

Significance
We performed single-cell transcriptomics in young versus aged mouse brains to identify mechanisms governing an age-dependent decline in cerebral vasculature and white matter repair/regrowth after stroke. Unique subsets of endothelial cells and oligodendrocyte progenitors emerged 3 days post-stroke in young mice but lost proregenerative features in aged mice. Aging impaired the ability of microglia (MG)/macrophages (MΦ) to boost angiogenesis and oligodendrogenesis through paracrine cell-to-cell cross talk. Permanent depletion of MG/MΦ impeded angiogenesis and oligodendrogenesis and hindered long-term stroke recovery irrespective of age. Transplantation of MG/MΦ from young brains into aged stroke brains partially restored angiogenesis and oligodendrogenesis and rejuvenated neurological functions. These observations reveal molecular and cellular processes that can be targeted to improve stroke recovery in the aged.
Keywords: ischemic stroke, angiogenesis, oligodendrogenesis, myeloid cells, stroke therapy
Abstract
Aging compromises the repair and regrowth of brain vasculature and white matter during stroke recovery, but the underlying mechanisms remain elusive. To understand how aging jeopardizes brain tissue repair after stroke, we performed single-cell transcriptomic profiling of young adult and aged mouse brains at acute (3 d) and chronic (14 d) stages after ischemic injury, focusing a priori on the expression of angiogenesis- and oligodendrogenesis-related genes. We identified unique subsets of endothelial cells (ECs) and oligodendrocyte (OL) progenitors in proangiogenesis and pro-oligodendrogenesis phenotypic states 3 d after stroke in young mice. However, this early prorepair transcriptomic reprogramming was negligible in aged stroke mice, consistent with the impairment of angiogenesis and oligodendrogenesis observed during the chronic injury stages after ischemia. In the stroke brain, microglia and macrophages (MG/MΦ) may drive angiogenesis and oligodendrogenesis through a paracrine mechanism. However, this reparative cell–cell cross talk between MG/MΦ and ECs or OLs is impeded in aged brains. In support of these findings, permanent depletion of MG/MΦ via antagonism of the colony-stimulating factor 1 receptor resulted in remarkably poor neurological recovery and loss of poststroke angiogenesis and oligodendrogenesis. Finally, transplantation of MG/MΦ from young, but not aged, mouse brains into the cerebral cortices of aged stroke mice partially restored angiogenesis and oligodendrogenesis and rejuvenated sensorimotor function and spatial learning and memory. Together, these data reveal fundamental mechanisms underlying the age-related decay in brain repair and highlight MG/MΦ as effective targets for promoting stroke recovery.
Stroke is the leading cause of long-term disability and has limited treatment options (1). Aging is a nonmodifiable risk factor for stroke (2), and approximately 75% of all strokes occur in people aged 65 y or older (3). Experimental models of stroke generally use young rodents as research subjects to save on cost, but young subjects may benefit from superior brain tissue repair and functional recovery processes compared to the aged. Thus, pharmaceutical interventions might be able to engage natural recovery mechanisms more readily in the young, contributing to a lack of pharmacologic translation from young animals to elderly stroke victims.
Endogenous brain repair mechanisms are crucial for spontaneous recovery after stroke and consist of inducible processes such as angiogenesis and oligodendrogenesis (4). Angiogenesis is an elaborate activity involving the migration, growth, and differentiation of endothelial cells (ECs), resulting in the formation of new blood vessels. Oligodendrogenesis includes the proliferation of oligodendrocyte progenitor cells (OPCs) and their subsequent differentiation into functional myelinating oligodendrocytes (OLs), which can promote white matter (WM) repair.
Emerging evidence suggests that angiogenesis and oligodendrogenesis vary across the lifespan (5–8) and are impeded in older animals after experimental stroke. For example, aged rats (19 to 20 mo old) have lower vascular densities in the brain 14 d after stroke compared to young rats and display weakened expression of angiogenesis-associated molecules at the bulk transcriptomic level (9). Middle-aged (8 mo old) mice also display greater WM injury after stroke compared to young mice, with fewer OL precursors and newborn OLs (10). However, there is a gap in our understanding of the endogenous mechanisms whereby aging impedes these repair processes after stroke. A detailed mechanistic understanding of such age-related factors may shed light on how to rejuvenate the injured as well as the normal aged brain.
Here, we deployed single-cell RNA sequencing to analyze the transcriptome of young and aged mouse brains at 3 and 14 d after stroke or 3 d after sham surgery. We surveyed repair processes in depth to elucidate the following: i) transcriptomic changes of major cell types during poststroke repair periods and ii) mechanisms underlying age-related deterioration of repair processes in the poststroke brain.
Results
Single-Cell Transcriptomic Profiling of Young and Aged Mouse Brains after Ischemic Stroke.
To investigate the dynamic transcriptomes of single cells in the poststroke brain and the impact of aging on this variable, we performed single-cell RNA sequencing on a total of 12 brain hemispheres from young adult (12 to 16 wk old) versus aged (20 mo old) mice under three different conditions: 3 days (d3) after sham surgery, 3 d after ischemic stroke induced by middle cerebral artery occlusion (MCAO), and 14 days (d14) after MCAO (n = 2 mice per group; Fig. 1A). Using T2-weighted MRI, we confirmed that cerebral infarction and edema were successfully induced in each mouse in post-MCAO groups (SI Appendix, Fig. S1).
Fig. 1.

Single-cell transcriptomic profiling of young and aged mouse brains after ischemic stroke. (A) Experimental design for single-cell RNA transcriptomic profiling of young and aged mouse brains after ischemic stroke. tMCAO or sham surgical operations were performed on young and aged mice. Ischemic left hemispheres were harvested at d3 and d14 after tMCAO, and left hemispheres were also collected from sham mice at d3 after tMCAO. (B) UMAP visualization of a total of 32,446 cells from all 11 nonhemorrhagic sample hemispheres (including sham), clustered into 18 cell types based on core markers shown in Dataset S1. (C) Violin plots showing logarithmic-scale expression levels of prototypic markers for each color-coded cell type after clustering was performed in (B). (D) UMAP visualization showing the color-coded major subsets after subclustering of Pecam1+ ECs and Olig1+ oligodendrocyte lineage cells, including OPCs and OLs. See markers for each subclusters in SI Appendix, Fig.S3 and Dataset S2.
After unsupervised clustering of all cells that had passed quality control, we checked the within-group differences of all six groups (i.e., young sham, young d3, young d14, aged sham, aged d3, and aged d14) (SI Appendix, Fig. S2A). The two biological replicates of each group demonstrated similar transcriptomic profiles, except for the aged d3 group (SI Appendix, Fig. S2B), of which one sample expressed high levels of hemoglobin transcripts, including Hba-a1 and Hba-a2 (SI Appendix, Fig. S2 C and D), likely due to postischemic hemorrhage transformation in the brain parenchyma. Thus, we excluded this sample from downstream analyses to avoid contamination of our results with blood cell transcripts.
We then reclustered the 32,446 cells from all nonhemorrhagic samples and identified 18 cell types according to their core markers (Fig. 1 B and C and Dataset S1), including microglia (MG), macrophages (MΦ), ECs, OLs, dendritic cells (DCs), neutrophils, T cells (T), astrocytes (ASCs), choroid plexus epithelial cells, pericytes (PCs), B cells (B), border-associated MΦ, OPCs, neuroblasts (NEUB), ependymal cells (EPEN), fibroblast-like cells (FB), neurons (NEUR), and olfactory ensheathing glial cells.
Pecam1+ ECs and Olig1+ OL lineage cells (including OPCs and OLs) play central roles in angiogenesis and oligodendrogenesis, respectively. These cell types were particularly heterogeneous compared to the 18 cell types mentioned above and were composed of multiple subsets (Fig. 1D, SI Appendix, Fig. S3, and Dataset S2). Based on the established functions of these lineage cells and their heterogeneity, we focused on these two repair-related cell types for a comprehensive analysis of their transcriptomic profiles at the single-cell level.
A Special Subset of ECs with Complex Angiogenic Regulation Features Emerges at Early Stages after Stroke.
Three major subsets (EC0, EC1, and EC3) accounted for 91.4% of all ECs from the aggregated young samples. The EC0 subset accounted for ~90% of the cells present in the young sham group (Fig. 2A) and expressed genes encoding prototypic EC markers, such as Cldn5 (encoding claudin 5) and Flt1 [encoding vascular endothelial growth factor receptor 1 (VEGFA)] (Fig. 2B). Thus, EC0 cells were designated as the “homeostatic ECs” that predominated under age-matched, baseline (nonischemic) conditions.
