

Significance
Dendritic cells (DCs) orchestrate the adaptive immune response against pathogens and cancerous cells. Despite intensive research, the subcellular architecture and molecular composition of the MHC I peptide-loading complex (PLC) in DCs remain unknown. Here, we examine the dynamic interactome of the PLC during differentiation and maturation of DCs generated from blood-derived monocytes. We identified ER proteins constituting ER exit- and membrane contact sites as PLC interaction partners located in nanometer proximity to the PLC. Although the role of the interaction partners remains enigmatic in the context of MHC I surface presentation, our data show plasticity in PLC composition during the differentiation and maturation of DCs that have not been identified previously, which will advance our understanding of antigen presentation.
Keywords: antigen processing, membrane organization, antigen presentation, primary cells, membrane proteins
Abstract
Dendritic cells (DCs) orchestrate immune responses by presenting antigenic peptides on major histocompatibility complex (MHC) molecules to T cells. Antigen processing and presentation via MHC I rely on the peptide-loading complex (PLC), a supramolecular machinery assembled around the transporter associated with antigen processing (TAP), which is the peptide transporter in the endoplasmic reticulum (ER) membrane. We studied antigen presentation in human DCs by isolating monocytes from blood and differentiating them into immature and mature DCs. We uncovered that during DC differentiation and maturation, additional proteins are recruited to the PLC, including B-cell receptor-associated protein 31 (BAP31), vesicle-associated membrane protein-associated protein A (VAPA), and extended synaptotagmin-1 (ESYT1). We demonstrated that these ER cargo export and contact site–tethering proteins colocalize with TAP and are within 40 nm proximity of the PLC, suggesting that the antigen processing machinery is located near ER exit- and membrane contact sites. While CRISPR/Cas9-mediated deletion of TAP and tapasin significantly reduced MHC I surface expression, single-gene deletions of the identified PLC interaction partners revealed a redundant role of BAP31, VAPA, and ESYT1 in MHC I antigen processing in DCs. These data highlight the dynamics and plasticity of PLC composition in DCs that previously was not recognized by the analysis of cell lines.
Dendritic cells (DCs) are essential in linking the innate and the adaptive arm of the immune system. They are the main professional antigen-presenting cells in vivo by displaying processed antigenic peptides via the major histocompatibility class I antigen complex (MHC I). DCs prime adaptive immune responses by stimulation of different subsets of T lymphocytes (1, 2). Due to their antigen-presentation capacity, DCs are important for personalized anticancer immunotherapies (3). However, only approximately 1% of peripheral blood mononuclear cells (PBMCs) are DCs, making research of this scarce cell population challenging (4). In contrast, monocytes make up approximately 10% of peripheral blood cells (5, 6). Monocytes are the major source of inflammatory myeloid cells including DCs. In vivo, monocytes recirculate in the blood, and they are recruited to sites of inflammation where in a first step they differentiate into immature DCs (imDCs). After additional inflammatory stimuli, imDCs develop into mature DCs (mDCs) by upregulating various costimulatory molecules required for T-cell stimulation and chemokine receptors that enable them to migrate to draining lymph nodes (7–10). Monocyte-derived dendritic cells (moDCs), including imDCs and mDCs, are a key model for studying human DCs. However, personalized DC therapy remains to be optimized in terms of DC preparation and antigen loading (11–14).
In the canonical MHC I pathway, the peptide-loading complex (PLC) confers the transport, editing, and loading of proteasomal degradation products onto MHC I molecules from the cytosol into the endoplasmic reticulum (ER) lumen. This mechanism is crucial for effective antigen presentation on the cell surface (15, 16). The PLC is a macromolecular machinery consisting of the heterodimeric transporter associated with antigen processing (TAP1/2), MHC I molecules, and the chaperones ERp57, calreticulin, and tapasin (17–19). Professional antigen-presenting cells can also present exogenously acquired antigens by MHC I molecules, a process that is referred to as cross-presentation. The extent of the PLC involvement in cross-presentation is still under debate (20). The PLC is arranged in homogeneous nanometer clusters located in the tubular ER network that increase in density upon maturation into mDCs (21). This suggests that the antigen-presentation capacity is locally adapted to activate T cells throughout the elongated morphology of moDCs.
Organelles are not individually separated compartments, but they are highly interconnected and organized through the so-called membrane contact sites (MCSs), allowing organelle interaction (22). For instance, the ER communicates with the plasma membrane (PM) (23–25) and other organelles such as mitochondria, endosomes, peroxisomes, and the Golgi apparatus (26–28). The importance of organelle interaction for the trafficking of antigen-loaded MHC molecules has received particular attention in recent years. MCSs between the ER and phagosomes are critical for cross-presentation and infection with intracellular pathogens (29–33). The PLC relocates from early endosomes to the ER, which is defined as a calnexin-positive compartment (34). Therefore, understanding the subcellular organization and identifying interaction partners of the PLC is important to elucidate PLC-dependent MHC I antigen presentation pathways in DCs.
Here, we deciphered dynamic changes of the PLC at the molecular level upon differentiation and maturation of DCs. In this study, we identified interaction partners of the PLC during maturation of monocytes into DCs, namely B-cell receptor-associated protein 31 (BAP31), a cargo receptor, vesicle-associated membrane protein-associated protein A (VAPA), which is involved in the formation of MCSs, and extended synaptotagmin-1 (ESYT1), which is found at contact sites between the ER and the plasma membrane. We detected cell type– and DC status–dependent alterations of the PLC composition, which correlated with the colocalization of the PLC with MCS tethering proteins. CRISPR/Cas9-mediated gene disruption of PLC core components and of the peripheral interaction partners in primary human DCs revealed that MHC I surface expression was only affected by deletion of the PLC core subunits TAP1 and tapasin. Deletion of the ER cargo receptor BAP31 and MCS–tethering proteins VAPA and ESYT1 without altering MHC I surface expression indicated that DCs have compensatory mechanisms to ensure MHC I antigen presentation. This study provides evidence of the high plasticity of the PLC in primary human DCs, which was not identified before by the analysis of cell lines.
Results
Dynamic Changes in PLC Composition during Differentiation of Monocytes into DCs.
Antigen-specific T cells are primed by DCs (35) that express costimulatory molecules and high levels of MHC I (36). As MHC I surface expression is largely dependent on PLC function, we sought to study the molecular composition of the PLC during monocyte to DC differentiation. In brief, monocytes were isolated from healthy donors and in vitro incubated in medium enriched with interleukin (IL)-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF) for 5 d. The resulting imDCs were cultivated for another 3 d in medium supplemented with IL-1β, tumor necrosis factor (TNF)-α, IL-6, and prostaglandin E2 (PGE2) to obtain mDCs (37) (Fig. 1A). During the initial monocyte-to-imDC differentiation process, the well-characterized monocytic marker CD14 was down-regulated, while MHC class II and the costimulatory molecule CD86 were up-regulated (SI Appendix, Fig. S1). The maturation of imDCs into mDCs was characterized by upregulation of CD86 and CD83 (SI Appendix, Fig. S1). Besides these changes in surface marker expression and alterations in the cell morphology, the TAP1/2 relocates from early endosomes to calnexin-positive compartments, i.e., the ER (34). Given this high plasticity, we aimed at analyzing the PLC composition during differentiation and maturation of human DCs.
Fig. 1.

Composition of PLC assembly changes upon differentiation and maturation of moDCs. (A) Scheme of moDC differentiation. Monocytes incubated with GM-CSF and IL-4 for 5 d into imDC. After addition of IL-6, IL-1β, TNF-α, and PGE2 and incubation for further 3 d, imDCs mature into mDCs. (B) Experimental setup. Isolated monocytes as well as imDCs and mDCs were lysed. Subsequently, TAP and associated proteins were isolated by co-IP using TAP1-specific monoclonal antibody mAb 148.3. Isotype antibody was used as control. Protein complexes were separated by SDS-PAGE, and proteins were cleaved by in-gel trypsin digestion. Derived peptides were analyzed by LC-MS/MS, and protein intensities were used for label-free quantification. (C) Volcano plots of coimmunoprecipitated, TAP-specific samples in comparison to control. Upper graph: monocytes; middle graph: imDCs; bottom graph: mDCs. Plots were generated by PERSEUS software and display the difference (fold change) and statistical significance (−log p) of the peptide intensities between TAP-specific and control co-IP samples. Proteins to the right and above the significance line (based on the applied FDR of 0.05 and an S0 of 1) are considered as enriched. Protein IDs in red are known members of the TAP complex. The identified interaction partners BAP31, VAPA, and ESYT1 are highlighted in blue. (D) Monocytes (mono), imDCs, mDCs, and Raji cells were solubilized in 1% (w/v) digitonin and TAP1 (mAb 148.3) coimmunoprecipitated. A corresponding isotype antibody (iso) was used as control. Proteins were analyzed by SDS-PAGE and subsequent immunoblotting. To avoid donor dependencies, two to three donors were pooled for each primary cell type. The left panel shows cell lysate material after solubilization and ultracentrifugation (solubilizate). On the right panel, the eluate samples after co-IP are presented. TAP1, BAP31, VAPA, and ESYT1 are detected by immunoblotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. *Heavy and light chains of anti-TAP1 and isotype antibodies.
