

Abstract
Herein, we investigated the thermodynamic stability and opto-electronic properties of a newly BN-doped azulene. The gas-phase formation enthalpies of 11 BN-doped azulene were calculated by the atomization energy method using three computational models (CBS-APNO, CBS-QB3, and G3MP2). The results suggest that AZ-1N9B exhibits the highest stability among the studied isomers. On the other hand, AZ-1B9N and AZ-9B10N display nearly equal stability with relative energies of 19.36 and 19.82 kcal/mol at CBS-QB3, respectively. These two isomers are considered the least stable among the investigated compounds. The frontier molecular orbitals (FMO), ionization energies (IE), and electron affinities (EA) of these isomers were discussed. Additionally, the electronic absorption spectra of the BN-doped azulenes were computed using the TD-B3LYP/6–31 + G(d,p) and TD-CAM-B3LYP level of theories, which using a long-range corrected hybrid functional in acetone. The computational results obtained in this research are align closely with the existing literature, thereby reinforcing the credibility and reliability of our findings.
Subject terms: Chemistry, Theoretical chemistry
Introduction
The presence of isosterism, where compounds with the same number of atoms and electrons, can lead to the formation of unique heterocylic aromatic hydrocarbons. One example of such isosterism is the BN/CC isosterism, where BN bond replaces the CC bond1,2. The atomic displacement can lead to a change in electronic, optical and reactivity properties relative to their parent hydrocarbons1–6. This phenomenon has been used in drug design to improve the pharmacokinetics and pharmacodynamics of drugs7. It has also been used in materials science to enhance materials' chemical and physical properties for potential use in specific applications1,2. A dopant atom, such as boron and nitrogen, allows for the tunability of electronic and structural properties8–10.
Dewar et al. in 1958 reported a single CC/BN substitution in an aromatic compound11 and synthesized a good library of mono-12 and polycyclic13 compounds Recently, Liu et al.14 synthesized the least stable isomer of BN-doped naphthalene for optoelectronic properties and supported their findings with quantum chemical calculations. However, the slow progress in this area can be attributed to the lack of suitable synthetic methods15.
Azulene is an aromatic non-alternant isomer of naphthalene, consisting of a pentagonal and a heptagonal fused ring. Since its discovery, it has attracted significant interest. Azulene has unique properties like a dipole moment of 1.08 Debye16 compared to zero dipole moment for naphthalene. It also exhibits a narrow highest occupied molecular orbital (HOMO) –lowest unoccupied molecular orbital (LUMO) energy gap resulting from the non-mirror-symmetric orbitals17. Azulene derivatives have been used in the development of new advanced materials, including anion receptors/sensors18, molecular switches19, electrochromic materials20, conductive charge-transfer complexes21, liquid crystals22, organic/polymeric conductors23, and near-infrared (NIR) resonance materials24.
To the best of our knowledge, this work represents the first attempt to study a novel class of BN-doped aromatics, namely BN-doped azulene, compared to the recently reported experimental investigation of BN-doped naphthalene analogs14. The gas-phase enthalpies of formation for eleven isomers of BN-substituted azulene were computed using three ab initio composite methods, G3MP2, CBS-QB3, and CBS-APNO. In addition, naphthalene and its six BN-doped species were calculated for comparison. Furthermore, the electronic absorption ultraviolet–visible (UV–Vis) spectra in the gas phase and in acetone were calculated using the time-dependent density functional theory (TD-DFT)25. The frontier molecular orbital energies, ionization energies, electron affinities, and the density of states (DOS) have also been discussed.
Computational details
All calculations were carried out using Gaussian 16 program26 using the multi-level ab initio composite methods G3MP2, CBS-QB3, and CBS-APNO developed for accurate thermochemical data27–31. Visualization of different structures was performed by chimeraX software32, while DOS is calculated using the GaussSum 3.0 program33.
Geometry optimization and frequency calculations at CBS-QB3 were performed with DFT/B3LYP34,35 using 6–311G(d,p) basis sets. Then, single-point energy calculations were performed at CCSD(T), MP4SDQ, and MP2 with different basis sets. Finally, CBS extrapolation is employed to get the final energy. However, CBS-APNO starts with geometry and frequency calculations at the HF/6–311G(d,p) level, followed by further optimization at the QCISD/6–311G(d,p). This high-level optimization step is used for four single-point energy calculations at the QCISD(T), MP2(full), HF, and MP2 levels. The G3MP2 combined method falls between CBS-QB3 and CBS-APNO regarding computational cost. It begins with geometry optimization at MP2(full)/6–31G(d) and zero-point vibrational energy from HF/6−31G(d). Similar to other composite methods, this optimization process is followed by a series of single-point energy calculations with QCISD(T)/6–31G(d,p), the most time-demanding step, followed by some corrections through basis set extensions at MP2 which eliminate the costly MP4 process in the G2MP2 method. The gas phase enthalpy of formation was computed using the atomization energy approach described by Ochterski36. The adiabatic electron affinity and ionization potentials for the considered systems were calculated from G3MP2 energies. Enthalpies of formation can be estimated from Eq. (1):
| 1 |
where Ee(S) and Ee(Xi) are the calculated energy of molecule S and ith atom X, respectively. [H298(S) – H0(S)] and [H298 (Xi) – H0(Xi)] are the thermal corrections to the enthalpy for the molecule S and the individual atoms X, respectively. ZPVE is the zero-point vibrational energy of molecule S. The separated atomic enthalpies ΔH°f(Xi) can be obtained from the NIST WebBook37. Ionization energy38,39 was calculated from Eq. (2):
| 2 |
where E (N+) and E (N) are zero-point corrected energies of the molecular cation and the neutral molecule, respectively, at the same level of theory. Electron affinity (EA) 39 was estimated from Eq. (3):
| 3 |
where E (N-) is the zero-point corrected energy of the molecular anion. Time-dependent density functional theory with the B3LYP functional (TD-B3LYP/6–31 + G(d,p)) and with the long-range corrected hybrid Coulomb attenuating method CAM-B3LYP40 (TD-CAM-B3LYP/6–31 + G (d,p)) were used to compute electronic absorption spectra of the considered compounds in acetone utilizing non-equilibrium polarizable continuum solvation model (PCM) at the B3LYP/6–31 + G (d,p) optimized gas phase geometry. HUMOs, LUMOs distribution and ESP were sketched using Gaussview41, while the ultraviolet–visible (UV–Vis) spectra were simulated using GaussSum 3.0 program33.
