

Abstract
The gold-catalyzed intermolecular oxyarylation of alkene is reported. This work employed the oxidative addition of aryl iodide to Me-DalphosAu+ for the formation of Au(III)-Ar intermediate. The better binding ability of alkene over O nucleophiles ensured the success of intermolecular oxyarylation, giving desired products with broad substrate scope and high efficiency (>50 examples with up to 95% yield). “One-pot” converting of methoxy group into other nucleophiles allowed achieving alkene difunctionalization with C-N, C-S, and C-C bonds construction under mild conditions.
Keywords: gold redox catalysis; alkenes; oxidative addition; oxyarylation; hemilabile (P,N) ligand
Graphical Abstract
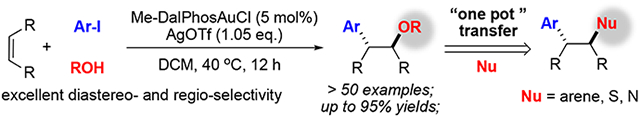
Homogeneous gold catalysis has witnessed tremendous development over the past two decades.[1] Comparing to Au(I), Au(III) complexes are stronger π-acids to promote reactions of less reactivity substrates, such as alkenes and internal alkynes.[2] Also, with a high oxidative potential, facile reductive elimination on Au(III) enables challenging bonds formation.[3] Therefore, the gold redox catalysis possesses an inherent capability to promote new transformations with high efficiency.[4] Among those reported examples, direct oxidation of Au(I)-Ar using strong oxidants, photocatalytic activation of diazonium, and ligand enabled gold(I) oxidative addition to aryl halide have received considerable attention as an easy access to active Au(III)-Ar intermediates, which is feasible for further arylation (Scheme 1A).[5] One route is to react with nucleophiles to give coupling products through reductive elimination. Both carbon (electron rich arene and terminal alkyne) and heteroatom nucleophiles have been reported as feasible coupling partner.[6] Additionally, the better π-acid properties of gold cations ensure stronger C-C unsaturated bond coordination.[7] As shown in Scheme 1A, nucleophilic addition to AuIII activated alkene B, followed by reductive elimination, could reach 1,2-difunctionalization of the alkene in high efficiency with excellent regio- (Markovnikov addition) and diastereoselectivity (trans-addition). [8] However, an intramolecular fashion is dominated to avoid unrenderable reaction pathways, including cross-coupling and protodeauration. Very few examples have been reported via an intermolecular “three-component” reaction. The only successful example is gold-catalyzed intermolecular alkene oxyarylation. The pioneering studies by Toste and Russell have demonstrated the feasible approach of using strong oxidants, such as Selectfluor or hypervalent iodine.[9] Recently, one elegant example by Glorius showed the feasibility of photoactivation of the aryl diazonium salt approach (Scheme 1B).[10] However, one common limitation of these transformations is that the electron-deficient aryl precursor is necessary to be compatible with more reactive electrophilic oxidants in the system. In the presence of EDG-modified aryl precursor, almost no product was observed. In some cases, a large excess amount of nucleophile is required (MeOH as solvent). Therefore, developing new intermolecular alkene difunctionalization methods bearing a broader substrate scope is highly desired. Herein, we report a first example of intermolecular oxyarylation of alkene through ArI directed oxidative addition of Au(I), bearing a broad substrate scope and good functional group tolerance (Scheme 1C).
Scheme 1.

Alkene difunctionalization via gold redox catalysis.
Over the past decade, our group has focused on developing new synthetic methods using homogenous gold catalysis.[11] Recently, we reported a series of transformations on base promoted diazonium activation for gold redox catalysis.[12] Compared with the photo-activation conditions, this base activation approach is much more robust with no need of strict degassing. With the interest in achieving the combination of gold π-acid and redox catalysis under intermolecular fashion, we have explored reactions between alkene, diazonium and nucleophiles under various reaction conditions. As shown in Figure 1A, our initial attempt to achieve the desired transformation was less successful after a systematic screening. As a result, we are seeking using a stable Au(III) intermediate to achieve this transformation.