Fig. 2.

A transient subset of angiogenesis-associated endothelial cells with complex angiogenic regulation features emerges within days after stroke. (A) UMAP visualization (Left) of 3,208 ECs within the total Pecam1+ EC subcluster (defined in Fig. 1D) from all young groups. Bar plots (Right) showing the proportions of the major EC subsets (accounting for >10% in all groups) in each group. (B) Interpretation of the compositional transition (curved colored arrows) of major young EC subsets over time after ischemic injury. Representative DEGs between two groups are listed, along with the direction of change (black arrows). (C) Volcano plot showing 346 down-regulated (red) and 452 up-regulated (green) DEGs (|log2 FC| > 0.25 and Bonferroni-adjusted P value < 0.05) of EC3 compared to EC0. (D) Scatter plot showing the up-regulated DEGs (log2FC > 0.25) of EC3 compared to all other ECs, with the percentage difference (defined as % of EC3s that express a given gene—the % of all other ECs that express a given gene) along the y axis and log2FC along the x axis. The combined z score of percentage difference and log2 FC were color coded, and the genes with high combined z scores (combined z score > 6) are annotated. (E) Violin plots and UMAP visualization showing the expression level of Lrg1 in all EC subtypes (Left) and groups (Right). (F) Representative images (Left) of CD31 and LRG1 immunofluorescence staining in indicated brain regions at 3 d after MCAO. [Scale bars, 50 μm (low magnification) and 10 μm (high magnification).] Quantification (Right) of the numbers of LRG1+ CD31+ cells in the ipsilateral and contralateral hemisphere. n = 4. *P < 0.05 and **P < 0.01, two-tailed, paired t test. White dashed lines: border of the external capsule. Yellow dashed lines indicate inset magnified for 3D rendering.
The EC1 group was the dominant subset of ECs in both young d3 and d14 stroke groups, accounting for ~70% in the young d3 group and ~85% in the young d14 group (Fig. 2A). Compared to homeostatic ECs, EC1 from the injured young d3 group transitioned (blue curved arrow) to higher expression of immediate early genes (IEGs; e.g., Fos, Fosb, Jun, and Junb) but lower levels of circadian genes such as Dbp, Nr1d1, Per3, and Tef (Fig. 2B and Dataset S3). EC1 in the young d14 group expressed similar levels of IEGs and circadian genes as EC1 in the young d3 group but had also up-regulated (blue curved arrow) major histocompatibility complex class I genes (e.g., H2-K1, H2-D1, and B2m) and interferon-induced genes (e.g., Psmb8, Psmb9, Ifi27l2a, and Ifit3; Fig. 2B and Dataset S3). These results suggest that most ECs remain in a stressed state even at the chronic stage (d14) after stroke and that immunoinflammatory responses may become prominent in ECs at d14.
Notably, the EC3 subset appeared transiently at d3 after stroke, and its proportion decreased by 92.6% at d14 after stroke (green curved arrow; Fig. 2 A and B). The transcriptomic profile of EC3 dramatically differed from that of homeostatic ECs, wherein EC3 cells had 452 up-regulated (88 ribosomal) and 346 down-regulated (3 ribosomal) differentially expressed genes (DEGs) compared to homeostatic ECs (Fig. 2C and Dataset S3). We sought to determine the potential markers of EC3 by screening the up-regulated DEGs, among which Lrg1 (encoding leucine-rich alpha-2-glycoprotein 1, LRG1) displayed both high expression and superior specificity (Fig. 2D and Dataset S3). Lrg1 was expressed in EC3 at d3 after stroke (Fig. 2E). Immunofluorescence staining identified LRG1+CD31+ blood vessels in the infarct area but not in the uninjured contralateral side at d3 after stroke (Fig. 2F). LRG1 exerts proangiogenic effects on ECs by modulating TGF-β signaling in choroidal and retinal neovascularization models in mice (11). Furthermore, the mRNA and protein expression levels of LRG1 peaked at 3 d after MCAO in the ischemic rat brain and were positively correlated with the proangiogenesis factors VEGF and Ang-2 (12), supporting the generalizability of Lrg1 induction across two rodent species.
The increased expression of proangiogenic LRG1 in EC3 3 d after stroke motivated us to explore the biological functions of EC3. Gene ontology (GO) enrichment analysis of 707 nonribosomal DEGs of EC3s compared to homeostatic ECs showed that the top 10 activated and inhibited biological processes (i.e., with the smallest P values) were angiogenesis related, such as “angiogenesis”, “blood vessel development”, and “epithelial migration” (SI Appendix, Fig. S4A and Dataset S3). These findings suggest that the EC3 phenotype is indeed associated with angiogenesis, but also that EC3 cells may up-regulate or down-regulate angiogenesis. To explore the effects of EC3 transcripts on angiogenesis, we examined all angiogenesis-associated DEGs of EC3 cells in detail, based on the GO database and previous studies (Dataset S3). As expected, up-regulated and down-regulated angiogenesis-associated DEGs of EC3 included both pro- and antiangiogenic genes (SI Appendix, Fig. S4B), further supporting complex and bidirectional angiogenic regulation by the transient EC3 subclass.
These findings reveal that a special subset of LRG1+ angiogenesis-associated ECs (AAECs) selectively emerge in the infarct area at d3 after stroke, but their numbers subside within 2 wk. The transcriptomic profile of AAECs suggests involvement in the complex regulation of the angiogenesis process.
Aged ECs Activate Strong Interferon Responses but Have Weaker Proangiogenic Features after Stroke.
ECs in the aged brains displayed remarkably different transcriptomic profiles from the young groups. The EC2s were the primary homeostatic subset in the aged brain, accounting for 82.9% of all ECs in the aged sham group (Fig. 3A). Compared to young homeostatic ECs (EC0), EC2 cells up-regulated 128 genes, including apoptosis-related Depp1 and Cdkn1a genes, while down-regulating 119 genes, including circadian Dbp and Nr1d1 genes (Fig. 3B and Dataset S3). These results suggest aging-associated increases in apoptotic processes and potential blunting or phase changes in circadian processes in homeostatic EC2 cells under sham-injured conditions.
Fig. 3.