Results and discussion
We have started our investigation by doing a benchmark calculation on the parent molecules (naphthalene and azulene). Our calculations of the zero-point corrected enthalpy of formations predicted that naphthalene is more stable than azulene, which can be attributed to the resonance stabilization energy. The calculated relative enthalpy of formation is 33.08 kcal/mol agrees well with the reported experimental value of 32.93 kcal/mol based on solid-state measurements42, with a deviation of only 0.15 kcal/mol. For naphthalene, the average experimental value was reported43 to be 35.98 ± 0.36 kcal/mol, as given in Table 1. This value is 3.58 and 5 kcal/mol higher than the values calculated by G3MP2 and CBS-APNO methods, respectively. However, the CBS-QB3 value lies at the upper limit of the experimental value.
Table 1.
Enthalpy of formation (kcal/mol) for naphthalene, BN-doped naphthalene, azulene and BN-doped azulene.
| Structures\Method | G3MP2 | G3MP2[6] | Difference | CBS-APNO | CBS-QB3 | Experiment |
|---|---|---|---|---|---|---|
| Napth | 32.25 | – | – | 30.83 | 37.24 | 35.98 ± 0.3643 |
| Napth-1B2N | 13.64 | 13.00 | 0.64 | 10.04 | 16.89 | – |
| Napth-1N2B | 13.23 | 12.60 | 0.63 | 9.40 | 16.49 | – |
| Napth-1N9B | 22.34 | 21.70 | 0.64 | 19.76 | 26.43 | – |
| Napth-2B3N | 24.39 | 23.80 | 0.59 | 21.25 | 28.00 | – |
| Napth-9N10B | 26.12 | 25.50 | 0.62 | 23.71 | 29.98 | – |
| Napth-1B9N | 31.88 | 31.30 | 0.58 | 28.20 | 35.28 | – |
| AZ | 66.12 | – | – | 64.38 | 71.95 | 66.8945, 73.58 44 |
| AZ-1N2B | 44.48 | – | – | 41.36 | 48.16 | – |
| AZ-1B2N | 53.44 | – | – | 50.94 | 57.50 | – |
| AZ-1N9B | 40.33 | – | – | 38.13 | 44.52 | – |
| AZ-1B9N | 59.71 | – | – | 57.39 | 63.88 | – |
| AZ-6N7B | 51.17 | – | – | 48.90 | 55.31 | – |
| AZ-6B7N | 47.12 | – | – | 44.60 | 51.03 | – |
| AZ-7N8B | 46.66 | – | – | 44.14 | 50.62 | – |
| AZ-7B8N | 53.51 | – | – | – | 57.67 | – |
| AZ-8N9B | 53.95 | – | – | 52.58 | 58.86 | – |
| AZ-8B9N | 44.96 | – | – | 41.80 | 48.26 | – |
| AZ-9B10N | 60.96 | – | – | 57.96 | 64.34 | – |
The gas-phase formation enthalpy of azulene at standard conditions was determined experimentally by Roth et al.44 and Kovats et al.45 as 73.61 and 66.92 kcal/mol, respectively. The calculated values at G3MP2 and CBS-QB3 levels agree with the experimental counterparts (Table 1). Although there is no experimental data for the BN-doped azulenes investigated in this study, we can discuss their stabilities with some confidence based on the above benchmark performed on naphthalene and azulene. Further, the reliability of the three methods employed in our study is well known to reproduce high accurate thermodynamic data.
Here, we studied the effect of different vicinal doping positions of BN on the enthalpy of the formation of azulene. Table 1 lists the calculated enthalpies of formation (kcal/mol) for 11 BN-doped azulene and 6 BN-doped naphthalene obtained from the atomization energy method using the G3MP2, CBS-QB3, and CBS-APNO methods.
The optimized structures of 11 BN-doped azulene isomers at B3LYP/6−311G(d,p) (optimization level of CBS-QB3 method) are depicted in Fig. 1, and the associated coordinates are reported in Table S1 in the supplementary information (SI). Figure 2 sketches the relative energies of the 11 BN-doped azulene isomers referenced to the most stable isomer AZ-1N9B using the three composite methods CBS-APNO, CBS-QB3, and G3MP2. These relative energies are also reported in Table S2. Figure 2 shows that AZ-1N9B is the most stable isomer, followed by AZ-1N2B and AZ-8B9N isomers with almost the same energies (relative energies of 3.64 and 3.74 kcal/mol), respectively. The least stable isomers are AZ-1B9N and AZ-9B10N with relative energies of 19.36 and 19.82 kcal/mol, respectively, at the CBS-QB3 method.
Figure 1.

Optimized structures of BN-doped azulene isomers at B3LYP/6–311G(d,p) (optimization level of CBS-QB3 method) performed by chimeraX software (version 1)32.
Figure 2.

(a) Zero-point corrected relative energies of 11 BN-doped azulene isomers (kcal/mol) at CBS-APNO, CBS-QB3, and G3MP2 methods. (b) Zero-point corrected relative energies of 6 BN-doped naphthalene isomers (kcal/mol) at CBS-APNO, CBS-QB3, and G3MP2 methods.
