

Abstract
Molecular modeling and simulation play important roles in biomedical research as they provide molecular-level insight into the underlying mechanisms of biological functions that are difficult to elucidate only with experiments. CHARMM-GUI (https://charmm-gui.org) is a web-based cyberinfrastructure that is widely used to generate various molecular simulation systems and input files and thus facilitates and standardizes the usage of common and advanced simulation techniques. In particular, PDB Manipulator provides various chemical modification options as the starting point for most input generation modules in CHARMM-GUI. Here, we discuss recent additions to PDB Manipulator, such as non-standard amino acids / RNA substitutions, ubiquitylation and SUMOylation, Lys / Arg post-translational modifications, lipidation, peptide stapling, and improved parameterization options of small molecules. These additional features are expected to make complex PDB modifications easy for biomolecular modeling and simulation.
Keywords: All-atom Modeling, Molecular Dynamics Simulation, Nonstandard Amino Acid/RNA, Post Translational Modifications, Ubiquitylation/SUMOylation
Graphical Abstract
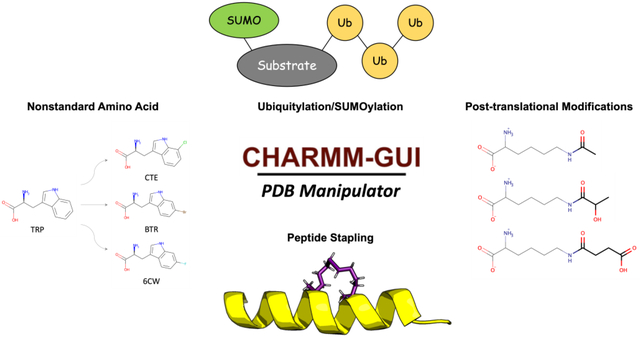
Introduction
Biomolecular modeling and simulation have been widely applied to explore biological systems and provide insight into their functional mechanism (Schlick et al., 2021). In particular, all-atom molecular dynamics (MD) simulation has been extensively used to study biomolecular structure and dynamics, their interactions with ligands, and drug discovery/design together with various free energy calculation methods (Nerenberg & Head-Gordon, 2018). The accuracy of such all-atom simulations is greatly dependent on the molecular mechanic force field (FF) that describes the energy and forces acting on each atom in the system (Croitoru et al., 2021). There are popular and well-optimized FFs such as the AMBER, CHARMM, OPLS, and GROMOS FFs for proteins, nucleic acids, lipids, carbohydrates, and small molecules. However, applying these FFs to and setting up complex biological systems for MD simulation often require a complicated set of submodules and scripts, ranging from construction of an initial structure to system equilibration and production (Lee et al., 2020).
CHARMM-GUI (https://charmm-gui.org) (Jo et al., 2008) is a web-based cyberinfrastructure that aims to standardize and simplify the processes to prepare MD system and input files for popular simulation software like CHARMM (Brooks et al., 2009), NAMD (Phillips et al., 2005), GROMACS (Jung et al., 2015), AMBER (Case et al., 2005), GENESIS (Jung et al., 2015), TINKER (Rackers et al., 2018), LAMMPS (Plimpton S, 1995), Desmond (Bowers et al., 2006), and OpenMM (Eastman et al., 2017). Since 2006, CHARMM-GUI has been developed to support all-atom modeling and simulation of complex biomolecular systems (Lee et al., 2016) with polymer systems (Choi et al., 2021), nanomaterial structures (Choi et al., 2022), ligands (Kim et al., 2017), and glycans (Jo et al., 2011; Park et al., 2017, 2019) in solution and membrane environments (Jo et al., 2007, 2009; Lee et al., 2019; Wu et al., 2014). Especially, PDB Reader & Manipulator is the starting point for the most input generation modules in CHARMM-GUI and allows users to seamlessly manipulate biomolecules for mutation, protonation, phosphorylation, ligand parameterization, etc.
