

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal and treatment-refractory cancer. Molecular stratification in pancreatic cancer remains rudimentary and does not yet inform clinical management or therapeutic development. Here we construct a high-resolution molecular landscape of the cellular subtypes and spatial communities that compose PDAC using single-nucleus RNA-seq and whole-transcriptome digital spatial profiling (DSP) of 43 primary PDAC tumor specimens that either received neoadjuvant therapy or were treatment-naïve. We uncovered recurrent expression programs across malignant cells and fibroblasts, including a newly-identified neural-like progenitor malignant cell program that was enriched after chemotherapy and radiotherapy and associated with poor prognosis in independent cohorts. Integrating spatial and cellular profiles revealed three multicellular communities with distinct contributions from malignant, fibroblast, and immune subtypes: classical, squamoid-basaloid, and treatment-enriched. Our refined molecular and cellular taxonomy can provide a framework for stratification in clinical trials and serve as a roadmap for therapeutic targeting of specific cellular phenotypes and multicellular interactions.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is increasingly treated with neoadjuvant chemotherapy and/or radiotherapy1, yet remains largely a treatment-refractory disease2. Thus, there is an urgent need to decipher the impact of preoperative treatment on residual cancer cells and stroma to identify additional therapeutic vulnerabilities. PDAC molecular subtyping remains nascent and does not yet inform clinical management or therapeutic development3,4. Bulk RNA profiling of PDAC tumors5–8 has identified two subtypes: (1) classical/epithelial, encompassing a spectrum of pancreatic lineage precursors, and (2) basal-like/squamous/quasi-mesenchymal, exhibiting loss of endodermal identity and genetic aberrations in chromatin modifiers, poorer responses to chemotherapy9, and worse survival. Additional proposed subtypes may reflect microenvironmental features and do not further stratify patient survival3,8. Moreover, most prior studies profiled tumors from untreated patients. Finally, while the tumor microenvironment (TME) impacts the effect of cytotoxic treatments10, motivating the use of adjunctive therapies such as losartan11,12, our understanding of the spatial architecture and multicellular interactions in the TME remains limited.
Single-cell RNA-seq (scRNA-seq) can reveal the diversity of malignant and non-malignant cells in tumors13–15, but has been challenging to apply in PDAC given high intrinsic nucleases and dense desmoplastic stroma16. Single-nucleus RNA-seq (snRNA-seq) provides a compelling alternative compatible with frozen samples17–19 and with better recovery of malignant and stromal cells20. Moreover, prior spatial proteotranscriptomic analyses of the PDAC TME were limited in molecular multiplexing21,22,23 or spatial resolution24.
Here, we optimized snRNA-seq for banked frozen PDAC specimens stored up to five years, profiled 224,988 nuclei across 43 tumors (18 untreated, 25 treated), and recovered similar overall cellular compositions to those from multiplex protein profiling in situ. We discovered treatment-associated changes in cellular composition and expression programs, including enrichment of a distinct neural-like progenitor (NRP) malignant program in residual tumor and patient-derived organoids after treatment. By integrating cell type signatures and expression programs with whole-transcriptome digital spatial profiles of matched specimens25, we identified several distinct multicellular communities, including a treatment-enriched community that features the NRP malignant program and CD8+ T cells. Furthermore, we discovered spatially-defined intercellular receptor-ligand interactions specifically enhanced in post-treatment residual disease, which are potential targets for improving neoadjuvant and adjuvant therapy in pancreatic cancer.
RESULTS
Single-nucleus RNA-seq for banked frozen human PDAC tumors
We collected 224,988 high-quality snRNA-seq profiles from frozen, histologically-confirmed, primary PDAC specimens from 43 patients (out of 48 in the study) who underwent surgical resection with (n=25) or without (n=18) neoadjuvant treatment (Figure 1a; Supplementary Table 1; Methods). Most treated patients received multi-cycle chemotherapy (FOLFIRINOX) followed by multi-fraction radiotherapy with concurrent 5-FU/capecitabine (CRT; n=14). Five additional patients were enrolled on a clinical trial (NCT01821729) investigating neoadjuvant CRT with the addition of losartan (CRTL). Another six received other forms of neoadjuvant treatment, including two on a phase 2 randomized clinical trial (NCT03563248) who received FOLFIRINOX, stereotactic body radiotherapy, and nivolumab with (n=1) or without losartan (n=1).
Figure 1 |. Single-nucleus RNA-seq of untreated and treated PDAC captures representative diversity of cell types.
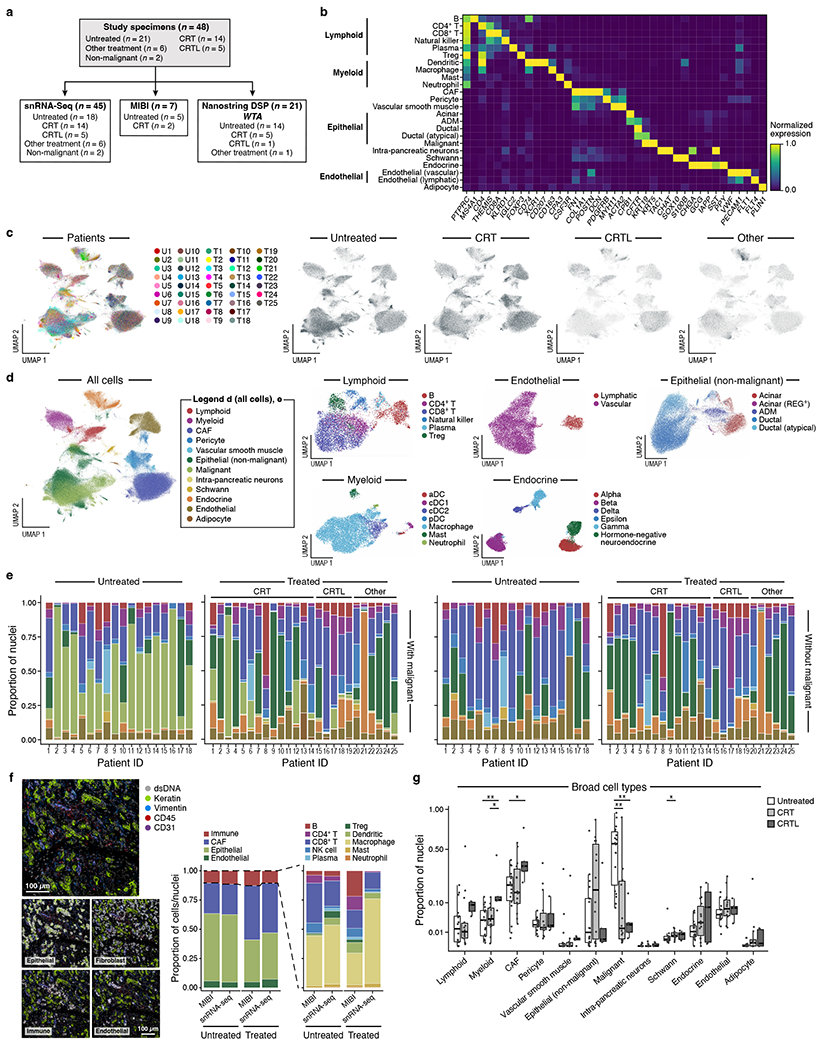
a, Experimental workflow of human PDAC tumors for snRNA-seq, Multiplex Ion Beam Imaging (MIBI), and digital spatial profiling (DSP; NanoString GeoMx). Three patient tumors were analyzed by DSP and not snRNA-seq and two specimens profiled by snRNA-seq were non-malignant pancreatic tissue. b, snRNA-seq captures diverse malignant, epithelial, immune and other stromal cell subsets. Mean normalized expression (color bar) of selected marker genes (columns) across annotated cell subsets (rows) of different compartments (labels, left). c, Distinctions between patients or treatment status. UMAP embedding of single nucleus profiles (dots) of PDAC tumors colored by patient ID (color legend, left) or treatment status (right). d, Cell subsets in each compartment. UMAP embeddings of single nucleus profiles of all cells (left, as in c) or in each compartment (right panels) colored by post hoc cell type annotations (color legend, shared with e). e, Cell type distributions across tumors. Proportions (y axis) of cell subsets (color legend, shared with d) across untreated (n=18) (left) vs. treated (n=25) tumors (right) either with (left) or without (right) malignant cells. Treated patients are further classified by specific treatment type. f, snRNA-seq captures representative cell types distributions compared to in situ assessment. Left: Representative MIBI images and segmentation showing staining with antibodies against cytokeratin (green), vimentin (blue), CD45 (red), CD31 (purple) and double-stranded DNA (gray). Right: Proportion of cells (y axis) in each of the four major compartments (left subpanel, color legend) or in each of the immune subsets (right subpanel, color legend) as estimated by snRNA-seq or MIBI (x axis) in aggregate across all untreated (two left bars; n=5) or treated (two right bars; n=2) tumors. g, Remodeling of tumor composition by treatment. For the untreated, CRT, and CRTL groups, there were n=18, 14, and 5 biologically independent tumor specimens, respectively. Proportions (y axis) of each cell subset (x-axis) among all nuclei. Pairwise comparisons were performed using the two-sided Mann-Whitney U test (* Bonferroni adjusted p < 0.05; ** p < 0.01; *** p < 0.001).
Unsupervised clustering of single nucleus profiles identified 33 cell subsets, which we annotated post hoc by known gene signatures (Figure 1b–e; Methods). We confirmed malignant cells by inferred Copy Number Alterations (CNAs), which were comparable to those derived from The Cancer Genome Atlas (TCGA) PDAC cohort8,13, and generally clustered by patient (adjusted mutual information (AMI) = 0.87 in malignant cells vs. 0.18 in non-malignant cells) (Extended Data Figure 1). Other cell types included non-malignant epithelial cells, immune, endocrine, and diverse stromal cells (cancer-associated fibroblasts/CAFs, endothelial cells, vascular smooth muscle cells, pericytes, intra-pancreatic neurons, Schwann cells, and adipocytes). A small subset of acinar cells (acinar-REG+) expressed high levels of regenerating family member genes (e.g., REG1A, REG3A), which have been implicated in promoting pancreatic inflammation, ADM and PanIN19,26–28 (Figure 1d). Cell types like CAFs, previously under-represented in scRNA-seq, were well-represented in our samples (Figure 1b,d,e; Methods). snRNA-seq captured representative distributions of epithelial, fibroblast, endothelial, and immune cell type proportions compared to estimates from multiplexed ion beam imaging (MIBI) across and within individual tumors, but with some differential capture within certain immune cell subsets (Figure 1f; Extended Data Figure 1d; Methods)20.
Among clinical subgroups with at least five patients (untreated, CRT, CRTL), malignant cell proportions were significantly lower in tumors treated with neoadjuvant therapy (CRT/CRTL vs. untreated; padj=1.16x10−3/3.81x10−3, Mann-Whitney; Figure 1g; Methods), consistent with histology. Several non-malignant cell subsets differed quantitatively and qualitatively across treatment groups (Figure 1g; Extended Data Figure 2). For example, within the immune compartment, there was a higher fraction of CD8+ T cells in neoadjuvant CRTL vs. CRT (padj=3.51x10−2; Mann-Whitney), and a lower proportion of regulatory T cells in CRT vs. untreated (padj=2.27x10−2) (Extended Data Figure 2). Moreover, CD8+ T cells in CRTL tumors expressed higher levels of effector function genes (e.g., IL2, CCL4, CCL5)29 and lower levels of quiescence and dysfunction genes (e.g., TIGIT, TCF7, KLF2, LEF1)30–32 vs. untreated and CRT tumors (Extended Data Figure 3a; Methods). These results are consistent with losartan-mediated increase in intra-tumoral cytotoxic T cell activity12.
Acinar-to-ductal metaplasia (ADM) and atypical ductal cells
Within the epithelial compartment, there were low CNA nuclei co-expressing markers of ductal and acinar lineages (Figure 1b,1d,2c,2d) that may reflect acinar-to-ductal metaplasia (ADM), which plays an initiating role in mouse pancreatic tumorigenesis33,34. These profiles had a typical number of transcripts and are unlikely to be doublets. Chronic inflammation and activating KRAS mutations are linked to persistence of the ADM state and progression to pancreatic intraepithelial neoplasia (PanIN)34,35. ADM cells had higher expression of the HALLMARK_KRAS_SIGNALING_UP signature36 compared to acinar cells (Figure 2a). Moreover, a distinct subset of ductal cells expressed high levels of both ductal (e.g., CFTR) and malignant (e.g., KRT5, KRT19) markers without elevated CNAs, which we termed atypical ductal cells (Figure 1b,1d,2c,2d). Atypical ductal cells featured genes (e.g., KRT17)37 that are expressed as early as the PanIN2/3 stage and had higher levels of the HALLMARK_KRAS_SIGNALING_UP signature than ADM cells, suggesting a progression from ADM to precursor lesions such as PanINs (Figure 1b,1d,2c,2d). Indeed, a partition-based graph abstraction inferred a dominant pseudotemporal trajectory from acinar to ADM to ductal to atypical ductal to malignant cells, paralleling a monotonic increase in the HALLMARK_KRAS_SIGNALING_UP signature, supporting ADM and atypical ductal cells as relevant intermediate states in PDAC tumorigenesis (Figure 2a,b).
Figure 2 |. Epithelial cell type composition and inferred pseudotemporal trajectory includes putative acinar-ductal metaplasia and atypical ductal intermediates.
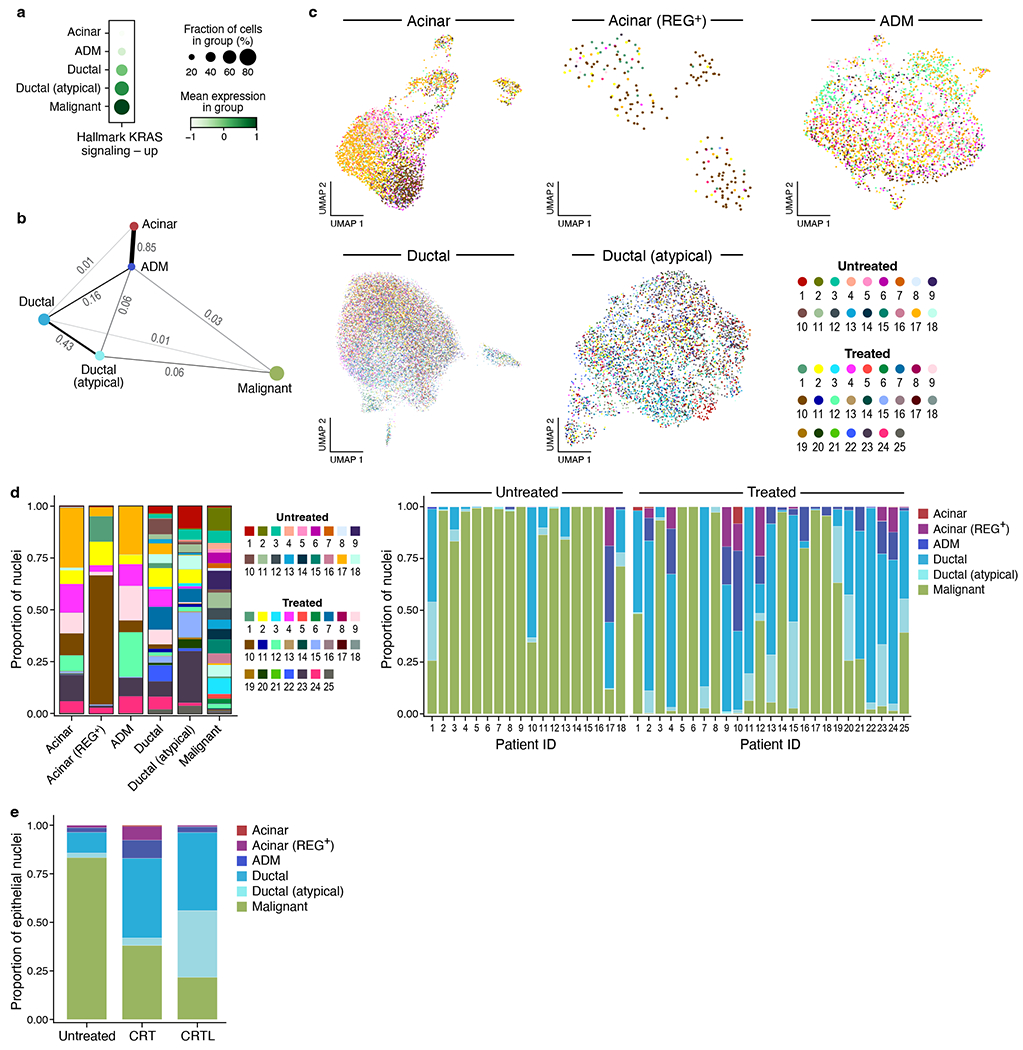
a,b, Inferred differentiation states in pre-malignant and malignant cells. a, Proportion of cells (dot size) with non-zero expression of gene set HALLMARK_KRAS_SIGNALING_UP in each epithelial cell subset and normalized mean expression (dot color) in expressing cells. b, Partition-based graph abstraction (PAGA) of an inferred pseudotemporal trajectory among epithelial cell subsets (nodes) connected by edges with numerical weights (line thickness). c, UMAP embeddings of single nucleus profiles (dots) for different epithelial cell subsets (panels) colored by patient ID (color legend). d, Left: proportions (y axis) of cells in each tumor (color legend) for each epithelial cell subset (x axis); Right: proportions (y axis) of epithelial cell subsets (color legend) for each tumor (x axis). e, Proportions (y axis) of epithelial cell subsets (color legend) summed across all tumors for each treatment category (x axis).