Human monocytes, imDCs, and mDCs, as well as the B-cell line Raji that we used as a control, were subjected to coimmunoprecipitation (co-IP) of the key PLC subunits using the anti-TAP1 monoclonal antibody (mAb) 148.3. Affinity-purified complexes were separated by SDS-PAGE followed by in-gel tryptic digestion. The resulting proteolytic fragments were analyzed by reverse-phase chromatography combined with tandem mass spectrometry (LC-MS/MS) (Fig. 1B and SI Appendix, Fig. S2). As a proof of principle, we first studied the composition of the PLC isolated from primary monocytes and Raji cells and mapped the results in volcano plots with statistically significant interaction partners that are shown above the significance threshold. In this analysis, the PLC core subunits, including the ATP-binding cassette (ABC) transporters TAP1 and TAP2, the ER chaperones tapasin, ERp57, and calreticulin, as well as MHC I molecules composed of the heavy chain and the β2m, were identified (Fig. 1C and SI Appendix, Fig. S3). Furthermore, in addition to the classical MHC alleles HLA-A, -B, and -C, the nonclassical HLA-E and -G alleles were found as components of the PLC, which further confirmed previous results of PLC constituents that were determined in human cell lines (17, 18, 38). We next examined the PLC composition after monocyte differentiation and maturation into imDCs and mDCs, as defined by their characteristic profiles of cell surface markers (SI Appendix, Fig. S1). Similar to the PLC interactome in monocytes, our analysis revealed all core PLC subunits in imDCs and mDCs. Strikingly, the volcano plots highlighted a higher number of statistically significant interaction partners of the PLC in DCs than in monocytes (Fig. 1C). Specifically, in addition to the well-described PLC subunits, BAP31, VAPA, and ESYT1 were identified in DCs as prominent interaction partners of the PLC.
Therefore, we next addressed the interaction of BAP31, VAPA, and ESYT1 with the PLC of monocytes, imDC, mDC, and Raji cells by co-IP and immunoblotting. Anti-TAP1 co-IP revealed the presence of all PLC core subunits, including TAP1/2, tapasin, ERp57, calreticulin, and the MHC I heavy chain (SI Appendix, Fig. S4), plus the absence of Sec61α, which is an ER marker and therefore verified the specificity of the co-IP for TAP1-interaction partners (SI Appendix, Fig. S5). It is noteworthy that comparable amounts of all core PLC components were detected in the cell lysates (solubilizate) of the cell types analyzed. A similar signal intensity of TAP1 served as a reference for comparing the signal intensities of the other components. Of note, in these assays, BAP31 was significantly more abundant in the PLC of imDC (SI Appendix, Fig. S6), while the control B-cell lymphoma cell line Raji only showed low levels of BAP31 and ESYT1 in the PLC. Furthermore, ESYT1 was detected at significantly higher levels in imDCs than in monocytes. These data indicated that BAP31 and ESYT1 are PLC interaction partners in primary DCs and that they are less prominent in human cell lines. VAPA was detected at slightly higher levels in the PLC of imDCs and mDCs than in monocytes and was also identified in Raji cells (Fig. 1D), although no significant differences were observed. The coelution levels of BAP31 and ESYT1 are similar in monocytes and mDCs (SI Appendix, Fig. S6). However, an enrichment of PLC interaction partners as identified by LC-MS/MS and immunoblotting indicated that the interactome of the PLC changes toward enhanced complexity during differentiation of monocytes into imDC.
BAP31, VAPA, and ESYT1 Are Interaction Partners of the Fully Assembled PLC in DCs.
To validate the results above, we performed an affinity isolation of the PLC using the viral factor ICP47 that carried a C-terminal streptavidin-binding peptide tag (ICP47SBP). ICP47 is encoded by herpes simplex virus and binds with high affinity to the fully assembled heterodimeric transport complex TAP but not to preassembled TAP1 or TAP2 subunits (39–42). In the presence of ICP47SBP, we solubilized the cells, purified the PLC complex, and analyzed the PLC components by immunoblotting. In the solubilizate of monocytes, imDC, mDC, and Raji cells, the core PLC components and the peripheral interaction partners, BAP31, VAPA, and ESYT1, were detected. Using the ICP47SBP pull-down approach, all core PLC subunits, including TAP1, TAP2, tapasin, ERp57, calreticulin, and MHC I, were affinity purified specifically in monocytes, imDC, mDC, and Raji cells (Fig. 2 A and B). The ER control marker Sec61α was not detected among ICP47SBP pull-down proteins, despite its abundance in the solubilizate (SI Appendix, Fig. S7). ICP47SBP pull-down using monocyte cell lysates revealed low levels of BAP31, VAPA, and ESYT1. BAP31 was detected to a significantly higher extent in the ICP47SBP pull-down of the DC subsets than in monocytes. VAPA and ESYT1 levels were slightly increased, although not significantly, in imDCs and mDCs when compared with monocytes, suggesting that they are interaction partners of the fully assembled PLC in imDCs and mDCs (SI Appendix, Fig. S8). ICP47SBP pull-down of the PLC in Raji cells showed low levels of BAP31 and ESYT1, whereas VAPA was detected at levels comparable to moDCs (Fig. 2C). In conclusion, BAP31, VAPA, and ESYT1 specifically interact with the virally arrested PLC of imDCs and mDCs; however, they are less prevalent in the PLC of monocytes.
Fig. 2.
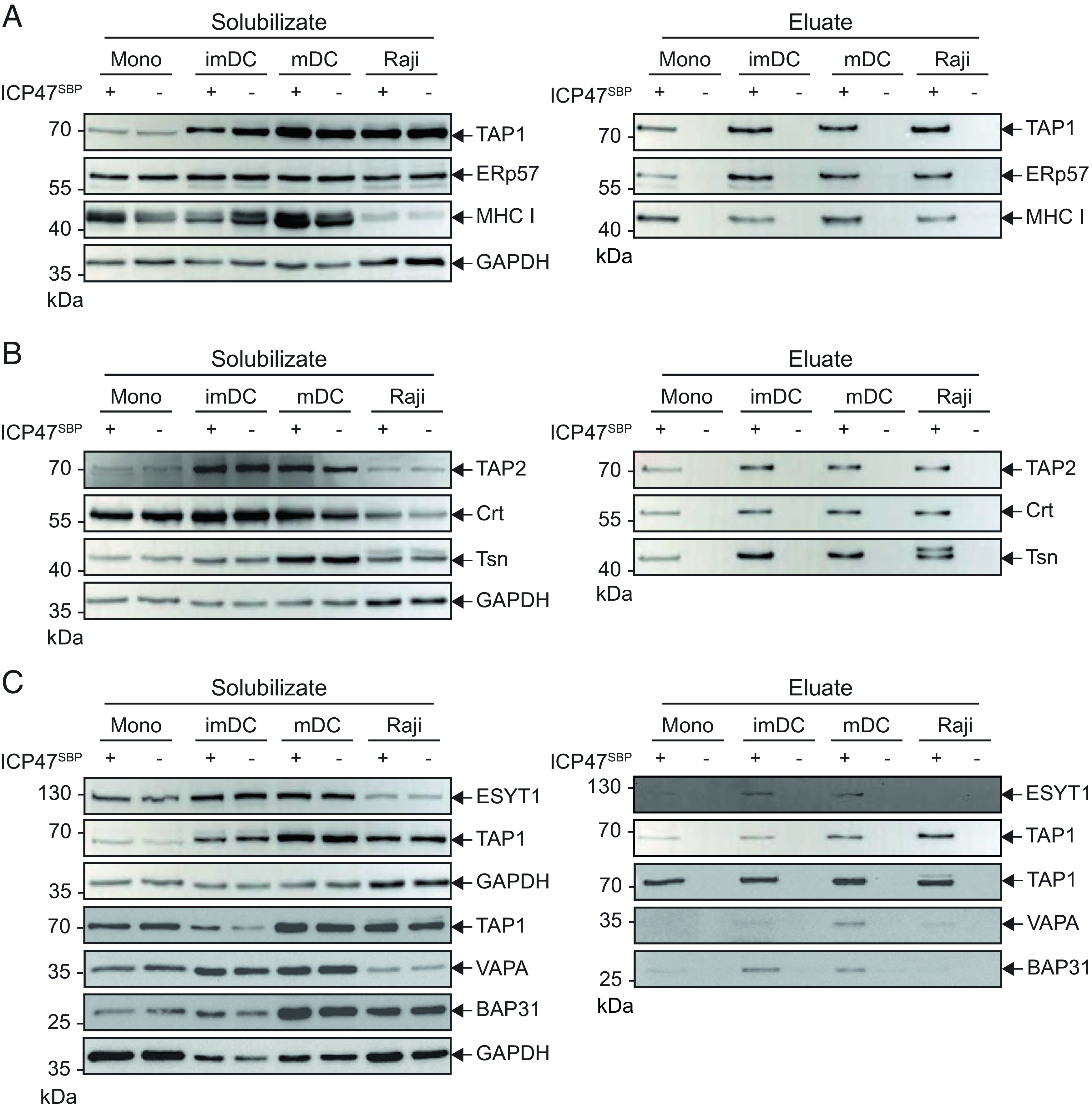
BAP31, VAPA, and ESYT1 are interaction partners of ICP47SBP-arrested PLCs in moDCs. Monocytes (mono), imDCs, mDCs, and Raji cells were solubilized in 1% (w/v) digitonin and affinity purified in the presence (+) or absence (−) of ICP47SBP. Proteins were separated on SDS-PAGE and subsequently immunoblotted. To avoid donor dependencies, two to three donors were pooled for each primary cell type. The left panel shows cell lysate material after solubilization and ultracentrifugation (solubilizate). On the right panel, the eluates after pulldown are presented. Classical components of the PLC (A) TAP1, ERp57, and MHC I and (B) TAP2, calreticulin (Crt), and tapasin (Tsn) are detected in the ICP47SBP pull-down. (C) ESYT1, VAPA, and BAP31 are detected by immunoblotting. GAPDH served as a loading control.