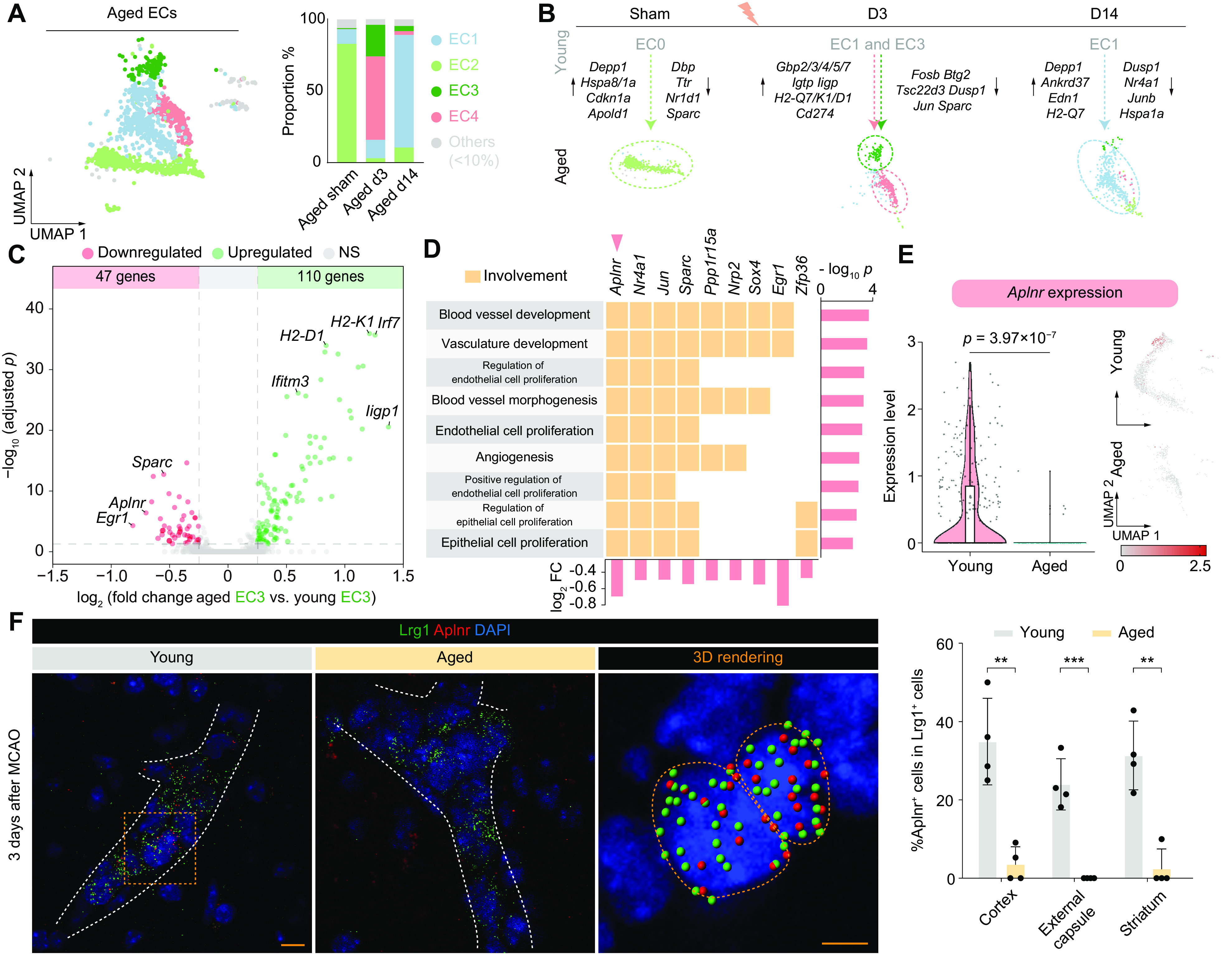
Proangiogenic features found in endothelial cells after stroke are lost in the aged brain. (A) UMAP visualization (Left) of 1,914 ECs from all aged groups. Stacked bar plots (Right) showing the proportions of the major subsets (accounting for >10% in all groups) in each group. (B) An overview of the aging-induced compositional and transcriptome transitions (colored arrows) in all groups. Representative DEGs between two groups are listed (black arrows). (C) Volcano plot showing 47 down-regulated (red) and 110 up-regulated (green) DEGs (|log2 FC| > 0.25 and Bonferroni-adjusted P < 0.05) of aged EC3 compared to young EC3. (D) Tile plot showing the relationship between all nine inhibited angiogenesis-associated biological processes and nine contributing genes. Bar plots show-log10 P of all terms (Right) and log2 fold change of all involved genes (below). (E) Violin plot (Left) and UMAP visualization (Right) showing the expression level of Aplnr in EC3 of young and aged groups as a natural log transformation of the raw count data. (F) Representative images of Aplnr and Lrg1 fluorescence in situ hybridization (FISH) in peri-infarct areas of young and aged mice at 3 d after MCAO. White dashed lines show the outlines of blood vessels based on the distribution of nuclei. Yellow lines in 3D rendering indicate boundaries of individual DAPI+ nuclei. Color-coded spheres show the puncta of FISH signals. The proportion of Aplnr+ cells in Lrg1+ cells in the peri-infarct areas was quantified. n = 4. **P < 0.01, two-tailed, unpaired t test. [Scale bars, 10 μm (low magnification) and 5 μm (3D rendering).]
After stroke, the major subsets of ECs in the aged d3 group were EC4 and EC3 (Fig. 3A). Numerous interferon-induced genes were strongly expressed in aged EC4 (Fig. 3B and SI Appendix, Fig. S5 A and B) and were also up-regulated in aged EC3 compared to young EC3 (Fig. 3 B and C and Dataset S3). These results suggest that a comprehensive interferon response is evoked in ECs of the aged brain after stroke.
Despite being in an inflammatory state, aged EC3 cells retained expression of core markers of AAECs, including Lrg1, Tmem252, and Litaf (SI Appendix, Fig. S5C). However, GO enrichment analysis of the DEGs of aged EC3 compared to young EC3 subsets (Fig. 3C and Dataset S3) showed that nine angiogenesis-related biological processes were inhibited (Fig. 3D and Dataset S3). Moreover, this inhibition was associated with downregulation of nine genes (Aplnr, Nr4a1, Jun, Sparc, Ppp1r15a, Nrp2, Sox4, Egr1, and Zfp36) (Fig. 3D), among which Aplnr had the second strongest decline in expression levels (Fig. 3 C and D). Aplnr encodes the apelin receptor (also named APJ) and was involved in all nine of the inhibited angiogenesis-related biological processes in aged EC3 (Fig. 3D). These results suggest that the proangiogenic features of AAECs decline in the aged poststroke brain and that the downregulation of Aplnr may play a role in this deterioration.
The proportion of cells expressing Aplnr decreased by 89.8%, and the average expression level of Aplnr was down-regulated (Bonferroni-adjusted P = 3.97 × 10−7) in aged EC3 compared to young EC3 (Fig. 3E). Fluorescence in situ hybridization (FISH) confirmed that Aplnr+ expression in Lrg1+ cells declined with age in the peri-infarct areas (Fig. 3F). Several studies demonstrate proangiogenic effects of apelin/APJ signaling in ECs of multiple species (13, 14). Thus, the dramatic lowering of Aplnr expression might deprive aged ECs of the ability to sense signals from apelin-releasing cells after stroke and impair proangiogenic efforts in aged ECs.
Poststroke Oligodendrogenesis: From OL Precursor Cell Proliferation to Transcriptomic Recovery of OLs.
Eight major subsets of OL lineage cells were identified, including four subsets of OPCs and four subsets of OLs (Fig. 4A). OPC0 accounted for 86.8% of all OPCs in the young sham brain (Fig. 4A) and expressed high levels of homeostatic markers, such as Pdgfra and Hes5 (SI Appendix, Fig. S6A), both of which prevent the excessive expansion of OLs by controlling OPC maturation (15, 16). The homeostatic OPC0 subset decreased rapidly in numbers after stroke, only accounting for 8.9% of all OPCs in the young d3 group (Fig. 4A). OPC2 cells became the major subset and constituted 66.1% of all OPCs 3 d after stroke (Fig. 4A). OPC2 cells expressed high levels of cell cycle markers, such as Mki67, Birc5, Ube2c, and Top2a (SI Appendix, Fig. S6B). Cell cycle scoring based on Seurat suggested that 32.1% of OPCs were in the G2/M phase at d3 after stroke (Fig. 4B). These results suggest that large numbers of OPCs abandoned the resting state and commenced proliferation within 3 d after stroke.
Fig. 4.

Decoding poststroke oligodendrogenesis: From oligodendrocyte precursor cell proliferation to transcriptomic recovery of oligodendrocytes. (A) UMAP visualization (Left) of oligodendrocyte lineage cells including 924 OLs and 235 OPCs from all young groups and with individual visualizations split by groups. Stacked bar plots (Right) showing the proportions of the major subsets (accounting for >10% in all groups) in each group. (B) UMAP visualization of young OPCs over time, colored by the cell cycle stages inferred by CellCycleScoring R function. Pie charts showing the proportion of cells belonging to different cell cycle stages. (C) Heatmap showing the degree of correlation (Pearson correlation coefficient) between OPC subsets and five stages in oligodendrocyte lineage differentiation reported by Marques et al. (17). (D) Violin plots showing the expression levels of markers for five differentiation stages in OPCs and OLs, as a natural log transformation of the raw count data. (E) Representative images of APC and BrdU immunofluorescence staining on brain sections of mice at 14 d after MCAO. [Scale bar, 1 mm (lower magnification) and 200 μm (higher magnification).] Note that images are purposefully captured at long exposures to view entire range of staining intensities from the noninjured to injured hemisphere; for this reason, background is bright in contralesional tissues. (F) The distribution of OLs on pseudotemporal axes in indicated young groups. Arrowheads, peak of pseudotemporal distribution density. Color-coded bars indicated the positions of OLs on pseudotemporal axes. (G) Line charts (Left) showing seven clusters of genes with distinct temporal kinetics in young groups and the numbers of included genes. Heatmap (Middle) showing the expression levels of top 10 genes for each temporal pattern. Representative predicted functions and genes involved in each cluster are annotated on the right.
