


Abstract
The deubiquitinating enzyme Ataxin-3 (ATXN3) contains a polyglutamine (PolyQ) region, the expansion of which causes spinocerebellar ataxia type-3 (SCA3). ATXN3 has multiple functions, such as regulating transcription or controlling genomic stability after DNA damage. Here we report the role of ATXN3 in chromatin organization during unperturbed conditions, in a catalytic-independent manner. The lack of ATXN3 leads to abnormalities in nuclear and nucleolar morphology, alters DNA replication timing and increases transcription. Additionally, indicators of more open chromatin, such as increased mobility of histone H1, changes in epigenetic marks and higher sensitivity to micrococcal nuclease digestion were detected in the absence of ATXN3. Interestingly, the effects observed in cells lacking ATXN3 are epistatic to the inhibition or lack of the histone deacetylase 3 (HDAC3), an interaction partner of ATXN3. The absence of ATXN3 decreases the recruitment of endogenous HDAC3 to the chromatin, as well as the HDAC3 nuclear/cytoplasm ratio after HDAC3 overexpression, suggesting that ATXN3 controls the subcellular localization of HDAC3. Importantly, the overexpression of a PolyQ-expanded version of ATXN3 behaves as a null mutant, altering DNA replication parameters, epigenetic marks and the subcellular distribution of HDAC3, giving new insights into the molecular basis of the disease.
Graphical Abstract
Graphical Abstract.
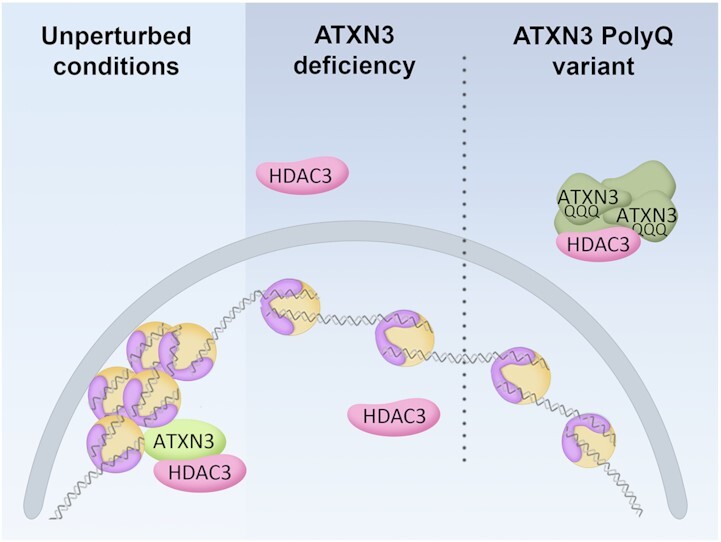
Wild-type ATXN3 controls chromatin organization by recruiting HDAC3, thereby increasing heterochromatin and controlling DNA replication and transcription, whereas lack of or PolyQ-expanded ATXN3 triggers mislocalization of HDAC3 and euchromatin prevalence.
INTRODUCTION
Mutations in the ATXN3 gene cause spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease, the most common inherited spinocerebellar ataxia. The ATXN3 protein (Ataxin-3/ATX3/AT3/MJD1) contains a polyglutamine (PolyQ) tract in its C-terminus, the expansion of which results in SCA3, one of the several polyglutamine neurodegenerative diseases. Various hypotheses have been proposed to explain the pathogenic role of ATXN3 PolyQ-expanded (ATXN3-PolyQ) variant in SCA3 but the principal molecular mechanism by which this ATXN3 variant leads to neurotoxicity remains elusive (1–4).
ATXN3 is a ubiquitin hydrolase that interacts with the ATPase VCP/p97, various ubiquitin ligases and other ubiquitin-related proteins, thereby controlling the stability or activity of several proteins in diverse cellular pathways (1,5–11). Additionally, ATXN3 is involved in transcriptional control. ATXN3-wild type (WT), but not ATXN3-PolyQ, is able to repress the expression of certain genes, in SCA3 cell and mouse models as well as in human SCA3 brains (12–15). Importantly, ATXN3 contributes to maintaining genome integrity by regulating various crucial DNA damage response proteins (16–18).
Chromatin is folded inside the nucleus in a fluid-like state and contains different genomic functional regions. Euchromatin is associated with actively transcribed genes, while constitutive and facultative heterochromatin are generally linked to a lack of gene expression (19–21). As heterochromatin mainly localizes at the nuclear and nucleolar periphery, these compartments are essential for the location and regulation of repressive genomic domains (22–24). Nucleoli are principally committed to polymerase I-dependent transcription of ribosomal genes and the assembly of pre-ribosomal particles (25,26). Due to its liquid–liquid phase separation nature, nucleoli may fuse as a consequence of chromosomal organization rearrangements or loss of appropriate proximal or distal chromatin anchoring with other nuclear compartments, such as lamins (26–28). The chromatin status controls the accessibility and sequential recruitment of molecular machineries to the DNA and therefore regulates processes such as DNA replication or transcription (19,20,29–31).
Here, we describe a novel role for ATXN3 in regulating chromatin structure, which affects global DNA replication and transcription in unperturbed cells. Interestingly, ATXN3-PolyQ triggers abnormal DNA replication and transcription, thereby uncovering a new mechanism that could explain the molecular basis of SCA3.
MATERIALS AND METHODS
Cell lines, cell culture and inhibitors
HEK293T, U2OS and CSM14.1 cells were grown in DMEM (Lonza) supplemented with 10% FBS and 1% penicillin-streptomycin (Biowest). CMS14.1 cells were additionally grown in 0.1 mg/ml neomycin, 0.1 mg/ml hygromycin (Alfa Aesar), 4 μg/ml puromycin (Alfa Aesar) and 1 μg/ml doxycycline to maintain selection the CSM14.1 clonal cell lines. SCA3-Q23 or SCA3-Q70 expression was induced by the withdrawal of doxycycline (Cayman Chemical). ΔATXN3 U2OS cells were described before (16).
G1/S boundary synchronization was performed by treating cells with 2.5 mM thymidine (Merk-Millipore) for 24 h. Cells were treated with 60 μM CDC7i (PHA767491, Cayman Chemical Company) for 4 h, 80 mM sucrose (Merk-Millipore) for 30 min or 1 h, HDAC3 inhibitor RGFP966 2 μM (HY-13909, MedChemExpress) for 24 h and 20 μg/ml cycloheximide (CHX, Merck Millipore) for the indicated times.
Antibodies
Antibodies against Ku86 (sc-1484 C-20, RRID:AB_2218751) and Lamin A/C (sc-7292 636, RRID:AB_627875) were purchased from Santa Cruz Biotechnology; anti-DNA-RNA Hybrid clone S9.6 (MABE1095, RRID:AB_ 2861387), anti-DNA G-quadruplex (MABE1126, RRID:AB_2924428), anti-NPM1 (B0556, RRID:AB_2154872), anti-H3K27me3 (07–449, RRID:AB_310624), anti-H3K9me3 (07–442, RRID:AB_310620) and anti-H4K16Ac (07–329, RRID:AB_310525) were from Merck Millipore; anti-HDAC3 for immunofluorescence (NB500-126, RRID:AB_10078274) was from Novus Biologicals, anti-HDAC3 for Western blot (PA5-85378, RRID:AB_2792520) from Thermo Fisher Scientific and the anti-BrdU antibody (A01650, RRID:AB_2622180) for flow cytometry was from GenScript. Antibodies against human ATXN3 and mCherry were generated by injecting rabbits with a His-tagged antigen (full length ATXN3 and mCherry). Antigens were obtained cloning the corresponding cDNAs in pET-30 (Novagen), followed by expression in Escherichia coli and purification with a Ni-NTA resin (Qiagen) following manufacturers recommendations. The antibodies against GFP (32) and RIF1 (33) were described before.
Western blotting
For whole cell extracts, cells were lysed in urea-SDS buffer (6 M urea, 1% SDS, 125 mM NaCl, 25 mM Tris pH 8). Lysates containing equal amounts of protein, measured by the BCA method (Thermo Fisher Scientific) were loaded in gels and SDS-PAGE was performed, followed by transfer using nitrocellulose membranes (Amersham™ Protran® Premium 0.45, Cytiva). Membranes were blocked with TBS-T + 5% non-fat milk, incubated with primary and HRP conjugated secondary antibodies (Jackson Immunoresearch), and subsequently with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). Chemiluminescent images were obtained using the ImageQuant LAS 4000 mini (GE Healthcare).
Cloning, plasmids, siRNA oligos and transfection
Plasmid DNA was transfected into cells using Lipofectamine 3000 (Thermo Fisher Scientific) or the standard calcium phosphate method (34). Full length human ATXN3 wild type and catalytic inactive (containing the mutation Cys14Ala) were amplified from plasmids previously described (16), as well as ATXN3 with expanded polyQ, containing an 80-glutamine track (kindly provided by Dr Henry L. Paulson, Addgene plasmid 22129), and cloned in the pEGFP-C1 vector (Clontech, TaKaRa). Human HDAC3 cDNA was synthetized (Genestrings, Thermo Fisher Scientific) and cloned with mCherry into the pcDNA3.1 myc-His A. The vector expressing histone H1.5-GFP was described before (35) and the GFP-HDAC3 plasmid was a kind gift from Dr James M. Holaska (Rowan University, NJ, USA).