Subcellular Organization of BAP31, VAPA, ESYT1, and the PLC in DCs.
The PLC mostly resides in the ER membrane (34), and during DC maturation, the tubular ER network extends to the tips of mDC protrusions, where TAP localizes at high density (21). We therefore investigated the subcellular colocalization of the PLC and the interaction partners during differentiation of moDCs. In monocytes, we observed no colocalization for BAP31, VAPA, and ESYT1 with TAP1 by confocal laser scanning microscopy (CLSM) as documented by the respective negative Pearson correlation coefficients. In contrast, after differentiation into imDCs and maturation into mDCs, we detected an increased colocalization of TAP1 in the PLC with BAP31, VAPA, and ESYT1 (Fig. 3). Similar to TAP1, the interaction partners were distributed over the whole cell. After maturation into mDC, BAP31, VAPA, and ESYT1 accumulated in the tips of mDC protrusions, as analogously observed for TAP1. Compared to monocytes, imDCs showed enhanced colocalization of the protein pairs TAP1-BAP31, TAP1-VAPA, and TAP1-ESYT1 (Fig. 3). After mDC maturation, the Pearson coefficients for TAP1-BAP31 and TAP1-VAPA were further enhanced when compared with imDCs. As the Pearson coefficient did not increase following mDC maturation, presumably TAP1 and ESYT1 colocalized already to a maximal extent at the imDC stage. Control images with secondary antibodies only showed no unspecific signal in the acquisition settings used (SI Appendix, Fig. S9). These results indicate a higher colocalization of TAP1 in the PLC with BAP31, VAPA, and ESYT1 in DCs than in monocytes.
Fig. 3.

Increased colocalization of BAP31, VAPA, and ESYT1 with TAP during maturation and differentiation into moDCs. On the left panel, monocytes (mono), imDCs, and mDCs were chemically fixed with 3% (v/v) formaldehyde/phosphate-buffered saline (PBS) and immune-labeled for TAP1 (magenta, mAb 148.3) and in green (A) BAP31, (B) VAPA, or (C) ESYT1. Nuclei are visualized by DAPI staining (blue). (Scale bar, 10 μm.) On the right, the corresponding Pearson’s correlation coefficients were calculated for the antibody pair in monocytes, imDCs, and mDCs. Mean ± SD is shown in magenta. At least four donors were analyzed. Statistical analysis was performed with the Kruskal–Wallis test using post hoc Dunn’s test for multiple comparisons. ****P ≤ 0.0001 and ***P ≤ 0.001, ns: not significant.
To analyze protein–protein interactions in a nanometer distance range, we performed in situ proximity ligation assay (PLA) in monocytes, imDCs, and mDCs by rolling circle DNA synthesis (Fig. 4A). Using CLSM and flow cytometry, BAP31, VAPA, and ESYT1 were found in ≤40 nm proximity to the PLC, which was immunolabeled by TAP1, in imDC and mDC (Fig. 4 B and C and SI Appendix, Fig. S10). In contrast, in monocytes and in experiments with isotype controls, no proximity signal was observed for the antibody pairs TAP1-BAP31, TAP1-VAPA, and TAP1-ESYT1. In imDCs, an 8- to 20-fold signal increase of TAP1 and the PLC interaction partners was detected, while in mDCs, this signal was increased 30- to 35-fold. TAP1-BAP31 and TAP1-VAPA proximity were significantly enhanced from monocytes to mDCs. Again, TAP1-ESYT1 was detected at similar levels in imDCs and mDCs. Simultaneously with the morphological changes that were induced during monocytes-to-DC differentiation, the colocalization of the PLC with the ER cargo export and contact site–tethering proteins BAP31, VAPA, and ESYT1 increased. Taken together with the increased number of signals for local proximity in imDCs and mDCs, the results suggest a relocalization of the antigen processing machinery to membrane exit- and contact sites.
Fig. 4.

BAP31, VAPA, and ESYT1 are in 40 nm proximity to TAP in imDCs and mDCs. (A) Schematic representation of proximity ligation assay (Duolink). Briefly, target proteins are immunolabeled with primary antibody raised in two different species; then, incubation with secondary antibodies coupled to an oligonucleotide sequence “PLUS” or “MINUS” follows. If target proteins are in 40 nm proximity, complementary oligo sequences are ligated, and the polymerase proceeds to a concatemeric amplification of the DNA template finalized with the hybridization of fluorescently labeled oligos to the amplified sequence. (B) For microscopy, moDCs were chemically fixed with 3% (v/v) formaldehyde/PBS, and a proximity ligation assay was performed with TAP1 (mAb 148.3) and BAP31, VAPA, or ESYT1. As control, corresponding isotype antibodies were used. Nuclei were stained with DAPI for visualization (blue). (Scale bar, 10 μm.) (C) For flow cytometry, moDCs were fixed and semipermeabilized, and a proximity ligation assay was performed with TAP1 (mAb 148.3) and BAP31, VAPA, or ESYT1. Median fluorescence intensity increase ± SD was plotted as x-fold over background (dash line). Two donors per experimental setup were analyzed. Statistical analysis was performed using one-way ANOVA with Turkey post hoc. *P ≤ 0.05 and **P ≤ 0.005, ns: not significant.
CRISPR/Cas9-mediated Deletion of PLC Components in imDC and mDC.
To study the impact of the identified PLC components of DC on MHC I surface expression, we established a CRISPR/Cas9-mediated gene deletion approach in imDCs and mDCs (43) for the PLC core components and the PLC interaction partners. We used a preassembled ribonucleoparticle (RNP) of guide RNA (gRNA)-Cas9, targeting TAP1, tapasin, ERp57, BAP31, VAPA, and ESYT1 at the genomic level, and applied nucleofection to deliver the RNPs in monocytes. To ensure efficient gene disruption, we employed a mixture of up to three gRNAs per target gene. After RNP nucleofection, cells were differentiated into imDCs and mDCs as described before. To confirm that CRISPR/Cas9 nucleofection did not alter DC differentiation and/or maturation, we examined the cell surface expression of CD14, CD83, CD86, and MHC II by flow cytometry of CRISPR/Cas9 gene–edited cells. In untreated cells, we observed the expected downregulation of the monocytic marker CD14, and a moderate upregulation of CD86 in imDCs, followed by a stronger upregulation of the activation markers CD83 and CD86 in mDCs (Fig. 5 A and B). Importantly, RNP treatment did not significantly alter the respective surface receptor expression of imDC and mDCs, except for a slight upregulation of MHC II in mDCs (Fig. 5 A and B). Nevertheless, the classical phenotype of moDCs remained largely unaffected, and the distinct stages of DC differentiation and maturation could be distinguished.
Fig. 5.