Prior work indicates that the proliferation of OPCs after stroke starts within 3 d (18), but if and how these OPCs subsequently differentiate and mature into myelinating OLs is not fully understood. To determine the specific states of different poststroke OPC subsets with greater precision, we compared our data with published scRNA-seq datasets of OL lineage cells (17). Remarkably, we found that the transcriptomic profile of OPC3, a subset accounting for 24.6% of all OPCs in the young d14 group, was highly consistent (Pearson correlation coefficient = 0.97) with that of differentiation-committed OL precursors (COPs) (Fig. 4C), which were previously reported to be a transitional subtype between OPCs and OLs (19). OPC3 cells barely expressed OPC markers Pdgfra or Cspg4, but they expressed high levels of COP markers Gpr17 and Bmp4 (17) (Fig. 4D). Moreover, trajectory and pseudotime analyses suggested that OPC3 was the intermediate stage of the OPC-to-OL transition (SI Appendix, Fig. S6C). FISH quantification validated the existence of Bmp4+Sox10+ cells in the peri-infarct area 14 d after ischemic stroke (SI Appendix, Fig. S6D). In addition, the fraction of Bmp4+ cells among the Sox10+ OL lineage cell population was higher in the peri-infarct areas than in corresponding brain regions on the contralateral side (SI Appendix, Fig. S6D). Thus, we identified a subset of Bmp4+Sox10+ cells committed to differentiate toward myelinating OLs in the peri-infarct areas 14 d after stroke.
OLs are highly vulnerable to ischemic injury, and replenishment of damaged OLs depends on the efficacy of oligodendrogenesis (19). To measure the extent to which poststroke oligodendrogenesis can replace damaged OLs within 2 wk, we injected 5-bromo-2′-deoxyuridine (BrdU) intraperitoneally 3 to 6 d after MCAO and performed immunofluorescence staining on mouse brain sections harvested 14 d after stroke. Colocalization of BrdU and mature OL marker adenomatous polyposis coli (APC) showed abundant APC+BrdU+ newly generated OLs accumulating in the peri-infarct area (Fig. 4E).
The temporal distribution of OLs revealed a dynamic shift of the OL transcriptome from a homeostatic to pathological state at d3 and partial recovery at d14 after stroke (Fig. 4F). We then dissected the OL transcriptome changes during this recovery process. All genes with significant changes in expression levels across groups were classified by their shifting patterns and divided into seven gene clusters (clusters 1 to 7, each including at least 10 genes) (Fig. 4G and Dataset S4). Clusters 1 to 6 reflect complete, incomplete, or no recovery of gene expression levels, and the remaining cluster 7 reflected upregulation patterns unique to d14 (Fig. 4G). For example, patterns I and IV consisted of 247 genes and 94 genes, respectively, and these genes displayed downregulation or upregulation on d3 but returned to homeostatic expression levels at d14 (Fig. 4G).
We performed GO enrichment analyses of genes within each pattern and annotated the most overrepresented biological processes and listed the genes involved (Fig. 4G and Dataset S4). The results suggest a prioritization of the steps involved in OL functional recovery. OLs might first alleviate the impairments in cell morphogenesis, overactive translation, and overactive ATP metabolism after stroke (clusters 1 and 4, Fig. 4G), followed by impaired myelination (cluster 2, Fig. 4G). Impaired organic acid biosynthesis, impaired cellular junction organization, and overactive regulation of transcription in response to stress were the last functions to be restored (clusters 3 and 6, Fig. 4G). In addition, phospholipid transport was selectively activated at 14 d after stroke (cluster 7, Fig. 4G).
In conclusion, OL lineage cells are remarkably dynamic after stroke. A large fraction of OPCs abandon the resting state and enter the cell cycle, leading to robust proliferation in the peri-infarct area within 3 d. The latter OPCs appear to undergo a transition toward OLs (i.e., oligodendrogenesis) within 14 d. Bmp4+ Sox10+ cells might lie in the intermediate state of this transition. Newly generated BrdU+APC+ OLs account for ~30% of all APC+ mature OLs in the peri-infarct area at 14 d after stroke. With robust replacement of OLs after OPC proliferation and maturation, the transcriptome measured in the OL population eventually shifts partly back toward baseline. The latter shift toward homeostatic OL expression profiles may facilitate the restoration of WM in young stroke mice.
Aging Impedes Poststroke Oligodendrogenesis and Alters the Pattern of OL Recovery.
In the aged poststroke brain, the number of G2/M OPCs decreased dramatically compared to the young poststroke brain (Figs. 4A and 5 A and B). Immunofluorescence quantification showed an age-related decline in the density of PDGFRα+Ki67+ cells (proliferative OPCs) in peri-infarct areas at d3 after stroke (Fig. 5C). By d14 after stroke, the proportion of resting OPC0 (among all OPCs) had risen by 156.9% (12.3% in the young d14 group and 31.6% in the aged d14 group) (SI Appendix, Fig. S7A), and the proportion of Bmp4+ OPC3 decreased by 18.2% (23.6% in the young d14 group and 19.3% in the aged d14 group) (SI Appendix, Fig. S7A), suggesting that the OL differentiation potential is weaker in the aged poststroke brain. Furthermore, the density of APC+BrdU+ newly generated, mature OLs in the peri-infarct areas was reduced with aging (Fig. 5D). Taken together, these results indicate that oligodendrogenesis is attenuated in the aged poststroke brain, and this likely results from a decline in OPC proliferation and differentiation.
Fig. 5.

Aging impedes poststroke oligodendrogenesis and alters the pattern of oligodendrocyte recovery. (A) (Left) UMAP visualization of oligodendrocyte lineage cells including 1,289 OLs and 101 OPCs from all aged groups and with individual visualizations split by groups. (B) Bar plots showing the proportions of the major subsets (accounting for >10% in all groups) in each group. (B) UMAP visualizations of OPCs in young and aged groups over time, colored by the cell cycle stages inferred by CellCycleScoring R function. (C) Representative images (Left) of PDGFRα and Ki67 immunofluorescence staining on brain sections of young and aged mice at 3 d after MCAO. Quantification (Right) of the numbers PDGFRα+ Ki67+ cells in the peri-infarct areas. n = 6. *P < 0.05, two-tailed, unpaired t test. Arrowheads, PDGFRα+Ki67+ cells. (Scale bar, 100 μm.) (D) Representative images (Left) of APC and BrdU immunofluorescence staining on brain sections of young and aged mice at 14 d after MCAO. Dashed lines show the position of the external capsule. Quantification (Right) of the numbers of APC+ BrdU+ cells in the ipsilateral infarct areas and parallel areas on the contralateral side. n = 5 to 6. *P < 0.05, two-tailed, unpaired t test. (Scale bar, 50 μm.) (E) The distribution of OLs on pseudotemporal axes in the indicated aged groups. Arrowheads, peak of pseudotemporal distribution density. Asterisks, age-specific peaks. Gray dashed lines, the density curves of young groups. Color-coded bars indicated the positions of OLs on pseudotemporal axes. (F) Line charts (Left) showing eight clusters of genes with distinct temporal kinetics in aged groups and the numbers of included genes. Heatmap (Middle) showing the expression level of top 10 genes in each pattern. Representative predicted functions and genes involved in each cluster are annotated on the right.
OLs in the aged brain also displayed a dynamic shift with time after stroke, which resembled the responses of young brains (Figs. 4G and 5E). These observations suggest that aged OLs undergo partial recovery of the transcriptome between d3 and d14 after stroke—despite weakened oligodendrogenesis. After classifying all significantly changed genes in the aged groups by their change patterns, eight clusters were acquired (Fig. 5F and Dataset S4). GO enrichment analysis of the genes contained in each cluster showed that the predicted functional restoration patterns of OLs in the aged brain were partially distinct from the young brain (Figs. 4G and 5F and Dataset S4). Overactive translation and impaired cell morphogenesis might be two of the initially altered functions that are first restored after stroke in the aged brain, while impaired organic acid biosynthesis and overactive regulation of transcription in response to stress might be two of the altered functions that are restored at later stages after stroke in the aged brain. Furthermore, phospholipid transport remained selectively activated at 14 d postinjury (cluster 7, Fig. 5F). Notably, impaired myelination was one of the earliest restored functions of OLs in the aged poststroke brain (cluster 1, Fig. 5F), suggesting that the aged brain might have a higher need for remyelination than the young brain after stroke.
The shifting OL recovery patterns could be a consequence of multiple factors altered by aging. First, aged OLs differed from young OLs in ribosomal activity, impaired myelination, and actin polymerization (SI Appendix, Fig. S7B), in accordance with previous studies (20, 21). Second, attenuated oligodendrogenesis may have impeded the regeneration of OLs in the aged brain. Last, the poststroke microenvironment around OLs in the aged brain differs dramatically from that in the young brain. For example, MG and ASCs foster a more proinflammatory microenvironment in the aged brain (22, 23), which may contribute to differential reprogramming of the transcriptome of aged vs. young OLs.
