

Abstract
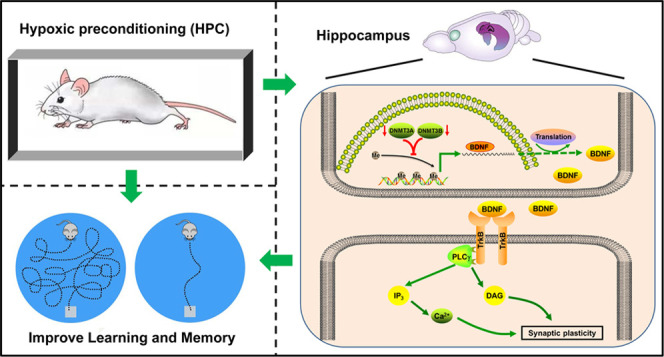
Hypoxic preconditioning (HPC) as an endogenous mechanism can resist hypoxia/ischemia injury and exhibit protective effects on neurological function including learning and memory. Although underlying molecular mechanisms remain unclear, HPC probably regulates the expression of protective molecules by modulating DNA methylation. Brain-derived neurotrophic factor (BDNF) activates its signaling upon binding to the tropomyosin-related kinase B (TrkB) receptor, which is involved in neuronal growth, differentiation, and synaptic plasticity. Therefore, this study focused on the mechanism by which HPC regulates BDNF and BDNF/TrkB signaling through DNA methylation to influence learning and memory. Initially, the HPC model was established by hypoxia stimulations on ICR mice. We found that HPC downregulated the expression of DNA methyltransferase (DNMT) 3A and DNMT3B. Then, the upregulation of BDNF expression in HPC mice was generated from a decrease in DNA methylation of the BDNF gene promoter detected by pyrophosphate sequencing. Subsequently, upregulation of BDNF activated BDNF/TrkB signaling and ultimately improved learning and spatial memory in HPC mice. Moreover, after mice were intracerebroventricularly injected with the DNMT inhibitor, the restraint of DNA methylation accompanied by an increase of BDNF and BDNF/TrkB signaling was also discovered. Finally, we observed that the inhibitor of BDNF/TrkB signaling prevented HPC from ameliorating learning and memory in mice. However, the DNMT inhibitor promoted spatial cognition in mice. Thus, we suggest that HPC may upregulate BDNF by inhibiting DNMTs and decreasing DNA methylation of the BDNF gene and then activate BDNF/TrkB signaling to improve learning and memory in mice. This may provide theoretical guidance for the clinical treatment of cognitive dysfunction caused by ischemia/hypoxia disease.
Keywords: hypoxic preconditioning, brain-derived neurotrophic factor, DNA methylation, DNA methyltransferase, BDNF/TrkB signaling, learning and memory
Introduction
Hypoxia is a common stress state in developmental and physiological processes, which is a fundamental pathological process and causes death in some diseases.1 However, tissues and cells may actively mobilize endogenous protective mechanisms to adapt to hypoxia, and hypoxic preconditioning (HPC) is one of these mechanisms. HPC refers to the administration of a sublethal hypoxic stimulus to a tissue or cell that can be made tolerant to the subsequent lethal hypoxic stimulus.2 Studies have shown that HPC is an effective endogenous neuroprotective strategy against neurological diseases. The mechanism involves processes such as hypoxia signaling, anti-inflammation, oxidative stress, and autophagy.3 In addition, HPC has also been found to have neuroprotective effects by affecting learning and memory, which are important functions of the brain. HPC may not only enhance spatial learning and memory in mice4 but also prevent cognitive dysfunction caused by age-related diseases.5 However, underlying molecular mechanisms still need to be investigated.
The neuroprotective effect of HPC is associated with some key factors in the nervous system. Brain-derived neurotrophic factor (BDNF) is the most abundant and common neurotrophin in the brain.6 BDNF binds to its specific receptor, tropomyosin-related kinase B (TrkB), which activates the BDNF/TrkB signaling to promote neuronal development, differentiation, maturation, and survival.7 Furthermore, BDNF may also resist brain damage derived from hypoxia/ischemia.8 For example, upregulation of BDNF via the specially developed viral constructs may increase survival after hypoxic injury in animals.9 However, the effects of BDNF, TrkB receptor, and BDNF/TrkB signaling in neuroprotection of the HPC remain unclear.
Notably, BDNF can modulate synaptic plasticity, which is a molecular mechanism of learning and memory in development and adulthood via activation of BDNF/TrkB signaling.10 In particular, through the phospholipase C γ (PLCγ)-Ca2+ pathway, calmodulin-dependent protein kinases (CaMK) and the cAMP-response element binding protein (CREB) are activated and involved in the formation and maintenance of long-term potentiation (LTP).11 Meanwhile, a study revealed that intermittent hypoxia training might upregulate BDNF and alleviate cognitive impairment in Alzheimer’s disease mice.12 Therefore, it is suggested that the improvement of learning and memory by HPC may be related to BDNF and its signaling.
The protective effect of HPC is endogenous and may be due to changes in gene expression when cells are under extreme conditions.13 The regulation of gene expression involves epigenetics. DNA methylation is an important mode of epigenetic modification, which is accomplished by DNA methyltransferase (DNMT) that modifies the methylation of genomic DNA to regulate gene expression.14 Changes in the function of DNMTs always cause alterations of DNA methylation and target gene expression. Interestingly, we found that HPC downregulated the expression and activity of DNMTs in mouse hippocampal neuronal cells.15 In addition, CpG islands are present in the promoter of BDNF genes. It has been demonstrated that DNMTs were decreased by 2% hydrogen gas intake and reduced DNA methylation of the BDNF gene promoter to increase the expression of BDNF, which ultimately relieved cognitive impairment caused by sepsis-related encephalopathy.16 Thus, we hypothesize that HPC may influence DNA methylation to regulate BNDF and its signaling to promote learning and memory.
Therefore, this study established the HPC mice model to explore the molecular mechanisms of HPC affecting learning and memory. The effects and regulation mechanisms of BDNF in the HPC mice model were determined by detecting the expressions of BDNF, DNMTs, and pyrophosphate sequencing of CpG islands in BDNF gene promoters. Then, we examined spatial learning and memory of the HPC mice model after DNMT inhibitor (5-Aza-2′-deoxycytidine, 5-Aza) and BDNF/TrkB signaling inhibitor (K-252a) treatment in order to identify whether HPC may regulate BDNF and its signaling through DNA methylation to promote learning and memory in mice.
Results
HPC Upregulates BDNF Expression in the Hippocampus of Mice
In the process of preparing an HPC model by four consecutive hypoxia stimulations, we found that the hypoxic tolerance time of mice showed a linear increase with the number of hypoxia stimulations (Figure 1A, P < 0.05), which on the one hand confirmed that HPC could enhance the hypoxic tolerance of mice and on the other hand indicated the success of our model-making. To verify the role of BDNF as an important brain protective molecule in the endogenous neuroprotection of HPC, we detected the expression of BDNF in the hippocampus of mice from 0–4 days after the HPC treatment. The results showed the upregulation of BDNF at the mRNA (Figure 1B) and protein (Figure 1C) levels in the HPC group compared with the control group, and the changes at both the mRNA and protein levels were the most pronounced and statistically significant on days 1 and 2 after the HPC treatment (P < 0.05). In addition, immunofluorescence showed that the BDNF protein was located in the CA3 region of the hippocampus (Figure 1D). Consistent with the enhanced expression of BDNF from western blot, significantly increased BDNF protein immunofluorescence was detected in the hippocampus of HPC mice as compared to the control group (Figure 1E, P < 0.05). Based on our findings, HPC might upregulate the BDNF expression in the hippocampus of mice.