Malignant and fibroblast programs shared across tumors
Prior expression signatures of epithelial- or CAF-enriched PDAC tumors only partially aligned with the partitioning of our single nucleus profiles (Extended Data Figure 4; Methods). Although most tumors featured malignant cells of the basal-like/squamous/quasi-mesenchymal and classical subtypes (Extended Data Figure 4a)38,39, these states overlapped in some malignant cells39,40, or were absent in others. Moreover, myofibroblastic and inflammatory signatures were expressed in somewhat distinct subsets of CAFs, but the antigen-presenting signature was not clearly identified41, and cross-tissue signatures42 only partially segregated our CAF profiles (Extended Data Figure 4b).
We therefore learned recurrent expression programs de novo across malignant cells and CAFs of different tumors, using consensus non-negative matrix factorization (cNMF). We selected the number of cNMF programs by stability and error (Extended Data Figure 5a), focused on those shared across cells from multiple patients (Figure 3a,3b,4; Supplementary Tables 2–4), and annotated each by its top-200 weighted genes (Methods).
Figure 3 |. Molecular stratification of malignant cells in PDAC reveals a neural-like progenitor program that is enriched in genes associated with the nervous system and perineural invasion.
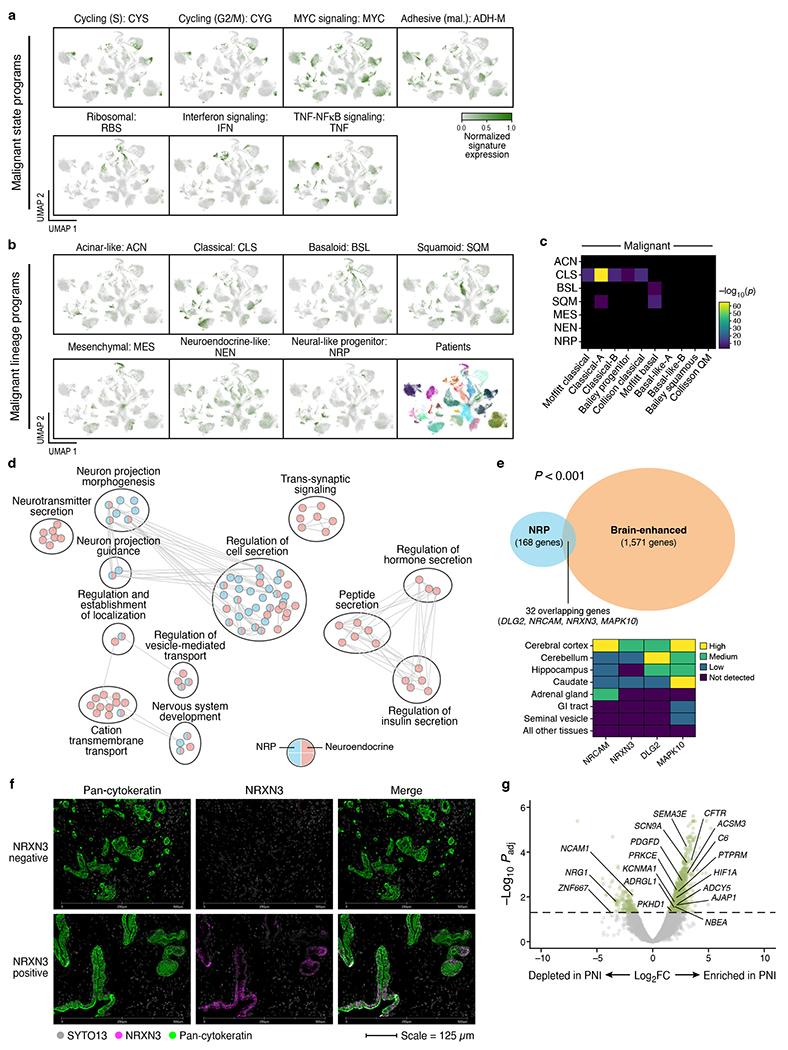
a-b, Expression programs in malignant cells based on consensus non-negative matrix factorization (cNMF). UMAPs of single nucleus profiles (dots) of malignant cells (a, state programs; b, lineage programs) from all tumors, colored by patient (b bottom right subpanel) or by the normalized expression score of each program (Methods). c, Similarity of de novo cNMF annotated programs (labels, rows) compared to prior malignant signatures (labels, columns)5–7,39. Statistical significance of overlap (−log10(p-value), two-sided hypergeometric test, color bar) for each pair of de novo cNMF annotated program and prior signature with significant overlap based on Bonferroni adjusted p-value < 0.05. Pairs with non-significant overlap are shown in black. d, Distinctions between the neural-like progenitor and neuroendocrine-like programs. Overlap of each gene set (colored pie charts) with the neural-like progenitor (blue) and neuroendocrine-like (red) programs. Circles (black outlines) encompass clusters of related gene sets. Edges represent overlaps between distinct gene sets based on an overlap coefficient threshold (>0.85, Cytoscape). e, The neural-like progenitor program includes ‘brain tissue enhanced’ genes from the Human Protein Atlas (HPA)84. Left: Overlap between the program (blue) and HPA brain enhanced (orange) genes (p=1.29x10−4; two-sided hypergeometric). Right: HPA expression categories (color code) for select genes (columns) across brain regions (rows). f, Representative multiplexed immunofluorescence images of untreated PDAC (n=3 independent tumors) showing absence of NRXN3 expression (top) and heterogeneous NRXN3 expression (bottom) in malignant cells/glands from two separate regions of the same tumor. Color legend indicates target of fluorophore-conjugated antibodies. g, Differential expression (log2(fold-change), x axis) and its significance (−log10(adjusted p-value), y axis, DESeq2) of TCGA PDAC patients with (right; n=134) and without (left; n=25) perineural invasion (PNI). P-values were adjusted for the false discovery rate using a Benjamini-Hochberg correction. The dashed horizontal line corresponds to Padj=0.05. Labeled genes are present in the neural-like progenitor program signature.
Figure 4 |. Molecular stratification of cancer-associated fibroblasts in PDAC.

a, Expression programs in CAFs based on cNMF. UMAPs of single nucleus profiles (dots) of CAFs from all tumors, colored by patient (bottom left subpanel) or by the normalized expression of each program (Methods). b, Similarity of de novo cNMF annotated programs (labels, rows) compared to prior fibroblast signatures (labels, columns)41,42.
We identified 14 malignant cell programs that reflected either lineage (classical-like, squamoid, basaloid, mesenchymal, acinar-like, neuroendocrine-like, neural-like progenitor) or cell state (cycling-S, cycling-G2/M, MYC, interferon, TNF-NFκB, ribosomal, adhesive), and four CAF programs (myofibroblastic progenitor, neurotropic, immunomodulatory, adhesive) (Figure 3a,3b,4; Supplementary Tables 2–4). Subsampling of tumors showed we recovered all 14 malignant programs when using 80% of samples and all four fibroblast programs at 50% subsampling (Extended Data Figure 5b).
Refined malignant classification reveals distinct programs
In addition to the classical-like program that strongly overlapped with previously defined classical signatures5–7,39 (Figure 3a–c), three programs corresponded to squamoid, basaloid, and mesenchymal states separately, in lieu of a joint basal-like/squamous/quasi-mesenchymal program5–7 from bulk expression profiles (Figure 3a–c; Supplementary Tables 2–3). We further identified in bona fide high CNA malignant cells an acinar-like program and a neuroendocrine-like program, reminiscent of neuroendocrine-like differentiation states in other tumor types43,44. Prior bulk studies often ascribed endocrine- or exocrine-like profiles to non-malignant cells in these less pure tumor subtypes3,5,7,8, but our data show that these are present in malignant cells.
The basaloid, squamoid and mesenchymal programs were each enriched in relevant genes (Supplementary Tables 2–3): epidermis development/proliferation, keratinocyte differentiation, and cornification in the squamoid program45–47; stemness, ribosomal proteins, rRNA processing, cell migration/invasion, cell-cell and cell-extracellular matrix (ECM) junctions, epithelial-mesenchymal transition (EMT), and metallothioneins in the basaloid program48,49; and EMT, matrisome, ECM production, and stemness in the mesenchymal program50. The squamoid and basaloid programs overlapped significantly with the Moffitt basal-like signature6, but the squamoid and mesenchymal programs did not exhibit significant overlap with the bulk-defined squamous7 and quasi-mesenchymal subtypes5, respectively (Figure 3c).
A distinct neural-like progenitor (NRP) program was simultaneously enriched for pathways and genes involved in neuronal development/migration/adhesion (e.g., CNTN4, CTNND2, NRXN3, RELN, SEMA5A, NRCAM, AUTS2)51–55, and in tissue stem cell modules and hepatocyte nuclear factor activity (Supplementary Table 3), known to function in embryonic and adult organ morphogenesis56. Notably, our data decouples the neural-like progenitor and neuroendocrine-like programs, which have been challenging to distinguish in studies of other malignancies57 (Figure 3b,d; Supplementary Tables 2–3). The NRP program was also significantly enriched in ‘brain tissue enhanced’ genes in the Human Protein Atlas (p=1.30x10−4, hypergeometric; Figure 3e; Methods). We validated the program in situ by multiplexed immunofluorescence, showing that a subset of malignant cells/glands co-express cytokeratins and NRXN3, a program gene typically expressed in neuronal and glial cells of the cerebral cortex and caudate (Figure 3e,f; Extended Data Figure 3b; Methods). Thus, neural-related proteins can be expressed within invasive epithelia. The features of this program are consistent with the frequent and diverse somatic aberrations in genes linked to axonal guidance, tumor-nerve crosstalk, and the high prevalence of perineural invasion (PNI) observed in PDAC58. For example, class 3 semaphorins, such as the program gene SEMA3E, are amplified or mutated in >20% of PDAC59, and genes that are differentially expressed in an independent cohort of untreated PDAC with (n=134) versus without (n=25) PNI60 have significantly higher program weights (p=0.006, Kolmogorov-Smirnov; Figure 3g; Methods).
There were seven additional ‘cell state’ programs in malignant cells (Figure 3a; Supplementary Tables 2–3). These spanned cell cycle programs in S and G2/M phases; ribosomal program; program enriched in type 1/2 interferon response genes; TNF-NFkB signaling program; MYC signaling program; and cell adhesion/motility program. Consistent with prior work21,48,61, inter-program correlation scores showed strong associations between the ribosomal and basaloid; MYC and classical; adhesive and neural-like progenitor; and cycling and MYC programs (Extended Data Figure 6).
Expanded CAF classification identifies four programs
Among the four CAF programs, the ACTA2-enriched myofibroblastic progenitor program overlapped with a published myCAF signature41 (Figure 4a,b; Supplementary Tables 2,4) but was differentiated by an enrichment of genes involved in embryonic mesodermal development and Wnt signaling (e.g., RUNX1, RUNX2, LEF1, SALL4, WNT5A, NKD1, FOXP1)62–66. The neurotropic, immunomodulatory, and adhesive programs all overlapped with the single-cell iCAF signature but not the myCAF signature41, suggesting they may reflect different iCAF subsets (Figure 4b). None of the programs significantly overlapped with the single-cell apCAF signature41. In addition, the CAF programs overlapped with various cross-tissue fibroblast signatures42 in a non-specific manner (Figure 4b).
Moreover, the immunomodulatory fibroblast program was enriched in pathways involving cytokine production/response, inflammation, and TNF-α/NF-κB signaling, and included CXCL12, CCL19 and CCL21, which play roles in pancreatic cancer pathogenesis67–69; IL34, which induces proliferation and differentiation in monocytes/macrophages70; and members of the complement pathway that may affect neutrophil recruitment71 (Figure 4a; Supplementary Tables 2,4). The cell adhesion program featured pathways involved in cell-cell/-ECM adhesion (e.g., CDH2), cytoskeletal remodeling, and motility. The neurotropic program was enriched for genes involved in neurogenesis, neuron differentiation, and neuronal projections (Figure 4a; Supplementary Tables 2,4). CAFs have been linked to neurotropic phenomena in pancreatic cancer58.
Treatment-associated malignant and CAF program expression
Neoadjuvant therapy was associated with significant differences in the expression of malignant and CAF programs at the patient level (Figure 5a,b). The malignant neural-like progenitor (padj=6.98x10−3, Mann-Whitney) and neuroendocrine-like (padj=1.39x10−2) programs were significantly higher in CRT vs. untreated, whereas the classical (padj=1.33x10−3) and squamoid (padj=3.34x10−3) programs were lower (Figure 5a,b). Neoadjuvant CRTL vs. untreated also showed higher expression of the neural-like progenitor program (padj=2.30x10−3; Mann-Whitney) and lower expression of the classical (padj=7.78x10−3), squamoid (padj=1.37x10−3), and basaloid (padj=1.52 x 10−2) programs (Figure 5a,b). Using snRNA-seq, we validated that the neural-like progenitor program was increased in a patient-derived (PDAC_U_12) organoid line that was ex vivo treated vs. untreated (p=1.33x10−15, Mann-Whitney; Figure 5c; Methods).
Figure 5 |. The neural-like progenitor program is enriched in residual tumor and patient-derived organoids after cytotoxic therapy and is associated with poor clinical outcomes.
