXON’ and ‘PATTERN’ analyses were merged to obtain a single list. Splice site scores were defined using MaxEntScan (20). Polypyrimidine tract length, score and distance of the branch point from the acceptor site were evaluated using SVM-BPfinder (21). The number and score of poly(A) sites were defined using APARENT (22).
Correlation of CDK7/12/13 expression with target genes in pancreatic adenocarcinoma [The Tumor Genome Atlas (TCGA), Firehose Legacy] was performed using the cBioPortal database (https://www.cbioportal.org/).
Reverse transcription–polymerase chain reaction (RT–PCR) and quantitative PCR analysis
Total RNA was extracted using Trizol reagent (Life Technologies-Thermo Fisher Scientific) or with the ‘Total RNA Mini Kit’ (Geneaid). After digestion with RNase-free DNase (Life Technologies-Thermo Fisher Scientific), RNA was resuspended in RNase-free water (Sigma Aldrich) and reverse transcribed (1 μg) using M-MLV reverse transcriptase (Promega) and oligo(dT) primers (Roche). 20 ng of cDNA was used as template for both conventional PCR analysis (GoTaq, Promega) and quantitative real-time PCR (qPCR) analysis (SYBR Green method, Roche). PCR primers are listed in Supplementary Table S1.
Protein extraction and western blot
Cell pellets were resuspended in RIPA buffer [50 mM Tris–HCl, 1% NP-40, 150 mM NaCl, 0.5% Na-deoxycholate, 2 mM EDTA and 0.1% sodium dodecylsulphate (SDS)] supplemented with 2 mM Na-orthovanadate, 0.5 mM sodium fluoride and Protease Inhibitor Cocktail (Sigma Aldrich), 1 mM dithiothreitol (DTT) or 1 mM TCEP [Tris(2-carboxyethyl)phosphine] for immunoprecipitation. After 10 min incubation on ice, extracts were sonicated at maximum intensity for 5 s, centrifuged for 10 min at 13 000 rpm (4°C) and supernatant fractions were resuspended in SDS–polyacrylamide gel electrophoresis (PAGE) sample buffer and boiled for 10 min.
For co-immunoprecipitation assays, nuclear extracts were obtained by lysing MiaPaCa-2 cells in RSB10 buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 2.5 mM MgCl2, 15 mM β-glycerophosphate, 1 mM TCEP, phosphatase and protease inhibitors) followed by 15 min incubation on ice. Cell lysates were then centrifuged for 8 min at 700 g at 4°C. The supernatant (cytoplasm) fraction was discarded, and the nuclear pellet was lysed in RSB100 buffer [10 mM Tris–HCl pH 7.4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% (v/v) Triton X-100, 15 mM β-glycerophosphate, 1 mM TCEP, phosphatase and protease inhibitors], sonicated, layered on a 30% sucrose pad in RSB100 and centrifuged for 15 min at 7000 g at 4°C. The supernatant (nuclear) fraction was collected, quantified and incubated at 4°C with the indicated antibodies or control mouse/rabbit IgG and Protein G magnetic beads (Life Technologies-Thermo Fisher Scientific). Beads were washed three times with RBS100 buffer, eluted in SDS–sample buffer and analysed by SDS–PAGE.
Cell fractionation to isolate chromatin-associated, nucleoplasmic and cytoplasmic proteins was performed as previously described (23). The isolated fractions were separated by SDS–PAGE, transferred to polyvinylidene fluoride (PVDF) membranes and incubated with primary antibodies (overnight at 4°C) for the following proteins: RNAPII (Santa Cruz, 8WG16; Cell Signaling, 14958), SF3b155/SAP155 (Bethyl, A300-996A), phospho-ser2 RNAPII (Millipore, 04–1571), phospho-ser5 RNAPII (Millipore, 04–1572), TUBULIN (Cell Signaling, 2146), H3 (Abcam, ab1791), U1-70k (Santa Cruz, sc-390899), CDK12 (Cell Signaling, 11973S), CDK13 (Merck, sab2700810), CDK7 (Cell Signaling, 2916), PRP6 (Santa Cruz, sc-13253), PRP8 (Santa Cruz, sc-30207), U1A (Santa Cruz, sc-376027), PARP1 (Cell Signaling, 9542), γH2AX (Cell Signaling, BK9718S) and HSP90 (Santa Cruz, sc-13119). Secondary anti-mouse or anti-rabbit IgGs conjugated to horseradish peroxidase (Amersham, UK) were incubated for 1 h at room temperature (1:5000). Immunostained bands were detected by the chemiluminescence method (24).
Isolation of nascent transcripts
Transcription of cells was blocked by treatment with 100 μM 5,6-dichlorobenzimidazole riboside (DRB) for 6 h. In the last hour, 200 nM THZ531 was added and maintained for 30 min after DRB removal. In these last 30 min, nascent RNAs were labelled with 1000 μM 4-thiouridine (4sU). For isolation of 4sU-labelled RNA, 50 μg of total RNA extracted with Trizol (Life Technologies-Thermo Fisher Scientific) was biotinylated with EZ-Link Biotin-HPDP (Life Technologies-Thermo Fisher Scientific) and isolated using the μMacs Streptavidin Kit (Miltenyi) according to the manufacturer's instructions. RNA was resuspended in RNase-free water (Sigma Aldrich) and reverse transcribed using M-MLV reverse transcriptase (Promega) and random primers (Roche).
Cell viability and cytotoxicity assays
Cells were seeded in a 96-well plate and imaged at ×10 magnification in an IncuCyte SX5 Live-content imaging system (Essen Bioscience) at 37 °C with 5% CO2. For cell viability assay, the images were acquired every 6 h for 4 days (four images/well) and analysed using IncuCyte Cell-by-Cell analysis software to detect and quantify live cells. For cytotoxicity assays, cells were labelled with the Cytotox Green Dye (Sartorius) to detect the dying cells, and with Nuclight Rapid NIR (Sartorius) to detect the nuclei of all cells in the dish. Four images/well were acquired after 96 h and analysed using the IncuCyte Cell-by-Cell software.
Cell cycle analysis
Cells were fixed in 70% ethanol–phosphate-buffered saline (EtOH-PBS) and stored at –20°C overnight. The cell cycle was evaluated by flow cytometry using 7-aminoactinomycin D (7-AAD; 2.5 μg/ml, Biotium) in the presence of RNase A (1 μg/ml). A total of 20 000 events were counted with a CytoFlex flow cytometer (Beckman) and analysed using FlowJo v.10 software (Becton Dickinson).