Figure 1.

Effects of HPC on hypoxia tolerance time and BDNF expression in the hippocampus of ICR mice. (A) Relationship between hypoxia tolerance time and number of hypoxia stimulations in ICR mice. (B) After 0–4 days of hypoxia or HPC treatment, changes in BDNF expression at the mRNA level were analyzed by qPCR. (C) After 0–4 days of hypoxia or HPC treatment, changes in BDNF expression at the protein level were analyzed by western blot. (D) Representative confocal images of BDNF immunoreactivity in the hippocampus of control, hypoxia, and HPC mice. BDNF was stained green. The nucleus was stained with DAPI and manifested as blue. The scale bar is 50 μm. (E) Fluorescence intensity of BDNF in control, hypoxia, and HPC groups. The data are presented as mean ± standard error of the mean (SEM), n = 6 for each group. Compared with the control group: *P < 0.05.
HPC Is Not Able to Regulate TrkB Receptor Expression in the Hippocampus of Mice
BDNF needs to bind to its specific TrkB receptor to perform functions; we also examined the expression of the TrkB receptor in the hippocampus of mice from 0–4 days after the HPC treatment. The results indicated that the expression of the TrkB receptor did not change significantly before and after HPC treatment, at both the mRNA (Figure 2A, P > 0.05) and protein levels (Figure 2B, P > 0.05). Therefore, we speculated that HPC regulates BDNF expression without affecting the BDNF-specific receptor TrkB in the hippocampus of mice.
Figure 2.

Effects of HPC on TrkB receptor expression in the hippocampus of ICR mice. (A) After 0–4 days of hypoxia or HPC treatment, qPCR analysis of changes in TrkB receptor expression at the mRNA level. (B) After 0–4 days of hypoxia or HPC treatment, western blot analysis of changes in the TrkB receptor expression at the protein level. The data are presented as mean ± SEM, n = 6 for each group.
HPC and 5-Aza Downregulate DNMT Expression in the Hippocampus of Mice
Our previous research suggests that DNA methylation may be responsible for gene expression modulated by HPC. However, DNA methylation regulates gene expression via completing the genomic DNA modification catalyzed by DNMTs. Thus, we analyzed the mechanism of HPC upregulation of BDNF from the perspective of DNA methylation. We investigated the expression of three kinds of DNMTs in the hippocampus of mice. Compared with the control group, DNMT3A that is responsible for de novo DNA methylation was diminished on days 0, 1, 2, 3, and 4 after HPC treatment at the mRNA (Figure 3C, P < 0.05) and protein levels (Figure 3D, P < 0.05); DNMT3B, which also undertakes de novo DNA methylation, showed downregulation of mRNA (Figure 3E, P < 0.05) and protein (Figure 3F, P < 0.05) expressions on days 0, 1, 2, 3, and 4 after HPC treatment. However, there was no significant change in DNMT1 that administers maintenance of DNA methylation among the control, hypoxia, and HPC groups (Figure 3A,B, P > 0.05). These results suggested that DNMT3A and DNMT3B were downregulated immediately after HPC treatment, while upregulation of BDNF appeared on the first day after HPC treatment. Therefore, HPC might influence the de novo DNA methylation of the BDNF gene through downregulation of DNMT3A and DNMT3B from the beginning and then upregulate the BDNF. Since BDNF, DNMT3A, and DNMT3B expressions were all significantly and statistically changed on the first day after HPC treatment, the later PCR, western blot, and immunofluorescence experiments were performed on this day.
Figure 3.

Effects of HPC on DNMT expression in the hippocampus of ICR mice. The qPCR and western blot validated that DNMT1 did not change mRNA (A) and protein (B) expressions in response to HPC. DNMT3A downregulated mRNA (C) and protein (D) expressions in response to HPC after 0, 1, 2, 3, and 4 days. DNMT3B also downregulated mRNA (E) and protein (F) expressions in response to HPC after 0, 1, 2, 3, and 4 days. The data are presented as mean ± SEM, n = 6 for each group. Compared with the control group: *P < 0.05.
As a DNMT-specific inhibitor, 5-Aza can affect the activity and expression of DNMTs and is one of the ways to prove the mechanism of DNA methylation regulating molecular expression. We also exogenously inhibited the function of DNMTs through intracerebroventricular injection of 5-Aza to mimic the effects of HPC and determine whether HPC regulates BDNF through DNA methylation. The results exhibited that the expression of DNMT1 was downregulated in the control and HPC groups after 5-Aza treatment (Figure 4A,B, P < 0.05); DNMT3A was attenuated in the control group after 5-Aza treatment (Figure 4C,D, P < 0.05); and DNMT3B was decreased in the control, hypoxia, and HPC groups after 5-Aza treatment (Figure 4E,F, P < 0.05). Based on the above results, DNMTs might be restrained by either an endogenous pathway of HPC or an exogenous pathway of 5-Aza. However, we also should explore the DNA methylation status of the BDNF gene promoter region to determine whether downregulation of DNMTs may activate the expression of BDNF.
Figure 4.

Effects of 5-Aza on DNMT expression in the hippocampus of ICR mice. After 5-Aza treatment, changes in DNMT1 (A), DNMT3A (C), and DNMT3B (E) mRNA expressions of control, hypoxia, and HPC mice were analyzed by qPCR and changes in DNMT1 (B), DNMT3A (D), and DNMT3B (F) protein expressions of control, hypoxia, and HPC mice were analyzed by western blot. The data are presented as mean ± SEM, n = 6 for each group. Compared with the control group: *P < 0.05. Compared with the hypoxia group: #P < 0.05. Compared with the HPC group: &P < 0.05.
HPC and 5-Aza Reduce DNA Methylation Levels in the Promoter of the BDNF Gene
We identified a CpG island with a sequence length of 212 bp in the BDNF gene promoter region (−456 to −667), which contains a total of 18 CpG sites. We divided this CpG island into three segments (CpG island 1: −456 to −469; CpG island 2: −514 to −552; CpG island 3: −636 to −667) and used pyrophosphate sequencing to determine the DNA methylation of 14 CpG sites for three CpG islands in different groups (Figure 5A).
Figure 5.

Analysis of DNA methylation measured by pyrophosphate sequencing in CpG island 1 of the BDNF gene promoter. (A) Distribution of CpG islands in the promoter of the BDNF gene. (B) Representative pyrophosphate sequencing analysis of control, hypoxia, and HPC groups. (C) Global DNA methylation of CpG island 1 in the different groups. (D) DNA methylation at three sites of CpG island 1 in control, hypoxia, and HPC groups. Before and after 5-Aza treatment, DNA methylation at three sites of CpG island 1 in the control group (E), hypoxia group (F), and HPC group (G). The data are presented as mean ± SEM, n = 3 for each group. Compared with the control group: *P < 0.05. Compared with the hypoxia group: #P < 0.05. Compared with the HPC group: &P < 0.05.