This may be attributed to the orbital amplitude being mostly concentrated at positions 1 and 3 of the azulene, favoring atoms with higher electronegativity at these positions. However, positions 2, 4, 6, and 8 have smaller orbital amplitudes and represent positions that would prefer nucleophilic substitution. Therefore, isomers with N substitutions at positions 1 or 3 are more stable than isomers with N at positions 2, 6, and 8. On the contrary, isostere with B at positions 2, 6, and 8 are more stable than isomers with B at positions 1 and 7. As shown in Table 1, the calculated formation enthalpies using G3MP2 and CBS-QB3 are close to those of the more expensive CBS-APNO method. Therefore, the discussion will be limited to the former two methods.
Frontier molecular orbital (FMO) energies provide useful information on electronic, optical, and electrical properties, reactivity, and kinetic stability. Figure 3 shows the HOMO–LUMO energy gap at the G3MP2 level. The corresponding plots of these orbitals are also shown in Fig. 4. The trend with respect to the HOMO energy is as follows: AZ-1B2N (− 4.99 eV) > AZ-8N9B (− 5.18 eV) > AZ-9B10N (− 5.29 eV) > AZ-6N7B (− 5.34 eV) > AZ-1B9N (− 5.38 eV) > AZ-7B8N (− 5.48 eV) > AZ-1N2B (− 5.59 eV) > AZ-6B7N (− 5.60 eV) > AZ-8B9N (− 5.69 eV) > AZ-1N9B (− 5.72 eV) > AZ-7N8B (− 5.73 eV) and the LUMO energy level ranges from − 1.84 to − 2.61 eV.
Figure 3.

Orbital energies of azulene and 11 BN-doped isomers at G3MP2.
Figure 4.
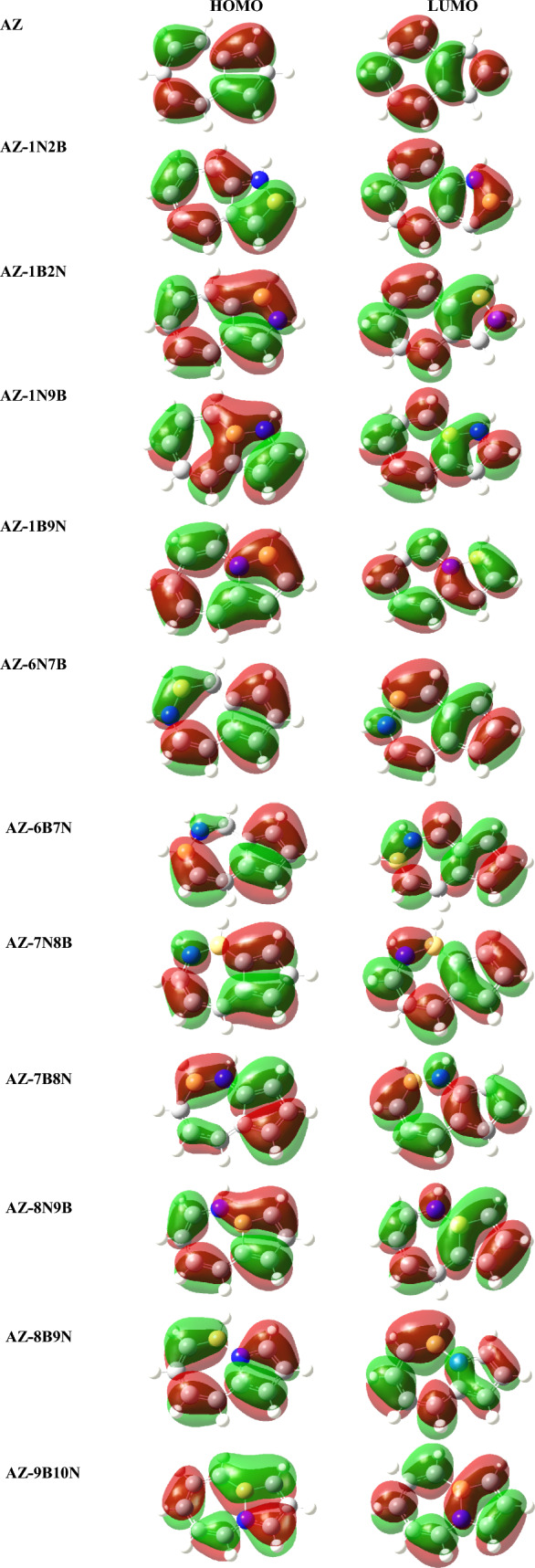
Plots of HOMOs and LUMOs distribution of BN-doped azulene using Gaussview package (version 5)41.
As can be noticed from Fig. 4, HOMO electron density distributions vary among the BN-substituted azulene isomers. The HOMO–LUMO energy gap is another useful parameter characteristic of the electronic and optical properties of molecules46,47 and provides qualitative information about the kinetic stability of the molecules48. Generally, a small energy gap results in a maximum absorption wavelength49. As shown in Fig. 3, BN substitution in azulene induces a change in the band gap. The position of BN in azulene plays a major and effective role in determining the energy gap of the molecule. Nitrogen substitution at positions 2, 6, and 8 and boron substitution at positions 1 and 7 make the molecule thermodynamically less stable and decrease the energy gap. Indeed, a large band gap in a material or molecule is often an indicator of its stability. The largest band gap calculated for the Az-1N9B isomer indicates that the molecule is very stable.
The molecular variations before and after HOMO and LUMO energy levels were displayed using DOS diagrams at the G3MP2 level for all the studied isomers (Fig. S1). The DOS provides information about the available states for the localization of charges in the energy range of − 15 to 5 eV. Figure S1 clearly shows the larger energy gap of the most stable B–N azulene-based isomers than the azulene. The less stable isomers have a smaller energy gap than azulene. Thus, azulene's B–N substitution has a significant impact on the stability.