RNA immunoprecipitation (RIP) and UV-cross-linking and RNA immunoprecipitation (CLIP) experiments
RNA immunoprecipitation (RIP) assays were performed as previously described (25) with some modifications. Briefly, MiaPaCa-2 nuclear extracts were pre-cleared for 1 h with Dynabeads Protein G (Life Technologies-Thermo Fisher Scientific) under rotation at 4°C. A total of 1 mg of nuclear extract was incubated with 3 mg of specific primary antibody (anti-SF3b155/SAP155, Bethyl) or IgGs (negative control, Sigma Aldrich) overnight at 4°C under rotation. Next, Dynabeads Protein G were added to the extracts and incubated for 2 h at 4°C under rotation. Immunoprecipitates were washed three times with RIPA buffer supplemented with Protease Inhibitor Cocktail (Sigma Aldrich) and RNase inhibitor 40 U/ml (Promega), resuspended in 100 μl of TE buffer (10 mM Tris, 1 mM EDTA pH 8.0) supplemented with 1% SDS, 10 mM DTT and 40 U/ml RNase inhibitor, and incubated for 1 h at 70°C to reverse cross-linking. After treatment with proteinase K (Roche) for 1 h at 55°C, RNA was isolated (miRNeasy micro kit, Qiagen), DNase treated (Qiagen), reverse transcribed (M-MLV reverse transcriptase, Promega) and used for qPCR analysis (SYBR Green method, Roche). Each sample was normalized with respect to its input.
CLIP experiments were performed as described (26). After irradiation with UV (400 mJ/cm2) on ice in PBS and preparation of the extracts (26), 1 mg/ml of extract from MiaPaCa-2 cells treated with 200 nM THZ531 or DMSO for 3 h was pre-cleared for 1 h and then immunoprecipitated using 3 μg of SF3b155/SAP155 (Bethyl, A300-996A), or IgGs as negative control, in the presence of Protein G Dynabeads (Thermo Fisher Scientific, Invitrogen) and 10 μl of RNase I (Ambion) diluted 1:1000. Then 20% of cell extract (0.2 mg) was collected and used as input. After 2 h of incubation, samples were washed and treated for 1 h with proteinase K (50 μg) at 55°C. RNA was isolated using the miRNeasy Micro Kit (Qiagen), reverse transcribed with random primers and used for qPCR analysis. Primers used are listed in Supplementary Table S1.
RESULTS
Inhibition of transcriptional CDKs causes widespread intron retention
CDK7, CDK12 and CDK13 have been recently shown to play a role in regulation of RNA processing (6–8). To further investigate this issue, we analysed a dataset of pancreatic ductal adenocarcinoma (PDAC) cells (MiaPaCa-2) treated with the CDK7/12/13 inhibitor THZ1 (6 h, 100 nM; GSE121273), as this cell line was shown to be highly sensitive to this drug (27). Bioinformatics analysis highlighted thousands of genes regulated by THZ1 at the alternative splicing (AS) level in MiaPaCa-2 cells. Most of these splicing-regulated genes (68,9%) were also regulated at the overall gene expression (GE) level (Figure 1A; Supplementary Table S2). In addition to the expected changes in the alternative first exon (19% of total events), which are related to transcriptional regulation, the most represented THZ1-regulated splicing patterns were ALEs (10%), IR (9%) and exon cassettes (8%) (Figure 1B; Supplementary Table S3). It is noteworthy that while THZ1-regulated alternative first exons were under-represented with respect to their expected frequency in the reference FAST-DB database (18), ALE, IR and exon cassette patterns were over-represented (Figure 1C). We also noted that more than half (53%) of the THZ1-modulated AS events were not annotated as alternative events (referred to as unannotated) in the FAST-DB database (Figure 1B). Manual inspection revealed that 90% of the unannotated events are intronic (Figure 1D; Supplementary Table S3). Thus, collectively, IR regulation accounted for >55% of total THZ1-modulated events (Figure 1E), indicating that CDK7/12/13 inhibition prevalently affects the splicing of alternative and constitutive introns.
Figure 1.

Inhibition of transcriptional CDKs causes widespread intron retention. (A) Venn diagram showing the overlap between genes regulated at the gene expression (GE) and alternative splicing (AS) level by THZ1 (6 h, 100 nM). Statistical analyses were performed by Fisher's exact test. (B) Pie chart showing percentages of the indicated splicing patterns affected by THZ1. (C) Bar graph showing percentages of events annotated in FAST-DB (white columns) and of those regulated by THZ1 treatment (grey columns) within each AS pattern. Statistical analyses of comparisons between THZ1-regulated events and their expected representation in the reference database were performed by modified Fisher's test. (D) Pie charts showing percentages of intronic or exonic events in unannotated events. (E) Pie charts showing percentages of intronic or exonic events in total events. (F) Pie charts showing percentages of up- and down-regulated introns in THZ1-treated cells. (G and H) Gene Ontology of up- and down-regulated IR events performed by the g-profiler tool.
Most IR events (70%) were down-regulated in cells treated with THZ1 (Figure 1F), suggesting that the widespread transcriptional block exerted by THZ1 (27) allows post-transcriptional splicing of these weaker introns. However, a substantial number of IR events (30%) were up-regulated (Figure 1F), indicating that inhibition of CDK7/12/13 impaired their splicing. Functional annotation of genes characterized by increased IR highlighted key tumorigenic pathways, such as ‘hippo signalling’, ‘cell cycle’ and ‘apoptosis’ (Figure 1G), whereas genes whose introns were down-regulated by THZ1 treatment were enriched in terms related to RNA transport and translation and to metabolic pathways (Figure 1H). qPCR analysis of eight arbitrarily selected genes (CDK12, TEAD1, SERPINB9, E2F3, CCNB1, LMF2, COMTD1 and DEF6) in independent MiaPaCa-2 cell samples indicated the reliability and reproducibility of the RNA-seq data and of our bioinformatics analysis (Supplementary Figure S1).
THZ1 treatment leads to widespread retention of introns 5′ of the regulated genes
Metagene analysis showed that up-regulated IR events were mainly distributed in the promoter-proximal region of genes, particularly in the first and second introns (45.6% of all up-regulated introns; Supplementary Table S4), whereas down-regulated introns tended to accumulate toward the distal region of genes (Figure 2A). The genes displaying up-regulation of the first or second intron were significantly more regulated at the GE level (higher fold difference) with respect to genes regulated in all other introns (Figure 2B). Moreover, they also showed a higher percentage of overlap with GE-regulated genes (Figure 2C, left panel). The larger extent of overlap with GE regulation was not due to their closer proximity to the transcription start site, because it was not observed in proximal introns that were down-regulated by THZ1 treatment (Figure 2C, right panel). These results suggest that increased retention of proximal introns upon treatment with THZ1 may impair expression of the full-length transcript. Indeed, visual inspection of select genes in this group highlighted the decrease in read coverage beyond the retained intron. As exemplified by the POLH and SOGA1 genes, read coverage in the first exon was almost unchanged in THZ1-treated cells, indicating that transcription started correctly, whereas it was significantly reduced beyond intron 1 in POLH and intron 2 in SOGA1 (Figure 2D). Analysis by qPCR confirmed that short-term treatment (6 h) with THZ1 leads to concomitant increase in the proximal intron and reduction in downstream portions of the transcript in both POLH and SOGA1 (Figure 2E–H), as well as in other genes (PCF11, NUFIP2, CDK12, CDK13, AEBP2 and DDX21) that shared these features (Supplementary Figure S2). These observations suggest that retention of proximal introns leads either to stalling of the RNAPII within the transcription unit or to premature termination of the transcript, thus causing the overall down-regulation of gene expression.
