

Abstract

Froth flotation is the most versatile process in mineral beneficiation, extensively used to concentrate a wide range of minerals. This process comprises mixtures of more or less liberated minerals, water, air, and various chemical reagents, involving a series of intermingled multiphase physical and chemical phenomena in the aqueous environment. Today’s main challenge facing the froth flotation process is to gain atomic-level insights into the properties of its inherent phenomena governing the process performance. While it is often challenging to determine these phenomena via trial-and-error experimentations, molecular modeling approaches not only elicit a deeper understanding of froth flotation but can also assist experimental studies in saving time and budget. Thanks to the rapid development of computer science and advances in high-performance computing (HPC) infrastructures, theoretical/computational chemistry has now matured enough to successfully and gainfully apply to tackle the challenges of complex systems. In mineral processing, however, advanced applications of computational chemistry are increasingly gaining ground and demonstrating merit in addressing these challenges. Accordingly, this contribution aims to encourage mineral scientists, especially those interested in rational reagent design, to become familiarized with the necessary concepts of molecular modeling and to apply similar strategies when studying and tailoring properties at the molecular level. This review also strives to deliver the state-of-the-art integration and application of molecular modeling in froth flotation studies to assist either active researchers in this field to disclose new directions for future research or newcomers to the field to initiate innovative works.
Keywords: Froth flotation, Molecular modeling, Density functional theory, Molecular dynamics, Ab initio, Rational design, Minerals, Reagents
1. Introduction
Froth flotation is a widely used mineral processing method that can be custom designed to recover valuable minerals from a wide range of ores. With a century-long technological tradition in the field, it is undoubtedly the “via regia” of mineral separation.1 In this process, desirable minerals are separated from undesirable ones by direct or reverse flotation based on inherent differences in wettability, although these are often artificially induced by the addition of chemical reagents to force the emergence of hydrophobic contrasts. However, this contrast is elicited, it is the introduction of tiny air bubbles into the pulp that provides the hydrophobic mineral carrier and enables mineral separation. The interfacial chemistry that governs flotation involves numerous complex phenomena, including adsorption and desorption of reagents, conformation and rearrangement of reagents on the mineral surface, translocation, dissolution, surface modification and reconstruction, precipitation, and so on.2,3 These interactions between water, air, and minerals make the process a multifaceted and intrinsically complex phenomenon.2,3
As in any engineering field, the evolutionary dynamics of knowledge that helped to establish flotation as a distinct discipline has essentially and traditionally relied on macroscopic experimental observations, empirical recipes, and more or less reliable analytical protocols, long before the modern “atomistic” view as we manipulate it today could open up new breaches and perhaps initiate a paradigm shift.1,2 As insightful as these traditional approaches have been, they cannot keep up with the increasing complexity of today’s exploitable resources. This is primarily due to the ever-decreasing amounts and quality of target minerals in the matrices of newly discovered and mined deposits. Direct measurement of the microphenomena at the interfaces of these mineral matrices, which are confined to the immediate atomic neighborhood of the target elements, is difficult, if not impossible, without first having an atomic resolution to map flotation phenomena at the Angstrom or nanometer scale.4 Ores containing trace amounts of rare earth elements, gold in arsenopyrite solid solutions, niobium in pyrochlore or columbite are some prominent examples, to name a few.5−8
A notable advance in mineral froth flotation studies to date has been the popularity of tools such as computational chemistry and molecular modeling to represent the minute geometric details at the atomic scale that can account for the specificity of thermodynamic, (electro)kinetic, and colloidal phenomena of mineral surfaces.4,9,10 Since its first application to mineral froth flotation in 1993,11 this tool has contributed to a better understanding of reagent adsorption on mineral surfaces, reagent conformation and desorption, mineral crystal and surface chemistry, mineral surface modification and dissolution, and interfacial phenomena at mineral/water/gas interfaces.4,9,12−18 However, advances in the near future are also a priority, as easily processed ores are rapidly depleted, leaving only very complex ores that are much more difficult to beneficiate.19 In addition to the depletion of high-grade deposits, the negative environmental impact of reagents used in ore beneficiation has become a concern that can no longer be ignored. Therefore, developing highly selective reagents is essential for the sustainable development of mineral beneficiation processes. Just as the advent of computational fluid dynamics tools has benefited chemical reactors and separations to simulate as many different scales of flow as possible with sufficient spatial and temporal resolution,20 molecular modeling is emerging as an indispensable tool for atomic resolution simulations needed to control and understand fluid-mineral interface phenomena and, ultimately, to tailor environmentally friendly reagents.4
Despite a large number of papers and several recent reviews on flotation, there are many conceptual and practical gaps in the coverage of the application of molecular modeling approaches in this field. From available literature it is clear that mineral processing community has not yet benefited from the advantages of molecular modeling techniques to the extent that other fields, such as pharmaceutical and polymer sciences, have. The advent of commercial packages has reduced attention to the fundamental aspects of molecular modeling and limited the level of postprocessing to those available in the graphical user interface. Worryingly, even in some applications, the basic theories and statistical concepts have been overlooked, and the interpretation of results has been fraught with many uncertainties. Scrutiny in the recent review papers further revealed a lack of emphasis on the postprocessing scenarios applicable to flotation processes, particularly within the molecular dynamics (MD) framework.14−17 For example, the recent review by Liu et al.14 focused on the theory behind various molecular modeling techniques and a recension of published work on the design of flotation collectors. Zhao et al.15 assessed the theories and characterization approaches of first-principle calculations, chiefly density functional theory (DFT), in adsorption studies of organic molecules on mineral surfaces at liquid–solid or gas–solid interfaces. Foucaud et al.16 reviewed the recently published articles using molecular modeling techniques in an illustrative-themed, focusing on liquid–solid adsorption phenomena. The most recent review article that comprehensively addressed the role of molecular modeling was limited exclusively to low-rank coal flotation, targeting coal-water, coal-reactive, coal-bubble, and coal-clay interactions, but without details for postanalysis.17 In this presentation, it was deemed appropriate to devote a review of the advanced approaches and critical postprocessing concepts and methods that are widely applicable in the flotation field. Further, we enumerate the molecular modeling approaches that have been applied in flotation studies to date, with emphasis on their potential from a practical point of view. Knowledge of the directions, prospects, and challenges for future applications of molecular modeling in mineral processing needs further attention to maximize the benefits from the development of computational approaches.
To this end, this contribution aims to provide froth flotation researchers with the insight needed to use molecular modeling to address the challenges of this discipline and to identify new research directions. To this end, this review is organized as a diptych. In the first part, we provide the descriptive framework of the principles underlying the molecular modeling approaches used in flotation by focusing on the types of data mining strategies related to flotation. In the second part, several representative examples of molecular modeling applications in froth flotation studies are discussed under the five headings reflecting current research priorities. Our presentation strategy in this section is more illustrative, but critical comments and suggestions are also included whenever necessary for clarification. Finally, we conclude this review with a brief summary and outlook on open problems in the field of flotation related to minerals computation.
2. Molecular Modeling Approaches Emphasizing Flotation Studies
Molecular modeling techniques and their rise in popularity have taken a quantum leap over the past two decades. Nowadays, several techniques and software (both commercial or open-source) are available to perform computational chemistry calculations,15 and advances in high-performance computing (HPC) infrastructure have expanded their applications. Molecular modeling approaches, in general, are proving to be powerful tools to shed light on different ranges of structural and dynamic properties, starting from the fundamental physics associated with a given microstructure.
Froth flotation involves three phases, namely, liquid, gas, and solid, as well as interfacial phenomena governing the solid–liquid and gas–liquid interfaces.1 In the flotation process, it is mainly the interfacial interactions between the dissolved reagents and minerals on the one hand, and the bubbles, on the other hand, that are at the center of the modeling at the molecular or atomic scale. The simulation results provide practical guidance for identifying the most effective mechanisms and facilitate the development of chemicals with improved performance rationally and systematically (applications discussed in depth in section 3).
The most common classification of molecular modeling methods labels them according to the time and length scales they are designed to represent. Figure 1 schematically aggregates the categories of simulation methods at the corresponding length and time scales.
Figure 1.

Schematic representation of the various molecular modeling techniques and the corresponding length and time scales (QM = Quantum Mechanics, HF = Hartree–Fock, DFT = Density Functional Theory, AIMD = Ab initio Molecular Dynamics, ReaxFF = Reactive Force field, MM = Molecular Mechanics, MD = Molecular Dynamics, MC = Monte Carlo).
Not surprisingly, the more accurate the method, the higher the computational cost, the smaller the accessible size scale, and the shorter the time scale. Quantum mechanical (QM) methods, particularly density functional theory (DFT), which includes electronic structure calculations, can routinely handle a few hundred atoms in the simulation. In contrast, methods that do not explicitly include electrons, such as force field methods like classical molecular dynamics (MD), can be applied to thousands of atoms and track their evolution over a few hundreds of nanoseconds.
The molecular modeling methods can be further classified according to the type of properties they can handle. QM methods provide nearly exact descriptions of system energy, adsorption energy, chemical bond cleavage/formation, band structure, bond population, etc., taking into account the electronic structure of constituent atoms.21 In contrast, in force field approaches, intra- and intermolecular interactions are expressed by analytical functions, taking into account the energy contributions of valence and nonbond interactions.4,21 Broadly speaking, molecular mechanics (MM) is well suited to the study of dynamic processes such as thermal properties, adhesion, diffusion, and wettability. Compared to QM methods, the effect of charge transfer, polarization, and bond breakage/formation cannot be modeled in classical MM techniques.
Hybrid atomistic methods were introduced to include the advantages of both QM and MM theories in a simulation. Ab initio molecular dynamics (AIMD)22−26 and the hybrid QM/MM algorithm27−29 are the two well-known methods in this category. The former method falls in the QM region, while the latter includes the overlap region between QM and MM simulations (Figure 1). Combining DFT and MD in AIMD, chemical molecular processes and reactions, as well as the effect of finite temperature and pressure, can be included in the simulations.25,26 For the QM/MM method, the general idea is to encompass the merits of both QM and MM so that the electronically important region (i.e., reactive region) of the system is treated with the more accurate QM methods, while for the surrounding region, lower-level simulation methods (e.g., classical MD) are allowed.27 These assumptions would considerably speed up the simulation.27
The reactive molecular dynamics method (also called ReaxFF) is another route to bridge the gap between QM and MM approaches.30−35 In the ReaxFF method, dynamic bonds and polarization effects are incorporated into the force field formulations through preset ab initio calculations and stored in the force field file.30,35 As a result, the bond formation/breakage can be modeled continuously in ReaxFF simulations. The primary challenge here is the accessibility of an efficient and scalable reactive force field for the system of interest. Otherwise, the ReaxFF method is limited to significantly larger lengths and time scales than the QM techniques.
This section briefly summarizes molecular modeling related to the flotation process, including DFT, MD (classical and reactive), and some hybrid methods. For hybrid simulation methods, AIMD and QM/MD techniques would be targeted. Without dwelling on the founding theories described competently elsewhere,36 the primary focus here is to discuss and compare the general features, application areas, and advantages/disadvantages of each technique.
2.1. Quantum Mechanical Calculations: Density Functional Theory
In quantum mechanical calculations, the nuclei and the (multi)electron interactions, and the system’s energy are described by the well-known Schrödinger equation.37,38 The complete solution of Schrödinger’s equation, if it is computable at all, is equivalent to a formidable complex many-body problem.39,40
Two independent steps are often used to ease the computational burden instead of solving the Schrödinger equation. The first, known as the Born–Oppenheimer approximation,41 assumes that the nuclei are immobile, given that the mass of a proton is 3 orders of magnitude greater than that of an electron. The nuclei merely act as a source of electrostatic potential for the electrons in the electronic structure calculations. The second, which in practice is a combination of several approximations, is often classified as a first principle or ab initio method.21 The ab initio approaches can be divided into two main branches, including wave function-based methods, which all start from the Hartree–Fock method,42,43 and density-based methods, which are mainly based on density functional theory.44 Density-based methods are the most commonly used electronic structure methods.
2.1.1. Simulation Setups in DFT
In all molecular modeling approaches, constructing the initial simulation box is a crucial step that must be handled with careful consideration. The simulation cell must be large enough to (a) freely allow all necessary changes in the structure (e.g., surface reconstruction) and (b) eliminate any undesirable interactions between the periodic images of any asymmetrical feature in the structure (e.g., defects). In most cases, the simulation cell consists of a slab model representing a surface of the adsorbate. In slab models, additional precautions must be taken in the nonperiodic direction by considering sufficient atomic layers in the surface and vacuum size. Verification of the system size (including slab surface area, number of atomic layers, and vacuum size) is an essential step in the preprocessing, usually performed by monitoring the convergence of the surface energy and/or the energy change caused by the nonperiodic feature (e.g., defect formation energy).
Previous investigations revealed that the initial arrangements of a complex system also directly affect the adsorption studies in DFT problems.45−51 In order to find the most stable adsorption mode in a complex, combinations of all potential initial relative positions of adsorbents and adsorbates must be constructed and evaluated. For small linear molecules, a manual initialization is possible.47 However, for complexes containing larger adsorbate molecules and/or surfaces with local disorder (such as a deficiency), it is highly plausible to miss a key initial mode, leading to inaccurate prediction of the globally optimized adsorption configuration. Alizadeh Sahraei and Larachi49−51 suggested that the grand canonical Monte Carlo (GCMC) method could be a robust framework for searching all the potential initial configurations in DFT calculations. Their procedure was tested on the adsorption of a molecule, a monolayer and a multilayer of water, and a solvated or nonsolvated amino acid on sphalerite surfaces with and without defects. It should be noted that the reliability of GCMC simulations depends on the availability of accurate force fields for the system; otherwise, the GCMC pretreatment would converge to unrealistic configurations.
The key to the DFT method is to combine the nuclei and the effective potentials into a single external potential for each electron, known as the exchange-correlation functional.40,52 Among the simulation parameters, the choice of the exchange-correlation functional is key for the accuracy of DFT calculations. The main difficulty with DFT is that there is no exact mathematical expression for the exchange-correlation functionals, so some approximations are needed in the first place. The local density approximation (LDA) is the simplest approximation for the exchange-correlation functional. It assumes that the energy is only related to the value of the electronic density at each point in space.53 Previous investigations have shown that LDA does not produce reliable results for structures with chemical bond breakage/formation (as this is a crucial topic in flotation modeling).54,55 The electron density gradient is included in the exchange-correlation functional by introducing a new family of functionals known as generalized gradient approximation (GGA).56 The most popular functionals in the GGA class are Perdew–Burke–Enzerhof (PBE), PBEsol, PW91, and BLYP. Meta-GGA and hybrid functionals are the next-generation functionals56−60 (i.e., further discussion and the amendments the exchange-correlation functionals continually undergo can be found in the literature.)40,44 In general, the richer a function is in terms of physical description, the more expensive the simulation is in terms of computation, although the results are more accurate. Perdew and Schmidt have suggested the idea of “Jacob’s Ladder” to select functionals in DFT problems.61 The ladder starts with LDA and then gradually rises in accuracy to GGA, meta-GGA, hybrid, and ultimate exact functionals.
In standard DFT, dispersion or van der Waals interactions are not accurately taken into account by the exchange-correlation functional. One efficient solution is to add an analytical correction term to the exchange-correlation functional.62−65 Although this comes at the expense of a more laborious calculation, the results indicate that the corrections would include the physics necessary for capturing most phenomena reliably.66−69
The choice of the basis set and k-points should also be considered carefully.21,40 Basis sets are the representation of wave functions, and their quality is determined by the number and type of basis functions in their formulations. The k-points are defined for periodic systems and are the sampling points in the first Brillouin zone of the model in reciprocal space. The widely used method for k-point sampling is the Monkhorst–Pack mesh. In general, it is essential to pre-evaluate the convergence of the energy, electronic structure, and target properties of the system based on different functionals, basis sets, and k-point sizes. Parameter verification tests can be very time-consuming; however, the prejudice of rerunning all simulations caused by choosing inappropriate input parameters will be much greater. Finally, it is important to emphasize that there is no prescription for parameter selection in DFT, and caution against blind reliance on the recommendations of others or the default values offered by simulation codes is to be exercised. Overall, a lack of understanding or the likening of modern simulation codes to a black box can be tendentious, but if the results are apparently convergent.
2.1.2. DFT Analysis
Energy Calculations in DFT
Depending on the system, many different relative energies can be calculated and applied to interpret the results. The apparent adsorption energy or affinity is frequently used to comment on the most stable adsorption configuration in a complex after DFT geometry optimization (applications discussed in section 3.1). On the basis of the definition:
| 1 |
where Etotal is the total energy of the system containing both the adsorbent and the adsorbates, Eadsorbent represents the energy of the optimized bare adsorbent surface, and Eadsorbate is the optimized energy of an isolated adsorbate molecule(s). Note that the energy calculations for all the systems and subsystems must be performed with the same convergence parameters. The relative energy obtained by eq 1 is called “interaction energy” when Eadsorbent and Eadsorbate are the single point energy of the naked adsorbent surface and the adsorbate, retaining the structure as in the adsorbed situation.49,50 The more negative values of Eads suggest stronger adsorption of the adsorbate on the adsorbent surface.
Vacancies and dislocations are common defects within bulk materials, especially minerals70−72 (applications discussed in section 3.5.2). The defect formation energy per defect can be calculated as21
| 2 |
where Edefective is the energy of the system with the defect, Evacant_atom represents the energy of the isolated atom removed to create the defect, and Edefect-free is the energy of the defect-free system. It is suggested to calculate the defect formation energy in two stages: (1) without further relaxation and (2) after relaxation of the defective surface. By comparing these two energies, the contribution of the relaxation to defect stabilization can be characterized.
The surface formation energy, or in short, the surface energy, is typically calculated with the intention of classifying the cleavage plane of a crystal structure. The surface energy in a vacuum is defined as73,74
| 3 |
where Eslab is the energy of the slab with two surfaces, A is the surface area, Ebulk is the energy of the bulk unit cell structure, and N represents the number of unit cells in the slab. To build a surface, the starting bulk structure should be thick enough to guarantee the convergence of the surface energy.
In the case of a hydrated surface, the surface energy of the surface is calculated as follows:75,76
| 4 |
where EslabNH2O is the energy of the optimized slab in the presence of explicit water molecules on the surface, NH2O is the number of water molecules on the surface, and EH2O is the energy of a single water molecule using the same calculation parameters. Gcorr = −Hvap – TSliq is the experimental free energy correction for the reference state of bulk water, where Hvap is the water evaporation enthalpy and Sliq° is the absolute entropy of liquid water. In most cases, the entropy of strongly adsorbed water can be ignored.75,76
The activation energy determines the minimum energy to be supplied to the reactants for a chemical reaction to occur, and it cannot be evaluated on the sole basis of the total energies of the products and reactants. A transition state calculation is required to find the transition state configuration. The activation energy is then calculated as the energy difference between the reactants and the obtained transition state.40 The activation Gibbs free energy and, consequently, the reaction rate constant can also be obtained through transition state simulations.
Population Analysis in DFT
Population analysis, also known as charge distribution, is a postprocessing approach often included in DFT studies. However, one must be aware of differences in the formulations used to predict atomic partial charges. Ambiguity in the methods stems from how regions of space to atoms are assigned and how areas of overlap between atoms are treated.21 The two best-known methods are the Hirshfeld77 and Becke78 schemes, which operate based on devising the overlapping area between atoms by assigning them different weights. The weights in the Hirshfeld scheme are assigned according to the contribution of each atom in the charge density, while for the Becke scheme, the atomic region shares are defined by a generalization of Voronoi polyhedra.79
The atoms-in-molecules Bader approach is another widely used method,80 which determines the position of transitions between atoms by analyzing the charge density topology. This method is more computationally expensive than the first two methods since it requires a full electron density analysis to calculate the atomic partial charges. The Mulliken method,81 which is applicable with localized basis sets, is also commonly used for charge calculations. The Mulliken method, in particular, is highly dependent on the basis set; thus, the output can be overly unreliable. Overall, extra caution would be warranted when implementing the Mulliken method for population analysis.79
Bond population analysis is further implemented to quantify the possibility of bond formation/breakage in a system, considering the overlap of charge density between atom pairs. The higher the bond population value, the higher the bond covalency levels. Note that most visualization codes can predict bonds in a system using a heuristic algorithm based on the distance between atoms and their van der Waals radii. One has to be aware however that the method is a purely distance-based prediction, so care must be taken to avoid misinterpretations.
Atom Reactivity in DFT
The most important orbitals for the reactivity of molecules are the frontier orbitals, called the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).21,40 The energy difference between the HOMO and LUMO is termed the band gap or HOMO–LUMO gap. In general, molecules with a larger band gap are less reactive. Note that functionals suffering from a self-interaction error do not provide an accurate result for the calculation of the HOMO–LUMO gap.21 The Fukui functions further characterize the reactivity of atoms in DFT calculations.82−86 In this description, the distribution of electron densities in the HOMO and LUMO is recognized as the factor defining the reactivity of an atom. The explicit expression of Fukui indices, using finite difference approximation, is given by
| 5 |
| 6 |
where f+ and f– functions measure the reactivity per nucleophilic and electrophilic attacks, respectively, and ρK(r) (K = N – 1, N, N + 1) denotes the electron density at a point r of a K-electron system. The value of f+ reflects the ability of an atom to accommodate an extra electron while f– signifies the ability of an atom to cope with the loss of an electron.86 The larger the values of f+ and f–, the more reactive the region during electrophilic and nucleophilic attacks, respectively.
The density of states (DOS) measures the proportion of states to be occupied in an energy window. DOS results can be applied to characterize the valence bandwidth and energy gap in insulators and to qualitatively interpret the reactivity by comparing the location and intensity of orbital peaks.21 For systems with local discontinuities, such as surfaces and defects, the DOS of the whole system is often not informative.21 In these cases, the projected density of states (PDOS) of a particular atom or group of atoms would be more instructive, although one should be aware of the illusory character of projecting the DOS onto atoms due to the delocalized nature of the wave functions in DFT.
Vibrational Spectroscopy in DFT
Vibrational spectroscopies, such as infrared (IR), Raman, and NMR, can also be extracted from DFT calculations. In DFT, the calculated frequencies are sometimes rescaled for convenience of comparisons with experimental spectra, as provided by Alecu et al.87 Frequency analysis is also included in the transition state search, in which a configuration with only one imaginary frequency manifests the genuine transition state mode. In geometry optimization, the absence of imaginary frequencies confirms the obtained structure as a globally optimized structure. Optical spectroscopies could be addressed by time-dependent DFT (TD-DFT) simulation,88−90 which is outside the scope of this review.
2.2. Molecular Dynamics Simulations
Molecular dynamics (MD) is a simulation of a time-dependent motion of the particles, atoms, or molecules dynamically framed according to Newton’s equation of motion under different ensembles, each fixes some thermodynamic variables in order to simulate the process that occurs in reality.91 The number of atoms, volume, pressure, temperature, and energy are variables that can be fixed at the cost of additional equations coupled to the equations of motion. Fixing thermodynamic variables relates to a specific ensemble like NVT, NVE, NPT, etc.
This computational approach ignores the explicit presence of electrons in the calculation and assigns the properties to the nucleus of the atoms. Therefore, classical molecular dynamics is not able to estimate the properties that contribute to the electron effect. Newton’s equation of motion requires the force between the atoms in the simulation box, which brings us to the core of molecular dynamics: the force field.92−94
Force fields are known as the engine of MD simulations and are sets of (analytical) equations that can describe the potential energy between constituent atoms.93 There are different types of force fields, and each has advantages and disadvantages that need to be considered. Each force field has bonded and nonbonded parts comprised of different sets of interactions. The bonded terms in the force field attempt to incorporate covalent bonds into the calculations by defining the potential energy contributed to the stretching of two-atom bonds, the bending of three-atom angle, and the deformation of four-atom dihedra. Nonbonded force field terms cover electrostatic and van der Waals interactions between atoms. van der Waals interaction occurs at a much shorter distance between the atoms. A cutoff radius is assigned to the van der Waals interaction to ignore the force calculation beyond this distance. For long-range electrostatic forces, more complicated approaches such as Ewald summation and particle–particle particle-mesh (PPPM) are employed to calculate electrostatic forces at much larger distances.93 Other potential interactions, especially bonded potential, have been proposed, taking into account the cross-terms of bond–bond, bond-angle, etc. COMPASS95 and PCFF96 are examples of this family of force fields. By adding more terms to the force field formulation, the accuracy of the force calculation is expected to increase, as well as the computational cost. CHARMM,97 OPLS,98,99 GROMOS,100 and AMBER101 are also among the well-known force fields used in molecular simulation methods.
In contrast to the rich availability of force fields for organic molecules, inorganic compounds (e.g., silicates, oxides, aluminates, metals, etc.) suffer from a lack of accurate and compatible force field parameters. Even if they existed, the simulation of organic–inorganic interfaces is hardly feasible when the energy expressions of the compound are dissimilar. In this regard, an early attempt to develop specialized force fields was modifying CSHFF or ClayFF102 to be compatible with harmonic energy expressions in PCFF, CHARMM, AMBER, COMPASS, and OPLS-AA, which allowed reliable MD simulations of interfaces of chemical admixtures and cement-related minerals.103−106 However, the energy expressions were limited in transferability and could not apply to the large, diverse family of minerals. More recently, Heinz et al.107,108 introduced a collection of all-atom parameters for more than 20 common minerals and metals within the INTERFACE force field compatible with PCFF, CVFF, and CHARMM. With all their achievements, developing a universal and concerted force field to describe the inorganic–organic systems has remained a work in progress.
Among all the substances and materials, the aforementioned force fields could be applied to, special attention must be given to water as the working fluid in the flotation process. There are numerous models of water, each attempting to describe the properties of water in a more realistic way. The existing water force fields attempt to model the molecules based on assumptions such as rigidity, partial atomic charge, and the number of sites. Among the water models, SPC/E, TIP3P, 4-site models, and higher accuracy models are the most recommended ones.109,110
All the force fields mentioned above are classical, meaning that they are not able to describe bond breaking/formation during the simulation. In this regard, reactive force fields (ReaxFF) were developed for a narrower range of atom types and molecules to overcome this limitation. Reactive force fields can be categorized as methods that lie between quantum calculation and classical molecular dynamics.34 First introduced by Van Duin30 in 2001, reactive force fields have opened up promising glades in the approach to molecular simulations. ReaxFF potentials incorporate more electron-based behaviors into the force field, making it more robust in monitoring systems that include bond dissociation/formation phenomena. More details about the force field and its contributed equations can be found in ref.30 Implementing MD simulations with reactive force fields would increase the computational cost because the force field needs a much shorter time step for convergence. To date, many parameter sets are published for different types of atoms and materials (such as silica)111−113 that could also be applicable to mineral flotation simulations.114−116 Note that since the ReaxFF parameters are found based on DFT calculations and fit with predefined equations, the transferability of the force field to any other system of interest should be checked according to the phenomena studied.
2.2.1. MD Analysis
Wettability (Contact Angle)
The quality of mineral separation in the flotation process depends strongly on the adsorption phenomena and, thus, on the properties of the mineral surfaces.1 Contact angle measurements are a simple and convenient way to infer valuable details about surface characteristics.117 Liquid–solid interaction, roughness, surface functional groups and their polarity, and surface hydrophobicity/hydrophilicity all contribute to the contact angle.118 It is customary to relate hydrophobicity/hydrophilicity to the measurement of the contact angle of water on a surface or to the free energy of hydration.119 The contact angle results are translated into the degree of hydrophobicity/hydrophilicity of the materials, providing some insight into the total solvent-solid interaction at the macroscopic scale.120 In contrast, doing the same in nanoscale surface probing still faces experimental and instrumental limitations.120 To this end, atomistic simulations can overcome such limitations by identifying events occurring at the nanoscale that are reflected in the contact angle calculation. Computational efforts to calculate contact angle can be categorized into two general approaches.121,122 The first approach, which is easy to implement, is known as sessile droplet. In this approach, a water nanodroplet is placed on the material’s surface, and the droplet equilibrates by adapting its shape during the simulation time. The second approach is based on free energy calculations, which may not be as easy as the first approach, but provides a more accurate contact angle determination.
The calculation of the contact angle in both theoretical and experimental approaches is based on Young’s fundamental equation of a planar geometry123 given as follows:
| 7 |
where θ is the contact angle of the droplet on the surface and γSV, γSL, and γVL are, respectively, the solid–vapor, solid–liquid, and vapor–liquid surface tensions. Some challenges have been addressed in the sessile droplet approach regarding the equilibration time for reshaping the droplet, the size of droplet, and systematic errors in contact angle calculation. In this approach, the isochoric vapor–liquid interface is needed for the calculation of the contact angle, which is the contour whose density is equal to half the bulk density.124,125 The contact angle is determined by drawing, from a point on the triple line, a tangent to the liquid–vapor interface and taking the angle inside the body of the drop bearing on the solid surface.121 Furthermore, contact angle calculation can be performed by fitting a spherical shape to the droplet124 or comparing the center of mass of the droplet with the ideal droplet on the surface.126,127 Both methodologies deal with the droplet geometry, which requires the droplet to be well equilibrated. However, some recent investigations suggest that droplet formation may take a much longer simulation time (∼100 ns) than is usually taken for granted (∼4–5 ns).128,129 Another issue with the sessile droplet simulation technique is the use of the macroscopic Young’s equation for the microscopic droplet. In this case, the modified Young’s equation is used, which includes the line tension terms in the original equation. In that case, one needs to simulate the system for different sizes of droplets and extrapolate the modified equation to an infinite size. However, the inadequacy of the approach for small nanodroplets continues to be debated.130 Thus, the contribution of system size to line tension values is not fully understood, and additional work is needed to further clarify what is happening at this scale.125 Some systematic precautions addressed in ref (122) must be considered when the sessile droplet is chosen as the primary method for contact angle determination.
The second simulation approach employs free energy calculation to determine the contact angle by forming a vapor–liquid interface, where the grand canonical transition-matrix Monte Carlo method is used. In this method, the vapor–liquid interfacial surface tension is calculated with and without the surface. The fluid is sandwiched between the surface and a hard wall that may have an attractive or repulsive affinity with the solvent. The determination of the contact line is no longer followed in this approach, and all calculations are based on the drying/spreading of the solvent on the surface. Drying potentials (attractive walls) are helpful for solvophobic surfaces, while spreading potentials (repulsive walls) are used for solvophilic surfaces. The GCMC is used to investigate the dry/spreading potentials in which the number of molecules and chemical potential of the system can be tuned. Then, the values are related to the modified version of Young’s contact angle equation.131−134 Errington and co-workers recently proposed the same methodology based on the MC/MD combination, which gave good results for drying potentials, but not for spreading interface potentials.135
A similar approach devised in molecular dynamics simulation is introduced as the phantom-wall method.136 In this approach, a liquid film is placed above the solid surface, while a repulsive hard wall lies between the liquid film and the solid. Removing the hard wall will make a gap or vacuum between the solid and liquid. The vacuum free energy can be approximated as solid–vapor free energy difference, which is valid only for weak solid–vapor interactions. In the calculations, the free energy is studied relative to the film thickness of the solvent in the system. The difference between the minimum free energy at the small thickness and the free energy plateau at the higher solvent thickness will be directly related to the vapor–liquid surface tension and contact angle. Free energy calculation can be performed using GCMC and MD. MD is computationally efficient for large and complex systems, but more simulations are needed for different densities of the solvent to construct the profile of free energy-thickness. For a simple system, the GCMC is more straightforward because it can change its number of molecules and its chemical potential, while it may not be efficient for large and complex systems.122
Another promising approach for contact angle calculation is based on enhanced sampling methods, which could be implemented through MD or GCMC. This efficient method uses the biased potential to change the number of solvent molecules to gradually wet the surface. The free energy difference relative to the wetting change is calculated and related to the contact angle. The approach simulates the Wilhelmy plate method of exposing two plates to the solvent but calculates the free energy rather than the capillary force.137 This approach also allows accurate calculation of the vapor–liquid surface tension for a given contact angle.137 The introduction of hydrophobic and hydrophilic patches is an alternative to grasp the mineral surface-reagent contact, especially in the context of hydrophobicity/hydrophilicity of complex molecules such as proteins.138
Surface Tension
In addition to measuring the contact angle, surface tension can also play a key role in the flotation and solvation of reagents, especially frothers, by helping to simulate the phenomenon on the vapor–liquid interface side.139 This section provides an alternative mechanical and thermodynamic insight into the above section on the calculation of wettability.
The surface tension can also be viewed as a quantity emerging from the mechanical equilibrium of forces at the interface known as the Irvin-Kirkwood equation:140
| 8 |
where PN and PT are the normal and tangential pressure tensors relative to the normal direction of the interface (z-direction) when the system is in a state of equilibrium. The main drawbacks of the method are the large fluctuation of the surface tension and its limitation of implementation at high temperatures.
The thermodynamic approach for surface tension calculation is based on the following fundamental equation:
| 9 |
where F is the Helmholtz free energy and A denotes the interfacial area. In essence, the variation of the free energy or potential energy relative to the variation of the interfacial area at a constant number of molecules, volume, and temperature gives the interfacial surface tension.141,142 The primary challenge of the method is the calculation of the Helmholtz free energy, which is far from trivial in molecular dynamics simulation. The test-area method is based on the perturbation of the interfacial area by then calculating the change in the free energy of the system as follows:143
| 10 |
In this method, rescaling is done at constant volume while each dimension of the simulation box can change. Similar concepts with different implementations for surface tension calculation are described elsewhere.144
Hydrogen Bonding
Interactions induced by water molecules
are a factor that should not be ignored when describing aqueous mineral
pulps in froth flotation processes. For instance, hydrogen bonding
with the water itself, the reactant, and polar sites on mineral surfaces
is about 5 to 10 times stronger than van der Waals interactions.118 In molecular dynamics simulation, geometrical
criteria are used to detect hydrogen bonds between polar atoms (N,
O, F, and Cl) connected to at least one hydrogen atom. A hydrogen
bond is comprised of a donor atom (D) (i.e., the polar atom connected
to a hydrogen atom), a hydrogen atom (H), and an acceptor (A) (i.e.,
the polar atom interacting with the hydrogen atom). The hydrogen bond
forms when the donor–acceptor distance (DA) and  angle are lower than critical values, respectively,
called cutoff distance and maximum angle. For water, the cutoff distance
on DA is 3.5 Å and the maximum
angle are lower than critical values, respectively,
called cutoff distance and maximum angle. For water, the cutoff distance
on DA is 3.5 Å and the maximum 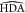 angle is 30°,145,146 but slightly different values can be found in other works for different
molecules.147 The number of hydrogen bonds
can change the solvation of reagents or prevent their adsorption on
the mineral surface due to competition with water hydrogen bonds and
minerals. The dynamics of the hydrogen bond and its lifetime is also
a feature to be accounted for. Usually, such dynamics is evaluated
by the normalized temporal autocorrelation function (CHB):148
angle is 30°,145,146 but slightly different values can be found in other works for different
molecules.147 The number of hydrogen bonds
can change the solvation of reagents or prevent their adsorption on
the mineral surface due to competition with water hydrogen bonds and
minerals. The dynamics of the hydrogen bond and its lifetime is also
a feature to be accounted for. Usually, such dynamics is evaluated
by the normalized temporal autocorrelation function (CHB):148
| 11 |
where hij is 1 or 0 depending on whether or not there is a hydrogen bond between a pair of atoms i and j. Two formulations exist for the hydrogen bond autocorrelation function: one for continuous and another for intermittent hydrogen bond lifetimes. In the first case, only if the H-bond breaks does the function cancel; otherwise, the lifetime is quantified as the continuous time of hydrogen bonds between atoms. In the second, intermittent hydrogen bonding considers a different time window of H-bond formation, regardless of whether H-bonds are broken beforehand. Obviously, a continuous lifetime would be shorter than an intermittent one.149 Fitting the hydrogen bond autocorrelation function by an exponential gives access to the retardation time, i.e., how long a hydrogen bond lasts between specific pairs of atoms. The case of a nonexponential behavior is interpreted by the contribution of molecular diffusion in the hydrogen bonding dynamics.145
Hydrogen Shell
The hydrophobic regions do not form hydrogen bonds with the water molecules, which instead cluster as hydration shells.150 The number of hydration shells around ions, reagents, or minerals is estimated using the radial distribution function. The first minimum of the radial distribution function of water-solvated molecules or ions gives the thickness of the first or primary hydration shell.151 The other detected hydration shells are of lesser intensity than the first one. Since the number of water molecules around the solvated species allows estimation of the degree of molecule solvation, the lifetime of water molecules remaining in the shell should be considered. To calculate the lifetime of water molecules remaining in the hydration shell, the normalized autocorrelation function is used:150
| 12 |
where N is the hydration number, with fi = 1 or 0 depending on whether or not the water molecule i is in the hydration shell. The average lifetime of the water molecules in the hydration shell is calculated by integrating R until the function approaches to 0.99. Through residence time calculations, if the molecules do not return to the hydration shell for 2 ps, they are considered noncontributing to the hydration residence time.
Although the effects of ion charge and diameter on the hydration shell are well established, the new flotation bioreagents can be better characterized by examining their solvation and adhesion to the mineral surface.152
Diffusion Coefficient
The mobility of the reagent and ion in the solution or close to the mineral reflects their interaction with the solvent and the solid-solvent interface. The stronger the affinity of a molecule with a mineral, the more reduced its normal mobility at the expense of diffusion, mainly on the plane parallel to the solid surface. The mean square displacement (MSD) is used to calculate the diffusion coefficient as follows:
| 13 |
where ri(t) is the coordinate vector for species i at the specific time of simulation and d is the diffusion dimension (3 for bulk and 2 for surface diffusion).
2.3. Hybrid Methods
Classified as hybrid methods, they stem: (1) in the temporal sense by combining QM and MM, and (2) in the spatial sense by applying QM and MM methods to different physical regions of a system. The AIMD simulation is an example of the first category, while the QM/MM method belongs to the second class. In what follows, these two methods are introduced with their application scenarios.
2.3.1. Ab Initio Molecular Dynamics
The description of the interatomic interaction forces in a given system is at the heart of MD simulations. In classical MD, these forces are computed from predefined force fields or interatomic potentials. However, the available force fields are often unable to afford acceptable accuracy, and, more importantly, chemical reactions cannot be modeled by MD.
The advent of ab initio molecular dynamics (AIMD) has provided a new route to calculate the interatomic forces directly from electronic structure and reduce its dependence on force fields.22−24 In AIMD, finite-temperature dynamical trajectories are obtained by including the forces collected from the “on the fly” electronic structure calculations as MD simulation proceeds.23 Therefore, the effect of temperature-induced thermal agitation, on the one hand, and electronic polarization and bond formation/cleavage, on the other hand, can be described in AIMD.
Ab initio electronic structure calculations are exceedingly computationally expensive; therefore, the electronic calculation portion of AIMD is usually DFT-coded to trade off accuracy and computational cost.24,25 In this case, the quality of the exchange-correlation functional is the key control on the overall accuracy of the simulation.
To date, AIMD has been successfully applied to a wide variety of problems and is already beginning to percolate through the mineral community as well.8,72,153−156 With the help of this tool, new physical phenomena could be unveiled, taking a new step forward compared to classical MD or ground-state DFT calculations. With advances in HPC, AIMD simulations can routinely reach total simulation times of a few hundred picoseconds for systems comprising a few hundred atoms.
2.3.2. Quantum Mechanics (QM)/Molecular Mechanics (MM) Method
The first step in the QM/MM method is to divide the input system into two distinct regions. The smaller “active site” region, where the force field parameters are unknown or where chemical reactions or polarization effects play an important role, must be treated by QM calculations. The remaining region is described by lower-class MM methods.27 In practice, due to a favorable trade-off between computational cost and accuracy, many problems are solved using DFT and classical MD to, respectively, represent the QM and MM components.
The various QM/MM approaches differ in the way they: a) describe the interaction between the QM and MM regions and b) compute the system energy.27−29 Interactions between QM and MM comprise long-range electrostatic interactions and local short-range ones (e.g., binding and van der Waals interactions). Electrostatic interactions are usually dominant; accordingly, depending on the treatment between QM and MM, QM/MM schemes can be classified as mechanical or electronic embedding.157 In the mechanical embedding scheme, the QM calculation is performed without considering the polarization effect caused by the atoms in the MM region. However, for the MM simulation, the Coulombic interactions of the QM region are accounted for by providing partial charges for the atoms in the QM region at the force field level. In the electronic embedding, the QM region is polarized by the atomic charges of the MM region, and similarly, the Coulomb forces on the MM atoms include the QM density share acting on the MM point partial charges. Of course, the electronic embedding provides a more accurate description with increased computational demands.27
In many cases, it is inevitable to split a molecule to clarify the QM and MM domains. With cleavage bonds at the boundary, a modification of the QM and MM calculations is necessary since the unpaired electrons caused by the broken bonds significantly influence the electronic structure in the QM region.158 A typical solution is the so-called link atom approach, in which the dangling bonds at the boundary are saturated by introducing capping (hydrogen) link atoms to the QM system. Regardless of the type of embedding, the total system total energy cannot be calculated simply as the sum of the subsystem energies due to the strong QM-MM interactions.27−29,157 Therefore, the energy expressions in QM/MM can be evaluated under different assumptions, either by an additive or subtractive expression.
In the subtractive schemes, the QM/MM energy is given by
| 14 |
where EMM and EQM are the energy values calculated by MM and QM methods, respectively. In eq 14, S represents the entire system, and C corresponds to a combination of QM regions and the link atom region. It should be noted that the subtractive embedding scheme requires proper force field parameters for the atoms of the entire system, which limits the overall range of applicability of the approach.159
Additive approaches for energy description, on the other hand, avoid MM calculation on the whole system and region C. This makes these methods particularly attractive since the unavailability of force field parameters for QM region would not be a concern. The total energy of the system is calculated as
| 15 |
As can be seen in eq 15, the MM calculation is performed only on the MM region. EQM–MM refers to an explicit coupling term, which collects the interaction terms between MM and QM subsystems. The EQM–MM coupling energy term is the most elaborate term in the additive formulation and includes bonded, van der Waals, and electrostatic interactions between QM and MM atoms.
QM/MM approaches are now widely established for modeling biomolecular, inorganic/organometallic, and solid-state systems, as well as for studying processes in explicit solvents.27,28 In mineral processing, QM/MM applications are still a relatively untouched area of research, while increased attention to these approaches can enrich future investigations.
2.4. Summary
In this section, the most important simulation techniques within the category of molecular modeling have been reviewed. The basic theories have been initially described for each method, while the primary focus has been on property calculations relevant to the flotation process. Representative examples of flotation applications will be reviewed in section 3. Before jumping in, and to get a better overview of the pros and cons of various simulation techniques, brief descriptions of all the methods reviewed are listed in Table 1. The motivation for performing every simulation should start with a straightforward question and clearly stated expectations. To select an appropriate simulation method, the ability of the different techniques to calculate the desired properties should be considered while accounting for the limitations and computational cost.
Table 1. Comparison of Different Molecular Modeling Techniques.
| molecular modeling techniques | merit | limitations |
|---|---|---|
| DFT | • more accurate calculation based on the electronic state | • high computational cost |
| • capable of simulating: | • small simulation size | |
| - charge polarization/transfer | • incapable of calculating dynamic properties | |
| - bond formation/cleavage | • incapable of including temperature effect (all simulations are performed at 0 K) | |
| - electronic-based properties, e.g., band structure, DOS, etc. | ||
| - chemical reactions | ||
| - transition state | ||
| AIMD | • includes finite temperature effect in DFT calculations | • high computational cost |
| • small simulation size | ||
| QM/MM | • takes advantage of both DFT and MM methods | • complexity in the implementation, especially at QM-MM boundary region |
| • suitable for larger systems | ||
| ReaxFF | • includes charge polarization to the classical force field | • force field availability and transferability |
| • adds bond formation/cleavage simulation into MD | • shorter time- and length scales than MD | |
| MD | • includes the effect of temperature, pressure, volume, etc. in the simulations | • incapable of simulating chemical reactions and bond formation/cleavage |
| • larger time (up to μs) and length (up to μm) scale | • lower accuracy compared to DFT calculations | |
| • capable of calculating: | • accuracy and feasibility depend on the availability of proper force fields | |
| - dynamic properties, e.g. wettability, diffusion, etc. | ||
| - thermodynamic properties |
In general, from a technical point of view, the unavailability of accurate force fields and atomic partial charges for complex mineral systems is the most critical limitation in the application of MD simulations in flotation systems. The danger of oversimplifying the interactions by applying generic force fields comes to the fore, which inevitably leads to underestimating the properties of interest. Even if all technical shortcomings are overcome, the limitations of molecular modeling techniques, mainly the size of the system and the length of the possible simulation, are still prohibitive. The computational cost of molecular simulations overgrows with the system size and is limited to a few hundred atoms for DFT calculations and several tens of thousands of atoms for MD simulations. The routine MD simulation time is currently in the order of a few hundred of nanoseconds. While this is adequate to address some scientific challenges, it is not for many others, especially those involving biological phenomena such as protein folding or crystal growth in mineral systems, which occur on the time scale of seconds in the real world. The nanosecond time limit further restricts the configurational sampling space accessible to MD, which is critical for calculating thermodynamic properties. It is worth mentioning, however, that with the rapidly growing HPC facilities, substantial headway toward larger scales and longer time scales is expected. As evidence, we already find in the literature DFT and MD simulations of systems with millions of atoms.160−165
3. Applications of Molecular Modeling to Froth Flotation
Once the ore is milled, the minerals of which it is composed, released with a sufficient degree of liberation, are separated from each other by exploiting the natural or artificial propensity of their surfaces to be wetted by water. The degree of hydrophobicity/aerophobicity of the mineral determines whether it ends up in the overflow (hydrophobic) or the underflow (aerophobic/hydrophilic) depending on the reagents, e.g. collectors, used. Therefore, all phenomena that alter or adjust the surface properties of minerals significantly contribute to the efficiency of the mineral separation procedure.166 A nonexhaustive sketch (Figure 2) of these phenomena focuses on the mineral surface and the interface with the bubble, namely, surface hydration, adsorption of reagents and their consequence on the hydrophilic/hydrophobic split at the gas–liquid interface, surface modification by the dissolution of leachable entities or (re)precipitation of species, and to some extent, the presence of surface defects or impurities. Having summarized in the foregoing section some of the principles of molecular modeling relevant to our topic, let us now turn to its applications in flotation by unpicking some of the phenomena illustrated in the preceding figure.
Figure 2.