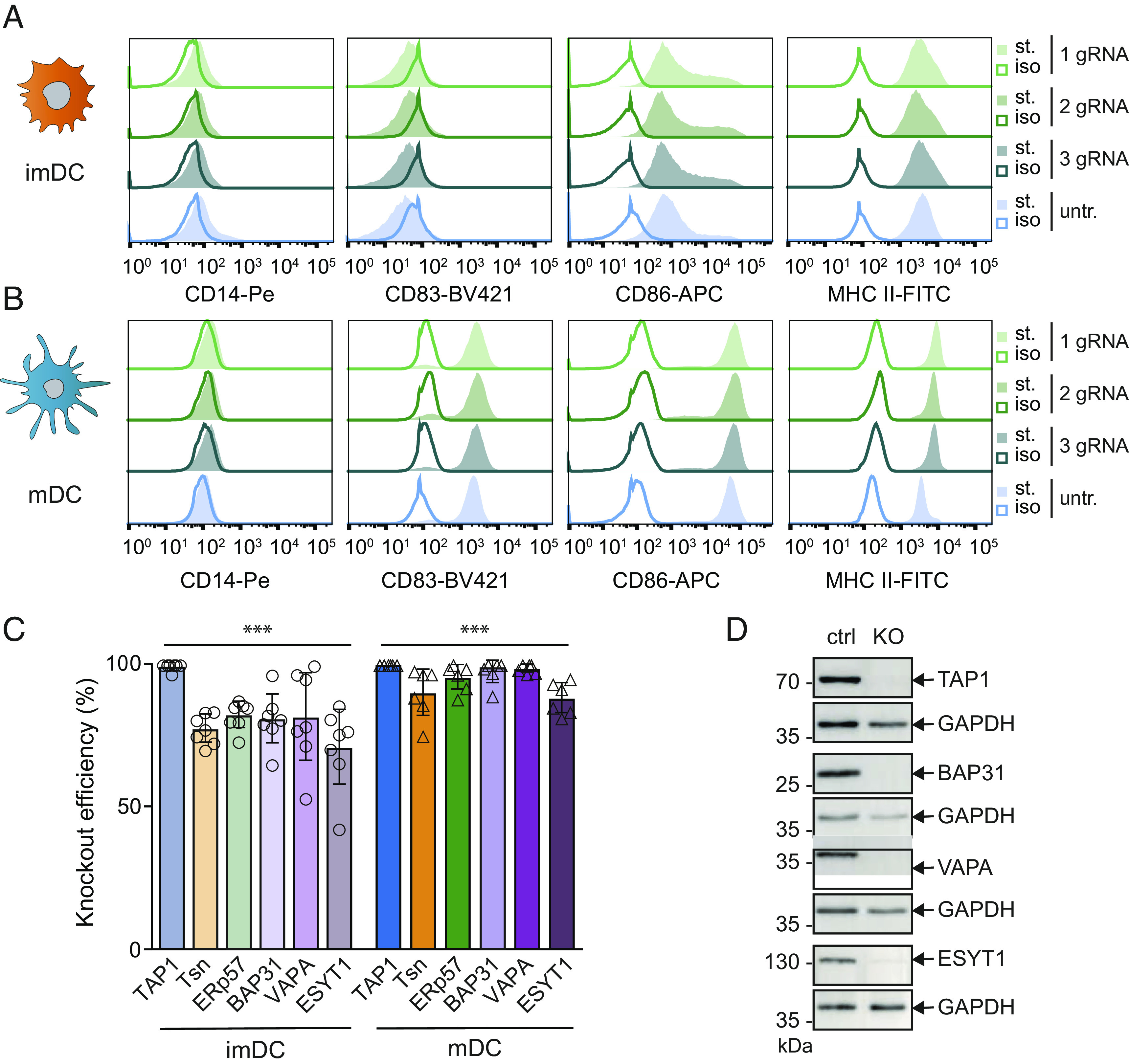
CRISPR/Cas9 knockouts of PLC subunits and interaction partners in moDC. Monocytes were nucleofected with up to three gRNAs in Cas9-RNPs and then differentiated into imDCs and mDCs. Differentiation markers CD14, CD83, CD86, and MHC II were analyzed by flow cytometry at the stage of (A) imDCs or (B) mDCs of CRISPR/Cas9 treated or untreated cells, respectively. Differentiation markers stained (st.) samples were compared to isotype controls (iso) of cells treated with one or mixtures of two or three gRNAs. (C) imDCs (light coloring) and mDCs (strong coloring) were lysed, and the knockout efficiency of TAP1, tapasin (Tsn), ERp57, BAP31, VAPA, and ESYT1 was analyzed by immunoblotting against control gRNA treated cells. Immunoblot results were first normalized to an internal loading control (GAPDH or β-actin) and subsequently normalized to control treated cells. Mean ± SD was plotted; imDCs are depicted in circles (n = 7) and mDCs in triangles (n = 6). Statistical analysis of quantification data compared to control gRNA moDC was performed with the t test with correction for multiple comparisons. ***P ≤ 0.0001. (D) Exemplary immunoblots of control gRNA (ctrl) or knockout (KO) mDCs against TAP1, BAP31, VAPA, or ESYT1. GAPDH was used as loading control.
We next examined the knockout efficiency at the protein level in imDCs and mDCs. At the imDC stage, 5 d post RNP nucleofection, the protein levels of TAP1, tapasin, ERp57, BAP31, VAPA, and ESYT1 were reduced by 70 to 100% (Fig. 5C). Of note, almost all targets showed a further enhanced knockout efficiency after 8 d post CRISPR/Cas9 nucleofection. For TAP1, BAP31, and VAPA deletion, no signal of the protein target was detected in mDCs, while only a very weak signal for ESYT1 remained (Fig. 5D). These data suggest that the lower knockout efficacy in imDCs can be attributed to protein turnover and/or mRNA stability. In conclusion, efficient deletion of the PLC core subunits and identified interaction partners were established in imDCs and mDCs.
MHC I Surface Expression Altered by Deletion of PLC Components in DCs.
We next examined the impact of individual gene knockouts for TAP1, tapasin, ERp57, BAP31, VAPA, and ESYT1 on MHC I surface presentation. We first analyzed MHC I protein levels by immunoblotting of lysates of imDCs and mDCs (Fig. 6A and SI Appendix, Fig. S11). In tapasin-deficient mDCs, a significant decrease of MHC I signals was detected, further verifying that tapasin expression is important for stabilization of nascent MHC I molecules before loading with peptides. In contrast, genomic deletion of TAP1, ERp57, BAP31, VAPA, or ESYT1 did not affect the total expression levels of MHC I molecules. We then determined the HLA-A, -B, and -C surface expression in imDCs and mDCs by flow cytometry (Fig. 6 B and C). No significant downregulation of HLA was observed in imDCs for any of the six knockouts, even though tapasin decreases whole cell MHC I protein levels. Notably, in mDCs, the deletion of TAP1 or tapasin caused a significant MHC I downregulation by up to 50% compared to cells that were nucleofected with control CRISPR/Cas9 gRNA RNPs.
Fig. 6.

Downregulation of surface MHC I molecules after specific knockouts in imDCs and mDCs. Monocytes were subject to CRISPR/Cas9 knockout of TAP1, tapasin, ERp57, VAPA, BAP31, or ESYT1, which were differentiated into imDCs and maturated into mDCs. (A) Exemplary immunoblots of imDCs (Left) or mDCs (Right) TAP1 or tapasin (Tsn) knockouts. Relative protein abundance of MHC I in control and knockout cells is shown. GAPDH was used as loading control. (B and C) Knockout or control DCs were stained with anti-HLA-A, -B, -C antibody, and the surface presentation was analyzed as median fluorescence intensity (FI) by flow cytometry. Normalized median FI ± SD was plotted. (B) imDCs are shown in light coloring and (C) mDCs in strong coloring. Median FI was normalized to control moDCs. Normalized median FI ± SD was plotted. Symbols represent results of one donor; imDCs are depicted in circles (n = 8) and mDCs in triangles (n = 6). (D) HLA-A2 [imDCs are depicted in circles (n = 5) and mDCs in triangles (n = 4)] or (E) HLA-A3 [imDCs are depicted in circles (n = 4) and mDCs in triangles (n = 4)] specific donors were treated with ribonucleoprotein complexes, and the surface presentation of the knockouts (KO) was analyzed by flow cytometry at imDC (light coloring) and mDC (strong coloring) stages. Results were normalized to control treated cells. Statistical analysis of all flow cytometry data was performed by the t test with correction for multiple comparisons. ***P ≤ 0.0001 and **P ≤ 0.001 and *P ≤ 0.01.
HLA-A molecules can be further subdivided into distinct alleles. We specifically analyzed the expressions of the most abundant HLA-A alleles, HLA-A2 and HLA-A3. These alleles are of particular interest for this study, since the expression of HLA-A2 alleles is partially TAP-dependent, while HLA-A3 represent fully TAP-dependent alleles (44). In HLA-A2-positive imDCs, deletion of TAP1 led to a significant downregulation of MHC I surface expression (Fig. 6D). In addition, tapasin-deficient imDCs showed the tendency to express diminished levels of HLA-A2 on the cell surface, whereas all other knockouts did not show downregulation of HLA-A2 surface levels. Interestingly, the deletion of ESYT1 in imDCs seemed to slightly up-regulate HLA-A2. Upon differentiation into mDCs, HLA-A2 was further downregulated by approximately 50% in the case of TAP1 and tapasin knockouts, while no significant effect was detected upon ERp57, BAP31, VAPA, or ESYT1 deletion.
imDCs from HLA-A3-positive donors also showed a significant reduction of MHC I surface expression upon deletion of TAP1 (Fig. 6E). In imDCs, tapasin deletion showed again a minor effect on HLA-A3 expression. Once more, no significant effect on HLA-A3 surface expression was detected upon deletion of ERp57, BAP31, VAPA, or ESYT1 in imDCs. After differentiation of HLA-A3-positive mDCs, a substantial reduction of MHC I surface expression was observed upon TAP1 and tapasin deletion. Here again, no effect on the knockouts of the other targets was seen. Of note, tapasin deletion resulted in a decrease of TAP1 protein levels (SI Appendix, Fig. S12), as previously reported similarly in the murine system (45, 46). Therefore, MHC I downregulation in tapasin-deficient cells cannot be exclusively linked with an effect of tapasin on its MHC I chaperone function.
The differences observed when studying the HLA-A alleles HLA-A2 and HLA-A3 in knockout imDCs remained similar as for the global HLA-A, -B, -C surface presentation. Nevertheless, when TAP1- or tapasin-deficient cells were differentiated into mDCs, global HLA-A, -B, -C, HLA-A2, and HLA-A3 surface expression was drastically diminished by approximately 50% in comparison to cells nucleofected with control CRISPR/Cas9 gRNA. However, ERp57, BAP31, VAPA, and ESYT1 knockouts did not significantly influence the level of MHC I surface expression. These data suggest resilience of MHC I antigen processing in professional presenting cells required for their effective function.