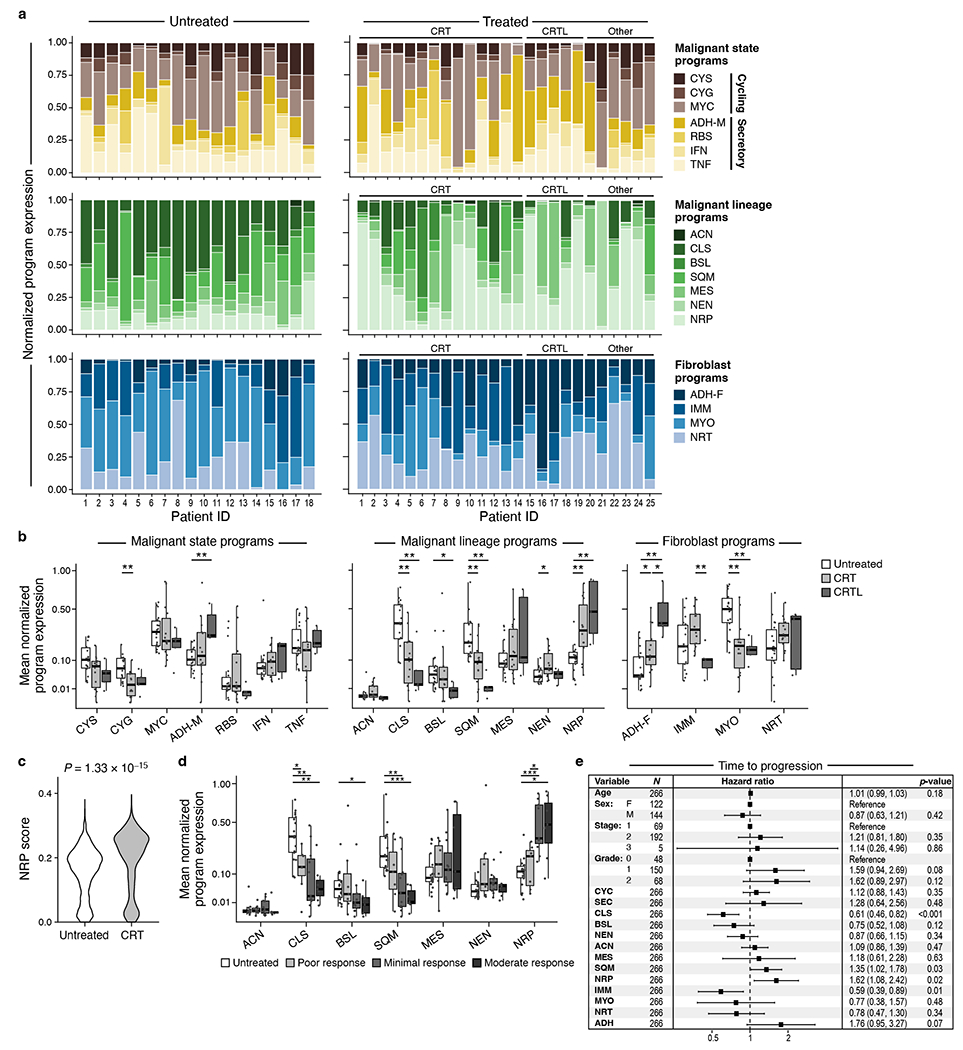
a, Intra-tumoral and inter-tumoral heterogeneity of malignant and fibroblast expression programs. Normalized expression scores (y axis) of malignant state (top), malignant lineage (middle) and CAF (bottom) programs (color legend) in each untreated (n=18, left) or treated (n=25, right) tumor (x axis). Treated patients are further ordered by treatment regimen. b, Malignant cell and CAF programs associated with treatment status. Mean normalized program expression (y axis) of malignant cell state (top), malignant cell lineage (middle), and CAF (bottom) programs (x axis) in untreated (n = 18), CRT (n=14), and CRTL (n=5) tumors. * Bonferroni adjusted p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, two-sided Mann-Whitney U test with Bonferroni correction. c, The neural-like progenitor program increases in organoids following CRT treatment. Distribution of mean expression of the top 200 cNMF-weighted genes from the neural-like progenitor program (y axis) across individual cells from matched untreated and CRT-treated organoids (x axis) derived from patient PDAC_U_12 (p=1.33x10−15, two-sided Mann-Whitney; untreated=2607 cells; CRT=341 cells). d, Expression of malignant lineage programs in residual neoplastic cells varies by patients’ treatment response. Distribution of mean normalized expression scores in each tumor (y axis) for each pathological treatment response grade (grayscale legend; untreated: n=18, poor: n=7, minimal: n=11, moderate: n=7) for each malignant lineage program (x axis) regardless of treatment group. * Bonferroni adjusted p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, two-sided Mann-Whitney U test with Bonferroni correction. e, Program expression and clinicopathologic parameters associated with time to disease progression using multivariable Cox regression analysis of bulk RNA-seq data from two independent cohorts of untreated, resected primary PDAC (TCGA and PanCuRx/ICGC; n=266). Data are presented as hazard ratio (HR) ± 95% confidence interval. Malignant lineage: NRP=neural-like progenitor, SQM=squamoid, MES=mesenchymal, ACN=acinar-like, NEN=neuroendocrine-like, BSL=basaloid, CLS=classical. Malignant state (aggregate): CYC=cycling, SEC=secretory. Fibroblast: IMM=immunomodulatory, NRT=neurotropic, ADH-F=adhesive, MYO=myofibroblastic progenitor.
In the CAF compartment, treatment was associated with lower myofibroblastic progenitor program expression (CRT/CRTL vs. untreated, padj=1.52x10−3/4.86x10−3, Mann-Whitney) and higher adhesive program expression (CRT/CRTL vs. untreated, padj=1.93x10−2/2.97x10−3; CRTL vs. CRT, padj=3.50x10−2) (Figure 5a,b). The immunomodulatory CAF program was higher in CRT vs. CRTL (padj=9.47 x 10−3; Mann-Whitney). These differences were consistent with differential gene expression among untreated, CRT, and CRTL malignant cells and CAFs (Extended Data Figure 3b,7; Methods).
Expression of programs post-treatment was also associated with clinical response. We annotated each of the 25 treated samples based on the patient’s surgical pathology treatment response grade (Methods) irrespective of treatment regimen and scored their remaining malignant cells for the seven malignant lineage programs. The residual malignant cells in patients with moderate response were enriched in the neural-like progenitor program and depleted in the classical and squamoid programs relative to untreated tumors (Figure 5d).
Neural-like progenitor malignant program has poor prognosis
To assess the prognostic relevance of the malignant and CAF programs, we scored them in clinically-annotated bulk RNA-seq data from patients with untreated, resected primary PDAC from TCGA8 and PanCuRx/ICGC9,39 (n=266; Methods). We performed a multivariable Cox regression analysis of the time to progression (TTP) and overall survival (OS) endpoints with age, sex, stage, grade, and the CAF and malignant programs as covariates. Age, sex, stage, and grade were not prognostic for TTP (Figure 5e). The neural-like progenitor (HR 1.62, 95%CI: 1.08-2.42, p=0.02) and squamoid programs (HR 1.35, 95%CI: 1.02-1.78, p=0.03) were associated with shorter TTP, whereas the classical (HR 0.61, 95%CI: 0.46-0.82, p<0.001) and immunomodulatory programs (HR 0.59, 95%CI: 0.39-0.89, p=0.01) were associated with longer TTP (Figure 5e). Notably, the neural-like progenitor and squamous programs remained significantly associated with shorter TTP even when the bulk classical and quasi-mesenchymal signatures5 were included as co-variates (Supplementary Table 5). For OS, age (HR 1.02, 95%CI: 1.00–1.04, p=0.02) and the adhesive CAF program (HR 1.84, 95%CI: 1.04–3.25, p=0.04) were associated with shorter survival, while the classical program (HR 0.73, 95%CI: 0.56–0.95, p=0.02) was associated with longer survival. The neural-like progenitor (HR 1.32, 95%CI: 0.90–1.96, p=0.16) and squamoid programs (HR 1.20, 95%CI: 0.93–1.55, p=0.15) trended towards a negative association with OS but did not reach significance (Extended Data Figure 8). These findings parallel an association between the neuronal subtype and poor outcomes in bladder cancer57. We caution, however, that applying snRNA-seq based programs to bulk profiles may be confounded by non-malignant cells that express some of the programs at relatively high levels.
Mapping cell types and programs to tumor architecture
To decipher how cells and expression programs are spatially organized in multicellular communities, we performed digital spatial profiling (DSP) with the NanoString GeoMx human whole transcriptome atlas (WTA; 18,269 genes). We hybridized UV-photocleavable barcode-conjugated RNA ISH probes on FFPE sections to capture and profile mRNA counts from regions of interest (ROIs) (Figure 6a; Extended Data Figure 9a; Methods)25. We used four-color immunofluorescence to select ROIs with diverse patterns of neoplastic cells, CAFs, and immune cells; created custom areas of illumination (AOI) for each cell type segment within an ROI; cleaved and collected barcodes from each AOI; and quantified barcode abundance by sequencing (Methods). We analyzed 21 tumors by DSP (18 with matching snRNA-seq) and deconvolved the data with snRNA-seq cell type signatures. The epithelial, CAF, and immune AOIs clustered appropriately by cell type (Extended Data Figure 9b). We then mapped the expression of each malignant and CAF program onto the ROIs, with 54% of the top 200-weighted program genes detected above background (Figure 6b).
Figure 6 |. Spatial mapping of malignant and CAF programs reveals program-specific associations with intra- versus inter-tumor heterogeneity.
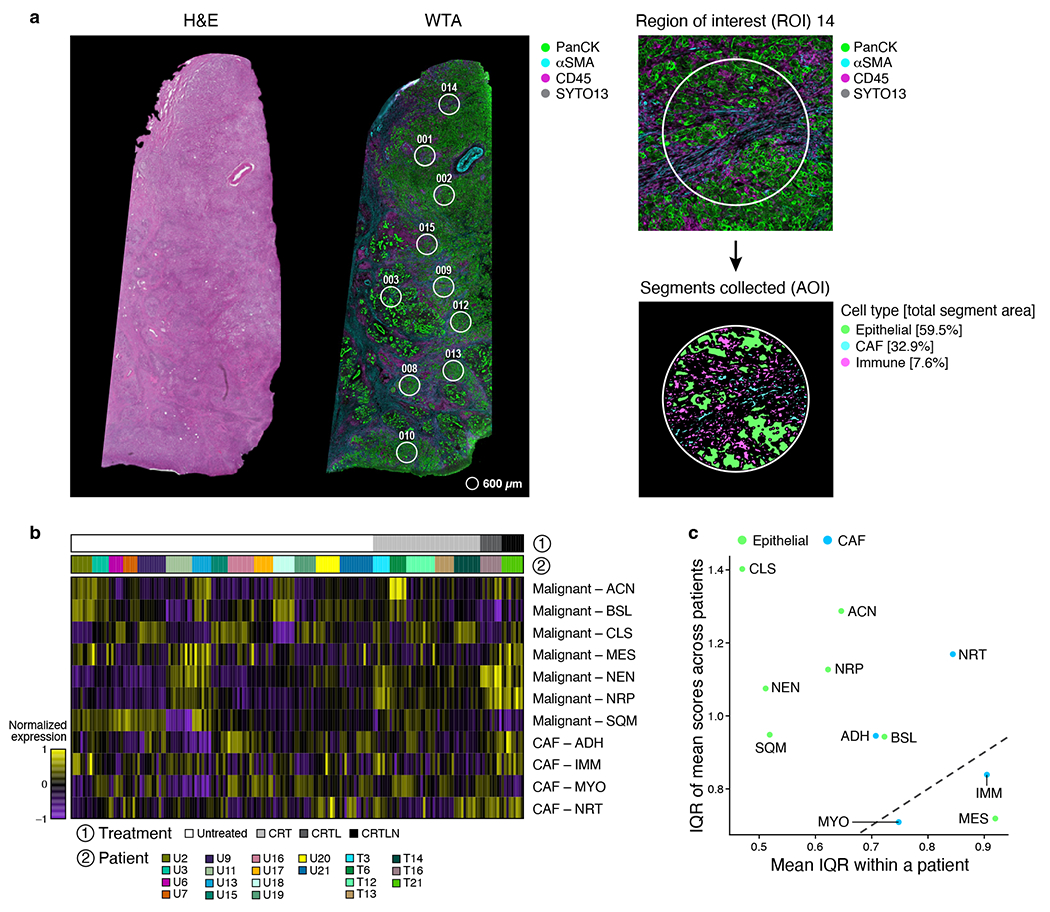
a, Whole Transcriptome Digital Spatial Profiling (WTA DSP). Left: Representative hematoxylin and eosin (H&E)-stained FFPE sections (5 μm thickness, left) and immunofluorescence image (GeoMx DSP, right) of consecutive sections from the same tumor FFPE block, showing selected regions of interest (ROIs, circles). WTA DSP was performed on 21 independent human PDAC tumors. Gray=SYTO13 (nuclear stain), green=anti-panCK, magenta=anti-CD45, cyan=anti-αSMA. Right: Example ROI (circle, 600 μm diameter) with segmentation masks used to enrich for the epithelial, CAF, and immune compartments and percent of total segment area occupied by each compartment. b,c, Higher variation across tumors than within tumor ROIs. b, Normalized expression (color scale) of malignant cell (top rows) and fibroblast (bottom rows) programs in each AOI (columns) across patients (color bar 2, color legend) and treatment status (grayscale bar 1 and color legend). c, Program expression variation between patients (y axis, interquartile range/IQR of the mean program score for each tumor) and within patients (x axis, mean of IQR across all ROIs within a tumor). Dotted line x=y.
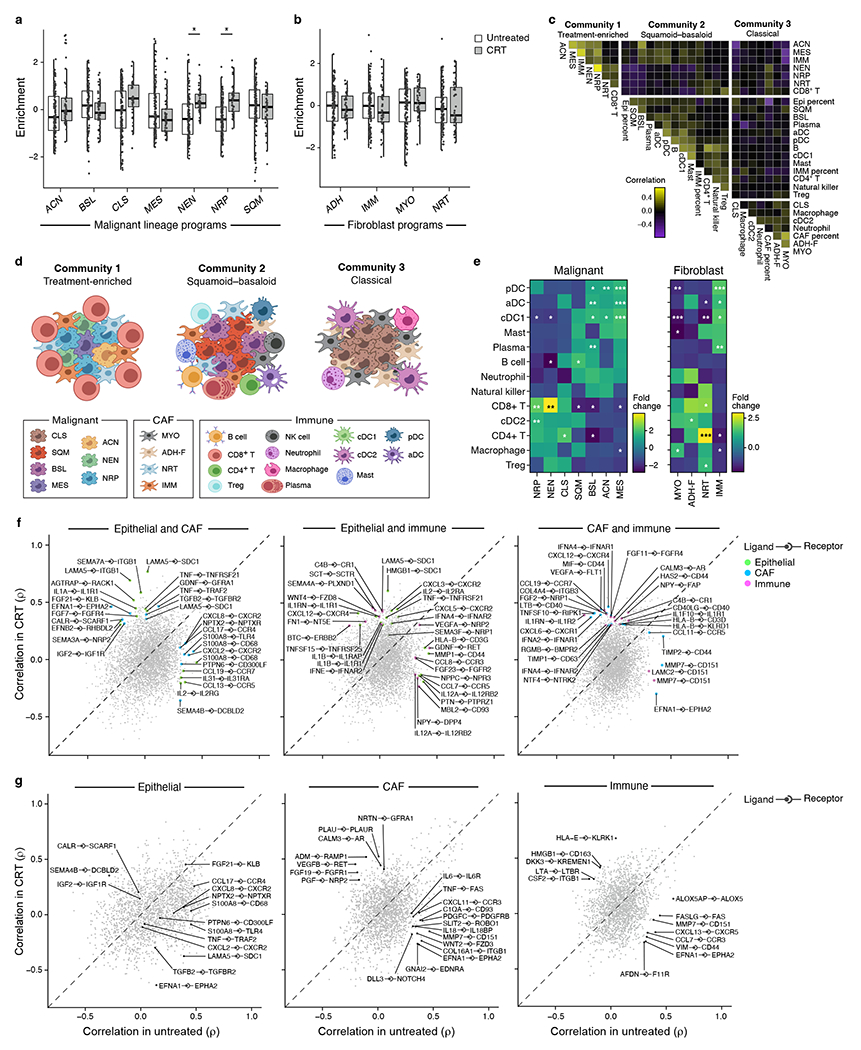
Most snRNA-seq programs were more variable between independent patient tumors (inter-patient dispersion) than across different ROIs from the same tumor (intra-patient dispersion), except for the mesenchymal, immunomodulatory, and myofibroblastic progenitor programs (Figure 6c). The neural-like progenitor and neuroendocrine-like malignant programs were enriched in ROIs from CRT vs. untreated samples (p=1.07x10−2/4.98x10−2, linear mixed-effects model; Figure 7a,b), consistent with our snRNA-seq analysis (Figure 5b).