General picture of the phenomena that govern the froth flotation of minerals.
3.1. Mineral-Reagent Interactions in Froth Flotation and Molecular Modeling Insights
As Wills and Finch mentioned: “true flotation exploits the differences in surface properties of particles of various minerals. After treatment with reagents, such differences in surface properties between the minerals within the flotation pulp become apparent.”166 Therefore, the mineral-reagent interaction is one of the key features of flotation, through which the surface properties of minerals can be tuned in a targeted way. Notably, the overwhelming majority of molecular modeling studies on froth flotation fall under the characterization of mineral-reagent interactions.
3.1.1. Principle Features of Interactions
In this section, the application of molecular modeling techniques to reveal the nature of the interaction (physical and chemical adsorptions) and the conformations of reagents on the mineral surface is reviewed.
Interaction Mechanism and Adsorption Conformation
While it is possible to infer the nature of the interactions by quantum computation, predictions of the covalent mechanism by MD simulations are always fraught with uncertainty. As an example in the rare earth element (REE) category, Chapleski et al.167 analyzed the adsorption mechanism and configuration of several salicylic (SA), salicylhydroxamic (SHA), and benzohydroxamic (BHA) acid–based collectors on bastnäsite (100) and calcite (104) surfaces using DFT calculations (Figure 3). For bastnäsite, the aromatic ring moieties of BHA and SHA (SA) are predominantly oriented face parallel (face perpendicular) to the mineral surface. While for the calcite surface, all dominant conformations are oriented almost normally with minimal shadowing on the surface. The orientation maximizing the shadowing of the aromatic rings (BHA, SHA) on the bastnäsite surface promotes multilayer adsorption of the collector. On the contrary, carboxylic collectors would lack this property suggesting a lower efficiency in promoting the increased accumulation of bastnäsite in the overflow.168 The uptake of the collector on the bastnäsite and calcite surfaces occurs either by the formation of covalent bonds (bidentate or monodentate) with the metal cations or by hydrogen bonding involving the reagent hydroxyl groups and the carbonates (Figure 3).167
Figure 3.

DFT structures of the most stable adsorption configurations of various hydroxamic bearing collectors on bastnäsite (100) and calcite (104) surfaces adapted from ref (167).
Wanhala et al.169 employed a combined experimental-computational approach to clarify the alkyl hydroxamic collector adsorption behavior on the bastnäsite surface. The study showed that the effect of collector concentration is bivalent. First, it affects the inclination of the collector alkyl chains relative to the surface, ranging from tight/straight to almost lying down. Second, the covalent bond can evolve from monodentate to bidentate. In another study, Sarvaramini et al.170 investigated the adsorption mechanism of alkyl-hydroxamic acids on monazite and bastnäsite surfaces by emphasizing the interactions between the collector and solvated REE species susceptible to leach and consisting of Ce(OH)2+ and Ce(OH)2+. Despite the intriguing findings, one should be aware that excluding solvation and surface complexation in an aqueous environment could cause uncertainties in the results, thereby compromising their practical applicability.50,51
Sutton et al.171 ranked the interactions of 20 bis-phosphinate ligands toward calcite (104) and Ce-bastnäsite (100) surfaces using a combination of DFT and MD calculations. Covalent bonds were revealed for four ligand-surface configurations, namely, bidentate, repeated monodentate, staggered monodentate, and neighboring monodentate. The prediction of Ce-bastnäsite-selective ligands was experimentally verified by vibrational sum-frequency (vSFG) spectroscopy, attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy, and isothermal titration calorimetry (ITC).
Based on DFT and AIMD calculations and diffuse reflectance infrared Fourier-transform (DRIFT) measurements, Foucaud et al.172,173 examined the adsorption conformations of carboxylate collectors on the fluorite surface. The results indicate that the adsorption of the carboxylate anion takes place through monodentate or bidentate covalent bonds with the fluorite or fluorapatite174 surface, while the sodium carboxylate counterion plays a role in the adsorption sequence of the collector. These studies demonstrate the power of molecular modeling when combined with experimental analysis to facilitate material characterization and improve the interpretation of experimental observations, ultimately leading to a more complete understanding of the phenomena under study.
Kumar et al.175 confirmed by DFT calculations the observed selectivity of 2-mercaptobenzothiazole (MBT) toward pyrite at the expense of chalcopyrite. This result is explained by stronger MBT adsorption on pyrite due to the formation of two Fe–S bonds with the endo- and exocyclic S atoms of MBT with Fe of the pyrite (110) surface. The MBT interactions with the chalcopyrite surfaces involve a single Cu-exocyclic bond. These different mechanisms would account for the greater MBT selectivity toward pyrite.
Molecular modeling techniques can also help shed light on the influence of depressants on minerals in flotation systems. As an example, the adsorption mechanism of pyrogallol (a tannic derivative), as an eco-friendly depressant, in the flotation of bismuth–molybdenum sulfide ore was studied by DFT.176 Chemisorption of pyrogallol on the bismuthinite surface involves its alcohol group in covalent bonds (monodentate and bidentate) with the mineral surface. Similar calculations on the adsorption of a new m-nitrobenzoate depressant with arsenopyrite,177 suggest the involvement of the -NOO residue of the depressant and not its −COO residue. The adsorption mechanism of sodium hexametaphosphate (NaHMP) depressant from the kaolinite surface was analyzed by combining DFT, MD, and Monte Carlo simulations.178 The anionic form of NaHMP adsorbs through hydrogen binding, although the effect of water molecules and solution pH was neglected. Neglecting the inclusion of counterions would not lead to an infinite charge of the simulation box in DFT calculations since the overall electroneutrality can be preset in the calculations; however, the partial charges assigned and, consequently, the electrostatic interactions would forcibly deviate from reality. This shortcoming could also affect the predictability of surface species, as shown by MD simulations of sylvite (KCl) and halite (NaCl) surfaces during flotation of potash ores by cationic collectors if water molecules were to be ignored in the phenomenology.179 More recently, Alizadeh Sahraei and Larachi51 revealed the importance of explicit water modeling in the DFT calculations. Their results suggest that implicit water modeling fails to predict the formation of solvated mineral-reagent complexes. The critical role of the explicit water model over implicit models was further highlighted in the DFT calculations of Chapleski et al.180 for separating rare-earth elements from gangue materials. For the description of such complexes, the inclusion in the simulations of water molecules in the form of a multilayer is essential and highly recommended.
Effect of pH on Reagent Interactions
Some researchers also highlighted the capability of molecular modeling to determine the effect of pH on collector–mineral interaction. For this purpose, they considered different species of collectors corresponding to a specific range of pH.181−184 Liu et al.184 used MD and a semiempirical quantum chemical method to compare the interactions with the bastnäsite surface of three possibly occurring species of lauryl phosphate, namely, C12H25OPO3H–, C12H25OPO3H2, and C12H25OPO32–. Adsorption of carboxylic acid-bearing collector species on spodumene in proton-impoverished solutions at pH 8 was DFT-simulated185 to examine the plausible interactions of ionic–molecular complexes consisting of (a) head–head, (b) head–middle, but not (c) head–tail on the spodumene surface.
At this stage, the value of these simulations of the pH and its impact on the speciation of the collector and, thus, on the reagent-mineral interactions is to be taken cum grano salis because all the criteria to account for the pH in the flotation are not there. The consideration of protonation/deprotonation states of adsorbates with pH-sensitive functional groups at different pKa is not sufficient to model adsorption phenomena at different pHs.186 For solid molecular modeling (particularly molecular dynamics simulations) at different pH, the mineral surface and solution must also be carefully constructed. The type and area density of surface groups upon solvation, the likely ionization degree as a function of pH, the ionic strength and type of cations, etc. are crucial parameters that must be considered in model constructions for accurate modeling of flotation systems. Knowledge is typically provided by experimental characterization via BET adsorption isotherms, spectroscopies (e.g., solid-state NMR, infrared, Raman, and X-ray photoelectron spectroscopy), potentiometric titration, and ζ-potential measurements. For instance, a fairly comprehensive database already exists for the silica family at different pH levels.108,187−189
In DFT calculations, however, the construction of the realistic formulation of the solution at different pHs is prohibitive due to the size limitation of the system. In this regard, the (de)protonation state of the adsorbate per pKa values of the functional groups should include the appropriate counterions to ensure that the physical system is neutral. While the explicit solvent model is not affordable for routine DFT calculations, one alternative is to use a cluster model. In this approach, the adsorbate molecule and counterions are surrounded by several water molecules to explicitly include the solvent effect, and an implicit solvation model (i.e., replacing the explicit solvent with a dielectric continuum) is applied for the remaining space. When the cluster model is not affordable, implicit models based on the polarized continuum solvation model are recommended for (de)protonated systems at different pHs.190
Interaction of Reagent Mixtures
Molecular modeling is proven to be instructive in developing a microscopic picture of mixed surfactants at mineral/water/gas interfaces. Chemisorption (covalent bonds) with the hematite surface is predicted by DFT upon adsorption of oleate alone or starch alone on iron oxide. On the other hand, in the presence of an oleate-starch-hematite system, starch inhibits the adsorption of oleate, leading to a significant reduction in its recovery by flotation.191 The mechanism of interaction of mixed cationic/anionic surfactants or polar/nonpolar collectors with the surface of low-rank coals can be further analyzed by means of MD simulations based on the ratio of their concentrations and the ability of their hydrophobic carbon chain to repel water molecules from adsorption on the coal surface.192−195 Sequential adsorption of cationic surfactants onto deprotonated oxygen functional groups dangling from the carbon surface, followed by adsorption of anionic surfactants onto preadsorbed cationic molecules, has been presented as a dominant mechanism for these surfactant systems. Experimental observations revealed that the process leads to a significant reduction in collector consumption and an improvement in flotation efficiency.196 In another study, MD simulations showed that mixed anionic/cationic collectors could significantly boost the hydrophobicity of the muscovite surface. The interaction between polar groups of cationic and anionic collectors helps reduce the electrostatic repulsion if cationic collectors were to act alone, improving the adsorption of the mixed collectors on the muscovite surface.197,198 Similar findings have been reported in the case of galena flotation using MD simulations.199
Wang et al.200 took advantage of MD simulations to shed insight into the adsorption behavior of mixed dodecylamine (DDA)/sodium oleate (NaOL) (cationic/anionic collectors) in quartz flotation. Prior to this study, it was assumed that adsorption of most DDA on the surface occurs first, and then NaOL can adsorb via hydrophobic interaction among alkyl chains. The MD results, however, demonstrated that most DDA and NaOL species form hydrophilic spherical-like micelles near the quartz surface (Figure 4a), and only a few DDA molecules adsorb directly on the quartz via hydrogen binding bridging the spherical micelles and the mineral surface (Figure 4a). On the contrary, using MD simulations, Shen et al.201 captured a different adsorption mechanism for the same reagent mixture in the case of kaolinite flotation. The computational results suggested that coadsorption of DDA and NaOL on the surface of (001) kaolinite may take place via hydrogen binding (Figure 4b), where the anionic molecules were intertwined among NaOL ions via a hydrophobic interaction between the alkyl chains on the mineral surface.201 Thus, MD simulations predict two different types of assembly for the same DDA/NaOL couple on quartz (Figure 4a) and kaolinite (Figure 4b) surfaces.
Figure 4.