siRNA oligonucleotides (Microsynth AG) targeting the indicated proteins were transfected into cells using Lipofectamine RNAiMax (Thermo Fisher Scientific) according to the manufacturer's protocol for 48 h, unless stated otherwise. Sequences of oligonucleotides were as follows:
| siRNA control | CGUACGCGGAAUACUUCGAdTdT |
| siATXN3-1 | GCACUAUUCUUGGCUCAAUdTdT |
| siATXN3-2 | GCAGGGCUAUUCAGCUAAGdTdT |
| siHDAC3-1 | CCGCCAGACAAUCUUUGAAdTdT |
| siHDAC3-2 | CGGGAUGGCAUUGAUGACCAGAGUUdTdT |
| siASF/SF2 -1 | CCAACAAGAUAGAGUAUAAdTdT |
| siASF/SF2 -2 | UUGGCAGUAUUGACCUUAdTdT |
DNA fiber analysis
Exponentially growing cells were pulsed with 30 μM CldU (Merk-Milipore) for 30 min and then with 250 μM IdU (Merk-Milipore) for another 30 min. Cells were then lysed on the slide by adding spreading buffer (0.5% SDS, 200 mM Tris pH 7.4, 50 mM EDTA) and incubated for 6 min at RT in humidity chamber. DNA fibers were stretched by tilting the slide ∼30° and, after air drying, fixed for with ice-cold 3:1 methanol:acetic acid solution. Slides were then incubated in 2.5 HCl for 30 min at RT, and blocked with PBS + 1% BSA + 0.1% Triton X-100 before incubation with anti-CldU (anti-BrdU, sc-56258 BU 1/75 ICR1 from Santa Cruz Biotechnology, RRID:AB_305426) and anti-IdU (anti-BrdU, B44 from BD Bioscience, RRID:AB_2313824) antibodies overnight at 4°C. The slides were then incubated with anti-rat IgG Alexa Fluor 555 (Thermo Fisher Scientific) and anti-mouse IgG1 Alexa Fluor 488 (Thermo Fisher Scientific). Finally, they were incubated with anti-ssDNA (MAB3034 from Merck Millipore) and anti-mouse IgG2a Alexa Fluor 647 (Thermo Fisher Scientific) and mounted with Prolong (Thermo Fisher Scientific). Visual acquisition of the DNA fibers was done with a Zeiss Cell Observer fluorescent microscope equipped with a 40× NA 1.3 oil immersion objective and ZEN imaging software. Images were analyzed with the image processing program FIJI software (36).
Immunoprecipitations
Immunoprecipitations were carried out with mCherry antibody bound to Protein A Sepharose CL-4B (Cytiva). Cells were lysed in lysis buffer 1 (20 mM Tris pH 8, 300 mM NaCl, 1 mM EDTA, 1% TX100, mM DTT) for 20 min on ice. After centrifugation 1600 rpm for 20 min, extracts were incubated with the antibody-linked beads for 2 h at 4 °C, followed by three washes with lysis buffer 1.
Immunofluorescence and microscopy
Cells were fixed with 4% PFA for 20 min at RT followed by permeabilization with ice-cold methanol. When indicated, pre-extraction was performed for 5 min with ice-cold pre-extraction buffer (10 mM HEPES pH 7, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100) prior to fixation. Cells were blocked with 1% FBS at RT and incubated with the indicated antibodies, overnight at 4°C. After washing, cells were incubated with secondary antibodies. DNA was stained with DAPI and coverslips were mounted using ProLong (Thermo Fisher Scientific). Images were taken with the same exposure time for each fluorescent channel using a Zeiss Cell Observer fluorescence microscope equipped with a 20× NA 0.8 air immersion objective or a 63× NA 1.3 water immersion objective and ZEN imaging software. Analysis was performed using FIJI software (36).
Quantitative real-time RT-PCR
Total RNAs were isolated from cells using Ribozol Extraction Reagent (VWR International). cDNA synthesis and PCR amplification were carried out in the same tube using the qScript One-Step SYBR Green RT-qPCR Kit (Quantbio). Primers used for RT-qPCR in this study:
| Sat-2-F | 5′-CATCGAATGGAAATGAAAGGAGTC-3′ |
| Sat-2-R | 5′-ACCATTGGATGATTGCAGTCAA-3′ |
FRAP
Live-cell microscopy was performed at 37 °C with a Leica SP8 inverted confocal microscope with a Leica HC PL APO CS2 63× NA 1.4 oil immersion lens. For GFP excitation, a 488-nm argon laser line was used. Scanning was bidirectional at the highest possible rate with a 7.5× zoom and a pinhole of 1 airy unit. Regions of interest (ROI) were selected within the nucleus from the areas strongly or weakly labelled with Hoechst 33342 (Thermo Fisher Scientific) for heterochromatin or euchromatin regions, respectively. Five single prebleach images were acquired before application of a laser pulse, lasting 0.5 s at a 95% power without scanning. After bleaching, single-section images were collected at 1-s intervals for the first 15 s and at 10-s intervals for the next 100 s. The average intensity values of ROIs, unbleached-reference regions, and background fluorescence were obtained from FRAP images. Background values were subtracted from those of ROI and reference regions for all time-points (corr_1). Subsequently, the recovery curve was plotted with the ROI value in relation to the reference value over the time-course (corr_2). Finally, the curve was normalized to the mean value of prebleached time-points, taken as 1 (corr_3). The data were fitted to an exponential recovery curve 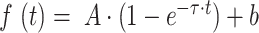 . Complete recovery would mean
. Complete recovery would mean 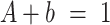 (37). Thus, the mobile and immobile fractions were calculated as:
(37). Thus, the mobile and immobile fractions were calculated as:
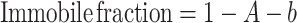 |
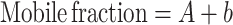 |
The recovery time of FRAP curves is reported as 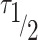 , which is the time to reach half intensity of full recovery. According to the exponential recovery fitting, is obtained as:
, which is the time to reach half intensity of full recovery. According to the exponential recovery fitting, is obtained as:
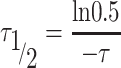 |
All quantitative values represent averages from at least 10 cells from three independent experiments.
Micrococcal nuclease sensitivity assay
Cells were resuspended in buffer A (10 mM Tris pH 7.5, 100 mM KCl, 3 mM MgCl2, 1 mM CaCl2, 0.5 mM PMSF) at a concentration of 26 × 106 cells/ml. After washing twice, the pellet was resuspended in 500 μl of buffer A supplemented with 0.6% NP-40. After a 5 min incubation on ice, samples were washed twice with micrococcal nuclease (MNase) buffer (15 mM Tris pH 7.5, 15 mM NaCl, 60 mM KCl, 1 mM CaCl2, 0.25 mM PMSF). Aliquots were digested with different amounts of MNase (New England Biolabs) at 30 °C for 5 min. The digestion reaction was stopped by the addition of TEES/Proteinase K buffer (10 mM Tris pH 7.5, 10 mM EGTA, 10 mM EDTA, 1% SDS, 50 mg/ml proteinase K) and incubated at 37 °C overnight.
After MNase digestion, DNA was extracted following a classic phenol-chloroform protocol. DNA was resuspended in TE buffer and samples were prepared with loading buffer (peqGOLD) and analyzed by electrophoresis in a 2% agarose gel prepared in TBE buffer with ethidium bromide (Merck Millipore) to identify DNA. Ethidium bromide detection was subsequently carried out using a Gel Doc EZ Imaging System (Bio-Rad). Densiometric profiles were obtained using FIJI software (36) and normalized to total DNA content in the lane.
FISH
Cells were fixed using 4% formaldehyde and permeabilized with 0.1% Triton X-100. Poly(A)-RNA was detected by FISH using an ATTO 565-labeled oligo-dT probe (Microsynth AG) prepared in FISH buffer (0.3 M potassium citrate, 0.03M NaCl, 10% formamide) at a final concentration of 0.5 μM, incubated overnight at 37 ºC. After hybridization of the probe and washing steps, cells were mounted using SlowFade Gold (Thermo Fisher Scientific) containing DAPI. Imaging was performed using a Zeiss Cell Observer fluorescence microscope equipped with a 63× NA 1.3 water immersion objective and ZEN imaging software. The image analysis was performed using FIJI software (36). Mean nuclear fluorescence (N), determined with DAPI signal, and mean cytoplasmic fluorescence (C) were measured for each cell. The ratio of mean nuclear fluorescence to mean cytoplasmic fluorescence (N/C) was calculated for each cell.
Flow cytometry
For cell cycle analysis, cells were incubated with 10 μM BrdU (Merck Millipore) 15 min prior to their harvesting. BrdU and PI staining were analyzed using a MACSQuant Analyzer flow cytometer and MACSQuantify software (Miltenyi Biotec) as described (38).
Nascent DNA and RNA labelling
For nascent DNA labelling, cells were incubated with 40 μM EdU for 15 min before collection, fixed in 3.7% formaldehyde and permeabilized with 0.5% Triton X-100. EdU incorporation was revealed by a click-iT reaction: 4 mM CuSO4 (Merck Millipore), 2.5 μM Sulfo-cyanine 5 Azide (Lumiprobe), 100 mM Sodium Ascorbate (Merck Millipore). DNA was stained with DAPI, and coverslips were mounted.
For nascent RNA labelling, cells were incubated with 1 mM EU for 1 h and processed as above. EU incorporation was revealed with Click-iT® RNA Imaging kits (Thermo Fisher Scientific) using when indicated either Alexa Fluor 488 (Thermo Fisher Scientific) or a Cy3 (Lumiprobe) dyes Azides according to manufacturer's instructions. EU incorporation in the nucleolar compartment (identified by NPM1 staining) was assessed by multiplying the EU-positive nucleolar area (number of pixels) by the EU signal intensity (mean) in the NPM1-positive nucleolar compartment normalized to the EU signal intensity (mean) in the surrounding nucleoplasm, giving the so-called Integrated Density (IntDen).
Statistical analysis
Appropriate statistical tests were performed for each experiment, and statistical significance calculated using Prism (GraphPad Software). Unless state otherwise, to determine statistical significance of two independent conditions without assuming Gaussian distribution (WT and ΔATXN3 U2OS cells), Mann Whitney test was applied. Whereas to compare three or more independent conditions with non-Gaussian distribution (siRNA-depleted U2OS, CSM14.1 cells), Kruskal–Wallis analysis, followed by Dunn's multiple comparisons test were performed. When Gaussian distribution was assumed, one-way analysis of variance [ANOVA], followed by Sidak's multiple comparisons test were performed. A P value <0.05 was considered statistically significant, ns = not significant. Statistical significance was noted as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Unless stated otherwise, representative experiments of at least three independent ones are shown and depicted is the mean ± standard deviation (SD). Boxplots span the interquartile range, and the inside segments show the median value. Whiskers above and below the box show the max. and min., respectively. N = number of replicates, n = number of individual values. Additional details are listed in the figure legends.