Figure 7 |. Spatial analysis of malignant programs, CAF programs and immune cell composition reveals three distinct multicellular communities and treatment-associated receptor-ligand interactions.
a,b, Digital spatial profiling shows enrichment of neural-like progenitor and neuroendocrine-like program after neoadjuvant CRT. Distribution of z-score normalized ssGSEA enrichment scores (y axis) of malignant (a) and fibroblast (b) programs (x axis) in AOIs from CRT (gray; n=5) and untreated (white; n=14) tumors. Box depicts interquartile range (IQR) with median marked as horizontal line. The whiskers correspond to 1.5 x IQR. * p < 0.05, mixed-effects model (Methods). c,d, Three multicellular communities with distinct malignant, CAF, and immune features. c, Pearson correlation coefficient (color bar) of the scores/proportions of each malignant, CAF, and immune feature (rows, columns) across ROIs. Rows and columns are ordered by hierarchical clustering. d, Schematic of key features of each multicellular community as defined in c. Malignant lineage: NRP=neural-like progenitor, SQM=squamoid, MES=mesenchymal, ACN=acinar-like, NEN=neuroendocrine-like, BSL=basaloid, CLS=classical. Fibroblast: IMM=immunomodulatory, NRT=neurotropic, ADH-F=adhesive, MYO=myofibroblastic progenitor. Created with BioRender.com. e, Malignant and CAF programs associated with immune cell composition. Fold change (color bar) of inferred immune subset proportions (rows) between the top quartile scoring ROIs and the bottom quartile scoring ROIs for each malignant (columns; left) or fibroblast (columns; right) program. f,g, Spatially correlated receptor-ligand pairs across (f) and within (g) compartments. Spearman rank correlation coefficient of expression of receptor-ligand pairs (gray dots) across paired epithelial:CAF (f, left), epithelial:immune (f, middle), CAF:immune (f, right), epithelial:epithelial (g, left), CAF:CAF (g, middle), and immune:immune (g, right) segments within the same ROI across all ROIs in CRT-treated (y axis) or untreated (x axis) tumors. Selected receptor-ligand pairs that were differentially correlated in CRT-treated or untreated tumors are labeled and colored based on the segment expressing the ligand (f, color legend). Dotted line: x=y.
Distinct multicellular communities
To identify multicellular spatial associations, we correlated each pair of features across the ROIs to yield a spatial co-variation matrix of the malignant lineage programs, CAF programs, proportions of immune cell types, and percent ROI area occupied by the malignant, fibroblast, and immune segments (Figure 7c; Methods). Unsupervised clustering identified three multicellular communities with distinct malignant, stromal and immune features (Figure 7c,d). Community 1 (“treatment-enriched”) was characterized by an association among the neural-like progenitor and neuroendocrine-like malignant programs, neurotropic CAF program, and CD8+ T cells – which were all enriched with treatment in snRNA-seq – as well as the mesenchymal and acinar malignant programs and the immunomodulatory CAF program. Community 2 (“squamoid-basaloid”) featured an association of the squamoid and basaloid malignant programs with a diverse set of lymphoid and myeloid cell types, higher epithelial and immune content, and lower CAF content. Community 3 (“classical”) exhibited an association among the classical malignant program, the myofibroblastic progenitor and adhesive CAF programs, macrophages, neutrophils, and conventional type 2 dendritic cells (cDC2s), as well as higher CAF and lower immune proportions. Clustering of patient-level features in the snRNA-seq data recapitulated some of these associations (Extended Data Figure 9c).
The three communities highlight broad canonical features, but finer pair-wise associations were recovered by more granular analysis. For example, the classical malignant program was not strongly correlated with macrophage and neutrophil prevalence despite being in the same community (Figure 7c). We therefore computed the fold-change of each deconvolved immune cell type proportion between the highest quartile-scoring and the lowest quartile-scoring ROIs for each malignant and CAF program (Figure 7e; Methods). Consistent with community 1 “treatment-enriched”, ROIs with high neural-like progenitor and/or neuroendocrine-like program scores were significantly enriched with CD8+ T cells and depleted of conventional type 1 dendritic cells (cDC1s); the former was also enriched in cDC2s (Figure 7c–e). In contrast, high-scoring squamoid, basaloid, or mesenchymal ROIs were depleted of CD8+ T cells; the squamoid program associated with B cells and the basaloid and mesenchymal programs associated with all DC subsets except cDC2s. ROIs with high classical program scores were enriched with CD4+ T cells. Similarly, the neurotropic CAF program was positively associated with CD8+, CD4+, and regulatory T cells and negatively with activated DCs and cDC1s; the myofibroblastic progenitor and adhesive programs were only positively associated with macrophages and cDC2s, respectively; and the immunomodulatory program was positively associated with activated DCs, cDC1s, plasmacytoid DCs, and plasma cells and negatively with CD4+ T cells and macrophages. Thus, spatial associations involved both broad multicellular communities, reflected in the clustering analysis, and finer features related to pairs of specific cell types and programs (Figure 7c–f).
Finally, to uncover interactions among the malignant, CAF, and immune compartments that may facilitate therapeutic resistance, we identified spatially-defined receptor-ligand (RL) pairs co-expressed across ROIs in either CRT or untreated samples (Figure 7f,g; Supplementary Tables 6,7; Methods). Although some RL pairs were well-correlated in both untreated and CRT specimens, many pairs were differentially correlated by treatment status. A subset of intercellular RL interactions enriched in the treated specimens may facilitate therapeutic resistance and serve as candidates for intervention.
DISCUSSION
In this study, we used snRNA-seq and digital spatial profiling of a large cohort of primary PDAC to construct a detailed classification of tumor composition, malignant and CAF programs, immune milieu, and clinical outcomes (Figure 8). Our snRNA-seq approach was compatible with untreated and heavily pre-treated frozen specimens and may yield better in situ cell type representation than scRNA-seq (Methods)72, albeit with some differential immune subset capture (Figure 1f; Extended Data Figure 1d). Analysis of some immune subsets may benefit from further application-specific optimization and complementary in situ approaches.
Figure 8 |. Refined molecular taxonomy of pancreatic ductal adenocarcinoma.
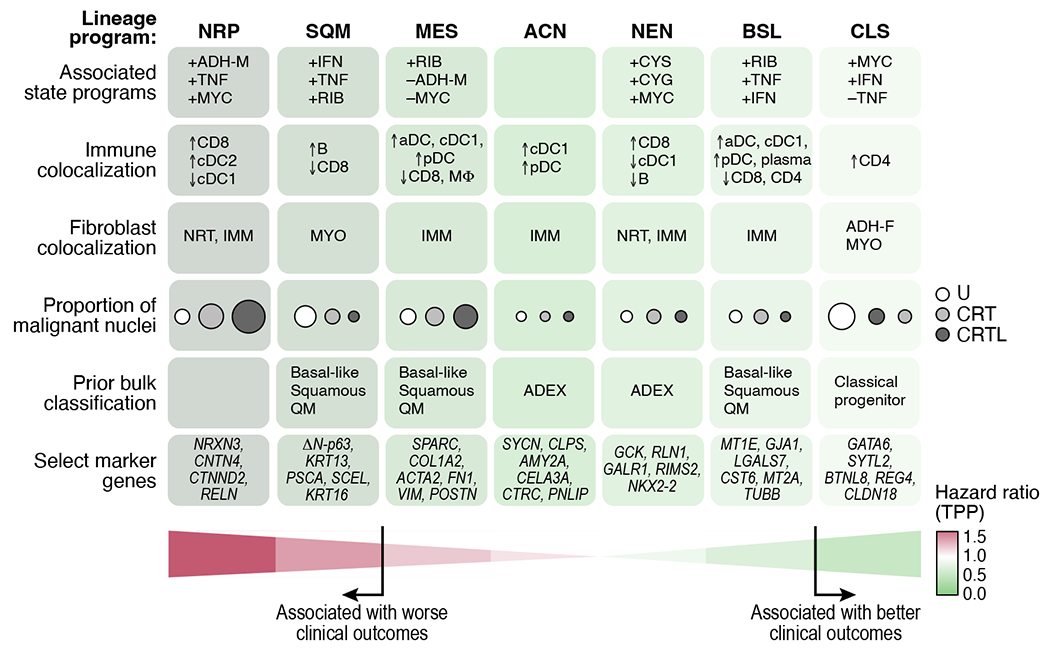
Cell intrinsic, clinical, and spatial associations for malignant lineage programs (columns). Malignant lineage: NRP=neural-like progenitor, SQM=squamoid, MES=mesenchymal, ACN=acinar-like, NEN=neuroendocrine-like, BSL=basaloid, CLS=classical. Malignant state: CYS=cycling (S), CYG=cycling (G2/M), MYC=MYC signaling, ADH-M=adhesive, RIB=ribosomal, IFN=interferon signaling, TNF=TNF-NFκB signaling. Fibroblast: IMM=immunomodulatory, NRT=neurotropic, ADH-F=adhesive, MYO=myofibroblastic progenitor.
Evidence of putative intermediate states (ADM, atypical ductal) supports a path of transformation from acinar to ADM to ductal to atypical ductal to malignant cells in patient tumors (Figure 1d,2). In some cases, the co-existence of precursor and malignant states in the same tumors may be secondary to field cancerization; elucidating this would require studies focused on precursor lesions and non-malignant tissue adjacent to tumors.
Our de novo expression programs provide a refined and expanded cell taxonomy of malignant cells and CAFs in PDAC (Figure 3,4). In addition to previously reported subtypes such as myCAFs41 and malignant classical5–7, our analysis partitioned an aggregate “basal-like”/“squamous”/“quasi-mesenchymal” subtype5–7 into discrete squamoid, basaloid, and mesenchymal programs; revealed neuroendocrine-like and acinar-like programs that support the existence of the aberrantly-differentiated endocrine exocrine (ADEX) subtype3,7, and uncovered a distinct neural-like progenitor program, which we validated in situ (Figure 3e,f).
While we expanded on prior studies by dissecting cellular ecosystems in both untreated and treated tumors, we were limited by the lack of matched pre- and post-treatment specimens, treatment heterogeneity, sample size for certain treatment groups, and technical challenges associated with single nucleus extractions. Nevertheless, with a large cohort and statistical adjustments to account for patient origin of each cell, the neural-like progenitor program was enriched in all post-treatment groups, including organoids treated ex vivo (Figure 5b–d), and was associated with worse clinical outcomes in two independent cohorts (Figure 5e).
The mechanisms through which neural-like progenitor cells may resist treatment remain open-ended, including whether they are cancer-intrinsic, derived from TME interactions, or both. Several program genes are involved in drug efflux, negative regulation of cell death, and chemoresistance (e.g., ABCB1, BCL2, PDGFD, SPP1)73–76. Moreover, neuronal migration and axonal guidance genes (e.g., SEMA3E, RELN, SEMA5A)53,77,78 and PNI-associated genes may reflect tumor-nerve crosstalk (Figure 3g), which has been associated with dissemination, post-treatment recurrence, and metabolic support58. The program includes NFIB, a transcription factor that promotes pro-metastatic neuronal gene expression programs in other cancer types79. Combining snRNA-seq with spatial whole transcriptome profiling identified how the different malignant and stromal programs relate to each other and to immune cell composition (Figure 8). Colocalization of the neural-like progenitor malignant program, neurotropic CAF program, and CD8+ T cells in one multicellular community suggest a functional interplay among these cell types/states.
Spatially-defined receptor-ligand interactions, especially those that are differentially correlated between untreated and CRT tumors may functionally underpin these communities (Figure 8; Supplementary Tables 6,7). The CXCL12-CXCR4 interaction80 is the most differentially correlated RL pair between epithelial and immune cells, and supports investigation of AMD3100, a small-molecule CXCR4 inhibitor80,81, as a potential adjunct to neoadjuvant CRT. Within epithelial ROIs in CRT tumors, several correlated RL pairs involved ERBB2—consistent with the role of HER2 signaling in chemoresistance in PDAC cell lines82 and suggests HER2 inhibition may augment neoadjuvant CRT, despite previous challenges in clinical trials83.
The lower expression of the squamoid program in post-treatment residual malignant cells was unexpected, as prior studies have associated squamous/basal-like tumors with poor treatment response and outcomes3,5–7 and the squamoid program was associated with poor prognosis in our analysis of bulk profiles from untreated patients (Figure 5b,c,e). However, prior classification based on untreated tumors did not account for potential reprogramming of expression profiles after treatment. Furthermore, previous reports have suggested that the basal-like A phenotype, distinguished by higher expression of squamous differentiation programs, is enriched in metastatic disease, which offers a potential explanation for the poor prognosis seen in our analyses39. We did not observe a significant depletion of the classical and squamoid programs in the DSP data (Figure 7a,b), but this may have been due to selection of tumors with poor treatment response to facilitate recovery of ROIs with adequate epithelial content.
The post-treatment enrichment and depletion of specific malignant and CAF programs may result from selection of pre-existing phenotypes and/or treatment-induced plasticity. The presence of the neural-like progenitor program in untreated specimens, albeit at lower prevalence, and its monotonic increase at the expense of the classical and squamoid programs with increasing treatment response support a model wherein treatment-mediated selection of pre-existing phenotypes shapes residual disease (Figure 5). However, these patterns are also present when comparing tumors with poor treatment response and abundant residual disease to those from untreated patients, suggesting a potential role for phenotypic plasticity. Future studies comparing matched pre- and post-treatment specimens and preclinical models with genetic tracing should provide further insights.
In conclusion, our high-resolution molecular framework sheds light on the inter- and intra-tumoral diversity of pancreatic cancer; spatial organization into discrete communities; treatment-associated remodeling; and clinically-relevant prognostication. These findings can be harnessed to augment precision oncology efforts in pancreatic cancer.
METHODS
Ethics
All patients in this study were consented without compensation to excess tissue biobank protocol 2003P001289 (principal investigator: CFC; co-investigators: ASL, WLH), which was reviewed and approved by the Massachusetts General Hospital (MGH) Institutional Review Board. This study is compliant with all relevant ethical regulations. Seven patients were also enrolled in interventional clinical trials (NCT03563248, NCT01821729). This study was not formally a part of these clinical trials, and no primary outcomes were reported. However, some excess samples from these trial patients were separately banked on protocol 2003P001289 for use in this study.
Human patient specimens
For inclusion in this study, patients had non-metastatic PDAC and went to surgical resection with or without prior neoadjuvant treatment in the form of chemotherapy and radiotherapy (Supplementary Table 1). An institutional clinical standard for grading neoadjuvant therapy response in surgical pathology was employed with the following scale: score of 1 is “moderate response”; 2 is “minimal response”; and 3 is “poor response.” Most treated patients received several cycles of FOLFIRINOX chemotherapy followed by multi-fraction conformal radiotherapy with concurrent capecitabine or 5-FU. Four patients received other forms of chemotherapy such as cisplatin, gemcitabine, or nab-paclitaxel. Seven patients received additional neoadjuvant therapy in the form of losartan, an angiotensin II receptor type 1 antagonist, and/or nivolumab, a PD-1 inhibitor, on two clinical trials (NCT03563248, NCT01821729). The most common radiotherapy regimens included 30 Gy in 10 fractions, 50.4 Gy in 28 fractions, and stereotactic body radiotherapy 36 Gy in 6 fractions.
Resected primary tumor samples were examined to confirm neoplastic content by a board-certified pathologist (MMK) and then snap frozen and stored at −80°C for up to 5 years prior to processing. Specimens were screened for an RNA integrity number (RIN; Agilent RNA 6000 Pico Kit, cat. no. 5067-1513) greater than an empirically determined threshold of 6; only specimens with RIN > 6 were processed further. Matched formalin-fixed paraffin-embedded (FFPE) blocks were used for multiplexed ion beam imaging (MIBI, Ionpath) and digital spatial profiling (DSP, Nanostring) in 7 and 21 cases, respectively.