Different adsorption behaviors of mixed cationic/anionic collectors (DDA/NaOL) on the surface of (a) quartz (promoting depression effect) and (b) kaolinite (promoting activation effect). Adapted with permission from refs (200) (a) and (201) (b). Copyright 2016 and 2017, respectively, Elsevier.
The muscovite (001) surface exposed to mixed DDA/NaOL collectors was also simulated using MD simulations.202 Alone, DDA adsorbs as an organized monolayer on the muscovite surface, while NaOL alone is unable to hold attached to the surface. However, for the DDA/NaOL surfactants mixture, the ammonium head groups of DDA remain adhered to the surface, and the NaOL molecules intertwine among the DDA molecules instead of being repelled by the muscovite surface (a mechanism similar to that discussed in the case of kaolinite201). The electrostatic interaction between the ionic head groups of the collectors as well as the hydrophobic interaction between their tails, was recognized as the dominant adsorption mechanism in this system.202 Other studies have also confirmed the possibility of forming micelle-like spherical structures (Figure 4a) in mixed surfactant systems, while such manifestation in single-surfactant systems is rare.202,203
When mixed with monohydric alcohols, DDA adsorption on magnesite and dolomite surfaces can be increased according to simulations.204 MD simulations with COMPASS force field205 elucidated the adsorption mechanism of mixed surfactants of citric acid, pregelatinized starch, and NaOL on hematite, quartz, and siderite surfaces. Coadsorption of all three surfactants occurs on the siderite surface (Figure 5). Citric acid and pregelatinized starch are densely distributed over the entire hematite surface preventing the adsorption of NaOL directly onto the mineral (Figure 5b). Neither pregelatinized starch nor NaOL is able to adsorb onto the quartz surface (Figure 5c), in contrast to citric acid. Such behaviors suggest that siderite, easily flocculated, is then separable by flotation, as the experimental results corroborate.205
Figure 5.

Adsorption patterns and relative concentration profiles of citric acid, pregelatinized starch, and NaOL during their interactions with (a) siderite, (b) hematite, and (c) quartz. Adapted with permission from ref (205). Copyright 2020 Elsevier.
MD simulations using COMPASS have examined the effects of mixing aluminosilicate polymorphs (andalusite, sillimanite, kyanite) with cationic (octadecylamine) and anionic (sodium hexadecanesulfonate) collectors.206 The two collectors acted according to different adsorption mechanisms. For the former, electrostatic attraction and hydrogen binding between the amine cation and the surface oxygens predominated. For the second, the chemisorption of sulfonate anions on Al atoms prevailed. Because classical MD cannot model chemical interactions, X-ray photoelectron spectroscopy (XPS) has validated this last mechanism. A clathrate-type interaction between DDA and starch was simulated by MD in a vacuum and aqueous solution using the generic Universal force field.207 This rationalized the observed decrease in the contact angle with water on the quartz surface by concealing the hydrophobic tail of DDA (Figure 6).
Figure 6.

Snapshot of a six glucose ring starch molecule and DDA before and after clathrate interaction, resulting in masking the DDA hydrophobic tail. Adapted with permission from ref (207). Copyright 2017 Elsevier.
Section Outlook
Overall, the molecular-scale representation afforded by molecular modeling provides first-hand information about mineral-reactant interactions. This knowledge is crucial for the design, control, and operation of flotation chemistry to improve process performance. A myriad of similar molecular modeling studies (by DFT and MD) have recently been performed to unveil the interaction mechanisms of various reagents with pure gold,208−212 hydrophobic mineral surfaces (e.g., talc, graphite),213,214 aluminas,215−218 sulfide minerals,175,219−223 Ca and Mg bearing minerals,224−229 and more. Such a wide range of case studies using molecular modeling is a testimony to the versatility of the tool that can address just about any mineral system of interest for froth flotation. Proportionally, however, there are fewer studies based on molecular dynamics simulations due to a lack of appropriate force fields for the various mineral families. Even in the case of using class II force fields (e.g., COMPASS and PCFF), the accuracy of the force field parameters of the minerals remain indecisive. Presently, the simulations implemented with the INTERFACE or specialized force fields produce the most reliable MD results.
3.1.2. Minerals–Reagents Adsorption Energy and Affinity
The apparent adsorption energy of a reactant on a mineral surface is a valuable indicator for the affinity ranking of reagent-mineral pairs in flotation systems. The affinity calculation provides insight into the magnitude of surface-reactant interactions and the feasibility of adsorbate configurations.
Different definitions in use have been explained in section 2.1.2, e.g., adsorption or interaction energy, and many more defined according to the requirements of the problem. Mineral surfaces offer a vast diversity of atomic termination and electronic structures depending on the cleavage directions (especially in oxide minerals). Therefore, one has to expect different interactions between the reagents and cleavage planes. In simulation studies, these interactions are often determined for the most stable cleavage plane. In theory, the lower energy surface has a higher probability of occurrence on particle facets. However, other cleavage directions also exist on the mineral facets to form a particle. In this regard, for more accurate simulation of reagent-mineral interactions, it is preferable to apply adsorption studies on a set of cleavage planes rather than a single stable plane. The Wulff construction theory230 is an effective tool for recognizing the preferential cleavage planes of minerals and for predicting their crystal morphology. Therefore, performing surface energy calculations of the different cleavage planes of the mineral and subsequently searching for the Wulff construction would be a preliminary step before initiating adsorption studies. Note that Wulff’s theory of surface construction is founded on thermodynamic equilibrium assumptions,231 whereas grinding prior to froth flotation could be an out-of-equilibrium process, especially for high-energy grinding.232,233 Therefore, expecting further disagreements between simulation models and reality is not exaggerated.231Table 2 summarizes the studies involving adsorption energy calculations, highlights as well as limitations found in these studies.
Table 2. Examples of Molecular Modeling Applications to Compare Interaction Affinity of Various Reagent–Mineral Systems.
| minerals | reagents (type and name) | molecular modeling approach | comparative results | main limitations | refs |
|---|---|---|---|---|---|
| bastnäsite, calcite | collector: salicylhydroxamic acid (SHA), benzohydroxamic acid (BHA), salicylic acid (SA) | DFT (GGA/PBE functional) | collector affinity toward bastnäsite: BHA > SHA > SA | 1. mineral surface charges and protonation states of surface ligands at studied pH not defined specified; 2. explicit water-solvated forms of models not studied | (167) |
| collector affinity toward calcite: SHA > SA > BHA | |||||
| sylvite (KCl), halite (NaCl) (potash ores) | collector: octadecylamine | MD (COMPASS force field) | mineral affinity toward collector: KCl > NaCl | 1. mineral surface complexations at the studied pH not considered in the simulations; 2. simulation box size too small for an MD simulation; 3. number of water molecules in the model very low, preventing collector solvation to occur | (234) |
| magnesite, calcite, dolomite | collector: potassium cetyl phosphate | MD (COMPASS and Universal, force fields) | mineral affinity toward phosphate-based collector: magnesite > calcite and dolomite | 1. mineral surface complexation at the studied pH not considered in the simulations; 2. no verification of reaching equilibrium state; 3. no verification of the force field parameters, particularly when using a generic force field like Universal | (235), (236) |
| quartz, magnesite, dolomite | Collector: isopropanol amine | MD (force field unknown) | mineral affinity toward amine-based collectors: quartz > dolomite > magnesite | 1. mineral surface complexation at studied pH not considered in the simulations; 2. number of water molecules in the model very low, preventing collector solvation to occur; 3. no information on force field parameters reported, so the accuracy of parameters cannot be verified | (237) |
| bastnäsite, calcite | collector: bisphosphinate ligands | DFT (PBEsol-D3) + AIMD (γ point) | 1. mineral affinity toward reagents: bastnäsite > calcite | (171) | |
| brucite, kaolinite, quartz | flocculant: hydrolyzed polyacrylamides | MD (AMBER + CLAYFF-MOH force field) | mineral affinity toward reagents: brucite > kaolinite > quartz | (238) | |
| kaolinite, montmorillonite | depressant: nine types of organic carboxylates | DFT (GGA/PBE functional) | 1. depressant affinity toward kaolinite: hydrogen phthalate > acetate > hydrogen succinate > hydrogen citrate > gallate > salicylate > hydrogen oxalate > bitartrate > lactate | 1. calculations carried out in gas phase, no water effect included; 2. effect of initial configurations of adsorbates not comprehensively investigated | (239) |
| 2. depressant affinity toward montmorillonite: hydrogen phthalate > acetate > hydrogen succinate > hydrogen citrate > gallate > salicylate > hydrogen oxalate > bitartrate > lactate | |||||
| chalcopyrite: (100) and (110) cleavage planes | collector: thiophosphorus collector, including diethyl dithiophosphate (DEDTPA), diethyl dithiophosphinate (DEDTPI), diethyl monothiophosphate (DEMTPA), and diethyl monothiophosphinate (DEMTPI) | DFT (GGA/RBPE functional) | 1. collector affinity toward (100) surface: DEDTPI > DEDTPA > DEMTPI > DEMTPA | 1. effect of water molecules not considered | (240) |
| 2. collector affinity toward (100) surface: DEDTPI > DEDTPA > DEMTPI > DEMTPA | |||||
| 3. (110) surface more strongly adsorbs collectors than (100) surface | |||||
| calcite, dolomite, ankerite | collector: alkyl hydroxamic acid bearing collector; depressant: sodium silicate | DFT (GGA/PBEsol functional) | 1. collector affinity toward minerals: ankerite > calcite > dolomite | 1. water effect not included in the calculations; 2. effect of initial configurations on adsorption modes not comprehensively investigated | (241) |
| 2. depressant affinity toward minerals: ankerite > dolomite > calcite | |||||
| 3. interaction energy of collector with minerals higher than depressant | |||||
| fluorapatite, dolomite | collector: N-hexadecanoylglycine | DFT (GGA/PBE functional) | collector affinity toward minerals: fluorapatite > dolomite | 1. water effect not included in the calculations; 2. effect of initial configurations on adsorption modes not comprehensively investigated | (242) |
| sphalerite, pyrite | collector: tert-dodecyl mercaptan (TDM) | DFT (GGA/PBE functional) | collector affinity toward mineral: sphalerite > pyrite | 1. water effect not included in the calculations; 2. vacuum thickness not tuned for DFT calculations: 10 Å vacuum insufficient to prevent relatively long reagent from seeing other slab side | (243) |
3.2. Flotation Reagents
“Without reagents, there would be no flotation, and without flotation, the mining industry, as we know it today, would not exist”.139 Numerous types of reagents and surfactants are used in froth flotation, including collectors (which selectively make mineral surfaces hydrophobic), depressants (which prevent flotation of unwanted minerals), modifiers (which deliberately change the properties of mineral surfaces), activators (which activate mineral surfaces for reagents uptake) and frothers (which prevent bubble coalescence).139,166 The selection of appropriate reagents for flotation has always been a challenge, especially for complex orebodies. As will be seen through the following examples, the advent of molecular modeling puts more than one arrow in the quiver to help deliberately tune flotation reagents’ properties.
3.2.1. Structure–Property Relationships
Effect of Polar and Nonpolar Groups
Liu et al.244 took advantage of DFT calculation to investigate the relationship between the structure of mercaptobenzoheterocyclic compounds and their potential to interact with the pyrite surface for flotation separability. Not unexpectedly, it is the O, N, and S heterocycle atoms that induce chemical bonding with the iron atoms on the pyrite surface. In particular, the surface complexes induced by the thione tautomers exhibit higher reactivity than the thiol tautomers. In another study on the selective separation of pyrite and sphalerite, thiophosphorus collectors were analyzed using a similar molecular modeling approach to reveal the chemical reactivity of their P = S group and affinity toward surfaces depending on the solvent-mediated ionization of their thione and thiol functions.245 The hydrophobic tail of collectors is another concerning issue. This aspect was addressed with fatty acid collectors adsorbing on the surface of fluorapatite (0 0 1), specifically in terms of carbon chain length and isomerism and number of C=C bonds.246 Several highlights emerge. First, the strongest interactions with the mineral surface stem from the collectors with the longest chains. Second, the number of C=C double bonds has a negligible impact on the collectors’ reactivity. Third, the isomerism of the collector carbon chains significantly modifies the adsorption energies.246
If polar groups are the crux of flotation collectors, their multiplication in a single collector should contribute to improving the collection capacity, as confirmed by DFT simulations when increasing the number of polar groups of hemimorphite-carboxylic acid collectors.247 A combination of DFT and AIMD simulations further underlined the importance of the number of polar groups in the bastnäsite flotation system using bis- and monophosphinate collectors.171 Indeed, the former would outperform the latter due to overall surface coverage and adsorption interactions.
MD simulations were employed to elucidate the role of secondary amino groups on the adsorption behavior of amine-based reagents on the quartz surface by comparing N-dodecylethylenediamine (ND) and dodecyl amine (DDA) adsorption configurations.248 ND, comprising both primary (−NH2) and secondary (−NH) amino groups in its structure, develops a stronger interaction with quartz as compared to DDA with only one primary amino group. Similarly, another MD simulation249 indicated that bis(2-hydroxyethyl)dodecyl amine (BHDA), a tertiary amine surfactant, has better selectivity toward quartz than DDA due to the weaker polar group electronegativity and stronger steric hindrance of BHDA. The superior adsorption behavior of BHDA compared to DDA was ascribed to the presence of the – CH2CH2OH group in BHDA, increasing the possibility of hydrogen binding with the quartz surface. The interactions of three amine-based collectors, including DDA, N-dodecyliso-propanolamine (DIPA, with one iso-propanol substituent group), and N,N-dodecyl-diiso-propanolamine (DDIPA, with two iso-propanol substituent groups) with quartz and magnesite were also compared within MD framework.250 The presence of isopropanol substituents in the structure of DIPA and DDIPA improves selectivity by enhancing the spatial position barrier effect and increasing the hydrogen binding site on the collector.
The influence of the number of ethylene oxide (EO) groups in the nonionic dodecyl ethoxyl ethers (C12EOn) on the efficiency of low-rank coal flotation was investigated using a combination of DFT and MD calculations.251 The simulation results confirmed the greater selectivity of the surfactant toward coal than the (gangue mineral) kaolinite upon increasing the EO number in C12EOn. Based on a more recent MD study,252 the polarity of a nonionic surfactant was shown to have a substantial role in its selectivity performance during the flotation of low-rank coal and kaolinite.
MD simulations on muscovite (001) by Xu et al.253 demonstrate that the nature of the interaction and the adsorption conformation of primary ammonium ions are not a function of the collector alkyl chain length. This is unlike the carbon chain length in low molecular weight carboxylic collectors where this would affect the adsorption of the collector on muscovite.254 The adsorption energy of carboxylic acid collectors with longer carbon chains (C4) is higher than the (C1, C2, and C3) ones, primarily due to the improvement in carboxylic acid diffusion at the water-muscovite interface.254 DFT simulations on the affinity of carboxylic acid collectors for smithsonite or of xanthate collectors for pyrite demonstrate similar behavior in terms of (alkyl) chain length.255,256
Reagent Mixtures
NaOL/alcohol mixtures by varying the alcohol carbon number have been studied in the flotation of dolomite and magnesite.257 Two key observations emerged from the MD results: first, the mixture of collectors works better than if NaOL acted alone; second, the higher the carbon number of the alcohol, the higher the flotation efficiency. The effect of carbon chain length and isomeric structure of monohydric alcohols was further investigated by DFT for the same system of the magnesite–dolomite system when coadsorbed with DDA.258 By compressing the hydration layer on mineral surfaces and diluting it, the adsorption of mixed collectors with longer alcohol chains is more stabilized on mineral surfaces.
The flotation of quartz with DDA assisted by nonpolar oil in an aqueous pulp was examined using MD simulations.259 The interaction between the hydrophobic alkyl tail of DDA and the nonpolar oil improves the diffusivity of the latter contributing to a greater thickness of the interface. The addition of DDA expands the oil–water interface by spontaneous cross displacement: the oil components by the DDA hydrophobic tail and the water molecules by its hydrophilic headgroup. On the other hand, the performance of quartz flotation with DDA–aromatic hydrocarbon mixtures seems to outperform that with (un)saturated aliphatic hydrocarbons.
A mixture of dodecane/2-octanol and dodecane/ether alcohol reagents were tested by MD simulations for improved performance in coal flotation systems. The results suggested that while low concentrations of 2-octanol and ether frothers would promote synergism in both the spreading and adsorption of dodecane and frothers on the coal surface, the excessive frother concentration would not show a synergistic effect, thus deteriorating dodecane adsorption on the surface.260
3.2.2. Reagents Rational Design
Molecular modeling techniques provide a powerful framework for upgrading common, time-consuming, and costly trial-and-error experimental methodologies for reagent design to a more robust, rational approach for the design and development of new, high-performance reagents for froth flotation. In recent years, the gradual depletion of easy-to-process ores has been accentuated in large part by the shift to mineral sources from complex and highly disseminated ore deposits. Without detailed knowledge of such complex ores, it is almost certain that the beneficiation of target minerals contained therein would be doomed to failure if one were to rely solely on recipes that have worked in the past, such as conventional chemicals and flotation methodologies. Indeed, conventional flotation reagents have shown low efficiency in the flotation of these complex ores since they have been designated for less refractory ore deposits.261,262 It becomes necessary to develop new reagents adapted to the flotation of complex ores but also to the urgency of sustainable development; otherwise, the development of the mining and metals sectors could be seriously compromised in the near future.261,262 Molecular modeling approaches would be a promising pathway toward the discovery and development of these new flotation reagents.10,13−15,121,263−267
Five different azolethione derivatives as novel collectors for copper mineral flotation were evaluated by DFT.268 The heptyl-substituted derivatives demonstrated exceptional recovery for chalcopyrite and malachite due to the chemisorption of azolethione at the Cu site. A new synthetic collector, S-benzoyl-N,N-diethyldithiocarbamate (BEDTC), was compared by DFT to conventional thio collectors, e.g., sodium diethyl dithiocarbamate (SEDTC) and sodium isobutyl xanthate (SIBX), for the selective flotation of galena over sphalerite.269 The DFT simulation predicts a bidentate chemisorption complex involving the BEDTC thiol group sulfur and carbonyl oxygen with the lead surface atoms of galena. The experiments confirmed the superior selectivity of the new synthetic collector as compared to the conventional ones.
Glucan, as a new depressant for the flotation separation of pyrite and pyrophyllite, was the subject of MD simulations.270 Due to its hydrophilic −OH group, the depressant prefers to act differentially with respect to the Al atoms of pyrophyllite and Fe of FeS2.
An AIMD-assisted design has resulted in new hydroxamate-based collectors for the flotation separation of rare earth minerals.271 The surface lanthanides associated with bastnäsite and xenotime form bi- and monodentate surface complexes with the hydroxamate ligands, respectively. Therefore, the emerging strategy for improving the selectivity of, for example, xenotime over gangue minerals is to design a collector in which a single chelating group is replaced by multiple monodentate groups to complement and encompass the simultaneous interactions of the collector with multiple adjacent sites on the surface.
Overall, the application of computational chemistry to the development of new reagents for various flotation systems is a trending topic in the mineral community, as summarized in Table 3.
Table 3. Examples of Molecular Modeling Investigations Focusing on Rational Design of Various Reagents for Mineral Froth Flotation.
| molecular modeling approach | reagent type and application | key findings | ref |
|---|---|---|---|
| DFT (B3LYP/6-3 functional) | polyamine-based cationic collector for goethite and kaolinites system | 1. design and synthesis of novel amine-based collector by DFT following lab-scale verification; 2. selectivity and collecting capability of collectors determined by interaction energy calculation | (272) |
| MD (Universal force field) | sodium hexametaphosphate (SHMP) dispersant for serpentine | 1. anionic components of SHMP adsorbed on Mg and Si surface sites; 2. dispersant interact with serpentine through chemi/physisorption; 3. SHMP dominantly adsorbed on basal plane of serpentine rather than edge surface due to presence of rich Mg atoms with broken bonds | (273) |
| MD (COMPASS force field) | butane-3-heptyloxy-1,2-diamine (BHLD) collector for quartz flotation | 1. electrostatic and hydrogen bond formation recognized as dominant binding mechanism of BHLD to quartz; 2. BHLD cations outperformed BHDL molecule as indicated by higher interaction energy values | (274) |
| DFT (B3LYP functional) | sodium O-benzythioethyl xanthate (SBEX) collector for chalcopyrite flotation | 1. SBEX features higher affinity toward chalcopyrite compared to conventional xanthate sodium isobutyl xanthate (SIBX) and sodium phenylethyl xanthate (SPEX); 2. superior performance of SBEX collector ascribed to presence of thioether structure in structure | (275) |
| DFT (GGA/PW91 functional) | polymaleamide-propyl dithiocarbamate (PMA-PDTC) depressant for galena | 1. study compared interactions and selectivity of DTC (depressant) and O-isopropyl-N-ethyl thionocarbamate (IPETC) (collector) with galena surface; 2. DTC shown to have higher affinity to galena surface; 3. based on DFT calculations, DTC (as galena-avid group) attach to hydrophilic PMA to form a new depressant for galena | (276) |
| DFT (B3LYP-D3 functional) | 2-cyano-N-ethylcarbamoyl acetamide (CEA) collector for fluorite, calcite, and scheelite flotation | 1. CEA collector outperform BHA and fatty acid collectors in mineral flotation due to presence of two functional groups interacting with both cations and anions on mineral surface; 2. based on DFT results, CEA affinity to minerals followed the order fluorite > calcite > scheelite | (277) |
| DFT (GGA/PBEsol functional) | 2-(carbamoylamino) lauric acid (2-CLA) collector for quartz flotation | 1. adsorption energy of main species in the flotation solution follows the order Ca2+ > Ca (OH)+ > OH– > H2O > 2-CLA–, meaning that quartz surfaces should be activated by metal cations or hydroxyl group prior to collector adsorption (especially in the case of 2-CLA–); 2. DFT calculations revealed superior performance of 2-CLA– due to chemical bonding and hydrogen binding abilities of amide group −CONH2 | (278) |
| DFT (GGA/PBE functional) | Evodiae fructus oil (EV-1) collector for fluorite flotation | 1. results suggest oleic acid anion/oleamide anion mixture result in stronger interaction with fluorite surface | (279) |
| DFT (GGA/PBE functional) | Rhodanine-3-acetic acid (3-Rd) as chalcopyrite depressant in molybdenite flotation | 1. DFT results reveal that 3-Rd collector prefers to adsorb on Fe rather than on Cu for chalcopyrite surface; 2. dominant binding mechanism in 3-Rd-chalcopyrite is chemisorption of – COO– and −CSS– groups of 3-Rd bind with Fe sites on chalcopyrite surface; 3. 3-Rd-molybdenite interactions mainly through physisorption via hydrogen bonding and electrostatic interactions | (280) |
| DFT (GGA/PBE functional) | l-cysteine as chalcopyrite depressant in molybdenite flotation | 1. l-cysteine dominantly adsorbed on chalcopyrite surface via chemisorption of −SH and/or −NH2 groups of cysteine with Cu atoms rather than Fe sites on the surface; 2. based on DFT results, formation of l-cysteine dimer is predominant species on chalcopyrite surface | (281) |
| MD (COMPASS force field) | dodecyltrimethylammonium bromide (DTAB) collectors for flotation of low-rank coal | 1. DTAB adsorption on coal surface improves hydrophobicity of coal by excluding water molecules from surface and reducing adsorption thickness; 2. adsorption configuration of DTAB, in which its headgroup faces coal surfaces and alkyl chain is exposed to water phase, is favorable for repelling water molecules | (282) |
| DFT (GGA/PBEsol functional) | ethylenediamine tetra(methylene phosphonic acid) sodium (EDTMPS) as magnesite depressant for quartz reverse flotation | 1. DFT result suggests that preadsorption of EDTMPS weakens DDA-magnesite interactions due to favorable adsorption of EDTMPS on magnesite surface; 2. preadsorption of EDTMPS showed negligible effect of DDA adsorption on quartz surface | (283) |
| DFT (GGA/PBEsol functional) | O-butyl S-(1 chloroethyl)carbonodithioate (GC-1) collector for flotation of copper sulfide ore | 1. DFT results confirm GC-1 interactions with chalcopyrite and pyrite are through chemi/physisorption, respectively; 2. adsorption energy calculations show that GC-1 collector forms more stable and stronger interactions with chalcopyrite compared to butyl xanthate collector | (284) |
| DFT (GGA/PBEsol functional) MD (COMPASS force field) | N-dimethyl-N′-dodecyl-1,3-propanediamine (DPDA) collector for quartz flotation in hematite–quartz systems | 1. DFT and MD simulation results confirm DPDA has better collectivity and selectivity toward quartz than conventional amine-based collectors (e.g., DDA); 2. DPDA-quartz interactions predominantly tuned by electrostatic attraction and hydrogen bonding | (285) |
| DFT (GGA/PBE functional) | Cupferron collector for cassiterite flotation in cassiterite–quartz systems | 1. DFT results confirm selective adsorption of Cupferron on cassiterite surface than quartz before activating surface by Pb2+; 2. Pb2+ activation shown to be more energetically favorable on cassiterite than quartz surface; 3. Interaction study further suggests that Pb2+ activation promotes Cupferron adsorption on hydrated quartz surface, and Cupferron interacts slightly more stronger with activated quartz than with cassiterite surface; 4. based on simulation results, for efficient separation of cassiterite from quartz, dosage of Pb2+ should be strictly controlled to prevent quartz activation | (286) |
| DFT (GGA/BFGS functional) | N-(carboxymethyl)-N-tetradecylglycine (NCNT) collector for fluorapatite flotation | 1. Based on DFT study, NCNT has stronger interaction with fluorapatite than oleic acid collector; 2. adsorption configurations suggest that NCNT adsorption mechanism on fluorapatite is through bidentate covalent bond with surface-exposed Ca atoms | (174) |
3.2.3. Rational Design of Novel Ecofriendly Collectors
Peptide-Based Collectors
Molecular modeling approaches have been put forward to develop a new family of environmentally friendly reagents by rationally tuning peptides for application in froth flotation as novel green collectors.49,51,287−290 In one of the earliest efforts to model peptide–mineral interactions, DFT calculations were utilized to determine the interaction between acetic acid and methylamine on clay mineral surfaces.291 The study revealed that the formation of the clay mineral-peptide bond occurs via an asynchronous mechanism by initiating a dative N–C bond, followed by proton transfer from N to O atoms. Another AIMD-MD study investigated the adsorption mode and mechanism of alanine and two dipeptides, including alanine-glutamic acid and alanine-lysine, on the rutile (110) surface.292 Chemisorption involves a bidentate complex by the two carboxyl O atoms of the amino acids with two adjacent Ti atoms on the rutile surface.292 Similarly, molecular modeling studies on the interaction nature of various peptide structures with minerals and metal surfaces, including gold,210,293,294 palladium,293 calcium oxalate monohydrate,154 platinum,295 hydroxyapatite,296,297 and rutile46 further confirmed that peptide-mineral interactions would predominantly occur as chemisorption. Lately, the promising potential of peptides as collectors in the flotation of sulfidic minerals was scrutinized through systematic molecular modeling studies on the amino acids, as the peptides’ building blocks. The combination of DFT calculation and Monte Carlo simulations has allowed examining the adsorption mechanism of alanine on the sphalerite surface.49,51 The interaction of all 20 natural amino acids on pyrite, chalcopyrite, and sphalerite surfaces has been studied recently.288 PDOS and Hirshfeld population charge analyses, site density calculations of the mineral surfaces, HOMO and LUMO distributions, and interaction energies were used to rank the amino acids against these model sulfides over a wide pH range. The affinity of pyrite toward amino acids is higher than that of chalcopyrite and sphalerite, with in all cases, the amino acids being adsorbed by chemisorption. The amino acid acid–base speciation as a function of pH also plays a role in their level of interaction with the sulfide surfaces. Very recently, a robust simulation scheme combining classical MD, replica exchange MD, and steered MD was synergized to characterize the quartz-selective peptide sequences initially identified by phage display at pH 9 as alternative reagents for direct flotation of quartz mineral.186 The simulation results revealed that the simultaneous presence of positively and negatively charged residues in peptide sequences are favorable for stronger binding of peptides to the surface due to two main reasons. First, the negatively charged residues have strong affinities toward abundant positively charged moieties on quartz surface at basic pH, and second, the repulsive self-interactions in the peptide chain containing dissimilarly charged residues keep the extended conformation of the peptide, guaranteeing the accessibility of peptide side groups to the surface. The investigation further highlighted the capability of molecular modeling to study mechanistic details and to accelerate the rational design of novel reagents for mineral processing applications.
Ionic Liquid-Based Collectors
The extension of ionic liquids (ILs) has not gone unnoticed by the molecular modeling field, which has been sharpened by the interest in new “green” collectors in minerals’ flotation. Although traditionally considered “designer solvents,” ILs have recently found applications in froth flotation168,298−300 because of virtually limitless possibilities for combining cationic and anionic moieties,301 allowing them to tailor a new range of collectors. The potential application of quaternary ammonium-based ILs (C4–C12 NR4+) as flotation collectors would suggest, according to MD simulations, that the adsorption mechanism on the surface (101) is mainly controlled by electrostatic and van der Waals forces. A more localized look by DFT on the interactions of phosphonium/ammonium-based ILs with semisoluble minerals such as calcite, bastnäsite, and monazite allowed researchers to differentiate the roles of the anions and cations associated with these ILs: if the first one adsorbs directly on the metallic active centers of the minerals, the second one is rather involved in weak solvophobic interactions.302,303
3.3. Properties of Mineral Crystals and Surfaces in Flotation
Interfacial phenomena at the gas–solid–liquid interfaces control the separation efficiency, but where the mineral solid phase plays an essential role in the flotation process.1,9,166,304 In contrast to fluid mechanics of amorphous media, the success of molecular modeling approaches owes much to solid-state physics, which has made possible the calculation of crystalline and surface properties of minerals useful for mineral flotation.10,15−17,121,264 This section shows how molecular modeling approaches can describe the inherent properties of mineral structures and compositions in terms of mineral surface reactivity and then floatability.
3.3.1. Mineral Surfaces: Structure–Property Relationships
Molecular modeling can provide atomic-scale information on mineral surfaces and crystal structures. In an early attempt, Kundu et al.305 conducted DFT calculations to study the electronic properties of seven different wollastonite cleavage planes. Surface energy calculations revealed that the (100) plane is the most energetically favorable surface in wollastonite. Similarly, Kundu et al.227 carried out the same procedure to recognize the lowest energy cleavage planes of magnesite and reported that the (110) and (111) directions are the predominant growth planes for magnesite.
DFT was utilized to elucidate the surface reconstruction and relaxation of chalcopyrite surfaces with different terminations.306,307 Three mechanisms were captured: (1) surfaces with metal-exposed atoms form several metal–metal bonds, (2) those directions with relatively close sulfur atoms with lower coordination numbers form an S–S bond on the outermost layer, and (3) in some directions (e.g., (101), (110), and (112)), only a relaxation of metal atoms occurs by moving the metal atoms downward. The three DFT-reconstruction mechanisms of chalcopyrite surfaces are illustrated in Figure 7.240 The different surface speciations would lead to significant variations in the chemical reactivity of chalcopyrite surfaces in terms of mineral-collector interactions.
Figure 7.