Figure 2.

THZ1 treatment leads to widespread retention of proximal introns. (A) Metagene representation of IR location within the gene body. (B) Bar graph showing GE fold change between the first and second introns and all other introns. Statistical analysis was performed by Student's test. (C) Bar graph showing percentages of IR genes that were regulated at the GE level or not between the first and second up-regulated introns and all other introns (left panel) and between the first and second up-regulated and down-regulated introns (right panel). Statistical analyses were performed by Fisher's exact test. (D) Graphic representation of POLH and SOGA1 genes showing the IR event in the first intron and the reduction in read coverage in downstream portions of the transcript after THZ1 treatment. (E–H) Visualization of the RNA-seq reads profile of the intron-retaining region and PCR primer strategy used in POLH (E) and SOGA1 (G) genes. Bar graphs showing the results of qPCR analyses for the expression of the retained first intron (ex1-int1) and the reduction in downstream portions of the transcript (ex5-ex6) in POLH (F) and SOGA1 (H) genes relative to FKBP9. Data represent the mean of at least three independent experiments with relative standard devation (SD). Statistical analyses were performed by Student's test *P <0.05; **P <0.01; ***P <0.001.
CDK12/13 kinase activity is required for optimal splicing of THZ1-regulated proximal introns
THZ1 was initially shown to be highly selective for CDK7 (28). However, subsequent studies revealed that this drug also potently inhibits the highly related CDK12 and CDK13 kinases (29,30).To investigate which CDK(s) is responsible for the up-regulated IR events, we employed the specific CDK7 inhibitor SY-1365 (31) and the CDK12/13 inhibitor THZ531 (30) (Figure 3A). First, parallel dose–response curves identified the minimal concentration exerting the maximal effect on cell proliferation for SY-1365 (33 nM) and THZ531 (100 nM) (Figure 3B). Next, qPCR analyses on representative genes showed that inhibition of CDK12/13 by THZ531 was sufficient to induce proximal IR, whereas SY-1365 exerted mild (POLH, NUFIP2 andCCNB1) or no effect (SOGA1, DDX21,TEAD1,E2F3 and AEBP2) (Figure 3C; Supplementary Figure S3A). THZ531 elicited an effect stronger than THZ1 on these target introns (Supplementary Figure S3B). Moreover, retention of these THZ1-regulated introns was recapitulated by silencing of CDK12/13, but not of CDK7 (Figure 3D; Supplementary Figure S3C–E). It is noteworthy that in most cases the depletion of both kinases was more efficient than individual knockdown (Supplementary Figure S3F, G), indicating a partially redundant role for CDK12 and CDK13 in the regulation of these introns. Lastly, we observed a significant positive correlation between the expression of CDK12 and CDK13 (Figure 3E, F; Supplementary Figure S4A, B) and that of THZ1 target genes in PDAC patients from TCGA database (https://www.cbioportal.org/; PDAC Firehose Legacy). In contrast, CDK7 expression showed no or negative correlation with these genes (Supplementary Figure S4C). These results suggest that the widespread retention of proximal introns induced by THZ1 is mainly due to pharmacological inhibition of CDK12/13 kinase activity.
Figure 3.

CDK12/13 kinase activity is required for optimal splicing of THZ1-regulated proximal introns. (A) Schematic representation of the substrates inhibited by THZ1 (CDK7, CDK12 and CDK13), SY-1365 (CDK7) and THZ531 (CDK12/13). (B) Proliferation assays of MiaPaCa-2 cells after 72 h of THZ1, THZ531 and SY-1365 treatment at the indicated doses. Data represent the mean of at least three independent experiments with relative SD. (C and D) Bar graphs showing the results of qPCR analyses for the expression of the retained first intron (ex1-int1) in POLH and SOGA1 genes relative to FKBP9 in cells treated for 6 h with THZ531 (100 nM) or SY-1365 (33 nM) (C) or knocked down for CDK12/13 (shCDK12/13) or CDK7 (shCDK7) or transfected with empty vector (shCTRL) (D). Data represent the mean of at least three independent experiments with relative SD. Statistical analyses were performed by one-way analysis of variance (ANOVA), *P <0.05; **P <0.01, ns = not significant. (E and F) Correlation of the expression of CDK12 (E) or CDK13 (F) with that of POLH and SOGA1 in patients from the pancreatic adenocarcinoma project (TCGA, Firehose Legacy). Statistical analyses were performed by Pearson's correlation coefficient (E and F).
CDK12/13 inhibition impairs splicing of proximal introns in target genes
Recent evidence suggests that inhibition of CDK12, and possibly CDK13, causes premature termination of transcripts by leading to activation of cryptic IPA sites and transcript cleavage (6–8). Thus, we asked whether proximal introns retained upon THZ1 treatment are characterized by features that favour their IPA. To address this question, we compared all introns located in the proximal region of genes (introns 1 and 2) for all groups (Supplementary Table S5). Our analysis indicated that the density of pAs (number of pAs/kb of intron) was not significantly different between introns that are up- or down-regulated by THZ1, and for both groups was lower than that found in constitutive introns (introns always spliced in the reference database) or in introns of genes regulated by THZ1 only at the GE level (Supplementary Figure S5A). Likewise, pAs located in up-regulated introns were not characterized by increased strength with respect to constitutive and down-regulated introns, or to introns of GE-only-regulated genes (Supplementary Figure S5B). These observations suggest that proximal retained introns are not intrinsically prone to IPA.
To experimentally test whether the increase in intronic reads was primarily due to an IPA event in the intron or, rather, to defective splicing imposed by CDK12/13 inhibition, we analysed processing of nascent transcripts. To this end, transcription was blocked by treatment of cells for 6 h with DRB, a CDK9 inhibitor that stalls RNAPII at the beginning of the gene (32). THZ531 was added in the last hour of treatment and maintained after DRB removal, when transcription restarts and progressively proceeds within the transcription unit (Supplementary Figure S5C). Nascent RNAs were then labelled with 4sU in the first 30 min after cells were released from the DRB block (Figure 4A), and immunopurified (33). Analysis by qPCR of nascent transcripts from the POLH,SOGA1 and AEBP2 genes showed that transcription does not stop following retention of the intron, as downstream regions of the gene unit (exon 5–exon 6) were equally detected in control (DMSO) and THZ531-treated cells (Supplementary Figure S5D, E). However, THZ531 caused a splicing defect in the proximal introns, as highlighted by the significant accumulation of pre-mRNA containing both the 5′ and 3′ splice sites of the intron with respect to the spliced mRNA (Figure 4B, C; Supplementary Figure S5F). In contrast, the splicing efficiency of the downstream intron 5 was not significantly affected by the treatment (Figure 4B, C; Supplementary Figure S5F). Thus, our data suggest that CDK12/13 inhibition impairs splicing of these proximal introns, an effect that may then favour the premature cleavage of the transcript at cryptic IPA sites, as previously reported in other cell types (6,7).