RESULTS
ATXN3 controls DNA replication timing
Previous reports demonstrated the involvement of ATXN3 in controlling several DNA damage response factors (16,17).To investigate possible additional mechanisms of how ATXN3 contributes to genome stability, we analyzed DNA replication after knocking out ATXN3 (ΔATXN3) or depleting the protein by siRNA in U2OS cells (Supplementary Figure S1A). When measuring S phase progression by flow cytometry in synchronized cells, no significant changes were observed after knocking out ATXN3 (Supplementary Figure S1B). However, the absence of ATXN3 led to alterations in replication dynamics when studied at single cell level by DNA fiber assays (Supplementary Figure S1C). Both in ΔATXN3 and siATXN3 cells the replication track length and the inter-origin distance (IOD) decreased compared to control cells (Figure 1A–E). The lack of ATXN3 also increased the levels of newly fired origins (Supplementary Figure S1D), without affecting the amount of stalled replication forks (Supplementary Figure S1E). We considered possible explanations of how ATXN3 could control DNA replication. A decrease in track length could be due to collisions between the DNA replication and transcription machineries, which would lead to more DNA-RNA hybrids, or R-loops (39). However, no difference in R-loop signal was observed when comparing control cells to ATXN3-depleted cells (Supplementary Figure S1F, G). To study if the primary effect of the lack of ATXN3 is increased origin firing, cells were treated with a CDC7 inhibitor (CDC7i) that blocks the origin firing (40), after which DNA fibers were analyzed. Interestingly, inhibiting CDC7 in ΔATXN3 cells increased the track length (Figure 1F), without affecting the percentage of stalled forks, but reducing, as expected, the proportion of newly fired origins (Supplementary Figure S1H, I), indicating that the absence of ATXN3 promotes an increase in origin firing and as the cells compete for limiting replication factors (40), fork speed is reduced.
Figure 1.

ATXN3 controls the timing of DNA replication. (A) Representative images of replication forks in DNA fiber analysis (A–D), performed as indicated in the materials and methods section, of wild type (WT) and ATXN3 knockout (ΔATXN3) U2OS cells (upper panel) and U2OS cells depleted for ATXN3 using two different siRNA oligonucleotides (siATXN3) (lower panel). (B) Quantification of replication DNA track length in WT and ΔATXN3 U2OS cells (n = 350). (C) Quantification of replication DNA track length in control and siATXN3 U2OS cells (n = 300). (D) Quantification of the inter-origin distance (IOD) in WT and ΔATXN3 U2OS cells (n = 50). (E) Quantification of IOD in control and siATXN3 U2OS cells (n = 50). (F) Quantification of replication DNA track length in WT and ΔATXN3 U2OS cells, treated or not with CDC7 inhibitor (CDC7i, 60 μM) for 4 h (n = 300). (G, H) Schematic representation (top) and representative images (bottom) of the different EdU labelling patterns, normal (G) and abnormal (H). (I) Quantification of the percentage of cells with abnormal EdU patterns in control, ΔATXN3 and siATXN3 cells. (J) Quantification of replication DNA track length in WT and ΔATXN3 U2OS cells, untreated or treated with sucrose (80 mM) for 1 h (n = 250).
Sites of DNA synthesis can be detected as EdU-positive nuclear foci by immunofluorescence (IF). During S phase progression, the number and localization of these foci change due to the coordination of origin firing at distinct chromatin and nuclear regions (Figure 1G) (41). Abnormal patterns of DNA synthesis can be observed in cells with deficient spatiotemporal control of DNA replication (Figure 1H), as described in the absence of factors important for DNA replication timing control, such as Treslin or RIF1 (42–47). Interestingly, cells deficient for ATXN3 displayed an increase in abnormal EdU DNA replication patterns (Figure 1I), in addition to significant differences in some of the regular replication patterns (Supplementary Figure S1J). Together, these data suggest that ATXN3 controls DNA replication and the absence of ATXN3 leads to an increase in origin firing and a decreased replication rate, possibly as a compensatory mechanism. Spatial organization of the chromatin is known to affect replication timing. In this context, the involvement of RIF1 in chromatin three-dimensional organization is critical for replication timing (47–51). We therefore studied possible differences in RIF1 staining by IF in the absence of ATXN3. Interestingly, in unperturbed conditions, the lack of ATXN3 caused a reduction in the number of RIF1 foci per nucleus (Supplementary Figure S2A–C), without a drop in RIF1 protein levels (Supplementary Figure S1A).
A lower IOD and a reduction in RIF1 foci could be indicative of changes in chromatin organization, for example, the loss of chromatin loops, which would make dormant origins more accessible to the replicative machinery, leading to an increased origin usage. Importantly, the lower DNA replication fork progression observed in ΔATXN3 cells could be rescued by inducing a hypertonic shock by incubating cells with 80 mM sucrose for 1 h, which promotes chromatin compaction (Figure 1J) (52). Moreover, the reduction in RIF1 foci in ATXN3 deficient cells was also rescued by a brief hypertonic shock (Supplementary Figure S2D). Altogether these data suggest that ATXN3 controls the replication timing via changes in chromatin structure.
ATXN3 influences chromatin organization
We next investigated the chromatin status in cells deficient for ATXN3, studying several epigenetic marks, associated to different chromatin organizations (Supplementary Figure S2E). The absence of ATXN3 caused an increase in the euchromatin-associated mark H4K16Ac (Figure 2A, B) and led to a statistically significant decrease in the facultative heterochromatin mark H3K27me3 (Figure 2C, D), while the constitutive heterochromatin mark H3K9me3 remained unchanged (Supplementary Figure S2F, G). The elevated H4K16 acetylation in ΔATXN3 cells was accompanied by an increased signal of G-quadruplex (G4), a non-canonical DNA secondary structure, predominantly present in nucleosome-depleted regions in euchromatin (53–55) (Supplementary Figure S2H, I).
Figure 2.

ATXN3 modulates chromatin marks and the mobility of histone H1.5 in chromatin. (A, B) Fluorescence analysis of H4K16Ac staining in WT and ΔATXN3 cells (A) or cells treated with the indicated siRNA for 96 h (B). All samples were analyzed with the same exposure time. The mean intensity within the nuclear area (identified by the DAPI signal) was quantified (n = 400). (C, D) As (A, B), but for H3K27me3 staining. (E) Example of FRAP experiment in WT and ΔATXN3 cells, transiently transfected with GFP-Histone H1.5. GFP-H1.5 was bleached in the indicated regions of interest, indicated by yellow and magenta circles (heterochromatic and euchromatic areas, respectively). Shown is the fluorescence recovery after 3, 7 and 15 s. (F) FRAP analysis of GFP-H1.5 in WT (n = 13) and ΔATXN3 (n = 11) cells in heterochromatin (upper panel) or euchromatin (lower panel) regions. Data are represented as mean ± SD. (G) Percentage of mobile fraction calculated from the fitted data of the FRAP at heterochromatin (left panel) and euchromatin (right panel) regions in WT and ΔATXN3 cells. (H) Half-times of fluorescence recovery (τ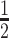 ) from the fitted data of FRAP in WT and ΔATXN3 cells at heterochromatin (left panels) and euchromatin (right panels) regions.
) from the fitted data of FRAP in WT and ΔATXN3 cells at heterochromatin (left panels) and euchromatin (right panels) regions.
Next, the mobility of the linker histone H1, which regulates higher-order chromatin folding, was studied as an indicator of chromatin organization (56–58). GFP-H1.5 was expressed in WT and ΔATXN3 U2OS cells and its mobility was studied by fluorescence recovery after photobleaching (FRAP). In these experiments, heterochromatin and euchromatin were defined as areas strongly or weakly labelled with Hoechst 33342, respectively (Supplementary Figure S2J). The absence of ATXN3 caused a change in GFP-H1.5 FRAP, with an increased mobile fraction in both heterochromatin and euchromatin regions (Figure 2E–G). However, no differences in the kinetics of recovery time, measured as the half time of fluorescence recovery, were detected (Figure 2H). These data of a lower amount of statically bound histone H1.5 in ΔATXN3 cells support the idea of an altered higher-order chromatin structure, in particular a less compacted state, in the absence of ATXN3.
As the lack of ATXN3 reduces facultative heterochromatin and increases histone H1 mobility, micrococcal nuclease (MNase) digestion was used to examine nucleosomal occupancy (59). Digestion of isolated nuclei with increasing concentrations of MNase demonstrated that chromatin from ΔATXN3 cells was more sensitive to nuclease digestion than WT chromatin, especially noticeable after treating the nuclei with low concentrations (10 units) of the enzyme (Figure 3A, B). Together, these results indicate that global chromatin structure is altered and less compact in ATXN3 deficient cells.
Figure 3.

ATXN3 deficiency increases chromatin accessibility and disrupts nuclear and nucleolar morphology and organization. (A) Nuclei from WT or ΔATXN3 U2OS cells were digested with indicated units (U) of MNase for 5 min. Digestion of the genomic DNA by agarose gel electrophoresis is shown. (B) Densitometric profiles of the 10 U MNase digestion product. Position of the nucleosomes (mono, di- and tri-nucleosomes) is schematically indicated. (C) Lamin A/C IF analysis in U2OS cells. Shown are examples of normal (top) and abnormal (bottom) nuclear shape, based on lamin A/C localization. (D) Quantification of abnormal nuclear shape (WT versus ΔATXN3 N = 3, siRNA N = 5). (E) NPM1 IF after pre-extraction. (F) Quantification of nucleolar area, as determined by NMP1 signal, in WT and ΔATXN3 cells (n = 600). (G) As (F), but control and siRNA ATXN3-depleted cells (n = 400). (H) Quantification of the number of nucleoli per nucleus in the indicated cells (n = 100).