Figure 1.

Intermolecular difunctionalization of alkene
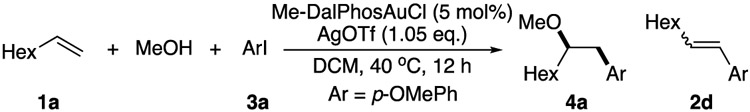
In 2017, Bourissou and co-workers first reported the application of hemilabile (P,N) ligand (Me-DalPhos) to facilitate ArI oxidative addition and stabilize the AuIII intermediate formed in-situ.[13] Using this strategy, cross coupling with electron rich arenes has been achieved.[14] Later, this method has also been applied in peptide modification through C-S bond construction.[15] In 2019, Patil group further extended this reaction's scope to direct C-N bond coupling.[16] Encouraged by these results, we wondered whether this new [AuIII-Ar] forming strategy could be applied to achieve the challenging intermolecular nucleophilic addition. It is important to note here that during our investigations, Patil and Bourissou simultaneously reported the combination of gold oxidation and alkene π-activation via intramolecular nucleophilic addition (C-C by Patil and C-O/C-N by Bourissou).[17] These two seminal works highlighted the great excitement for this gold redox catalysis strategy and implied the potential challenge for the intermolecular reaction. When we conducted the intermolecular reaction among TsNH2, aryl iodide, and simple alkene 1a under their reported optimal conditions, the desired amino arylation product 2a was only obtained in < 8% yields for both cases (Figure 1B).
Analyzing the reaction mixture revealed C-N coupling compound 2b as the major product under basic conditions. To avoid this competing reaction, we carried out the reactions under neutral or acidic conditions. We were pleased to find the aminoarylation product 2a in 38% yield with the addition of 5.0 eq. MeOH at 70 °C. However, after carefully monitoring the reaction process (2 h vs 6 h), we found that aryl iodide was completely consumed within two hours with the formation of 4a, extension of reaction time gave continuous formation of 2a and the disappearance of 4a. This result strongly suggested that the amino arylation product 2a was likely formed from methoxy arylation product 4a under the reaction conditions. Based on this result, we put our efforts on the condition screening for the oxyarylation. After exploring different reaction factors, an optimal condition was identified with Me-DalPhos-AuCl (5%) in DCM at 40 °C, giving the desired oxyarylation product 4a in 95% isolated yield. Some alternative conditions are summarized in Table 1.
Table 1.
| ||||
|---|---|---|---|---|
| Entry | Variation from “standard conditions” | conv. (3a) | 4a | 2d |
| 1 | none | 100% | 95% (93%) | <5% |
| 2 | AgSbF6 | 100% | 73% | 25% |
| 3 | 60 °C | 100% | 65% | 30% |
| 4 | rt | 20% | 15% | <5% |
| 5 | DCB as solvent | 100% | 89% | 10% |
| 6 | MeOH as solvent | 50% | 45% | <5% |
| 7 | Add 1 eq. K3PO4 | 100% | 87% | 10% |
| 8 | Add 1 eq. KOAc | <5% | 0% | 0% |
| 9 | Mor-DalphosAuCl 5 mol% | 20% | 15% | <5% |
| 10 | No [Au] | 0% | - | - |
| 11 | No [Ag] | 0% | - | - |
| 12 | No ArI | No conversion on 1a | ||
| 13 | dppm(AuBr)2 10 mol%, ArB(OH)2, Selectfluor, MeCN/MeOH, 50 °C | 20% | ||
| 14 | PPh3AuCl 10 mol%, ArN2BF4, [Ir] cat. MeOH, rt, hv | 0% | ||
Conditions: 1a (0.4 mmol), MeOH (0.6 mmol), 3a (0.2 mmol), Au cat. (0.01 mmol), AgOTf (0.21 mmol), DCM (2 mL), 40 °C, 12 h.
1H NMR yields using 1,3,5-tribromobenzene as an internal standard (isolated yields).