Schematic of surface reconstruction evolution of different chalcopyrite surfaces: (a) metal-exposed (100) surface, (b) sulfur-exposed (100) surface, and (c) (110) surface (Fe: purple, Cu: red, S: yellow). Adapted with permission from ref (240). Copyright 2017 Elsevier.
DFT calculations were employed to disclose more information about the surface reactivity of bastnäsite, considering its complex surface chemistry behavior in flotation.298,308 DFT was able to successfully capture the reconstruction and hydration behavior of different cleavage directions (as suggested by Wulff’s construction theory) in aqueous media as a crucial step to explain the complex flotation behavior of bastnäsite. Molecular modeling has provided insight into the flotation behavior observed with aluminosilicates (e.g., spodumene and albite) by specifying the directional reactivities of the minerals’ surfaces.304 The surface energy calculated by DFT was related to the cleavage properties of spodumene and albite and their impact on the reactivity of their surface atoms as well as their flotation behavior.309 The broken bond density was used to account for the differences in reactivity of different cleavage directions. As an example, the number of broken bonds on different cleavage planes of feldspar was determined after surface optimization (Figure 8).310 Three types of broken bonds, including Si–O, Na–O, and Al–O, are present on the various cleaved surfaces but differ in number on each surface. The feldspar (110), (001), and (110) cleaved planes have 4 (Figure 8a), 8 (Figure 8b), and 10 (Figure 8c) broken bonds, respectively. Al atoms, as the most favorable sites for chemisorption of the collectors on feldspar,311,312 are more abundant on the (001) and (110) surfaces. Similar work has been done on fluorite and spodumene.313−315 This information could be put to good use by controlling the grinding conditions to generate mineral particles that favor particular reactive cleavage planes.
Figure 8.

Schematic illustration of the broken bonds presents on different crystal planes of feldspar: (a) (110), (b) (001), and (c) (100). (O: red, Si: yellow, Al: pink, Na: purple). Adapted with permission from ref (310). Copyright 2017 Elsevier.
In the case of the bastnäsite surface, bond population analysis by DFT calculations showed that the C–O bonds in the mineral structure feature covalent bonds, while the Ce–O and Ce–F consist of strong ionic bonds. Consequently, Ce–O and Ce–F bonds are more prone to be ruptured from the bastnäsite cleaved planes, leaving ≡Ce3+, ≡CO32-, and ≡F– dangling bonds to interact with water molecules in the solution, thus resulting in high hydrophilicity of bastnäsite.298,316
DFT calculations were carried out to characterize the hydrophilicity level of coal as a function of surface functional groups.317 X-ray photoelectron spectroscopy (XPS) of the samples revealed the existence of four oxygen-containing functional groups, namely, −OH, C–O, C=O, and −COOH, on the coal surface. The hydrogen binding energy between these functional groups with water molecules was DFT-calculated to be in the order −COOH > −OH > −O– > C=O, where more hydrophilic −COOH groups form two hydrogen bonds. The observations suggest that the complexity of lignite flotation is predominately due to the availability of hydrophilic oxygen-containing function groups on its surfaces. Shen et al.318 investigated the effect of surface porosity on the flotation of low-rank coal using the MD framework. The results showed that surface porosity would stabilize the hydration layer on the coal surface, thus disrupting bubbles-surface attachment. On the contrary, the reduction in pore content would lead to a decrease in water-surface interactions and would possibly facilitate the expulsion of water molecules from the surface by bubbles. The coalification degree of low-rank coal, and thus its wettability, was qualified by MD simulations as being controlled by the amount of hydrogen binding between water molecules and coal surfaces.319 These atomic/molecular level insights into the wettability of coal surfaces provide significant guidance toward improving the flotation efficiency of coal slime.
3.3.2. Effect of Surface Vacancy on Mineral Surface Properties
Real minerals almost always contain defects in the form of impurities or vacancies in their crystal lattice or surfaces. Quantum-based molecular modeling techniques are able to describe the electronic properties of deficient mineral surfaces.49−51,69,175,320−328 The role of vacancies on the wettability of sphalerite depends on the type of surface vacancies (Zn or S) as demonstrated by DFT calculations integrated with the GCMC method on the (110) cleavage plane of sphalerite (Figure 9).49−51 The S (Zn) vacancy produces a hydrophobic (hydrophilic) region around the deficient site, thereby potentially changing the dominant adsorption pattern of a reagent on the surface.49 Nonstochiometric sites on the pyrite surface would also be involved in the chemical evolution of glycine from zwitterionic form to an ionic species, according to a DFT study.325
Figure 9.

Optimized structures of ZnS (110) slab with (a) no defect, (b) single sulfur vacancy defect, (c) single zinc vacancy defect, and (d) with double congruent vacancies (Zn: purple and S: yellow). Adapted with permission from ref (49). Copyright 2021 Elsevier.
Atomistic details about the evolution of O2 by DFT calculations captured distinct dominant adsorption configurations on defect-free and defective pyrite surfaces.322 While dioxygen dissociative chemisorption on sulfur atoms was more favorable than on iron atoms for a defect-free surface, molecular oxygen adsorption on sulfur vacancies predominantly took place on defective pyrite surfaces. More recently, a DFT study further confirmed that the dissociative adsorption of water on the sulfur-deficient pyrite surface is less favorable.329 Zn-vacancies repressed water adsorption on the smithsonite surface;327 i.e., pure molecular adsorption on the Zn-deficient surface replaced the energetically more favorable dissociative adsorption of H2O on the defect-free surface.
3.3.3. Effect of Lattice Impurities on Mineral Properties
The presence of impurities in the natural mineral crystal lattice is another common phenomenon modulating reagent adsorption in froth flotation, chiefly in the case of solid solutions.330−332 In this regard, Chen et al.333 conducted DFT calculations to determine the effect of lattice impurities on the electronic structures and floatability of sphalerite. Their results indicated that the lattice parameter, semiconductor type, Fermi level, and density of state of sphalerite can significantly vary in the presence of various elemental impurities such as Cd, Hg, Ga, Ge, In, Ag, Sn, Pb, and Sb. Furthermore, the HOMO/LUMO analysis shows that the impurities would even alter the electrophilicity of the sphalerite surface, so that the presence of Mn, Fe, Co, Ni, Cu, Hg, and Pb would increase the affinity of the sphalerite toward the xanthate collector, thus improving its flotation recovery. The sensitivity to oxygen and xanthate during flotation of pyrite when it contains impurities such as As, Se, Te, Co, or Ni can be well understood in light of a DFT calculation.334 By means of frontier orbitals, while the tendency of pyrite is to oxidize when it contains As, Co, or Ni compared to Se and Te, it has a greater affinity for xanthate in the presence of Co or Ni. DFT calculations further confirmed the possible occurrence of Au in the structure of pyrite and captured the changes in electronic properties and reactivity of pyrite, as already reported by experiments.335,336
The influence of impurity incorporation on minerals’ wettability and adsorption properties was also studied through MD simulations. For instance, using the sessile drop contact angle measurement concept, it was revealed that replacing Si4+ with Al3+ in the talc crystal structure can reduce talc’s natural hydrophobicity.337 MD simulations were utilized to compare the swelling pattern of Na- and Ca-montmorillonites and suggested that Ca-montmorillonites have a stronger hydration swelling than Na-montmorillonites, most probably due to a larger hydration radius of Ca2+.338 The isomorphous substitution effect of Fe, Cr, and Mn for Al and Mn for Si in the spodumene structure was investigated with respect to the wetting characteristics and NaOL adsorption using DFT and MD simulations.339 The results showed that spodumene would keep its natural hydrophilicity with and without elemental substitution; however, such impurities would promote NaOL adsorption on the surface. Based on DFT calculations, the surface with Mn substituting for Si showed the highest affinity toward NaOL. The molecular-scale insights would provide beneficial information for interpreting the complex flotation of spodumene as a solid-solution mineral occurring in pegmatite ores.340
3.3.4. Effect of Mineral–Mineral Close Contacts on Mineral Properties
Galvanic Contacts in Sulfidic Minerals
In addition to impurities in the crystal structure, mineral–mineral close contacts encompass changes in mineral reactivities and electronic properties. In this regard, galvanic coupling in sulfide–sulfide systems would significantly affect the floatability of sulfide minerals.341−343 DFT was implemented to determine the galvanic interactions between pyrite and galena surfaces, as well as the xanthate electrochemical adsorption on galena.344,345 Galvanic coupling prompts electron transfer from galena to pyrite, as a result, the collector-surface adsorption modes would differ from the single mineral-collector complexes. DFT calculations revealed that pyrite–arsenopyrite contacts decrease the band gap at the interface, thus facilitating electron transfer and curbing the pyrite oxidation rate.346
Nonsulfide Minerals Contacts
DFT calculations were implemented to shed insights into the coating mechanism of kaolinite on low-rank coal.347 The results divulged that van der Waals attraction between low-rank coal and the kaolinite (001) surface causes kaolinite coating on low-rank coal flotation. The finding is in line with the results reported by Chen et al.348 for the kaolinite interactions with fine coal particles.
3.4. Mineral Surface Modification and Activation in Flotation
Mineral surface modification and activation are critical subjects in mineral flotation, routinely used to promote mineral-reagent interactions. Surface alterations may also occur by some uncontrolled phenomena (e.g., surface precipitation and dissolution) within the process, sometimes resulting in a reduction of flotation recovery.1,166 Molecular modeling techniques, however, are capable of evaluating mineral-reagent interactions, foreseeing surface speciations, and assessing mineral surface reactivity as part and parcel of tackling the challenges of froth flotation.
3.4.1. Surface Activation by Metal Cations
Metal cations and metal complex species, intervening deliberately or not on minerals’ surfaces, alter the surface properties via surface-reagent interactions. Precipitation of Ca(OH)+ and Ca(OH)2 hydroxide species on the spodumene surface boosts surface activation and mineral-reagent interactions, according to a DFT study.349 Ca atoms of the hydroxide species form covalent bonds with O atoms on the mineral surface, yielding new active sites on the spodumene (110) surface. DFT-simulated Ca(OH)+ are shown to act as a bridge for α-bromolauric acid (α-BLA–) anionic collectors to interact with quartz (101) surfaces,350 while α-BLA– is unable to adsorb on the quartz surface due to a shielding hydration shell. Quartz surface activation with Ca2+ or Ca(OH)+ repels water molecules from the surface, bridging α-BLA– to it and leading to a significant promotion of quartz floatability. DFT calculations further revealed that the preactivation of hydroxylated quartz surface with Ca2+ introduces favorable active sites for subsequent adsorption of sodium oleate, making direct flotation of quartz industrially feasible.351
MD simulations based on the Universal force field parameters and Mulliken partial charges were conducted to assess the hydrophilicity of Cu2+ activated sphalerite surface.352 The wettability of the pristine and the Cu2+-activated sphalerite was compared by calculating the contact angle, water number density distribution, water dipole orientation, and water residence time, among other things. Although using a generic force field combined with Mulliken partial charges should be treated cautiously, the MD results confirmed that activating the sphalerite surface with Cu2+ increases the mineral hydrophobicity due to decreased surface polarity. The different hydrophobic characteristic between the naked and the Cu-activated sphalerite was confirmed by DFT calculations.48
A DFT study of the activation of stibnite (010) surface by Pb2+ indicated that lead cation cannot substitute to lattice Sb3+ on the stibnite surface;353 Pb-surface interactions of covalent nature being more likely. Five energetically favorable Pb2+-adsorption configurations enable activating as many adsorptions for xanthate on the stibnite (010) surface (Figure 10). Adsorption energy calculations confirmed that xanthate adsorption on the Pb-activated stibnite surface was more favorable than on the inactivated surface. Tian et al.354 discerned the importance of sequential activation for the sake of higher flotation efficiencies for oxide minerals. Their DFT calculations confirmed that the complexation of benzohydroxamic acid (BHA) collector with Pb2+ hydroxide species prior to its introduction in the flotation solution is remarkably more energetically favorable than activating mineral surface (cassiterite in this case) in the first place and using BHA afterward. A similar improving mechanism was also reported by DFT calculations for the complexation of BHA with Fe (III) for the flotation of cassiterite mineral.355
Figure 10.

Stibnite (010) surface activation by Pb2+ at five different possible surface sites as well as butyl xanthate collector interaction with activated surfaces at these five Pb-activated sites. Adapted with permission from ref (353). Copyright 2018 Elsevier.
The adsorption of different alkali and earth-alkali cations on the spodumene surface can be followed by MD/DFT integrated surface charge simulations to change the reactivity of the mineral surface with respect to surfactants.356 Below the zero-charge point, adsorption of anionic collectors occurs primarily via electrostatic interactions with the abundant cations on the spodumene surface. However, above the zero-charge point, the anionic collectors continue to adsorb onto the spodumene cationic sites albeit with lesser numbers on the surface, leading to more negative potential ζ values.
3.4.2. Surface Sulfidization and Oxidation
Surface Sulfidization
Surface sulfidization is a surface modification approach in froth flotation to promote recovery, especially in the case of oxide minerals.357 A density functional based tight binding (DFTB+) study was performed to clarify the sulfidization-amine flotation mechanism of the smithsonite surface.358 The study demonstrated two different mechanisms based on the concentration of hydrosulfide (HS–) on the surface: (1) per lower concentrations of HS–, Zn-SH-SH structure is formed on the surface, preventing the N atom of amine collectors from interacting with Zn on the surface but rather with the S atom in the HS–, and (2) per higher HS– concentrations, ZnS is formed on the surface, promoting the adsorption of amine collectors on the Zn atoms of the smithsonite surface. In another study, DFT calculations of cerussite (PbCO3) surface sulfidization revealed that HS– ions can readily adsorb onto the surface through hybridization of S atom of HS– and the surface-exposed Pb atoms on the PbCO3 surface.359 DFTB+ calculations of the sulfidization of the same PbCO3 mineral suggest that Pb2+ on the activated surface interacts more readily with xanthate but less with water, based on the adsorption energy values.360 Surface sulfidization, however, is not always favorable for collector adsorption. For instance, a DFT study showed that chalcopyrite and galena surface sulfidization by Na2S prevents the adsorption of xanthate collectors on the mineral surfaces and thus reduces flotation recovery.361 The higher interaction of sodium sulfide with the chalcopyrite and galena surfaces was introduced as an indication of a significant reagent-removal effect of Na2S in the chalcopyrite-xanthate and galena-xanthate systems.
Surface Oxidation
Surface oxidations (i.e., surface–oxygen interaction) would strongly modify the electronic properties of mineral surfaces, leading to a different adsorption behavior of collectors.47,322,362,363 Liu et al.364 applied DFT to determine how O2 adsorption alters the reactivity of gold (211) surface. A transition state study captured four possible pathways for surface activations of the gold surface, all give almost equivalent free energy barriers in an aqueous environment, indicating the substantial role of surface H and H2O coverage on the reaction mechanism. Another DFT study implied that the adsorption of water molecules on galena (100) favorably occurs upon coadsorption with O2 or after galena surface oxidation.365
3.4.3. Surface Precipitation of Solution Components
The presence of a whole “fauna” of ions in the real solutions of flotation pulps makes the controlled solutions studied in the laboratory more the exception than the norm. These species can precipitate on the mineral surfaces, altering their surface properties and, ultimately, the flotation behavior of the minerals. DFT/experimental studies have shown that the presence in the flotation solution of species from gangue leaching (e.g., calcite, dolomite) present together with the rare earth minerals would spoil the efficiency of the hydroxamic collectors through complex formation, negatively affecting the flotation recovery of rare earth (RE) minerals.366,367 Specifically, the DFT results revealed two dominant decaying mechanisms for the lower floatability of RE-minerals in these complex systems: (1) adsorption energy calculation confirmed that the complexation of dolomite supernatant species with the hydroxamic collector is energetically favorable, causing the depletion of free collectors in the solution and, subsequently, the reduction of collector adsorption on the target minerals, and 2) dissolved dolomite and calcite species, including Ca(OH)2, Mg(OH)2, CaCO3, and MgCO3, can readily be precipitated on the RE-minerals’ surfaces (through chemisorption), reducing the surface affinity toward hydroxamic acid collectors by preventing the collector from directly interacting with the mineral surface (Figure 11). As seen in Figure 11, ion surface precipitation would critically change the adsorption mode of hydroxamic acid on the RE-mineral: bidentate adsorption of hydroxamic acid on rare-earth atoms on the monazite surface would change to monodentate adsorption with Ca when CaCO3 has been priorly precipitated on the surface. The adsorption energy of the former configuration was calculated to be more than two times lower than the latter one.
Figure 11.

Interaction of benzohydroxamic collector with (a) directly monazite (100) surface and (b) CaCO3 species precipitated on monazite (100) surface, (Ce: beige, O: red, P: purple, C: gray, Mg: orange, H; white, Ca; green, N: blue). Adapted with permission from ref (367). Copyright 2018 Elsevier.
3.5. Minerals/Water/Gas Interfaces in Flotation
The froth flotation process is a three-phase system bounded by up to as many interfaces (Figure 2). In this section, we will discuss complementary aspects of molecular modeling techniques to those covered in the previous sections but with an emphasis on the interfaces themselves.
3.5.1. Mineral/Water Interface
Mineral Surface Electronic Properties and Reagent Adsorption
The electronic properties and reactivity of minerals’ surfaces can readily be altered in an aqueous environment, leading to different adsorption properties compared to dry surfaces.50,73−75,173,363−365,368−372 For instance, DFT calculations showed that the water hydration layer on the smithsonite weakens the hybridization of s and p orbitals and lowers the electrophilicity of Zn atoms on the outermost mineral layer, subsequently reducing the interaction levels of smithsonite with carboxyl collectors.255 As further confirmed by DFT, hydration of sphalerite significantly alters the mineral surface properties through localization of Zn 3d, 4s, and 4p orbitals, resulting in a reduction in the reactivity of Zn atoms and, ultimately, the Zn-xanthate collector interactions.373 The multilayer hydration of the hydrophobic galena and hydrophilic pyrite surfaces via DFT calculations captured different dominant interaction mechanisms for each surface; while water-surface interactions in hydrophobic surfaces mainly occur through hydrogen bonds, covalent bonding between surface-exposed metal atoms and oxygens of water molecules dominantly occurs for hydrophilic surfaces.372
Ai et al.234 used MD simulations to compare the flotation separation of KCl and NaCl in the presence of a hydration layer using the octadecylamine (ODA) collector. The ODA collector is able to cross the hydration layer to adsorb on the KCl surface. This is due to the adsorption energy of ODA/KCl being greater than the cohesive energy between water molecules in the hydration layer. On the contrary, the cohesive energy in the hydration layer is higher than that of the ODA/NaCl, preventing the collector from interacting with the NaCl surface. Thus, only KCl would be collected from the potash ores upon addition of ODA collectors.
DFT-simulated adsorption of BHA on the hydrated cassiterite (110) surface demonstrated that direct adsorption of BHA on the tin sites of the pristine surface is not favorable.374 However, activation of the cassiterite surface by Pb2+ was shown to assist in the selective adsorption of BHA by allowing the departure of two water molecules from the lead ion coordination shell. Other studies confirmed that Pb2+ activation of cassiterite,374 and smithsonite surface sulfidization by means of HS– and S2– would repel the hydration-layer water molecules,375 facilitating collector uptake by the mineral surface. A DFT study on the adsorption of alanine on the ZnS surface further suggested that solvated alanine cannot be chemically adsorbed on the defect-free ZnS surface, while surface sulfur vacancy prompts alanine adsorption on the ZnS surface through the formation of monodentate bond with the undercoordinated Zn atoms around the vacancy site.51 Alteration of the adsorption modes, from chemisorption in the vacuum to physisorption in the presence of water molecules, was also reported in the case of sodium dimethyl dithiocarbamate (SDD) and butyl xanthate (BX) collectors adsorption on the sphalerite (110) surface.368 All of these examples emphasize the need to consider water molecules to reflect true interactions in modeling flotation systems.
Mineral Surface Wettability
The microscopic wettability characteristics of a surface are mainly inferred from contact angle determinations using MD simulations.216,313,376−381 For an accurate prediction of the contact angle, the vapor–liquid border on the outer surface of the water droplet must be carefully recognized (see section 2.2.1 for explanations and references therein). The literature review points to a gap in MD simulations of the water-vapor boundary and determination of the contact angle on mineral surfaces of interest for flotation. The degree of hydrophobicity of the bastnäsite surface was simulated by MD from the viewpoint of a change in the concentration of hydroxamate collectors.382 Snapshots of the simulation after introducing a water droplet on the bastnäsite surface show this concentration effect for octyl hydroxamate: 8.3% (Figure 12a), 25% (Figure 12b), and 50% (Figure 12c). The contact angle or, in other words, the hydrophobicity of the surface increases with the concentration of the collector.
Figure 12.

MD simulation snapshot of the initial configuration (0 ps) and the configuration after 40 ps for a water droplet on the bastnäsite surface with different levels of octyl hydroxamate coverage: (a) 8.3% hydroxamate coverage, (b) 25% hydroxamate coverage, and (c) 50% hydroxamate coverage (red: oxygen; blue: cerium; green: fluorine; cyan: carbon; white: hydrogen, pink: hydroxamate oxygen; gray: nitrogen). Adapted with permission from ref (382). Copyright 2014 Elsevier.
As an alternative to estimating the contact angle, the wettability of a mineral surface can be assessed qualitatively by means of the hydration energy, hydration shell, orientation, degree of segregation of water molecules, and radial distribution of water molecules on the surface.308,383−389 Combining DFT with Wulff’s construction theory for the calculation of water adsorption energy demonstrates that a Ca-bearing bastnäsite surface is more hydrophilic than the same surface but bearing La.384
3.5.2. Water/Gas Interface
Frother at the Interface
Chong et al.390 employed MD simulations to simulate the frothers/air/oily saltwater interface with three surfactant frothers, namely, methyl isobutyl carbinol (MIBC), terpineol, and ethyl glycol butyl ether (EGBE), through a binary combination of liquid–air and liquid–liquid interfaces. The MIBC, terpineol, and EGBE molecules were representative of the alcohol, cyclic alcohol, and glycol ether chemical classes of common flotation frothers. The surface tension calculation at the frother/air interfaces provided good agreement with experimental results for MIBC and terpineol. While for EGBE, the value was lower than experimentally measured, possibly due to the reduced charges assigned to EGBE molecules. The authors further extended the work to study the effect of frother on saltwater/air interfaces.391 Simulation results indicate that the polar alcohol groups of the frother molecules orient themselves to adsorb onto the saltwater. Surface tensions were found to decrease upon the addition of MIBC and terpineol, while for the EGBE, comparatively small changes in surface tensions were observed.
Collector at the Interface
MD simulations were utilized to better understand the adsorption and packing of octadecylamine hydrochloride (ODA) collector molecules at the air/water interface in the presence of various ions in the solution.392 Some anions (e.g., I– and SO42–) are shown to penetrate the ODA monolayer at the air/water interface due to their larger size and greater polarizability and disrupt the arrangement of the ODA molecule. Furthermore, the interactions between the anions and the -NH3 headgroup of the ODA revealed that the SO42– anion has a denser distribution around ODA, while no anion layer for NO3– and SCN– was observed. Wang et al.393 investigated the surface activity and self-aggregation behavior of DDA and four alcohol mixtures at the water/air interface using MD simulations. Tighter surfactant aggregation takes place at the water/air interface for longer alkyl chain alcohols. Comparison of the radial distribution function (RDF) between the DDA and alcohol headgroups pairs revealed that the alcohol positions between the DDA headgroups leading to an increased rigidity of the surfactants’ adsorbed monolayer. The new configuration reduces the surface tension of water, confirming the auxiliary role of alcohols in DDA/alcohol mixture.
Effect of External Electric Field
The MD method has also demonstrated its ability to simulate the effect of an external electric field on water/mineral/gas interfaces.394−397 Air flotation of hydrophobic particles immersed in saline solutions with different concentrations of NaCl and in the presence of an electric field396 shows that the latter would deteriorate the adhesion of bubbles to hydrophobic particles. The application of an external electric field could also lead to a reduction in the size of the bubbles, thus promoting better attachment of hydrophobic particles during flotation.395 Furthermore, the application of an electric field can weaken the restriction of the aqueous solvent on the surfactant (e.g., dodecyl trimethylammonium chloride, DTAC), reduce the surface tension, inhibit the coalescence of small bubbles, and thus promote the adsorption of DTAC at the gas–liquid interface.397 Controlling the direction of the electric field would also manipulate the distribution of the DTAC surfactant helping to improve the stability and adsorption capacity of the bubbles.
3.5.3. Mineral/Gas Interface
The mineral-air bubble attachment is the main objective that must be achieved for an efficient flotation process (Figure 2).17 Scrutiny of the dynamics of bubble attachment at the molecular scale would be an undeniable asset to the scope of MD simulations. The mechanism of nitrogen nanobubble attachment on molybdenite and quartz surfaces has been recently unraveled.398 The chain of events on a hydrophobic surface unfolds through thinning the water film on the mineral surface, rupturing the thinned water film, film displacement, and, finally, direct bubble-surface interactions. Water film rupture does not occur at the (hydrophilic) quartz surface, keeping a stable hydration layer (Figure 13a). Film thinning and breakup are clearly reproduced by simulation on the molybdenite face surface (Figure 13d), forming a hemispherical gas bubble at the interface. The nitrogen bubble retains its spherical shape for zigzag- and armchair-edged molybdenite surfaces (Figure 13c, b), while thinning and rupture of the film occur to a lesser degree. These surfaces show a more modest hydrophobic character compared to the molybdenite face surface. Specifically, analysis of the interfacial water region captures a “water exclusion zone” at the hydrophobic molybdenite face surfaces, promoting spontaneous attachment of bubbles to the surface by attractive van der Waals forces. This exclusion zone is relatively smaller on the zigzag-edge and armchair-edge molybdenite surfaces, restricting surface interactions with gas molecules.
Figure 13.