Ionization energy (IE) and electron affinity (EA) are crucial parameters as they are used to calculate electronegativity, softness, hardness, and electrophilicity indices38,39. The ionization energy and electron affinity of a molecule refer to its ability to lose or accept an electron, and hence are good measures of the stability of the investigated systems toward oxidation and reduction, respectively. According to Koopmans theorem50, the ionization energy and electron affinity are calculated as the negative values of the HOMO and LUMO energies, respectively. In the case of DFT methods, the ionization energy calculated from orbital energies is of low accuracy, with an error of more than 2 eV depending on the used exchange–correlation functionals51,52. The LUMO energies for electron affinities are even worse53. Table 2 lists ionization energies and electron affinities from HOMO and LUMO energies for 11 isomers of BN-doped azulene, as well as the energies from the adiabatic approach which takes into account the geometrical rearrangement of the molecular cation and anion after electron loss and acceptance, respectively. The vertical ionization energy (VIE) can be obtained by the energy difference between a neutral molecule and its cation with the molecular cation at the same geometry as the neutral molecule. Electron affinity is similar to ionization energy, but adiabatic electron affinity (AEA) and vertical electron affinity (VEA) can be obtained from molecular anions. The higher value of AIE indicates the higher stability of the molecule against oxidation. The electron affinities calculated from Koopmans theory for azulene and its isosteric isomers were all negative. However, positive electron affinities have been reported for azulene in experiments. Therefore, the AEAs for azulene and all BN-doped structures giving positive values were considered. The agreement between the experimental and calculated adiabatic electron affinity for the parent azulene is very good. It is obvious that the most stable isostere, AZ-1N9B, has the greatest AIE (7.85 eV), which decreases to 7.25 eV in the least stable isostere (AZ-9B10N). On the other hand, the lowest AEA value of AZ-1N9B is 0.50 eV, which increases to 0.86 eV in the AZ-9B10N. That is proven by the hardness, the least reactivity, and the stability of AZ-1N9B isostere.
Table 2.
Ionization energies, electron affinities (Koopmans IE, EA), adiabatic ionization energies, and electron affinities (AIE, AEA) calculated using G3MP2 method.
| Systems | IE (eV) | AIE (eV) | Exp. IE (eV) | EA (eV) | AEA (eV) | Exp. EA (eV) |
|---|---|---|---|---|---|---|
| AZ | 7.12 | 7.66 | 7.38 ± 0.0354, 7.32 ± 0.0555 | − 1.00 | 0.68 | 0.79 ± 0.00856, 0.69 ± 0.1057, 0.80 ± 0.1058 |
| AZ-1N2B | 7.13 | – | – | − 0.96 | 0.61 | – |
| AZ-1B2N | 6.58 | 7.15 | – | − 1.01 | 0.79 | – |
| AZ-1N9B | 7.36 | 7.85 | – | − 1.02 | 0.50 | – |
| AZ-1B9N | 7.10 | 7.56 | − 0.92 | 0.93 | – | |
| AZ-6N7B | 7.10 | 7.39 | – | − 0.88 | 0.89 | – |
| AZ-6B7N | 7.36 | 7.60 | – | − 0.87 | 0.46 | – |
| AZ-7N8B | 7.56 | 7.79 | – | − 0.87 | 0.79 | – |
| AZ-7B8N | 7.43 | 7.44 | – | − 0.57 | 1.18 | – |
| AZ-8N9B | 6.85 | 7.35 | – | − 0.95 | 0.59 | – |
| AZ-8B9N | 7.35 | 7.77 | – | − 1.01 | 0.63 | – |
| AZ-9B10N | 6.51 | 7.25 | – | − 0.57 | 0.86 | – |
The reactivity of a given molecule can be monitored by plotting the electrostatic potential (ESP). Figures 5 and 6 display the ESP surfaces of azulene, naphthalene, and their BN-doped isosteres at B3LYP/6–31 + G(d,p). ESP maps depict the charged regions in the molecule with different colors, which reflect values of the electrostatic potential. The red color in ESP maps represents the most negative electrostatic potential, while the blue indicates the most positive electrostatic potential regions. The azulene compound has a negative charge on the five-membered ring and a positive charge on the seven-membered ring (see Fig. 5). ESP of the most stable structure, AZ-1N9B, shows the localization of a significant negative charge on the seven-membered ring and the appearance of the blue color around the H atom bonded to the nitrogen. The higher electron-donating ability of the heptagonal ring, as opposed to the electron-accepting ability of 1N-H, might provide evidence of the stability of the AZ-1N9B structure. In AZ-1B9N, AZ-8B9N, and AZ-9B10N, one fused carbon is substituted with a nitrogen atom, resulting in the dispersion of the positive charge around the structure and consequently the negative charge as well. This charge dispersion explains the lower stability of AZ-1B9N, AZ-8B9N, and AZ-9B10N.
Figure 5.

Molecular ESP surfaces of azulene and its BN-doped isosteres at B3LYP/6–31 + G(d,p) level using Gaussview package (version 5)41.
Figure 6.

Molecular ESP surfaces of naphthalene and its BN-doped isosteres at B3LYP/6–31 + G(d,p) level using Gaussview package (version 5)41.