Modeling and simulation of protein structures that contain nonstandard amino acids or post-translational modifications (PTMs) have received less attention (Khoury et al., 2013). For example, although ubiquitylation is one of the most abundant PTMs in nature, to the best of our knowledge, there is no available computational tool to easily set up ubiquitylation in MD simulation. In addition, covalent connection of acyl tails to protein, known as lipidation, is an essential class of PTMs, but it is a nontrivial process to include lipidation in MD simulation. In this work, we describe the most recent developments in PDB Manipulator to cover various structural modifications such as 1) nonstandard amino acids and RNA substitution, 2) ubiquitylation and SUMOylation, 3) Lys and Arg PTMs, 4) lipidation, 5) peptide stapling, and 6) improved parameterization of small molecules. In addition, we also introduce an automatic correction of user-made PDB structure formats.
Nonstandard Amino Acids / RNA Substitution
Nonstandard amino acids are common in nature and essential to many cellular activities. Nonstandard amino acids can be artificially introduced to specific sites to produce proteins with new functionalities or increased catalytic activity and to explore protein structure, dynamics, and function (W. Gao et al., 2019). Like nonstandard amino acids, post-transcriptionally modified nucleotides are common in RNA. More than 90% of the so-far-reported 112 naturally occurring nucleotide modifications are found in tRNA with significant distinctions between archaea, prokaryotes, and eukaryotic organisms, and roughly 25% of the nucleotides in eukaryotic tRNAs are modified (Carell et al., 2012).
To support all-atom molecular modeling and simulation of post-translational modifications in amino acids and RNAs, a number of FFs have been developed. In particular, the CHARMM36 additive FF has been expanded to nonstandard amino acid (Croitoru et al., 2021) and nonstandard RNA (Xu et al., 2016), with their parameterization focusing on the rotatable torsion angles. Even with these FFs, it is still nontrivial to manually model these nonstandard amino acids / RNA in RCSB PDB. To resolve this problem, a new feature has been added to PDB Manipulator to automatically detect and model a total 354 nonstandard amino acids and 49 nonstandard nucleic acids that are parameterized in the CHARMM FF (Fig 1B). For example, Table S1 shows the supported 39 out of the top 50 most prevalent nonstandard amino acids in RCSB PDB (see Table S2 for all 354 supported non-standard amino acids). In addition, with the “Nonstandard amino acid / RNA substitution” option on the PDB manipulation page, one can specify a standard residue by choosing “SEGID”, ‘RESID”, and residue name (Fig. 1A) and substitute it with an available nonstandard residue from the dropdown menu. To facilitate structure visualization of available nonstandard residues, a “select image” option is provided to display a new popup window in which one can select a nonstandard residue by clicking its chemical structure image (Fig. 1C,1D). This popup window supports two filter options in terms of 1) parent normal amino acid / RNA and 2) SMILES strings used for the substructure search or the fingerprint similarity search (Landrum G, 2022) based on a given Tanimoto coefficient cut-off value. The selected nonstandard residues are then modeled with their internal (optimized) coordinate information (Fig. 1E).
Figure 1. Web implementation of nonstandard amino acid / RNA substitution.

(A) Snapshot of nonstandard amino acid / RNA substitution option in PDB Manipulator. (B) Automatic detection of nonstandard amino acid / RNA in a PDB file (C) All available nonstandard amino acids are displayed on the popup window. Filtering options are provided for given parent amino acids. Also, a substructure search or fingerprint similarity search with a Tanimoto coefficient is possible. (D) Nonstandard nucleic acid substitution option. (E) An example (PDB ID 6KUG) with several nonstandard amino acid substitutions.
Ubiquitylation and SUMOylation
PTMs are one of the essential regulation mechanisms of proteins with various biological functions. Ubiquitin is a small heat-stable ubiquitous protein composed of 76 residues with a mixed α/β secondary structure. Ubiquitination is a well-studied PTM that governs numerous biological processes, such as immunological responses, apoptosis, and cancer (Jafari et al., 2018). SUMOylation is a PTM in which a member of the small ubiquitin-like modifier (SUMO) family is attached to Lys residues of target proteins. There is evidence that SUMOylation regulates subcellular protein localization, protein–DNA binding, protein–protein interactions, transcriptional control, DNA repair, and genome organization. Although SUMO and ubiquitin have almost same size, their biological functions are quite different. For example, poly-ubiquitylation can cause degradation, whereas SUMO cannot.