Thin sections of initial and final states (after 1 ns) of nitrogen bubbles at (a) quartz (001), (b) molybdenite zigzag-edge, (c) molybdenite armchair-edge, and (d) molybdenite face surfaces, adopted from ref (398).
Implementing the same approach to study the thinning and breakage of the wetting film during bubble attachment on charcoal surfaces with different degrees of coalifaction demonstrates a higher rate of bubble attachment on surfaces with higher hydrophobicity.399 During film thinning, the decay rate of hydrogen bond number is the fastest on the bituminous coal (BC) surface and the slowest on the low-rank coal (LRC) surface.
4. Summary, Outlook, and Recommendations
This review of molecular modeling applied to flotation attests to the extent to which this tool contributes to advancing fundamental knowledge in mineral processing. Perhaps if Fuerstenau, Jameson, and Yoon were to write a new edition of their book “Froth Flotation: A Century of Innovation,”1 they would endorse the important role of molecular modeling approaches in the ongoing rational design of flotation chemicals. In order for this review to be useful, we felt it was necessary to highlight directions in the study of flotation using molecular modeling for researchers who are actively using this tool in their flotation studies or for those new to the research field who have begun to use this tool. Hence, the first part (section 2) aimed to review the key principles of a wide range of molecular modeling approaches, whereas emerging trends of their applications to the froth flotation process were the subject of the second part (section 3).
Considering the promising avenue depicted in this review, it can be concluded that molecular modeling techniques are applicable to support or refute hypotheses that are difficult to prove through experimentation alone. Thus, this tool must be seen for what it is, i.e. to complement the analytical and experimental work on froth flotation. As highlighted throughout this review, molecular modeling techniques are capable of providing scientists with first-hand atomic information at solid–liquid, solid–gas, and liquid–gas interfaces. The quest for the Holy Grail in the application of molecular modeling simulations is to build a rational framework to (i) describe the reactivity of the mineral surface and its interactions with different classes of flotation reagents and (ii) use the accumulated knowledge to design suitable reagents based on the mimicry of the phenomena occurring at the atomic scale at mineral/water/gas interfaces. However, the authors caution researchers to carefully study the theory and technical details before embarking on hands-on practice and to consult the available references in other disciplines to familiarize themselves with standard implementation and analysis approaches. This comprehensive review shows remarkable excitement about the application and implementation of molecular modeling in the mineral processing community. However, the road remains somewhat bumpy in some respects; for example, the majority of published work on water contact angle evaluation has neglected the basic statistical concepts of molecular modeling.
The following is a collection of key recommendations for future directions of molecular studies in the field of froth flotation:
The next step would be to address the dynamic behaviors occurring during flotation. The reagent adsorption and surface hydration are dynamic behaviors, meaning that the adsorption could occur with different molecular configurations and the effect of temperature is nontrivial. Adding temperature to the simulation would decrease the energy barrier between multiple minima, most probably leading to different stable adsorption configurations overlooked by geometry optimizations at 0 K. Dynamic simulations allow us to explain the reagent solubility, the (self)diffusion of the adsorbates, the orientational parameters, the configurational transition, and the phenomenological and mechanistic details of the adsorption phenomena in the process. The competitive adsorptions of adsorbates (i.e., reagents, water, and solution ions) are also trackable with dynamic simulations. Hereupon, investigating dynamic properties is inevitable for the sake of more realistic conclusions.
Extra attention must be given to developing accurate force fields for mineral systems. The scarcity of concerted force fields, particularly for minerals with transition metals, is the most critical bottleneck in the application of MD simulations in flotation studies. More recently, the INTERFACE force field has been developed for several minerals, but the development remains a work in progress for the large portfolios of mineral classes.
A key step toward the reliability of atomistic models of flotation systems is the accurate definition of solution environments and mineral surface complexations in contact under different pH levels and ionic strengths. To this end, systematic attempts comprising coupled experiments and ab initio calculations for various minerals at different pH must be pursued to (i) build realistic surface models that are essential for predictive simulations and (ii) find entry into molecular models to resolve the shortcomings of conventional force fields.
Despite the lack of accurate force fields, AIMD and QM/MM techniques have demonstrated descriptive power to simulate dynamic behaviors in mineral systems. AIMD simulations still cannot afford a swarm of water molecules required for appropriate solvation of reagents; however, QM/MM is capable of modeling larger-size systems, where the solid/reagent/water are taken care of with quantum calculations, and beyond that, molecular mechanics plays a role. The applications of these two methods are still relatively unknown; attention to these dynamic processes would benefit from being studied in order to fertilize and enrich future research.
Rather than using commercially available simulation software, open-source packages should be further employed to perform MD simulations at higher length scales along with more prolonged simulation time to explore the dynamics of the system thoroughly.
More attention must be paid to important flotation topics that remain unexplored by molecular modeling tools. For example, the number of studies on the characteristics of water/mineral/gas interfaces (in particular, the gas–mineral interface) remains limited, while the accumulated knowledge could be beneficial in the rational design of flotation reagents.
The environmental risks of flotation have been critically brought to the fore by pointing fingers at the broad portfolio of nonbenign and biohazardous reagents used in industrial processes. In this situation, principles of green chemistry in terms of designing biodegradable and sustainable alternatives must become an imperative part of future studies. Along with the rapid advances in green synthetic methodologies and computational toxicology, molecular modeling techniques can significantly accelerate the transition toward greener solutions, i.e., designing ecofriendly and nontoxic reagents with improved efficiency and selectivity.
Acknowledgments
The authors gratefully acknowledge the Natural Sciences and Engineering Research Council of Canada for its lead financial support in the preparation of this review.
Author Contributions
CRediT: Abolfazl Alizadeh Sahraei conceptualization (equal), data curation (equal), formal analysis (equal), writing-original draft (equal); Dariush Azizi conceptualization (equal), data curation (equal), formal analysis (equal), writing-original draft (equal); Abdol Hadi Mokarizadeh conceptualization (equal), data curation (equal), formal analysis (equal), writing-original draft (equal); Daria C. Boffito funding acquisition (equal), project administration (equal), supervision (equal), writing-review & editing (supporting); Faïçl Larachi funding acquisition (equal), project administration (equal), supervision (equal), writing-review & editing (supporting).
The authors declare no competing financial interest.
References
- Fuerstenau M. C.; Jameson G. J.; Yoon R.-H.. Froth flotation: a century of innovation; SME, 2007. [Google Scholar]
- Somasundaran P.; Wang D.. Solution chemistry: minerals and reagents; Elsevier, 2006. [Google Scholar]
- Rao S. R.Surface chemistry of froth flotation: Vol. 1: Fundamentals; Springer Science & Business Media, 2013. [Google Scholar]
- Rai B.Molecular Modeling for the Design of Novel Performance Chemicals and Materials; Taylor & Francis Group, 2019. [Google Scholar]
- Fougerouse D.; Micklethwaite S.; Tomkins A. G.; Mei Y.; Kilburn M.; Guagliardo P.; Fisher L. A.; Halfpenny A.; Gee M.; Paterson D.; Howard D. L. Gold remobilisation and formation of high grade ore shoots driven by dissolution-reprecipitation replacement and Ni substitution into auriferous arsenopyrite. Geochim. Cosmochim. Acta 2016, 178, 143–159. 10.1016/j.gca.2016.01.040. [DOI] [Google Scholar]
- Pokrovski G. S.; Escoda C.; Blanchard M.; Testemale D.; Hazemann J. L.; Gouy S.; Kokh M. A.; Boiron M. C.; Parseval F. d.; Aigouy T.; Menjot L.; Parseval P. d.; Proux O.; Rovezzi M.; Béziat D.; Salvi S.; Kouzmanov K.; Bartsch T.; Pöttgen R.; Doert T. An arsenic-driven pump for invisible gold in hydrothermal systems. Geochemical Perspectives Letters 2021, 17, 39–44. 10.7185/geochemlet.2112. [DOI] [Google Scholar]
- Dholabhai P. P.; Perriot R.; Uberuaga B. P. Atomic-Scale Structure and Stability of the Low-Index Surfaces of Pyrochlore Oxides. J. Phys. Chem. C 2016, 120 (19), 10485–10499. 10.1021/acs.jpcc.6b02847. [DOI] [Google Scholar]
- Alizadeh Sahraei A.; Larachi F. DFT and ab-initio molecular dynamics of pyrochlore surface dissolution in aqueous media – Effect of cleavage plane and surface atomic vacancies. Appl. Surf. Sci. 2023, 615, 156425. 10.1016/j.apsusc.2023.156425. [DOI] [Google Scholar]
- Krasowska M.; Zawala J.; Bradshaw-Hajek B. H.; Ferri J. K.; Beattie D. A. Interfacial characterisation for flotation: 1. Solid-liquid interface. Curr. Opin. Colloid Interface Sci. 2018, 37, 61–73. 10.1016/j.cocis.2018.06.004. [DOI] [Google Scholar]
- Rai B.; Pradip Modeling self-assembly of surfactants at interfaces. Current opinion in chemical engineering 2017, 15, 84–94. 10.1016/j.coche.2016.12.003. [DOI] [Google Scholar]
- Tossell J.; Vaughan D. Theoretical studies of xanthates, dixanthogen, metal xanthates, and related compounds. J. Colloid Interface Sci. 1993, 155 (1), 98–107. 10.1006/jcis.1993.1015. [DOI] [Google Scholar]
- Ramachandran K.; Deepa G.; Namboori K.. Computational chemistry and molecular modeling: principles and applications; Springer Science & Business Media, 2008. [Google Scholar]
- Fuerstenau D. W.; Pradip A century of research leading to understanding the scientific basis of selective mineral flotation and design of flotation collectors. Mining, Metallurgy & Exploration 2019, 36 (1), 3–20. 10.1007/s42461-018-0042-6. [DOI] [Google Scholar]
- Liu G.; Yang X.; Zhong H. Molecular design of flotation collectors: A recent progress. Adv. Colloid Interface Sci. 2017, 246, 181–195. 10.1016/j.cis.2017.05.008. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Yang Y.; Shu X.; Wang Y.; Ran Q. Adsorption of organic molecules on mineral surfaces studied by first-principle calculations: A review. Adv. Colloid Interface Sci. 2018, 256, 230–241. 10.1016/j.cis.2018.04.003. [DOI] [PubMed] [Google Scholar]
- Foucaud Y.; Badawi M.; Filippov L.; Filippova I.; Lebègue S. A review of atomistic simulation methods for surface physical-chemistry phenomena applied to froth flotation. Minerals Engineering 2019, 143, 106020. 10.1016/j.mineng.2019.106020. [DOI] [Google Scholar]
- Xia Y.; Zhang R.; Cao Y.; Xing Y.; Gui X. Role of molecular simulation in understanding the mechanism of low-rank coal flotation: A review. Fuel 2020, 262, 116535. 10.1016/j.fuel.2019.116535. [DOI] [Google Scholar]
- Wang C.; Man X.; Ou L. Molecular dynamics study of the effect of temperature on the flotation behavior of sodium oleate on the surface of diaspore. Minerals Engineering 2023, 192, 108004. 10.1016/j.mineng.2023.108004. [DOI] [Google Scholar]
- Fuerstenau D. W.; Pradip Pradip, A Century of Research Leading to Understanding the Scientific Basis of Selective Mineral Flotation and Design of Flotation Collectors. Mining, Metallurgy & Exploration 2019, 36 (1), 3–20. 10.1007/s42461-018-0042-6. [DOI] [Google Scholar]
- Wang M.; Wang Y.; Tian W.; Qiu S.; Su G. H. Recent progress of CFD applications in PWR thermal hydraulics study and future directions. Annals of Nuclear Energy 2021, 150, 107836. 10.1016/j.anucene.2020.107836. [DOI] [Google Scholar]
- Brázdová V.; Bowler D. R.. Atomistic computer simulations: a practical guide; John Wiley & Sons, 2013. [Google Scholar]
- Tuckerman M. E.; Ungar P. J.; von Rosenvinge T.; Klein M. L. Ab Initio Molecular Dynamics Simulations. J. Phys. Chem. 1996, 100 (31), 12878–12887. 10.1021/jp960480+. [DOI] [Google Scholar]
- Tuckerman M. E. Ab initio molecular dynamics: basic concepts, current trends and novel applications. J. Phys.: Condens. Matter 2002, 14 (50), R1297–R1355. 10.1088/0953-8984/14/50/202. [DOI] [Google Scholar]
- Iftimie R.; Minary P.; Tuckerman M. E. Ab initio molecular dynamics: Concepts, recent developments, and future trends. Proc. Natl. Acad. Sci. U.S.A. 2005, 102 (19), 6654–6659. 10.1073/pnas.0500193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet E.; Viktor H. L. Computational Methods for Ab Initio Molecular Dynamics. Advances in Chemistry 2018, 2018, 9839641. 10.1155/2018/9839641. [DOI] [Google Scholar]
- Rocca D.; Dixit A.; Badawi M.; Lebègue S.; Gould T.; Bučko T. Bridging molecular dynamics and correlated wave-function methods for accurate finite-temperature properties. Physical Review Materials 2019, 3 (4), 040801 10.1103/PhysRevMaterials.3.040801. [DOI] [Google Scholar]
- Lin H.; Truhlar D. G. QM/MM: what have we learned, where are we, and where do we go from here?. Theor. Chem. Acc. 2007, 117 (2), 185. 10.1007/s00214-006-0143-z. [DOI] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48 (7), 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- da Silva L. F. M.; Öchsner A.; Adams R. D.. Handbook of adhesion technology; Springer: Heidelberg, 2011; Vol. 1. 10.1007/978-3-642-01169-6. [DOI] [Google Scholar]
- van Duin A. C. T.; Dasgupta S.; Lorant F.; Goddard W. A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105 (41), 9396–9409. 10.1021/jp004368u. [DOI] [Google Scholar]
- Chenoweth K.; van Duin A. C. T.; Goddard W. A. ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Hydrocarbon Oxidation. J. Phys. Chem. A 2008, 112 (5), 1040–1053. 10.1021/jp709896w. [DOI] [PubMed] [Google Scholar]
- Russo M. F.; van Duin A. C. T. Atomistic-scale simulations of chemical reactions: Bridging from quantum chemistry to engineering. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2011, 269 (14), 1549–1554. 10.1016/j.nimb.2010.12.053. [DOI] [Google Scholar]
- Aktulga H. M.; Pandit S. A.; Duin A. C. T. v.; Grama A. Y. Reactive Molecular Dynamics: Numerical Methods and Algorithmic Techniques. SIAM Journal on Scientific Computing 2012, 34 (1), C1–C23. 10.1137/100808599. [DOI] [Google Scholar]
- Senftle T. P.; Hong S.; Islam M. M.; Kylasa S. B.; Zheng Y.; Shin Y. K.; Junkermeier C.; Engel-Herbert R.; Janik M. J.; Aktulga H. M.; Verstraelen T.; Grama A.; van Duin A. C. T. The ReaxFF reactive force-field: development, applications and future directions. npj Computational Materials 2016, 2 (1), 15011. 10.1038/npjcompumats.2015.11. [DOI] [Google Scholar]
- Meuwly M. Reactive molecular dynamics: From small molecules to proteins. WIREs Computational Molecular Science 2019, 9 (1), e1386. 10.1002/wcms.1386. [DOI] [Google Scholar]
- Levine I. N.Quantum Chemistry; Pearson, 2014. [Google Scholar]
- Schrödinger E. An Undulatory Theory of the Mechanics of Atoms and Molecules. Phys. Rev. 1926, 28 (6), 1049–1070. 10.1103/PhysRev.28.1049. [DOI] [Google Scholar]
- Chung K. L.; Zhao Z.. From Brownian motion to Schrödinger’s equation; Springer Science & Business Media, 2012; Vol. 312. 10.1007/978-3-642-57856-4. [DOI] [Google Scholar]
- Dirac P. A. M.The principles of quantum mechanics; Oxford university press, 1981. [Google Scholar]
- Griffiths D. J.; Schroeter D. F.. Introduction to quantum mechanics; Cambridge University Press, 2018. [Google Scholar]
- Combes J.-M.; Duclos P.; Seiler R.. The born-oppenheimer approximation. In Rigorous atomic and molecular physics; Springer: 1981; pp 185–213. [Google Scholar]
- Slater J. C. Note on Hartree’s Method. Phys. Rev. 1930, 35 (2), 210–211. 10.1103/PhysRev.35.210.2. [DOI] [Google Scholar]
- Hartree D. R.; Hartree W. Self-consistent field, with exchange, for beryllium. Proceedings of the Royal Society of London. Series A - Mathematical and Physical Sciences 1935, 150 (869), 9–33. 10.1098/rspa.1935.0085. [DOI] [Google Scholar]
- Sholl D.; Steckel J. A.. Density functional theory: a practical introduction; John Wiley & Sons, 2022. [Google Scholar]
- Kang Y.; Li X.; Tu Y.; Wang Q.; Ågren H. On the Mechanism of Protein Adsorption onto Hydroxylated and Nonhydroxylated TiO2 Surfaces. J. Phys. Chem. C 2010, 114 (34), 14496–14502. 10.1021/jp1037156. [DOI] [Google Scholar]
- Guo Y.-n.; Lu X.; Zhang H.-p.; Weng J.; Watari F.; Leng Y. DFT Study of the Adsorption of Aspartic Acid on Pure, N-Doped, and Ca-Doped Rutile (110) Surfaces. J. Phys. Chem. C 2011, 115 (38), 18572–18581. 10.1021/jp200598t. [DOI] [Google Scholar]
- Li Y.; Chen J.; Chen Y.; Zhao C.; Zhang Y.; Ke B. Interactions of Oxygen and Water Molecules with Pyrite Surface: A New Insight. Langmuir 2018, 34 (5), 1941–1952. 10.1021/acs.langmuir.7b04112. [DOI] [PubMed] [Google Scholar]
- Li Y.; Chen J.; Chen Y.; Zhu Y.; Liu Y. DFT Simulation on Interaction of H2O Molecules with ZnS and Cu-Activated Surfaces. J. Phys. Chem. C 2019, 123 (5), 3048–3057. 10.1021/acs.jpcc.8b12273. [DOI] [Google Scholar]
- Alizadeh Sahraei A.; Larachi F. Chemical transformation and dissociation of amino acids on metal sulfide surface: Insights from DFT into the effect of surface vacancies on alanine-sphalerite system. Appl. Surf. Sci. 2021, 540, 148304. 10.1016/j.apsusc.2020.148304. [DOI] [Google Scholar]
- Sahraei A. A.; Larachi F. How Do Surface Defects Change Local Wettability of the Hydrophilic ZnS Surface? Insights into Sphalerite Flotation from Density Functional Theory Calculations. J. Phys. Chem. C 2021, 125 (1), 998–1009. 10.1021/acs.jpcc.0c09034. [DOI] [Google Scholar]
- Alizadeh Sahraei A.; Larachi F. Electronic simulations of alanine and water coadsorption over Defect-free and Sulfur-depleted sphalerite surfaces. Appl. Surf. Sci. 2022, 576, 151899. 10.1016/j.apsusc.2021.151899. [DOI] [Google Scholar]
- Parr R. G., Density Functional Theory of Atoms and Molecules. In Horizons of Quantum Chemistry; Fukui K., Pullman B., Eds.; Académie Internationale Des Sciences Moléculaires Quantiques/International Academy of Quantum Molecular Science; Springer: Dordrecht, 1980; Vol 3, pp 5–15. [Google Scholar]
- Bagayoko D. Understanding density functional theory (DFT) and completing it in practice. AIP Advances 2014, 4 (12), 127104. 10.1063/1.4903408. [DOI] [Google Scholar]
- Perdew J. P.; Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23 (10), 5048–5079. 10.1103/PhysRevB.23.5048. [DOI] [Google Scholar]
- Ceperley D. M.; Alder B. J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45 (7), 566–569. 10.1103/PhysRevLett.45.566. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38 (6), 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Chevary J. A.; Vosko S. H.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46 (11), 6671–6687. 10.1103/PhysRevB.46.6671. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Perdew J. P.; Ruzsinszky A.; Csonka G. I.; Vydrov O. A.; Scuseria G. E.; Constantin L. A.; Zhou X.; Burke K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100 (13), 136406. 10.1103/PhysRevLett.100.136406. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Schmidt K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577 (1), 1–20. 10.1063/1.1390175. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Grimme S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25 (12), 1463–1473. 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27 (15), 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Tkatchenko A.; Scheffler M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102 (7), 073005 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- Haider S.; Roldan A.; de Leeuw N. H. Catalytic Dissociation of Water on the (001), (011), and (111) Surfaces of Violarite, FeNi2S4: A DFT-D2 Study. J. Phys. Chem. C 2014, 118 (4), 1958–1967. 10.1021/jp409522q. [DOI] [Google Scholar]
- Dzade N. Y.; Roldan A.; de Leeuw N. H. DFT-D2 Study of the Adsorption and Dissociation of Water on Clean and Oxygen-Covered {001} and {011} Surfaces of Mackinawite (FeS). J. Phys. Chem. C 2016, 120 (38), 21441–21450. 10.1021/acs.jpcc.6b06122. [DOI] [Google Scholar]
- Dzade N. Y.; Roldan A.; de Leeuw N. H. Surface and shape modification of mackinawite (FeS) nanocrystals by cysteine adsorption: a first-principles DFT-D2 study. Phys. Chem. Chem. Phys. 2016, 18 (47), 32007–32020. 10.1039/C6CP05913A. [DOI] [PubMed] [Google Scholar]
- Dzade N. Y.; Roldan A.; de Leeuw N. H. DFT-D2 simulations of water adsorption and dissociation on the low-index surfaces of mackinawite (FeS). J. Chem. Phys. 2016, 144 (17), 174704. 10.1063/1.4947588. [DOI] [PubMed] [Google Scholar]
- Holt D. B. Defects in the sphalerite structure. J. Phys. Chem. Solids 1962, 23 (10), 1353–1362. 10.1016/0022-3697(62)90188-9. [DOI] [Google Scholar]
- Maeda T.; Wada T. First-principles calculation of defect formation energy in chalcopyrite-type CuInSe2, CuGaSe2 and CuAlSe2. J. Phys. Chem. Solids 2005, 66 (11), 1924–1927. 10.1016/j.jpcs.2005.09.067. [DOI] [Google Scholar]
- Stirling A.; Bernasconi M.; Parrinello M. Defective pyrite (100) surface: An ab initio study. Phys. Rev. B 2007, 75 (16), 165406. 10.1103/PhysRevB.75.165406. [DOI] [Google Scholar]
- Srinivasan S. G.; Shivaramaiah R.; Kent P. R. C.; Stack A. G.; Navrotsky A.; Riman R.; Anderko A.; Bryantsev V. S. Crystal Structures, Surface Stability, and Water Adsorption Energies of La-Bastnäsite via Density Functional Theory and Experimental Studies. J. Phys. Chem. C 2016, 120 (30), 16767–16781. 10.1021/acs.jpcc.6b04747. [DOI] [Google Scholar]
- Goverapet Srinivasan S.; Shivaramaiah R.; Kent P. R. C.; Stack A. G.; Riman R.; Anderko A.; Navrotsky A.; Bryantsev V. S. A comparative study of surface energies and water adsorption on Ce-bastnäsite, La-bastnäsite, and calcite via density functional theory and water adsorption calorimetry. Phys. Chem. Chem. Phys. 2017, 19 (11), 7820–7832. 10.1039/C7CP00811B. [DOI] [PubMed] [Google Scholar]
- Roy S.; Wu L.; Goverapet Srinivasan S.; Stack A. G.; Navrotsky A.; Bryantsev V. S. Hydration structure and water exchange kinetics at xenotime–water interfaces: implications for rare earth minerals separation. Phys. Chem. Chem. Phys. 2020, 22 (15), 7719–7727. 10.1039/D0CP00087F. [DOI] [PubMed] [Google Scholar]
- Gibson L. D.; Jayanthi K.; Yang S.; Thiele N.; Anovitz L. M.; Sacci R. L.; Navrotsky A.; Bryantsev V. S. Characterization of Lanthanum Monazite Surface Chemistry and Crystal Morphology through Density Functional Theory and Experimental Approaches. J. Phys. Chem. C 2022, 126 (44), 18952–18962. 10.1021/acs.jpcc.2c06308. [DOI] [Google Scholar]
- Hirshfeld F. L. Bonded-atom fragments for describing molecular charge densities. Theoretica chimica acta 1977, 44 (2), 129–138. 10.1007/BF00549096. [DOI] [Google Scholar]
- Becke A. D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88 (4), 2547–2553. 10.1063/1.454033. [DOI] [Google Scholar]
- Fonseca Guerra C.; Handgraaf J.-W.; Baerends E. J.; Bickelhaupt F. M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25 (2), 189–210. 10.1002/jcc.10351. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W.Atoms in Molecules: A Quantum Theory; Clarendon Press, 1990. [Google Scholar]
- Carbó-Dorca R.; Bultinck P. Quantum Mechanical Basis for Mulliken Population Analysis. J. Math. Chem. 2004, 36 (3), 231–239. 10.1023/B:JOMC.0000044221.23647.20. [DOI] [Google Scholar]
- Fukui K.; Yonezawa T.; Nagata C.; Shingu H. Molecular Orbital Theory of Orientation in Aromatic, Heteroaromatic, and Other Conjugated Molecules. J. Chem. Phys. 1954, 22 (8), 1433–1442. 10.1063/1.1740412. [DOI] [Google Scholar]
- Parr R. G.; Yang W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106 (14), 4049–4050. 10.1021/ja00326a036. [DOI] [Google Scholar]
- Michalak A.; De Proft F.; Geerlings P.; Nalewajski R. F. Fukui Functions from the Relaxed Kohn–Sham Orbitals. J. Phys. Chem. A 1999, 103 (6), 762–771. 10.1021/jp982761i. [DOI] [Google Scholar]
- Oláh J.; Van Alsenoy C.; Sannigrahi A. B. Condensed Fukui Functions Derived from Stockholder Charges: Assessment of Their Performance as Local Reactivity Descriptors. J. Phys. Chem. A 2002, 106 (15), 3885–3890. 10.1021/jp014039h. [DOI] [Google Scholar]