Before the absorption wavelengths calculation for the investigated 11 isomers BN-doped azulene in the gas phase and acetone, we have carried out benchmark calculations to assess the impact of the basis set, functionals, and dispersion corrections. We have examined B3LYP35, CAM-B3LYP40, wB97XD59, HSEH1PBE (HSE06)60, PBE0DH60, MN1561, MN12-SX62, and B97D63 functionals. These are the recently developed and most accurate functionals reported to date. The dispersion corrections GD3 of Grimme with the original D3 damping parameter64, and GD3BJ with the Becke–Johnson damping D3 parameter65. We also tested several basis sets; 6−311G(d,p), 6–311 + G(d,p), cc-pVTZ, cc-pVQZ, and Def2TZVPP. The results of these benchmark calculations are shown in Fig. S2, while the optimized structures are collected in Table S3 in SI file. We did not observe any impact of using different basis sets, functionals, or dispersion corrections on the optimized geometries of the most stable isomer AZ-1N9B. Therefore, we limit the optimization level to B3LYP/6–31 + G(d,p), and the λmax values were calculated at TD-B3LYP/6–31 + G(d,p) and summarized in Tables 3 and 4, respectively. According to Tawada et al.66, long-range corrected DFT should be used for computing the charge transfer excited states in TD-DFT. Therefore, Table S4 lists the λmax values, and Fig. S3 depicts the absorption spectra of the different isomers calculated using TD-CAM-B3LYP/6–31 + G(d,p) level. Table S5 reported the calculated and previously studied transition wavelengths for S0 → S1 and S0 → S2 electronic transitions for azulene. Our calculated S0 → S1 and S0 → S2 electronic transitions for azulene at CAM-B3LYP are in good agreement with those previously calculated at TD-PBE0/6–31G(d)67. For S0 → S1 and S0 → S2, the computed wavelength in acetone with TD-B3LYP are longer by 9 and 16 nm with respect to the TD-CAM-B3LYP estimates, however, TD-B3LYP shows great accordance with the S0 → S2 experimental result68 (within 2 nm eV). In the case of S0 → S1, both TD-B3LYP and TD-CAM-B3LYP have close wavelengths being both underestimated by up to 60 nm with respect to the experimental values. Therefore, the TD-B3LYP method will be used through the discussion of the UV absorption spectra of the studied isomers.
Table 3.
Computed absorption spectra (λmax, nm/eV) of azulene and BN-doped azulene at TD-DFT(PCM)/B3LYP/6–31 + G(d,p) in acetone.
| Compound | Absorption energy | F | Transitions | |
|---|---|---|---|---|
| (nm) | (eV) | |||
| AZ | 264 | 4.70 | 1.2649 | H-1 → L (55%), H → L + 1 (44%) |
| AZ-1N2B | 249 | 4.99 | 0.7499 | H-1 → L (12%), H-1 → L + 1 (70%) |
| AZ-1B2N | 250 | 4.96 | 0.3154 | H-2 → L (10%), H-1 → L (38%), H → L + 5 (39%) |
| AZ-1N9B | 213 | 5.82 | 0.5386 | H-2 → L (73%), H-1 → L + 1 (15%) |
| AZ-1B9N | 278 | 4.46 | 0.8577 | H-1 → L (52%), H → L + 1 (39%) |
| AZ-6N7B | 269 | 4.61 | 0.6006 | H-2 → L (29%), H-1 → L (34%), H → L + 1 (30%) |
| AZ-6B7N | 263 | 4.72 | 1.0129 | H-1 → L (66%), H → L + 1 (20%) |
| AZ-7N8B | 252 | 4.93 | 0.6146 | H-1 → L + 1 (80%) |
| AZ-7B8N | 284 | 4.37 | 0.8134 | H-1 → L (31%), H → L + 1 (65%) |
| AZ-8N9B | 250 | 4.96 | 0.4542 | H-1 → L + 1 (89%) |
| AZ-8B9N | 259 | 4.79 | 0.5616 |
H-2 → L (10%), H-1 → L (53%), H → L + 1 (33%) |
| AZ-9B10N | 287 | 4.32 | 0.5006 | H-1 → L (58%), H → L + 1 (31%) |
Table 4.
Absorption spectra (λmax, nm/eV) of azulene and BN-doped azulene calculated at TD-DFT/B3LYP/6–31 + G(d,p) in the gas phase.
| Compound | Absorption energy | F | Transitions | |
|---|---|---|---|---|
| (nm) | (eV) | |||
| AZ | 252 | 4.92 | 1.0782 | H-1 → L (53%), H → L + 1 (45%) |
| AZ-1N2B | 242 | 5.13 | 0.7340 | H-1 → L (22%), H-1 → L + 1 (53%), H → L + 1 (11%) |
| AZ-1B2N | 235 | 5.27 | 0.3766 | H-2 → L (32%), H-1 → L (22%), H-1 → L + 1 (32%) |
| AZ-1N9B | 247 | 5.02 | 0.3534 | H-2 → L (22%), H-1 → L + 1 (64%) |
| AZ-1B9N | 268 | 4.63 | 0.6860 | H-1 → L (47%), H → L + 1 (41%) |
| AZ-6N7B | 241 | 5.14 | 0.5392 | H-2 → L (41%), H-1 → L (26%), H → L + 1 (17%) |
| AZ-6B7N | 253 | 4.89 | 0.8507 | H-1 → L (62%), H → L + 1 (23%) |
| AZ-7N8B | 248 | 5.01 | 0.5734 | H-1 → L + 1 (65%), H → L + 1 (18%) |
| AZ-7B8N | 275 | 4.51 | 0.6441 | H-1 → L (33%), H → L + 1 (59%) |
| AZ-8N9B | 246 | 5.04 | 0.3888 | H-1 → L + 1 (86%) |
| AZ-8B9N | 254 | 4.88 | 0.4067 | H-2 → L (16%), H-1 → L (44%), H → L + 1 (34%) |
| AZ-9B10N | 281 | 4.41 | 0.3568 | H-1 → L (53%), H → L + 1 (30%) |
Results of the absorption wavelengths reported in Tables 3 and 4, reveal a red shift of about 4–15 nm in all calculated wavelengths except for the AZ-1N9B isomer, in which we observed a blue shift. This is expected as the molecule exhibits the highest stability and the highest band gap. The UV–Vis absorption spectra are shown in Fig. 7 for S0–S1 transition calculated at TD-B3LYP and TD-CAM-B3LYP levels, respectively. As shown in Fig. 7, the electronic transition can be referred to as that of π → π* and n → π* transitions. The most intense absorption peaks of azulene are in a lower wavelength region, mostly below 300 nm. All BN-doped azulene isomers show an extension of the absorption regions above 300 nm, to be in the UV and visible regions of the electromagnetic spectrum. The AZ-1N9B isomer gives the shortest λmax but the highest extension at the visible region by having a peak centered at 578 nm. AZ-8N9B, AZ-9B10N, and AZ-6N7B exhibit intense peaks centered at 495, 479, and 560 nm, respectively.