ATXN3 regulates nuclear and nucleolar morphology and organization
Next, the possible control of nuclear morphology by ATXN3 was studied using IF for lamins A/C, key components of the nuclear lamina, a fibrillar network close to the inner nuclear membrane involved in the regulation of many nuclear activities (60–62). With this staining, cells with disrupted nuclear morphology can be distinguished (Figure 3C). The absence of ATXN3 caused a significative increase in the percentage of cells with abnormal morphology without changes in lamin A/C protein levels (Figure 3D, Supplementary figure S3A).
Global chromatin organization impacts on the nucleoli, which are formed around specific chromosomal regions called nucleolar organizer regions that contain arrays of ribosomal DNA gene clusters. To examine a possible effect on nucleoli morphology and organization after ATXN3 depletion, IF for NPM1, the major nucleolar protein, was performed (Figure 3E). ATXN3 loss caused an increase in nucleolar area (Figure 3E–G), whereas no consistently significant differences in NPM1 protein levels or in nuclear size were observed (Supplementary Figure S3A–C). Moreover, ATXN3 deficiency led to a significant decrease in the number of nucleoli per nucleus, compared to control cells (Figure 3H). Given the liquid–liquid phase properties of nucleoli, these data suggest that multiple nucleoli are fused in the absence of ATXN3, probably due to a less compacted state of the chromatin. Indeed, when comparing the proportion of total nucleolar area to nuclear area, an increase was observed in ATXN3 deficient cells (Supplementary Figure S3D, E), indicating that nucleolar fusion is not the only cause for these effects and suggesting that also relaxation of nucleolar chromatin takes place. Indeed, under hypertonic conditions, a decrease in nucleolar area was observed in ATXN3-depleted cells (Supplementary Figure S3F, G).
ATXN3 depletion leads to increased global transcription
The observed changes in nucleolus morphology and number upon ATXN3 depletion could be associated with alterations in transcription and the amount of ribosomal RNA (rRNA) (63). To study RNA polymerase I-dependent transcription (rRNA), EU incorporation in the nucleolar compartment was analyzed. ATXN3-depleted cells showed a significant increase in EU signal (IntDen) within the NPM1-positive nucleolar compartment (Figure 4A–C). This higher nucleolar EU signal could be due to increased rRNA transcription, but also to a defect in nuclear export of RNA as a consequence of nuclear morphology disruptions (64,65). Therefore, to evaluate RNA export, messenger RNA (mRNA) accumulation in the nucleus and in the cytoplasm was quantified using an oligo-(dT) FISH assay (Figure 4D). The nuclear/cytoplasmic ratios in the absence of ATXN3 do not support RNA-retention in the nucleus as explanation for the observed increase in nucleolar EU signal (Figure 4E, F). However, loss of ATXN3 caused an increase in poly(A)-RNA in both nuclear and cytoplasmic compartments (Figure 4G–J), indicating that RNA polymerase-II-dependent transcription is also increased, and therefore suggesting that ATXN3 controls not only rRNA, but global transcription, instead of alterations in nuclear-cytoplasm transport processes. In addition, the transcript levels of pericentromeric satellite 2 Sat-2, a marker of heterochromatin structure, were increased upon ATXN3 depletion (Supplementary Figure S3H). Together these data suggest that the absence of ATXN3 leads to augmented global transcription, likely due to a less compacted chromatin status, which facilitates the accessibility of the transcriptional machinery.
Figure 4.

ATXN3 depletion increases global transcription. (A) Control, ΔATXN3 and siATXN3 U2OS cells were incubated with EU prior to fixation, followed by EU detection and NPM1 IF. Representative images are shown (B). Quantification of EU Integrated Density (IntDen) in the nucleolar compartment of WT and ΔATXN3 cells. For calculation of the IntDen, see materials and methods (n = 200). (C) As (B), but for control and siATXN3 cells (n = 400). (D) Representative images showing poly(A)-RNA (red) FISH in WT and ΔATXN3 cells. Yellow lines show nuclear and plasmatic compartments. (E) The distribution of poly(A)-RNA was depicted as the ratio of the quantified nuclear to cytoplasmic mean intensities in WT and ΔATXN3 cells from (D) (n = 240). (F) As (E), but for control and ATXN3 siRNA cells (n = 300). (G, H) Quantification of nuclear (G) and cytoplasmic (H) poly(A)-RNA levels in WT or ΔATXN3 cells (n = 240). (I, J) Quantification of nuclear (I) and cytoplasmic (J) poly(A)-RNA levels in control or siATXN3 cells (n = 300).
Transcriptional control by ATXN3 is independent of its ubiquitin hydrolase activity
ATXN3 harbors a Josephin domain with deubiquitinase activity (66–68). To investigate the molecular mechanism of ATXN3 action, we studied if the catalytic activity is required for the described transcriptional regulation. For this, ΔATXN3 cells were complemented with a WT or a catalytic inactive (CI) version of the protein, followed by analysis of EU IntDen incorporation. Interestingly, the anomaly in rRNA transcription caused by knockout of ATXN3 could be restored by both versions of ATXN3 (Figure 5A, B, Supplementary Figure S3I). When using EU staining to evaluate nucleolar morphology (69), an increase in the nucleolar EU-positive area was observed in the absence of ATXN3 (Supplementary Figure S3J), in accordance with our data on NPM1 staining (Figure 3E–G). As for transcription, the expression of both WT and CI ATXN3 also restored the nucleolar area values (Supplementary Figure S3J). Altogether, these results indicate that the ubiquitin hydrolase activity is not required for the role of ATXN3 in nucleolar morphology and transcription.
Figure 5.

ATXN3 functions independently of its enzymatic activity and cooperates with HDAC3 to control chromatin structure. (A) WT or ΔATXN3 cells were transfected with empty vector (EV), wild type (WT) or catalytic inactive (CI) Flag-tagged ATXN3. 36 h after transfection cells were incubated with EU and stained for EU and Flag. Representative images are shown. (B) Quantification of EU IntDen in the nucleolar compartment of Flag-positive cells of (A). Dash red line indicates the mean of the reference condition (WT cells with EV) (n = 400). (C) Quantification of the nucleolar area of (Supplementary Figure S4B) based on NPM1 staining (n = 400). (D) Quantification of replication DNA track length in WT and ΔATXN3 U2OS cells transfected with the indicated siRNA and left untreated or treated with sucrose (80 mM) for 1 h (n = 300). (E) Quantification of nucleolar area based on NPM1 staining (Supplementary Figure S5B) in WT or ΔATXN3 transfected with the indicated siRNAs (n = 400). (F) Quantification of EU IntDen in the nucleolar compartment (Supplementary Figure S5C) of WT and ΔATXN3 cells transfected with EV or GFP-HDAC3 (n = 270). (G) Representative HDAC3 IF images after pre-extraction in WT and ΔATXN3 U2OS cells. (H) Quantification nuclear HDAC3 intensity from (G) (n = 200). (I) WT or ΔATXN3 cells were transfected with GFP-HDAC3. Representative images are shown. (J) The distribution of HDAC3 was depicted as the ratio of the quantified nuclear to cytoplasmic mean intensities in WT and ΔATXN3 cells from (I) (n = 100).
ATXN3 and histone deacetylase HDAC3 cooperate in regulating chromatin organization
In general, histone acetylation correlates with gene activation, while deacetylation is associated with repression (70). ATXN3 was described to control the transcription of the MMP-2 gene through the recruitment of the histone deacetylase HDAC3 (14). Moreover, HDAC3 was previously reported to regulate DNA replication and chromatin structure, and the absence or inhibition of HDAC3 results in similar phenotypes to those described here in the absence of ATXN3 (71–73). Therefore, we investigated if ATXN3 could control some aforementioned functions via HDAC3.
Depletion of HDAC3 in WT U2OS cells (Supplementary Figure S4A) caused changes in nuclear and nucleolar morphology. Specifically, an increase in nuclei with abnormal nuclear shape was observed, when evaluated with lamin A/C staining (Supplementary Figure S4B), together with an increase in the nucleolar area (Figure 5C, Supplementary figure S4C). In addition, treatment with an HDAC3 inhibitor mimicked the nucleolar phenotype observed after HDAC3 depletion, demonstrating that the deacetylation activity was required for this process (Supplementary Figure S4D, E). Furthermore, HDAC3 depletion increased H4K16 acetylation and reduced the levels of H3K27me3 (Supplementary Figure S4F-H), similar to what observed in the absence of ATXN3 (Figure 2A, B). Additional results regarding replication dynamics demonstrated that depletion of HDAC3 in WT cells caused a drop in the DNA replication track length whereas it had no additional effect in ΔATXN3 cells (Figure 5D). Similar to what was observed before for cells deficient for ATXN3 (Figure 1J), the track length of HDAC3-depleted cells increased upon treatment with a short hypertonic shock (Figure 5D).
As HDAC inhibition was shown to lead to enhanced G4 formation (74), G4 intensity upon inhibition of HDAC3 was studied in our cells (Supplementary Figure S4I). As expected, this treatment resulted in an increased G4 signal (Supplementary Figure S4J). Although HDAC3i in the absence of ATXN3 slightly elevated the G4 intensity, the increase of G4 signal was much higher when inhibiting HDAC3 in WT cells (Supplementary Figure S4J), indicating that this effect is partially epistatic.
Interestingly, downregulating HDAC3 in the absence of ATXN3 led to the same increase in nucleolar area as the single depletion of the proteins (Figure 5E, Supplementary figure S5A, B). Expression of GFP-HDAC3 additionally decreased EU IntDen incorporation in both WT and ΔATXN3 cells (Figure 5F, Supplementary figure S5C). Altogether, these data indicate that ATXN3 regulates euchromatin and facultative heterochromatin via HDAC3.