Aging Modifies the Molecular Cross Talk between ECs/OL Lineage Cells and MG/Infiltrating MΦ or ASCs.
As the central target cells of poststroke repair processes such as angiogenesis and oligodendrogenesis, ECs and OL lineage cells (EC/OL lineage cells) are the subject of extensive regulation by other cells (24–26). Emerging evidence shows that MG and infiltrating MΦ (MG/MΦ) are crucial for angiogenesis and oligodendrogenesis during poststroke recovery (27, 28). Accordingly, we showed that depletion of MG/MΦ worsened poststroke recovery outcomes in young mice at d35 after MCAO (SI Appendix, Fig. S8), including 1) sensorimotor functions assessed by rotarod and adhesive removal tests (SI Appendix, Fig. S8 B and C), 2) angiogenesis measured by the density of CD31+BrdU+ cells (SI Appendix, Fig. S8F), and 3) oligodendrogenesis measured by the density of APC+BrdU+ cells (SI Appendix, Fig. S8G). On the other hand, the mechanisms whereby MG/MΦ interact with EC/OL lineage cells to facilitate tissue repair processes and the molecular processes underlying the impact of aging on proreparative functions of MG/MΦ remain unclear. Hence, we took advantage of CellChat (29) to infer cell-to-cell communication and explore potential cross talk between MG/MΦ and EC/OL lineage cells (Fig. 6A). Based on our single-cell transcriptome data, CellChat recognized 71 ligand-receptor (L-R) pairs consisting of ligands expressed by MG/MΦ and receptors on EC/OL lineage cells, with various probabilities under different conditions (Fig. 6 B and C).
Fig. 6.
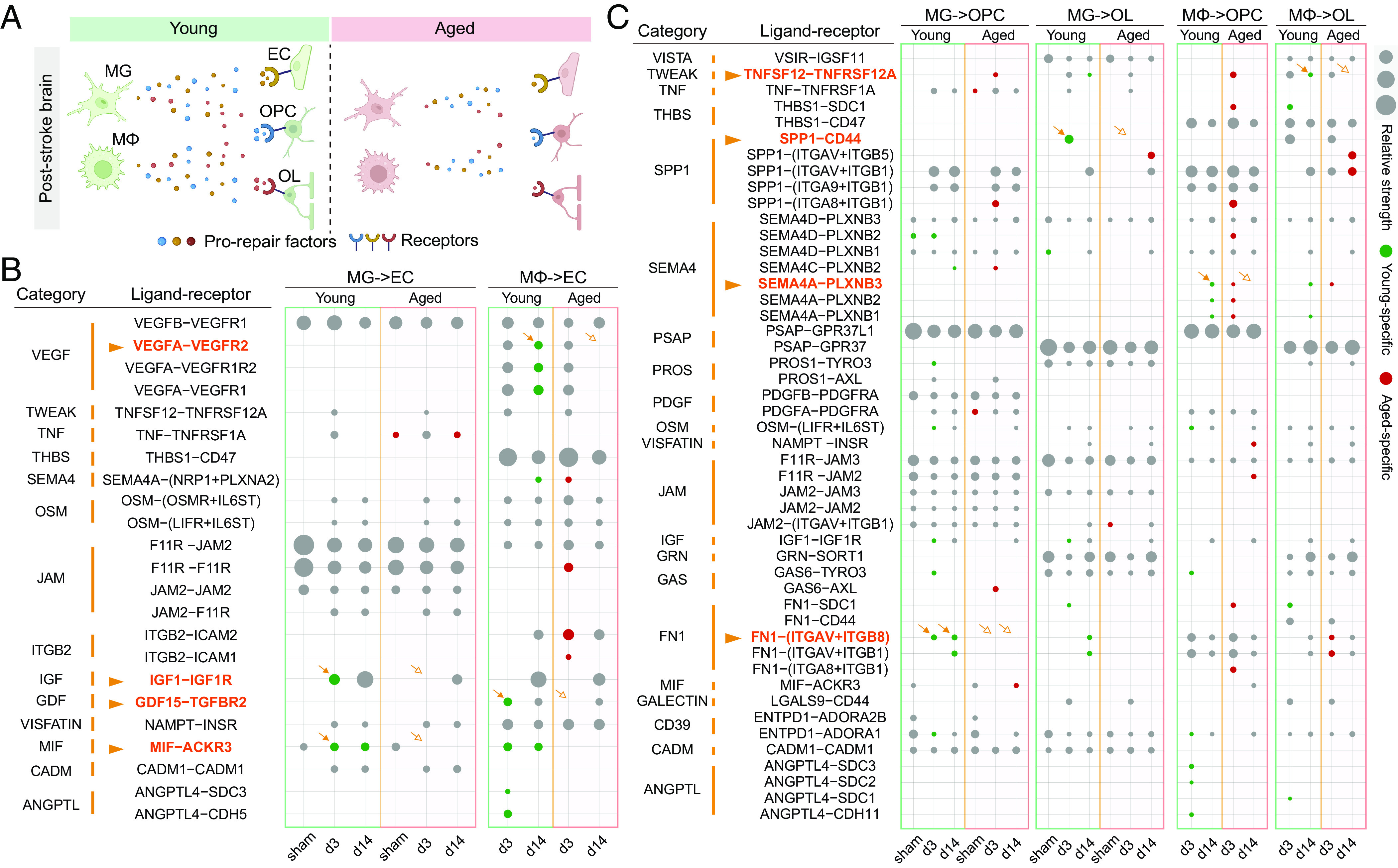
Unveiling the age-sensitive contribution of microglia/infiltrating macrophages to poststroke repair processes. (A) Overview of age-sensitive alterations in repair-related inferred interactions between MG/MΦ and EC/OL/OPCs in poststroke brains. (B and C) Shown are the predicted effects that MG/MΦ exert on ECs (B) or on OL/OPCs (C) inferred by the CellChat R package. All probable ligand–receptor (L–R) pairs and their categories are listed. The sizes of the dots represent the probability of the L–R pairs. The L–R pairs specific to the young and aged groups are colored in green and red, respectively.
Notably, 62.0% L-R pairs (44 out of 71) were predicted to emerge specifically in young or in aged groups at certain time points (Fig. 6 B and C), and among these pairs, multiple repair-associated proteins/cytokines drew our interest, including VEGFA, insulin-like growth factor 1 (IGF1), growth/differentiation factor 15 (GDF15), macrophage migration inhibitory factor (MIF), fibronectin (FN), semaphorin-4A, secreted phosphoprotein 1 (SPP1), and tumor necrosis factor ligand superfamily member 1 (TNFSF12) with their receptors (Fig. 6 B and C). Thus, we examined the expression levels of these L-R pairs across young vs. old mice and across the two timepoints after stroke (SI Appendix, Fig. S9 A–C).
VEGFA, GDF15, IGF1, and MIF were inferred to mediate the interaction of MG/MΦ with ECs at both d3 and d14 after stroke in young mice, but this cross talk relationship disappeared in the aged mice at specific times described below (Fig. 6B). VEGFA (encoded by Vegfa) exerts proangiogenic functions in a variety of diseases, including cerebral ischemia (30). VEGFA was predicted to be secreted by MΦ and act on EC via VEGF receptors in poststroke young brains, but this interaction was suppressed in aged brains at d14 after stroke (Fig. 6B), mainly due to a decrease of Vegfa expression in MΦ at d14 after stroke (SI Appendix, Fig. S9A). GDF15 (encoded by Gdf15) is another angiogenesis-promoting cytokine (31) and is predicted to act on TGF-beta receptor type-2 (TGFBR2) on ECs (Fig. 6B). Aging delayed the onset of the GDF15-TGFBR2 interaction (Fig. 6B), as MΦ expression of Gdf15 was lower in aged brains at d3 after stroke (SI Appendix, Fig. S9A). IGF1 (encoded by Igf1) can promote angiogenesis by binding the IGF1 receptor (IGF1R) (32). Expression levels of Igf1 in MG were decreased in aged brains compared to young brains at d3 and d14 after stroke (SI Appendix, Fig. S9A), thereby predicting a decrease or loss of the IGF1-IGF1R interaction (Fig. 6B). MIF (encoded by Mif), a ligand for atypical chemokine receptor 3 (ACKR3), has displayed proangiogenic features (33). The poststroke MIF-ACKR3 interaction between MG and EC was absent in aged brains, as the ECs in the aged poststroke brains almost completely lost Ackr3 expression (Fig. 6B). Thus, ECs unlikely could receive MIF signals released by MG in aged brains after stroke.