Figure 4.

CDK12/13 inhibition causes a splicing defect of proximal introns in target genes. (A) Schematic representation for the isolation of nascent RNAs labelled with 4sU. Transcription of cells was blocked by treatment with 100 μM DRB for 6 h. In the last hour, 200 nM THZ531 or DMSO was added; then DRB was removed by washing the cells with PBS. In the last 30 min, nascent RNAs were labelled with 4sU in the presence or not of THZ531. (B and C) Bar graphs showing the results of qRT–PCR analysis of POLH (B) and SOGA1 (C) to evaluate the level of IR in nascent RNA (4sU labelled) using primers spanning the 5′ (ex1-i1 5′) and 3′ (i1/2 3′-ex2/3) splice site regions of the regulated intron with respect to the spliced mRNA (ex1-ex2/3) and primers spanning the 5′ splice site region of a non-regulated intron (ex5-i5 5′) with respect to the spliced mRNA (ex5-ex6). A graphical representation of the genomic regions analysed and of the PCR primer strategy used is illustrated. Data represent the mean of at least three independent experiments with relative SD. Statistical analyses were performed by Student's test *P <0.05; **P <0.01, ns = not significant.
Proximal introns regulated by CDK12/13 activity are characterized by weak 3′ splice sites
Next, we asked whether the proximal introns whose splicing is susceptible to CDK12/13 inhibition display specific structural features that distinguish them from down-regulated introns or other non-regulated alternative introns (reference IR). Bioinformatics analyses of proximal introns (Supplementary Table S5) from genes expressed in MiaPaCa-2 cells indicated that up-regulated introns are longer than down-regulated and reference introns, but shorter than constitutively spliced introns (reference constitutive) and introns from genes regulated by THZ1 only at the GE level (reference GE-regulated; Supplementary Figure S6A). This result suggested that intron length is not sufficient to cause susceptibility to CDK inhibition. Likewise, while up-regulated introns were characterized by higher GC content than constitutive introns and those of reference GE-regulated genes, they were less GC-rich than reference IR introns (Supplementary Figure S6B). However, up-regulated introns were characterized by a significantly weaker 3′ splice site with respect to all other intron categories (Figure 5A). In contrast, the strength of the 5′ splice site was comparable with that of constitutive introns and stronger than that of reference IR (Figure 5A).
Figure 5.

Proximal introns regulated by CDK12/13 activity are characterized by weak 3′ splice sites. (A) Box plots representing comparison of the 5′ and 3′ splice site strength (MaxEnt score) between proximal introns (first and second) up- and down-regulated by THZ1 treatment, reference IR events not affected by THZ1 treatment (Ref. IR), constitutively spliced introns (Ref. const.) and introns from genes regulated by THZ1 only at the GE level (Ref. GE-reg). (B and C) Boxplots showing comparison between the groups described in (A) for distance (B) and percentage of pyrimidine (C) between the branchpoint (BP) and the 3′ splice site. Statistical analyses were performed by Student's test *P <0.05; **P <0.01; ***P <0.001, ns = not significant.
Activation of the 3′ splice site requires recognition of the branchpoint by the U2 snRNP, which is aided by binding of the U2 auxiliary factors U2AF65 and U2AF35, respectively, to the polypyrimidine tract and the invariant AG dinucleotide at the 3′ end of the intron (34). The strength of the branchpoint and of the polypyrimidine tract of up-regulated introns was similar to that of down-regulated and reference IR introns (Supplementary Figure S6C, D). However, the distance between the branchpoint and the polypyrimidine tract was significantly larger in up-regulated introns (Supplementary Figure S6E), thus causing a wider distance and a lower percentage of pyrimidines between the branchpoint and the 3′ splice site (Figure 5B, C). The same features (i.e. weaker 3′ splice site, greater distance from the branchpoint and 3′ splice site, and a lower percentage of pyrimidines between the branchpoint and the 3′ splice site) were also observed in proximal introns retained by THZ531 treatment of ovarian cancer cells (GSE222493; Supplementary Figure S6F–H; Supplementary Table S6). Together, these results suggest that inhibition of CDK12/13 impairs productive splicing of introns characterized by weak 3′ splice sites that are more distant from the branchpoint.
CDK12/13 inhibition impairs the physical interaction of SF3B1 with RNAPII
A key step for the recognition of the 3′ splice site by the U2 snRNP is represented by binding of SF3B1 to the branchpoint sequence at the early stage of spliceosome assembly (35). We found that SF3B1 tightly interacts with RNAPII in an RNA-independent manner in MiaPaCa-2 cells (Figure 6A). Since interaction of RNAPII with RNA processing factors is strongly regulated by post-translational modifications of its CTD, we then asked whether SF3B1 associates with RNAPII phosphorylated on Ser2 or Ser5, the two main modifications of the CTD associated with transcription elongation and splicing in the gene body (2,3). To this end, we immunoprecipitated SF3B1 and quantified the enrichment of total, phospho-Ser2 and phospho-Ser5 RNAPII co-immunoprecipitated with this factor. Our analysis indicated that phospho-Ser2 RNAPII was co-immunoprecipitated much more efficiently by SF3B1 than total or phospho-Ser5 RNAPII, as measured by enrichment with respect to the input of the immunoprecipitated nuclear extract (Figure 6B). Since co-transcriptional splicing is favoured by the interaction between phosphorylated RNAPII and spliceosome components (36), we asked whether inhibition of CDK12/13 affected this interaction. As expected, treatment with THZ531 (200 nM, 3 h) preferentially inhibited Ser2 phosphorylation of RNAPII (Figure 6C). Strikingly, this effect was accompanied by a strong repression of the interaction between SF3B1 and RNAPII (Figure 6D), indicating that it relies on Ser2 phosphorylation of the CTD. The interaction between RNAPII and SF3B1 was also compromised at an earlier time point of CDK12/13 inhibition (1.5 h; Supplementary Figure S7A, B), when transcription of new pre-mRNAs is not suppressed (Supplementary Figure S5D, E). Moreover, the interaction between RNAPII and the U1 snRNP protein U1A was not affected by treatment with THZ531 for 90–180 min (Figure 6D; Supplementary Figure S7B), ruling out a general impairment of spliceosome recruitment by the RNAPII upon CDK12/13 inhibition. Uncoupling of the SF3B1–RNAPII interaction by inhibition of CDK12/13 for 90–180 min resulted in release of SF3B1 from the chromatin, where splicing of most introns occurs (37), and its accumulation in the nucleoplasm (Figure 6E; Supplementary Figure S7D). Interestingly, the chromatin-bound fraction of other core proteins such as PRP6, PRP8 and U170K was not significantly affected by the treatment (Figure 6E; Supplementary Figure S7C), ruling out indirect effects due to transcription inhibition and overt spliceosome disassembly from the chromatin. In support of this hypothesis, inhibition of CDK7 (1.5 h) by treatment with SY-1365, which predominantly affected Ser5 phosphorylation, did not trigger release of SF3B1 from the chromatin (Supplementary Figure S7E). More importantly, RIP and CLIP assays indicated that treatment with THZ531 significantly reduced binding of SF3B1 to the 3′ splice site region of POLH and SOGA1 intron 1 (Figure 6F, G). Collectively, these results suggest that CDK12/13-mediated Ser2 phosphorylation of RNAPII promotes its association with SF3B1 and the recruitment of this splicing factor to proximal introns characterized by weak 3′ splice sites.