We next investigated how ATXN3 could control HDAC3. Although ATXN3 was described to control HDAC3 stability (75), no changes in HDAC3 stability in absence of ATXN3 were detected in our system (Supplementary Figure S5D). These data are in agreement with our earlier observations that the ATXN3 catalytic activity is not required for this phenotype. We then hypothesized that ATXN3 could control chromatin organization by recruiting HDAC3 to the nucleus/chromatin. Analysis of chromatin-bound HDAC3 by IF after removal of the soluble proteins by extraction with a mild detergent demonstrated a decrease in chromatin-bound HDAC3 upon ATXN3 depletion (Figure 5G, H). Moreover, when GFP-HDAC3 was expressed in WT and ΔATXN3 cells, the ratio of the nuclear versus cytoplasmic GFP-HDAC3 signal was lower in the ATXN3 null cells, suggesting that ATXN3 regulates the efficient recruitment of HDAC3 to the nuclei (Figure 5I, J). Subsequent immunoprecipitation experiments confirmed the described interaction between HDAC3 and ATXN3 and ATXN3-CI retained its ability to bind HDAC3 (Figure 6A) (14). Also, the expanded form of ATXN3 (ATXN3-PolyQ), responsible for SCA3, interacted with HDAC3 (Figure 6A).
Figure 6.

ATXN3-PolyQ fails in recruiting HDAC3 into the nucleus. (A) HEK293T cells were transfected with GFP-tagged ATXN3-WT, CI or PolyQ (80Q) and mCherry-HDAC3. mCherry was immunoprecipitated (IP) and analyzed by western blot using the indicated antibodies. (B) WT or ΔATXN3 cells were transfected with GFP-ATXN3-WT, -CI or -PolyQ (80Q) together with mCherry-HDAC3 and imaged for fluorescence. Representative images are shown. (C) The distribution of mCherry-HDAC3 was depicted as the ratio of the quantified nuclear to cytoplasmic mean intensities of mCherry in GFP-positive WT cells from (B) (n = 150). (D) As (C) but in GFP-positive ΔATXN3 cells from (B) (n = 100). (E) WT or ΔATXN3 cells were transfected with EV or GFP-ATXN3-PolyQ (80Q), incubated with and stained for EU. Representative images are shown. (F) Quantification of EU IntDen in the nucleolar compartment of GFP-positive cells of (E). Dash red line indicates the mean of the reference condition (WT cells with EV) (n = 350). (G) Quantification of nucleolar area (areas with higher EU intensity) from (E). Dash red line indicates the mean of the reference condition (WT with EV) (n = 350).
The pathogenic form of ATXN3 acts as a dysfunctional mutant
Although ATXN3-PolyQ interacted with HDAC3, expansion of the PolyQ could still have functional consequences for transcription and DNA replication. Studying this could shed light on the molecular mechanisms of SCA3. First the effect of ATXN3-PolyQ on HDAC3 localization was investigated by expressing GFP-ATXN3-WT, -CI or -PolyQ together with mCherry-HDAC3 in WT or ΔATXN3 cells (Figure 6B). The nuclear/cytoplasmic ratios of the mean mCherry-HDAC3 signal in GFP-positive cells showed reduced values in cells expressing ATXN3-PolyQ both in WT (Figure 6C) and ΔATXN3 cells (Figure 6D), indicating a dominant negative effect of ATXN3-PolyQ on HDAC3 localization. In contrast, the ATXN3-CI expression did not reduce the nuclear localization of HDAC3, which is in agreement with our previous data showing that the catalytic activity of ATXN3 was not required to rescue nucleolar function and morphology. ATXN3-PolyQ expression additionally was not able to rescue the increased nucleolar transcription observed in ΔATXN3 cells and led to an increase in transcription in WT cells, mimicking the effect of ATXN3 depletion (Figure 6E, F). Moreover, the EU high intensity incorporation areas were enlarged after expression of ATXN3-PolyQ in control cells and its expression in ΔATXN3 cells did not restore the nucleolar area to the values observed in control cells (Figure 6G).
To study the effect of expanded ATXN3 on chromatin structure, rat mesencephalic CSM14.1 cells expressing human full length ATXN3 with a nonexpanded ATXN3-WT (SCA3-Q23) or an expanded glutamine track ATXN3-PolyQ (SCA3-Q70) using a Tet-Off system (Supplementary Figure S5E) were used as cell culture model of SCA3 (16). Upon expression of SCA3-Q70 but not SCA3-Q23, abnormal shaped nuclei, evaluated by DAPI staining, were observed (Figure 7A, B). Also, a statistically significant increase in the euchromatin-associated mark H4K16Ac was observed after SCA3-Q70 expression, compared to SCA3-Q23 (Figure 7C, D). This increase was accompanied by a decrease in the facultative heterochromatin-associated mark H3K27me3 (Figure 7C, E).
Figure 7.

ATXN3-PolyQ acts as a dysfunctional mutant. (A) Representative images of DAPI staining of CSM14.1 cells expressing empty vector (mock), human ATXN3-WT (SCA3-Q23) or ATXN3-PolyQ (SCA3-Q70) to evaluate nuclear morphology. (B) Quantification of abnormal nuclear shape from (A) (N = 4). (C) CSM14.1 cells expressing the indicated forms of ATXN3 were stained for H4K16Ac and H3K27me3. Representative images are shown. (D) Quantification of H4K16Ac signal from (C). All samples were analyzed with the same exposure time. The mean intensity within the nuclear area (identified by the DAPI signal) was quantified (n = 300). (E) As (D) but for H3K27me3 staining (n = 300). (F) Representative images of replication forks in DNA fiber analysis of CSM14.1 cells expressing the indicated forms of ATXN3. (G) Quantification of DNA replication track length from cells in (F) (n = 300). (H) CSM14.1 cells expressing SCA3-Q23 or SCA3-Q70 human ATXN3 were incubated with EU prior to fixation, followed by EU staining. (I). Quantification of EU IntDen of (H) (n = 430).
Surprisingly, and similar to what we observed in the absence of ATXN3, SCA3-Q70 expression decreased the DNA replication track length and IOD compared to control cells and those expressing SCA3-Q23 (Figure 7F, G, Supplementary figure S5F). Moreover, SCA3-Q70 expression led to a higher EU IntDen when compared to control or SCA-Q23-expressing cells (Figure 7H, I). Together these data demonstrate that the expanded form of ATXN3 is dysfunctional in controlling DNA replication and transcription, a similar phenotype as observed in the absence of ATXN3.
DISCUSSION
ATXN3 was recently described to be involved in maintaining genomic stability upon DNA damage by controlling several proteins in the DNA damage response (16–18). Previous studies have also suggested that ATXN3 is a transcriptional regulator, as the lack of ATXN3 leads to deregulated expression of certain genes. However, up to date, this function has been associated to specific control in specific gene promoters (2,14,76). Here we demonstrate a novel function of ATXN3: regulation of global chromatin organization in unperturbed conditions with effects on DNA replication and transcription. We show that the lack of ATXN3 increases the mobility of histone H1 and triggers the conversion of facultative heterochromatin into euchromatin, with elevated H4K16 acetylation and diminished trimethylation of H3K27, data indicative of a more open status of the chromatin, as reinforced with the MNase sensitivity assay. Interestingly, nuclear and nucleolar morphologies were affected, specifically the nucleolar area was enlarged in cells with lower levels of ATXN3 and we speculate that this effect is due to a fusion of multiple nucleoli as a consequence of chromatin relaxation, which allows nucleoli to get closer to each other and fuse due to their liquid–liquid phase properties (77,78). In accordance, nucleolar area values in ATXN3 deficient cells were rescued by a brief hypertonic treatment, which triggers chromatin compaction (52).
The tight coordination of replication origin firing at different chromatin regions demonstrates that the timing of DNA replication heavily depends on chromatin organization. A variation in chromatin organization will therefore affect DNA replication (43,47). Indeed, lack of ATXN3 leads to an increased replication fork speed and reduced IOD, indicative of higher origin firing. Our data show that the primary cause of this abnormality in DNA replication is increased origin activation, described to result in competition for limiting replication factors, since inhibiting origin firing in ΔATXN3 cells leads to an augmented fork speed. These data are in accordance with previous studies that show that more open chromatin could lead to higher origin usage (79,80). Alterations in chromatin structure would also lead to a dysregulation of replication timing, in accordance with the observed increase in abnormal replication patterns. Moreover, the observed increase in euchromatin and chromatin accessibility is compatible with higher transcription levels in certain genes reported in cells lacking ATXN3 (2) and indeed, we demonstrated an enhancement in both polymerase I and II-dependent transcription in cells deficient for ATXN3. By regulating global chromatin structure, ATXN3 could also control origin firing in rDNA, regions of acrocentric chromosomes or other heterochromatin regions such as lamin-associated domains (LADs) (23,81,82).
How does ATXN3 control chromatin structure? Surprisingly, and in contrast to many previously described functions of ATXN3 (10,75,83–87), the catalytic activity seems dispensable for this process, as both the morphological and functional nucleolar alterations observed in ATXN3-deficient cells can be restored by a CI version of the enzyme. Instead, HDAC3, a known ATXN3 interactor and regulator of transcriptional repression, DNA replication and chromatin structure, was identified as mediator of the effect of ATXN3 on chromatin structure. HDAC3 depletion or enzymatic inhibition phenocopies the effects detected upon the lack of ATXN3. Furthermore, ATXN3 and HDAC3 co-depletion did not show an additional effect on nucleolar area or DNA replication dynamics, and the expression of HDAC3 was able to rescue the transcriptional defects shown in ΔATXN3 cells, indicating that the effects of ATXN3 upon chromatin organization occur with the involvement of HDAC3. Most importantly, the abnormalities in DNA replication shown in ATXN3 or HDAC3 deficient cells could be alleviated by a short hypertonic treatment, which triggers chromatin condensation. We confirmed the previously described ATXN3 and HDAC3 interaction, and interestingly, cells lacking ATXN3 show less chromatin-bound HDAC3 together with a decreased ratio of nuclear/cytoplasmic HDAC3. Expression of ATXN3-WT or -CI in ΔATXN3 cells moreover promotes HDAC3 to localize in the nucleus. These data together suggest that ATXN3 regulates chromatin compaction by recruiting HDAC3 to the chromatin in a manner that is independent of the ubiquitin hydrolase activity and therefore is acting in this context as a scaffold protein (Figure 8).