- Bultinck P.; Carbó-Dorca R.; Langenaeker W. Negative Fukui functions: New insights based on electronegativity equalization. J. Chem. Phys. 2003, 118 (10), 4349–4356. 10.1063/1.1542875. [DOI] [Google Scholar]
- Alecu I. M.; Zheng J.; Zhao Y.; Truhlar D. G. Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries. J. Chem. Theory Comput. 2010, 6 (9), 2872–2887. 10.1021/ct100326h. [DOI] [PubMed] [Google Scholar]
- Cheeseman J. R.; Trucks G. W.; Keith T. A.; Frisch M. J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104 (14), 5497–5509. 10.1063/1.471789. [DOI] [Google Scholar]
- Onida G.; Reining L.; Rubio A. Electronic excitations: density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 2002, 74 (2), 601–659. 10.1103/RevModPhys.74.601. [DOI] [Google Scholar]
- Botti S.; Schindlmayr A.; Sole R. D.; Reining L. Time-dependent density-functional theory for extended systems. Rep. Prog. Phys. 2007, 70 (3), 357–407. 10.1088/0034-4885/70/3/R02. [DOI] [Google Scholar]
- McQuarrie D. A.Statistical mechanics; Sterling Publishing Company, 2000. [Google Scholar]
- Allen M. P.; Tildesley D. J.. Computer simulation of liquids; Oxford university press, 2017. [Google Scholar]
- Frenkel D.; Smit B.. Understanding Molecular Simulation: From Algorithms to Applications; Elsevier, 2001; Vol. 1. [Google Scholar]
- Sadus R. J.Molecular Simulation of Fluids: Theory, Algorithms and Object-Orientation; Elsevier Science, 2023. [Google Scholar]
- Sun H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase ApplicationsOverview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102 (38), 7338–7364. 10.1021/jp980939v. [DOI] [Google Scholar]
- Sun H.; Mumby S. J.; Maple J. R.; Hagler A. T. An ab Initio CFF93 All-Atom Force Field for Polycarbonates. J. Am. Chem. Soc. 1994, 116 (7), 2978–2987. 10.1021/ja00086a030. [DOI] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110 (6), 1657–1666. 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118 (45), 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Gunsteren W. F. v.Biomolecular simulation: the GROMOS96 manual and user guide; Vdf Hochschulverlag AG an der ETH Zürich: Groningen; Zürich, 1996. [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25 (9), 1157–74. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Shahsavari R.; Pellenq R. J. M.; Ulm F.-J. Empirical force fields for complex hydrated calcio-silicate layered materials. Phys. Chem. Chem. Phys. 2011, 13 (3), 1002–1011. 10.1039/C0CP00516A. [DOI] [PubMed] [Google Scholar]
- Mishra R. K.; Flatt R. J.; Heinz H. Force Field for Tricalcium Silicate and Insight into Nanoscale Properties: Cleavage, Initial Hydration, and Adsorption of Organic Molecules. J. Phys. Chem. C 2013, 117 (20), 10417–10432. 10.1021/jp312815g. [DOI] [Google Scholar]
- Mishra R. K.; Fernández-Carrasco L.; Flatt R. J.; Heinz H. A force field for tricalcium aluminate to characterize surface properties, initial hydration, and organically modified interfaces in atomic resolution. Dalton Transactions 2014, 43 (27), 10602–10616. 10.1039/C4DT00438H. [DOI] [PubMed] [Google Scholar]
- Mishra R. K.; Mohamed A. K.; Geissbühler D.; Manzano H.; Jamil T.; Shahsavari R.; Kalinichev A. G.; Galmarini S.; Tao L.; Heinz H.; Pellenq R.; van Duin A. C. T.; Parker S. C.; Flatt R. J.; Bowen P. cemff: A force field database for cementitious materials including validations, applications and opportunities. Cem. Concr. Res. 2017, 102, 68–89. 10.1016/j.cemconres.2017.09.003. [DOI] [Google Scholar]
- Androniuk I.; Landesman C.; Henocq P.; Kalinichev A. G. Adsorption of gluconate and uranyl on C-S-H phases: Combination of wet chemistry experiments and molecular dynamics simulations for the binary systems. Physics and Chemistry of the Earth, Parts A/B/C 2017, 99, 194–203. 10.1016/j.pce.2017.05.005. [DOI] [Google Scholar]
- Heinz H.; Lin T.-J.; Kishore Mishra R.; Emami F. S. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field. Langmuir 2013, 29 (6), 1754–1765. 10.1021/la3038846. [DOI] [PubMed] [Google Scholar]
- Choi Y. K.; Kern N. R.; Kim S.; Kanhaiya K.; Afshar Y.; Jeon S. H.; Jo S.; Brooks B. R.; Lee J.; Tadmor E. B.; Heinz H.; Im W. CHARMM-GUI Nanomaterial Modeler for Modeling and Simulation of Nanomaterial Systems. J. Chem. Theory Comput. 2022, 18 (1), 479–493. 10.1021/acs.jctc.1c00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark P.; Nilsson L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105 (43), 9954–9960. 10.1021/jp003020w. [DOI] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Fogarty J. C.; Aktulga H. M.; Grama A. Y.; van Duin A. C. T.; Pandit S. A. A reactive molecular dynamics simulation of the silica-water interface. J. Chem. Phys. 2010, 132 (17), 174704. 10.1063/1.3407433. [DOI] [PubMed] [Google Scholar]
- Yeon J.; van Duin A. C. T. ReaxFF Molecular Dynamics Simulations of Hydroxylation Kinetics for Amorphous and Nano-Silica Structure, and Its Relations with Atomic Strain Energy. J. Phys. Chem. C 2016, 120 (1), 305–317. 10.1021/acs.jpcc.5b09784. [DOI] [Google Scholar]
- Hahn S. H.; van Duin A. C. T. Surface Reactivity and Leaching of a Sodium Silicate Glass under an Aqueous Environment: A ReaxFF Molecular Dynamics Study. J. Phys. Chem. C 2019, 123 (25), 15606–15617. 10.1021/acs.jpcc.9b02940. [DOI] [Google Scholar]
- Zhang Q.; Çaǧın T.; van Duin A.; Goddard W. A.; Qi Y.; Hector L. G. Adhesion and nonwetting-wetting transition in the Al/α– Al2O3 interface. Phys. Rev. B 2004, 69 (4), 045423 10.1103/PhysRevB.69.045423. [DOI] [Google Scholar]
- Narayanan B.; van Duin A. C. T.; Kappes B. B.; Reimanis I. E.; Ciobanu C. V. A reactive force field for lithium–aluminum silicates with applications to eucryptite phases. Modell. Simul. Mater. Sci. Eng. 2012, 20 (1), 015002 10.1088/0965-0393/20/1/015002. [DOI] [Google Scholar]
- Kim S.-Y.; Kumar N.; Persson P.; Sofo J.; van Duin A. C. T.; Kubicki J. D. Development of a ReaxFF Reactive Force Field for Titanium Dioxide/Water Systems. Langmuir 2013, 29 (25), 7838–7846. 10.1021/la4006983. [DOI] [PubMed] [Google Scholar]
- Law K.-Y.; Zhao H.. Surface wetting: characterization, contact angle, and fundamentals; Springer International Publishing: Basel, Switzerland, 2015. [Google Scholar]
- Israelachvili J. N.Intermolecular and surface forces; Academic Press, 2011. [Google Scholar]
- van Oss C. J.Interfacial Forces in Aqueous Media; CRC Press, 2006. [Google Scholar]
- Law K.-Y. Definitions for Hydrophilicity, Hydrophobicity, and Superhydrophobicity: Getting the Basics Right. J. Phys. Chem. Lett. 2014, 5 (4), 686–688. 10.1021/jz402762h. [DOI] [PubMed] [Google Scholar]
- Andreotti B.; Snoeijer J. H. Statics and Dynamics of Soft Wetting. Annu. Rev. Fluid Mech. 2020, 52 (1), 285–308. 10.1146/annurev-fluid-010719-060147. [DOI] [Google Scholar]
- Jiang H.; Patel A. J. Recent advances in estimating contact angles using molecular simulations and enhanced sampling methods. Current Opinion in Chemical Engineering 2019, 23, 130–137. 10.1016/j.coche.2019.03.012. [DOI] [Google Scholar]
- de Gennes P. G. Wetting: statics and dynamics. Rev. Mod. Phys. 1985, 57 (3), 827–863. 10.1103/RevModPhys.57.827. [DOI] [Google Scholar]
- de Ruijter M. J.; Blake T. D.; De Coninck J. Dynamic Wetting Studied by Molecular Modeling Simulations of Droplet Spreading. Langmuir 1999, 15 (22), 7836–7847. 10.1021/la990171l. [DOI] [Google Scholar]
- Ravipati S.; Aymard B.; Kalliadasis S.; Galindo A. On the equilibrium contact angle of sessile liquid drops from molecular dynamics simulations. J. Chem. Phys. 2018, 148 (16), 164704. 10.1063/1.5021088. [DOI] [PubMed] [Google Scholar]
- Hautman J.; Klein M. L. Microscopic wetting phenomena. Phys. Rev. Lett. 1991, 67 (13), 1763–1766. 10.1103/PhysRevLett.67.1763. [DOI] [PubMed] [Google Scholar]
- Fan C. F.; Caǧin T. Wetting of crystalline polymer surfaces: A molecular dynamics simulation. J. Chem. Phys. 1995, 103 (20), 9053–9061. 10.1063/1.470016. [DOI] [Google Scholar]
- Shih C.-J.; Wang Q. H.; Lin S.; Park K.-C.; Jin Z.; Strano M. S.; Blankschtein D. Breakdown in the Wetting Transparency of Graphene. Phys. Rev. Lett. 2012, 109 (17), 176101. 10.1103/PhysRevLett.109.176101. [DOI] [PubMed] [Google Scholar]
- Włoch J.; Terzyk A. P.; Gauden P. A.; Wesołowski R.; Kowalczyk P. Water nanodroplet on a graphene surface—a new old system. J. Phys.: Condens. Matter 2016, 28 (49), 495002. 10.1088/0953-8984/28/49/495002. [DOI] [PubMed] [Google Scholar]
- Scocchi G.; Sergi D.; D’Angelo C.; Ortona A. Wetting and contact-line effects for spherical and cylindrical droplets on graphene layers: A comparative molecular-dynamics investigation. Phys. Rev. E 2011, 84 (6), 061602 10.1103/PhysRevE.84.061602. [DOI] [PubMed] [Google Scholar]
- Errington J. R.; Wilbert D. W. Prewetting Boundary Tensions from Monte Carlo Simulation. Phys. Rev. Lett. 2005, 95 (22), 226107. 10.1103/PhysRevLett.95.226107. [DOI] [PubMed] [Google Scholar]
- Grzelak E. M.; Errington J. R. Computation of interfacial properties via grand canonical transition matrix Monte Carlo simulation. J. Chem. Phys. 2008, 128 (1), 014710 10.1063/1.2812285. [DOI] [PubMed] [Google Scholar]
- Rane K. S.; Kumar V.; Errington J. R. Monte Carlo simulation methods for computing the wetting and drying properties of model systems. J. Chem. Phys. 2011, 135 (23), 234102. 10.1063/1.3668137. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Errington J. R. The Use of Monte Carlo Simulation to Obtain the Wetting Properties of Water. Physics Procedia 2014, 53, 44–49. 10.1016/j.phpro.2014.06.024. [DOI] [Google Scholar]
- Jain K.; Schultz A. J.; Errington J. R. Application of the interface potential approach for studying wetting behavior within a molecular dynamics framework. J. Chem. Phys. 2019, 150 (20), 204118. 10.1063/1.5096362. [DOI] [PubMed] [Google Scholar]
- Leroy F.; dos Santos D. J. V. A.; Müller-Plathe F. Interfacial Excess Free Energies of Solid–Liquid Interfaces by Molecular Dynamics Simulation and Thermodynamic Integration. Macromol. Rapid Commun. 2009, 30 (9–10), 864–870. 10.1002/marc.200800746. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Fialoke S.; Vicars Z.; Patel A. J. Characterizing surface wetting and interfacial properties using enhanced sampling (SWIPES). Soft Matter 2019, 15 (5), 860–869. 10.1039/C8SM02317D. [DOI] [PubMed] [Google Scholar]
- Rego N. B.; Xi E.; Patel A. J. Protein Hydration Waters Are Susceptible to Unfavorable Perturbations. J. Am. Chem. Soc. 2019, 141 (5), 2080–2086. 10.1021/jacs.8b11448. [DOI] [PubMed] [Google Scholar]
- Bulatovic S. M.Handbook of flotation reagents: chemistry, theory and practice: Vol. 1: flotation of sulfide ores; Elsevier, 2007. [Google Scholar]
- Irving J. H.; Kirkwood J. G. The Statistical Mechanical Theory of Transport Processes. IV. The Equations of Hydrodynamics. J. Chem. Phys. 1950, 18 (6), 817–829. 10.1063/1.1747782. [DOI] [Google Scholar]
- Buff F. P. Some considerations of surface tension. Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für physikalische Chemie 1952, 56 (4), 311–313. 10.1002/bbpc.19520560413. [DOI] [Google Scholar]
- Buff F. P. Spherical Interface. II. Molecular Theory. J. Chem. Phys. 1955, 23 (3), 419–427. 10.1063/1.1742005. [DOI] [Google Scholar]
- Gloor G. J.; Jackson G.; Blas F. J.; de Miguel E. Test-area simulation method for the direct determination of the interfacial tension of systems with continuous or discontinuous potentials. J. Chem. Phys. 2005, 123 (13), 134703. 10.1063/1.2038827. [DOI] [PubMed] [Google Scholar]
- Errington J. R.; Kofke D. A. Calculation of surface tension via area sampling. J. Chem. Phys. 2007, 127 (17), 174709. 10.1063/1.2795698. [DOI] [PubMed] [Google Scholar]
- Luzar A.; Chandler D. Effect of Environment on Hydrogen Bond Dynamics in Liquid Water. Phys. Rev. Lett. 1996, 76 (6), 928–931. 10.1103/PhysRevLett.76.928. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. HBonanza: A computer algorithm for molecular-dynamics-trajectory hydrogen-bond analysis. Journal of Molecular Graphics and Modelling 2011, 31, 5–9. 10.1016/j.jmgm.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torshin I. Y.; Weber I. T.; Harrison R. W. Geometric criteria of hydrogen bonds in proteins and identification of `bifurcated’ hydrogen bonds. Protein Engineering, Design and Selection 2002, 15 (5), 359–363. 10.1093/protein/15.5.359. [DOI] [PubMed] [Google Scholar]
- Luzar A.; Chandler D. Structure and hydrogen bond dynamics of water–dimethyl sulfoxide mixtures by computer simulations. J. Chem. Phys. 1993, 98 (10), 8160–8173. 10.1063/1.464521. [DOI] [Google Scholar]
- Luzar A. Resolving the hydrogen bond dynamics conundrum. J. Chem. Phys. 2000, 113 (23), 10663–10675. 10.1063/1.1320826. [DOI] [Google Scholar]
- Impey R. W.; Madden P. A.; McDonald I. R. Hydration and mobility of ions in solution. J. Phys. Chem. 1983, 87 (25), 5071–5083. 10.1021/j150643a008. [DOI] [Google Scholar]
- Chandler D.; Percus J. K. Introduction to Modern Statistical Mechanics. Phys. Today 1988, 41 (12), 114–118. 10.1063/1.2811680. [DOI] [Google Scholar]
- García A. E.; Stiller L. Computation of the mean residence time of water in the hydration shells of biomolecules. J. Comput. Chem. 1993, 14 (11), 1396–1406. 10.1002/jcc.540141116. [DOI] [Google Scholar]
- Stirling A.; Bernasconi M.; Parrinello M. Ab initio simulation of water interaction with the (100) surface of pyrite. J. Chem. Phys. 2003, 118 (19), 8917–8926. 10.1063/1.1566936. [DOI] [Google Scholar]
- Hug S.; Hunter G. K.; Goldberg H.; Karttunen M. Ab initio simulations of peptide-mineral interactions. Physics Procedia 2010, 4, 51–60. 10.1016/j.phpro.2010.08.008. [DOI] [Google Scholar]
- Bittarello E.; Bruno M.; Aquilano D. Ab Initio Calculations of the Main Crystal Surfaces of Baryte (BaSO4). Cryst. Growth Des. 2018, 18 (7), 4084–4094. 10.1021/acs.cgd.8b00460. [DOI] [Google Scholar]
- Geneyton A.; Foucaud Y.; Filippov L. O.; Menad N. E.; Renard A.; Badawi M. Synergistic adsorption of lanthanum ions and fatty acids for efficient rare-earth phosphate recovery: Surface analysis and ab initio molecular dynamics studies. Appl. Surf. Sci. 2020, 526, 146725. 10.1016/j.apsusc.2020.146725. [DOI] [Google Scholar]
- Bakowies D.; Thiel W. Hybrid Models for Combined Quantum Mechanical and Molecular Mechanical Approaches. J. Phys. Chem. 1996, 100 (25), 10580–10594. 10.1021/jp9536514. [DOI] [Google Scholar]
- Yang Z.; Zhao Y.-P. QM/MM and classical molecular dynamics simulation of histidine-tagged peptide immobilization on nickel surface. Materials Science and Engineering: A 2006, 423 (1), 84–91. 10.1016/j.msea.2005.10.078. [DOI] [Google Scholar]
- Lu X.; Fang D.; Ito S.; Okamoto Y.; Ovchinnikov V.; Cui Q. QM/MM free energy simulations: recent progress and challenges. Mol. Simul. 2016, 42 (13), 1056–1078. 10.1080/08927022.2015.1132317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino P. L.; Arkhipov A. S.; Larson S. B.; McPherson A.; Schulten K. Molecular Dynamics Simulations of the Complete Satellite Tobacco Mosaic Virus. Structure 2006, 14 (3), 437–449. 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Gumbart J.; Trabuco L. G.; Schreiner E.; Villa E.; Schulten K. Regulation of the Protein-Conducting Channel by a Bound Ribosome. Structure 2009, 17 (11), 1453–1464. 10.1016/j.str.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepeis J. L.; Lindorff-Larsen K.; Dror R. O.; Shaw D. E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009, 19 (2), 120–127. 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Schulz R.; Lindner B.; Petridis L.; Smith J. C. Scaling of Multimillion-Atom Biological Molecular Dynamics Simulation on a Petascale Supercomputer. J. Chem. Theory Comput. 2009, 5 (10), 2798–2808. 10.1021/ct900292r. [DOI] [PubMed] [Google Scholar]
- Bowler D. R.; Miyazaki T. Calculations for millions of atoms with density functional theory: linear scaling shows its potential. J. Phys.: Condens. Matter 2010, 22 (7), 074207 10.1088/0953-8984/22/7/074207. [DOI] [PubMed] [Google Scholar]
- Evans J. D.; Fraux G.; Gaillac R.; Kohen D.; Trousselet F.; Vanson J.-M.; Coudert F.-X. Computational Chemistry Methods for Nanoporous Materials. Chem. Mater. 2017, 29 (1), 199–212. 10.1021/acs.chemmater.6b02994. [DOI] [Google Scholar]
- Wills B. A.; Finch J.. Wills’ mineral processing technology: an introduction to the practical aspects of ore treatment and mineral recovery; Butterworth-Heinemann, 2015. [Google Scholar]
- Chapleski R. C.; Chowdhury A. U.; Wanhala A. K.; Bocharova V.; Roy S.; Keller P. C.; Everly D.; Jansone-Popova S.; Kisliuk A.; Sacci R. L.; Stack A. G.; Anderson C. G.; Doughty B.; Bryantsev V. S. A Molecular-Scale Approach to Rare-Earth Beneficiation: Thinking Small to Avoid Large Losses. iScience 2020, 23 (9), 101435. 10.1016/j.isci.2020.101435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion C.; Li R.; Waters K. E. A review of reagents applied to rare-earth mineral flotation. Adv. Colloid Interface Sci. 2020, 279, 102142. 10.1016/j.cis.2020.102142. [DOI] [PubMed] [Google Scholar]
- Wanhala A. K.; Doughty B.; Bryantsev V. S.; Wu L.; Mahurin S. M.; Jansone-Popova S.; Cheshire M. C.; Navrotsky A.; Stack A. G. Adsorption mechanism of alkyl hydroxamic acid onto bastnäsite: Fundamental steps toward rational collector design for rare earth elements. J. Colloid Interface Sci. 2019, 553, 210–219. 10.1016/j.jcis.2019.06.025. [DOI] [PubMed] [Google Scholar]
- Sarvaramini A.; Azizi D.; Larachi F. Hydroxamic acid interactions with solvated cerium hydroxides in the flotation of monazite and bastnäsite—experiments and DFT study. Appl. Surf. Sci. 2016, 387, 986–995. 10.1016/j.apsusc.2016.07.044. [DOI] [Google Scholar]
- Sutton J. E.; Roy S.; Chowdhury A. U.; Wu L.; Wanhala A. K.; De Silva N.; Jansone-Popova S.; Hay B. P.; Cheshire M. C.; Windus T. L.; et al. Molecular Recognition at Mineral Interfaces: Implications for the Beneficiation of Rare Earth Ores. ACS Appl. Mater. Interfaces 2020, 12 (14), 16327–16341. 10.1021/acsami.9b22902. [DOI] [PubMed] [Google Scholar]
- Foucaud Y.; Lainé J.; Filippov L. O.; Barrès O.; Kim W. J.; Filippova I. V.; Pastore M.; Lebègue S.; Badawi M. Adsorption mechanisms of fatty acids on fluorite unraveled by infrared spectroscopy and first-principles calculations. J. Colloid Interface Sci. 2021, 583, 692–703. 10.1016/j.jcis.2020.09.062. [DOI] [PubMed] [Google Scholar]
- Foucaud Y.; Lebègue S. b.; Filippov L. O.; Filippova I. V.; Badawi M. l. Molecular insight into fatty acid adsorption on bare and hydrated (111) fluorite surface. J. Phys. Chem. B 2018, 122 (51), 12403–12410. 10.1021/acs.jpcb.8b08969. [DOI] [PubMed] [Google Scholar]
- Nan N.; Zhu Y.; Han Y.; Liu J. Molecular modeling of interactions between N-(Carboxymethyl)-N-tetradecylglycine and fluorapatite. Minerals 2019, 9 (5), 278. 10.3390/min9050278. [DOI] [Google Scholar]
- Kumar D.; Goverapet Srinivasan S.; Jain V.; Rai B. Understanding flotation processes at the atomic scale using density functional theory – A case study on adsorption of 2-Mercaptobenzothiazole on chalcopyrite and pyrite surfaces. Appl. Surf. Sci. 2022, 579, 152112. 10.1016/j.apsusc.2021.152112. [DOI] [Google Scholar]
- Lin S.; He J.; Liu R.; Hu Y.; Sun W. Depression behavior and mechanism of pyrogallol on bismuthinite flotation. Journal of Cleaner Production 2021, 281, 125322. 10.1016/j.jclepro.2020.125322. [DOI] [Google Scholar]
- Sun X.; Wu B.; Qiu H.; Chen J.; Hu M.; Hu K. Interaction of a novel depressant m-nitrobenzoate with arsenopyrite surface: DFT and experimental studies. Colloids Surf., A 2022, 650, 129613. 10.1016/j.colsurfa.2022.129613. [DOI] [Google Scholar]
- Han Y.; Liu W.; Zhou J.; Chen J. Interactions between kaolinite AlOH surface and sodium hexametaphosphate. Appl. Surf. Sci. 2016, 387, 759–765. 10.1016/j.apsusc.2016.07.002. [DOI] [Google Scholar]
- Cao Q.; Du H.; Miller J. D.; Wang X.; Cheng F. Surface chemistry features in the flotation of KCl. Minerals Engineering 2010, 23 (5), 365–373. 10.1016/j.mineng.2009.11.010. [DOI] [Google Scholar]
- Chapleski R. C. Jr.; Chowdhury A. U.; Wanhala A. K.; Gibson L. D.; Stamberga D. n.; Jansone-Popova S.; Sacci R. L.; Meyer H. M. III; Stack A. G.; Bocharova V.; Doughty B.; Bryantsev V. S. Improving Rare-Earth Mineral Separation with Insights from Molecular Recognition: Functionalized Hydroxamic Acid Adsorption onto Bastnäsite and Calcite. Langmuir 2022, 38 (18), 5439–5453. 10.1021/acs.langmuir.1c03422. [DOI] [PubMed] [Google Scholar]
- Peng C.; Min F.; Liu L. Effect of pH on the adsorption of dodecylamine on montmorillonite: Insights from experiments and molecular dynamics simulations. Appl. Surf. Sci. 2017, 425, 996–1005. 10.1016/j.apsusc.2017.07.085. [DOI] [Google Scholar]
- Liu A.; Fan J.-c.; Fan M.-q. Quantum chemical calculations and molecular dynamics simulations of amine collector adsorption on quartz (0 0 1) surface in the aqueous solution. Int. J. Miner. Process. 2015, 134, 1–10. 10.1016/j.minpro.2014.11.001. [DOI] [Google Scholar]
- Zhao G.; Zhong H.; Qiu X.; Wang S.; Gao Y.; Dai Z.; Huang J.; Liu G. The DFT study of cyclohexyl hydroxamic acid as a collector in scheelite flotation. Minerals Engineering 2013, 49, 54–60. 10.1016/j.mineng.2013.04.025. [DOI] [Google Scholar]
- Liu W.; Wang X.; Xu H.; Miller J. Lauryl phosphate adsorption in the flotation of Bastnaesite,(Ce, La) FCO3. J. Colloid Interface Sci. 2017, 490, 825–833. 10.1016/j.jcis.2016.11.016. [DOI] [PubMed] [Google Scholar]
- Yu F.; Wang Y.; Zhang L.; Zhu G. Role of oleic acid ionic– molecular complexes in the flotation of spodumene. Minerals Engineering 2015, 71, 7–12. 10.1016/j.mineng.2014.10.001. [DOI] [Google Scholar]
- Alizadeh Sahraei A.; Mejia Bohorquez B.; Tremblay D. M.; Moineau S.; Garnier A.; Larachi F.; Lague P. Insight into the Binding Mechanisms of Quartz-Selective Peptides: Toward Greener Flotation Processes. ACS Appl. Mater. Interfaces 2023, 15, 17922. 10.1021/acsami.3c01275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan S. V.; Emami F. S.; Berry R. J.; Jones S. E.; Naik R. R.; Deschaume O.; Heinz H.; Perry C. C. Chemistry of Aqueous Silica Nanoparticle Surfaces and the Mechanism of Selective Peptide Adsorption. J. Am. Chem. Soc. 2012, 134 (14), 6244–6256. 10.1021/ja211307u. [DOI] [PubMed] [Google Scholar]
- Emami F. S.; Puddu V.; Berry R. J.; Varshney V.; Patwardhan S. V.; Perry C. C.; Heinz H. Force Field and a Surface Model Database for Silica to Simulate Interfacial Properties in Atomic Resolution. Chem. Mater. 2014, 26 (8), 2647–2658. 10.1021/cm500365c. [DOI] [Google Scholar]
- Emami F. S.; Puddu V.; Berry R. J.; Varshney V.; Patwardhan S. V.; Perry C. C.; Heinz H. Prediction of Specific Biomolecule Adsorption on Silica Surfaces as a Function of pH and Particle Size. Chem. Mater. 2014, 26 (19), 5725–5734. 10.1021/cm5026987. [DOI] [Google Scholar]
- Caricato M. Coupled cluster theory with the polarizable continuum model of solvation. Int. J. Quantum Chem. 2019, 119 (1), e25710. 10.1002/qua.25710. [DOI] [Google Scholar]
- Li L.; Zhang C.; Yuan Z.; Xu X.; Song Z. AFM and DFT study of depression of hematite in oleate-starch-hematite flotation system. Appl. Surf. Sci. 2019, 480, 749–758. 10.1016/j.apsusc.2019.02.224. [DOI] [Google Scholar]
- Li W.; Wang H.; Li X.; Liang Y.; Wang Y.; Zhang H. Effect of mixed cationic/anionic surfactants on the low-rank coal wettability by an experimental and molecular dynamics simulation. Fuel 2021, 289, 119886. 10.1016/j.fuel.2020.119886. [DOI] [Google Scholar]
- Liu Z.; Xia Y.; Lai Q.; An M.; Liao Y.; Wang Y. Adsorption behavior of mixed dodecane/n-valeric acid collectors on low-rank coal surface: Experimental and molecular dynamics simulation study. Colloids Surf., A 2019, 583, 123840. 10.1016/j.colsurfa.2019.123840. [DOI] [Google Scholar]
- Xia Y.; Rong G.; Xing Y.; Gui X. Synergistic adsorption of polar and nonpolar reagents on oxygen-containing graphite surfaces: Implications for low-rank coal flotation. J. Colloid Interface Sci. 2019, 557, 276–281. 10.1016/j.jcis.2019.09.025. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Ren H.; Yang Z.; Liao Y.; Cao Y. Effect of mixed collector addition sequence on the adsorption behavior of low-rank coal surface: Experimental and molecular dynamics simulation study. Powder Technol. 2022, 397, 117119. 10.1016/j.powtec.2022.117119. [DOI] [Google Scholar]
- Zhang R.; Xing Y.; Xia Y.; Guo F.; Ding S.; Tan J.; Che T.; Meng F.; Gui X. Synergistic Adsorption Mechanism of Anionic and Cationic Surfactant Mixtures on Low-Rank Coal Flotation. ACS omega 2020, 5 (32), 20630–20637. 10.1021/acsomega.0c02948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Li C.; An H.; Wang G.; Zhao X.; Zhang J. Flotation and molecular dynamics simulation of muscovite with mixed anionic/cationic collectors. Physicochemical Problems of Mineral Processing 2020, 56, 313. [Google Scholar]
- Wang L.; Hu Y.; Liu J.; Sun Y.; Sun W. Flotation and adsorption of muscovite using mixed cationic–nonionic surfactants as collector. Powder technology 2015, 276, 26–33. 10.1016/j.powtec.2015.02.019. [DOI] [Google Scholar]