Despite their biological importance, to the best of our knowledge, there is no all-atom modeling tool for in silico ubiquitylation and SUMOylation. Therefore, a new functionality called “Ubiquitylation/SUMOylation” has been added to PDB Manipulator to support in silico ubiquitylation and SUMOylation, i.e., attaching a predefined ubiquitin or SUMO (Fig. 2A) chain to a target protein chain. In nature, mono-ubiquitination has occurred via the isopeptide bond between ubiquitin’s C-terminal carboxylate group and Lys side chain on the substrate protein. In the case of poly-ubiquitylation, the secondary ubiquitin chain is connected to any of the seven Lys residues or the N-terminus of the previously added ubiquitin chain. SUMOylation is also attached in the same manner as ubiquitylation. When ubiquitylation and SUMOylation are detected from the LINK header information in a PDB entry or an uploaded PDB file, the ubiquitin and SUMO chains are displaced under “Ubiquitylation/SUMOylation” (Fig. 2B). Our implementation could detect all RCSB PDB entries that contain covalent linkages between ubiquitin oligomers (Table S3).
Figure 2. Structural modeling of ubiquitylation/SUMOylation.

(A) The predefined ubiquitin (PDB ID 1UBQ) and SUMO (PDB ID 1A5R) models are attached to the target lysine residues. (B) Existing covalent linkages between protein-ubiquitin and between ubiquitin chains are automatically detected (top, PDB ID 6OQ1). Users can add ubiquitin chains via the user interface window (bottom). (C) Output models of ubiquitin chains (yellow) that are attached to substate chain (blue). Rigid body torsional searches for linkages between ubiquitin chains are conducted to avoid any bad contact.
By clicking the “Add Ubiquitylation” button, one can add a new ubiquitin chain to a specific protein chain (Fig. 2B top) regardless of a degree of ubiquitylation; one can do the same for SUMOylation, although not shown in Fig. 2B. The “edit” button allows users to open a new popup window on which the “+” button can be used to add any further ubiquitylation at any specific Lys residue (Fig. 2B bottom). In order to build branched ubiquitin chains, “+” button should be pressed at the branching point. As users modify the ubiquitin link sequence, the sequence diagram is updated accordingly to provide a visual guidance to the user. Finally, user-defined poly-ubiquitin chains are built through conformational sampling using rigid body rotations of ubiquitin conjugations until there is no steric clash between ubiquitin chains (Fig. 2C).
Lys and Arg PTMs
Protein PTMs alter the physicochemical properties of the amino acid side chains to increase their functional diversity. Among the 20 standard amino acids, Lys is the most post-translationally modified residue that plays important regulatory roles in gene expression, protein–protein interactions, protein processing and degradation (Zencheck et al., 2012). Therefore, a new feature called “Lys / Arg PTMs” has been added to PDB Manipulator to support the Lys and Arg PTMs (Fig. S1A), such as succinylation, lactylation, acetylation, crotonylation, carbamylation, citrullination, and methylation (Fig. S1B).