Figure 8.
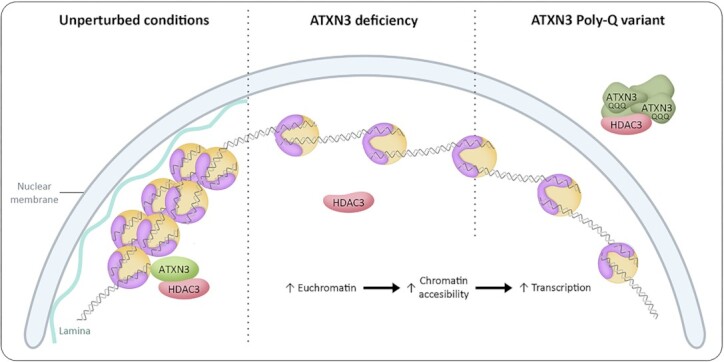
Schematic model of ATXN3 controlling chromatin structure. In unperturbed conditions, ATXN3 promotes HDAC3 chromatin binding and heterochromatin formation. ATXN3 deficiency leads to mislocalization of HDAC3, causing an increase in euchromatin prevalence. In an SCA3 context, the ATXN3-PolyQ variant sequesters HDAC3 impeding correct HDAC3 recruitment to the chromatin, thereby generating a situation equivalent to the absence of ATXN3. An increase in the less condensed euchromatin could explain the described phenotypes, with special consideration to transcriptional dysregulation, which could have an important role in SCA3 pathogenesis.
In order to explain the molecular basis of SCA3, we investigated how the expression of the ATXN3-PolyQ variant affected the phenotypes linked to this novel ATXN3 function. Expression of ATXN3-PolyQ was not able to rescue the imbalanced nucleolar morphology and function due to ATXN3 depletion, and similar defects upon expression of ATXN3-PolyQ in WT cells suggest a dominant negative effect. ATXN3-PolyQ expression additionally caused abnormalities in nuclear shape, which could be explained by induced alterations in chromatin organization, possibly by affecting the contact between the chromatin and nuclear-insoluble structures, such as lamins and nucleoli. Moreover, the expression of this version of ATXN3 was also described to induce transcriptional changes in certain genes (14,15,88,89), which is consistent to our observations regarding transcription in cells lacking ATXN3. Interestingly, however, ATXN3-PolyQ and HDAC3 were shown to interact. We believe that this pathogenic version of ATXN3 might affect DNA replication, transcription and nuclear morphology possibly due to the formation of aggregates that could sequester HDAC3 (Figure 8), as suggested for many pathological extended PolyQ proteins (90–93). Indeed, the expression of ATXN3-PolyQ causes the mislocalization of exogenous HDAC3, and HDAC3 high-intensity signal regions were detected overlapping with the ATXN3-PolyQ higher signal (Figure 6B). In accordance, a decrease in ATXN3-PolyQ chromatin binding was demonstrated before (13,14), which could affect HDAC3 recruitment to the chromatin. ATXN3-PolyQ was reported to maintain its hydrolase activity (86), confirming once more that the catalytic activity of ATXN3 is not involved in the control of DNA replication, transcription and nuclear/nucleolar morphology. Altogether, this adds an extra layer of complexity to the transcriptional defect on MMP2 observed by Evert and co-workers (14), as this loss of function in SCA3 could lead to generally dysregulated transcription profiles, thereby not only affecting specific genes involved in certain pathways, such as inflammatory cascade (13), but promoting the expression of physiologically repressed genes in a larger-scale way. Indeed, microarray analysis of the cerebellum of SCA3 model mice detected transcriptional changes in genes involved in a range of pathways (15).
Interestingly, the importance of the acetylation balance for neuronal viability is widely accepted. The levels and enzymatic activity of histone acetyltransferases (HATs) and HDACs, the enzymes responsible of regulating acetylation homeostasis within the nucleus, play therefore a critical role in neurodegenerative conditions. The observation that treatment of neurons with HDACs inhibitors, or overexpression of HATs in these cells, leads to cell death, underscores the hypothesis of the need of a precise balance in histone acetylation for neuronal survival in non-pathological conditions (94–98). Hence, the observed dysregulation of HDAC3 localization upon ATXN3-PolyQ expression could contribute to the onset or progression of SCA3. We believe that our findings shed light on the molecular pathogenesis of SCA3, and although more research needs to be done in this field, we speculate that treatments that could counteract HDAC3 dysfunction might contribute to alleviate SCA3 symptoms.
DATA AVAILABILITY
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs James Holaska and Henry Paulson for sharing reagents.
Contributor Information
Esperanza Hernández‐Carralero, Unidad de Investigación, Hospital Universitario de Canarias, La Laguna, Santa Cruz de Tenerife, Spain; Escuela de Doctorado y Estudios de Posgrado, Universidad de la Laguna, Santa Cruz de Tenerife, Spain; Instituto de Tecnologías Biomédicas, Centro de Investigaciones Biomédicas de Canarias, Facultad de Medicina, Campus Ciencias de la Salud, Universidad de La Laguna, Santa Cruz de Tenerife, Spain.
Elisa Cabrera, Unidad de Investigación, Hospital Universitario de Canarias, La Laguna, Santa Cruz de Tenerife, Spain.
Gara Rodríguez-Torres, Unidad de Investigación, Hospital Universitario de Canarias, La Laguna, Santa Cruz de Tenerife, Spain; Escuela de Doctorado y Estudios de Posgrado, Universidad de la Laguna, Santa Cruz de Tenerife, Spain; Instituto de Tecnologías Biomédicas, Centro de Investigaciones Biomédicas de Canarias, Facultad de Medicina, Campus Ciencias de la Salud, Universidad de La Laguna, Santa Cruz de Tenerife, Spain.
Yeray Hernández-Reyes, Unidad de Investigación, Hospital Universitario de Canarias, La Laguna, Santa Cruz de Tenerife, Spain; Escuela de Doctorado y Estudios de Posgrado, Universidad de la Laguna, Santa Cruz de Tenerife, Spain; Instituto de Tecnologías Biomédicas, Centro de Investigaciones Biomédicas de Canarias, Facultad de Medicina, Campus Ciencias de la Salud, Universidad de La Laguna, Santa Cruz de Tenerife, Spain.
Abhay N Singh, MRC Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Oxford, UK.
Cristina Santa-María, Centro de Biología Molecular Severo Ochoa (CBMSO), Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid (CSIC/UAM), Madrid, Spain.
José Miguel Fernández-Justel, Centro de Biología Molecular Severo Ochoa (CBMSO), Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid (CSIC/UAM), Madrid, Spain.
Roel C Janssens, Department of Molecular Genetics, Oncode Institute, Erasmus MC, University Medical Center Rotterdam, Rotterdam, The Netherlands.
Jurgen A Marteijn, Department of Molecular Genetics, Oncode Institute, Erasmus MC, University Medical Center Rotterdam, Rotterdam, The Netherlands.
Bernd O Evert, Department of Neurology, University Hospital Bonn, Bonn, Germany.
Niels Mailand, Protein Signaling Program, Novo Nordisk Foundation Center for Protein Research, University of Copenhagen, Copenhagen, Denmark; Center for Chromosome Stability, Department of Cellular and Molecular Medicine, University of Copenhagen, Copenhagen, Denmark.
María Gómez, Centro de Biología Molecular Severo Ochoa (CBMSO), Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid (CSIC/UAM), Madrid, Spain.
Kristijan Ramadan, MRC Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Oxford, UK.
Veronique A J Smits, Unidad de Investigación, Hospital Universitario de Canarias, La Laguna, Santa Cruz de Tenerife, Spain; Instituto de Tecnologías Biomédicas, Centro de Investigaciones Biomédicas de Canarias, Facultad de Medicina, Campus Ciencias de la Salud, Universidad de La Laguna, Santa Cruz de Tenerife, Spain; Universidad Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain.
Raimundo Freire, Unidad de Investigación, Hospital Universitario de Canarias, La Laguna, Santa Cruz de Tenerife, Spain; Instituto de Tecnologías Biomédicas, Centro de Investigaciones Biomédicas de Canarias, Facultad de Medicina, Campus Ciencias de la Salud, Universidad de La Laguna, Santa Cruz de Tenerife, Spain; Universidad Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Agencia Estatal de Investigación [PID2019-109222RB-I00/AEI/10.13039/501100011033]; European Union Regional Funds (FEDER) (to R.F., V.A.J.S.); Agencia Canaria de Investigación, Innovación y Sociedad de la Información [ProID2020010109]; FEDER (to R.F.); Agencia Canaria de Investigación, Innovación y Sociedad de la Información de la Consejería de Economía, Industria, Comercio y Conocimiento and the European Social Fund integrated Operational programme of the Canary Islands 2014–2020, Eje 3 Tema Prioritario 74 (85%) (to E.H.C.); Medical Research Council Programme [MR/X006409/1 to K.R.]; Breast Cancer Now [2019DecPR1406 to K.R.]. Funding for open access charge: Agencia Canaria de Investigación, Innovacióny Sociedad de la Información [ProID2020010109]; European Union RegionalFunds (FEDER).
Conflict of interest statement. None declared.