CpG island 1 (−456 to −469) contains three CpG sites in the promoter of the BDNF gene. We observed a significant difference in the global DNA methylation between the HPC group and the control group in CpG island 1 (Figure 5C, P < 0.05). The DNA methylation of CpG sites 1, 2, and 3 was also significantly decreased in the HPC group compared to the control group (Figure 5D, P < 0.05). In addition, after 5-Aza treatment, the global DNA methylation and DNA methylation of each site in CpG island 1 reduced in the control (Figure 5E, P < 0.05), hypoxia (Figure 5F, P < 0.05), and HPC (Figure 5G, P < 0.05) groups.
CpG island 2 (−514 to −552) contains seven CpG sites in the promoter of the BDNF gene. Compared with the control group, the global DNA methylation of CpG island 2 declined in the HPC group (Figure 6B, P < 0.05). There was a decrease in the DNA methylation of the HPC group at 1, 2, and 4 CpG sites when compared with the control group (Figure 6C, P < 0.05). The global DNA methylation of CpG island 2 reduced in the control, hypoxia, and HPC groups after 5-Aza treatment (Figure 6B, P < 0.05). However, these decreasing trends were only statistically different at individual sites, such as 1, 4, and 6 in the control group (Figure 6D, P < 0.05); 1 in the hypoxia group (Figure 6E, P < 0.05); and 1, 2, 5, 6, and 7 in the HPC group (Figure 6F, P < 0.05).
Figure 6.

Analysis of DNA methylation measured by pyrophosphate sequencing in CpG island 2 of the BDNF gene promoter. (A) Representative pyrophosphate sequencing analysis of control, hypoxia, and HPC groups. (B) Global DNA methylation of CpG island 2 in the different groups. (C) DNA methylation at seven sites of CpG island 2 in control, hypoxia, and HPC groups. Before and after 5-Aza treatment, the DNA methylation at seven sites of CpG island 2 in the control group (D), hypoxia group (E), and HPC group (F). The data are presented as mean ± SEM, n = 3 for each group. Compared with the control group: *P < 0.05. Compared with the hypoxia group: #P < 0.05. Compared with the HPC group: &P < 0.05.
CpG island 3 (−636 to −667) contains four CpG sites in the promoter of the BDNF gene. The global DNA methylation and two sites (1 and 2) methylation of CpG island 3 diminished in the HPC group when compared with the control group (Figure 7B,C, P < 0.05). The downward trend appeared in the global and site methylation of each group treated with 5-Aza. However, these trends were not statistically significant (Figure 7D,E,F, P > 0.05), except for the control group (Figure 7B, P < 0.05).
Figure 7.

Analysis of the DNA methylation measured by pyrophosphate sequencing in CpG island 3 of the BDNF gene promoter. (A) Representative pyrophosphate sequencing analysis of control, hypoxia, and HPC groups. (B) Global DNA methylation of CpG island 3 in the different groups. (C) DNA methylation at four sites of CpG island 3 in control, hypoxia, and HPC groups. Before and after 5-Aza treatment, DNA methylation at four sites of CpG island 3 in the control group (D), hypoxia group (E), and HPC group (F). The data are presented as mean ± SEM, n = 3 for each group. Compared with the control group: *P < 0.05. Compared with the hypoxia group: #P < 0.05. Compared with the HPC group: &P < 0.05.
In conclusion, the results suggested that HPC and 5-Aza treatment might mitigate DNA methylation in the CpG islands of the BDNF gene promoter and subsequently regulate the expression of BDNF.
HPC Upregulates BDNF Expression in Mice by Downregulating DNMTs and Reducing DNA Methylation of the BDNF Gene Promoter
The above results revealed that HPC downregulated DNMT3A and DNMT3B and decreased the DNA methylation in the CpG islands of the BDNF gene promoter, accompanied by an upregulation of BDNF. If alteration of BDNF expression under 5-Aza treatment appeared derived from the changes in DNMTs and DNA methylation of the BDNF gene promoter, we may eventually uncover the mechanism of HPC regulating the BDNF expression through DNA methylation. As expected, we found that 5-Aza treatment ameliorated the mRNA (Figure 8A, P < 0.05) and protein (Figure 8B, P < 0.05) expressions of BDNF in the control group. In addition, HPC mice treated with 5-Aza also showed an increase in BDNF expression, but there was no statistical difference. After being treated with 5-Aza, the immunofluorescence intensity of BDNF in the hippocampus was significantly enhanced in the control and HPC groups (Figure 8C,D, P < 0.05). Finally, we analyzed the correlation between the BDNF expression and the DNA methylation levels in the CpG islands of the BDNF gene promoter; it showed a typical negative correlation with a −0.7805 correlation coefficient (Figure 8E, P < 0.05). After all, HPC might upregulate BDNF in the hippocampus of mice via the decrease in DNA methylation of the BDNF gene promoter modulated by downregulation of DNMTs.
Figure 8.

HPC modulates DNA methylation of the BDNF gene to upregulate its expression in mice. (A) The results from qPCR indicated that HPC upregulated BDNF mRNA expression in the hippocampus of mice, and 5-Aza treatment increased BDNF in control groups. (B) The results from western blot indicated that HPC enhanced BDNF protein expression in the hippocampus of mice, and 5-Aza treatment augmented BDNF in control groups. (C) Representative confocal images of BDNF immunoreactivity in the hippocampus of different groups. BDNF was stained green. The nucleus was stained with DAPI and manifested as blue. The scale bar is 50 μm. (D) Fluorescence intensity of BDNF in different groups. (E) Correlation coefficients provided for the association between BDNF expression and DNA methylation. The data are presented as mean ± SEM, n = 6 for each group. Compared with the control group: *P < 0.05. Compared with the hypoxia group: #P < 0.05. Compared with the HPC group: &P < 0.05.
HPC Activates BDNF/TrkB Signaling and Downstream PLCγ Pathway in Mice through the Upregulation of BDNF Modulated by DNA Methylation
The BDNF/TrkB signaling may be activated by the upregulation of BDNF and involved in the neuroprotection of HPC. The effect of HPC on BDNF/TrkB signaling was calculated as the ratio of phosphorylated TrkB (p-TrkB) to the total amount of TrkB, and the p-TrkB/total TrkB ratio was significantly increased in the HPC group when compared with the control group (Figure 9A, P < 0.05). It meant that HPC could activate the BDNF/TrkB signaling. In addition, 5-Aza treatment also upregulated the p-TrkB/total TrkB ratio in the control groups (Figure 9A, P < 0.05), and the ratio tended to increase after 5-Aza treatment in HPC mice. Thus, HPC might activate BDNF/TrkB signaling via the upregulation of BDNF modified by DNA methylation. However, the BDNF/TrkB signaling inhibitor K-252a reduced the p-TrkB/total TrkB ratio in all groups (Figure 9B, P < 0.05). Some studies disclosed that the downstream PLCγ pathway of BDNF/TrkB signaling may take part in synaptic plasticity. We also examined the effect of HPC on the PLCγ pathway and found that HPC was also able to increase the p-PLCγ/total PLCγ ratio compared with the control group (Figure 9C, P < 0.05). Therefore, we hypothesized that HPC might activate BDNF/TrkB signaling and the downstream PLCγ pathway to impact learning and memory of mice.
Figure 9.