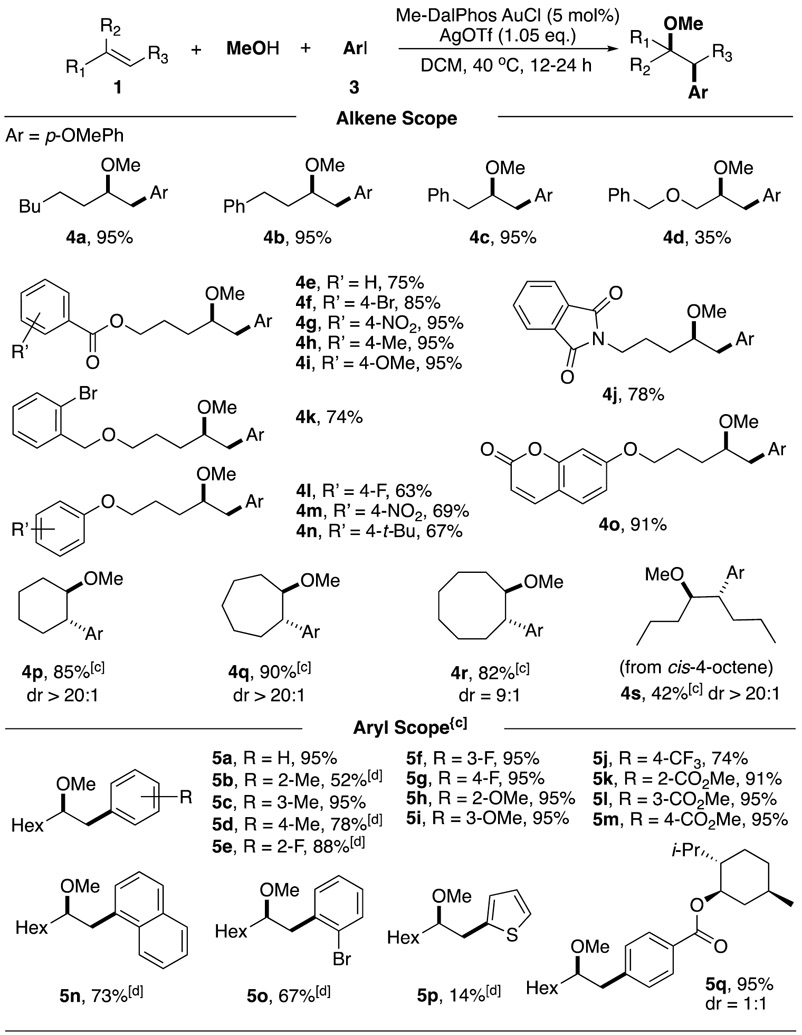
As shown in Table 1, with the addition of 2.0 eq. alkene 1a, 1.05 eq. AgOTf, and 3.0 eq. MeOH, the aryl iodide 1a was completely converted to the desired product 4a in 95% yield (entry 1). Conducting reaction using AgSbF6 as iodide scavenger led to a lower yield with more elimination product 2d, likely caused by the increased acidity (entry 2). Similarly, a higher temperate (60 °C) also resulted in the increasing of elimination product (entry 3). The conversion dropped to 20% at room temperature (entry 4). DCB was also the suitable solvent for this transformation (entry 5). Interestingly, when methanol was used as the solvent, slow reaction kinetic was observed, which might due to the competing coordination of OMe with alkene towards Au(III) intermediate (entry 6). Base is not necessary in the reaction (entries 7 and 8). Only 20% conversion was observed when using Mor-DalphosAuCl (entry 9). Finally, we performed the control experiments, showing that Au catalyst, Ag salt and aryl iodide were all necessary for this transformation (entries 10-12). With the optimal condition in hand, we then explored the substrate scopes, and the results are summarized in Table 2.
Table 2.
|
Conditions: 1 (0.4 mmol), MeOH (0.6 mmol), 3a (0.2 mmol), Au cat. (0.01 mmol), AgOTf (0.21 mmol), DCM (2 mL), 40 °C.
isolated yields.
use DCM:MeOH = 1:1 (0.5 M) as solvent instead.