Figure 6.

CDK12/13 inhibition impairs the physical interaction of SF3B1 with RNAPII. (A) Representative western blot analysis of the co-immunoprecipitation between RNAPII and SF3B1 from MiaPaCa-2 nuclear extracts treated or not with RNase A. Rabbit non-immune IgGs were used as negative control. To assess degradation, RNA was isolated from both RNase A-untreated and -treated cell lysates and run on a 1% agarose gel to determine the integrity of the 28S:18S rRNA. The bar graph represents densitometric analysis of the co-immunoprecipitated SF3B1 with respect to the levels of immunoprecipitated RNAPII (mean of three independent experiments with relative SD). Statistical analyses were performed by Student's test, ns = not significant. (B) Representative western blot and densitometric analyses of RNAPII phosphorylated on Ser2, Ser5 or total RNAPII that co-immunoprecipitates with SF3B1 from MiaPaCa-2 nuclear extracts (mean of three independent experiments with relative SD). Statistical analyses were performed by one-way ANOVA, ***P <0.001. (C) Representative western blot and densitometric analyses of phospho-Ser2, phospho-Ser5 and total RNAPII in MiaPaCa2 cells treated or not with 200 nM THZ531 for 3 h. HSP90 was used as loading control (mean of three independent experiments with relative SD). Statistical analyses were performed by Student's test, **P <0.01, ns = not significant. (D) Representative western blot and densitometric analyses of SF3B1 and U1A co-immunoprecipitated with RNAPII in MiaPaCa-2 cells treated or not with 200 nM THZ531 for 3 h (mean of at least three independent experiments with relative SD). Statistical analyses were performed by Student's test, **P <0.01. (E) Representative western blot of SF3B1, RNAPII, PRP6 and PRP8 in cytosol, nucleoplasm and chromatin fractions of MiaPaCa-2 cells treated for 3 h with 200 nM THZ531. TUBULIN, U1-70K and H3 expression were evaluated as loading controls of the indicated fractions. For densitometric analyses, SF3B1 levels were normalized to U170K for the nucleoplasm fraction, and SF3B1 levels were normalized to H3 for the chromatin fraction. Data represent the mean of at least three independent experiments with relative SD. Statistical analyses were performed by Student's test, *P <0.05, ns = not significant. (F and G) qPCR analyses of RIP (F) and CLIP (G) assays of the binding of SF3B1 to the 3′ splice site of POLH intron 1 and SOGA1 intron 2 from cells treated for 3 h with DMSO or THZ531 (200 nM). Non-immune IgGs were used as control of the assays (mean of three independent experiments with relative SD). Statistical analyses were performed by one-way ANOVA, *P <0.05, **P <0.01, ***P <0.001.
THZ531 synergizes with spliceosome inhibition in IR regulation and suppression of cell proliferation
Since CDK12/13 inhibition affected the recruitment of SF3B1 to the target introns, we asked whether pharmacological inhibition of SF3B1 would also affect splicing of these proximal introns. In line with our hypothesis, treatment with PdB (10 nM, 6 h), an SF3B1 inhibitor that impairs recognition of the branchpoint site by the U2 snRNA (38,39), increased retention of THZ531 target introns (Supplementary Figure S8A). These results indicated that optimal SF3B1 function is required for efficient splicing of CDK12/13 target introns.
THZ531 (interaction with RNAPII) and PdB (recognition of branchpoint) target different SF3B1 features. Thus, we asked whether they could synergize in promoting retention of proximal introns in these model genes. To this end, MiaPaCa-2 cells were treated with suboptimal doses of THZ531 (50 nM; Figure 3B) and PdB (3.3 nM; Supplementary Figure S8B), either alone or in combination. At these doses, THZ531 and PdB caused mild or no effect on POLH, SOGA1, AEBP2 and DDX21 proximal introns, whereas combined administration restored their strong retention (Figure 7A; Supplementary Figure S8C). Similar results were also observed in MDA-MB-231 cells, a triple-negative breast cancer (TNBC) cell line previously shown to be sensitive to CDK12/13 inhibition (40) (Supplementary Figure S9A). These results suggest that cooperation between CDK12/13 and SF3B1 is required for the efficient splicing of THZ531-sensitive introns.
Figure 7.

THZ531 synergizes with spliceosome inhibition in IR regulation and suppression of cell proliferation. (A) Results of qPCR analyses for the expression of the retained first intron in POLH and SOGA1 genes relative to FKBP9 in MiaPaCa-2 cells treated with suboptimal doses of THZ531 (50 nM) and PdB (3.3 nM) either alone or in combination for 6 h (mean of at least three independent experiments with relative SD). Statistical analyses were performed by one-way ANOVA, ***P <0.001. (B) Cell proliferation analysis of MiaPaCa-2 cells treated with suboptimal doses of THZ531 (25 nM) and PdB (1 nM) either alone or in combination (mean of at least three independent experiments with relative SD). Statistical analyses are reported in light blue when referring to 25 nM THZ531 treatment, in green when referring to 1 nM PdB treatment and in grey when referring to DMSO. Statistical analyses were performed by two-way ANOVA *P <0.05; **P <0.01; ***P <0.001. (C) Representative images and bar graph of MiaPaCa-2 cells labelled with cytotoxic green and NIR dyes for live cells after 96 h of treatment with suboptimal doses of THZ531 and PdB either alone or in combination analysed by the IncuCyte SX5 technology to evaluate the percentage of dead cells in each sample (mean of three independent experiments with relative SD). Statistical analyses were performed by one-way ANOVA, ***P <0.001. Scale bar 150 μm. (D) Representative western blot analysis of PARP1, γH2AX, H3 and HSP90 in MiaPaCa-2 cells treated for 48 h as indicated. (E–H) Fluorescence-activated cell sorting (FACS) analyses showing DNA content (7-AAD) to evaluate the cell cycle state of MiaPaCa2 cells after 24 h at optimal (E) or suboptimal doses (G) of the indicated drugs. Bar graphs (F and H) show the percentage of cells in G1, S and G2 phase. Data represent the mean of four independent experiments with relative SD. Statistical analyses were performed by one-way ANOVA, *P <0.05; **P <0.01; ***P <0.001.