HPC activates BDNF/TrkB signaling and PLCγ pathway via inhibition of DNA methylation and upregulation of BDNF. (A) The results from western blot showed that HPC upregulated phosphorylated TrkB to activate BDNF/TrkB signaling, and 5-Aza treatment also increased the p-TrkB/total TrkB ratio in the control groups. (B) The results from western blot showed that K-252a suppressed the activation of BDNF/TrkB signaling in control, hypoxia, and HPC groups. (C) The results from western blot showed that HPC enhanced phosphorylated PLCγ to activate the PLCγ pathway. The data are presented as mean ± SEM, n = 6 for each group. Compared with the control group: *P < 0.05. Compared with the hypoxia group: #P < 0.05. Compared with the HPC group: &P < 0.05.
HPC Improves Learning and Memory of Mice via Regulating DNA Methylation and Activating BDNF/TrkB Signaling
The Morris water maze test was used to evaluate the effect of HPC on learning and spatial memory in mice. The results of the visible platform test showed no significant changes in escape latency among all groups (Figure 10B, P > 0.05), suggesting that HPC, 5-Aza, and K-252a treatment did not affect mouse vision. In the hidden platform test, the HPC group demonstrated shorter escape latencies on days 2, 3, 4, and 5 of the test period than those of the control group (Figure 10C, P < 0.05). In the probe test, HPC mice also showed reduced escape latency compared with control mice, but there was an increase in the number of target crossings (Figure 10F, P < 0.05) and the percentage of time spent in the target quadrant (Figure 10I, P < 0.05). It indicated that HPC might improve the learning and memory of mice. However, both the control and HPC groups subjected to K-252a exhibited a significant augment of latency times over the hidden platform test (Figure 10D,E, P < 0.05). During the probe test, K-252a treatment enhanced the escape latency of mice in the control and HPC groups (Figure 10D,E, P < 0.05) and reduced the time in the target quadrant and number of target crossings (Figure 10G,H,J,K, P < 0.05). The improvement of learning and memory in mice might be generated from BDNF/TrkB signaling activated by HPC. Furthermore, throughout the hidden platform test, mice treated with 5-Aza spent less time in reaching the platform compared to control mice (Figure 10D, P < 0.05). Moreover, HPC mice treated with 5-Aza also traveled less time to reach the platform on day 1 of the test (Figure 10E, P < 0.05). In the probe test, the increase of time in the target quadrant and numbers of target crossings, respectively, emerged after 5-Aza was injected into the control and HPC mice (Figure 10G,H,J,K, P < 0.05). Finally, we illustrated that HPC might upregulate BDNF expression by suppressing the DNMTs and DNA methylation of the BDNF gene and then activate BDNF/TrkB signaling to promote learning and memory in mice.
Figure 10.

HPC promotes learning and memory in mice by inhibiting DNA methylation and activating BDNF/TrkB signaling. (A) Typical swimming trajectory of mice in the Morris water maze. (B) Escape latency of each group during the visible platform test. (C) Escape latency of HPC mice reduced during the hidden platform and probe tests. The escape latency of control (D) and HPC (E) mice increased by K-252 during the hidden platform and probe tests and decreased by 5-Aza during the hidden platform test. (F) Number of target crossings raised in HPC mice during the probe period. The target crossings of control (G) and HPC (H) mice were attenuated by K-252 during the probe period and increased by 5-Aza. (I) Percentage of time spent in the target quadrant increased in HPC mice during the probe period. The time in the target quadrant of control (J) and HPC (K) mice diminished by K-252 during the probe period and increased by 5-Aza. The data are presented as mean ± SD, n = 10 for each group. Compared with the control group: *P < 0.05. Compared with the HPC group: #P < 0.05.
Discussion
The protective effect of HPC on the nervous system is evident. HPC has been found to be able to regulate the process of HIF signaling,3 neurogenesis,17 and autophagy18 through various mechanisms, thus exerting an obviously protective effect on neurons. More importantly, HPC may also protect the overall function of the nervous system, such as learning and memory. Cognitive dysfunction is one of the common symptoms of hypoxia/ischemia diseases, yet HPC may prevent learning and memory impairment caused by hypoxia/ischemia diseases.5 The studies indicate that HPC improves spatial cognition of mice4 and zebrafish.19 Similarly, our results showed that the HPC mice had a decrease in the escape latency compared to the control group and presented enhanced cognitive function in the hidden platform test of water maze (Figure 10C). This promoted learning and memory as also manifested by the fact that the HPC mice needed a shorter time to first traverse the location where the platform was previously located during the probe test (the escape latency of the probe test). However, the platform was removed in the probe test, and the mice could only try to find the original location of the platform with the memory acquired through previous training. Therefore, HPC mice might repeatedly enter and cross the target quadrant several times to find the nonexistent platform during the probe test due to long-lasting memory persistence, which eventually led to an increase in the number of target quadrant crossings and the time spent in the target quadrant compared with the control group (Figure 10F,I). In conclusion, the trend of change in escape latency and the crossings, as well as dwell time, was opposite, but both reflect an improvement of learning and memory in HPC mice. Although HPC did not focus on a certain disease to rescue the loss of memory caused by hypoxia/ischemia injury in the present study, we aim to look for an intrinsic mechanism by which HPC targeted the preinjury via reinforcing learning and memory. Our results may provide an prospective application for HPC in the clinical treatment of neurological dysfunction originated from hypoxia/ischemia diseases.
A possible endogenous neuroprotective mechanism of HPC is the downregulation of “bad genes” with damage effects and the upregulation of “good genes” with protection effects through regulation of the gene expression.2,13 BDNF is a typical “good gene” that regulates many neurological functions and plays a critical role in the recovery of the brain from injury.20 Our results showed that BDNF was significantly upregulated at the mRNA and protein levels in the hippocampus of HPC mice (Figure 1). Other studies have also reported that overexpression of BDNF alleviated neuron damage after HPC treatment.21 In addition, neural stem cells treated with HPC increase BDNF expression and promote functional recovery after spinal cord injury in rats through transplantation.22 These results suggest that BDNF may be involved in the neuroprotective effect of HPC. However, we did not observe the change in TrkB, that is, the receptor of BDNF (Figure 2). Thus, HPC subsequently activated the BDNF/TrkB signaling to exert neuroprotective effects mainly by upregulating BDNF but not TrkB. Interestingly, a significant increase in BDNF expression was achieved around 24 h after HPC treatment (1 day) and was still present at 48 h (2 day) but began to decline by 72 h (3 day), which was expected of a typical epigenetic response. Then, we tried to explore the reasons for the upregulation of BDNF by HPC in terms of DNA methylation.
However, our previous and present studies demonstrated that HPC downregulated DNMT3A and DNMT3B expressions in mouse hippocampal neurons and human SH-SY5Y cells but had no effect on DNMT1.15,23 Therefore, the endogenous protective mechanism of HPC may be achieved by upregulated “good genes” through DNA methylation. In addition to restraint of the DNMT expression (Figure 3), we also found that HPC reduced DNA methylation of CpG islands in the mouse BDNF gene promoter. The above results implied that HPC might ameliorate the expression of BDNF by attenuating the DNMTs and DNA methylation of CpG islands in the BDNF gene. However, the negative example also confirms that hypobaric hypoxia downregulated the BDNF expression by upregulating DNMTs, resulting in neuronal loss and spatial memory impairment in rats.24 Especially in our results, DNMT3A and DNMT3B were reduced on day 0 after HPC, while the changes in BDNF were later than in DNMTs, which was increased on day 1 after HPC. Thus, clearly there was a mechanistic “connect” between DNA methylation being responsible for upregulation of BDNF following HPC.