60 °C and DCE instead.
We first examined the scope of alkenes. In general, monosubstituted terminal alkene substrates worked well in all cases, giving desired products in good to excellent yields (4a-4o). Substrates containing various functional groups, such as ester (4e-4i), benzyl ether (4k), phthalimide (4j), and coumarin (4o) were tolerated under this condition. Electron-rich phenyl ether groups (4l-4o) were also compatible, giving moderate yields. Notably, no aromatic electrophilic addition product was observed under this condition, indicating that OMe is a better nucleophile. For internal alkene substrates, slower reaction kinetics was observed under standard condition (<20% conversion in 24 h). To our delight, cyclic alkenes run smoothly with an increased concentration (0.5 M), affording the desired oxyarylation products in excellent yield and good diastereoselectivity (4p-4r). As for acyclic internal alkenes, only cis-1,2-disubstituted alkene was suitable for this transformation, though with relatively lower conversion (50%) and yield (42%) in 24 h (4s). Trans-4-octene remained unreactive under the same condition. These results suggested that reducing steric hindrance will be benefit for both alkene coordination and OMe addition.
The reaction scopes of aryl iodide were also investigated. All these substrates were treated at a higher concentration (0.5 M) due to slower reaction kinetics. The presence of both electron-withdrawing/-donating groups on the aromatic ring of aryl iodide did not show influence on the reaction outcome, and the products were formed in good to excellent yields. The ortho-substituted aryl iodides usually required a harsher condition (60 °C) with a decreased yield (5b, 5e, 5n, 5o), which might be associated with less feasible oxidative addition under the steric influence. Comparing with Pd, Au cation exhibited a good selectivity, which exclusively underwent oxidative addition into C-I bond (5o). This result highlighted an orthogonal reactivity of gold redox catalysis. Menthol ester modified aryl iodide was also explored, giving the corresponding oxy-arylation product in good yield but 1:1 dr selectivity (5q). Overall, this approach presented a broader scope than gold redox catalysis using aryl diazonium salt, highlighting the mild condition and synthetic potential for more complex system.
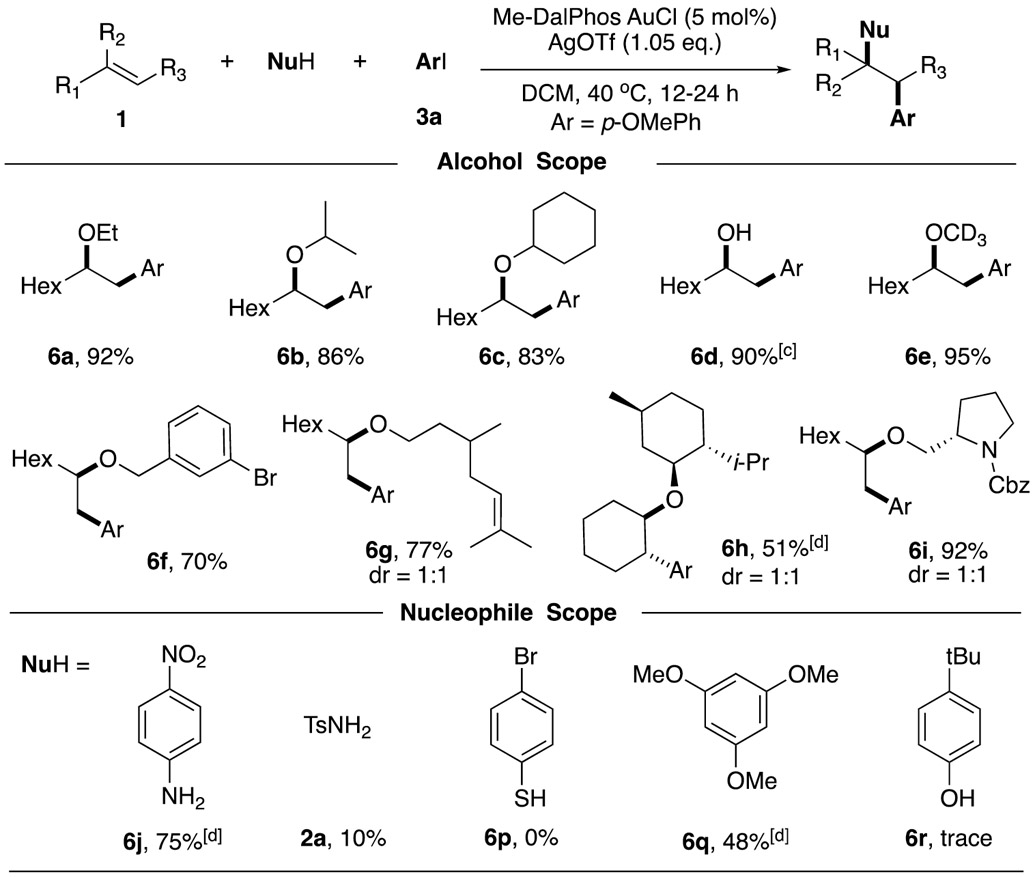