The suboptimal doses of THZ531 and PdB that were ineffective in inducing IR also elicited mild effects on cell proliferation (Figure 7B). However, consistent with their synergic effects on IR, combined treatment with suboptimal doses of the two inhibitors significantly reduced cell proliferation with respect to either drug alone (Figure 7B). Reduced growth by combined THZ531/PdB treatment was accompanied by enhanced cell death (Figure 7C) and by increased levels of cleaved PARP1 and phosphorylated H2Ax (γH2AX) (Figure 7D), two markers of apoptosis. Similar results were also observed in MDA-MB-231 cells (Supplementary Figure S9B, C).
Retained proximal introns were enriched in genes involved in the cell cycle and cell proliferation (Figure 1G; Supplementary Figure S1), such as E2F3, which encodes a transcription factor that promotes proliferation, and CCNB1, which encodes the mitotic cyclin B1. Thus, we tested whether the synergic effect of THZ531 and PdB on IR regulation resulted in a similar effect on cell cycle progression. As previously reported for other cell lines (7,41), treatment of MiaPaCa-2 cells with optimal concentrations of either drug caused a significant increase in the G2/M phase and a concomitant reduction of cells in the G1 phase (Figure 7E, F). In contrast, suboptimal doses of either THZ531 or PdB had minimal effects on cell cycle distribution (Figure 7G, H). However, their combined administration recapitulated the dysregulation of cell cycle progression observed with either drug at optimal concentration (Figure 7G, H), further confirming their synergic effect on cell proliferation and survival. These results support the notion that CDK12/13 inhibition impairs splicing of proximal introns by interfering with SF3B1 function at the 3′ splice site, with strong consequences on cell proliferation and survival.
DISCUSSION
In this study, we report that inhibition of CDK7/12/13 activity by the covalent inhibitor THZ1 leads to widespread splicing dysregulation, primarily by affecting the regulation of intron splicing. We particularly focused on a subset of promoter-proximal introns (introns 1 and 2) that are significantly retained upon THZ1 treatment, because they were enriched in genes with strong relevance for cancer. By using specific inhibitors and knockdown approaches that distinguish between these CDKs, we show that efficient splicing of these proximal introns relies on the activity of CDK12 and CDK13, whereas selective inhibition of CDK7 exerted mild or no effect. Interestingly, retention of these proximal introns leads to significant reduction of gene expression, probably due to premature termination of the transcript. Indeed, we observed a clear reduction in read coverage downstream of the retained introns and a significant overlap between genes characterized by promoter-proximal IR and GE changes. These results suggest that CDK12/13 are required for the efficient splicing of a specific class of introns, which are enriched in the 5′ region of the transcription unit.
Previous data suggested that CDK12/13 promote the elongation rate of RNAPII within the gene body by phosphorylating Ser2 in its CTD (4). The fast elongation rate is also required to prevent usage of cryptic pA sites present in introns, which would cause premature termination of the transcript if actioned. Accordingly, inhibition of CDK12/13 was shown to induce widespread IPA, particularly in genes characterized by long introns (6,7). Although the mechanism was not fully elucidated, these observations suggested that the slow pace of RNAPII within long introns extends the window of opportunity for recognition and usage of cryptic pA sites by the cleavage and polyadenylation machinery before the 3′ splice site is transcribed at the other end of the intron. However, our analysis of pulse-labelled nascent transcripts showed that transcription proceeds beyond the retained intron in cells treated with THZ531, suggesting that CDK12/13 inhibition imposes a specific splicing defect in a select class of introns. Thus, IPA of CDK12/13 target transcripts may represent the consequence of prolonged IR due to inefficient splicing.
To understand why some introns are particularly susceptible to CDK12/13 inhibition, we analysed their sequence features. Since the retained introns were mainly in the proximal region of the target genes, and intron 1 is generally longer than downstream introns, we performed comparative analyses with other proximal introns (introns 1 and 2) that are not affected by the treatment. In line with previous data (7), we observed that up-regulated introns are significantly larger than non-regulated and down-regulated IR events. However, increased size does not appear to be sufficient, as constitutive proximal introns not affected by the treatment are significantly larger than THZ1-up-regulated introns. Moreover, up-regulated introns were also significantly shorter than proximal introns of other genes that are regulated by THZ1 only at the GE level. Thus, although large size may contribute to IR, it does not appear to represent the primary cause. On the other hand, we found that up-regulated proximal introns display a weak 3′ splice site with respect to all other intron categories tested. Up-regulated introns were also characterized by a greater distance of the 3′ splice site from the branchpoint, a feature that also diminishes the strength of the splice site (42). Importantly, the same structural features were also found in proximal introns up-regulated by treatment of ovarian cancer cells with THZ531, supporting the hypothesis that their impaired splicing is caused by CDK12/13 inhibition and that the activity of these kinases is required for the efficient recognition of weak 3′ splice sites by the U2 snRNP. Accordingly, these introns were also sensitive to PdB, an inhibitor of the U2 snRNP component SF3B1 that recognizes the branchpoint sequence and favors U2 snRNP recruitment to the 3′ splice site (35). Mechanistically, we found that SF3B1 preferentially associates with RNAPII phosphorylated on Ser2, and that THZ531 suppresses this interaction by inhibiting Ser2 phosphorylation. This regulation reduces the association of SF3B1 with the chromatin and its recruitment to the 3′ splice site of CDK12/13 target introns. Our data also indicate that efficient coupling between SF3B1 and RNAPII specifically requires Ser2 phosphorylation of the CTD, and not optimal transcription efficiency. Indeed, reduction of Ser5 phosphorylation by CDK7 inhibition, which also impacts on transcription (14,31), did not impair the association of SF3B1 with the chromatin and did not induce retention of these proximal introns. Thus, we suggest that inhibition of CDK12/13 specifically impairs splicing of introns with a weak 3′ splice site by uncoupling SF3B1 from RNAPII and by reducing the efficiency of its recruitment to the pre-mRNA. In this scenario, the premature cleavage of the transcript at cryptic IPAs observed in other cell types (6,7) may be the consequence of the accumulation of intron-containing transcripts and impairment of spliceosome assembly at the 3′ end of the intron.