As a case study to highlight the functionality of these PTMs and to explore the effect of Lys succinylation, acetylation, and lactylation on the peptide, we generated and simulated the MD systems of Tau peptide (171IPAKTPPAPK180). In previous work (Min et al., 2015), in vitro and in vivo studies showed that acetylation of Tau at K174 is the most important pathogenic change that affects Tau homeostasis and toxicity in the early stages of Alzheimer’s disease. As a following computational research, Shah et al. (Shah et al., 2022) showed that Lysine acetylation significantly contributed to the large aggregation of the Tau peptide using MD simulation and Markov state model analysis. In order to validate our parameters for Lys PTMs against their simulation result, we built the peptide models with the above Tau sequence via PEP-FOLD server (Thévenet et al., 2012). The peptide models were placed within a rectangular box of explicit TIP3P water molecules at least 10 Å from the water box’s boundary. 150 mM KCl was added to simulate the bulk ion concentration. Particle-mesh Ewald summation was utilized to calculate the long-range electrostatic interactions. All five replicas of each system’s production runs were executed in the NPT for 1 μs at 303.15 K. During the production runs, hydrogen mass repartitioning was used to boost the simulation timestep to 4 fs (Y. Gao et al., 2021). All simulations were performed by the OpenMM package (Eastman et al., 2017) and the CHARMM36m additive FF (Huang et al., 2016). For the conformational analysis of these peptide models, dihedral angle distributions of peptide backbone and sidechain rotamers over whole trajectories were utilized. As a result, Janin plots on K174 show clear distinctions between all replicas of WT K174 systems and other Lys PTMs systems (Fig. S1C). Both Ramachandran and Janin plots from one replica of the WT and K174su systems are depicted in Fig. S1D. The Ramachandran plot shows the similar distributions of backbone dihedral angles each other, whereas the Janin plot shows different χ2 distributions. Overall, our parameters of Lys PTMs are reasonable in that distinct structural behaviors of Tau peptides with different Lys PTMs are observed along the MD simulations, which is similar to the previous study (Shah et al., 2022).
Lipidation
Lipidation is an important PTM process to covalently link acyl chains to a specific protein residue. Lipidated proteins are directly localized to membranes and serve key functions in membrane signaling and trafficking (Ray et al., 2017). PDB Manipulator supports six lipidation types (Fig. 3A) such as palmitoylation, myristoylation, farnesylation, geranylgeranylation, and triacylation (for bacterial lipoproteins to Cys residues). With “Add Lipid-tail”, one can specify a target residue that is replaced by CYSP (for Cys palmitoylation), GLYM (for Gly myristoylation), LYSM (for Lys myristoylation), CYSF (for Cys farnesylation), CYSG (for Cys geranylgeranylation), and CYSL (for Cys triacylation). The initial lipid tail model is built by their internal coordinate information in the CHARMM FF. To avoid any bad contact, we then use an iterative conformational search that consists of random rotations of the dihedral angles between the target amino acid and lipid tail and short minimization steps. In the case of a membrane protein, the selected lipid tail(s) is placed in the hydrophobic core of a bilayer centered at Z = 0, so it is important to pre-orient the protein in a bilayer before using this functionality. For the attachment of the glycosylphosphatidylinositol (GPI) anchor, one can generate a GPI-linked protein by using “GPI anchor” in PDB Manipulator. The GPI core sequence (Man(1–2)Man(1–6)Man(1–4)GlcN(1–6)PI-DAG) is covalently attached to the C-terminus of a protein through ethanolamine phosphate as a default (Fig. 3B).
Figure 3. Web implementation of lipidation option.

(A) Available lipidation types in PDB Manipulator. (B) Snapshot of attaching a GPI-anchor.
Stapling
Peptide stapling is a set of techniques to covalently link the side chains of two amino acids within a peptide sequence. Many staple structures exist, where they differ in length and composition of the resulting staple moieties. PDB Manipulator implements three categories of staple structures such as (1) ring-closing metathesis, (2) cysteine bis-alkylation, and (3) click reaction.
In ring-closing metathesis, two selected amino acid residues are mutated to alanine, and an alkene segment replaces the Cα hydrogen atoms (Fig. S2). The metathesis options differ in the alkane staple’s length and whether the chosen amino acids are mutated to L-alanine (META3–7) or D-alanine (RMETA3–7). The initial alkane coordinates are positioned using the internal coordinates in their patch residues. Any unrealistically long bonds and bad contacts are resolved by minimization of the two amino acids and their staple with dihedral restraints maintaining the chirality of the cis double bond and Cα carbon.