- Wang Z.; Peng Y.; Zheng Y.; Ding W.; Wang J.; Xu L. Improved flotation of artificial galena using a new catanionic mixture. Minerals Engineering 2020, 148, 106206. 10.1016/j.mineng.2020.106206. [DOI] [Google Scholar]
- Wang L.; Liu R.; Hu Y.; Liu J.; Sun W. Adsorption behavior of mixed cationic/anionic surfactants and their depression mechanism on the flotation of quartz. Powder Technol. 2016, 302, 15–20. 10.1016/j.powtec.2016.08.043. [DOI] [Google Scholar]
- Shen L.; Zhu J.; Liu L.; Wang H. Flotation of fine kaolinite using dodecylamine chloride/fatty acids mixture as collector. Powder technology 2017, 312, 159–165. 10.1016/j.powtec.2017.02.032. [DOI] [Google Scholar]
- Di Y.; Jiang A.; Huang H.; Luo Q.; Wei W.; Wang R.; Chen S. Molecular dynamics simulations of adsorption behavior of DDAH, NaOL and mixed DDAH/NaOL surfactants on muscovite (001) surface in aqueous solution. Journal of Molecular Graphics and Modelling 2022, 113, 108161. 10.1016/j.jmgm.2022.108161. [DOI] [PubMed] [Google Scholar]
- Wang L.; Hu Y.; Sun W.; Sun Y. Molecular dynamics simulation study of the interaction of mixed cationic/anionic surfactants with muscovite. Appl. Surf. Sci. 2015, 327, 364–370. 10.1016/j.apsusc.2014.11.160. [DOI] [Google Scholar]
- Zhang H.; Liu W.; Han C.; Wei D. Intensify dodecylamine adsorption on magnesite and dolomite surfaces by monohydric alcohols. Appl. Surf. Sci. 2018, 444, 729–738. 10.1016/j.apsusc.2018.03.124. [DOI] [Google Scholar]
- Li L.; Hao H.; Yuan Z.; Liu Z.; Li C. Regulating effects of citric acid and pregelatinized starch on selective flocculation flotation of micro-fine siderite. J. Mol. Liq. 2020, 315, 113726. 10.1016/j.molliq.2020.113726. [DOI] [Google Scholar]
- Jin J.; Gao H.; Chen X.; Peng Y.; Min F. The flotation of aluminosilicate polymorphic minerals with anionic and cationic collectors. Minerals Engineering 2016, 99, 123–132. 10.1016/j.mineng.2016.08.005. [DOI] [Google Scholar]
- Shrimali K.; Yin X.; Wang X.; Miller J. D. Fundamental issues on the influence of starch in amine adsorption by quartz. Colloids Surf., A 2017, 522, 642–651. 10.1016/j.colsurfa.2017.03.031. [DOI] [Google Scholar]
- Andreoni W.; Curioni A.; Grönbeck H. Density functional theory approach to thiols and disulfides on gold: Au (111) surface and clusters. Int. J. Quantum Chem. 2000, 80 (4–5), 598–608. . [DOI] [Google Scholar]
- Fajín J. L.; Cordeiro M. N. D.; Gomes J. R. Adsorption of atomic and molecular oxygen on the Au (321) surface: DFT study. J. Phys. Chem. C 2007, 111 (46), 17311–17321. 10.1021/jp073796y. [DOI] [Google Scholar]
- Irrera S.; Portalone G.; De Leeuw N. H. Chemisorption of uracil on gold surfaces via density functional theory. Surface science 2013, 614, 20–23. 10.1016/j.susc.2013.03.006. [DOI] [Google Scholar]
- Sun Q.; Deng T.-Y.; Chen J.-J.; Liu J.-Y.; Lu X.; Zhang Z.-X.; Li J.-H. Insights into the gold(i)-catalyzed intermolecular annulations of alkynes with N-allenamides: a mechanistic DFT study. Dalton Transactions 2022, 51 (9), 3734–3739. 10.1039/D1DT04028F. [DOI] [PubMed] [Google Scholar]
- Liu C.; Li Y.; Cheng Q.; Zhao Y. Atomic Model of Gold Adsorption onto the Pyrite Surface with DFT Study. Minerals 2022, 12 (3), 387. 10.3390/min12030387. [DOI] [Google Scholar]
- Du H.; Miller J. D. Adsorption states of amphipatic solutes at the surface of naturally hydrophobic minerals: a molecular dynamics simulation study. Langmuir 2007, 23 (23), 11587–11596. 10.1021/la701604u. [DOI] [PubMed] [Google Scholar]
- Sime F. M.; Jin J.; Wang X.; Wick C. D.; Miller J. D. Characterization and simulation of graphite edge surfaces for the analysis of carbonaceous material separation from sulfide ores by flotation. Minerals Engineering 2022, 182, 107590. 10.1016/j.mineng.2022.107590. [DOI] [Google Scholar]
- Gocmez H. The interaction of organic dispersant with alumina: A molecular modelling approach. Ceramics international 2006, 32 (5), 521–525. 10.1016/j.ceramint.2005.04.005. [DOI] [Google Scholar]
- Luo J.; Liu M.; Xing Y.; Gui X.; Li J. Investigating agglomeration of kaolinite particles in the presence of dodecylamine by force testing and molecular dynamics simulation. Colloids Surf., A 2022, 645, 128930. 10.1016/j.colsurfa.2022.128930. [DOI] [Google Scholar]
- Abbaspour Tamijani A.; Augustine L. J.; Bjorklund J. L.; Catalano J. G.; Mason S. E. First-principles characterisation and comparison of clean, hydrated, and defect α-Al2O3 and α-Fe2O3 (110) surfaces. Mol. Simul. 2022, 48 (3), 247–263. 10.1080/08927022.2021.2009117. [DOI] [Google Scholar]
- Ou S.; Heimann J. E.; Bennett J. W. A Density Functional Theory (DFT) Investigation of Sulfur-Based Adsorbate Interactions on Alumina and Calcite Surfaces. Clays and Clay Minerals 2022, 70 (3), 370–385. 10.1007/s42860-022-00194-5. [DOI] [Google Scholar]
- Hung A.; Yarovsky I.; Russo S. P. Density-functional theory of xanthate adsorption on the pyrite FeS2 (100) surface. Philosophical magazine letters 2004, 84 (3), 175–182. 10.1080/09500830310001628248. [DOI] [Google Scholar]
- Hung A.; Yarovsky I.; Russo S. P. Density-functional theory studies of xanthate adsorption on the pyrite FeS 2 (110) and (111) surfaces. J. Chem. Phys. 2003, 118 (13), 6022–6029. 10.1063/1.1556076. [DOI] [Google Scholar]
- Chen J.; Lan L.; Chen Y. Computational simulation of adsorption and thermodynamic study of xanthate, dithiophosphate and dithiocarbamate on galena and pyrite surfaces. Minerals Engineering 2013, 46, 136–143. 10.1016/j.mineng.2013.03.015. [DOI] [Google Scholar]
- Liu Y.; Li Y.; Chen J. Competitive adsorption of water and collector molecules on sulfide mineral surfaces. Chem. Phys. 2022, 563, 111681. 10.1016/j.chemphys.2022.111681. [DOI] [Google Scholar]
- Dai P.; Chen H.; Chen L.; Liu Y.; Wei Z. Depression mechanism of peracetic acid for flotation separation of chalcopyrite from arsenopyrite based on coordination chemistry. Minerals Engineering 2022, 186, 107757. 10.1016/j.mineng.2022.107757. [DOI] [Google Scholar]
- Pradip; Rai B.; Rao T.; Krishnamurthy S.; Vetrivel R.; Mielczarski J.; Cases J. Molecular modeling of interactions of diphosphonic acid based surfactants with calcium minerals. Langmuir 2002, 18 (3), 932–940. 10.1021/la010625q. [DOI] [Google Scholar]
- Cooper T. G.; de Leeuw N. H. A computer simulation study of sorption of model flotation reagents to planar and stepped {111} surfaces of calcium fluoride. J. Mater. Chem. 2004, 14 (13), 1927–1935. 10.1039/b402088j. [DOI] [Google Scholar]
- He J.; Hu Y.; Sun W.; Zhang C.; Zhang C.; Lin S.; Zou J. Computational insights into the adsorption mechanism of gallic acid-bearing reagents on calcium-bearing mineral surfaces. Minerals Engineering 2020, 156, 106485. 10.1016/j.mineng.2020.106485. [DOI] [Google Scholar]
- Kundu T.; Rao K. H.; Parker S. Atomistic simulation studies of magnetite surface structures and adsorption behavior in the presence of molecular and dissociated water and formic acid. J. Colloid Interface Sci. 2006, 295 (2), 364–373. 10.1016/j.jcis.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Fa K.; Nguyen A. V.; Miller J. D. Interaction of calcium dioleate collector colloids with calcite and fluorite surfaces as revealed by AFM force measurements and molecular dynamics simulation. Int. J. Miner. Process. 2006, 81 (3), 166–177. 10.1016/j.minpro.2006.08.006. [DOI] [Google Scholar]
- Semmeq A.; Foucaud Y.; El Yamami N.; Michailovski A.; Lebègue S.; Badawi M. Hydration of magnesite and dolomite minerals: new insights from ab initio molecular dynamics. Colloids Surf., A 2021, 631, 127697. 10.1016/j.colsurfa.2021.127697. [DOI] [Google Scholar]
- Wulff G. XXV. Zur Frage der Geschwindigkeit des Wachsthums und der Auflösung der Krystallflächen. Zeitschrift für Kristallographie - Crystalline Materials 1901, 34 (1–6), 449–530. 10.1524/zkri.1901.34.1.449. [DOI] [Google Scholar]
- Dobrushin R. L.; Kotecký R.; Shlosman S.. Wulff construction: a global shape from local interaction; American Mathematical Society: Providence, 1992; Vol. 104. [Google Scholar]
- Suryanarayana C.Non-equilibrium processing of materials; Elsevier, 1999. [Google Scholar]
- Feng D.; Aldrich C. A comparison of the flotation of ore from the Merensky Reef after wet and dry grinding. Int. J. Miner. Process. 2000, 60 (2), 115–129. 10.1016/S0301-7516(00)00010-7. [DOI] [Google Scholar]
- Ai Z.; Li S.; Zhao Y.; Yi H.; Chen L.; Chen P.; Nie G.; Song S. Atomic insights into flotation separation of KCl and NaCl from a new viewpoint of hydration layer: A molecular dynamic study. Colloids Surf., A 2020, 602, 125071. 10.1016/j.colsurfa.2020.125071. [DOI] [Google Scholar]
- Tang Y.; Kelebek S.; Yin W. Surface chemistry of magnesite and calcite flotation and molecular dynamics simulation of their cetyl phosphate adsorption. Colloids Surf., A 2020, 603, 125246. 10.1016/j.colsurfa.2020.125246. [DOI] [Google Scholar]
- Tang Y.; Yin W.; Kelebek S. Molecular dynamics simulation of magnesite and dolomite in relation to flotation with cetyl phosphate. Colloids Surf., A 2021, 610, 125928. 10.1016/j.colsurfa.2020.125928. [DOI] [Google Scholar]
- Liu W.; Liu W.; Zhao B.; Zhao L.; Li D.; Fang P.; Liu W. Novel insights into the adsorption mechanism of the isopropanol amine collector on magnesite ore: A combined experimental and theoretical computational study. Powder Technol. 2019, 343, 366–374. 10.1016/j.powtec.2018.11.063. [DOI] [Google Scholar]
- Quezada G. R.; Jeldres M.; Toro N.; Robles P.; Toledo P. G.; Jeldres R. I. Understanding the flocculation mechanism of quartz and kaolinite with polyacrylamide in seawater: A molecular dynamics approach. Colloids Surf., A 2021, 608, 125576. 10.1016/j.colsurfa.2020.125576. [DOI] [Google Scholar]
- Luo Q.; Liu W.; Zhuo Q.; Han Y. Interaction mechanism of organic carboxylate with kaolinite and montmorillonite: A density functional theory study. Colloids Surf., A 2021, 613, 126047. 10.1016/j.colsurfa.2020.126047. [DOI] [Google Scholar]
- Sarvaramini A.; Larachi F. Understanding the interactions of thiophosphorus collectors with chalcopyrite through DFT simulation. Comput. Mater. Sci. 2017, 132, 137–145. 10.1016/j.commatsci.2017.02.027. [DOI] [Google Scholar]
- Azizi D.; Larachi F. Surface interactions and flotation behavior of calcite, dolomite and ankerite with alkyl hydroxamic acid bearing collector and sodium silicate. Colloids Surf., A 2018, 537, 126–138. 10.1016/j.colsurfa.2017.09.054. [DOI] [Google Scholar]
- Cao Q.; Zou H.; Chen X.; Wen S. Flotation selectivity of N-hexadecanoylglycine in the fluorapatite–dolomite system. Minerals Engineering 2019, 131, 353–362. 10.1016/j.mineng.2018.11.033. [DOI] [Google Scholar]
- Jiao F.; Wu J.; Qin W.; Wang X.; Liu R. Interactions of tert dodecyl mercaptan with sphalerite and effects on its flotation behavior. Colloids Surf., A 2016, 506, 104–113. 10.1016/j.colsurfa.2016.05.095. [DOI] [Google Scholar]
- Liu G.; Zeng H.; Lu Q.; Zhong H.; Choi P.; Xu Z. Adsorption of mercaptobenzoheterocyclic compounds on sulfide mineral surfaces: A density functional theory study of structure–reactivity relations. Colloids Surf., A 2012, 409, 1–9. 10.1016/j.colsurfa.2012.04.036. [DOI] [Google Scholar]
- Liu G.; Xiao J.; Zhou D.; Zhong H.; Choi P.; Xu Z. A DFT study on the structure-reactivity relationship of thiophosphorus acids as flotation collectors with sulfide minerals: Implication of surface adsorption. Colloids Surf., A 2013, 434, 243–252. 10.1016/j.colsurfa.2013.05.050. [DOI] [Google Scholar]
- Xie J.; Li X.; Mao S.; Li L.; Ke B.; Zhang Q. Effects of structure of fatty acid collectors on the adsorption of fluorapatite (0 0 1) surface: A first-principles calculations. Appl. Surf. Sci. 2018, 444, 699–709. 10.1016/j.apsusc.2018.03.105. [DOI] [Google Scholar]
- Zhao L.; Liu W.; Duan H.; Wang X.; Fang P.; Liu W.; Zhou X.; Shen Y. Design and selection of flotation collectors for zinc oxide minerals based on bond valence model. Minerals Engineering 2021, 160, 106681. 10.1016/j.mineng.2020.106681. [DOI] [Google Scholar]
- Duan H.; Liu W.; Wang X.; Liu W.; Zhang X. Effect of secondary amino on the adsorption of N-Dodecylethylenediamine on quartz surface: A molecular dynamics study. Powder Technol. 2019, 351, 46–53. 10.1016/j.powtec.2019.04.009. [DOI] [Google Scholar]
- Liu W.; Liu W.; Wang B.; Zhao Q.; Duan H.; Chen X. Molecular-level insights into the adsorption of a hydroxy-containing tertiary amine collector on the surface of magnesite ore. Powder Technol. 2019, 355, 700–707. 10.1016/j.powtec.2019.07.098. [DOI] [Google Scholar]
- Liu W.; Peng X.; Liu W.; Wang X.; Zhao Q.; Wang B. Effect mechanism of the iso-propanol substituent on amine collectors in the flotation of quartz and magnesite. Powder Technol. 2020, 360, 1117–1125. 10.1016/j.powtec.2019.10.060. [DOI] [Google Scholar]
- Li B.; Liu S.; Fan M.; Zhang L. The effect of ethylene oxide groups in dodecyl ethoxyl ethers on low rank coal flotation: An experimental study and simulation. Powder Technol. 2019, 344, 684–692. 10.1016/j.powtec.2018.12.063. [DOI] [Google Scholar]
- Li B.; Guo J.; Albijanic B.; Liu S.; Zhang L.; Sun X. Understanding flotation mechanism of nonionic surfactants with different polarity on kaolinite as a gangue mineral: An experimental and simulation study. Minerals Engineering 2020, 148, 106226. 10.1016/j.mineng.2020.106226. [DOI] [Google Scholar]
- Xu Y.; Liu Y.-L.; Liu G.-S. Molecular dynamics simulation of primary ammonium ions with different alkyl chains on the muscovite (001) surface. Int. J. Miner. Process. 2015, 145, 48–56. 10.1016/j.minpro.2015.07.003. [DOI] [Google Scholar]
- Xue X.; Xu Z.; Pedruzzi I.; Li P.; Yu J. Interaction between low molecular weight carboxylic acids and muscovite: Molecular dynamic simulation and experiment study. Colloids Surf., A 2018, 559, 8–17. 10.1016/j.colsurfa.2018.09.033. [DOI] [Google Scholar]
- Liu M.; Chen J.; Chen Y.; Zhu Y. Interaction between smithsonite and carboxyl collectors with different molecular structure in the presence of water: A theoretical and experimental study. Appl. Surf. Sci. 2020, 510, 145410. 10.1016/j.apsusc.2020.145410. [DOI] [Google Scholar]
- Yang X.; Albijanic B.; Liu G.; Zhou Y. Structure–activity relationship of xanthates with different hydrophobic groups in the flotation of pyrite. Minerals Engineering 2018, 125, 155–164. 10.1016/j.mineng.2018.05.032. [DOI] [Google Scholar]
- Zhang H.; Liu W.; Han C.; Hao H. Effects of monohydric alcohols on the flotation of magnesite and dolomite by sodium oleate. J. Mol. Liq. 2018, 249, 1060–1067. 10.1016/j.molliq.2017.11.148. [DOI] [Google Scholar]
- Han C.; Zhang H.; Tan R.; Shen Y.; Wei D.; Liu W. Effects of monohydric alcohols of varying chain lengths and isomeric structures on magnesite and dolomite flotation by dodecylamine. Powder Technol. 2020, 374, 233–240. 10.1016/j.powtec.2020.07.048. [DOI] [Google Scholar]
- Liu A.; Fan M.-q.; Li Z.-h.; Fan J.-c. Non-polar oil assisted DDA flotation of quartz II: Effect of different polarity oil components on the flotation of quartz. Int. J. Miner. Process. 2017, 168, 25–34. 10.1016/j.minpro.2017.09.003. [DOI] [Google Scholar]
- Cheng Y.; Chen J.; Min F.; Wang L.; Shu Q. Research on the flotation efficiency of alcohol/ether alcohol frothers for common collectors: Insight of molecular dynamics simulations. Appl. Surf. Sci. 2023, 614, 156233. 10.1016/j.apsusc.2022.156233. [DOI] [Google Scholar]
- Nagaraj D.; Farinato R. Evolution of flotation chemistry and chemicals: a century of innovations and the lingering challenges. Minerals Engineering 2016, 96, 2–14. 10.1016/j.mineng.2016.06.019. [DOI] [Google Scholar]
- Pradip; Fuerstenau D. W. Design and development of novel flotation reagents for the beneficiation of Mountain Pass rare-earth ore. Mining, Metallurgy & Exploration 2013, 30 (1), 1–9. 10.1007/BF03402335. [DOI] [Google Scholar]
- Liu W.; Wang X.; Miller J. D. Collector chemistry for bastnaesite flotation–recent developments. Mineral Processing and Extractive Metallurgy Review 2019, 40 (6), 370–379. 10.1080/08827508.2019.1647840. [DOI] [Google Scholar]
- Abdalla M. A.; Peng H.; Wu D.; Abusin L.; Mbah T. J. Prediction of hydrophobic reagent for flotation process using molecular modeling. ACS omega 2018, 3 (6), 6483–6496. 10.1021/acsomega.8b00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greathouse J. A.; Johnson K. L.; Greenwell H. C. Interaction of natural organic matter with layered minerals: recent developments in computational methods at the nanoscale. Minerals 2014, 4 (2), 519–540. 10.3390/min4020519. [DOI] [Google Scholar]
- Rai B.; Pradip Pradip, Design of highly selective industrial performance chemicals: a molecular modelling approach. Mol. Simul. 2008, 34 (10–15), 1209–1214. 10.1080/08927020802350927. [DOI] [Google Scholar]
- Pradip; Rai B. Molecular modeling and rational design of flotation reagents. Int. J. Miner. Process. 2003, 72 (1–4), 95–110. 10.1016/S0301-7516(03)00090-5. [DOI] [Google Scholar]
- Yang X.; Huang Y.; Liu G.; Liu J.; Ma L.; Niu X.; Qu X. A DFT prediction on the chemical reactivity of novel azolethione derivatives as chelating agents: Implications for copper minerals flotation and copper corrosion inhibition. Journal of the Taiwan Institute of Chemical Engineers 2018, 93, 109–123. 10.1016/j.jtice.2018.09.022. [DOI] [Google Scholar]
- Ma X.; Hu Y.; Zhong H.; Wang S.; Liu G.; Zhao G. A novel surfactant S-benzoyl-N, N-diethyldithiocarbamate synthesis and its flotation performance to galena. Appl. Surf. Sci. 2016, 365, 342–351. 10.1016/j.apsusc.2016.01.048. [DOI] [Google Scholar]
- Zhao K.; Wang X.; Wang Z.; Yan W.; Zhou X.; Xu L.; Wang C. A novel depressant for selective flotation separation of pyrite and pyrophyllite. Appl. Surf. Sci. 2019, 487, 9–16. 10.1016/j.apsusc.2019.04.252. [DOI] [Google Scholar]
- Roy S.; Wu L.; Goverapet Srinivasan S.; Stack A. G.; Navrotsky A.; Bryantsev V. S. Hydration structure and water exchange kinetics at xenotime–water interfaces: implications for rare earth minerals separation. Phys. Chem. Chem. Phys. 2020, 22 (15), 7719–7727. 10.1039/D0CP00087F. [DOI] [PubMed] [Google Scholar]
- Patra A. S.; Nulakani N. V. R.; Pavan Kumar Y.; Subramanian V.; Dash J.; Mukherjee A.K. Design and synthesis of novel polyamine collector to recover iron values from iron ore slimes. Powder Technol. 2018, 325, 180–191. 10.1016/j.powtec.2017.10.045. [DOI] [Google Scholar]
- Lu J.; Sun M.; Yuan Z.; Qi S.; Tong Z.; Li L.; Meng Q. Innovative insight for sodium hexametaphosphate interaction with serpentine. Colloids Surf., A 2019, 560, 35–41. 10.1016/j.colsurfa.2018.09.076. [DOI] [Google Scholar]
- Cheng Z.; Zhu Y.; Li Y.; Han Y. Flotation and adsorption of quartz with the new collector butane-3-heptyloxy-1, 2-diamine. Mineralogy and Petrology 2019, 113 (2), 207–216. 10.1007/s00710-018-0639-y. [DOI] [Google Scholar]
- Huang X.; Jia Y.; Wang S.; Ma X.; Cao Z.; Zhong H. Novel sodium O-benzythioethyl xanthate surfactant: synthesis, DFT calculation and adsorption mechanism on chalcopyrite surface. Langmuir 2019, 35 (47), 15106–15113. 10.1021/acs.langmuir.9b03118. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Qian Z.; Zheng G.; Zhu Y.; Wu W. The design of a macromolecular depressant for galena based on DFT studies and its application. Minerals Engineering 2017, 112, 50–56. 10.1016/j.mineng.2017.07.007. [DOI] [Google Scholar]
- Zhang W.; Feng Z.; Yang Y.; Sun W.; Pooley S.; Cao J.; Gao Z. Bi-functional hydrogen and coordination bonding surfactant: A novel and promising collector for improving the separation of calcium minerals. J. Colloid Interface Sci. 2021, 585, 787–799. 10.1016/j.jcis.2020.10.059. [DOI] [PubMed] [Google Scholar]
- Guo W.; Han Y.; Zhu Y.; Li Y.; Tang Z. Effect of amide group on the flotation performance of lauric acid. Appl. Surf. Sci. 2020, 505, 144627. 10.1016/j.apsusc.2019.144627. [DOI] [Google Scholar]
- Jong K.; Paek I.; Kim Y.; Li I.; Jang D. Flotation mechanism of a novel synthesized collector from Evodiaefructus onto fluorite surfaces. Minerals Engineering 2020, 146, 106017. 10.1016/j.mineng.2019.106017. [DOI] [Google Scholar]
- Wang C.; Liu R.; Wu M.; Xu Z.; Tian M.; Yin Z.; Sun W.; Zhang C. Flotation separation of molybdenite from chalcopyrite using rhodanine-3-acetic acid as a novel and effective depressant. Minerals Engineering 2021, 162, 106747. 10.1016/j.mineng.2020.106747. [DOI] [Google Scholar]
- Yin Z.; Chen S.; Xu Z.; Zhang C.; He J.; Zou J.; Chen D.; Sun W. Flotation separation of molybdenite from chalcopyrite using an environmentally-efficient depressant L-cysteine and its adsoption mechanism. Minerals Engineering 2020, 156, 106438. 10.1016/j.mineng.2020.106438. [DOI] [Google Scholar]
- Xia Y.; Yang Z.; Zhang R.; Xing Y.; Gui X. Enhancement of the surface hydrophobicity of low-rank coal by adsorbing DTAB: An experimental and molecular dynamics simulation study. Fuel 2019, 239, 145–152. 10.1016/j.fuel.2018.10.156. [DOI] [Google Scholar]
- Sun H.; Yang B.; Zhu Z.; Yin W.; Sheng Q.; Hou Y.; Yao J. New insights into selective-depression mechanism of novel depressant EDTMPS on magnesite and quartz surfaces: Adsorption mechanism, DFT calculations, and adsorption model. Minerals Engineering 2021, 160, 106660. 10.1016/j.mineng.2020.106660. [DOI] [Google Scholar]
- Chi X.; Guo Y.; Zhong S.; Li G.; Lv X. Molecular modelling and synthesis of a new collector O-butyl S-(1-chloroethyl) carbonodithioate for copper sulfide ore and its flotation behavior. RSC Adv. 2020, 10 (6), 3520–3528. 10.1039/C9RA09648E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Liu W.; Zhao Q.; Peng X.; Wang B.; Zhou S.; Zhao L. Investigating the performance of a novel polyamine derivative for separation of quartz and hematite based on theoretical prediction and experiment. Sep. Purif. Technol. 2020, 237, 116370. 10.1016/j.seppur.2019.116370. [DOI] [Google Scholar]
- Tian M.; Khoso S. A.; Wang L.; Sun W.; Zhang C.; Hu Y. Selective separation behavior and its molecular mechanism of cassiterite from quartz using Cupferron as a novel flotation collector with a lower dosage of Pb2+ ions. Appl. Surf. Sci. 2019, 486, 228–238. 10.1016/j.apsusc.2019.05.039. [DOI] [Google Scholar]
- Costa D.; Savio L.; Pradier C.-M. Adsorption of amino acids and peptides on metal and oxide surfaces in water environment: a synthetic and prospective review. J. Phys. Chem. B 2016, 120 (29), 7039–7052. 10.1021/acs.jpcb.6b05954. [DOI] [PubMed] [Google Scholar]
- Azizi D.; Larachi F.; Garnier A.; Lagüe P.; Levasseur B. Sorption of aqueous amino acid species on sulphidic mineral surfaces—DFT study and insights on biosourced-reagent mineral flotation. Canadian Journal of Chemical Engineering 2021, 99 (8), 1758–1779. 10.1002/cjce.23841. [DOI] [Google Scholar]
- daFonseca B. G.; Costa L. A. S.; Sant’Ana A. C. Insights of adsorption mechanisms of Trp-peptides on plasmonic surfaces by SERS. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2018, 190, 383–391. 10.1016/j.saa.2017.09.039. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Wang J.; Zhu S.; Yang X.; Jia Y.; Sun Q.; Guan S. A DFT study of the adsorption of short peptides on Mg and Mg-based alloy surfaces. Phys. Chem. Chem. Phys. 2018, 20 (5), 3602–3607. 10.1039/C7CP07431J. [DOI] [PubMed] [Google Scholar]
- Aquino A. J.; Tunega D.; Gerzabek M. H.; Lischka H. Modeling catalytic effects of clay mineral surfaces on peptide bond formation. J. Phys. Chem. B 2004, 108 (28), 10120–10130. 10.1021/jp049149d. [DOI] [Google Scholar]
- Carravetta V.; Monti S. Peptide– TiO2 surface interaction in solution by ab initio and molecular dynamics simulations. J. Phys. Chem. B 2006, 110 (12), 6160–6169. 10.1021/jp056760j. [DOI] [PubMed] [Google Scholar]
- Heinz H.; Farmer B. L.; Pandey R. B.; Slocik J. M.; Patnaik S. S.; Pachter R.; Naik R. R. Nature of molecular interactions of peptides with gold, palladium, and Pd– Au bimetal surfaces in aqueous solution. J. Am. Chem. Soc. 2009, 131 (28), 9704–9714. 10.1021/ja900531f. [DOI] [PubMed] [Google Scholar]
- Feng J.; Pandey R. B.; Berry R. J.; Farmer B. L.; Naik R. R.; Heinz H. Adsorption mechanism of single amino acid and surfactant molecules to Au {111} surfaces in aqueous solution: design rules for metal-binding molecules. Soft Matter 2011, 7 (5), 2113–2120. 10.1039/c0sm01118e. [DOI] [Google Scholar]
- Seker U. O. S.; Wilson B.; Dincer S.; Kim I. W.; Oren E. E.; Evans J. S.; Tamerler C.; Sarikaya M. Adsorption behavior of linear and cyclic genetically engineered platinum binding peptides. Langmuir 2007, 23 (15), 7895–7900. 10.1021/la700446g. [DOI] [PubMed] [Google Scholar]
- Almora-Barrios N.; de Leeuw N. H. Modelling the interaction of a Hyp-Pro-Gly peptide with hydroxyapatite surfaces in aqueous environment. CrystEngComm 2010, 12 (3), 960–967. 10.1039/B917179G. [DOI] [Google Scholar]
- Almora-Barrios N.; de Leeuw N. H. A density functional theory study of the interaction of collagen peptides with hydroxyapatite surfaces. Langmuir 2010, 26 (18), 14535–14542. 10.1021/la101151e. [DOI] [PubMed] [Google Scholar]
- Azizi D.; Larachi F.; Latifi M. Ionic-liquid collectors for rare-earth minerals flotation-Case of tetrabutylammonium bis (2-ethylhexyl)-phosphate for monazite and bastnäsite recovery. Colloids Surf., A 2016, 506, 74–86. 10.1016/j.colsurfa.2016.06.011. [DOI] [Google Scholar]