Direct (PdB) or indirect (THZ531) inhibition of SF3B1 strongly suppressed cell cycle progression and proliferation. Since the two inhibitors acted on SF3B1 by different mechanisms, we reasoned that they may cooperate. Indeed, suboptimal doses of these inhibitors exerted a synergic effect on IR of target genes, as well as on proliferation and cell death. Cell cycle progression was specifically blocked at the G2/M transition by the two drugs, and this effect may directly correlate with increased IR and down-regulation of genes involved in this phase, such as E2F3 and CCNB1. The synergic effect observed with combined inhibition of CDK12/13 and SF3B1 may have clinical relevance. Indeed, mounting evidence points to splicing dysregulation as a therapeutic vulnerability in human cancers, particularly for tumours driven by MYC amplification. It is noteworthy that both MiaPaCa-2 and MDA-MB231 cells used in our study express high levels of MYC (27,43), and MYC up-regulation is known to cause a transcriptional overload that imposes a stress on the splicing machinery, rendering the cell particularly vulnerable to splicing inhibitors. However, past clinical trials using the PdB derivative E7107 failed due to the adverse effects caused by the treatment (39,44). Novel and potentially less toxic PdB derivatives are currently being tested in clinical trials (45). Thus, the possibility to reduce the dose of splicing inhibitors by combined treatment with CDK12/13 inhibitors may enhance their clinical performance by limiting adverse toxic effects. This combined approach may be particularly relevant for tumours characterized by poor prognosis and lack of targeted approaches, such as PDAC and TNBCs. Notably, these cancers frequently display up-regulation of MYC (43,46) and were shown to be selectively sensitive to inhibition of transcriptional CDKs (27,40). Thus, our study suggests that combined inhibition of CDK12/13 and the spliceosome may represent a suitable therapeutic approach for cancers that display MYC amplification or that are driven by oncogenic dysregulation of transcription factors, such as Ewing sarcoma (47) or prostate cancer (48).
DATA AVAILABILITY
All data generated or analysed during this study are included in this article. The RNA sequences analysed are available in the Gene Expression Omnibus under accession number GSE121273 (MiaPaCa-2 cells, 6 h of 100 nM THZ1) (27) and GSE222493 (OVCAR-3 cells, 6 h of 200 nM THZ531).
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank all members of the Sette's laboratory for fruitful suggestions and discussions throughout this study.
Contributor Information
Valentina Panzeri, Department of Neuroscience, Section of Human Anatomy, Catholic University of the Sacred Heart, Rome, Italy.
Marco Pieraccioli, Department of Neuroscience, Section of Human Anatomy, Catholic University of the Sacred Heart, Rome, Italy; Gemelli Science and Technology Park (GSTeP)-Organoids Research Core Facility, Fondazione Policlinico Agostino Gemelli IRCCS, Rome, Italy.
Eleonora Cesari, Department of Neuroscience, Section of Human Anatomy, Catholic University of the Sacred Heart, Rome, Italy; Gemelli Science and Technology Park (GSTeP)-Organoids Research Core Facility, Fondazione Policlinico Agostino Gemelli IRCCS, Rome, Italy.
Pierre de la Grange, GenoSplice, Paris, France.
Claudio Sette, Department of Neuroscience, Section of Human Anatomy, Catholic University of the Sacred Heart, Rome, Italy; Gemelli Science and Technology Park (GSTeP)-Organoids Research Core Facility, Fondazione Policlinico Agostino Gemelli IRCCS, Rome, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Associazione Italiana Ricerca sul Cancro [AIRC IG23416 to C.S. and Fellowship 23938 to V.P.]; Italian Ministry of Health [‘Ricerca Corrente 2021 and 2022’ to IRCCS Fondazione Policlinico A. Gemelli]; and Università Cattolica del Sacro Cuore. Funding for open access charge: Associazione Italiana Ricerca sul Cancro; Catholic University of the Sacred Heart; Ministero della Salute Ricerca Corrente 2022.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cramer P. Organization and regulation of gene transcription. Nature. 2019; 573:45–54. [DOI] [PubMed] [Google Scholar]
- 2. Phatnani H.P., Greenleaf A.L.. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006; 20:2922–2936. [DOI] [PubMed] [Google Scholar]
- 3. Chen F.X., Smith E.R., Shilatifard A.. Born to run: control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018; 19:464–478. [DOI] [PubMed] [Google Scholar]
- 4. Chou J., Quigley D.A., Robinson T.M., Feng F.Y., Ashworth A.. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 2020; 10:351–370. [DOI] [PubMed] [Google Scholar]
- 5. Wong K.H., Jin Y., Struhl K.. TFIIH phosphorylation of the Pol II CTD stimulates mediator dissociation from the preinitiation complex and promoter escape. Mol. Cell. 2014; 54:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dubbury S.J., Boutz P.L., Sharp P.A.. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature. 2018; 564:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krajewska M., Dries R., Grassetti A.V., Dust S., Gao Y., Huang H., Sharma B., Day D.S., Kwiatkowski N., Pomaville M.et al.. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019; 10:1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fan Z., Devlin J.R., Hogg S.J., Doyle M.A., Harrison P.F., Todorovski I., Cluse L.A., Knight D.A., Sandow J.J., Gregory G.et al.. CDK13 cooperates with CDK12 to control global RNA polymerase II processivity. Sci. Adv. 2020; 6:eaaz5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tellier M., Zaborowska J., Caizzi L., Mohammad E., Velychko T., Schwalb B., Ferrer-Vicens I., Blears D., Nojima T., Cramer P.et al.. CDK12 globally stimulates RNA polymerase II transcription elongation and carboxyl-terminal domain phosphorylation. Nucleic Acids Res. 2020; 48:7712–7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naro C., Bielli P., Sette C.. Oncogenic dysregulation of pre-mRNA processing by protein kinases: challenges and therapeutic opportunities. FEBS J. 2021; 288:6250–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hertel K.J., Graveley B.R.. RS domains contact the pre-mRNA throughout spliceosome assembly. Trends Biochem. Sci. 2005; 30:115–118. [DOI] [PubMed] [Google Scholar]
- 12. Ko T.K., Kelly E., Pines J.. CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J. Cell Sci. 2001; 114:2591–2603. [DOI] [PubMed] [Google Scholar]
- 13. Tien J.F., Mazloomian A., Cheng S.-W.G., Hughes C.S., Chow C.C.T., Canapi L.T., Oloumi A., Trigo-Gonzalez G., Bashashati A., Xu J.et al.. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017; 45:6698–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rimel J.K., Poss Z.C., Erickson B., Maas Z.L., Ebmeier C.C., Johnson J.L., Decker T.-M., Yaron T.M., Bradley M.J., Hamman K.B.et al.. Selective inhibition of CDK7 reveals high-confidence targets and new models for TFIIH function in transcription. Genes Dev. 2020; 34:1452–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naro C., De Musso M., Delle Monache F., Panzeri V., de la Grange P., Sette C.. The oncogenic kinase NEK2 regulates an RBFOX2-dependent pro-mesenchymal splicing program in triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2021; 40:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Naro C., Jolly A., Di Persio S., Bielli P., Setterblad N., Alberdi A.J., Vicini E., Geremia R., De la Grange P., Sette C.. An orchestrated intron retention program in meiosis controls timely usage of transcripts during germ cell differentiation. Dev. Cell. 2017; 41:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mauger O., Lemoine F., Scheiffele P.. Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron. 2016; 92:1266–1278. [DOI] [PubMed] [Google Scholar]