Aging also altered the cross talk between MG/MΦ and OL lineage cells by regulating repair-associated cell–cell interactions (Fig. 6C). FN (encoded by Fn1), an extracellular matrix protein, is involved in the proliferation and maintenance of OPCs (34). Similar to aging-induced changes in Igf1, expression of Fn1 in MG was down-regulated at both time points after stroke (SI Appendix, Fig. S9B). SPP1, also known as osteopontin (encoded by Spp1), promotes OL differentiation during stroke recovery (35). Compared to young brains, aged brains expressed lower levels of MG-derived Spp1 at d3 after stroke (SI Appendix, Fig. S9C). The binding of TNFSF12 (encoded by Tnfsf12) to its receptor Fn14 (encoded by Tnfrsf12a) promotes cell regeneration and tissue repair (36). Expression levels of Tnfsf12 in MΦ and Tnfrsf12a in OLs were both lower in aged brains at d14 after stroke (SI Appendix, Fig. S9C).
Together, these findings suggest aging-induced alterations in cross talk between MG/MΦ and EC/OL lineage cells after stroke (Fig. 6A). Thus, aging may impede poststroke angiogenesis and oligodendrogenesis by down-regulating multiple proreparative factors released by MG/MΦ and/or by limiting the ability of EC/OL lineage cells to receive the proreparative signals.
In addition to MG and infiltrating MΦ, ASCs may contribute to poststroke recovery. Using unsupervised clustering, we discovered four ASC cell clusters (ASCs) in sham nonischemic brains, showing comparable compositions in young (ASC0, 44.9%; ASC1, 38.5%; ASC2, 11.8%; and ASC3, 4.9%) and aged (ASC0, 62.6%; ASC1, 24.6%; ASC2, 10.3%; and ASC3, 2.6%) mice (SI Appendix, Fig. S11 A and B). ASC0 appeared to be a homeostatic subset, enriched in genes related to gliogenesis, glial cell differentiation, and cell projection morphogenesis (SI Appendix, Fig. S11 C and D). Ischemic stroke forced ASCs to undergo dramatic alterations, in that the percentages of ASC0 were decreased in young (23.7% at 3 d and 35.6% at 14 d) and aged (30.2% at 3 d and 35.4% at 14 d) mice. In contrast, the percentages of ASC2 were markedly increased in young (35.59% at 3 d and 28.8% at 14 d) and aged (31.0% at 3 d and 43.4% at 14 d) mice after stroke. The ASC2 cluster is characterized by enhanced signaling pathways related to the innate immune response, regulation of TNF production, and cell activation (SI Appendix, Fig. S11 C and D). These results suggest that ASCs are persistently activated after stroke with a potential functional impact on neuroinflammation.
ASCs likely interact with ECs, OLs, and OPCs through various ligand–receptor pairs, including prorepair or proremodeling molecules such as SPP1, VEGF, PTN, FGF1, and LAMB2 (SI Appendix, Fig. S11 F–H). These pathways are activated after stroke, consistent with literature suggesting that ASC activation may impact stroke outcomes through various mechanisms (24, 37, 38), including the paracrine signaling pathways identified in the current scRNA-seq analysis. However, the overall differences in transcriptomic profiles between poststroke young and aged mice are modest (SI Appendix, Fig. S11E). While many ligand–receptor signaling pathways are activated after stroke, only the COL9A3-SDC1 signaling pathway associated with mature OLs is significantly down-regulated (P = 0.0026) in poststroke aged mice compared to young adult mice (SI Appendix, Fig. S12). These results suggest that alterations in cross talk between ASCs and EC/OL lineage cells are not the major mediator of impaired brain repair and functional recovery in aged mice after stroke.
Young MG/Infiltrating MΦ Alleviate Age-Related Impairments in Poststroke Repair Processes.
To validate our scRNA-seq analyses of age-associated impairments in the proreparative functions of MG/MΦ after stroke, we used fluorescence-activated cell sorting to isolate MG/MΦ (CD45+CD11b+Ly6G− cells) from young and aged ischemic hemispheres, respectively, 14 d after stroke. Recipient aged mice were subjected to MCAO and received ~50,000 cells via stereotactic transplantation in the ipsilateral cortex 6 d after MCAO (Fig. 7A). To create the spatial niches for the transplanted cells, a 6-d MG/MΦ depletion protocol was completed before MG/MΦ transplantation, via dietary administration of the CSF1R antagonist PLX5622 (39). Ten aged mice subjected to dMCAO with neither PLX5622 administration nor cell transplantation (no PLX), and nine aged mice with permanent MG/MΦ depletion up to day 35 after MCAO (permanent PLX) served as the control groups (Fig. 7A).
Fig. 7.

Replacing microglia/infiltrating macrophages in aged postischemic host mice with microglia/infiltrating macrophages from young postischemic donor brains promotes long-term stroke recovery. (A) Experimental design for validation of aging-induced impairment in prorepair effects of MG/MΦ. (B and C) Assessment of sensorimotor functions using the rotarod test (B, Left) and adhesive removal test (C, Left). Bar plots show the rotarod test (B, Right) and adhesive removal test (C, Right) at day 35 after MCAO. n = 8 to 10/group. Blue marks: Permanent PLX vs. No PLX, pink marks: PLX+Aged MG/MΦ vs. Permanent PLX, green marks: PLX+Young MG/MΦ vs. Permanent PLX, yellow marks: PLX+Young MG/MΦ vs. PLX+Aged MG/MΦ, ***P < 0.001, two-way repeated-measures ANOVA and Bonferroni test on data after 10 d after MCAO. (D and E) Evaluation of cognitive functions, including spatial learning (D) and memory (E) using the Morris water maze test. n = 8 to 10/group. Blue marks: Permanent PLX vs. No PLX, pink marks: PLX+Aged MG/MΦ vs. Permanent PLX, green marks: PLX+Young MG/MΦ vs. Permanent PLX, yellow marks: PLX+Young MG/MΦ vs. PLX+Aged MG/MΦ, *P < 0.05, **P < 0.01, and ***P < 0.001, two-way repeated-measures ANOVA and Bonferroni test (D) or one-way ANOVA and Bonferroni test (E). (F) Representative images of CD31 immunofluorescence staining in the peri-infarct areas from aged recipient mice at 35 d after stroke. Bar plot shows the quantification of CD31+ blood vessel densities. n = 8/group. (Scale bar, 100 μm.) *P < 0.05 and **P < 0.01, one-way ANOVA and Bonferroni test. (G) Representative images of Luxol fast blue staining in the peri-infarct areas from aged recipient mice at 35 d after stroke. Bar plot shows the quantification of the mean intensity decreases in ischemic hemispheres compared to intact hemispheres. n = 8/group. (Scale bar, 100 μm.) ***P < 0.001, one-way ANOVA and Bonferroni test. (H) Representative images (Left) of CD31 and BrdU immunofluorescence staining in the peri-infarct areas in aged recipient mice at 35 d after MCAO. (Scale bars, 100 μm.) Arrowhead, CD31+ BrdU+ cell. Quantification (Right) of CD31+ BrdU+ cells in the peri-infarct areas from aged recipient mice at 35 d after stroke. n = 8/group. ***P < 0.001, one-way ANOVA and Bonferroni test. (I) Representative images (Left) of APC and BrdU immunofluorescence staining in the peri-infarct areas in aged recipient mice at 35 d after MCAO. (Scale bars, 100 μm.) Arrowhead, APC+ BrdU+ cell. Quantification (Right) of APC+ BrdU+ cells in the peri-infarct areas from aged recipient mice at 35 d after stroke. n = 8/group. **P < 0.01 and ***P < 0.001, one-way ANOVA and Bonferroni test. (J) Two-tailed Pearson correlations of behavioral performances in the rotarod test with BV density (Upper Left), WM density loss (Lower Left), angiogenesis in ec (Upper Right), and oligodendrogenesis in ec (Lower Right) at day 35 postischemia. The 95% CIs are shown as dotted lines. n = 8/group. Abbreviationsns, not significant, MWM, Morris water maze; BV, blood vessel; WM, white matter; ctx, cortex; ec, external capsule; str, striatum.