Discussion
In this study, we delineated the dynamic interactome of the PLC during the differentiation and maturation of monocytes into imDCs and mDCs, respectively. By co-IP and LC-MS/MS analysis, BAP31, VAPA, and ESYT1 were identified as interaction partners of the PLC in moDCs. These findings were corroborated by pull-down experiments using the viral inhibitor ICP47SBP, which captures only fully assembled TAP complexes. In imDCs and mDCs, we detected a significantly enhanced colocalization of TAP1 with BAP31, VAPA, and ESYT1 when compared with monocytes. A proximity of ≤40 nm between the identified interaction partners and TAP was demonstrated by in situ proximity ligation assays in imDCs and mDCs. VAPA showed a higher tendency to interact with the PLC during differentiation and maturation into DCs than in monocytes. Our observations indicate a significantly increased interaction of BAP31 and ESYT1 with the PLC owing to the differentiation status of DCs rather than due to protein expression levels.
Among other functions, BAP31 acts as a cargo receptor at ER exit sites for intracellular transport of MHC I, which was analyzed in various different model cell lines (47, 48). Here, we uncovered the recruitment of BAP31 to the PLC during differentiation and maturation of moDCs, including the localization to the tips of the mDC protrusions. To the best of our knowledge, an association of VAPA with the PLC has not been reported so far. VAPA was linked to MHC molecules and the endocytic pathway by its involvement in intracellular trafficking, by positioning late endosomes and autophagosomes with the ER (49, 50). We hypothesize that VAPA might be implicated in antigen cross-presentation based on its ability to interact with vesicle SNARE proteins (51, 52) that mediate the transfer of ER-resident proteins such as TAP to phagosomes, where MHC I peptide-loading can occur (32, 53). Like VAPA, ESYT1 has not been linked to the PLC before. ESYT1 is localized in the ER membrane, specifically at microdomains that tether the ER to the PM. VAP and ESYT protein families have been shown to interact with each other (54, 55), as ESYTs are potential tethers to VAPs at ER-PM MCSs (56). However, no interaction between MHC molecules and the contact site proteins VAPA and ESYT1 has been reported so far. It is tempting to speculate that ER-PM MCSs regulate the direct ER-PM shuttling of MHC I molecules. Here, we report an interaction, colocalization, and ≤40-nm proximity of the PLC with BAP31, VAPA, and ESYT1 in imDCs and mDCs, suggesting that the PLC might be localized at the ER MCS. However, the supramolecular architecture and direct contact link between the interaction partners and the PLC needs to be determined in future studies.
To assess possible functional effects of the interaction partners on the MHC I antigen presentation pathway, we analyzed alterations in MHC I surface expression after BAP31, VAPA, and ESYT1 knockout besides deletion of the three core PLC components, TAP1, tapasin, and ERp57. First, we validated the generation of primary cell knockouts by CRISPR/Cas9 and its influence on moDC differentiation. RNP nucleofection of monocytes with up to three gRNAs did not disturb the differentiation and maturation into imDCs and mDCs, respectively. It is worth mentioning that sorting and subsequent expansion of single clones of nondividing primary cells per definition was not possible because unlike lymphocytes, myeloid cells do not proliferate upon stimulation. No double or triple knockout of BAP31, VAPA, and ESYT1 could be generated as the separation of single, double, and triple knockout cells from mixed populations would be very challenging. Since the interaction partners are not expressed on the cell surface, labeling of these proteins and subsequent enrichment of cells deficient for these markers is difficult. In general, we observed knockout efficiencies ranging from 70 to 90% in imDCs and from 85 to 100% in mDCs throughout different donors. This enhanced deletion efficacy in mDCs could be due to slower protein turnover in moDCs, as observed in case of CD86 and MHC II molecules in moDCs, where their ubiquitination and degradation are reduced, possibly to enhance immunogenicity (57).
Next, we focused on the functional phenotype of MHC I surface expression after loss of core PLC components and the identified peripheral interaction partners, BAP31, VAPA, and ESYT1. The effect on MHC I surface expression was examined on two levels: i) pan-specific HLA including all HLA-A, -B, and -C alleles and ii) focusing on selected alleles such as HLA-A2 and HLA-A3. At the stage of imDCs, no significant HLA-A, -B, -C downregulation was observed for any of the six single knockouts in comparison to cells nucleofected with control CRISPR/Cas9 gRNAs. After maturation into mDCs, we detected downregulation of MHC I surface expression in cells with genomic deletion of TAP1 or tapasin. In case of HLA-A2 and HLA-A3 alleles, this significant downregulation after TAP1 gene disruption was already observed at the imDC stage. In mDCs, both HLA-A alleles displayed decreased MHC I surface expression in TAP1- or tapasin-knockout cells. Notably, the significant downregulation observed in tapasin-deficient cells cannot be directly linked to the peptide editing and loading function of tapasin, as this chaperone is also required for TAP1 stabilization, and its ablation leads to TAP1 degradation (46, 58). The knockouts of the interaction partners BAP31, VAPA, and ESYT1, together with the core PLC component ERp57, failed to show an impact on MHC I surface expression in moDCs. This insinuates a dispensable role of the interaction partners and highlights the superior plasticity of MHC I surface presentation in DCs. Interestingly, even though BAP31 overexpression was reported to increase surface MHC I expression, BAP31 deficiency in HeLa cells, or individuals with a natural deletion across the BAP31 locus, did not show a reduced MHC I surface expression, suggesting a conditional or redundant function of the cargo receptor (59). Deletion of ERp57 has been reported to alter MHC I recruitment to the PLC and surface presentation in mice (46). However, our data do not corroborate these results in human moDCs. One interpretation could be that the peripheral interaction partners are not involved in the MHC I antigen presentation pathway. Nevertheless, we cannot rule out the possibility that our readout is not sensitive enough to detect the phenotype or kinetic disturbances of the knockout. Human DCs have many backup mechanisms to ensure antigen presentation, as even TAP dysfunction can be bypassed by noncanonical antigen presentation pathways (31).
In summary, we showed that the common human DC model tailors its PLC composition according to the differentiation status of monocytes into imDCs and mDCs. We revealed dynamic changes in the PLC composition during differentiation from monocytes into DCs. We identified interaction partners of the moDC PLC, specifically BAP31, VAPA, and ESYT1, suggesting that the antigen processing machinery is located near ER exit and MCSs. The colocalization and proximity of the PLC with the interaction partners was observed even at the tips of the elongated protrusions of mDCs, where higher PLC density has been described (21). This subcellular architecture could facilitate the transfer of TAP and the PLC into cross-presentation organelles (30, 60–62) and even the direct translocation of peptide-loaded MHC I molecules to the cell membrane (30), bypassing the Golgi apparatus. As DCs express high amounts of MHC I on the cell surface, this expedite transport could be beneficial for the formation of MHC I clusters, e.g., via tetraspanin-5, and thus for T-cell activation (63, 64). Moreover, we showed that DCs are not easily influenced in MHC I antigen presentation, as the absence of single PLC interaction partners left HLA surface expression unaffected. Corroboration of this redundancy and compensatory mechanisms could be achieved by intracellular studies of antigen presentation, for example, by using antibody-like tools that can follow the loading and trafficking of MHC I molecules. Whether changes in the PLC specifically apply only for differentiation and maturation of DCs or whether other antigen-presenting cells show similar phenomena remains to be investigated to better understand the biology behind PLC-dependent antigen presentation.
Materials and Methods
Mass Spectrometry.
Frozen cell samples (~1.5 × 107 cells per condition and IP) were thawed on ice and subsequently lysed in IP buffer (20 mM HEPES NaOH pH 7.5, 150 mM NaCl, 8.6% (v/v) glycerol, and 1% (w/v) digitonin). After 1 h solubilization on ice, the mixture was centrifuged for 15 min at 20,000g, 4 °C, to remove cell debris. The cleared lysate was transferred onto 50 µL Protein G Sepharose 4 Fast Flow resin (GE Healthcare) coupled either with 15 µg purified TAP1 antibody (mAb 148.3) or with 15 µg isotype control antibody (Abcam, ab170190). Samples were incubated for 3 h at 4 °C on an overhead rotator. Afterward, beads were washed three times with wash buffer [20 mM HEPES NaOH pH 7.5, 150 mM NaCl, 8.6% (v/v) glycerol, and 0.05% (w/v) digitonin] and then eluted with 25 µL NuPAGE 4 × LPS sample buffer (Thermo Fisher Scientific). Eluates were mixed with dithiothreitol to a final concentration of 100 mM, and samples were separated on NuPAGE 4 to 12% Bis–Tris gels (Thermo Fisher Scientific). Thereafter, in-gel digest of all proteins per lane was performed using standard methods (65). Notably, on-bead digestion was not used to avoid digitonin contaminations. Derived peptides were stage-tipped (66), eluted in buffer A* [0.5% (v/v) acetic acid, 0.2% (v/v) trifluoroacetic acid (TFA)], and analyzed by online LC-MS/MS with a nanoflow ultra-high-performance liquid chromatography instrument (Easy1000 nLC, Thermo Fisher Scientific) connected to a Q Exactive mass spectrometer (Thermo Fisher Scientific) as previously described (67). LC and MS conditions were as follows: Peptide samples were separated on a 15-cm analytical column (75 μm inner diameter) packed in-house with 3 µm C18 beads (Reprosil Pur-AQ, Dr Maisch) via a linear gradient of 6 to 40% ACN/H2O containing 0.5% acetic acid. Spray voltage was 2.0 kV with no sheath and auxiliary gas flow; heated capillary temperature was set to 275 °C. The Q Exactive was run with Xcalibur 2.2 and the LTQ Orbitrap Tune Plus Developers Kit version 2.6.0.1042. Software was operated in data-dependent mode to automatically switch between MS and MS2 acquisitions as previously described (68). Alternatively, operational parameters were monitored in real time by SprayQC software (69). Raw data were analyzed with MaxQuant (versions 1.4.1.1 and 1.6.1.0) (70). Parent ion (MS) and fragment (MS2) spectra were searched against the human Uniprot FASTA database to identify corresponding proteins. The search was performed with the integrated Andromeda search engine, using a target-decoy-based strategy to allow a maximum false discovery rate of <1%. Peptide identifications were filtered for length and mass error. Cysteine carbamidomethylation was searched as a fixed modification, whereas N-acetyl protein and oxidized methionine were searched as variable modifications. Relative quantification of protein abundancies was performed using the default “LFQ” setting in MaxQuant. Proteins were filtered for potential contaminants, reverse proteins, and proteins that are only identified by site. Statistical analysis of LFQ intensities was performed using the Perseus software platform (71), and data were visualized using GraphPad Prism V7 (GraphPad Software). FDR lines of Volcano plots are based on the two-sample t test.