- 20. Yeo G., Burge C.B.. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004; 11:377–394. [DOI] [PubMed] [Google Scholar]
- 21. Corvelo A., Hallegger M., Smith C.W.J., Eyras E.. Genome-wide association between branch point properties and alternative splicing. PLoS Comput. Biol. 2010; 6:e1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bogard N., Linder J., Rosenberg A.B., Seelig G.. A deep neural network for predicting and engineering alternative polyadenylation. Cell. 2019; 178:91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pandya-Jones A., Black D.L.. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009; 15:1896–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sette C., Paronetto M.P., Barchi M., Bevilacqua A., Geremia R., Rossi P.. Tr-kit-induced resumption of the cell cycle in mouse eggs requires activation of a src-like kinase. EMBO J. 2002; 21:5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naro C., Pellegrini L., Jolly A., Farini D., Cesari E., Bielli P., de la Grange P., Sette C.. Functional interaction between U1snRNP and Sam68 insures proper 3′ end pre-mRNA processing during germ cell differentiation. Cell Rep. 2019; 26:2929–2941. [DOI] [PubMed] [Google Scholar]
- 26. Pieraccioli M., Caggiano C., Mignini L., Zhong C., Babini G., Lattanzio R., Di Stasi S., Tian B., Sette C., Bielli P.. The transcriptional terminator XRN2 and the RNA-binding protein Sam68 link alternative polyadenylation to cell cycle progression in prostate cancer. Nat. Struct. Mol. Biol. 2022; 29:1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu P., Geng J., Zhang L., Wang Y., Niu N., Fang Y., Liu F., Shi J., Zhang Z.-G., Sun Y.-W.et al.. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene. 2019; 38:3932–3945. [DOI] [PubMed] [Google Scholar]
- 28. Kwiatkowski N., Zhang T., Rahl P.B., Abraham B.J., Reddy J., Ficarro S.B., Dastur A., Amzallag A., Ramaswamy S., Tesar B.et al.. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014; 511:616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeng M., Kwiatkowski N.P., Zhang T., Nabet B., Xu M., Liang Y., Quan C., Wang J., Hao M., Palakurthi S.et al.. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. Elife. 2018; 7:e39030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang T., Kwiatkowski N., Olson C.M., Dixon-Clarke S.E., Abraham B.J., Greifenberg A.K., Ficarro S.B., Elkins J.M., Liang Y., Hannett N.M.et al.. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016; 12:876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu S., Marineau J.J., Rajagopal N., Hamman K.B., Choi Y.J., Schmidt D.R., Ke N., Johannessen L., Bradley M.J., Orlando D.A.et al.. Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res. 2019; 79:3479–3491. [DOI] [PubMed] [Google Scholar]
- 32. Singh J., Padgett R.A.. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009; 16:1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garibaldi A., Carranza F., Hertel K.J.. Isolation of newly transcribed RNA using the metabolic label 4-thiouridine. Methods Mol. Biol. 2017; 1648:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kielkopf C.L., Rodionova N.A., Green M.R., Burley S.K.. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001; 106:595–605. [DOI] [PubMed] [Google Scholar]
- 35. Sun C. The SF3b complex: splicing and beyond. Cell. Mol. Life Sci. 2020; 77:3583–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nojima T., Rebelo K., Gomes T., Grosso A.R., Proudfoot N.J., Carmo-Fonseca M.. RNA polymerase II phosphorylated on CTD serine 5 interacts with the spliceosome during co-transcriptional splicing. Mol. Cell. 2018; 72:369–379.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haque N., Oberdoerffer S.. Chromatin and splicing. Methods Mol. Biol. 2014; 1126:97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonnal S., Vigevani L., Valcárcel J.. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012; 11:847–859. [DOI] [PubMed] [Google Scholar]
- 39. Sette C., Paronetto M.P.. Somatic mutations in core spliceosome components promote tumorigenesis and generate an exploitable vulnerability in human cancer. Cancers. 2022; 14:1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quereda V., Bayle S., Vena F., Frydman S.M., Monastyrskyi A., Roush W.R., Duckett D.R.. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell. 2019; 36:545–558. [DOI] [PubMed] [Google Scholar]
- 41. Sciarrillo R., Wojtuszkiewicz A., El Hassouni B., Funel N., Gandellini P., Lagerweij T., Buonamici S., Blijlevens M., Zeeuw van der Laan E.A., Zaffaroni N.et al.. Splicing modulation as novel therapeutic strategy against diffuse malignant peritoneal mesothelioma. EBioMedicine. 2019; 39:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiara M.D. Evidence that U5 snRNP recognizes the 3′ splice site for catalytic step II in mammals. EMBO J. 1997; 16:4746–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang E., Sorolla A., Cunningham P.T., Bogdawa H.M., Beck S., Golden E., Dewhurst R.E., Florez L., Cruickshank M.N., Hoffmann K.et al.. Tumor penetrating peptides inhibiting MYC as a potent targeted therapeutic strategy for triple-negative breast cancers. Oncogene. 2019; 38:140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bonnal S.C., López-Oreja I., Valcárcel J.. Roles and mechanisms of alternative splicing in cancer—implications for care. Nat. Rev. Clin. Oncol. 2020; 17:457–474. [DOI] [PubMed] [Google Scholar]
- 45. Steensma D.P., Wermke M., Klimek V.M., Greenberg P.L., Font P., Komrokji R.S., Yang J., Brunner A.M., Carraway H.E., Ades L.et al.. Phase I first-in-human dose escalation study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia. 2021; 35:3542–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hessmann E., Schneider G., Ellenrieder V., Siveke J.T.. MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene. 2016; 35:1609–1618. [DOI] [PubMed] [Google Scholar]
- 47. Paronetto M.P. Ewing Sarcoma protein: a key player in human cancer. Int. J. Cell Biol. 2013; 2013:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Constantin T.A., Greenland K.K., Varela-Carver A., Bevan C.L.. Transcription associated cyclin-dependent kinases as therapeutic targets for prostate cancer. Oncogene. 2022; 41:3303–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this article. The RNA sequences analysed are available in the Gene Expression Omnibus under accession number GSE121273 (MiaPaCa-2 cells, 6 h of 100 nM THZ1) (27) and GSE222493 (OVCAR-3 cells, 6 h of 200 nM THZ531).