Figure 7.

UV–Vis spectra for azulene and BN-doped azulene at TD-DFT/6–31 + G(d,p)(PCM)//B3LYP/6−31G(d,p) in acetone.
Conclusion
The thermodynamic stabilities and opt-electronic properties of new series of BN-doped azulene molecules are investigated. To do so, we have calculated formation enthalpies of 11 BN-substituted azulenes at three computational levels. Based on the available experimental data, the formation enthalpy obtained from G3MP2 and CBS-QB3 is more accurate than those estimated from the CBS-APNO model chemistry. Our calculations reveal that the most stable is AZ-1N9B isomer with the largest HOMO–LUMO gap and lowest formation enthalpy among all other isomers. Ionization energies and electron affinities were estimated by the vertical approach in the G3MP2 model. AZ-7N8B, AZ-7B8N, AZ-6B7N, and AZ-1N9B isomers were found to give higher ionization energies. Moreover, the electronic absorption spectra of these isomers have been determined in the gas phase and in acetone. The absorption wavelength in acetone displays a red shift compared with the gas phase except for the AZ-1N9B isomer which exhibits a blue shift. The BN vicinal doping of azulene populates the visible region with absorption peaks compared to the parent azulene, which increases the light-harvesting capacity of the doped molecules.
Supplementary Information
Author contributions
M.A.A.-R. Performed the calculations, collected the data, wrote the first draft of the manuscript, edit & review, and answered the reviewers' comments. K.A.S. performed the calculations, collected the data and wrote the first draft of the manuscript. S.A. performed the benchmark calculations, edit & review the paper and answered the reviewer's comments. A.M.E.-N. Supervision, while T.T. and T.N. edit & review. A.B.E.-M. Performed the calculations, collected the data, wrote the first draft of the manuscript, edit & review, and answered the reviewer's comments.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Data availability
All data generated through this study are included in this manuscript and the Supporting Information file.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mohamed A. Abdel-Rahman, Email: Mohamed.Abdel-Rahman@sci.suezuni.edu.eg, Email: mohammadadel2015@yahoo.com
Kamal A. Soliman, Email: kamal.soliman@fsc.bu.edu.eg
Asmaa B. El-Meligy, Email: asmaaphys@yahoo.com
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-37047-7.
References
- 1.Bosdet MJD, Piers WE. BN as a CC substitute in aromatic systems. Can. J. Chem. 2009;87:8–29. doi: 10.1139/v08-110. [DOI] [Google Scholar]
- 2.Wang X-Y, Wang J-Y, Pei J. BN heterosuperbenzenes: Synthesis and properties. Chem. Eur. J. 2015;21:3528–3539. doi: 10.1002/chem.201405627. [DOI] [PubMed] [Google Scholar]
- 3.Muz I, Atiş M. Structural transformations in the carborane series: CnB6−nH6 (n = 0–6) upon substitution of boron by carbon. Inorg. Chim. Acta. 2016;453:626–632. doi: 10.1016/j.ica.2016.09.035. [DOI] [Google Scholar]
- 4.Muz I, Kurban M. Ab initio study of structural and electronic properties of SinC5-nH8 (n = 0–5) series: Probing the 2D to 3D structural transition. Inorg. Chim. Acta. 2018;477:318–325. doi: 10.1016/j.ica.2018.03.008. [DOI] [Google Scholar]
- 5.Muz I. Structural transition of C6−nSinNH7 at n = 0–6 clusters upon substitution of carbon by silicon. Inorg. Chim. Acta. 2019;495:118950. doi: 10.1016/j.ica.2019.06.001. [DOI] [Google Scholar]
- 6.Muz I. Structural and electronic properties of B6-nCnHn (n = 0–6) series upon the substitution of boron atoms by the CH groups: A density functional theory study. Selcuk Univ. J. Eng. Sci. Technol. 2019;7(2):467–477. [Google Scholar]
- 7.Muz I, Göktaş F, Kurban M. A density functional theory study on favipiravir drug interaction with BN-doped C60 heterofullerene. Physica E. 2022;135:114950. doi: 10.1016/j.physe.2021.114950. [DOI] [Google Scholar]
- 8.Muz I, Kurban M. The electronic structure, transport and structural properties of nitrogen-decorated graphdiyne nanomaterials. J. Alloys Compd. 2020;842:155983. doi: 10.1016/j.jallcom.2020.155983. [DOI] [Google Scholar]
- 9.Muz I, Göktaş F, Kurban M. Size dependence in the electronic and optical properties of a BN analogue of two-dimensional graphdiyne: A theoretical study. Chem. Phys. 2020;539:110929. doi: 10.1016/j.chemphys.2020.110929. [DOI] [Google Scholar]
- 10.Muz I, Kurban M. Electronic structures and bonding of graphdiyne and its BN analogs: Transition from quasi-planar to planar sheets. J. Alloys Compd. 2020;846:155987. doi: 10.1016/j.jallcom.2020.155987. [DOI] [Google Scholar]
- 11.Dewar MJS, Kubba VP, Pettit R. Part I. 9-Aza-10-boraphenanthrene, new heteroaromatic compounds. J. Chem. Soc. 1958;1:3073–3076. doi: 10.1039/jr9580003073. [DOI] [Google Scholar]
- 12.Culling GC, Dewar MJS, Marr PA. New heteroaromatic compounds. XXIII. Two analogs of triphenylene and a possible route to borazarene. J. Am. Chem. Soc. 1964;86:1125–1127. doi: 10.1021/ja01060a034. [DOI] [Google Scholar]
- 13.Dewar MJS, Dietz R. Part III. 2, 1-Borazaro-naphthalene (1, 2-dihydro-1-aza-2-boranaphthalene), new heteroaromatic compounds. J. Chem. Soc. 1959;546:65–67. [Google Scholar]