Isolation of CD14+ Monocytes from Human Peripheral Blood.
PBMCs were isolated by gradient centrifugation. To this end, buffy coats were diluted 1:4 (v/v) with 37 °C prewarmed 1 × PBS. Twenty-five milliliters of diluted blood was pipetted onto 15 mL of Ficoll® (Biocoll, δ = 1.077 g/mL, isotonic) solution and centrifuged at 900g for 20 min at room temperature (RT) without break. The PBMC layer was washed twice with cold MACS buffer and centrifuged at 300g for 10 min at RT. Cells were incubated with 10 μL of CD14 magnetic beads (CD14 human MicroBeads) per 107 PBMCs for 20 min at 4 °C protected from light. Manual MACS CD14+ isolation was performed with QuadroMACS™ separator according to the manufacturer’s instructions using the LS column protocol. Automated CD14+ isolation was done with AutoMACS® pro separator using the program “posseld” for positive selection. Buffy coats were kindly provided by the DRK-Blutspendedienst Baden-Württemberg/Hessen or Blutspendedienst NSTOB Springe. All blood samples were de-identified prior to use in this study. Subject data were treated as confidential information protected by medical confidentiality.
Differentiation and Maturation of DCs.
The procedure was followed as previously described (34). In brief, isolated monocytes were differentiated into imDCs by the addition of each 1,000 units/mL interleukin (IL)-4 and GM-CSF (Miltenyi Biotec) in CellGro DC medium (Cell Genix). After 5 d, imDCs were stimulated into mDCs for further 3 d with 10 ng/mL tumor necrosis factor (TNF)-α (PeproTech), 1,000 U/mL IL-6 (PeproTech), 10 ng/mL IL-1β (PeproTech), and 1 mg/mL prostaglandin E2 (Cayman).
Coimmunoprecipitation.
Monocyte, imDC, mDC, and Raji cell pellets (~1.5 × 107 cells per cell type per condition) were thawed on ice and lysed for 1 h in 50 mM HEPES NaOH pH 7.4, 150 mM NaCl, 8.6% (v/v) glycerol supplemented with 1% (w/v) digitonin, 2.5 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride (PMSF). For primary cells, a pool of two to three different donors was mixed to avoid donor-to-donor variances. For the co-IP, 50 μg/condition Dynabeads™ M-280 sheep anti-mouse IgG was incubated with 0.1% (w/v) BSA/PBS for 15 min at RT, and subsequently, 10 μg/condition α-TAP1 (mAb 148.3) (72) or mouse IgG1, κ isotype antibody was added. Beads were incubated for 2 h at 4 °C, and the antibodies were cross-linked with 13.0 mg/ml dimethyl pimelimidate (Pierce™ DMP cross-linker) in 0.2 M sodium borate pH 8.8 for 20 min at RT. Beads were washed with 0.2 M triethanolamine/PBS. Cross-linking was repeated three times. At the end, the beads were washed with 1 M Tris–HCl pH 8.9. Antibody-cross-linked-beads were stored in PBS at 4 °C until usage.
Upon cell lysis, samples were centrifuged for 30 min at 100,000g at 4 °C. Solubilizates were precleared with 25 µg/condition uncoupled Dynabeads™ for 30 min at 4 °C with overhead rotation. Solubilizates were added to TAP1 antibody- or isotype antibody-bound Dynabeads™ and incubated for 3 h at 4 °C with a head-over-tail rotator. Beads were washed with 50 mM HEPES NaOH pH 7.4, 150 mM NaCl, 8.6% (v/v) glycerol, and 0.1% (w/v) digitonin. Proteins were eluted in 50 μL 2× oxidizing SDS buffer at 65 °C for 10 min. Afterward, β-mercaptoethanol was added to the eluate to a final concentration of 0.7 M. Aliquots of the solubilizate were resuspended in 2× reducing SDS buffer. All samples were stored at −20 °C.
Affinity Purification.
For primary cells, a pool of two to three different donors was mixed to avoid donor-to-donor variances. Cell pellets (~1.5 × 107 cells/condition) stored at −80 °C were thawed on ice for 10 min and resuspended in 50 mM HEPES NaOH pH 7.4, 150 mM NaCl, 8.6% (v/v) glycerol supplemented with 1% (w/v) digitonin, 2.5 mM benzamidine, and 1 mM PMSF. Ten micrograms of streptavidin-binding peptide (SBP) tagged ICP47 (ICP47SBP) was added per corresponding condition. Cells were solubilized at 4 °C for 1 h and centrifuged at 100,000g for 30 min at 4 °C. Solubilizate was transferred to Dynabeads™ M-280 streptavidin (Thermo Fisher Scientific) or high-capacity streptavidin agarose resin beads (Thermo Fisher Scientific) and incubated at 4 °C for 3 h. Protein-bound beads were washed with 50 mM HEPES NaOH pH 7.4, 150 mM NaCl, 8.6% (v/v) glycerol supplemented with 0.1% (w/v) digitonin, and eluted in 2.5 mM biotin. Samples were resuspended in 2x reducing SDS buffer and stored at −20 °C.
Immunoblotting.
Cells were resuspended in 50 mM HEPES NaOH pH 7.4, 150 mM NaCl, 8.6% (v/v) glycerol supplemented with 1% (w/v) digitonin, 2.5 mM benzamidine, and 1 mM PMSF and incubated for 1 h rotating at 4 °C. Samples were centrifuged for 15 to 20 min at 4 °C, 16,000g, and the supernatant was transferred into a new vessel. Samples were resuspended in reducing SDS sample buffer, denatured at 65 °C for 10 min, separated by 10% Tris–glycine gels, and blotted onto polyvinylidene fluoride membranes. Upon blocking with 5% (w/v) milk in TBS-T buffer, membrane incubated with primary antibodies: mouse-α-TAP1 (1:10, hybridoma, clone 148.3) (72), rabbit-α-BAP31 (1:2,000, Abcam, ab109304), rabbit-α-VAPA (1:5,000, Abcam, ab181067), rabbit-α-ESYT1 (1:500, Sigma, HPA016858), rat-α-tapasin (1:3,000, purified in-house, clone 7F6-1-1), mouse-α-MHC I (1:20, hybridoma, clone HC10), rabbit-α-calreticulin (1:2,000, Sigma, C4606), rabbit-α-ERp57 (1:2,000, Abcam, ab10287), and rabbit-α-Sec61α (1:2,000, Abcam, ab183046) for 2 h at RT or overnight at 4 °C. Subsequently, membrane incubated with α-mouse (A2554, Merck), α-rat (A9037, Sigma), and α-rabbit (AP307P, Merck) horseradish peroxidase (HRP) for 1 h at RT. For GAPDH, the membrane incubated with α-GAPDH-HRP (1:2,000, BioLegend, 607904) for 2 h at RT. Membranes were developed with Clarity Western ECL Reagent (BioRad) at Lumi™ F1 system (Roche) or Fusion FX (Vilber). Protein levels were quantified using Fiji (73), and the graphical results were generated with GraphPad Prism V5, and V8 (GraphPad Software).
Ribonucleoprotein Complex Formation.
CRISPR RNA (crRNA), transactivating RNA (tracrRNA), and Cas9 from S. pyogenes were purchased from Integrated DNA Technologies (IDT). crRNA sequences were selected from the IDT or GenScript® database (SI Appendix, Table S1). Single crRNA and tracrRNA were dissolved in nuclease-free duplex buffer (IDT) and mixed in a 1:1 molar ratio (210 pmol). The mix was heated to 95 °C and cooled down Δ0.1 °C/s until RT. The resulting gRNAs were either mixed individually or pooled together with Cas9 at a 3:1 molar gRNA:Cas9 ratio and incubated for 30 to 60 min at RT. Electroporation enhancer (IDT) was added to the ribonucleoprotein (RNP) complexes and nuclease-free duplex buffer, if required, to achieve a final volume of 10 µL per reaction. As negative control, RNPs containing crRNA#1 (IDT) were prepared as described above. If not used immediately, RNPs were stored at −20 °C.