Like shown in young poststroke mice (SI Appendix, Fig. S8 A–E), permanent depletion of MG/MΦ in aged poststroke mice hindered the long-term recovery of sensorimotor functions assessed by the rotarod test (Fig. 7B) and the adhesive removal test (Fig. 7C) but had no significant impact on recovery of spatial learning and memory measured by the Morris water maze tests (Fig. 7 D and E). Transplantation of aged MG/MΦ after PLX5622-induced depletion only rescued behavioral performance in the rotarod test (Fig. 7B), while transplantation of young MG/MΦ was able to rescue behavioral performance in both tests for sensorimotor functions (Fig. 7 B–E). Furthermore, postischemic aged mice transplanted with young MG/MΦ displayed superior long-term behavioral performances than the no PLX control group in all four behavioral tests (Fig. 7 B–E).
Compared to transplantation of aged MG/MΦ, transplantation of young MG/MΦ rejuvenated the behavioral performance of aged postischemic mice, including sensorimotor functions (Fig. 7 B and C), spatial learning (Fig. 7D), and spatial memory (Fig. 7E). Aged postischemic mice implanted with young MG/MΦ had higher blood vessel densities (Fig. 7F), less WM loss (Fig. 7G), and improved remyelination (denser and narrower nodes of Ranvier, SI Appendix, Fig. S10) in peri-infarct areas 35 d after MCAO. Furthermore, aged postischemic mice harboring young MG/MΦ cells showed higher numbers of CD31+BrdU+ cells (Fig. 7H) and APC+BrdU+ cells (Fig. 7I) per unit area, indicating that MG/MΦ harvested from young poststroke brains promote more angiogenesis and oligodendrogenesis than MG/MΦ from aged counterparts, fully consistent with predictions from the cell–cell interaction analyses (Fig. 6). Superior histologic recovery and more vigorous cell regeneration likely led to improvements in neurological outcomes, as suggested by correlations of behavior measures with histology/cell regeneration data (Fig. 7J).
Together, these results demonstrate that replacing MG/MΦ in aged postischemic hemispheres with MG/MΦ from young postischemic hemispheres alleviates aging-induced detrimental effects on brain vasculature and WM repair processes and rejuvenates multidimensional aspects of behavioral recovery.
Discussion
In the current study, we used high-throughput RNA sequencing at the single-cell level to explore molecular and cellular mechanisms underlying age-related impairments in poststroke brain repair. The major findings of this report are as follows: 1) Unique subsets of proliferating ECs and OL progenitors emerged 3 d after stroke in young mice but displayed weakened proregenerative transcriptomic features in aged mice. 2) The transcriptomics revealed age-related impairments in OPC proliferation and OL differentiation after stroke, which were verified with histological tools. 3) Mechanistically, MG/MΦ in aged mice displayed lower levels of paracrine prorepair molecules that target ECs and OL lineage cells, which likely contributed to the compromised ability of the MG/MΦ to facilitate poststroke angiogenesis, oligodendrogenesis, and behavioral recovery in aged stroke mice. 4) Permanent depletion of MG/MΦ impeded angiogenesis and oligodendrogenesis and hindered long-term stroke recovery in young and aged mice. 5) Transplantation of MG/MΦ from young brains into aged stroke brains partially restored angiogenesis and oligodendrogenesis and neurological functions. These results suggest that loss of homeostatic functions of aging MG/MΦ contributes to the age-related decay in poststroke brain repair and long-term functional recovery.
We discovered a group of LRG1+ AAECs that appeared in the peri-infarct regions 3 d after stroke. These AAECs displayed high expression of LRG1, which is known to have proangiogenic effects via TGF signaling in models of stroke and retinal neovascularization (12, 13). AAECs exhibit a complex angiogenesis-related profile, as they express multiple pro- and antiangiogenic molecules. A bidirectional regulation would be consistent with the previously reported complex feedback process of angiogenesis (40).
Poststroke angiogenesis is weakened in aged individuals (9). We found that aged AAECs down-regulated or even lost expression of a series of angiogenesis-related genes compared to young AAECs, including Aplnr, which encodes the apelin receptor (also named APJ). The apelin-APJ axis plays a proangiogenic role in various contexts, including stroke recovery (14, 41). Thus, aging-induced dysregulation of the apelin-APJ axis in AAECs may underlie impaired angiogenesis in aged poststroke subjects. In addition, aged ECs robustly up-regulated ~40 IFN-responsive genes 3 d after stroke, suggesting that ECs are one of the primary sources for strong activation of proinflammatory IFN pathways in the poststroke aged brain (42).
We performed an in-depth dissection of poststroke oligodendrogenesis processes, from the proliferation of OPCs to differentiation into mature OLs. Heterogeneity of the single-cell transcriptome in OL lineage cells has been reported during development (17). Using data matching, we found that Bmp4+ committed OL precursors, a type of intermediate cells, accounted for ~20% of Sox10+ cells 2 wk after stroke. This suggests that the transformation of OPCs to OLs progresses for at least 2 wk and that ~20% of the OL lineage cells have the potential to further differentiate into mature OLs.
Senescence has been reported to impede poststroke oligodendrogenesis (10). We observed that aging delayed the proliferation of OPCs and attenuated the differentiation of new OLs. Poststroke oligodendrogenesis leads to the replacement of damaged OLs by new OL populations, and our observations on the recovery of OL transcriptome patterns showed a modularized feature, suggesting that orders of priority exist in the recovery of OL functions. Aging changed the poststroke transcriptome recovery patterns of OLs. For example, the priority of the recovery of the expression of myelination-related genes is higher in aged brains than in young ones, suggesting a greater need to maintain myelin homeostasis in the aged brains after stroke.
MG and infiltrating MΦ are instrumental in brain repair processes after stroke (27, 43). Cell–cell interaction analyses showed that these two myeloid cell populations expressed various prorepair paracrine factors (VEGFA, IGF1, SPP1, etc., Fig. 6 and SI Appendix, Fig. S9) that can promote EC/OPC/OL regeneration (27, 30, 44). MG/MΦ can also assist poststroke brain repair through phagocytosis-mediated clearance of dead cells and broken myelin (45, 46). Supporting the role of MG/MΦ in stroke recovery, permanent depletion of MG/MΦ in either young (SI Appendix, Fig. S8 B–E) or aged (Fig. 7 B–E) mice hindered the long-term sensorimotor functional recovery and expression of brain repair processes, including angiogenesis and oligodendrogenesis, after stroke. Nevertheless, the poststroke expression of these proreparative factors was down-regulated in aged MG/MΦ, suggesting that the proreparative capacities of MG/MΦ were weakened with aging. Consistent with this finding, our previous RNA-seq study showed that tissue remodeling pathways are diminished in MG of aged mice 5 d after stroke (47). Using cell transplantation experiments, we found that MG/MΦ from the ipsilateral hemispheres of poststroke young mice were indeed able to promote angiogenesis and oligodendrogenesis in poststroke aged mice, thereby improving long-term behavioral recovery.
The present study has several limitations. First, NEUR-related repair processes such as neurogenesis and synaptic remodeling were not examined here due to inadequate numbers of intact NEURs for in-depth analyses. This may reflect shear stress–induced loss of fragile NEURs during single-cell suspension protocols. Future studies using single-nuclear sequencing or spatial transcriptome sequencing may help preserve and explore the neuronal transcriptome. Second, recent evidence suggests that the impact of circadian rhythms on ischemic brain injury and stroke recovery could be substantial (48, 49). Therefore, we need to explore the potential effects of circadian biology on MG/MΦ-mediated stroke recovery and brain repair in future studies. Third, functional differences between MG and infiltrating MΦ in poststroke repair need to be further dissected. Fourth, aged MG may have direct deleterious effects on brain functions by overproducing reactive oxygen species and neurotoxic proinflammatory cytokines (50). Thus, microglial transplantation could improve stroke outcomes by replacing the “sick” aged MG and mitigating their detrimental effects. Future studies are needed to investigate these alternative mechanisms. Fifth, spatial proteomic profiling at the single-cell levels and knockdown/knockout of cell-specific expression of individual genes in the future will help expand the current set of new observations.
In conclusion, aging impedes poststroke angiogenesis and oligodendrogenesis, at least partly by impairing the expression of prorepair factors in MG/MΦ. Thus, rejuvenation of MG/MΦ may be a therapeutic strategy to boost repair capacities in aged individuals after stroke.