- 14.Liu Z, et al. The least stable isomer of BN naphthalene: Toward predictive trends for the optoelectronic properties of BN acenes. J. Am. Chem. Soc. 2017;139:6082–6085. doi: 10.1021/jacs.7b02661. [DOI] [PubMed] [Google Scholar]
- 15.Morgan MM, Piers WE. Efficient synthetic methods for the installation of boron–nitrogen bonds in conjugated organic molecules. Dalton. Trans. 2016;45:5920–5924. doi: 10.1039/C5DT03991F. [DOI] [PubMed] [Google Scholar]
- 16.Anderson AG, Steckler BM. VIII a study of the visible absorption spectra and dipole moments of some 1- and 1,3-substituted azulenes. J. Am. Chem. Soc. 1959;81:4941–4946. doi: 10.1021/ja01527a046. [DOI] [Google Scholar]
- 17.Lemal DM, Goldman GD. Synthesis of azulene, a blue hydrocarbon. J. Chem. Educ. 1988;65:923–925. doi: 10.1021/ed065p923. [DOI] [Google Scholar]
- 18.Salman H, et al. 1,3-Di(2-pyrrolyl)azulene: An efficient luminescent probe for fluoride. Eur. J. Org. Chem. 2005;11:2207–2212. doi: 10.1002/ejoc.200500012. [DOI] [Google Scholar]
- 19.Murai M, et al. Modulating structure and properties in organic chromophores: influence of azulene as a building block. Chem. Sci. 2014;5:3753–3760. doi: 10.1039/C4SC01623H. [DOI] [Google Scholar]
- 20.Shoji T, et al. Synthesis and properties of azulene-substituted donor–acceptor chromophores connected by arylamine cores. Eur. J. Org. Chem. 2013;34:7785–7799. doi: 10.1002/ejoc.201301006. [DOI] [Google Scholar]
- 21.Scmitt S, Baumgarten M, Simon J, Hafner K. 2, 4, 6, 8-Tetracyanoazulene: A new building block for “organic metals”. Angew. Chem. Int. Ed. 1998;37:1077–1081. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1077::AID-ANIE1077>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 22.Ito S, et al. Synthesis of poly (6-azulenylethynyl) benzene derivatives as a multielectron redox system with liquid crystalline behavior. J. Am. Chem. Soc. 2003;125:1669–1680. doi: 10.1021/ja0209262. [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Lai Y, Han M. Stimuli-responsive conjugated copolymers having electro-active azulene and bithiophene units in the polymer skeleton: Effect of protonation and p-doping on conducting properties. Macromolecules. 2004;37:3222–3230. doi: 10.1021/ma035335q. [DOI] [Google Scholar]
- 24.Kurotobi K, Kim KS, Noh SB, Kim D, Osuka A. A quadruply azulene-fused porphyrin with intense near-IR absorption and a large two photon absorption cross section. Angew. Chem. Int. Ed. 2006;45:3944–3947. doi: 10.1002/anie.200600892. [DOI] [PubMed] [Google Scholar]
- 25.Perdew JP, et al. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. J. Chem. Phys. 2005;123:062201–62209. doi: 10.1063/1.1904565. [DOI] [PubMed] [Google Scholar]
- 26.Frisch MJ, et al. Gaussian 09. Gaussian Inc; 2009. [Google Scholar]
- 27.Curtiss LA, Raghavachari K, Trucks GW, Pople JA. Gaussian-2 theory for molecular energies of first- and second-row compounds. J. Chem. Phys. 1991;94:7221–7230. doi: 10.1063/1.460205. [DOI] [Google Scholar]
- 28.Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA. Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys. 1998;109:7764–7776. doi: 10.1063/1.477422. [DOI] [Google Scholar]
- 29.Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999;110:2822–2827. doi: 10.1063/1.477924. [DOI] [Google Scholar]
- 30.Ochterski JW, Petersson GA, Montgomery JA. A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 1996;104:2598–2619. doi: 10.1063/1.470985. [DOI] [Google Scholar]
- 31.Petersson GA, et al. Calibration and comparison of the Gaussian-2, complete basis set, and density functional methods for computational thermochemistry. J. Chem. Phys. 1998;109:10570–10579. doi: 10.1063/1.477794. [DOI] [Google Scholar]
- 32.Pettersen EF, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Boyle NM, Tenderholt AL, Langner KM. Cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008;29:839–845. doi: 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- 34.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 35.Lee C, Yang W, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 36.Ochterski JW. Thermochemistry in Gaussian. Gaussian Inc; 2000. p. 264. [Google Scholar]
- 37.Afeefy HY, Liebman JF, Stein SE. Neutral thermochemical data. In: Linstrom PJ, Mallard WG, editors. NIST Chemistry WebBook, NIST standard reference database number 69. National Institute of Standards and Technology; 2005. [Google Scholar]
- 38.Rayne S, Forest K. Estimated adiabatic ionization energies for organic compounds using the Gaussian-4 (G4) and W1BD theoretical methods. J. Chem. Eng. Data. 2011;56:350–355. doi: 10.1021/je100913f. [DOI] [Google Scholar]
- 39.Lewars EG. Computational Chemistry; Introduction to the Theory and Applications of Molecular and Quantum Mechanics. 3. Springer; 2011. [Google Scholar]
- 40.Yanai T, Tew PD, Handy NC. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem. Phys. Lett. 2004;393:51–57. doi: 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- 41.Dennington, R., Keith, T. & Millam, J. GaussView, Version 5 (Semichem Inc., 2009). https://gaussian.com/gaussview6/.