Organoid derivation and ex vivo chemoradiotherapy
Fresh patient-derived tumor tissue was minced with a razor blade in 1x PBS and incubated in digestion buffer (125 U/mL collagenase IV in 1x PBS; Worthington, cat. no. LS004189) for 30 minutes at 37°C with constant agitation in a hybridization oven. Tumor cell suspension was poured over a 70 μm filter, washed with 1x PBS, and centrifuged at 500 x g with slow deceleration. Cell pellets were resuspended in 85% growth-factor reduced Matrigel (Corning, cat. no. 356231) and 15% complete media (see below for details), plated as 50 μL plugs in a 24-well plate, and solidified at 37°C. Cells were cultured in complete media, monitored for outgrowth, and passaged with TrypLE Express (Life Technologies) for four passages to purify the malignant epithelial component from contaminating stromal cells. Complete media for pancreatic organoids was formulated based on L-WRN cell conditioned media as described previously (Supplementary Note)85–87.
Organoids were then subjected to a 10-day ex vivo chemoradiotherapy regimen as follows. Organoids were plated in complete media. Twenty-four hours later, organoids were treated with FOLFIRINOX-like chemotherapy (SN-38 substituted for irinotecan) for four days using a molar ratio similar to that given to patients (molar ratio of SN-38 adjusted to account for enhanced activity relative to irinotecan)88,89. Chemotherapy consisted of 34.4 μm 5-fluorouracil (Sigma-Aldrich, cat. no. F6627), 4 nm SN-38 (Sigma-Aldrich, cat. no. H0165-10MG), and 0.32 μm oxaliplatin (Sigma-Aldrich, cat. no. O9512). Complete media and drugs were replaced on Day 3. Chemotherapy was terminated on Day 5. On Day 5 and Day 7, radiotherapy (7.5 Gy) was administered using a dual 137Cs source irradiator (Gammacell 40 Exactor, Best Theratronics). After three days of rest, organoid plugs were flash frozen and nucleus isolation was performed using the same protocol as tumor specimens as described below. Matched untreated and post-treatment organoids were compared by snRNA-seq.
Nucleus isolation from frozen samples
The following protocol is an adaptation and optimization of prior nucleus isolation techniques20 specifically for pancreatic tumors. A 2x stock of STc buffer in nuclease-free water was prepared with a final concentration of 292 mM NaCl (ThermoFisher Scientific, cat. no. AM9759), 40 mM Tricine (VWR, cat. no. E170-100G), 2 mM CaCl2 (VWR, cat. no. 97062-820), and 42 mM MgCl2 (Sigma Aldrich, cat. no. M1028). For each specimen, 2 mL of NSTcPA buffer was prepared by combining 1 mL of 2x STc buffer, 40 μL of 10% Nonidet P40 Substitute (Fisher Scientific, cat. no. AAJ19628AP), 10 μL of 2% bovine serum albumin (New England Biolabs, cat. no. B9000S), 0.3 μL of 1M spermine (Sigma-Aldrich, cat. no. S3256-1G), 1 μL of 1M spermidine (Sigma-Aldrich, cat. no. S2626-1G), and 948.7 μL of nuclease-free water. For each specimen, 3 mL of 1x working STc buffer was made by diluting 2x STc 1:1 in nuclease-free water.
NSTcPA buffer (1 mL) was pipetted into one well of a 6-well plate (Stem Cell Technologies, cat. no. 38015) on ice. Fragments (20-50 mg) from several regions of the frozen tumor was processed for snRNA-seq. The remainder of the specimen was returned to −80°C. Selected tissue was transferred into the NSTcPA buffer and manually minced with fine straight tungsten carbide scissors (Fine Science Tools, cat. no. 14568-12) for 8 minutes. The homogenized tissue solution was then filtered through a 40 μm Falcon cell filter (Thermo Fisher Scientific, cat. no. 08-771-1) into a 50 mL conical tube. An additional 1 mL of NSTcPA buffer was used to rinse the well and filter. The total volume was brought up to 5 mL with 3 mL of 1x STc buffer and transferred into a 15 mL conical tube. The sample was spun for 5 min at 500xg, 4°C and the supernatant was removed. The pellet was resuspended in 100-200 μL 1x STc and then filtered through a 35 μm Falcon cell strainer (Corning, cat. no. 352235). Nuclei were quantified using a C-chip disposable hemocytometer (VWR, cat. no. 82030-468) and diluted in 1x STc as necessary to achieve a final concentration of 300-2,000 nuclei/μL.
Single-nucleus RNA-seq (snRNA-seq)
Approximately 8,000-10,000 nuclei per sample were loaded into each channel of a Chromium single-cell 3’ chip (V2 or V3, 10x Genomics) according to the manufacturer’s instructions. Single nuclei were partitioned into droplets with gel beads in the Chromium Controller to form emulsions, after which nuclear lysis, barcoded reverse transcription of mRNA, cDNA amplification, enzymatic fragmentation, and 5’ adaptor and sample index attachment were performed according to manufacturer’s instructions. The 45 snRNA-seq sample libraries (Figure 1a) were pooled 3-4 per flow cell lane and sequenced on the HiSeq X Version 2.5 (Illumina) with the following paired end read configuration: read 1, 26-28 nt; read 2, 96-98 nt; index read, 8 nt.
snRNA-seq data pre-processing
BCL files were converted to FASTQ using bcl2fastq2-v2.20. CellRanger v3.0.2 was used to demultiplex the FASTQ reads, align them to the hg38 human transcriptome (pre-mRNA) reference and extract the UMI and nuclei barcodes. The output of this pipeline is a digital gene expression (DGE) matrix for each sample, which has quantified for each nucleus barcode the number of UMIs that aligned to each gene.
First, we used CellBender remove-background90 to remove ambient RNA, enhancing cell distinction and marker specificity. CellBender (snapshot 11) remove-background was run (on Terra) to remove ambient RNA and other technical artifacts from the count matrices. The parameters “expected-cells” and “total-droplets-included” were chosen for each dataset based on the total UMI per cell vs. cell barcode curve in accordance with CellBender documentation. The false positive rate parameter “fpr” was set to 0.01, 0.05, and 0.1. For downstream analyses we used the ‘FPR_0.01_filtered.h5’ file. Nuclei with over 10,000 UMI counts were filtered. UMI counts were normalized by the total number of UMIs per nucleus and converted to transcripts-per-10,000 (TP10K) as the final expression unit.
Dimensionality reduction, clustering and annotation
Treatment-naïve and neoadjuvant-treated specimens were aggregated into a single dataset. The log2(TP10K+1) expression matrix was constructed for downstream analyses. We identified the top 2,000 highly-variable genes across the entire dataset using the Scanpy 1.7.291 highly_variable_genes function with sample ID as input for the batch. We then performed a Principal Component Analysis (PCA) over the top 2,000 highly variable genes and identified the top 40 principal components (PCs) beyond which negligible additional variance was explained in the data. Subsequently, we performed batch correction using Harmony-Pytorch v0.1.792 and built a k-nearest neighbors graph of nuclei profiles (k = 10) based on the top 40 batch corrected components and performed community detection on this neighborhood graph using the Leiden graph clustering method93 with resolution set to 1 to identify distinct cell population clusters. Individual nucleus profiles were visualized using the Uniform Manifold Approximation and Projection (UMAP)94. Doublets were identified and removed in part using Scrublet v0.2.3. Distinct cell populations identified from the previous steps were annotated using known cell type-specific gene expression signatures and representative gene markers19,26,95–97. We used the Adjusted Mutual Information (AMI) score to quantify the similarity in single cell assignments between the partitions imposed by the Leiden clustering labels and patient ID labels. The AMI was computed using the adjusted_mutual_info_score function in the scikit-learn v0.22.2 package.
While earlier scRNA-seq studies in PDAC did not fully capture the stromal milieu and necessitated enrichment strategies for CAFs such as fluorescence-activated cell sorting41,72,98,99, they were well-represented in our samples. Specifically, our snRNA-seq had a higher yield of high quality nuclei per patient in the untreated group (6,054 ± 1,529) than a recent scRNA-seq study of primary untreated PDAC72 (1,718 ± 773), despite comparable quantities of loaded cells/nuclei (p = 1.92 x 10−9, Mann-Whitney U test; Extended Data Figure 10), recovered six additional cell types absent in scRNA-seq, and captured significantly higher proportions of CAFs, pericytes, and endocrine cells and lower proportions of vascular smooth muscle cells, myeloid cells, lymphoid cells, and endothelial cells (p < 0.05; Mann-Whitney U Test; comparable results using Dirichlet-multinomial regression; Extended Data Figure 10).
Quantifying statistically significant changes in composition between cell populations
To compute the significance of changes in the cellular composition between untreated and treated (CRT and CRTL) samples, we used multiple statistical tests: (1) Dirichlet-multinomial regression, and (2) Mann-Whitney test. To account for dependencies among cell proportions, we used a Dirichlet-multinomial regression. This statistical test and its inclusion probabilities (pi) were calculated using the scCODA v0.1.2.post1 Python package100. We also performed a non-parametric Mann-Whitney U test (two-sided) on the proportions of each cell subset in untreated versus treated (CRT and CRTL) samples. Bonferroni corrections were applied in instances where multiple pairwise comparisons were made between treatment or response groups. These same statistical approaches were applied to quantify the differences in cells/nuclei captured by the snRNA-seq approach and a previously published scRNA-seq method72.
Inferring copy number aberrations from single nucleus profiles
A Python implementation of InferCNV v3.9 based on the InferCNV R implementation as provided at https://github.com/broadinstitute/inferCNV (inferCNV of the Trinity CTAT Project) was run jointly on all treated and untreated single nuclei profiles. To avoid circularity, we used a set of high confidence non-neoplastic cells as the reference that was derived from two non-malignant pancreas snRNA-seq samples. We used the default parameters for InferCNV including a 100-gene window in sub-clustering mode and a hidden Markov model to predict the copy number aberration (CNA) count and construct a CNA score for each nucleus based on the predicted CNAs in each cell. Annotated epithelial cells were subject to Leiden sub-clustering and those with an average CNA score greater than 0.01 were labeled as malignant epithelial cells.
Partition-based graph abstraction
The pseudotemporal orderings/trajectories of annotated epithelial cell types was estimated using the diffusion map and partition-based graph abstraction (PAGA v1.2) method101. The diffusion map was computed with 15 components and the cell neighborhood map utilized a local neighborhood of 15.
Multiplexed ion beam imaging (MIBI)
Formalin-fixed paraffin-embedded pancreatic tissue sections were cut onto gold MIBI slides (IONpath, cat. no. 567001) and stained at IONpath (Menlo Park, CA) with the internal Epithelial i-Onc isotopically-labelled antibody panel (IONpath). Quantitative imaging was performed using a beta unit MIBIscope (IONpath) equipped with a duoplasmatron ion source. Time-of-flight mass spectrometer was tuned to 1-200 m/z+ and mass resolution of 1000 m/Δm, operating at a 100 KHz repetition rate. The primary ion beam was aligned daily to minimize imaging astigmatism and ensure consistent secondary ion detection levels using a built-in molybdenum calibration sample.
For data collection, three fields of view were acquired for each sample by matching the secondary electron morphological signal to annotated locations on sequential H&E stained slides. The experimental parameters used in acquiring all imaging runs were as follows: pixel dwell time (12 ms), image size (500 μm2 at 1024 x 1024 pixels), primary ion current (5 nA O2+), aperture (300 μm), stage bias (+67 V).
MIBI image processing, segmentation and quantification
Mass spectrometer run files were converted to multichannel tiff images using MIB.io software (IONpath). Mass channels were filtered individually to remove gold-ion background and spatially uncorrelated noise. HLA Class 1 and Na/K-ATPase signals were combined into a single membrane marker. These image files (tiff) were used as a starting point for single cell segmentation, quantification, and interactive analysis using histoCAT (v1.76)102. We followed a similar approach for segmentation as proposed for Imaging Mass Cytometry data102–104. Briefly, we used Ilastik 1.3.3post2105 to manually train three classes (nuclei, cytoplasm and background) to improve subsequent watershed segmentation using CellProfiler v3.1.9106. Finally, the tiff images and masks were combined for histoCAT loading with a script optimized for MIBI image processing (code, classifiers and configuration files are available at https://github.com/DenisSch/MIBI).
Immune cells were partitioned into subsets by incorporating protein markers available along with the snRNA-seq data. First, we used the scvi-tools v0.8.1 gimVI variational autoencoder to train a model107 taking both spatial MIBI and snRNA-seq data modalities as well as the correspondence between genes and antibody markers as input and encoding both the MIBI and snRNA-seq datasets into a joint latent space. The gimVI model was trained for 10 epochs. The latent space representation of the snRNA-seq data was then extracted from this model and used as the features to build a random forest model for cell type classification. Subsequently, the latent space representation extracted for each MIBI image was then evaluated using our trained model to generate a predicted cell type for each segmented immune cell in the spatial data.
Differential gene expression analysis
For each annotated cell type detected in both untreated and treated tumors, a differential gene expression analysis using a mixed effects Poisson model was performed between cells in the two populations to identify upregulated and downregulated genes. We considered untreated, all treated, CRT, and CRTL treatment categories in this analysis. We constructed a mixed effects model with the sample ID as a random effect; treatment status, two principal components and sex were fixed effect covariates; and finally, the log total UMIs as an offset. The mixed effects model was implemented using the glmer v3.1-152 package108.
Scoring gene signatures for each nucleus profile and patient
A signature score for each nucleus profile was computed as the mean log2(TP10K+1) expression across all genes in the signature. Subsequently, to identify statistically significant gene expression patterns, we computed the mean log2(TP10K+1) expression across a background set of 50 genes randomly selected with matching expression levels to those of the genes in the signature iterated 25 times. The gene signature score was defined to be the excess in expression found across the genes in the signature compared to the background set. To score gene programs at the patient level, these gene program scores were normalized for each nucleus and then the mean of all nuclei from an individual tumor was computed for each program of interest. In box plots featuring program scores, the boxes indicate upper and lower quartiles, with the horizontal lines marking the means. The lines extending vertically from the boxes (whiskers) indicate the maximum and minimum values excluding outliers. Data points are plotted as circles.
Consensus non-negative matrix factorization
We formulated the task of dissecting gene expression programs as a matrix factorization problem where the input gene expression matrix is decomposed into two matrices. We utilized the non-negative matrix factorization implemented in scikit-learn 0.22 to derive the malignant and CAF expression programs across both untreated and treated samples. We repeated NMF 50 times per cell type category and computed a set of consensus programs by aggregating results from all 50 runs and computed a stability and reconstruction error. This consensus NMF was performed by making custom updates to the cNMF v1.1 python package109. To determine the optimal number of programs (p) for each cell type and condition, we struck a balance between maximizing stability and minimizing error of the cNMF solution, while ensuring that the resulting programs were biologically coherent and parsimonious. Each program was annotated utilizing a combination of GSEA110 and comparison to bulk expression signatures.
Measuring similarity between gene expression programs
To measure the similarity between cNMF-derived gene expression programs and pre-existing bulk derived gene sets representing PDAC subtypes or differentially-expressed genes associated with perineural invasion, we performed the hypergeometric test and Kolmogorov-Smirnov test, respectively, to quantify the overlap between the two gene sets. To measure the similarity among the cNMF-derived gene expression programs we computed the correlation of the cell by program vector for each program to identify which programs were found to be co-occurring across the same cells. Finally, we computed the patient-level statistical comparisons of program compositional changes by treatment type and response. This were performed by computing the average program weight over all cells for each patient and testing for changes to the program abundance using statistical tests as described previously.