Nucleofection of Monocytes.
After PBMC isolation, a sample was stained with APC α-human HLA-A2 (Miltenyi Biotec, 130-116-657) and PE-Vio 615 α-HLA-A3 (Miltenyi Biotec, 130-115-845) in FACS buffer (2% (v/v) FCS in PBS) and mixed with 5% (v/v) human FcR blocking reagent (Miltenyi Biotec) for 20 min at 4 °C. Samples were acquired with the BD FACSCelesta™ (BD) using the instrument-specific software. HLA-A2 or HLA-A3 positive donors were then selected for CD14+ monocyte isolation. Up to 107 monocytes/knockout target were centrifuged for 10 min at 90g and resuspended in 100 µL RT P3 buffer (Lonza). Nucleofection cuvettes (Amaxa Human Monocyte Nucleofector™ Kit, Lonza) were prepared with 10 µL of RNP, and the cell suspension was added. RNP was nucleofected with Nucleofector® 2b Device (Lonza) using the program Y-001. After RNP nucleofection, cells were further differentiated into moDCs as described above.
Flow Cytometry.
Cells were stained in FACS buffer and mixed with 5% (v/v) human FcR blocking reagent (Miltenyi Biotec) together with corresponding antibodies for 20 min at 4 °C. Cells were washed in FACS buffer and resuspended in 0.25% (v/v) formaldehyde/FACS buffer. For intracellular staining, cells were fixed and permeabilized with Cytofix/Cytoperm (BD) for 20 min at 4 °C in the dark. Staining was performed in 1x Permwash (BD) with 5% (v/v) human FcR blocking reagent (Miltenyi Biotec) together with corresponding antibodies (SI Appendix, Table S2) for 30 min at RT. The same staining procedure was performed with fluorophore-labeled secondary antibodies. Cells were washed in 1× Permwash and resuspended in 0.25% (v/v) formaldehyde/FACS buffer. Samples were acquired with the BD FACSCelesta™ (BD) using the instrument-specific software. Data analysis was performed with FlowJo™ V10 (Treestar). For each donor, the median fluorescence intensity (MFI) of every knockout sample was normalized to the MFI of the control gRNA cells (100%). All normalized knockouts were then pooled together for the statistical analysis.
Immunofluorescence Staining.
Monocytes were seeded in eight-well chambers (Nunc II, Lab-tek) in CellGro DC medium and incubated at 37 °C for 30 min for adherence. Cells were then chemically fixed with 3% (v/v) formaldehyde/PBS for 15 to 20 min, washed with PBS, and subsequently quenched with 50 mM glycine/PBS for 20 min. For semipermeabilization, monocytes were treated with 0.1% (v/v) TritonX-100/PBS for 15 min, washed with PBS, and incubated with 5% (w/v) BSA/PBS at RT for 2 h to block unspecific binding. Upon PBS washing, primary antibodies were added in 1% BSA/PBS overnight at 4 °C. Primary antibodies used were mouse-α-TAP1 (1.5 µg/mL, monoclonal, clone 148.3) (72), rabbit-α-BAP31 (1:200, monoclonal, Abcam, ab109304), goat-α-VAPA (1:250, Santa Cruz Technologies, sc-48698), and rabbit-α-ESYT1 (1:25, Sigma, HPA076926). Cells were washed again with PBS and incubated for 2 h at RT with a secondary antibody. Secondary antibodies used were donkey-α-mouseAF488 (1:1,000, Invitrogen, A-21202), goat-α-rabbitAF568 (1:1,000, Invitrogen, A-11011), goat-α-mouseAF647 (1:1,000, Invitrogen, A-21235), donkey-α-rabbitAF647(1:1,000, Invitrogen, A-31573), and donkey-α-goatAF647 (1:1,000, Invitrogen, A-21447). Monocytes were washed again and incubated with 1:5,000 4,6-diamidino-2-phenylindole (DAPI) (Thermo Scientific Fisher) for 2 min at RT. Upon extensive PBS washing, cells were fixed again in 3% (v/v) formaldehyde/PBS for 10 min. Samples were stored in fluorescence mounting medium (DAKO, S3023) at 4 °C.
For immunostaining of imDCs and mDC, monocytes were seeded upon isolation in eight-well chambers and differentiated and matured. moDCs were chemically fixed in 3% (v/v) formaldehyde/PBS for 15 to 20 min. Cells were blocked and permeabilized with 3% (w/v) BSA, 1.5% (w/v) glycine, and 0.01% (w/v) saponin in PBS for 1 h at RT. Antibodies were prepared in 0.1% (w/v) BSA and 0.01% (w/v) saponin in PBS, and the staining protocol was further followed as described for monocytes. Cells were imaged at LSM880 (Zeiss) with Plan-Appochromat 63×/1.4 oil objective (Olympus).
Proximity Ligation Assay.
For microscopy analysis, moDCs were chemically fixed with 3% (v/v) formaldehyde/PBS for 15 to 20 min at RT, and the manufacturer’s protocol of Duolink® in situ far red mouse/rabbit (Sigma) was followed. Primary antibodies rabbit-α-BAP31 (1:200, monoclonal, Abcam, ab109304), rabbit-α-VAPA (1:250, Sigma, HPA009174), rabbit-α-ESYT1 (1:25, Sigma, HPA076926), and mouse-α-TAP1 (1.5 µg/mL, clone 148.3) (72) were used. Isotype antibodies mouse IgG1 kappa monoclonal (ab18437) and rabbit monoclonal IgG (ab172730) were used, correspondingly. Primary antibodies were labeled with α-mouse or α-rabbit Duolink® PLA probes PLUS/MINUS (Sigma) or alternatively for α-TAP1 and mouse isotype with Duolink® PLA Probemaker PLUS (Sigma). Samples were stored in DuoLink® in situ mounting media containing DAPI at 4 °C in the dark. Samples were imaged at LSM880 (Zeiss) with Plan-Appochromat 63×/1.4 oil objective (Olympus).
For flow cytometry, moDCs were fixed and permeabilized with Cytofix/Cytoperm (BD) for 20 min at 4 °C in the dark. After washing, cells were blocked according to the manufacturer’s instructions (Duolink® flowPLA detection reagent far red, Sigma) and 105 cells stained per sample. The primary antibodies used are described in the previous paragraph. Samples were acquired with the BD FACSCelesta™ (BD) using the instrument-specific software. Data analysis was performed with FlowJo™ V10 (Treestar).
Statistical Analysis.
Statistical analysis was performed using GraphPad Prism V5 and V8 (GraphPad Software). For colocalization analysis, a previously generated R-based script was used for semiautomated computational analysis (34), and the nonparametric Kruskal–Wallis test was utilized with Dunn’s test for multiple comparisons. For immunoblot quantification and proximity ligation assays, statistical analysis was performed using one-way ANOVA with Tukey’s post hoc. For flow cytometry and immunoblot quantification, the data were analyzed with the t test using correction for multiple comparisons.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Dr. Simon Trowitzsch, Dr. Lukas Sušac, Jamina Brunnberg, Andrea Pott, Inga Nold, and all members of the Institute of Biochemistry for discussion and many helpful comments. This research was supported by the German Research Foundation (FOR 2830/P04 and CRC 900/B2 to U.K.; CRC 807/P16, the Reinhart Koselleck Project TA 157/12-1, CRC 1507/P18 to R.T.), Excellence Strategy EXC 2155 “RESIST” (Project ID39087428 to R.F. and U.K.), and by an ERC Advanced Grant (798121 to R.T.) of the European Research Council.