- 42.Sousa CCS, Matos MAR, Morais VMF. Energetics and stability of azulene: From experimental thermochemistry to high-level quantum chemical calculations. J. Chem. Thermodyn. 2014;73:101–109. doi: 10.1016/j.jct.2013.11.008. [DOI] [Google Scholar]
- 43.Roux MV, Temprado M, Chickos JS, Nagano Y. Critically evaluated thermochemical properties of polycyclic aromatic hydrocarbons. J. Phys. Chem. Ref. Data. 2008;37(4):1855–1996. doi: 10.1063/1.2955570. [DOI] [Google Scholar]
- 44.Roth WR, Bohm D, Lennartz HW, Vogel E. Resonance energy of bridged [10] annulenes. Angew Chem. Int. Ed. Engl. 1983;22:1007. doi: 10.1002/anie.198310071. [DOI] [Google Scholar]
- 45.Kovats E, Gunthard H, Plattner A. Katalytische dehydrierungen mit schwefelkohlenstoff. I. Dehydrierung von oktahydro-azulen. Helv. Chim. Acta. 1954;7:2123–2133. doi: 10.1002/hlca.19540370724. [DOI] [Google Scholar]
- 46.Pearson RG. Chemical Hardness. Springer; 1997. pp. 1–10. [Google Scholar]
- 47.Jagadeesan R, Velmurugan G, Venuvanalingam P. The nature of Pd–carbene and Pd–halogen bonds in (bisNHC)PdX2 type catalysts: Insights from density functional theory. RSC Adv. 2015;5:80661–80667. doi: 10.1039/C5RA16500H. [DOI] [Google Scholar]
- 48.Nagarajan N, et al. Highly emissive luminogens based on imidazo[1,2-a]pyridine for electroluminescent applications. Chem. Asian J. 2014;9:294–304. doi: 10.1002/asia.201301061. [DOI] [PubMed] [Google Scholar]
- 49.Solomon RV, Bella AP, Vedha SA, Venuvanalingam P. Designing benzosiloles for better optoelectronic properties using DFT & TDDFT approaches. Phys. Chem. Chem. Phys. 2012;14:14229–14237. doi: 10.1039/c2cp41554b. [DOI] [PubMed] [Google Scholar]
- 50.Koopmans T. Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica. 1934;1:104–113. doi: 10.1016/S0031-8914(34)90011-2. [DOI] [Google Scholar]
- 51.Politzer P, Abu-Awwad F. A comparative analysis of Hartree–Fock and Kohn–Sham orbital energies. Theor. Chem. Acta. 1998;99:83–87. doi: 10.1007/s002140050307. [DOI] [Google Scholar]
- 52.Hamel S, Duffy P, Casida ME, Salahub DR. Kohn–Sham orbitals and orbital energies: Fictitious constructs but good approximations all the same. J. Electron. Spectros. Relat. Phenomena. 2002;123:345–363. doi: 10.1016/S0368-2048(02)00032-4. [DOI] [Google Scholar]
- 53.Zhang G, Musgrave CB. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A. 2007;111:1554–1561. doi: 10.1021/jp061633o. [DOI] [PubMed] [Google Scholar]
- 54.Jochims H-W, Rasekh H, Ruhl E, Baumgartel H, Leach S. The photofragmentation of naphthalene and azulene monocations in the energy range 7–22 eV. Chem. Phys. 1992;168:159–184. doi: 10.1016/0301-0104(92)80118-F. [DOI] [Google Scholar]
- 55.Meot-Ner M, Nelsen SF, Willi MR, Frigo TB. Special effects of an unusually large neutral to radical cation geometry change: Adiabatic ionization energies and proton affinities of alkylhydrazines. J. Am. Chem. Soc. 1984;106:7384–7389. doi: 10.1021/ja00336a015. [DOI] [Google Scholar]
- 56.Schiedt J, Knott WJ, Le Barbu K, Schlag EW, Weinkauf R. Microsolvation of similar-sized aromatic molecules: Photoelectron spectroscopy of bithiophene-, azulene-, and naphthalene-water anion clusters. J. Chem. Phys. 2000;113:9470–9478. doi: 10.1063/1.1319874. [DOI] [Google Scholar]
- 57.Chowdhury S, Heinis T, Grimsrud EP, Kebarle P. Entropy changes and electron affinities from gas-phase electron-transfer equilibria: A-+ B = A+ B. J. Phys. Chem. 1986;90(12):2747–2751. doi: 10.1021/j100403a037. [DOI] [Google Scholar]
- 58.Ando N, Mitsui M, Nakajima A. Photoelectron spectroscopy of cluster anions of naphthalene and related aromatic hydrocarbons. J. Chem. Phys. 2008;128:154318. doi: 10.1063/1.2903473. [DOI] [PubMed] [Google Scholar]
- 59.Chai J-D, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections Phys. Chem. Chem. Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 60.Heyd J, Scuseria GE, Ernzerhof M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003;118:8207–8215. doi: 10.1063/1.1564060. [DOI] [Google Scholar]
- 61.Haoyu SY, He X, Li SL, Truhlar DG. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016;7:5032–5051. doi: 10.1039/C6SC00705H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peverati R, Truhlar DG. Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012;14:16187–16191. doi: 10.1039/c2cp42576a. [DOI] [PubMed] [Google Scholar]
- 63.Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 64.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 65.Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 66.Tawada Y, Tsuneda T, Yanagisawa S, Yanai T, Hirao K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004;120:8425–8433. doi: 10.1063/1.1688752. [DOI] [PubMed] [Google Scholar]
- 67.Veys K, Escudero D. Computational protocol to predict anti-kasha emissions: The case of azulene derivatives. J. Phys. Chem. A. 2020;124(36):7228–7237. doi: 10.1021/acs.jpca.0c05205. [DOI] [PubMed] [Google Scholar]
- 68.Shevyakov SV, et al. Orbital control of the color and excited state properties of formylated and fluorinated derivatives of azulene. J. Phys. Chem. A. 2003;107:3295–3299. doi: 10.1021/jp021605f. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated through this study are included in this manuscript and the Supporting Information file.