Materials and Methods
Key reagents or resources that are essential to reproduce the results are in SI Appendix, Table S1. All animal procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed following the Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and reduce animal suffering. Surgeries and all outcome assessments were performed by investigators blinded to experimental group assignments. All statistics are summarized in SI Appendix, Table S2. For further methodological details, please see SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Acknowledgments
We thank Lesley M. Foley for assistance with the MRI experiments and Patricia Strickler for administrative support. This project was supported in part by the VA Merit Review 1 I01BX005290 (to J.C.), American Heart Association grant TPA926806 (to J.C.), the University of Pittsburgh Medical Center Immune Transplant and Therapy Center grant (to J.C.), University of Pittsburgh Medical Center Endowed Chair fund (to J.C.) and the University of Pittsburgh School of Medicine. J.C. is the Richard King Mellon Professor of Neurology and a recipient of the VA Senior Research Career Scientist Award (# IK6 BX006298). M.V.L.B. is the Emeritus Sylvia and Robert S. Olnick Professor of Neuroscience.
Author contributions
C.J., Y.S., L.S., M.V.L.B., and J.C. designed research; C.J., Y.S., L.S., W.Z., K.C., Q.Y., S.H., and J.L. performed research; C.J., Y.S., L.S., R.K.L., W.Z., K.C., Q.Y., S.H., J.L., X.H., M.V.L.B., and J.C. analyzed data; and C.J., Y.S., L.S., R.K.L., X.H., R.A.S., M.V.L.B., and J.C. wrote the paper.
Competing interests
R.A.S. and J.A. coauthored a review article in 2021 (https://www.frontiersin.org/articles/10.3389/fnagi.2021.623751/full). The other authors declare no competing interest.
Footnotes
Reviewers: J.A.A., University of Texas Health Science Center at Houston; and E.H.L., Massachusetts General Hospital.
Contributor Information
Michael V. L. Bennett, Email: michael.bennett@einstein.yu.edu.
Jun Chen, Email: chenj2@upmc.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information. Previously published data were used for this work (GSE75330) (17, 51).
Supporting Information
References
- 1.Tsao C. W., et al. , Heart disease and stroke statistics-2022 update: A report from the American heart association. Circulation 145, e153–e639 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Boehme A. K., Esenwa C., Elkind M. S., Stroke risk factors, genetics, and prevention. Circ. Res. 120, 472–495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yousufuddin M., Young N., Aging and ischemic stroke. Aging (Albany NY) 11, 2542–2544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ergul A., Alhusban A., Fagan S. C., Angiogenesis: A harmonized target for recovery after stroke. Stroke 43, 2270–2274 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petcu E. B., Smith R. A., Miroiu R. I., Opris M. M., Angiogenesis in old-aged subjects after ischemic stroke: A cautionary note for investigators. J. Angiogenes. Res. 2, 26 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen D., et al. , Demyelinating processes in aging and stroke in the central nervous system and the prospect of treatment strategy. CNS Neurosci. Ther. 26, 1219–1229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engler-Chiurazzi E. B., Monaghan K. L., Wan E. C. K., Ren X., Role of B cells and the aging brain in stroke recovery and treatment. Geroscience 42, 1199–1216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baltan S., Shi Y., Keep R. F., Chen J., The effect of aging on brain injury and recovery after stroke. Neurobiol. Dis. 126, 1–2 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Buga A. M., et al. , Transcriptomics of post-stroke angiogenesis in the aged brain. Front. Aging Neurosci. 6, 44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyamoto N., et al. , Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke 44, 2573–2578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X., et al. , LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature 499, 306–311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng H., et al. , LRG1 promotes angiogenesis through upregulating the TGF-β1 pathway in ischemic rat brain. Mol. Med. Rep. 14, 5535–5543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyries M., et al. , Hypoxia-induced apelin expression regulates endothelial cell proliferation and regenerative angiogenesis. Circ. Res. 103, 432–440 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Helker C. S., et al. , Apelin signaling drives vascular endothelial cells toward a pro-angiogenic state. Elife 9, e55589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emery B., Lu Q. R., Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 7, a020461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Đăng T. C., et al. , Powerful homeostatic control of oligodendroglial lineage by PDGFRα in adult brain. Cell Rep. 27, 1073–1089.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Marques S., et al. , Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonfanti E., et al. , The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis. 8, e2871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R., Chopp M., Zhang Z. G., Oligodendrogenesis after cerebral ischemia. Front. Cell Neurosci. 7, 201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sams E. C., Oligodendrocytes in the aging brain. Neuronal Signal. 5, NS20210008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ximerakis M., et al. , Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, 1696–1708 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Candelario-Jalil E., Paul S., Impact of aging and comorbidities on ischemic stroke outcomes in preclinical animal models: A translational perspective. Exp. Neurol. 335, 113494 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin C., et al. , A unique type of highly-activated microglia evoking brain inflammation via Mif/Cd74 signaling axis in aged mice. Aging Dis. 12, 2125–2139 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xing C., Lo E. H., Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog. Neurobiol. 152, 181–199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arai K., Lo E. H., Wiring and plumbing: Oligodendrocyte precursors and angiogenesis in the oligovascular niche. J. Cereb. Blood Flow Metab. 41, 2132–2133 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ornelas S., et al. , Three-dimensional ultrastructure of the brain pericyte-endothelial interface. J. Cereb. Blood Flow Metab. 41, 2185–2200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi L., et al. , Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity 54, 1527–1542.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedragosa J., et al. , CCR2 deficiency in monocytes impairs angiogenesis and functional recovery after ischemic stroke in mice. J. Cereb. Blood Flow Metab. 40, S98–S116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin S., et al. , Inference and analysis of cell-cell communication using cell chat. Nat. Commun. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenberg D. A., Jin K., Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol. Life Sci. 70, 1753–1761 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S., et al. , Growth differentiation factor 15 promotes blood vessel growth by stimulating cell cycle progression in repair of critical-sized calvarial defect. Sci. Rep. 7, 9027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin S., et al. , IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif. 50, e12390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amin M. A., et al. , Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ. Res. 93, 321–329 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Lourenço T., et al. , Modulation of oligodendrocyte differentiation and maturation by combined biochemical and mechanical cues. Sci. Rep. 6, 21563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Velthoven C. T., Heijnen C. J., van Bel F., Kavelaars A., Osteopontin enhances endogenous repair after neonatal hypoxic-ischemic brain injury. Stroke 42, 2294–2301 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Girgenrath M., et al. , TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 25, 5826–5839 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park J. H., et al. , Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J. Cereb. Blood Flow Metab. 41, 1523–1535 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diaz A., et al. , Tissue-type plasminogen activator induces TNF-α-mediated preconditioning of the blood-brain barrier. J. Cereb. Blood Flow Metab. 42, 667–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Z., et al. , Efficient strategies for microglia replacement in the central nervous system. Cell Rep. 32, 108041 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Pepper M. S., Positive and negative regulation of angiogenesis: From cell biology to the clinic. Vasc. Med. 1, 259–266 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Wu L., Chen L., Li L., Apelin/APJ system: A novel promising therapy target for pathological angiogenesis. Clin. Chim. Acta. 466, 78–84 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Androvic P., et al. , Decoding the transcriptional response to ischemic stroke in young and aged mouse brain. Cell Rep. 31, 107777 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Lyu J., et al. , Microglial/macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 27, 515–527 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mason J. L., Xuan S., Dragatsis I., Efstratiadis A., Goldman J. E., Insulin-like growth factor (IGF) signaling through type 1 IGF receptor plays an important role in remyelination. J. Neurosci. 23, 7710–7718 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai W., et al. , STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight. 4, e131355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ting S. M., et al. , Brain cleanup as a potential target for poststroke recovery: The role of RXR (Retinoic X Receptor) in phagocytes. Stroke 51, 958–966 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi L., et al. , Genome-wide transcriptomic analysis of microglia reveals impaired responses in aged mice after cerebral ischemia. J. Cereb. Blood Flow Metab. 40, S49–S66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esposito E., et al. , Potential circadian effects on translational failure for neuroprotection. Nature 582, 395–398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo E. H., et al. , Circadian biology and stroke. Stroke 52, 2180–2190 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Marschallinger J., et al. , Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23, 194–208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marques S., Zeisel A., Linnarsson S., Castelo-Branco G., RNA-seq analysis of single cells of the oligodendrocyte lineage from nine distinct regions of the anterior-posterior and dorsal-ventral axis of the mouse juvenile central nervous system. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75330. Deposited 24 November 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Data Availability Statement
All study data are included in the article and/or supporting information. Previously published data were used for this work (GSE75330) (17, 51).