Author contributions
M.B., N.K., C.S., and R.T. designed research; M.B., N.K., C.S., V.D., B.B., J.B., M.D., and H.B. performed research; V.D., B.B., J.B., M.D., and H.B. contributed new reagents/analytic tools; M.B., N.K., C.S., R.F., U.K., and R.T. analyzed data; and M.B., N.K., R.F., U.K., and R.T. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All data are deposited to the Zenodo open repository (DOI/accession numbers: 10.5281/zenodo.7296464 (74), 10.5281/zenodo.7407085 (75), 10.5281/zenodo.7294549 (76), and 10.5281/zenodo.7250121 (77). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Jung S., et al. , In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17, 211–220 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman R. M., Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 30, 1–22 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Salah A., et al. , Insights into dendritic cells in cancer immunotherapy: From bench to clinical applications. Front. Cell Dev. Biol. 9, 686544 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mastelic-Gavillet B., Balint K., Boudousquie C., Gannon P. O., Kandalaft L. E., Personalized dendritic cell vaccines-recent breakthroughs and encouraging clinical results. Front Immunol. 10, 766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.León B., López-Bravo M., Ardavin C., Monocyte-derived dendritic cells. Semin Immunol. 17, 313–318 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Banchereau J., Palucka A. K., Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 5, 296–306 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Trombetta E. S., Mellman I., Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 23, 975–1028 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Banchereau J., Steinman R. M., Dendritic cells and the control of immunity. Nature 392, 245–252 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Collin M., Bigley V., Human dendritic cell subsets: An update. Immunology 154, 3–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohl L., et al. , CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21, 279–288 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Gardner A., de Mingo Pulido A., Ruffell B., Dendritic cells and their role in immunotherapy. Front Immunol. 11, 924 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wculek S. K., et al. , Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Laureano R. S., et al. , Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology 11, 2096363 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens D., Ingels J., Van Lint S., Vandekerckhove B., Vermaelen K., Dendritic cell-based immunotherapy in lung cancer. Front. Immunol. 11, 620374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trowitzsch S., Tampé R., Multifunctional chaperone and quality control complexes in adaptive immunity. Annu. Rev. Biophys. 49, 135–161 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Blum J. S., Wearsch P. A., Cresswell P., Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blees A., et al. , Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Domnick A., et al. , Molecular basis of MHC I quality control in the peptide loading complex. Nat. Commun. 13, 4701 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas C., Tampé R., Structural and mechanistic principles of ABC transporters. Annu. Rev. Biochem. 89, 605–636 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Cruz F. M., Colbert J. D., Merino E., Kriegsman B. A., Rock K. L., The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 35, 149–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koller N., et al. , Nanoscale organization of the MHC I peptide-loading complex in human dendritic cells. Cell Mol. Life Sci. 79, 477 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prinz W. A., Toulmay A., Balla T., The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saheki Y., De Camilli P., Endoplasmic reticulum-plasma membrane contact sites. Annu. Rev. Biochem. 86, 659–684 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Li C., et al. , Endoplasmic reticulum-plasma membrane contact sites: Regulators, mechanisms, and physiological functions. Front. Cell Dev. Biol. 9, 627700 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hewlett B., Singh N. P., Vannier C., Galli T., ER-PM contact sites - SNARING actors in emerging functions. Front. Cell Dev. Biol. 9, 635518 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips M. J., Voeltz G. K., Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King C., Sengupta P., Seo A. Y., Lippincott-Schwartz J., ER membranes exhibit phase behavior at sites of organelle contact. Proc. Natl. Acad. Sci. U.S.A. 117, 7225–7235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin S., et al. , Molecular machineries and physiological relevance of ER-mediated membrane contacts. Theranostics 11, 974–995 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nunes-Hasler P., Demaurex N., The ER phagosome connection in the era of membrane contact sites. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1513–1524 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Ackerman A. L., Kyritsis C., Tampé R., Cresswell P., Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc. Natl. Acad. Sci. U.S.A. 100, 12889–12894 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbet G., et al. , TAP dysfunction in dendritic cells enables noncanonical cross-presentation for T cell priming. Nat. Immunol. 22, 497–509 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cebrian I., et al. , Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147, 1355–1368 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Touret N., et al. , Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell 123, 157–170 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Döring M., et al. , Modulation of TAP-dependent antigen compartmentalization during human monocyte-to-DC differentiation. Blood Adv. 3, 839–850 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saibil S. D., Ohashi P. S., Targeting T cell activation in immuno-oncology. Curr. Oncol. 27, S98–S105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ackerman A. L., Cresswell P., Regulation of MHC class I transport in human dendritic cells and the dendritic-like cell line KG-1. J. Immunol. 170, 4178–4188 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Jonuleit H., et al. , Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 27, 3135–3142 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Sethumadhavan S., et al. , Viral immune evasins impact antigen presentation by allele-specific trapping of MHC I at the peptide-loading complex. Sci. Rep. 12, 1516 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Früh K., et al. , A viral inhibitor of peptide transporters for antigen presentation. Nature 375, 415–418 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Hill A., et al. , Herpes simplex virus turns off the TAP to evade host immunity. Nature 375, 411–415 (1995). [DOI] [PubMed] [Google Scholar]
- 41.Ahn K., et al. , Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 15, 3247–3255 (1996). [PMC free article] [PubMed] [Google Scholar]
- 42.Tomazin R., et al. , Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 15, 3256–3266 (1996). [PMC free article] [PubMed] [Google Scholar]
- 43.Duran V., et al. , Fucosylated lipid nanocarriers loaded with antibiotics efficiently inhibit mycobacterial propagation in human myeloid cells. J. Control Release 334, 201–212 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Doytchinova I., Hemsley S., Flower D. R., Transporter associated with antigen processing preselection of peptides binding to the MHC: A bioinformatic evaluation. J. Immunol. 173, 6813–6819 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Papadopoulos M., Momburg F., Multiple residues in the transmembrane helix and connecting peptide of mouse tapasin stabilize the transporter associated with the antigen-processing TAP2 subunit. J. Biol. Chem. 282, 9401–9410 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Garbi N., Tiwari N., Momburg F., Hämmerling G. J., A major role for tapasin as a stabilizer of the TAP peptide transporter and consequences for MHC class I expression. Eur. J. Immunol. 33, 264–273 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Paquet M. E., Cohen-Doyle M., Shore G. C., Williams D. B., Bap29/31 influences the intracellular traffic of MHC class I molecules. J. Immunol. 172, 7548–7555 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Spiliotis E. T., Manley H., Osorio M., Zuniga M. C., Edidin M., Selective export of MHC class I molecules from the ER after their dissociation from TAP. Immunity 13, 841–851 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Rocha N., et al. , Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 185, 1209–1225 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wijdeven R. H., et al. , Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat. Commun. 7, 11808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weir M. L., Xie H., Klip A., Trimble W. S., VAP-A binds promiscuously to both v- and tSNAREs. Biochem. Biophys. Res. Commun. 286, 616–621 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Murphy S. E., Levine T. P., VAP, a versatile access point for the endoplasmic reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta 1861, 952–961 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Alloatti A., et al. , Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J. Exp. Med. 214, 2231–2241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cabukusta B., et al. , Human VAPome analysis reveals MOSPD1 and MOSPD3 as membrane contact site proteins interacting with FFAT-related FFNT motifs. Cell Rep. 33, 108475 (2020). [DOI] [PubMed] [Google Scholar]
- 55.James C., Kehlenbach R. H., The interactome of the VAP family of proteins: An overview. Cells 10, 1780 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Helle S. C., et al. , Organization and function of membrane contact sites. Biochim. Biophys. Acta 1833, 2526–2541 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Tze L. E., et al. , CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 208, 149–165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leonhardt R. M., Abrahimi P., Mitchell S. M., Cresswell P., Three tapasin docking sites in TAP cooperate to facilitate transporter stabilization and heterodimerization. J. Immunol. 192, 2480–2494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ladasky J. J., et al. , Bap31 enhances the endoplasmic reticulum export and quality control of human class I MHC molecules. J. Immunol. 177, 6172–6181 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guermonprez P., et al. , ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425, 397–402 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Houde M., et al. , Phagosomes are competent organelles for antigen cross-presentation. Nature 425, 402–406 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Burgdorf S., Schölz C., Kautz A., Tampé R., Kurts C., Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol. 9, 558–566 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Colbert J. D., Cruz F. M., Baer C. E., Rock K. L., Tetraspanin-5-mediated MHC class I clustering is required for optimal CD8 T cell activation. Proc. Natl. Acad. Sci. U.S.A. 119, e2122188119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu X., et al. , Endogenous viral antigen processing generates peptide-specific MHC class I cell-surface clusters. Proc. Natl. Acad. Sci. U.S.A. 109, 15407–15412 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M., In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Rappsilber J., Mann M., Ishihama Y., Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Michalski A., et al. , Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteomics 10, M111 011015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kelstrup C. D., Young C., Lavallee R., Nielsen M. L., Olsen J. V., Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J. Proteome Res. 11, 3487–3497 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Scheltema R. A., Mann M., SprayQc: A real-time LC-MS/MS quality monitoring system to maximize uptime using off the shelf components. J. Proteome Res. 11, 3458–3466 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Cox J., et al. , Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 13, 2513–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tyanova S., et al. , The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Meyer T. H., van Endert P. M., Uebel S., Ehring B., Tampé R., Functional expression and purification of the ABC transporter complex associated with antigen processing (TAP) in insect cells. FEBS Lett. 351, 443–447 (1994). [DOI] [PubMed] [Google Scholar]
- 73.Schindelin J., et al. , Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schölz C., et al. , Mass spectrometry raw data monocytes, imDCs and mDCs. Zenodo. https://zenodo.org/record/7296464. Deposited 22 February 2023.
- 75.Schölz C., et al. , MaxQuant output data for monocytes, imDCs, and mDCs. Zenodo. https://zenodo.org/record/7407085. Deposited 22 February 2023.
- 76.Schölz C., et al. , Mass spectrometry and MaxQuant data of LFQ for monocytes, imDCs, and mDCs. Zenodo. https://zenodo.org/record/7294549. Deposited 22 February 2023.
- 77.Schölz C., et al. , Mass spectrometry and MaxQuant output data for Raji cells. Zenodo. https://zenodo.org/record/7250121. Deposited 22 February 2023. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All data are deposited to the Zenodo open repository (DOI/accession numbers: 10.5281/zenodo.7296464 (74), 10.5281/zenodo.7407085 (75), 10.5281/zenodo.7294549 (76), and 10.5281/zenodo.7250121 (77). All other data are included in the manuscript and/or SI Appendix.