5-Aza has been applied to reveal the mechanism of DNA methylation in neurological function by specifically inhibiting DNMTs.25 Therefore, we used 5-Aza to exogenously inhibit DNMTs and then found that the expression of DNMTs (Figure 4) and DNA methylation of CpG islands in the mouse BDNF gene promoter were decreased in the control, hypoxia, and HPC groups after 5-Aza treatment (Figures 5–7). Subsequently, the expression of BDNF was increased (Figure 8). It should be noted that 5-Aza further reduced DNMT1 and DNMT3B in HPC mice, suggesting that DNMT1 might be not, and DNMT3B was partially involved in the effect of HPC. In contrast, for DNMT3A, no further decrease was observed in 5-Aza-treated animals with HPC, suggesting that DNMT3A might be involved in the overall response of HPC. Finally, we supposed that HPC might upregulate BDNF expression via downregulating DNMT3A and DNMT3B, which use unmethylated DNA as the substrate to complete de novo DNA methylation of the BDNF gene. Additionally, 5-Aza may play a similar role to HPC.
Upregulation of BDNF can activate BDNF/TrkB signaling and then regulate neuronal growth, differentiation, and transcription via rasmitogen-activated protein kinase (MAPK) and phosphatidylinositide3-kinases (PI3-K)/Akt, and the PLCγ-Ca2+ pathway, in particular, acts as a key regulator of synaptic plasticity.26 Many studies have proven that activation of BDNF/TrkB signaling can promote the formation of learning and memory.27,28 Furthermore, cognitive dysfunction caused by hypoxia/ischemia brain injury may be alleviated by activating the BDNF/TrkB signaling.29,30 Remarkably, our experiments revealed that HPC activated the BDNF/TrkB signaling and the PLCγ pathway in the hippocampus (Figure 9) and then improved spatial cognition of mice.
Furthermore, to verify whether BDNF/TrkB signaling is involved in the process by which HPC affects cognition, we treated control and HPC mice with the signaling specific inhibitor K-252a. As a result, both groups took a longer time to find the platform in the water maze after K-252a treatment (increase in escape latency) (Figure 10D,E). Due to the fading of information about platform locations in memory, mice treated with K-252a random searched for the platform in other quadrants of the maze, thereby reducing the number of crossings and activity time in the target quadrant (Figure 10G,H,J,K). It confirmed that the improvement of learning and memory in HPC mice was suppressed by K-252a treatment that inhibited the BDNF/TrkB signaling. We speculate that activation of BDNF/TrkB signaling induces the appearance of inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) and diacylglycerol (DAG) with the PLCγ pathway. Ins (1,4,5)P3 stimulates the release of Ca2+ from intracellular calcium stores and activates Ca2+/CaM-dependent protein kinases (CaMK), followed by CaMK activating the cAMP-response element binding protein (CREB), a transcription factor that regulates LTP. Besides, DAG activates protein kinase C (PKC), which is also involved in the formation of synaptic plasticity.11,31 The above process may be one of the mechanisms by which HPC improves learning and memory through BDNF/TrkB signaling. In addition, BDNF/TrkB signaling may regulate calcium channels to promote Ca2+ inward flow and maintain learning and memory.32 However, detailed downstream pathways of BDNF/TrkB signaling related to the effect of HPC on learning and memory, such as the PLCγ pathway and calcium channels, are the focus of our subsequent research.
Since BDNF/TrkB signaling is engaged in the formation of cognition after governing synaptic plasticity, activating BDNF/TrkB signaling by upregulated BDNF through DNA methylation has the potential to ameliorate cognitive impairment. Such 5-Aza increased BDNF and activated its signaling via suppressed DNA methylation and then enhanced the learning performance.33,34 We also consider the effect of HPC on learning and memory after upregulating BDNF through DNA methylation. In HPC mice and control animals treated with 5-Aza, there was a downregulation of DNMTs accompanying activation of BDNF and its signaling. Subsequently, 5-Aza treatment resulted in control and HPC mice exhibiting a shortened escape latency while increasing the number of crossing and activity time in the target quadrant because of mice repeatedly entering the target quadrant to find the platform during the probe test (Figure 10). The results positively validated that HPC, similar to 5-Aza, might enhance cognition in mice by restraining DNA methylation and upregulating BDNF expression, which was also consistent with our previous study on the effect of 5-Aza.35,36 After all, the cognition amelioration caused by 5-Aza demonstrated that DNA methylation regulating BDNF expression engaged in the spatial learning and memory of mice improved by HPC, whereas the cognition impairment induced by K-252a suggested that the subsequently activated BDNF/TrkB signaling was associated with the above process.
In conclusion, our study manifested that HPC downregulated the expression of DNMT3A, DNMT3B, and the DNA methylation of CpG islands located in the BDNF gene promoter in the hippocampus of mice, which subsequently upregulated the BDNF expression and activated BDNF/TrkB signaling, and finally promoted learning and memory in mice. These results will refine the molecular mechanism of the endogenous neuroprotective effect of HPC and suggest that HPC, the novel endogenous protective approach that activates the body’s potential, may be an alternative to surgery or drugs and has the potential to be translated into a safe and convenient strategy for the prevention and treatment of human diseases. As many neurological hypoxia/ischemia diseases such as stroke, epilepsy, and depression are characterized by cognitive dysfunction and associated with epigenetic changes and downregulation of BDNF, HPC may have a preventive effect on these disorders through the above-mentioned pathways. Especially, remote ischemic conditioning, one of the types of HPC, has a significant contribution to neural repair after stroke in clinical practice.37 However, the design of drugs targeting DNA methylation and BDNF signaling may also be one of the therapeutic treatments for neurological diseases.
Materials and Methods
Animals
Male SPF-grade ICR mice weighing 18–22 g were purchased from SiPeiFu (Beijing) Biotechnology Co., Ltd. The mice were housed in sterile, professional cages and had free access to water and standard chow diet. The rearing environment was alternated between 12 h day and 12 h night, with the temperature controlled at 20–24 °C and humidity controlled at 50–60%. All experimental protocols were in accordance with the National Institutes of Health requirements for animal welfare and approved by the Medical Ethics Review Committee of Baotou Medical College (No. 201812. December 20, 2018).
Drugs and Intracerebroventricular Injection
Mice were anesthetized using sodium pentobarbital (Sigma-Aldrich, St Louis, MO) with 50 mg/kg through an intraperitoneal injection. After induction of anesthesia, the mice were fixed on a brain stereotaxic apparatus, the top of the head was debrided, the skin was disinfected, and an incision was made in the middle of the top of the head to expose the bregma. Using bregma as a reference point, according to the coordinates (AP, −0.5 mm; DV, −2.5 mm; ML, +1.0 mm, right) in the standard mouse brain stereotaxic atlas (the atlas of Paxinos and Franklin), the guide cannula was implanted for intracerebroventricular injection of 5 μL of 10 μM 5-Aza-cdR (Sigma-Aldrich, St. Louis, MO) as described in our previous study,35 or 5 μL of 0.6 μg/μL K-252a (Sigma-Aldrich, St. Louis, MO), or the same volume of PBS. The drugs were injected by a microinjection pump at the rate of 1 μL/min for 5 min, and the injector was left in the lateral ventricle for one minute to prevent reflux. Finally, the skin incision was sutured.