Multiplexed immunofluorescence validation of the neural-like progenitor program
For multiplexed immunofluorescence validation of the NRP program, we prepared FFPE sections from three independent PDAC tumor in the same manner as the DSP experiments described above except that probe hybridization and subsequent washes were omitted. We incubated the slide with 1:10 SYTO13 (ThermoFisher Scientific, cat. no. 57575), 1:40 anti-panCK-Alexa Fluor 532 (clone AE-1/AE-3; Novus Biologicals, cat. no. NBP2-33200AF532), and 1:50 anti-NRXN3 (rabbit polyclonal IgG; Invitrogen , cat. no. PA5-101708) in blocking buffer W (NanoString) for 1 hour at room temperature followed by secondary antibody staining with 1:1000 goat anti-rabbit IgG Alexa Fluor 647 (Invitrogen, A-21245) for 1 hour at room temperature. The slide was imaged on the NanoString GeoMx instrument in slide scanning mode with exposure times of 150 ms, 600 ms, and 75 ms in the SYTO 13, Alexa Fluor 532, and Alexa Fluor 647 channels, respectively.
Survival analysis of bulk RNA-seq data
Bulk RNA-seq data from two previously published resected primary PDAC cohorts with overall survival annotated were obtained (TCGA, n = 139; PanCuRx/ICGC, n = 168)8,9,39. Patients with metastases or those that received neoadjuvant therapy were excluded from this analysis, yielding a total of 266 patients for further analysis. Gene expression levels from RNA-seq data was estimated using RSEM v1.3.3111.
To score malignant and fibroblast cNMF programs in each tumor, we summed the expression of the top 200 genes for each program and z-score normalized the expression scores within the TCGA and PanCuRx/ICGC cohorts independently to account for batch effects. Age, sex, grade and stage were available for all patients. There were 154 progression events and 167 deaths. We consolidated some of the GEPs (TNF-NFκB, adhesive, interferon, and ribosomal into secretory; cycling (S), cycling (G2/M), and MYC into cycling) into aggregate programs to avoid overfitting a Cox proportional-hazards regression model with covariates. Multivariable Cox regression analyses was performed for time to progression (TTP) and overall survival (OS) (Stata/SE 15.1).
Digital spatial profiling experiments
We followed published experimental methods25 (Nanostring) with modifications as noted below. Briefly, serially-sectioned formalin-fixed paraffin-embedded (FFPE) sections (5 μm) of 21 specimens were prepared on the IRB-approved protocol (2003P001289) to generate consecutive sections that were processed for H&E and WTA, respectively. For WTA, slides were baked at 60°C for one hour, deparaffinized with CitriSolv (DECON), rehydrated, antigen-retrieved in 1x Tris-EDTA/pH 9 in a steamer for 15 min at 100°C, proteinase K (ThermoFisher Scientific, AM2548) digested at 0.1 ng/mL for 15 min at 37°C, post-fixed in neutral-buffered formalin for 10 min, hybridized to UV-photocleavable barcode-conjugated RNA in situ hybridization probe set (WTA with 18,269 gene targets) overnight at 37°C, washed to remove off-target probes, and then counterstained with morphology markers for 2 hours at room temperature.
The morphology markers consisted of: 1:10 SYTO13 (ThermoFisher Scientific, cat. no. 57575), 1:20 anti-panCK-Alexa Fluor 532 (clone AE-1/AE-3; Novus Biologicals, cat. no. NBP2-33200AF532), 1:100 anti-CD45-Alexa Fluor 594 (clone D9M8I; Cell Signaling Technology, cat. no. 13917S), and 1:100 anti-αSMA-Alexa Fluor 647 (clone 1A4; Novus Biologicals, cat. no. IC1420R) in blocking buffer W (NanoString). The anti-panCK and anti-αSMA antibodies were acquired pre-conjugated whereas the anti-CD45 antibody was conjugated using the Alexa Fluor 594 Antibody Labeling Kit (Invitrogen, A20185). These four morphology markers allowed delineation of the nuclear, epithelial, immune, and fibroblast compartments. Immunofluorescence images, region of interest (ROI) selection, segmentation into marker-specific areas of interest (AOI), and spatially-indexed barcode cleavage and collection were performed on a GeoMx Digital Spatial Profiling instrument (NanoString). Typical exposure times were 50 ms for SYTO13, 300 ms for anti-panCK-Alexa Fluor 532, 400-450 ms for anti-CD45-Alexa Fluor 594, and 50 ms for anti-αSMA-Alexa Fluor 647. Approximately 8-14 ROIs and 20-36 AOIs were collected per specimen. Library preparation was performed according to the manufacturer’s instructions and involved PCR amplification to add Illumina adapter sequences and unique dual sample indices. A minimum sequencing depth of 150-200 reads per square micron of illumination area was achieved by sequencing all WTA AOIs on a NovaSeq S2 (100 cycles, read 1: 27 nt, read 2: 27 nt, index 1: 8 nt, index 2: 8 nt).
Digital spatial profiling - data preprocessing
FASTQ files for DSP were aggregated into count matrices as described previously25. Briefly, deduplicated sequencing counts were calculated based on UMI and molecular target tag sequences. Single probe genes were reported as the deduplicated count value. The limit of quantitation (LOQ) was estimated as the geometric mean of the negative control probes plus 2 geometric standard deviations of the negative control probes. Targets were removed that consistently fell below the LOQ, and the datasets were normalized using upper quartile (Q3) normalization. Normalized expression was detrended to model cell-type specific expression by calculating an adjustment factor:
Where adjustment factors, A, are calculated for the expression, E, of a gene, g, within a given ROI, r, by comparing a given segment, S1, to the max expression observed in other segments, S2 and S3. The original expression was then detrended by calculating:
Digital spatial profiling - program scoring and correlation analysis
Statistical analysis was performed using R v4.1.1. Programs were scored for each DSP sample within each ROI using single-sample gene set enrichment analysis (ssGSEA, from GSVA v1.41.3)112, which were transformed using the z-score. For each program, intra-patient dispersion of program expression across ROIs was calculated as the patient-level mean of the interquartile range (IQR; difference between upper and lower quartiles) across all ROIs within each individual tumor: where n = number of patients; rj = number of ROIs in patient j; and pi,j = program score for ROI i in patient j. In contrast, inter-patient dispersion of program expression was computed as the IQR of the mean program score for each tumor: .
Unsupervised hierarchical clustering was performed on all features (malignant programs, CAF programs, deconvolved immune cell type proportions, compartment areas within ROI) using the Pearson correlation distance and average linkage. Cell deconvolution analysis was performed using the SpatialDecon v0.99.1 package (https://github.com/Nanostring-Biostats/SpatialDecon/). Analysis of expression or program scores used linear mixed effects models113 to control for multiple sampling within a slide, using Satterthwaite’s approximation114 for degrees of freedom for p-value calculation. Correlation coefficients were calculated using the Spearman rank correlation.
Digital spatial profiling - receptor ligand (RL) correlation across ROIs
Known receptor-ligand pairs were obtained from CellPhoneDB v2.0 with potential receptor-ligand pairs quantified using the Spearman rank correlation between paired segments within the same ROI across all ROIs with said pairs. Interactions were calculated for non-self (juxtacrine) and self (autocrine) occurring within the same segment. Receptor-ligand interactions were calculated separately for untreated and CRT specimens to determine interactions that are differential between conditions. All analyses were two-sided and used a significant level of p-value ≤0.05 and were adjusted for multiple testing where appropriate using the false discovery rate115.
Statistics and reproducibility
Multiple statistical methods are described above, and analyses yielding potentially variable results were repeated to ensure reproducibility (e.g., consensus non-negative matrix factorization). A p-value < 0.05 was generally used as a threshold for significance, except where otherwise indicated. Adjustments for multiple hypothesis testing were employed using FDR and/or Bonferroni correction. Overall, no statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized.
Data Availability
All figures are associated with raw data. Raw images for MIBI and Nanostring GeoMx experiments are publicly available on Science Data Bank (DOI 10.57760/sciencedb.01706). Raw and processed sequencing data (single-nucleus RNA-seq) for patient-derived tumors and organoids, and Nanostring GeoMx reads and count matrices have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE202051 and GSE199102, respectively. Raw single-nucleus RNA-seq data are also available in the controlled access repository DUOS (https://duos.broadinstitute.org/), under dataset ID 000139. Processed single-nucleus RNA-seq data are also available on the Single Cell Portal at: https://singlecell.broadinstitute.org/single_cell/study/SCP1089 (untreated) https://singlecell.broadinstitute.org/single_cell/study/SCP1096 (treated)
Code Availability
All code has been made available in Github at https://github.com/karthikj89/humanpdac116 and https://github.com/whwanglab/PDAC117.
Extended Data
Extended Data Fig. 1 |. Cell type and inferred CAN composition across PDAC tumors.

a, UMAP embeddings of single nucleus profiles (dots) from individual tumors (panels) from untreated (left) and treated (right) patients colored by post hoc cell type annotations (color legend). b, Example inferCNV analysis of the epithelial subset from a study specimen. Inferred amplifications (red) and deletions (blue) based on expression (color bar) of sliding 100-gene window in each chromosomal locus (columns) from each cell (rows) labeled by its annotated cell type (color code). c, Inferred CNA frequencies in the snRNA-seq cohort have similar distribution as those derived from TCGA genomic study1. Frequency (y axis) of CNAs on each chromosome arm (x axis) as inferred across the patients in the snRNA-seq cohort (light green bars) and from genome analysis of PDAC (dark green bars) from the TCGA cohort. d, Proportion of cells (y axis) in each of the four major compartments (color legend, top) or immune cell subsets (color legend, bottom) as estimated by snRNA-seq or MIBI (x axis) in each matched untreated (left; n = 5) or treated (right; n = 2) tumor.
Extended Data Fig. 2 |. Treatment associated with distinct cell type proportions across compartments.

Proportions (y axis) of cell types (x axis) in untreated (n = 18), CRT (n = 14), or CRTL (n = 5) tumors out of all non-malignant cells (top left) or in specific non-malignant cell compartments in the tumor. The boxes indicate upper and lower quartiles, with the horizontal lines marking the means. The lines extending vertically from the boxes (whiskers) indicate the maximum and minimum values excluding outliers. Data points are plotted as circles. * Bonferroni adjusted p < 0.05, ** p < 0.01, *** p < 0.001, two-sided Mann-Whitney U test with Bonferroni correction. Exact p-values for significant comparisons were non-malignant myeloid CRT-CRTL = 0.016344, epithelial (non-malignant) ductal CRT-CRTL = 0.031683, lymphoid Treg untreated-CRT = 0.033156, immune CD8+ T CRT-CRTL = 0.03507, immune Treg untreated-CRT = 0.022686.
Extended Data Fig. 3 |. Impact of treatment on differential gene expression in immune cells, malignant cells, and cancer-associated fibroblasts.
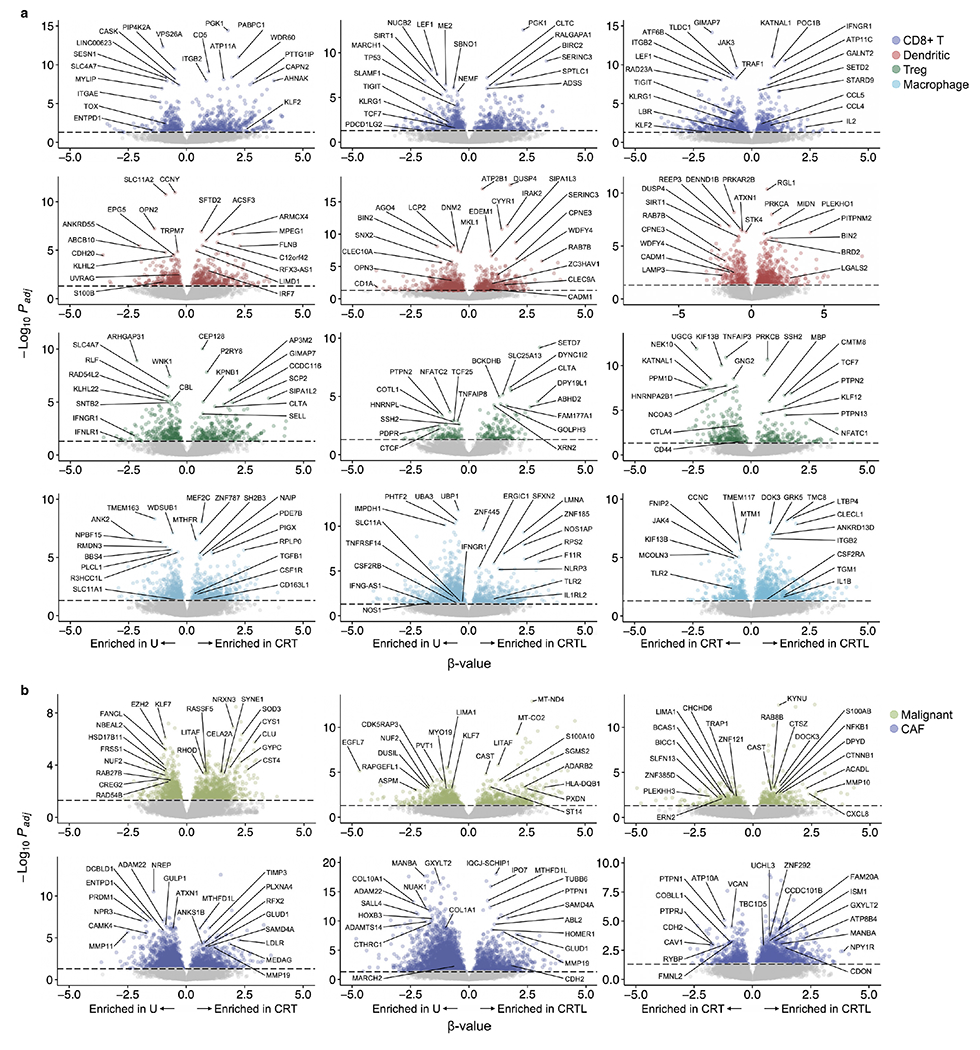
a, Differential expression (β-value, x axis, Poisson mixed-effect linear regression model, lme4 R package) and its significance (−log10(adjusted p-value), y axis) for CD8+ T cells (top row), dendritic cells (second row), Tregs (third row) and macrophages (bottom row, color legend) in CRT vs. untreated (left), CRTL vs. untreated (middle), and CRTL vs. CRT (right) tumors. Selected enriched or depleted genes are labeled. Bonferroni adjusted p-value < 0.05 is indicated with a dotted horizontal line. b, Differential expression (β-value, x axis, Poisson mixed-effect linear regression model, lme4 R package) and its significance (−log10(adjusted p-value), y axis) for malignant cells (top row) and CAFs (bottom row, color legend) in CRT vs. untreated (left), CRTL vs. untreated (middle), and CRTL vs. CRT (right) tumors. Selected enriched or depleted genes are labeled. Bonferroni adjusted p-value < 0.05 is indicated with a dotted horizontal line.
Extended Data Fig. 4 |. Prior signatures derived primarily from the bulk setting insufficiently delineate cells from snRNA-seq.

a, Malignant cell signatures. UMAP embeddings of single nucleus profiles (dots) from all tumor nuclei (top panels) or only malignant cells (bottom panels) colored by expression score (color bar, Methods) of signatures derived from the Bailey2, Collisson3, Moffitt4, and Chan-Seng-Yue5 studies. b, CAF signatures. UMAP embeddings of single nucleus profiles (dots) from all fibroblast nuclei colored by normalized expression score (color bar, Methods) of myCAF, apCAF, and iCAF signatures6 and well as cross-tissue fibroblast lineage signatures (COL3A1+ myofibroblast, LRRC15+ myofibroblast, CCL19+ colitis, ADAMDEC1+ colitis, NPNT+ alveolar, and PI16+ adventitial)7.
Extended Data Fig. 5 |. Stability and power in selection of programs in consensus NMF.

a, Estimated stability (blue, left y axis) and error (red, right y axis) in the cNMF solution learned with different numbers of programs (k, x axis) for malignant cells (left) and CAFs (right). b, Number of malignant (out of 14; left) and CAF (out of 4; right) programs recovered in the cNMF solution learned with a different proportion of samples (x axis) subsampled from our cohort.