- Sahoo H.; Rath S. S.; Das B. A review on the application of quaternary ammonium-based ionic liquids in mineral flotation. Mineral Processing and Extractive Metallurgy Review 2020, 41 (6), 405–416. 10.1080/08827508.2019.1635472. [DOI] [Google Scholar]
- Azizi D., Phosphonium/ammonium-based ionic liquids for rare earth minerals beneficiation: case of monazite and bastnäsite. Ph.D. Thesis, Université Laval, Québec, Canada, 2018. [Google Scholar]
- Plechkova N. V.; Seddon K. R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37 (1), 123–150. 10.1039/B006677J. [DOI] [PubMed] [Google Scholar]
- Azizi D.; Larachi F. Immiscible dual ionic liquid-ionic liquid mineral separation of rare-earth minerals. Sep. Purif. Technol. 2018, 191, 340–353. 10.1016/j.seppur.2017.09.061. [DOI] [Google Scholar]
- Azizi D.; Sarvaramini A.; Larachi F. Liquid-liquid mineral separation via ionic-liquid complexation of monazite and bastnäsite—An alternate route for rare-earth mineral beneficiation. Colloids Surf., A 2017, 520, 301–323. 10.1016/j.colsurfa.2017.01.079. [DOI] [Google Scholar]
- Xu L.; Tian J.; Wu H.; Fang S.; Lu Z.; Ma C.; Sun W.; Hu Y. Anisotropic surface chemistry properties and adsorption behavior of silicate mineral crystals. Adv. Colloid Interface Sci. 2018, 256, 340–351. 10.1016/j.cis.2018.02.004. [DOI] [PubMed] [Google Scholar]
- Kundu T.K; Hanumantha Rao K; Parker S.C Atomistic simulation of the surface structure of wollastonite and adsorption phenomena relevant to flotation. Int. J. Miner. Process. 2003, 72 (1–4), 111–127. 10.1016/S0301-7516(03)00092-9. [DOI] [Google Scholar]
- de Oliveira C. u.; de Lima G. F.; de Abreu H. A.; Duarte H. l. A. Reconstruction of the Chalcopyrite Surfaces— A DFT Study. J. Phys. Chem. C 2012, 116 (10), 6357–6366. 10.1021/jp300713z. [DOI] [Google Scholar]
- de Lima G. F.; de Oliveira C.; de Abreu H. A.; Duarte H. A. Water adsorption on the reconstructed (001) chalcopyrite surfaces. J. Phys. Chem. C 2011, 115 (21), 10709–10717. 10.1021/jp201106e. [DOI] [Google Scholar]
- Srinivasan S. G.; Shivaramaiah R.; Kent P. R.; Stack A. G.; Navrotsky A.; Riman R.; Anderko A.; Bryantsev V. S. Crystal structures, surface stability, and water adsorption energies of La-bastnäsite via density functional theory and experimental studies. J. Phys. Chem. C 2016, 120 (30), 16767–16781. 10.1021/acs.jpcc.6b04747. [DOI] [Google Scholar]
- Xu L.; Peng T.; Tian J.; Lu Z.; Hu Y.; Sun W. Anisotropic surface physicochemical properties of spodumene and albite crystals: Implications for flotation separation. Appl. Surf. Sci. 2017, 426, 1005–1022. 10.1016/j.apsusc.2017.07.295. [DOI] [Google Scholar]
- Xu L.; Tian J.; Wu H.; Deng W.; Yang Y.; Sun W.; Gao Z.; Hu Y. New insights into the oleate flotation response of feldspar particles of different sizes: Anisotropic adsorption model. J. Colloid Interface Sci. 2017, 505, 500–508. 10.1016/j.jcis.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Pollet-Villard M.; Daval D.; Ackerer P.; Saldi G.; Knauss K.; Wild B.; Fritz B. Experimental study of dissolution kinetics of K-feldspar as a function of crystal structure anisotropy under hydrothermal conditions. Procedia Earth and Planetary Science 2017, 17, 165–168. 10.1016/j.proeps.2016.12.042. [DOI] [Google Scholar]
- Tian J.; Xu L.; Deng W.; Jiang H.; Gao Z.; Hu Y. Adsorption mechanism of new mixed anionic/cationic collectors in a spodumene-feldspar flotation system. Chem. Eng. Sci. 2017, 164, 99–107. 10.1016/j.ces.2017.02.013. [DOI] [Google Scholar]
- Zhu G.; Wang X.; Li E.; Wang Y.; Miller J. D. Wetting characteristics of spodumene surfaces as influenced by collector adsorption. Minerals Engineering 2019, 130, 117–128. 10.1016/j.mineng.2018.10.010. [DOI] [Google Scholar]
- Zheng R.; Ren Z.; Gao H.; Chen Z.; Qian Y.; Li Y. Effects of crystal chemistry on sodium oleate adsorption on fluorite surface investigated by molecular dynamics simulation. Minerals Engineering 2018, 124, 77–85. 10.1016/j.mineng.2018.05.017. [DOI] [Google Scholar]
- Xu L.; Hu Y.; Wu H.; Tian J.; Liu J.; Gao Z.; Wang L. Surface crystal chemistry of spodumene with different size fractions and implications for flotation. Sep. Purif. Technol. 2016, 169, 33–42. 10.1016/j.seppur.2016.06.005. [DOI] [Google Scholar]
- Cao S.; Cao Y.; Ma Z.; Liao Y.; Zhang X. Structural and electronic properties of bastnaesite and implications for surface reactions in flotation. Journal of Rare Earths 2020, 38 (3), 332–338. 10.1016/j.jre.2019.04.020. [DOI] [Google Scholar]
- Wang J.; He Y.; Peng Z.; Ling X.; Wang S. Estimation of hydrophilicity of coals by using the quantum chemistry calculation. Int. J. Miner. Process. 2017, 167, 9–15. 10.1016/j.minpro.2017.07.015. [DOI] [Google Scholar]
- Shen L.; Wang C.; Min F.; Liu L.; Xue C. Effect of pores on the flotation of low-rank coal: An experiment and simulation study. Fuel 2020, 271, 117557. 10.1016/j.fuel.2020.117557. [DOI] [Google Scholar]
- Zhang R.; Xing Y.; Xia Y.; Luo J.; Tan J.; Rong G.; Gui X. New insight into surface wetting of coal with varying coalification degree: An experimental and molecular dynamics simulation study. Appl. Surf. Sci. 2020, 511, 145610. 10.1016/j.apsusc.2020.145610. [DOI] [Google Scholar]
- Chen J.; Chen Y. A first-principle study of the effect of vacancy defects and impurities on the adsorption of O2 on sphalerite surfaces. Colloids Surf., A 2010, 363 (1), 56–63. 10.1016/j.colsurfa.2010.04.013. [DOI] [Google Scholar]
- Nguyen H. T.; Nguyen M. T. Effects of Sulfur-Deficient Defect and Water on Rearrangements of Formamide on Pyrite (100) Surface. J. Phys. Chem. A 2014, 118 (23), 4079–4086. 10.1021/jp5013945. [DOI] [PubMed] [Google Scholar]
- Rozgonyi T.; Stirling A. DFT Study of Oxidation States on Pyrite Surface Sites. J. Phys. Chem. C 2015, 119 (14), 7704–7710. 10.1021/acs.jpcc.5b01943. [DOI] [Google Scholar]
- Dzade N. Y.; de Leeuw N. H. Adsorption and Desulfurization Mechanism of Thiophene on Layered FeS(001), (011), and (111) Surfaces: A Dispersion-Corrected Density Functional Theory Study. J. Phys. Chem. C 2018, 122 (1), 359–370. 10.1021/acs.jpcc.7b08711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Yano T.-a.; Hara M.; Ebisuzaki T. Cysteine and cystine adsorption on FeS2(100). Surf. Sci. 2018, 674, 6–12. 10.1016/j.susc.2018.03.011. [DOI] [Google Scholar]
- Galvez-Martinez S.; Escamilla-Roa E.; Zorzano M.-P.; Mateo-Marti E. Defects on a pyrite(100) surface produce chemical evolution of glycine under inert conditions: experimental and theoretical approaches. Phys. Chem. Chem. Phys. 2019, 21 (44), 24535–24542. 10.1039/C9CP03577J. [DOI] [PubMed] [Google Scholar]
- Yi H.; Zhang X.; Jia F.; Wei Z.; Zhao Y.; Song S. Competition of Hg2+ adsorption and surface oxidation on MoS2 surface as affected by sulfur vacancy defects. Appl. Surf. Sci. 2019, 483, 521–528. 10.1016/j.apsusc.2019.03.350. [DOI] [Google Scholar]
- Luo Y.; Ou L.; Chen J.; Zhang G.; Xia Y.; Zhu B.; Zhou H. Effects of defects and impurities on the adsorption of H2O on smithsonite (101) surfaces: Insight from DFT-D and MD. Colloids Surf., A 2021, 628, 127300. 10.1016/j.colsurfa.2021.127300. [DOI] [Google Scholar]
- Azizi D.; Larachi F. Surface Speciation of Brucite Dissolution in Aqueous Mineral Carbonation: Insights from Density-Functional Theory Simulations. J. Phys. Chem. A 2019, 123 (4), 889–905. 10.1021/acs.jpca.8b09140. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Li Y.; Chen J.; Kang D.; Yang X. Influence of sulfur vacancy on pyrite oxidization by water and oxygen molecules. Colloids Surf., A 2022, 634, 127954. 10.1016/j.colsurfa.2021.127954. [DOI] [Google Scholar]
- Ataman E.; Andersson M.; Ceccato M.; Bovet N.; Stipp S. Functional group adsorption on calcite: I. Oxygen containing and nonpolar organic molecules. J. Phys. Chem. C 2016, 120 (30), 16586–16596. 10.1021/acs.jpcc.6b01349. [DOI] [Google Scholar]
- Ataman E.; Andersson M.; Ceccato M.; Bovet N.; Stipp S. Functional group adsorption on calcite: II. Nitrogen and sulfur containing organic molecules. J. Phys. Chem. C 2016, 120 (30), 16597–16607. 10.1021/acs.jpcc.6b01359. [DOI] [Google Scholar]
- Escamilla-Roa E.; Sainz-Díaz C. I.; Huertas F. J.; Hernandez-Laguna A. Adsorption of molecules onto (1014) dolomite surface: An application of computational studies for microcalorimetry. J. Phys. Chem. C 2013, 117 (34), 17583–17590. 10.1021/jp404529e. [DOI] [Google Scholar]
- Chen Y.; Chen J.; Guo J. A DFT study on the effect of lattice impurities on the electronic structures and floatability of sphalerite. Minerals Engineering 2010, 23 (14), 1120–1130. 10.1016/j.mineng.2010.07.005. [DOI] [Google Scholar]
- Li Y.-Q.; Chen J.-H.; Chen Y.; Guo J. Density functional theory study of influence of impurity on electronic properties and reactivity of pyrite. Transactions of Nonferrous Metals Society of China 2011, 21 (8), 1887–1895. 10.1016/S1003-6326(11)60946-1. [DOI] [Google Scholar]
- Chen J.; Li Y.; Zhao C. First principles study of the occurrence of gold in pyrite. Computational materials science 2014, 88, 1–6. 10.1016/j.commatsci.2014.02.033. [DOI] [Google Scholar]
- Azizi D.; Larachi F. DFT simulations of pyrite galvanic interactions with bulk, solid-solution and nanoparticle Au occurrences–Insights into gold cyanidation. Minerals Engineering 2020, 149, 106239. 10.1016/j.mineng.2020.106239. [DOI] [Google Scholar]
- Atluri V.; Jin J.; Shrimali K.; Dang L.; Wang X.; Miller J. D. The hydrophobic surface state of talc as influenced by aluminum substitution in the tetrahedral layer. J. Colloid Interface Sci. 2019, 536, 737–748. 10.1016/j.jcis.2018.10.085. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yi H.; Zhao Y.; Min F.; Song S. Study on the differences of Na-and Ca-montmorillonites in crystalline swelling regime through molecular dynamics simulation. Advanced Powder Technology 2016, 27 (2), 779–785. 10.1016/j.apt.2016.03.005. [DOI] [Google Scholar]
- Zhu G.; Cao Y.; Wang Y.; Wang X.; Miller J. D.; Lu D.; Zheng X. Surface chemistry features of spodumene with isomorphous substitution. Minerals Engineering 2020, 146, 106139. 10.1016/j.mineng.2019.106139. [DOI] [Google Scholar]
- Tadesse B.; Makuei F.; Albijanic B.; Dyer L. The beneficiation of lithium minerals from hard rock ores: A review. Minerals Engineering 2019, 131, 170–184. 10.1016/j.mineng.2018.11.023. [DOI] [Google Scholar]
- Finkelstein N. The activation of sulphide minerals for flotation: a review. Int. J. Miner. Process. 1997, 52 (2–3), 81–120. 10.1016/S0301-7516(97)00067-7. [DOI] [Google Scholar]
- Van Deventer J. Deendence of Froth Behaviour on Galvanic Interactions. Frothing in Flotation II 2018, 337–364. 10.1201/9780203755457-11. [DOI] [Google Scholar]
- Gu G.-h.; Dai J.-p.; Wang H.; Qiu G.-z. Galvanic coupling and its effect on origin potential flotation system of sulfide minerals. Journal of Central South University of Technology 2004, 11 (3), 275–279. 10.1007/s11771-004-0056-8. [DOI] [Google Scholar]
- Ke B.; Li Y.; Chen J.; Zhao C.; Chen Y. DFT study on the galvanic interaction between pyrite (100) and galena (100) surfaces. Appl. Surf. Sci. 2016, 367, 270–276. 10.1016/j.apsusc.2016.01.063. [DOI] [Google Scholar]
- Ke B.; Chen J.; Li Y.; Chen Y. Galvanic contacting effect of pyrite on xanthate adsorption on galena surface: DFT simulation and cyclic voltammetric measurements. Physicochemical Problems of Mineral Processing 2018, 54 (3), 826–836. [Google Scholar]
- Dos Santos E. C.; Lourenço M. P.; Pettersson L. G.; Duarte H. l. A. Stability, Structure, and Electronic Properties of the Pyrite/Arsenopyrite Solid–Solid Interface–A DFT Study. J. Phys. Chem. C 2017, 121 (14), 8042–8051. 10.1021/acs.jpcc.7b02642. [DOI] [Google Scholar]
- Li B.; Liu S.; Guo J.; Zhang L. Interaction between low rank coal and kaolinite particles: A DFT simulation. Appl. Surf. Sci. 2018, 456, 215–220. 10.1016/j.apsusc.2018.06.121. [DOI] [Google Scholar]
- Chen J.; Min F.-f.; Liu L.-y. The interactions between fine particles of coal and kaolinite in aqueous, insights from experiments and molecular simulations. Appl. Surf. Sci. 2019, 467, 12–21. 10.1016/j.apsusc.2018.10.130. [DOI] [Google Scholar]
- Yu F.-S.; Wang Y.-H.; Wang J.-M.; Xie Z.-F.; Zhang L. First-principle investigation on mechanism of Ca ion activating flotation of spodumene. Rare Metals 2014, 33 (3), 358–362. 10.1007/s12598-014-0304-5. [DOI] [Google Scholar]
- Zhu Y.; Luo B.; Sun C.; Liu J.; Sun H.; Li Y.; Han Y. Density functional theory study of α-Bromolauric acid adsorption on the α-quartz (1 0 1) surface. Minerals Engineering 2016, 92, 72–77. 10.1016/j.mineng.2016.03.007. [DOI] [Google Scholar]
- Zhang H.; Sun W.; Chen D.; Lin S.; Zhang C. Effects of Interfacial Hydroxylation Microstructure on Quartz Flotation by Sodium Oleate. Langmuir 2023, 39 (6), 2182–2191. 10.1021/acs.langmuir.2c02651. [DOI] [PubMed] [Google Scholar]
- Jin J.; Miller J. D.; Dang L. X.; Wick C. D. Effect of Cu2+ activation on interfacial water structure at the sphalerite surface as studied by molecular dynamics simulation. Int. J. Miner. Process. 2015, 145, 66–76. 10.1016/j.minpro.2015.07.001. [DOI] [Google Scholar]
- Cao Q.; Chen X.; Feng Q.; Wen S. Activation mechanism of lead ion in the flotation of stibnite. Minerals Engineering 2018, 119, 173–182. 10.1016/j.mineng.2018.01.039. [DOI] [Google Scholar]
- Tian M.; Gao Z.; Sun W.; Han H.; Sun L.; Hu Y. Activation role of lead ions in benzohydroxamic acid flotation of oxide minerals: New perspective and new practice. J. Colloid Interface Sci. 2018, 529, 150–160. 10.1016/j.jcis.2018.05.113. [DOI] [PubMed] [Google Scholar]
- Tian M.; Zhang C.; Han H.; Liu R.; Gao Z.; Chen P.; Wang L.; Li Y.; Ji B.; Hu Y.; et al. Effects of the preassembly of benzohydroxamic acid with Fe (III) ions on its adsorption on cassiterite surface. Minerals Engineering 2018, 127, 32–41. 10.1016/j.mineng.2018.07.019. [DOI] [Google Scholar]
- Quezada G. R.; Toledo P. G. Structure of the Interface between Lithium-Rich Spodumene and Saltwater by Density Functional Theory Calculations and Molecular Dynamics Simulations. J. Phys. Chem. C 2020, 124 (2), 1446–1457. 10.1021/acs.jpcc.9b10074. [DOI] [Google Scholar]
- Ejtemaei M.; Gharabaghi M.; Irannajad M. A review of zinc oxide mineral beneficiation using flotation method. advances in Colloid and Interface Science 2014, 206, 68–78. 10.1016/j.cis.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Liu M.; Chen J.; Li Y.; Zhao C.; Mu X. A density functional based tight binding (DFTB+) study on the sulfidization-amine flotation mechanism of smithsonite. Appl. Surf. Sci. 2018, 458, 454–463. 10.1016/j.apsusc.2018.07.014. [DOI] [Google Scholar]
- Feng Q.; Wen S.; Deng J.; Zhao W. DFT study on the interaction between hydrogen sulfide ions and cerussite (110) surface. Appl. Surf. Sci. 2017, 396, 920–925. 10.1016/j.apsusc.2016.11.061. [DOI] [Google Scholar]
- Tang X.; Chen J.; Chen Y. A density functional based tight binding (DFTB+) study on the sulfidization-xanthate flotation mechanism of cerussite. Appl. Surf. Sci. 2023, 612, 155677. 10.1016/j.apsusc.2022.155677. [DOI] [Google Scholar]
- Liu R.; Lu H.; Xu Z.; Wang C.; Sun W.; Wu M.; Dong Y.; Bai L. New insights into the reagent-removal mechanism of sodium sulfide in chalcopyrite and galena bulk flotation: a combined experimental and computational study. Journal of Materials Research and Technology 2020, 9 (3), 5352–5363. 10.1016/j.jmrt.2020.03.062. [DOI] [Google Scholar]
- Xiong X.; Hua X.; Zheng Y.; Lu X.; Li S.; Cheng H.; Xu Q. Oxidation mechanism of chalcopyrite revealed by X-ray photoelectron spectroscopy and first principles studies. Appl. Surf. Sci. 2018, 427, 233–241. 10.1016/j.apsusc.2017.08.047. [DOI] [Google Scholar]
- Niu X.; Chen J.; Li Y.; Xia L.; Li L.; Sun H.; Ruan R. Correlation of surface oxidation with xanthate adsorption and pyrite flotation. Appl. Surf. Sci. 2019, 495, 143411. 10.1016/j.apsusc.2019.07.153. [DOI] [Google Scholar]
- Liu J.; Cao X.-M.; Hu P. Density functional theory study on the activation of molecular oxygen on a stepped gold surface in an aqueous environment: a new approach for simulating reactions in solution. Phys. Chem. Chem. Phys. 2014, 16 (9), 4176–4185. 10.1039/c3cp54384f. [DOI] [PubMed] [Google Scholar]
- Chen J.; Chen Y.; Long X.; Li Y. DFT study of coadsorption of water and oxygen on galena (PbS) surface: An insight into the oxidation mechanism of galena. Appl. Surf. Sci. 2017, 420, 714–719. 10.1016/j.apsusc.2017.05.199. [DOI] [Google Scholar]
- Espiritu E.; Da Silva G.; Azizi D.; Larachi F.; Waters K. Flotation behavior and electronic simulations of rare earth minerals in the presence of dolomite supernatant using sodium oleate collector. Journal of Rare Earths 2019, 37 (1), 101–112. 10.1016/j.jre.2018.04.016. [DOI] [Google Scholar]
- Espiritu E.; da Silva G.; Azizi D.; Larachi F.; Waters K. The effect of dissolved mineral species on bastnäsite, monazite and dolomite flotation using benzohydroxamate collector. Colloids Surf., A 2018, 539, 319–334. 10.1016/j.colsurfa.2017.12.038. [DOI] [Google Scholar]
- Liu J.; Wang Y.; Luo D.; Zeng Y.; Wen S.; Chen L. DFT study of SDD and BX adsorption on sphalerite (1 1 0) surface in the absence and presence of water molecules. Appl. Surf. Sci. 2018, 450, 502–508. 10.1016/j.apsusc.2018.04.194. [DOI] [Google Scholar]
- Long X.; Chen Y.; Chen J.; Xu Z.; Liu Q.; Du Z. The effect of water molecules on the thiol collector interaction on the galena (PbS) and sphalerite (ZnS) surfaces: A DFT study. Appl. Surf. Sci. 2016, 389, 103–111. 10.1016/j.apsusc.2016.07.084. [DOI] [Google Scholar]
- Liu L.; Qiao E.; Shen L.; Min F.; Xue C. Effect of Hydration Layer on the Adsorption of Dodecane Collector on Low-Rank Coal: A Molecular Dynamics Simulation Study. Processes 2020, 8 (10), 1207. 10.3390/pr8101207. [DOI] [Google Scholar]
- Fisicaro G.; Filice S.; Scalese S.; Compagnini G.; Reitano R.; Genovese L.; Goedecker S.; Deretzis I.; La Magna A. Wet Environment Effects for Ethanol and Water Adsorption on Anatase TiO2 (101) Surfaces. J. Phys. Chem. C 2020, 124 (4), 2406–2419. 10.1021/acs.jpcc.9b05400. [DOI] [Google Scholar]
- Chen J.; Long X.; Chen Y. Comparison of Multilayer Water Adsorption on the Hydrophobic Galena (PbS) and Hydrophilic Pyrite (FeS2) Surfaces: A DFT Study. J. Phys. Chem. C 2014, 118 (22), 11657–11665. 10.1021/jp5000478. [DOI] [Google Scholar]
- Long X.; Chen J.; Chen Y. Adsorption of ethyl xanthate on ZnS (110) surface in the presence of water molecules: A DFT study. Appl. Surf. Sci. 2016, 370, 11–18. 10.1016/j.apsusc.2016.02.094. [DOI] [Google Scholar]
- He J.; Zhang H.; Yue T.; Sun W.; Hu Y.; Zhang C. Effects of Hydration on the Adsorption of Benzohydroxamic Acid on the Lead-Ion-Activated Cassiterite Surface: A DFT Study. Langmuir 2021, 37 (6), 2205–2212. 10.1021/acs.langmuir.0c03575. [DOI] [PubMed] [Google Scholar]
- Liu J.; Zeng Y.; Ejtemaei M.; Nguyen A. V.; Wang Y.; Wen S. DFT simulation of S-species interaction with smithsonite (0 0 1) surface: Effect of water molecule adsorption position. Results in Physics 2019, 15, 102575. 10.1016/j.rinp.2019.102575. [DOI] [Google Scholar]
- Jin J.; Miller J. D.; Dang L. X. Molecular dynamics simulation and analysis of interfacial water at selected sulfide mineral surfaces under anaerobic conditions. Int. J. Miner. Process. 2014, 128, 55–67. 10.1016/j.minpro.2014.03.001. [DOI] [Google Scholar]
- Hosseini Anvari M.; Liu Q.; Xu Z.; Choi P. Line tensions of galena (001) and sphalerite (110) surfaces: A molecular dynamics study. J. Mol. Liq. 2017, 248, 634–642. 10.1016/j.molliq.2017.10.037. [DOI] [Google Scholar]
- Anvari M. H.; Liu Q.; Xu Z.; Choi P. Molecular Dynamics Study of Hydrophilic Sphalerite (110) Surface as Modified by Normal and Branched Butylthiols. Langmuir 2018, 34 (10), 3363–3373. 10.1021/acs.langmuir.7b04303. [DOI] [PubMed] [Google Scholar]
- Qi C.; Lei X.; Zhou B.; Wang C.; Zheng Y. Temperature regulation of the contact angle of water droplets on the solid surfaces. J. Chem. Phys. 2019, 150 (23), 234703. 10.1063/1.5090529. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Xing Y.; Li M.; Liu M.; Tan J.; Cao Y.; Gui X. Studying interactions between undecane and graphite surfaces by chemical force microscopy and molecular dynamics simulations. Fuel 2020, 269, 117367. 10.1016/j.fuel.2020.117367. [DOI] [Google Scholar]
- Zhang C.; Liu Z.; Deng P. Contact angle of soil minerals: A molecular dynamics study. Computers and Geotechnics 2016, 75, 48–56. 10.1016/j.compgeo.2016.01.012. [DOI] [Google Scholar]
- Zhang X.; Du H.; Wang X.; Miller J. Surface chemistry aspects of bastnaesite flotation with octyl hydroxamate. Int. J. Miner. Process. 2014, 133, 29–38. 10.1016/j.minpro.2014.08.009. [DOI] [Google Scholar]
- Yi H.; Zhang X.; Zhao Y.; Liu L.; Song S. Molecular dynamics simulations of hydration shell on montmorillonite (001) in water. Surf. Interface Anal. 2016, 48 (9), 976–980. 10.1002/sia.6000. [DOI] [Google Scholar]
- Goverapet Srinivasan S.; Shivaramaiah R.; Kent P. R. C.; Stack A. G.; Riman R.; Anderko A.; Navrotsky A.; Bryantsev V. S. A comparative study of surface energies and water adsorption on Ce-bastnäsite, La-bastnäsite, and calcite via density functional theory and water adsorption calorimetry. Phys. Chem. Chem. Phys. 2017, 19 (11), 7820–7832. 10.1039/C7CP00811B. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Zaoui A. Wetting and nanodroplet contact angle of the clay 2:1 surface: The case of Na-montmorillonite (001). Appl. Surf. Sci. 2017, 396, 717–722. 10.1016/j.apsusc.2016.11.015. [DOI] [Google Scholar]
- Stack A. G.; Stubbs J. E.; Srinivasan S. G.; Roy S.; Bryantsev V. S.; Eng P. J.; Custelcean R.; Gordon A. D.; Hexel C. R. Mineral–Water Interface Structure of Xenotime (YPO4){100}. J. Phys. Chem. C 2018, 122 (35), 20232–20243. 10.1021/acs.jpcc.8b04015. [DOI] [Google Scholar]
- Yi H.; Jia F.; Zhao Y.; Wang W.; Song S.; Li H.; Liu C. Surface wettability of montmorillonite (0 0 1) surface as affected by surface charge and exchangeable cations: a molecular dynamic study. Appl. Surf. Sci. 2018, 459, 148–154. 10.1016/j.apsusc.2018.07.216. [DOI] [Google Scholar]
- Chen J.; Min F.-f.; Liu L.-y.; Liu C.-f. Mechanism research on surface hydration of kaolinite, insights from DFT and MD simulations. Appl. Surf. Sci. 2019, 476, 6–15. 10.1016/j.apsusc.2019.01.081. [DOI] [Google Scholar]
- Shi Z.; Li P.; Liu L. Interactions between CTAB and montmorillonite by atomic force microscopy and molecular dynamics simulation. Colloids Surf., A 2023, 657, 130656. 10.1016/j.colsurfa.2022.130656. [DOI] [Google Scholar]
- Chong L.; Lai Y.; Gray M.; Soong Y.; Shi F.; Duan Y. Molecular Dynamics Study of the Bulk and Interface Properties of Frother and Oil with Saltwater and Air. J. Phys. Chem. B 2017, 121 (13), 2788–2796. 10.1021/acs.jpcb.6b13040. [DOI] [PubMed] [Google Scholar]
- Chong L.; Lai Y.; Gray M.; Soong Y.; Shi F.; Duan Y. Effects of Frothers and Oil at Saltwater–Air Interfaces for Oil Separation: Molecular Dynamics Simulations and Experimental Measurements. J. Phys. Chem. B 2017, 121 (27), 6699–6707. 10.1021/acs.jpcb.7b03313. [DOI] [PubMed] [Google Scholar]
- Li E.; Wang X.; Du Z.; Miller J. D.; Cheng F. Specific anion effects on adsorption and packing of octadecylamine hydrochloride molecules at the air/water interface. Colloids Surf., A 2017, 522, 544–551. 10.1016/j.colsurfa.2017.03.017. [DOI] [Google Scholar]
- Wang L.; Hu Y.; Liu R.; Liu J.; Sun W. Synergistic adsorption of DDA/alcohol mixtures at the air/water interface: A molecular dynamics simulation. J. Mol. Liq. 2017, 243, 1–8. 10.1016/j.molliq.2017.08.029. [DOI] [Google Scholar]
- Wu L.; Han Y.; Zhang Q.; Zhu L.; Zhang C.; Zhao R. Molecular Dynamics Simulation: Influence of External Electric Field on Bubble Interface in Air Flotation Process. Chemical Research in Chinese Universities 2018, 34 (6), 939–944. 10.1007/s40242-018-8195-x. [DOI] [Google Scholar]
- Wu L.; Han Y.; Zhang Q.; Zhao S. Effect of external electric field on nanobubbles at the surface of hydrophobic particles during air flotation. RSC Adv. 2019, 9 (4), 1792–1798. 10.1039/C8RA08935C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Wu L.; Jiao T.; Gao Q. Effect of an Electric Field on Surface Properties of Hydrophobic Particles during a Flotation Process in Salt Water. Langmuir 2020, 36 (30), 8922–8928. 10.1021/acs.langmuir.0c01376. [DOI] [PubMed] [Google Scholar]
- Ma S.; Han Y.; Zhang Y.; Guo X.; Jiao T. Electrically enhanced activity of cationic surfactant for the bubble surface modification of solvent sublation to remove acetaminophen from water. J. Mol. Liq. 2022, 362, 119700. 10.1016/j.molliq.2022.119700. [DOI] [Google Scholar]
- Jin J.; Dang L. X.; Miller J. D. Molecular dynamics simulations study of nano bubble attachment at hydrophobic surfaces. Physicochemical Problems of Mineral Processing 2018, 54 (1), 89–101. [Google Scholar]
- Zhang R.; Xing Y.; Luo J.; Xia Y.; Xu M.; Wang X.; Tan J.; Gui X. Molecular Dynamics Simulation Study of Bubble Attachment at the Coal Surface with Varying Coalification Degrees. ACS omega 2020, 5 (32), 20134–20140. 10.1021/acsomega.0c01754. [DOI] [PMC free article] [PubMed] [Google Scholar]