Animal Model
Hypoxic exposure was performed as described previously.18 Briefly, a mouse was placed in a 125 mL jar containing fresh air and immediately sealed with a rubber plug. When the first “gasping breath’’ phenomenon occurred in mouse, it was used as the index of tolerance limit and the end of hypoxic exposure. The mouse was then immediately removed from the jar and placed in another 125 mL jar with fresh air for the second hypoxic exposure, and so on for the third and fourth hypoxic exposures. Mice exposed to hypoxia once were categorized as the hypoxia group, and mice exposed to hypoxia four times in a row were categorized as the HPC group. Mice that were not treated with hypoxic exposure were categorized as the control group. The time period between the beginning of airtightness and the onset of the first gasping breath is referred to as the “tolerance time” for each hypoxic exposure.
Quantitative Real-Time PCR
Total RNA was extracted from mice hippocampus using the TRIzol reagent (Invitrogen, Carlsbad, CA). The cDNA was synthesized by reverse transcription of 1 μg of RNA using the RevertAid First Strand cDNA synthesis kit (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania) after determination of the RNA concentration. The expression of mRNA was measured using quantitative real-time PCR with a RealSYBR Mixture (CoWin Biosciences, Jiangsu, China) under the following conditions: 94 °C for 2 min, followed by 40 cycles of 94 °C for 15 s, 59 °C for 15 s, and 72 °C for 30 s, a final period at 50 °C for 2 min. The primers are as follows: BDNF forward primer: 5′-tctgacgacgacatcactggc-3′, reverse primer: 5′-ccagcagaaagagtagaggaggc-3′. TrkB forward primer: 5′-gagctgctgaccaacctcca-3′, reverse primer: 5′-gtccccgtgcttcatgtactca-3′. DNMT1 forward primer: 5′-cctggctaaagtcaagtccct-3′, reverse primer: 5′-gtgtgtgttccgttctccaag-3′. DNMT3A forward primer: 5′-ggccgaattgtgtcttggtg-3′, reverse primer: 5′-ccatctccgaaccacatgac-3′. DNMT3B forward primer: 5′-aagctcccggctgtctaaga-3′, reverse primer: 5′-ctgcgtgtaattcagaaggct-3′. β-actin forward primer: 5′-ggctgtattcccctccatcg-3′, reverse primer: 5′-ccagttggtaacaatgccatgt-3′. The experiments were performed with the Applied Biosystems 7900 system (Applied Biosystems, Foster City, CA). Relative expression was calculated using the 2–ΔΔCt method. All data were normalized to β-actin.
Western Blot
The proteins in the hippocampus of mice were extracted by RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, and 50 mM Tris–HCl with pH 7.4, Beyotime Institute of Biotechnology, Shanghai, China) supplemented with the protease inhibitor cocktail (1:100 dilution to samples, Gene-Protein Link, Beijing, China) or phosphatase inhibitor cocktail II (1:100 dilution to samples, MedChemExpress, Monmouth Junction, NJ). The concentration of proteins was measured using the BCA method (Pierce Biotechnology, Rockford, IL). Proteins were separated by SDS-polyacrylamide gels and transferred to PVDF membranes (Millipore, Billerica, MA). The membranes were incubated with corresponding primary antibodies overnight at 4 °C. The following primary antibodies were used: BDNF (1:1000, Abcam, MA), TrkB (1:200, Santa Cruz, CA), phosphorylated TrkB (1:1000, Cell Signaling Technology, MA), DNMT1 (1:100, Novus Biologicals, CO), DNMT3A (1:100, Novus Biologicals, CO), DNMT3B (1:100, Novus Biologicals, CO), and β-actin (1:1000, Santa Cruz, CA). Then, membranes were exposed to HRP-linked secondary antibodies (Santa Cruz, CA) (1:5000 for antirabbit and mouse IgG) for 1 h at room temperature. At last, the bands were luminous using Super ECL Plus+ (Applygen, Beijing, China). ImageJ was used to analyze the gray value. All data were normalized to β-actin.
Immunofluorescence Staining
The mice of each group were anesthetized and perfused with 30 mL of saline and 30 mL of 4% paraformaldehyde in turn. Subsequently, the whole brain was removed and put into 4% paraformaldehyde in order to fix overnight, followed by replacement with 30% sucrose for 72 h dehydration. After OTG embedding, the whole brain was cut into coronal frozen sections with 20 μm thickness using a Thermo Fisher Scientific Cyrostar NX50 (Thermo Fisher Scientific, Waltham, MA). Sections were blocked with 10% normal goat serum (Boster, Wuhan, China) at room temperature for 1 h and then incubated overnight at 4 °C with the primary antibody specific for BDNF (1:500, Abcam, MA). After washing with PBS, sections were incubated with an Alexa Fluor Plus 488 goat antirat IgG secondary antibody (Invitrogen, Carlsbad, CA) for 4 h at room temperature and DAPI (Invitrogen, Carlsbad, CA) for 10 min at room temperature. The images were captured by a Nikon A1R+ confocal microscopy system with 40× magnification (Nikon, Melville, NY) and processed by NIS Elements C confocal-specific software (Nikon, Melville, NY).
Pyrophosphate Sequencing
Pyrophosphate sequencing was performed to determine the methylation rate of the CpG islands of the BDNF promoter in mouse hippocampal tissues. Genomic DNA was extracted from mouse hippocampal tissue using the TIANamp Genomic DNA kit (Tiangen Biotech, Beijing, China). After measuring the concentration, the genomic DNA was methylated using EpiTect Bisulfite kits (Qiagen, Germantown, MD). The primers were designed for the promoter fragment of the BDNF gene using PyroMark Assay Design 2.0 software and synthesized with biotin-labeling. The primers were as follows: BDNF CpG island 1 forward primer: 5′-agtgggaagtgtagtggt-3′, reverse primer: 5′-biotin-cctccccaaaatccctaccctattttaa-3′, and sequencing primer: 5′-ggaagtgtagtggttag-3′. BDNF CpG island 2 forward primer: 5′-agggtagagattttggggaggaag-3′, reverse primer: 5′-biotin-aaaacctatacaaaccaaactctcaa-3′, and sequencing primer: 5′-attttggggaggaagta-3′. BDNF CpG island 3 forward primer: 5′-taaaatagggtagagattttggggaggaag-3′, reverse primer: 5′-biotin- caaaaacctatacaaaccaaactctcaa-3′, and sequencing primer: 5′-caaataaaaattaacaacctctat-3′. Subsequently, the promoter fragment of the BDNF gene was amplified by PCR using the PyroMark PCR Kit (Qiagen, Germantown, MD) under the following conditions: 95 °C for 3 min, followed by 40 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min, and a final period at 72 °C for 7 min. The experiments were performed with the Applied Biosystems 9700 system (Applied Biosystems, Foster City, CA). Finally, the product of PCR was sequenced and analyzed by PyroMark Q48 Autoprep (Qiagen, Germantown, MD) to detect methylation levels of the BDNF gene promoter.