Extended Data Fig. 6 |. Correlation among malignant cell or CAF expression programs.
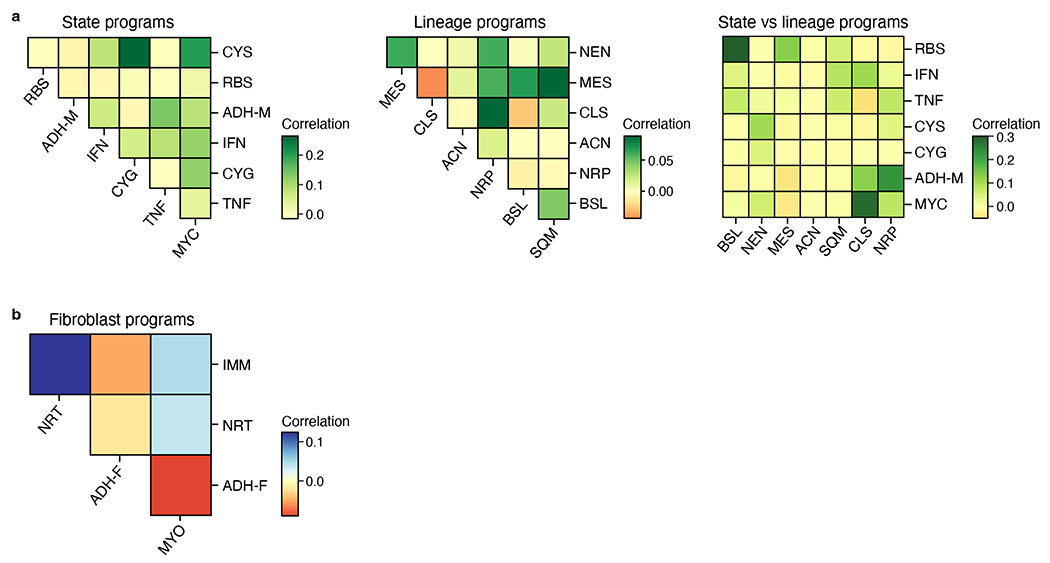
Correlation (color bar) among expression scores of malignant state and lineage programs across all malignant nuclei (a) or fibroblast programs across all fibroblast nuclei (b).
Extended Data Fig. 7 |. Enrichment of malignant cell and CAF programs in genes differentially expressed with treatment regimen.
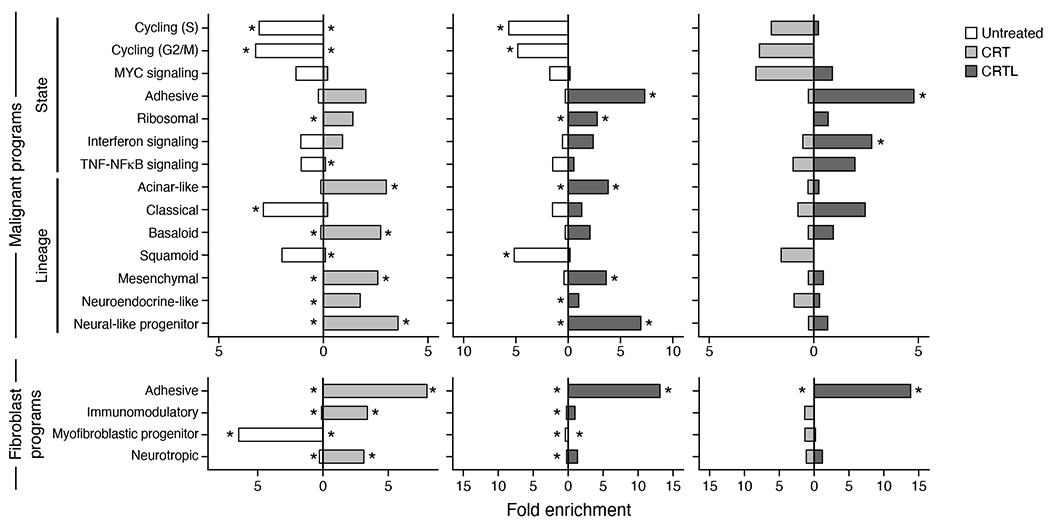
Fold enrichment of overlap (x axis) between gene program signatures (top 200 genes; rows) and genes differentially expressed (q < 0.05) in CRT (n = 14) vs. untreated (n = 18) (left), CRTL (n = 5) vs. untreated (middle), or CRTL vs. CRT (right). * Bonferroni adjusted p < 0.05, two-sided hypergeometric test. Exact p-values for significant comparisons were untreated-CRT: CYS-untreated = 8.54x10−6, CYS-CRT = 7.36x10−4, CYG-untreated = 9.53x10−7, CYG-CRT = 5.96x10−4, MYC-CRT = 3.72x10−2, ADH-M-CRT = 2.95x10−2, RBS-untreated = 2.25x10−2, TNF-CRT = 5.61x10−3, ACN-untreated = 2.87x10−2, ACN-CRT = 1.51x10−6, CLS-untreated = 4.77x10−5, CLS-CRT = 3.72x10−2, BSL-untreated = 2.44x10−2, BSL-CRT = 2.65x10−5, SQM-CRT = 5.10x10−3, MES-untreated = 2.16x10−3, MES-CRT = 1.09x10−4, NEN-untreated = 4.83x10−3, NRP-untreated = 2.58x10−3, NRP-CRT = 6.06x10−10, ADH-F-untreated = 1.03x10−8, ADH-F-CRT = 3.68x10−43, IMM-untreated = 6.12x10−6, IMM-CRT = 5.24x10−8, MYO-untreated = 3.66x10−72, MYO-CRT = 5.60x10−4, NRT-untreated = 4.56x10−4, NRT-CRT = 5.60x10−7; untreated-CRTL: CYS-untreated = 9.45x10−19, CYS-CRT = 4.91x10−3, CYG-untreated = 1.04x10−13, ADH-M-CRT = 1.10x10−21, RBS-untreated = 7.60x10−3, RBS-CRT = 4.91x10−3, IFN-CRT = 4.49x10−2, ACN-untreated = 9.62x10−3, ACN-CRT = 4.14x10−6, SQM-untreated = 2.65x10−16, MES-CRT = 9.59x10−6, NEN-untreated = 1.43x10−2, NRP-untreated = 6.79x10−3, NRP-CRT = 2.74x10−20, ADH-F-untreated = 7.60x10−21, ADH-F-CRT = 1.54x10−151, IMM-untreated = 2.58x10−9, MYO-untreated = 4.80x10−6, MYO-CRT = 2.09x10−5, NRT-untreated = 2.57x10−10; CRT-CRTL: MYC-CRT = 3.07x10−2, ADH-M-CRTL = 4.17x10−8, IFN-CRTL = 1.99x10−2, ADH-F-CRT = 7.16x10−7, ADH-F-CRTL = 5.88x10−60, IMM-CRTL = 2.56x10−2.
Extended Data Fig. 8 |. Multivariable Cox regression analysis for overall survival in TCGA and PanCuRx/ICGC PDAC cohorts.
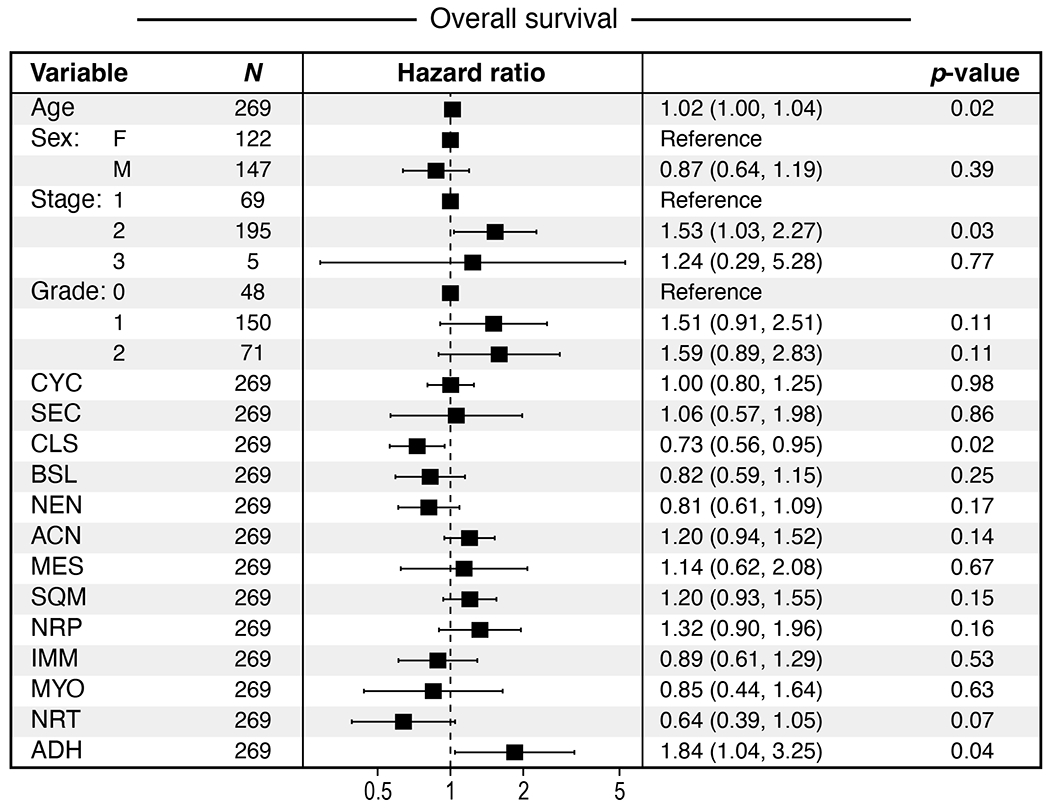
Hazard ratios ± 95% confidence interval (middle) and p-values (left) for each variable (clinicopathologic and program expression score in bulk RNA-seq, rows) in multivariable Cox regression model for overall survival (OS), based on a cohort of 269 patients with untreated, resected primary PDAC profiled by RNA-seq in TCGA and PanCuRx/ICGC.
Extended Data Fig. 9 |. Digital spatial profiling with whole transcriptome atlas (WTA) enables accurate mapping of cell type signatures in space as a complement to snRNA-seq.

a, Immunofluorescence images of FFPE sections from all PDAC specimens analyzed using whole transcriptome DSP (n = 21 independent tumors) separated by treatment status (top, untreated; bottom, treated). Color legend indicates target of fluorophore-conjugated antibodies. b, Expression (z-score of normalized counts across segments; purple/yellow color bar) of signature genes (rows) from different cell types (color legend 3 and left color bar 3) across segments (columns, color legend 2 and horizontal color bar 2) and treatment regimens (columns, grayscale legend 1 and horizontal grayscale bar 1) profiled by WTA, capturing epithelial (green), fibroblasts (blue) and immune (red) cells. Columns and rows are clustered by unsupervised hierarchical clustering. c, Pearson correlation coefficient (color bar) of the scores of each CAF, malignant, and immune feature in snRNA-seq (rows, columns) across patient tumors. Rows and columns are ordered by hierarchical clustering.
Extended Data Fig. 10 |. snRNA-seq captures a greater diversity and abundance of cell types relative to prior single-cell approaches.
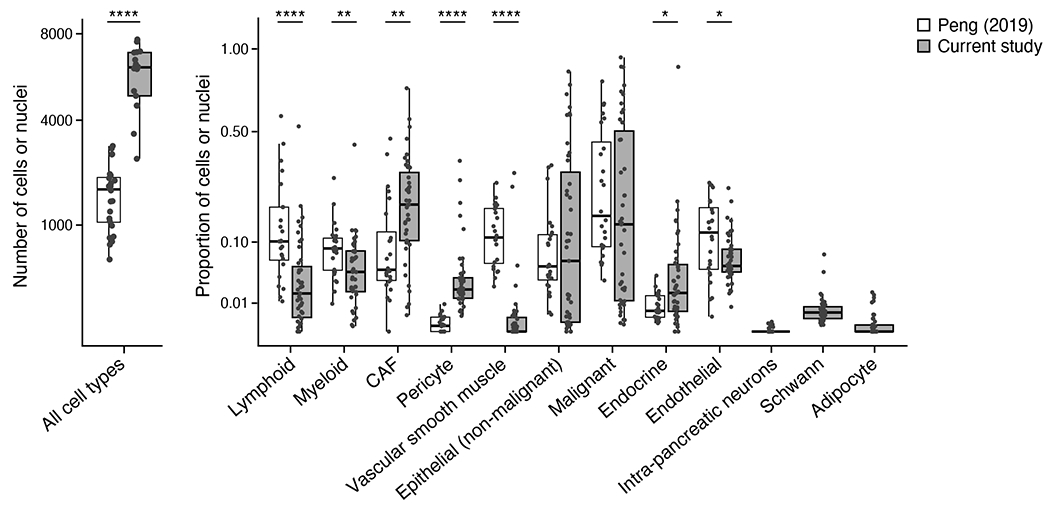
Number of nuclei/cells per untreated tumor that passed quality control filters (y axis) in our study (n = 18) vs. Peng et al. study (n = 24)69 (grayscale legend), in total (left) and partitioned by cell type (right). The boxes indicate upper and lower quartiles, with the horizontal lines marking the means. The lines extending vertically from the boxes (whiskers) indicate the maximum and minimum values excluding outliers. Data points are plotted as solid circles. *Bonferroni adjusted p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, two-sided Mann-Whitney U test with Bonferroni correction. Exact p-values for significant comparisons were all cell types = 5.76 x 10−9, lymphoid = 5.54 x 10−5, myeloid = 7.25 x 10−3, CAF = 3.70 x 10−3, pericyte = 5.22 x 10−11, vascular smooth muscle = 1.09 x 10−9, endocrine = 1.84 x 10−2, endothelial = 4.00 x 10−2.
Supplementary Material
Supplementary Table 1 | Patient cohort and clinicopathologic data.
Supplementary Table 2 | Top 200 weighted genes for malignant and fibroblast cNMF programs.
Supplementary Table 3 | Gene Set Enrichment Analysis for malignant cNMF programs ranked by increasing FDR q-value. Threshold FDR q-value < 0.05.
Supplementary Table 4 | Gene Set Enrichment Analysis for fibroblast cNMF programs ranked by increasing FDR q-value. Threshold FDR q-value < 0.05.
Supplementary Table 5 | Spearman rank correlation coefficients for receptor-ligand pairs (CellPhoneDB) expressed in all paired segments (epithelial, CAF, immune) within the same ROI across all ROIs with said segment pairs, stratified by treatment group (CRT vs. untreated).
Supplementary Table 6 | Select spatially defined receptor ligand pairs as a function of treatment. Cell-type specific Spearman’s rank correlation coefficients of expression (ρ) are provided based on treatment group.
Supplementary Table 7 | Multivariable Cox regression analysis for time-to-progression including the following covariates: age, sex, stage, grade, malignant lineage programs, CAF programs, Collisson classical signature, Collison quasi-mesenchymal signature.