In cysteine bis-alkylation, two selected amino acid residues are mutated to cysteine and linked via reaction with meta (DIBM) or para (DIBP) dibromo-xylene. Similarly, the click reaction method mutates two selected residues to L-alanine and links them via Azide-Alkyne (CR12) or Alkyne-Azide (CR21) Huisgen cycloaddition. In both cysteine bis-alkylation and click reaction stapling, the stapled amino acid structures then undergo unrestrained energy minimization.
Improved Ligand Parameterizations
MD simulations of drug-like compounds are becoming more prevalent for computer-aided drug discovery research. Therefore, it is crucial to accurately model ligands with a well-validated FF. The general AMBER force field (GAFF) (Wang et al., 2004), the OPLS all-atom force field (OPLS-AA), the CHARMM general force field (CGenFF) (Vanommeslaeghe et al., 2010), and the GROMOS automatic topology builder have been used for this purpose (Mortier et al., 2015). Recently, the Open Force Field (OpenFF) Initiative has used SMIRNOFF that assigns parameters based on an atom’s entire chemical surroundings via direct chemical perception (Wagner J et al., 2021).
PDB Manipulator supports modeling of ligands or small molecules (Jo et al., 2014) with four FF parameterization options such as CGenFF, GAFF2, OpenFF, and custom CHARMM FF (Fig. S3A). The simplest approach is to search the CHARMM FF for an identical molecule. In our previous work, a “CSML Search” option (Fig. S3B) was added to PDB Manipulator to make the search and residue/atom match process smooth. The reference structure file corresponding to a specified PDB residue name is obtained from RCSB chemical component dictionary and used for the CSML search. If custom CHARMM FF parameters are not available for a small molecule of interest, the CGenFF, GAFF, or OpenFF option can be used to build FF parameters.
Previously, the parameter generation option in CHARMM-GUI directly used a MOL2 or SDF file for molecular representation without verification and modification capabilities. In this update, one can now use the 2D sketchpad editor window (Fig. S3C) to verify and modify an existing ligand, allowing users to specify protonation states or bond orders easily. In the end, this option builds 3D ligand structures based on the original molecule’s 3D structure using the maximum common substructure (MCS) search between the user-drawn chemical structure and the uploaded ligand structure by using RDKit FMCS (Fig. S3D) (Dalke & Hastings, 2013). In this MCS search, hydrogen atoms are not considered and partial ring matches are also avoided. The 3D coordinates mapped by MCS are preserved while the other parts are generated with the RDKit EmbedMolecule command that uses a distance geometry approach with experimental torsion preference obtained from small-molecule crystallographic data (Landrum G, 2022). Finally, a small number of minimization steps are executed to optimize the ligand structure with MMFF94.
Automatic Correction of Input PDB Format
The Check/Correct PDB Format option in the front of PDB Reader & Manipulator has been developed to reduce the preprocessing necessary if a user wants to use a custom PDB. This option automatically checks and corrects mis-formatted custom PDB files. It first checks the connectivity section. If there is no CONECT section, it generates one based on interatomic distances. This process builds a CONECT section based on the official PDB format description. Atomic chain IDs are then corrected by looping through the CONECT record and building molecules from consecutive connected atoms. Two molecules that are connected by one or more disulfide bonds are assigned two different chain IDs. Finally, TER records are added directly after the terminus of each chain in the ATOM and HETATM sections. Note that this process does not work on PDBx/mmCIF files, so users should only check this option for a PDB format file. Users should double check that each chain is correctly identified in the subsequent page.
Conclusions
In this study, we have enhanced CHARMM-GUI PDB Manipulator’s functionality with features relevant to PTMs such as 1) automated identification and substitution of 354 nonstandard amino acids and 49 nonstandard nucleic acids in the official CHARMM36m additive FF, 2) in silico ubiquitylation and SUMOylation by attaching predefined ubiquitin or SUMO chains to a target protein chain, 3) Lys / Arg PTMs such as succinylation, lactylation, acetylation, crotonylation, carbamylation, citrullination, and methylation, 4) protein lipidations like palmitoylation, myristoylation, farnesylation, geranylgeranylation, and triacylation, 5) modeling of stapled peptides under three primary categories like ring-closing metathesis, cysteine bis-alkylation, and click reaction. In addition, we have carefully incorporated a number of user input choices to facilitate PTMs modeling for MD simulations. We expect that the enhanced CHARMM-GUI PDB Manipulator will be useful to the biomolecular simulation community in the construction of a wide range of more sophisticated models of biological systems.