Morris Water Maze Test
The maze consists of a circular pool with a diameter of 120 cm and a height of 40 cm, which is filled with nontoxic black water with a temperature of 20–24 °C and a depth of 30 cm. The water-filled maze was divided into four quadrants. The Morris water maze test was executed with a visible platform test, a hidden platform test, and a probe test in sequence. The visible platform test was conducted on the first day to verify the vision of the mice. The platform was placed in a quadrant and 1 cm above the water surface, with a visible marker placed on the platform to guide the mice to the platform. Each mouse was tested four times starting from a different quadrant. The time taken for the mice to reach the platform was recorded as the escape latency. If the mouse did not find the platform within 120 s, the mouse was guided to the platform and held there for 20 s and the time was recorded as 120 s. In the hidden platform test, the platform was fixed in the center of the third quadrant 1 cm below the water surface. The mice were tested for 5 days, once in each of the four quadrants of the maze, for a total of 4 times every day, each time at an interval of 1 h. At the beginning of the experiment, mice were gently placed in the maze, facing the wall of the maze, and allowed to swim freely and find the hidden platform within 120 s, after which they stayed on the platform for 10 s, and the time it took for the mice to find the platform was recorded as the escape latency. If the mice could not find the platform within 120 s, they were placed on the platform and left there for 20 s while the escape latency was recorded as 120 s. The probe test was performed on the sixth day; the mice were placed in the maze after the hidden platform was removed and allowed to swim freely for 120 s. The escape latency, the percentage of time spent in the target quadrant, and the number of target crossings were recorded to evaluate spatial learning and memory in mice. All data and the swimming paths of mice were collected and analyzed by the SMART 3.0 system (Panlab, Barcelona, Spain).
Statistical Analysis
Data are presented as mean ± SEM. Student’s t-tests or one-way ANOVA with LSD and S–N-K’s post hoc tests were applied for statistical analysis of the results, and P < 0.05 was considered statistically significant.
Acknowledgments
This research was funded by support from the National Natural Science Foundation of China (82071479 and 31860307 to W.X., 82060337 to G.S.), the Inner Mongolia Natural Science Foundation (2020MS08171 to W.X., 2020MS08006 to S.J., 2020MS08172 to X.L.), the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region of China (NJYT-18-B26 to W.X.), the Shenzhen Longgang District Economic and Technological Development Special Fund Medical, Health Technology Plan Project (LGKCYLWS2021000033 to G.S.), the Shenzhen Science and Technology Plan Project (JCYJ20220531092412028 to G.S.), the Key Projects of Scientific Research of Higher Education Institutions in Inner Mongolia Autonomous Region (NJZZ19189 to Y.X., NJZY17250 to X.L.), and the Scientific Research Foundation of Baotou Medical College (BYJJ-BSJJ201804 to Y.X.).
Author Contributions
⊥ S.Z. and W.F. contributed equally to this work. Conceived the research and drafted the manuscript: W.X. and S.J. Performed the experiments: S.Z. and W.F. Processed and analyzed the data: R.B., Y.X., and X.L. Designed the experiments: W.X., S.J., Y.X., G.S., and X.J. Checked and revised the manuscript: W.X., S.J., and G.S.
The authors declare no competing financial interest.
References
- Luo Z.; Tian M.; Yang G.; Tan Q.; Chen Y.; Li G.; Zhang Q.; Li Y.; Wan P.; Wu J. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduction Targeted Ther. 2022, 7, 218 10.1038/s41392-022-01080-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G. W.; Shao G. Hypoxic preconditioning: effect, mechanism and clinical implication (Part 1). Chin. J. Appl. Physiol. 2014, 30, 489–501. [PubMed] [Google Scholar]; From Nlm.
- Liu J.; Gu Y.; Guo M.; Ji X. Neuroprotective effects and mechanisms of ischemic/hypoxic preconditioning on neurological diseases. CNS Neurosci. Ther. 2021, 27, 869–882. 10.1111/cns.13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao G.; Zhang R.; Wang Z. L.; Gao C. Y.; Huo X.; Lu G. W. Hypoxic preconditioning improves spatial cognitive ability in mice. Neuro-Signals 2006, 15, 314–321. 10.1159/000121368. [DOI] [PubMed] [Google Scholar]; From Nlm.
- Correia S. C.; Alves M. G.; Oliveira P. F.; Casadesus G.; LaManna J.; Perry G.; Moreira P. I. Hypoxic Preconditioning Averts Sporadic Alzheimer’s Disease-Like Phenotype in Rats: A Focus on Mitochondria. Antioxid. Redox Signaling 2022, 37, 739–757. 10.1089/ars.2019.8007. [DOI] [PubMed] [Google Scholar]
- Li Y.; Li F.; Qin D.; Chen H.; Wang J.; Wang J.; Song S.; Wang C.; Wang Y.; Liu S.; et al. The role of brain derived neurotrophic factor in central nervous system. Front. Aging Neurosci. 2022, 14, 986443 10.3389/fnagi.2022.986443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordvall G.; Forsell P.; Sandin J. Neurotrophin-targeted therapeutics: A gateway to cognition and more?. Drug Discovery Today 2022, 27, 103318 10.1016/j.drudis.2022.07.003. [DOI] [PubMed] [Google Scholar]
- Azman K. F.; Zakaria R. Recent Advances on the Role of Brain-Derived Neurotrophic Factor (BDNF) in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6827 10.3390/ijms23126827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrish M. S.; Urazov M. D.; Mishchenko T. A.; Turubanova V. D.; Epifanova E. A.; Krut V. G.; Babaev A. A.; Vedunova M. V.; Mitroshina E. V. Overexpression of Brain- and Glial Cell Line-Derived Neurotrophic Factors Is Neuroprotective in an Animal Model of Acute Hypobaric Hypoxia. Int. J. Mol. Sci. 2022, 23, 9733 10.3390/ijms23179733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numakawa T.; Odaka H. The Role of Neurotrophin Signaling in Age-Related Cognitive Decline and Cognitive Diseases. Int. J. Mol. Sci. 2022, 23, 7726 10.3390/ijms23147726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Ryou M. G.; Chen X.; Cai M.; Wang H.; Jung M. E.; Metzger D. B.; Mallet R. T.; Shi X. Intermittent Hypoxia Training Prevents Deficient Learning-Memory Behavior in Mice Modeling Alzheimer’s Disease: A Pilot Study. Front. Aging Neurosci. 2021, 13, 674688 10.3389/fnagi.2021.674688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Ren C.; Stone C.; Chandra A.; Xu J.; Li N.; Han C.; Ding Y.; Ji X.; Shao G. Hamartin: An Endogenous Neuroprotective Molecule Induced by Hypoxic Preconditioning. Front. Genet. 2020, 11, 582368 10.3389/fgene.2020.582368. [DOI] [PMC free article] [PubMed] [Google Scholar]; From Nlm.