Next we explored the substrate scope of alcohols (Table 3). Both primary and secondary alcohols can serve as suitable nucleophile, generating corresponding products in excellent yields (6a-6c, 6e). Water can also work as a nucleophile here, achieving the alcohol product in 90% yield (6d). Benzyl alcohol containing Br on aromatic rings was also tolerated (6f). Some bio-compatible alcohols, including β-Citronellol (6g), Menthol (6h), and Prolinol (6l) proved successful for this transformation. However, the dr selectivity of corresponding products are all 1:1, indicating no steric control on nucleophilic addition step. Overall, various alcohol derivatives performed well with good functional group tolerability under this optimal condition, making it a practical late-stage functionalization strategy for complex molecule synthesis.
Table 3.
|
Conditions: 1 (0.4 mmol), NuH (0.6 mmol), 3a (0.2 mmol), Au cat. (0.01 mmol), AgOTf (0.21 mmol), DCM (2 mL), 40 °C.
isolated yields.
10 eq. of NuH instead.
0.5 M DCM instead.
Encouraged by the successful application of alcohol nucleophile, we then turned our attention to other nucleophiles. However, among all tested nucleophiles, only a few cases worked. 4-nitroaniline performed well at a higher concentration (0.5 M), generating the desired product in 75% yield (6l). 1,3,5-trimethoxybenzene can also react, giving a decreased yield due to a large amount of C-C bond cross coupling products. Other tested nucleophiles, such as tosyl amine, phenol, and thiophenol failed to yield our desired product (see SI for the detailed nucleophile screening). Inspired by our initial discovery of amino arylation (Figure 1B), we reasoned that OMe could be a good leaving/transformation group, therefore, other nucleophiles may still have their position on this alkene difunctionalization reaction. We then conducted the reaction in a “one-pot” fashion: oxyaryalation of alkene followed by various nucleophiles substitution.
As shown in Figure 2, to our delight, we were able to complete these transformations in good yields (2a, 6k-6s). The methoxy group can be transferred to various nucleophiles (N, C, S) with elimination product 2d as the only byproduct. Interestingly, control experiment revealed that the optimal yield can only be achieved under this “one-pot” fashion, highlighting the unique promotion effect in this reaction system. Notably, the thiophenol product (6m-6p) was obtained through this reaction protocol, showing the potential application on cysteine modification in the biological system with catalytic amount of gold.
Figure 2.

Synthetic utility of the products.
In conclusion, we reported an example of ligand enabled Au(I/III) catalyzed intermolecular alkene oxyarylation reaction with a broad substrate scope and good functional group tolerability. The one-pot nucleophile transfer strategy enabled diversed alkene 1, 2-difunctionalization, which provides a practical synthetic value and offers a valuable mechanistic insight.