REFERENCES
- 1. Costa M.C., Paulson H.L.. Toward understanding Machado-Joseph disease. Prog. Neurobiol. 2012; 97:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zeng L., Zhang D., McLoughlin H.S., Zalon A.J., Aravind L., Paulson H.L.. Loss of the spinocerebellar Ataxia type 3 disease protein ATXN3 alters transcription of multiple signal transduction pathways. PLoS One. 2018; 13:e0204438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., Akiguchi I.et al.. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994; 8:221–228. [DOI] [PubMed] [Google Scholar]
- 4. Evert B.O., Wüllner U., Schulz J.B., Weller M., Groscurth P., Trottier Y., Brice A., Klockgether T.. High level expression of expanded full-length ataxin-3 in vitro causes cell death and formation of intranuclear inclusions in neuronal cells. Hum. Mol. Genet. 1999; 8:1169–1176. [DOI] [PubMed] [Google Scholar]
- 5. Doss-Pepe E.W., Stenroos E.S., Johnson W.G., Madura K.. Ataxin-3 interactions with Rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol. Cell. Biol. 2003; 23:6469–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laço M.N., Cortes L., Travis S.M., Paulson H.L., Rego A.C.. Valosin-containing protein (VCP/p97) is an activator of wild-type ataxin-3. PLoS One. 2012; 7:43563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rao M.V., Williams D.R., Cocklin S., Loll P.J.. Interaction between the AAA atpase p97 and its cofactor ataxin3 in health and disease: nucleotide-induced conformational changes regulate cofactor binding. J. Biol. Chem. 2017; 292:18392–18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhong X., Pittman R.N.. Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 2006; 15:2409–2420. [DOI] [PubMed] [Google Scholar]
- 9. Burnett B., Li F., Pittman R.N.. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003; 12:3195–3205. [DOI] [PubMed] [Google Scholar]
- 10. Schmitt I., Linden M., Khazneh H., Evert B.O., Breuer P., Klockgether T., Wuellner U.. Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem. Biophys. Res. Commun. 2007; 362:734–739. [DOI] [PubMed] [Google Scholar]
- 11. Chai Y., Berke S.S., Cohen R.E., Paulson H.L.. Poly-ubiquitin binding by the polyglutamine disease protein ataxin-3 links its normal function to protein surveillance pathways. J. Biol. Chem. 2004; 279:3605–3611. [DOI] [PubMed] [Google Scholar]
- 12. Evert B.O., Vogt I.R., Kindermann C., Ozimek L., De Vos R.A.I., Brunt E.R.P., Schmitt I., Klockgether T., Wüllner U.. Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains. J. Neurosci. 2001; 21:5389–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evert B.O., Vogt I.R., Vieira-Saecker A.M., Ozimek L., De Vos R.A.I., Brunt E.R.P., Klockgether T., Wüllner U.. Gene expression profiling in ataxin-3 expressing cell lines reveals distinct effects of normal and mutant ataxin-3. J. Neuropathol. Exp. Neurol. 2003; 62:1006–1018. [DOI] [PubMed] [Google Scholar]
- 14. Evert B.O., Araujo J., Vieira-Saecker A.M., De Vos R.A.I., Harendza S., Klockgether T., Wüllner U.. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006; 26:11474–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chou A.H., Yeh T.H., Ouyang P., Chen Y.L., Chen S.Y., Wang H.L.. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis. 2008; 31:89–101. [DOI] [PubMed] [Google Scholar]
- 16. Singh A.N., Oehler J., Torrecilla I., Kilgas S., Li S., Vaz B., Guérillon C., Fielden J., Hernandez-Carralero E., Cabrera E.et al.. The p97–Ataxin 3 complex regulates homeostasis of the DNA damage response E3 ubiquitin ligase RNF8. EMBO J. 2019; 38:e102361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pfeiffer A., Luijsterburg M.S., Acs K., Wiegant W.W., Helfricht A., Herzog L.K., Minoia M., Böttcher C., Salomons F.A., van Attikum H.et al.. Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J. 2017; 36:1066–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tu Y., Liu H., Zhu X., Shen H., Ma X., Wang F., Huang M., Gong J., Li X., Wang Y.et al.. Ataxin-3 promotes genome integrity by stabilizing Chk1. Nucleic Acids Res. 2017; 45:4532–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turner B.M. Histone acetylation and an epigenetic code. Bioessays. 2000; 22:836–845. [DOI] [PubMed] [Google Scholar]
- 20. Li G., Reinberg D.. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011; 21:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trojer P., Reinberg D.. Facultative heterochromatin: is there a distinctive molecular signature?. Mol. Cell. 2007; 28:1–13. [DOI] [PubMed] [Google Scholar]
- 22. Bizhanova A., Kaufman P.D.. Close to the edge: heterochromatin at the nucleolar and nuclear peripheries. Biochim. Biophys. Acta Gene Regul. Mech. 2021; 1864:194666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gonzalez-Sandoval A., Gasser S.M.. On tads and lads: spatial control over gene expression. Trends Genet. 2016; 32:485–495. [DOI] [PubMed] [Google Scholar]
- 24. Harr J.C., Gonzalez-Sandoval A., Gasser S.M.. Histones and histone modifications in perinuclear chromatin anchoring: from yeast to man. EMBO Rep. 2016; 17:139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raška I., Shaw P.J., Cmarko D.. New insights into nucleolar architecture and activity. Int. Rev. Cytol. 2006; 255:177–235. [DOI] [PubMed] [Google Scholar]
- 26. Lafontaine D.L.J., Riback J.A., Bascetin R., Brangwynne C.P.. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021; 22:165–182. [DOI] [PubMed] [Google Scholar]
- 27. Matsumoto A., Sakamoto C., Matsumori H., Katahira J., Yasuda Y., Yoshidome K., Tsujimoto M., Goldberg I.G., Matsuura N., Nakao M.et al.. Loss of the integral nuclear envelope protein SUN1 induces alteration of nucleoli. Nucleus. 2016; 7:68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caragine C.M., Haley S.C., Zidovska A.. Nucleolar dynamics and interactions with nucleoplasm in living cells. Elife. 2019; 8:e47533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chagin V.O., Stear J.H., Cardoso M.C.. Organization of DNA replication. Cold Spring Harb. Perspect. Biol. 2010; 2:a000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Babokhov M., Hibino K., Itoh Y., Maeshima K.. Local chromatin motion and transcription. J. Mol. Biol. 2020; 432:694–700. [DOI] [PubMed] [Google Scholar]
- 31. Lubelsky Y., Prinz J.A., DeNapoli L., Li Y., Belsky J.A., MacAlpine D.M.. DNA replication and transcription programs respond to the same chromatin cues. Genome Res. 2014; 24:1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Refolio E., Cavero S., Marcon E., Freire R., San-Segundo P.A.. The Ddc2/ATRIP checkpoint protein monitors meiotic recombination intermediates. J. Cell Sci. 2011; 124:2488–2500. [DOI] [PubMed] [Google Scholar]
- 33. Warmerdam D.O., Alonso-de Vega I., Wiegant W.W., van den Broek B., Rother M.B., Wolthuis R.M., Freire R., van Attikum H., Medema R.H., Smits V.A. PHF6 promotes non-homologous end joining and G2 checkpoint recovery. EMBO Rep. 2020; 21:e48460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pérez-Castro A.J., Freire R.. Rad9B responds to nucleolar stress through ATR and JNK signalling, and delays the G1-S transition. J. Cell Sci. 2012; 125:1152–1164. [DOI] [PubMed] [Google Scholar]
- 35. Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T.K., Choudhary C., Bekker-Jensen S.et al.. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015; 527:389–393. [DOI] [PubMed] [Google Scholar]
- 36. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B.et al.. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carnell M., Macmillan A., Whan R.. Fluorescence recovery after photobleaching (FRAP): acquisition, analysis, and applications. Methods Mol. Biol. 2015; 1232:255–271. [DOI] [PubMed] [Google Scholar]
- 38. Cabrera E., Hernández-Pérez S., Koundrioukoff S., Debatisse M., Kim D., Smolka M.B., Freire R., Gillespie D.A.. PERK inhibits DNA replication during the Unfolded Protein Response via Claspin and Chk1. Oncogene. 2017; 36:678–686. [DOI] [PubMed] [Google Scholar]
- 39. García-Muse T., Aguilera A.. R loops: from physiological to pathological roles. Cell. 2019; 179:604–618. [DOI] [PubMed] [Google Scholar]
- 40. Zhong Y., Nellimootti T., Peace J.M., Knott S.R.V., Villwock S.K., Yee J.M., Jancuska J.M., Rege S., Tecklenburg M., Sclafani R.A.et al.. The level of origin firing inversely affects the rate of replication fork progression. J. Cell Biol. 2013; 201:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rhind N., Gilbert D.M.. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013; 5:a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O’Keefe R.T., Henderson S.C., Spector D.L.. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific α- satellite DNA sequences. J. Cell Biol. 1992; 116:1095–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sansam C.G., Pietrzak K., Majchrzycka B., Kerlin M.A., Chen J., Rankin S., Sansam C.L.. A mechanism for epigenetic control of DNA replication. Genes Dev. 2018; 32:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillespie P.J., Blow J.J.. Clusters, factories and domains: the complex structure of S-phase comes into focus. Cell Cycle. 2010; 9:3218–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leonhardt H., Rahn H.P., Weinzierl P., Sporbert A., Cremer T., Zink D., Cardoso M.C.. Dynamics of DNA replication factories in living cells. J. Cell Biol. 2000; 149:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakamura H., Morita T., Sato C.. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell. Res. 1986; 165:291–297. [DOI] [PubMed] [Google Scholar]
- 47. Yamazaki S., Ishii A., Kanoh Y., Oda M., Nishito Y., Masai H.. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012; 31:3667–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Foti R., Gnan S., Cornacchia D., Dileep V., Bulut-Karslioglu A., Diehl S., Buness A., Klein F.A., Huber W., Johnstone E.et al.. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell. 2016; 61:260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Renard-Guillet C., Kanoh Y., Shirahige K., Masai H.. Temporal and spatial regulation of eukaryotic DNA replication: from regulated initiation to genome-scale timing program. Semin. Cell Dev. Biol. 2014; 30:110–120. [DOI] [PubMed] [Google Scholar]
- 50. Alavi S., Ghadiri H., Dabirmanesh B., Moriyama K., Khajeh K., Masai H.. G-quadruplex binding protein Rif1, a key regulator of replication timing. J. Biochem. 2021; 169:1–14. [DOI] [PubMed] [Google Scholar]