Supplementary Material
Highlights.
Nonstandard amino acids and RNA substitution option: It is very hard to handle the parameterizations of the nonstandard amino acids and nucleic acids in RCSB PDB because of the vast number of possible topologies. PDB Manipulator can detect a total 354 nonstandard amino acids and 49 nonstandard nucleic acids, and substitute them with target amino acids and nucleic acids.
Ubiquitylation and SUMOylation option: Despite of the biological importance of these post-translational modifications (PTMs) in nature, to the best of our knowledge, there is no all-atom modeling tool for in silico ubiquitylation and SUMOylation. PDB Manipulator provides an option to attach ubiquitin/SUMO chains onto the user’s protein structure.
Lys and Arg PTMs option: PTMs of Lys and Arg residues are major regulators of biological processes such as gene expression and protein-protein interaction. Modifications for succinylation, lactylation, acetylation, crotonylation, carbamylation, citrullination, and methylation are supported in PDB Manipulator.
Ligand 2D sketch pad option: In previous work, we developed a CHARMM small molecule library (CSML) in CHARMM-GUI, so users can search for a molecule of interest in the CHARMM force field. CHARMM-GUI also supports various ligand parameterization tools such as CGenFF, GAFF, and OpenFF. The new functionality has been added to modify and edit the ligand structure for ligand parameterization.
Acknowledgments
The authors thank Han Zhang and Hugo Guterres for proofreading and critiques on this manuscript. We also thank Seonghoon Kim, Hwayoung Lee, and Andrew Zimmerly for the parameterization of Lys/Arg PTMs, and Key Won Wang for the implementation of the GAFF ligand parameterization option. This work was supported by the grants from NIH GM138472.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
W.I. is the co-founder and CEO of MolCube INC. J.L. is the co-founder and CTO of MolCube INC.
References
- Bowers KJ, Chow DE, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, & Shaw DE (2006). Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. ACM/IEEE SC 2006 Conference (SC’06), 43–43. [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, … Karplus M (2009). CHARMM: The biomolecular simulation program. Journal of Computational Chemistry, 30(10), 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carell T, Brandmayr C, Hienzsch A, Müller M, Pearson D, Reiter V, Thoma I, Thumbs P, & Wagner M (2012). Structure and function of noncanonical nucleobases. In Angewandte Chemie - International Edition (Vol. 51, Issue 29, pp. 7110–7131). 10.1002/anie.201201193 [DOI] [PubMed] [Google Scholar]
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, & Woods RJ (2005). The Amber biomolecular simulation programs. In Journal of Computational Chemistry (Vol. 26, Issue 16, pp. 1668–1688). 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Kern NR, Kim S, Kanhaiya K, Afshar Y, Jeon SH, Jo S, Brooks BR, Lee J, Tadmor EB, Heinz H, & Im W (2022). CHARMM-GUI Nanomaterial Modeler for Modeling and Simulation of Nanomaterial Systems. Journal of Chemical Theory and Computation, 18(1), 479–493. 10.1021/acs.jctc.1c00996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Park SJ, Park S, Kim S, Kern NR, Lee J, & Im W (2021). CHARMM-GUI Polymer Builder for Modeling and Simulation of Synthetic Polymers. Journal of Chemical Theory and Computation, 17(4), 2431–2443. 10.1021/acs.jctc.1c00169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croitoru A, Park SJ, Kumar A, Lee J, Im W, Mackerell AD, & Aleksandrov A (2021). Additive CHARMM36 Force Field for Nonstandard Amino Acids. Journal of Chemical Theory and Computation, 17(6), 3554–3570. 10.1021/acs.jctc.1c00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalke A, & Hastings J (2013). FMCS: a novel algorithm for the multiple MCS problem. Journal of Cheminformatics, 5(S1). 10.1186/1758-2946-5-s1-o6 [DOI] [Google Scholar]
- Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD, Wiewiora RP, Brooks BR, & Pande VS (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Computational Biology, 13(7). 10.1371/journal.pcbi.1005659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Cho E, Liu Y, & Lu Y (2019). Advances and challenges in cell-free incorporation of unnatural amino acids into proteins. In Frontiers in Pharmacology (Vol. 10, Issue MAY). Frontiers Media S.A. 10.3389/fphar.2019.00611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Lee J, Smith IPS, Lee H, Kim S, Qi Y, Klauda JB, Widmalm G, Khalid S, & Im W (2021). CHARMM-GUI Supports Hydrogen Mass Repartitioning and Different Protonation States of Phosphates in Lipopolysaccharides. Journal of Chemical Information and Modeling, 61(2), 831–839. 10.1021/acs.jcim.0c01360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, & MacKerell AD (2016). CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nature Methods, 14(1), 71–73. 10.1038/nmeth.4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari M, Mehrnejad F, Rahimi F, & Asghari SM (2018). The Molecular Basis of the Sodium Dodecyl Sulfate Effect on Human Ubiquitin Structure: A Molecular Dynamics Simulation Study. Scientific Reports, 8(1). 10.1038/s41598-018-20669-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Cheng X, Islam SM, Huang L, Rui H, Zhu A, Lee HS, Qi Y, Han W, Vanommeslaeghe K, MacKerell AD, Roux B, & Im W (2014). CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. In Advances in Protein Chemistry and Structural Biology (Vol. 96, pp. 235–265). Academic Press Inc. 10.1016/bs.apcsb.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, & Im W (2007). Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE, 2(9). 10.1371/journal.pone.0000880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, & Im W (2008). CHARMM-GUI: A web-based graphical user interface for CHARMM. Journal of Computational Chemistry, 29(11), 1859–1865. 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- Jo S, Lim JB, Klauda JB, & Im W (2009). CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophysical Journal, 97(1), 50–58. 10.1016/j.bpj.2009.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Song KC, Desaire H, MacKerell AD, & Im W (2011). Glycan reader: Automated sugar identification and simulation preparation for carbohydrates and glycoproteins. Journal of Computational Chemistry, 32(14), 3135–3141. 10.1002/jcc.21886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Mori T, Kobayashi C, Matsunaga Y, Yoda T, Feig M, & Sugita Y (2015). GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdisciplinary Reviews: Computational Molecular Science, 5(4), 310–323. 10.1002/wcms.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury GA, Thompson JP, Smadbeck J, Kieslich CA, & Floudas CA (2013). Forcefield-PTM: Ab initio charge and AMBER forcefield parameters for frequently occurring post-translational modifications. Journal of Chemical Theory and Computation, 9(12), 5653–5674. 10.1021/ct400556v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee J, Jo S, Brooks CL, Lee HS, & Im W (2017). CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. Journal of Computational Chemistry, 38(21), 1879–1886. 10.1002/jcc.24829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum G (2022). RDKit 2022.09.1 documentation. http://rdkit.org/docs/source/rdkit.Chem.rdmolops.html
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, & Im W (2016). CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. Journal of Chemical Theory and Computation, 12(1), 405–413. 10.1021/acs.jctc.5b00935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hitzenberger M, Rieger M, Kern NR, Zacharias M, & Im W (2020). CHARMM-GUI supports the Amber force fields. Journal of Chemical Physics, 153(3). 10.1063/5.0012280 [DOI] [PubMed] [Google Scholar]
- Lee J, Patel DS, Ståhle J, Park SJ, Kern NR, Kim S, Lee J, Cheng X, Valvano MA, Holst O, Knirel YA, Qi Y, Jo S, Klauda JB, Widmalm G, & Im W (2019). CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. Journal of Chemical Theory and Computation, 15(1), 775–786. 10.1021/acs.jctc.8b01066 [DOI] [PubMed] [Google Scholar]
- Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, … Gan L (2015). Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nature Medicine, 21(10), 1154–1162. 10.1038/nm.3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortier J, Rakers C, Bermudez M, Murgueitio MS, Riniker S, & Wolber G (2015). The impact of molecular dynamics on drug design: Applications for the characterization of ligand-macromolecule complexes. In Drug Discovery Today (Vol. 20, Issue 6, pp. 686–702). Elsevier Ltd. 10.1016/j.drudis.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Nerenberg PS, & Head-Gordon T (2018). New developments in force fields for biomolecular simulations. In Current Opinion in Structural Biology (Vol. 49, pp. 129–138). Elsevier Ltd. 10.1016/j.sbi.2018.02.002 [DOI] [PubMed] [Google Scholar]
- Park SJ, Lee J, Patel DS, Ma H, Lee HS, Jo S, & Im W (2017). Glycan Reader is improved to recognize most sugar types and chemical modifications in the Protein Data Bank. Bioinformatics, 33(19), 3051–3057. 10.1093/bioinformatics/btx358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee J, Qi Y, Kern NR, Lee HS, Jo S, Joung I, Joo K, Lee J, & Im W (2019). CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology, 29(4), 320–331. 10.1093/glycob/cwz003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, & Schulten K (2005). Scalable molecular dynamics with NAMD. In Journal of Computational Chemistry (Vol. 26, Issue 16, pp. 1781–1802). John Wiley and Sons Inc. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimpton S (1995). LAMMPS. Journal of Computational Physics, 117, 1–19. [Google Scholar]
- Rackers JA, Wang Z, Lu C, Laury ML, Lagardère L, Schnieders MJ, Piquemal JP, Ren P, & Ponder JW (2018). Tinker 8: Software Tools for Molecular Design. Journal of Chemical Theory and Computation, 14(10), 5273–5289. 10.1021/acs.jctc.8b00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Jatana N, & Thukral L (2017). Lipidated proteins: Spotlight on protein-membrane binding interfaces. In Progress in Biophysics and Molecular Biology (Vol. 128, pp. 74–84). Elsevier Ltd. 10.1016/j.pbiomolbio.2017.01.002 [DOI] [PubMed] [Google Scholar]
- Schlick T, Portillo-Ledesma S, Myers CG, Beljak L, Chen J, Dakhel S, Darling D, Ghosh S, Hall J, Jan M, Liang E, Saju S, Vohr M, Wu C, Xu Y, & Xue E (2021). Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field. In Annual Review of Biophysics (Vol. 50, pp. 267–301). Annual Reviews Inc. 10.1146/annurev-biophys-091720-102019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SJA, Zhong H, Zhang Q, & Liu H (2022). Deciphering the Effect of Lysine Acetylation on the Misfolding and Aggregation of Human Tau Fragment171IPAKTPPAPK180 Using Molecular Dynamic Simulation and the Markov State Model. International Journal of Molecular Sciences, 23(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, & Tufféry P (2012). PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Research, 40(W1). 10.1093/nar/gks419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, & Mackerell AD (2010). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of Computational Chemistry, 31(4), 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Thompson M, Dotson D, Jang D, Boothroyd H, & Rodríguez-Guerra J. (2021). Open Force Field. Sage. [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, & Case DA (2004). Development and testing of a general Amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, & Im W (2014). CHARMM-GUI membrane builder toward realistic biological membrane simulations. In Journal of Computational Chemistry (Vol. 35, Issue 27, pp. 1997–2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Vanommeslaeghe K, Aleksandrov A, MacKerell AD, & Nilsson L (2016). Additive CHARMM force field for naturally occurring modified ribonucleotides. Journal of Computational Chemistry, 37(10), 896–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zencheck WD, Xiao H, & Weiss LM (2012). Lysine post-translational modifications and the cytoskeleton. Essays in Biochemistry, 52(1), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.