Supplementary Material
Acknowledgements
We are grateful to the NSF (CHE-1665122) and NIH (1R01GM120240-01) for financial support. This work has been supported in part by University of South Florida Interdisciplinary NMR Facility and the Chemical Purification, Analysis, and Screening (CPAS) Core Facility.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Hashmi ASK, Hutchings GJ, Angew. Chem. Int. Ed 2006, 45, 7896–7936; [DOI] [PubMed] [Google Scholar]; b) Gorin DJ, Toste FD, Nature 2007, 446, 395–403; [DOI] [PubMed] [Google Scholar]; c) Gorin DJ, Sherry BD, Toste FD, Chem. Rev 2008, 108, 3351–3378; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sengupta S, Shi XD, Chemcatchem 2010, 2, 609–619; [Google Scholar]; e) Hashmi ASK, Acc. Chem. Res 2014, 47, 864–876; [DOI] [PubMed] [Google Scholar]; f) Wang YM, Lackner AD, Toste FD, Acc. Chem. Res 2014, 47, 889–901; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zi WW, Toste FD, Chem. Soc. Rev 2016, 45, 4567–4589.26890605 [Google Scholar]
- [2].Joost M, Amgoune A, Bourissou D, Angew. Chem. Int. Ed 2015, 54, 15022–15045. [DOI] [PubMed] [Google Scholar]
- [3].(a) For review, see: Zhang Y, Luo S, Zhu CJ, Chinese J. Org. Chem 2012, 32, 2073–2080; [Google Scholar]; b) Miro J, del Pozo C, Chem. Rev 2016, 116, 11924–11966. [DOI] [PubMed] [Google Scholar]; a) For selected examples, see: Winston MS, Wolf WJ, Toste FD, J. Am. Chem. Soc 2014, 136, 7777–7782; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wolf WJ, Winston MS, Toste FD, Nature Chem. 2014, 6, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Huang B, Hu M, Toste FD, Trends in Chemistry 2020. 10.1016/j.trechm.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye X, Zhao P, Zhang S, Zhang Y, Wang Q, Shan C, Wojtas L, Guo H, Chen H, Shi X, Angew. Chem. Int. Ed 2019, 58, 17226–17230. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang J, Zhang S, Xu C, Wojtas L, Akhmedov NG, Chen H, Shi X, Angew. Chem. Int. Ed 2018, 57, 6915–6920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Kramer S, Synthesis 2020, 52, 2017–2030. [Google Scholar]; b) Nijamudheen A, Datta A, Chem. Eur. J 2020, 26, 1442–1487. [DOI] [PubMed] [Google Scholar]; c) Akram MO, Banerjee S, Saswade SS, Bedi V, Patil NT, Chem. Commun 2018, 54, 11069–11083. [DOI] [PubMed] [Google Scholar]; d) Hopkinson MN, Tlahuext-Aca A, Glorius F, Acc. Chem. Res 2016, 49, 2261–2272. [DOI] [PubMed] [Google Scholar]
- [6].(a) For selected examples, see: Brand JP, Charpentier J, Waser J, Angew. Chem. Int. Ed 2009, 48, 9346–9349; [DOI] [PubMed] [Google Scholar]; b) Zhang GZ, Peng Y, Cui L, Zhang LM, Angew. Chem. Int. Ed 2009, 48, 3112–3115; [DOI] [PubMed] [Google Scholar]; c) de Haro T, Nevado C, J. Am. Chem. Soc 2010, 132, 1512–1513; [DOI] [PubMed] [Google Scholar]; d) Ball LT, Lloyd-Jones GC, Russell CA, J. Am. Chem. Soc 2014, 136, 254–264. [DOI] [PubMed] [Google Scholar]; e) Levin MD, Toste FD, Angew. Chem. Int. Ed 2014, 53, 6211–6215 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Hofer M, Genoux A, Kumar R, Nevado C, Angew. Chem. Int. Ed 2017, 56, 1021–1025 [DOI] [PubMed] [Google Scholar]; g) Xie J, Sekine K, Witzel S, Kramer P, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 2018, 57, 16648–16653; [DOI] [PubMed] [Google Scholar]; h) Liu K, Li N, Ning YY, Zhu CJ, Xie J, Chem 2019, 5, 2718–2730. [Google Scholar]