ACKNOWLEDGEMENTS
We are grateful to the patients and families who contributed their time and surgical specimens to this study. We thank Leslie Gaffney for assistance with preparing figures; Erroll Rueckert from NanoString for assistance with instrumentation; Jason Ptacek from IONpath for assistance with MIBI; and Karen Yee, Judy Teixeira, Katherine Anderson, Margaret Magendantz, Kim Mercer, William Rideout, Bo Li, Peter Westcott, Cristin McCabe, Nahien Sharif, and Jenna Pfiffner-Borges for administrative and technical support. This work was supported in part by the Ludwig Institute for Cancer Research (A.R.), Klarman Cell Observatory (A.R. and R.X.), Lustgarten Foundation (T.E.J.), American Society for Clinical Oncology/Conquer Cancer Foundation Young Investigator Award (W.L.H), Hopper-Belmont Foundation Inspiration Award (W.L.H.), American Cancer Society/MGH Institutional Research Grant (W.L.H.), UCSF Dean’s Yearlong Fellowship (J.A.G.), Early Postdoc Mobility Fellowship (no. P2ZHP3 181475) from the Swiss National Science Foundation (D.S.), SU2C-Lustgarten Foundation (T.S.H., D.T.T.) and the Robert L. Fine Cancer Research Foundation (D.T.T.). This study was also conducted with support of the Ontario Institute for Cancer Research (PanCuRx Translational Research Initiative) through funding provided by the Government of Ontario, the Wallace McCain Centre for Pancreatic Cancer supported by the Princess Margaret Cancer Foundation, the Terry Fox Research Institute, the Canadian Cancer Society Research Institute, and the Pancreatic Cancer Canada Foundation. W.L.H. is an Andrew L. Warshaw, M.D. Institute for Pancreatic Cancer Research Fellow. D.S. is a Damon Runyon Cancer Research Fellow (DRQ-03-20). R.K.J. receives support from an NCI Cancer Moonshot grant (U01-CA224348) and the Ludwig Cancer Center at Harvard. T.E.J. and A.R. were investigators of the Howard Hughes Medical Institute during the time this work was performed. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
COMPETING INTERESTS
A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov. From August 1, 2020, A.R. is an employee of Genentech. T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. Dr. Jacks’ laboratory also currently receives funding from Johnson & Johnson, but this fund did not support the research described in this manuscript. D.T.T. has received consulting fees from Nanostring Technologies, which was used in this work. D.T.T. has received consulting fees from ROME Therapeutics, Foundation Medicine, Inc., EMD Millipore Sigma, and Pfizer that are not related to this work. D.T.T. is a founder and has equity in ROME Therapeutics, PanTher Therapeutics and TellBio, Inc., which is not related to this work. D.T.T. receives research support from ACD-Biotechne, PureTech Health LLC, Ribon Therapeutics, which was not used in this work. M.M.K. has served as a compensated consultant for H3 Biomedicine and AstraZeneca and received a research grant (to institution) from Novartis that is not related to this work. R.K.J. received consultant fees from Elpis, Pfizer, SPARC, SynDevRx; owns equity in Accurius, Enlight, SynDevRx; serves on the Board of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla World Healthcare Fund and received a Research Grant from Boehringer Ingelheim all not related to this work. The interests of D.T.T., M.M.K., and R.K.J. were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. O.R.R. is a co-inventor on patent applications filed by the Broad Institute for inventions related to single cell genomics. ORR is an employee of Genentech since October 19, 2020. W.L.H., K.A.J., J.A.G., H.I.H., T.J., and A.R. are co-inventors on U.S. Provisional Patent Application No. 63/313,596 related to this work. All other authors declare no competing interests.
REFERENCES
- 1.Versteijne E et al. Neoadjuvant Chemoradiotherapy Versus Upfront Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Long-Term Results of the Dutch Randomized PREOPANC Trial. J. Clin. Oncol Online ahead of print (2022) doi: 10.1200/jco.21.02233. [DOI] [PubMed] [Google Scholar]
- 2.Mizrahi JD, Surana R, Valle JW & Schroff RT Pancreatic cancer. Lancet 395, 2008–2020 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Collisson EA, Bailey P, Chang DK & Biankin AV Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol 16, 207–220 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Guo JA et al. Refining the Molecular Framework for Pancreatic Cancer with Single-cell and Spatial Technologies. Clin. Cancer Res 27, 3825–3833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collisson EA et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med 17, 500–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moffitt RA et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet 47, 1168–1178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey P et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Raphael BJ et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aung KL et al. Genomics-driven precision medicine for advanced pancreatic cancer: Early results from the COMPASS trial. Clin. Cancer Res 24, 1344–1354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosein AN, Brekken RA & Maitra A Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol 17, 487–505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy JE et al. Total Neoadjuvant Therapy with FOLFIRINOX in Combination with Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 5, 1020–1027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H et al. Use of angiotensin system inhibitors is associated with immune activation and longer survival in nonmetastatic pancreatic ductal adenocarcinoma. Clin. Cancer Res 23, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (80-.) 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puram SV et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steele NG et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat. Cancer 1, 1097–1112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azevedo-Pouly ACP, Elgamal OA & Schmittgen TD RNA isolation from mouse pancreas: A ribonuclease-rich tissue. J. Vis. Exp e51779 (2014) doi: 10.3791/51779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drokhlyansky E et al. The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 182, 1606–1622.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosti L et al. Single-Nucleus and In Situ RNA–Sequencing Reveal Cell Topographies in the Human Pancreas. Gastroenterology 160, 1330–1344 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Slyper M et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ligorio M et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 178, 160–175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Väyrynen SA et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res 27, 1069–1081 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liudahl SM et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome. Cancer Discov. 11, 2014–2031 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grünwald BT et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 184, 1–16 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Merritt CR et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol 38, 586–599 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Muraro MJ et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 3, 385–394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q et al. Reg proteins promote acinar-to-ductal metaplasia and act as novel diagnostic and prognostic markers in pancreatic ductal adenocarcinoma. Oncotarget 7, 77838–77853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X et al. REG3A accelerates pancreatic cancer cell growth under IL-6-associated inflammatory condition: Involvement of a REG3A-JAK2/STAT3 positive feedback loop. Cancer Lett. 362, 45–60 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Eberlein J et al. Chemokine Signatures of Pathogen-Specific T Cells I: Effector T Cells. J. Immunol 205, 2169–2187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston RJ et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell 26, 923–937 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlson CM et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442, 299–302 (2006). [DOI] [PubMed] [Google Scholar]
- 33.De La O JP et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. U. S. A 105, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerra C et al. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 11, (2007). [DOI] [PubMed] [Google Scholar]
- 35.Morris IV JP, Cano DA, Sekine S, Wang SC & Hebrok M β-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Invest 120, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liberzon A et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roa-Peña L et al. Keratin 17 identifies the most lethal molecular subtype of pancreatic cancer. Sci. Rep 9, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Juiz N et al. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 34, 12214–12228 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Chan-Seng-Yue M et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet 52, 231–240 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Raghavan S et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elyada E et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buechler MB et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Dardenne E et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 30, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiu H et al. MEN1 deficiency leads to neuroendocrine differentiation of lung cancer and disrupts the DNA damage response. Nat. Commun 11, 1009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He H et al. Krüppel-like Factor 4 Promotes Esophageal Squamous Cell Carcinoma Differentiation by Up-regulating Keratin 13 Expression. J. Biol. Chem 290, 13567–13577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rorke EA et al. Structural and biochemical changes underlying a keratoderma-like phenotype in mice lacking suprabasal AP1 transcription factor function. Cell Death Dis. 6, e1647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Champliaud MF et al. Gene characterization of sciellin (SCEL) and protein localization in vertebrate epithelia displaying barrier properties. Genomics 70, 264–268 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Zhang D et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat. Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karasawa M et al. Localization of metallothionein in hair follicles of normal skin and the basal cell layer of hyperplastic epidermis: Possible association with cell proliferation. J. Invest. Dermatol 97, 97–100 (1991). [DOI] [PubMed] [Google Scholar]
- 50.Aiello NM et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 45, 681–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y-A, Lu I-L & Tsai J-W Contactin-1/F3 Regulates Neuronal Migration and Morphogenesis Through Modulating RhoA Activity. Front. Mol. Neurosci 11, 422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donato V. Di et al. An Attractive Reelin Gradient Establishes Synaptic Lamination in the Vertebrate Visual System. Neuron 97, 1049–1062 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Kantor DB et al. Semaphorin 5A Is a Bifunctional Axon Guidance Cue Regulated by Heparan and Chondroitin Sulfate Proteoglycans. Neuron 44, 961–975 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Hori K & Hoshino M Neuronal Migration and AUTS2 Syndrome. Brain Sci. 7, 54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sakurai T The role of NrCAM in neural development and disorders--beyond a simple glue in the brain. Mol. Cell. Neurosci 49, 351–363 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Lau HH, Ng NHJ, Loo LSW, Jasmen JB & Teo AKK The molecular functions of hepatocyte nuclear factors – In and beyond the liver. J. Hepatol 68, 1033–1048 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Robertson AG et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 171, 540–556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi DD et al. Therapeutic avenues for cancer neuroscience: translational frontiers and clinical opportunities. Lancet Oncol. 23, e62–e74 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biankin AV et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo JA et al. Pan-cancer Transcriptomic Predictors of Perineural Invasion Improve Occult Histopathologic Detection. Clin. Cancer Res 27, 2807–2815 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farrell AS et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun 8, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Komori T Runx2, a multifunctional transcription factor in skeletal development. J. Cell. Biochem 87, 1–8 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Roel G, YY G, J P-M, FJ V & O D Lef1 plays a role in patterning the mesoderm and ectoderm in Xenopus tropicalis. Int. J. Dev. Biol 53, 81–89 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Tahara N et al. Sall4 regulates neuromesodermal progenitors and their descendants during body elongation in mouse embryos. Development 146, dev177659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andre P, Song H, Kim W, Kispert A & Yang Y Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development 142, 1516–1527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cherubini A et al. FOXP1 circular RNA sustains mesenchymal stem cell identity via microRNA inhibition. Nucleic Acids Res. 47, 5325–5340 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sleightholm RL et al. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther 179, 158–170 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Cheng HW et al. CCL19-producing fibroblastic stromal cells restrain lung carcinoma growth by promoting local antitumor T-cell responses. J. Allergy Clin. Immunol 142, 1257–1271 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Hirth M et al. CXCL10 and CCL21 Promote Migration of Pancreatic Cancer Cells Toward Sensory Neurons and Neural Remodeling in Tumors in Mice, Associated With Pain in Patients. Gastroenterology 159, 665–681 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Boulakirba S et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep 8, 256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bettac L, Denk S, Seufferlein T & Huber-Lang M Complement in pancreatic disease-Perpetrator or savior? Front. Immunol 8, 15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng J et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 29, 725–738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maji S et al. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res 137, 37–75 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Zhang M et al. Platelet-derived growth Factor D is a prognostic biomarker and is associated with platinum resistance in epithelial ovarian cancer. Int. J. Gynecol. Cancer 28, 323–331 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Chen X et al. SPP1 inhibition improves the cisplatin chemo-sensitivity of cervical cancer cell lines. Cancer Chemother. Pharmacol 83, 603–613 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Christie EL et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun 10, 1295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chauvet S et al. Gating of Sema3E/PlexinD1 signaling by neuropilin-1 switches axonal repulsion to attraction during brain development. Neuron 56, 807–822 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wasser CR & Herz J Reelin: Neurodevelopmental Architect and Homeostatic Regulator of Excitatory Synapses. J. Biol. Chem 292, 1330–1338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Denny SK et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 166, 328–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biasci D et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. U. S. A 117, 28960–28970 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fearon DT & Janowitz T AMD3100/Plerixafor overcomes immune inhibition by the CXCL12-KRT19 coating on pancreatic and colorectal cancer cells. Br. J. Cancer 125, 149–151 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Skrypek N et al. The oncogenic receptor ErbB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget 6, 10853–10867 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harder J et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br. J. Cancer 106, 1033–1038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sjöstedt E et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science (80-.). 367, eaay5947 (2020). [DOI] [PubMed] [Google Scholar]
- 85.VanDussen KL, Sonnek NM & Stappenbeck TS L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res. 37, 101430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boj SF et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Freed-Pastor WA et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. In review, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Man FM, Goey AKL, van Schaik RHN, Mathijssen RHJ & Bins S Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018 5710 57, 1229–1254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Conroy T et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. in New England Journal of Medicine vol. 379 2395–2406 (Massachussetts Medical Society, 2018). [DOI] [PubMed] [Google Scholar]
- 90.Fleming SJ, Marioni JC & Babadi M CellBender remove-background: A deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv (2019) doi: 10.1101/791699. [DOI] [Google Scholar]
- 91.Wolf FA, Angerer P & Theis FJ SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv (2018). [Google Scholar]
- 95.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med 21, 938–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zilionis R et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schupp JC et al. Integrated Single Cell Atlas of Endothelial Cells of the Human Lung. Circulation 144, 286–302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bernard V et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin. Cancer Res 25, 2194–2205 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moncada R et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol 38, 333–342 (2020). [DOI] [PubMed] [Google Scholar]
- 100.Büttner M, Ostner J, Müller C, Theis F & Schubert B scCODA: A Bayesian model for compositional single-cell data analysis. bioRxiv 12.14.422688 (2020) doi: 10.1101/2020.12.14.422688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolf FA et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schapiro D et al. HistoCAT: Analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods 14, 873–876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zanotelli VRRT & Bodenmiller B ImcSegmentationPipeline: A pixelclassification based multiplexed image segmentation pipeline. (Zenodo, 2017). doi: 10.5281/zenodo.3841961. [DOI] [Google Scholar]
- 104.Zanotelli V, Ndamond & Strotton M BodenmillerGroup/ImcSegmentationPipeline: IMC Segmentation Pipeline. (2020) doi: 10.5281/zenodo.3841961. [DOI] [Google Scholar]
- 105.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 106.McQuin C et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, e2005970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lopez R et al. A joint model of unpaired data from scRNA-seq and spatial transcriptomics for imputing missing gene expression measurements. arXiv 1905.02269 (2019). [Google Scholar]
- 108.Muus C et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med 27, 546–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kotliar D et al. Identifying gene expression programs of cell-type identity and cellular activity with. Elife 8, e43803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li B & Dewey CN RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hänzelmann S, Castelo R & Guinney J GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bates D, Mächler M, Bolker BM & Walker SC Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48 (2015). [Google Scholar]
- 114.Kuznetsova A, Brockhoff PB & Christensen RHB lmerTest Package: Tests in Linear Mixed Effects Models . J. Stat. Softw 82, 1–26 (2017). [Google Scholar]
- 115.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 116.
- 117.
Extended Data References
- 1.Raphael BJ et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey P et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Collisson EA, Bailey P, Chang DK & Biankin AV Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol 16, 207–220 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Moffitt RA et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet 47, 1168–1178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan-Seng-Yue M et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet 52, 231–240 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Elyada E et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buechler MB et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 | Patient cohort and clinicopathologic data.
Supplementary Table 2 | Top 200 weighted genes for malignant and fibroblast cNMF programs.
Supplementary Table 3 | Gene Set Enrichment Analysis for malignant cNMF programs ranked by increasing FDR q-value. Threshold FDR q-value < 0.05.
Supplementary Table 4 | Gene Set Enrichment Analysis for fibroblast cNMF programs ranked by increasing FDR q-value. Threshold FDR q-value < 0.05.
Supplementary Table 5 | Spearman rank correlation coefficients for receptor-ligand pairs (CellPhoneDB) expressed in all paired segments (epithelial, CAF, immune) within the same ROI across all ROIs with said segment pairs, stratified by treatment group (CRT vs. untreated).
Supplementary Table 6 | Select spatially defined receptor ligand pairs as a function of treatment. Cell-type specific Spearman’s rank correlation coefficients of expression (ρ) are provided based on treatment group.
Supplementary Table 7 | Multivariable Cox regression analysis for time-to-progression including the following covariates: age, sex, stage, grade, malignant lineage programs, CAF programs, Collisson classical signature, Collison quasi-mesenchymal signature.
Data Availability Statement
All figures are associated with raw data. Raw images for MIBI and Nanostring GeoMx experiments are publicly available on Science Data Bank (DOI 10.57760/sciencedb.01706). Raw and processed sequencing data (single-nucleus RNA-seq) for patient-derived tumors and organoids, and Nanostring GeoMx reads and count matrices have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE202051 and GSE199102, respectively. Raw single-nucleus RNA-seq data are also available in the controlled access repository DUOS (https://duos.broadinstitute.org/), under dataset ID 000139. Processed single-nucleus RNA-seq data are also available on the Single Cell Portal at: https://singlecell.broadinstitute.org/single_cell/study/SCP1089 (untreated) https://singlecell.broadinstitute.org/single_cell/study/SCP1096 (treated)