- Wang K.; Liu H.; Hu Q.; Wang L.; Liu J.; Zheng Z.; Zhang W.; Ren J.; Zhu F.; Liu G. H. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduction Targeted Ther. 2022, 7, 374 10.1038/s41392-022-01211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N.; Zhang X. L.; Jiang S. Y.; Shi J. H.; Cui J. H.; Liu X. L.; Han L. H.; Gong K. R.; Yan S. C.; Xie W.; et al. Neuroprotective mechanisms of DNA methyltransferase in a mouse hippocampal neuronal cell line after hypoxic preconditioning. Neural Regener. Res. 2020, 15, 2362–2368. 10.4103/1673-5374.285003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Qin C.; Li P.; Zhang Y.; Wang Y.; Zhang J.; Li D.; Wang H.; Lu Y.; Xie K.; et al. Hydrogen gas alleviates sepsis-induced neuroinflammation and cognitive impairment through regulation of DNMT1 and DNMT3a-mediated BDNF promoter IV methylation in mice. Int. Immunopharmacol. 2021, 95, 107583 10.1016/j.intimp.2021.107583. [DOI] [PubMed] [Google Scholar]
- Li S.; Yang Y.; Li N.; Li H.; Xu J.; Zhao W.; Wang X.; Ma L.; Gao C.; Ding Y.; et al. Limb Remote Ischemic Conditioning Promotes Neurogenesis after Cerebral Ischemia by Modulating miR-449b/Notch1 Pathway in Mice. Biomolecules 2022, 12, 1137 10.3390/biom12081137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi R.; Xie Y.; Zhang X.; Jiang S.; Liu X.; Xie W.; Jia X.; Bade R.; Liu Y.; Gong K.; et al. Possible Involvement of DNA Methylation in TSC1 Gene Expression in Neuroprotection Induced by Hypoxic Preconditioning. Oxid. Med. Cell. Longevity 2022, 2022, 9306097 10.1155/2022/9306097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. H.; Lee K. S.; Kim Y. S.; Kim Y. H.; Kim J. H. Effects of hypoxic preconditioning on memory evaluated using the T-maze behavior test. Anim. Cells Syst. 2019, 23, 10–17. 10.1080/19768354.2018.1557743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng S.; Huang J.; Ren Y.; Zhi F.; Tian X.; Wen G.; Ding G.; Xia T. C.; Hua F.; Xia Y. Neuroprotection Against Hypoxic/Ischemic Injury: delta-Opioid Receptors and BDNF-TrkB Pathway. Cell. Physiol. Biochem. 2018, 47, 302–315. 10.1159/000489808. [DOI] [PubMed] [Google Scholar]
- Turovskaya M. V.; Gaidin S. G.; Vedunova M. V.; Babaev A. A.; Turovsky E. A. BDNF Overexpression Enhances the Preconditioning Effect of Brief Episodes of Hypoxia, Promoting Survival of GABAergic Neurons. Neurosci. Bull. 2020, 36, 733–760. 10.1007/s12264-020-00480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Wei H.; Du J.; Lu X.; Wang L. Hypoxic preconditioning neural stem cell transplantation promotes spinal cord injury in rats by affecting transmembrane immunoglobulin domain-containing. Hum. Exp. Toxicol. 2022, 41, 9603271211066587 10.1177/09603271211066587. [DOI] [PubMed] [Google Scholar]
- Cui J.; Liu N.; Chang Z.; Gao Y.; Bao M.; Xie Y.; Xu W.; Liu X.; Jiang S.; Liu Y.; et al. Exosomal MicroRNA-126 from RIPC Serum Is Involved in Hypoxia Tolerance in SH-SY5Y Cells by Downregulating DNMT3B. Mol. Ther.--Nucleic Acids 2020, 20, 649–660. 10.1016/j.omtn.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.; Jain V.; Kushwah N.; Dheer A.; Mishra K. P.; Prasad D.; Singh S. B. Role of DNA Methylation in Hypobaric Hypoxia-Induced Neurodegeneration and Spatial Memory Impairment. Ann. Neurosci. 2018, 25, 191–200. 10.1159/000490368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Q.; Xu W.; Lin Z.; Liu J.; Chen W.; Zhu H.; Lai M.; Zhuang D.; Xu Z.; Fu D.; et al. Role of GABRD Gene Methylation in the Nucleus Accumbens in Heroin-Seeking Behavior in Rats. Front. Pharmacol. 2020, 11, 612200 10.3389/fphar.2020.612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A. M.; Chauhan L.; Bhardwaj A.; Sharma A.; Fayaz F.; Kumar B.; Alhashmi M.; AlHajri N.; Alam M. S.; Pottoo F. H. Brain-Derived Neurotropic Factor in Neurodegenerative Disorders. Biomedicines 2022, 10, 1143 10.3390/biomedicines10051143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camuso S.; La Rosa P.; Fiorenza M. T.; Canterini S. Pleiotropic effects of BDNF on the cerebellum and hippocampus: Implications for neurodevelopmental disorders. Neurobiol. Dis. 2022, 163, 105606 10.1016/j.nbd.2021.105606. [DOI] [PubMed] [Google Scholar]
- Gao L.; Zhang Y.; Sterling K.; Song W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4 10.1186/s40035-022-00279-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Zhang M.; Yang L.; Zeng M. GPR68 Improves Nerve Damage and Myelination in an Immature Rat Model Induced by Sevoflurane Anesthesia by Activating cAMP/CREB to Mediate BDNF. ACS Chem. Neurosci. 2022, 13, 423–431. 10.1021/acschemneuro.1c00830. [DOI] [PubMed] [Google Scholar]
- Yu L.; Liu S.; Zhou R.; Sun H.; Su X.; Liu Q.; Li S.; Ying J.; Zhao F.; Mu D.; Qu Y. Atorvastatin inhibits neuronal apoptosis via activating cAMP/PKA/p-CREB/BDNF pathway in hypoxic-ischemic neonatal rats. FASEB J. 2022, 36, e22263 10.1096/fj.202101654RR. [DOI] [PubMed] [Google Scholar]
- Kim S.; Kim J.; Park Y. M.; Suh P. G.; Lee C. J. Visuosocial Preference Memory, but Not Avoidance Memory, Requires PLCgamma1 in the CA2 Hippocampus. Exp. Neurobiol. 2022, 31, 332–342. 10.5607/en22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furini C. R. G.; Nachtigall E. G.; Behling J. A. K.; Assis Brasil E. S.; Saenger B. F.; Narvaes R. F.; de Carvalho Myskiw J.; Izquierdo I. Molecular Mechanisms in Hippocampus Involved on Object Recognition Memory Consolidation and Reconsolidation. Neuroscience 2020, 435, 112–123. 10.1016/j.neuroscience.2020.03.047. [DOI] [PubMed] [Google Scholar]
- Ni C.; Qian M.; Geng J.; Qu Y.; Tian Y.; Yang N.; Li S.; Zheng H. DNA Methylation Manipulation of Memory Genes Is Involved in Sevoflurane Induced Cognitive Impairments in Aged Rats. Front. Aging Neurosci. 2020, 12, 211 10.3389/fnagi.2020.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Ma Q.; Dasgupta C.; Halavi S.; Hartman R. E.; Xiao D.; Zhang L. Inhibition of DNA Methylation in the Developing Rat Brain Disrupts Sexually Dimorphic Neurobehavioral Phenotypes in Adulthood. Mol. Neurobiol. 2017, 54, 3988–3999. 10.1007/s12035-016-9957-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Z.; Xu W.; Jiang S.; Liu X.; Zhu H.; Wang P.; Gao B.; Gong K.; Guo G.; Sun K.; et al. Effects of 5-Aza on neurogenesis contribute to learning and memory in the mouse hippocampus. Biomed. Pharmacother. 2022, 154, 113623 10.1016/j.biopha.2022.113623. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Xie Y.; Xu W.; Liu X.; Jiang S.; Bao M.; Xie W.; Jia X.; Bade R.; Gong K.; et al. Effects of 5-Aza on p-Y1472 NR2B related to learning and memory in the mouse hippocampus. Biomed. Pharmacother. 2019, 109, 701–707. 10.1016/j.biopha.2018.10.090. [DOI] [PubMed] [Google Scholar]
- Yu W.; Ren C.; Ji X. A review of remote ischemic conditioning as a potential strategy for neural repair poststroke. CNS Neurosci. Ther. 2023, 29, 516–524. 10.1111/cns.14064. [DOI] [PMC free article] [PubMed] [Google Scholar]