- 51. Kanoh Y., Matsumoto S., Fukatsu R., Kakusho N., Kono N., Renard-Guillet C., Masuda K., Iida K., Nagasawa K., Shirahige K.et al.. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015; 22:889–897. [DOI] [PubMed] [Google Scholar]
- 52. Richter K., Nessling M., Lichter P.. Experimental evidence for the influence of molecular crowding on nuclear architecture. J. Cell Sci. 2007; 120:1673–1680. [DOI] [PubMed] [Google Scholar]
- 53. Shen J., Varshney D., Simeone A., Zhang X., Adhikari S., Tannahill D., Balasubramanian S.. Promoter G-quadruplex folding precedes transcription and is controlled by chromatin. Genome Biol. 2021; 22:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Robinson J., Raguseo F., Nuccio S.P., Liano D., di Antonio M.. DNA G-quadruplex structures: more than simple roadblocks to transcription. Nucleic Acids Res. 2021; 49:8419–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mukherjee A.K., Sharma S., Chowdhury S.. Non-duplex G-quadruplex structures emerge as mediators of epigenetic modifications. Trends Genet. 2019; 35:129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Contreras A., Hale T.K., Stenoien D.L., Rosen J.M., Mancini M.A., Herrera R.E.. The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol. Cell. Biol. 2003; 23:8626–8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Misteli T., Gunjan A., Hock R., Bustin M., Brown D.T.. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000; 408:877–881. [DOI] [PubMed] [Google Scholar]
- 58. Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T.. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006; 10:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zaret K. Micrococcal nuclease analysis of chromatin structure. Curr. Protoc. Mol. Biol. 2005; Chapter 21:Unit 21.1. [DOI] [PubMed] [Google Scholar]
- 60. Burla R., Torre M.L., Merigliano C., Vernì F., Saggio I.. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus. 2018; 9:368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Camps J., Erdos M.R., Ried T.. The role of lamin Bl for the maintenance of nuclear structure and function. Nucleus. 2015; 6:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dechat T., Adam S.A., Goldman R.D.. Nuclear lamins and chromatin: when structure meets function. Adv. Enzyme. Regul. 2009; 49:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Berry J., Weber S.C., Vaidya N., Haataja M., Brangwynne C.P., Weitz D.A.. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5237–E5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dworak N., Makosa D., Chatterjee M., Jividen K., Yang C.S., Snow C., Simke W.C., Johnson I.G., Kelley J.B., Paschal B.M.. A nuclear lamina-chromatin-ran gtpase axis modulates nuclear import and DNA damage signaling. Aging Cell. 2019; 18:e12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Paul B., Montpetit B.. Altered RNA processing and export lead to retention of mRNAs near transcription sites and nuclear pore complexes or within the nucleolus. Mol. Biol. Cell. 2016; 27:2742–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matos C.A., de Almeida L.P., Nóbrega C.. Machado–Joseph disease/spinocerebellar ataxia type 3: lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2019; 148:8–28. [DOI] [PubMed] [Google Scholar]
- 67. Ichikawa Y., Goto J., Hattori M., Toyoda A., Ishii K., Jeong S.Y., Hashida H., Masuda N., Ogata K., Kasai F.et al.. The genomic structure and expression of MJD, the Machado-Joseph disease gene. J. Hum. Genet. 2001; 46:413–422. [DOI] [PubMed] [Google Scholar]
- 68. Harris G.M., Dodelzon K., Gong L., Gonzalez-Alegre P., Paulson H.L.. Splice isoforms of the polyglutamine disease protein ataxin-3 exhibit similar enzymatic yet different aggregation properties. PLoS One. 2010; 5:e13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dvořáčková M., Fajkus J.. Visualization of the nucleolus using ethynyl uridine. Front. Plant Sci. 2018; 9:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kornberg R.D., Lorch Y.. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999; 98:285–294. [DOI] [PubMed] [Google Scholar]
- 71. Wells C.E., Bhaskara S., Stengel K.R., Zhao Y., Sirbu B., Chagot B., Cortez D., Khabele D., Chazin W.J., Cooper A.et al.. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One. 2013; 8:e68915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bhaskara S., Knutson S.K., Jiang G., Chandrasekharan M.B., Wilson A.J., Zheng S., Yenamandra A., Locke K., Yuan J.L., Bonine-Summers A.R.et al.. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010; 18:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Summers A.R., Fischer M.A., Stengel K.R., Zhao Y., Kaiser J.F., Wells C.E., Hunt A., Bhaskara S., Luzwick J.W., Sampathi S.et al.. HDAC3 is essential for DNA replication in hematopoietic progenitor cells. J. Clin. Invest. 2013; 123:3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hänsel-Hertsch R., Beraldi D., Lensing S.v., Marsico G., Zyner K., Parry A., di Antonio M., Pike J., Kimura H., Narita M.et al.. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 75. Feng Q., Miao Y., Ge J., Yuan Y., Zuo Y., Qian L., Liu J., Cheng Q., Guo T., Zhang L.et al.. ATXN3 Positively regulates type I IFN antiviral response by deubiquitinating and stabilizing HDAC3. J. Immunol. 2018; 201:675–687. [DOI] [PubMed] [Google Scholar]
- 76. Rodrigues A.-J., Coppola G., Santos C., Costa M.C., Ailion M., Sequeiros J., Geschwind D.H., Mariel P.. Functional genomics and biochemical characterization of the C. Elegans orthologue of the Machado-Joseph disease protein ataxin-3. FASEB J. 2007; 21:1126–1136. [DOI] [PubMed] [Google Scholar]
- 77. Sen Gupta A., Sengupta K.. Lamin B2 modulates nucleolar morphology, dynamics, and function. Mol. Cell. Biol. 2017; 37:e00274-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mitrea D.M., Cika J.A., Stanley C.B., Nourse A., Onuchic P.L., Banerjee P.R., Phillips A.H., Park C.G., Deniz A.A., Kriwacki R.W.. Self-interaction of NPM1 modulates multiple mechanisms of liquid–liquid phase separation. Nat. Commun. 2018; 9:842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gindin Y., Valenzuela M.S., Aladjem M.I., Meltzer P.S., Bilke S.. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol. Syst. Biol. 2014; 10:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Boos D., Ferreira P.. Origin firing regulations to control genome replication timing. Genes (Basel). 2019; 10:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Németh A., Längst G.. Genome organization in and around the nucleolus. Trends Genet. 2011; 27:149–156. [DOI] [PubMed] [Google Scholar]
- 82. Vertii A., Ou J., Yu J., Yan A., Pagès H., Liu H., Zhu L.J., Kaufman P.D.. Two contrasting classes of nucleolus-associated domains in mouse fibroblast heterochromatin. Genome Res. 2019; 29:1235–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Herzog L.K., Kevei É., Marchante R., Böttcher C., Bindesbøll C., Lystad A.H., Pfeiffer A., Gierisch M.E., Salomons F.A., Simonsen A.et al.. The Machado–Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell. 2020; 19:e13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu H., Li X., Ning G., Zhu S., Ma X., Liu X., Liu C., Huang M., Schmitt I., Wü Llner U.et al.. The Machado–Joseph Disease Deubiquitinase ataxin-3 regulates the stability and apoptotic function of p53. PLoS Biol. 2016; 14:e2000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Reina C.P., Nabet B.Y., Young P.D., Pittman R.N.. Basal and stress-induced Hsp70 are modulated by ataxin-3. Cell Stress Chaperones. 2012; 17:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Winborn B.J., Travis S.M., Todi S., Scaglione K.M., Xu P., Williams A.J., Cohen R.E., Peng J., Paulson H.L.. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 2008; 283:26436–26443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Scaglione K.M., Zavodszky E., Todi S.V., Patury S., Xu P., Rodríguez-Lebrón E., Fischer S., Konen J., Djarmati A., Peng J.et al.. Ube2w and Ataxin-3 coordinately regulate the Ubiquitin Ligase CHIP. Mol. Cell. 2011; 43:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Haas E., Incebacak R.D., Hentrich T., Huridou C., Schmidt T., Casadei N., Maringer Y., Bahl C., Zimmermann F., Mills J.D.et al.. A novel SCA3 knock-in mouse model mimics the Human SCA3 disease phenotype including neuropathological, behavioral, and transcriptional abnormalities especially in oligodendrocytes. Mol. Neurobiol. 2022; 59:495–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Toonen L.J.A., Overzier M., Evers M.M., Leon L.G., Van Der Zeeuw S.A.J., Mei H., Kielbasa S.M., Goeman J.J., Hettne K.M., Magnusson O.T.et al.. Transcriptional profiling and biomarker identification reveal tissue specific effects of expanded ataxin-3 in a spinocerebellar ataxia type 3 mouse model. Mol. Neurodegener. 2018; 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Preisinger E., Jordan B.M., Kazantsev A., Housman D.. Evidence for a recruitment and sequestration mechanism in Huntington's disease. Philos. Trans. Roy. Soc. B: Biol. Sci. 1999; 354:1029–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yang H., Li J.J., Liu S., Zhao J., Jiang Y.J., Song A.X., Hu H.Y.. Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: implication in a loss-of-function pathology. Sci. Rep. 2014; 4:6410–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Suhr S.T., Senut M.C., Whitelegge J.P., Faull K.F., Cuizon D.B., Gage F.H.. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 2001; 153:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yang H., Yue H.W., He W.T., Hong J.Y., Jiang L.L., Hu H.Y.. PolyQ-expanded huntingtin and ataxin-3 sequester ubiquitin adaptors hHR23B and UBQLN2 into aggregates via conjugated ubiquitin. FASEB J. 2018; 32:2923–2933. [DOI] [PubMed] [Google Scholar]
- 94. Boutillier A.-L., Trinh E., Loeffler J.-P.. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J. Neurochem. 2003; 84:814–828. [DOI] [PubMed] [Google Scholar]
- 95. Dietz K.C., Casaccia P.. HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol. Res. 2010; 62:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rouaux C., Jokic N., Mbebi C., Boutillier S., Loeffler J.P., Boutillier A.L.. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003; 22:6537–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saha R.N., Pahan K.. HATs and hdacs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006; 13:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Salminen A., Tapiola T., Korhonen P., Suuronen T.. Neuronal apoptosis induced by histone deacetylase inhibitors. Mol. Brain Res. 1998; 61:203–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.