- [7].Zheng Z, Wang Z, Wang Y, Zhang L, Chem. Soc. Rev 2016, 45, 4448–4458. [DOI] [PubMed] [Google Scholar]
- [8].a) Brenzovich WE, Benitez D, Lackner AD, Shunatona HP, Tkatchouk E, Goddard WA, Toste FD, Angew. Chem. Int. Ed 2010, 49, 5519–5522; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) de Haro T, Nevado C, Angew. Chem. Int. Ed 2011, 50, 906–910; [DOI] [PubMed] [Google Scholar]; c) Shu XZ, Zhang M, He Y, Frei H, Toste FD, J. Am. Chem. Soc 2014, 136, 5844–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Melhado AD, Brenzovich WE, Lackner AD, Toste FD, J. Am. Chem. Soc 2010, 132, 8885–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Harper MJ, Emmett EJ, Bower JF, Russell CA, J. Am. Chem. Soc 2017, 139, 12386–12389. [DOI] [PubMed] [Google Scholar]
- [10].Hopkinson MN, Sahoo B, Glorius F, Adv. Synth. Catal 2014, 356, 2794–2800. [Google Scholar]
- [11].a) Duan HF, Sengupta S, Petersen JL, Akhmedov NG, Shi XD, J. Am. Chem. Soc 2009, 131, 12100–12102; [DOI] [PubMed] [Google Scholar]; b) Peng HH, Xi YM, Ronaghi N, Dong BL, Akhmedov NG, Shi XD, J. Am. Chem. Soc 2014, 136, 13174–13177; [DOI] [PubMed] [Google Scholar]; c) Xi YM, Dong BL, McClain EJ, Wang QY, Gregg TL, Akhmedov NG, Petersen JL, Shi XD, Angew. Chem. Int. Ed 2014, 53, 4657–4661; [DOI] [PubMed] [Google Scholar]; d) Cai R, Ye XH, Sun Q, He QQ, He Y, Ma SQ, Shi XD, ACS. Catal 2017, 7, 1087–1092; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ye XH, Peng HH, Wei CY, Yuan T, Wojtas L, Shi XD, Chem 2018, 4, 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Cai R, Lu M, Aguilera EY, Xi YM, Akhmedov NG, Petersen JL, Chen H, Shi XD, Angew. Chem. Int. Ed 2015, 54, 8772–8776; [DOI] [PubMed] [Google Scholar]; b) Peng HH, Cai R, Xu C, Chen H, Shi XD, Chem. Sci 2016, 7, 6190–6196; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jimoh AA, Hosseyni S, Ye XH, Wojtas L, Hu Y, Shi XD, Chem. Commun 2019, 55, 8150–8153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rodriguez J, Zeineddine A, Carrizo EDS, Miqueu K, Saffon-Merceron N, Amgoune A, Bourissou D, Chem. Sci 2019, 10, 7183–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zeineddine A, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D, Nature Commun. 2017, 8, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Messina MS, Stauber JM, Waddington MA, Rheingold AL, Maynard HD, Spokoyny AM, J. Am. Chem. Soc 2018, 140, 7065–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Akram MO, Das A, Chakrabarty I, Patil NT, Org. Lett 2019, 21, 8101–8105. [DOI] [PubMed] [Google Scholar]; b) Rodriguez J, Adet N, Saffon-Merceron N, Bourissou D, Chem. Commun 2020, 56, 94–97. [DOI] [PubMed] [Google Scholar]
- [17].a) Chintawar CC, Yadav AK, Patil NT, Angew. Chem. Int. Ed 2020, 59, 11808. [DOI] [PubMed] [Google Scholar]; b) Rigoulet M, Thillaye du Boullay O, Amgoune A, Bourissou D, Angew. Chem. Int. Ed, ASAP, 10.1002/anie.202006074 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.