

Summary
Minichromosome Maintenance 8 Homologous Recombination Repair Factor (MCM8) and Minichromosome Maintenance 9 Homologous Recombination Repair Factor (MCM9) are recently discovered minichromosome maintenance proteins and are implicated in multiple DNA-related processes and pathologies, including DNA replication (initiation), meiosis, homologous recombination and mismatch repair. Consistent with these molecular functions, variants of MCM8/MCM9 may predispose carriers to disorders such as infertility and cancer and should therefore be included in relevant diagnostic testing. In this overview of the (patho)physiological functions of MCM8 and MCM9 and the phenotype of MCM8/MCM9 variant carriers, we explore the potential clinical implications of MCM8/MCM9 variant carriership and highlight important future directions of MCM8 and MCM9 research. With this review, we hope to contribute to better MCM8/MCM9 variant carrier management and the potential utilization of MCM8 and MCM9 in other facets of scientific research and medical care.
Subject area(s): Cell biology
Graphical abstract
Cell biology
Introduction
Minichromosome Maintenance 8 Homologous Recombination Repair Factor (MCM8; OMIM 60817) and Minichromosome Maintenance 9 Homologous Recombination Repair Factor (MCM9; OMIM 610098) are the most recent minichromosome maintenance (MCM) proteins to be discovered, having been identified in 2003 and 2005, respectively.1,2,3 They show sequence homology with the MCM2-7 proteins (OMIM 116945; OMIM 602693; OMIM 602638; OMIM 602696; OMIM 601806; OMIM 600592), which form a stable hetero-hexamer that is a component of the replication initiation complex responsible for the initiation of DNA synthesis in all eukaryotic cells.4,5 Although MCM1 and MCM10 (OMIM 609357) are not members of this family, they are conserved in higher eukaryotes. MCM1 acts as a transcription factor, whereas MCM10 is also directly involved in the initiation of DNA synthesis.4,5
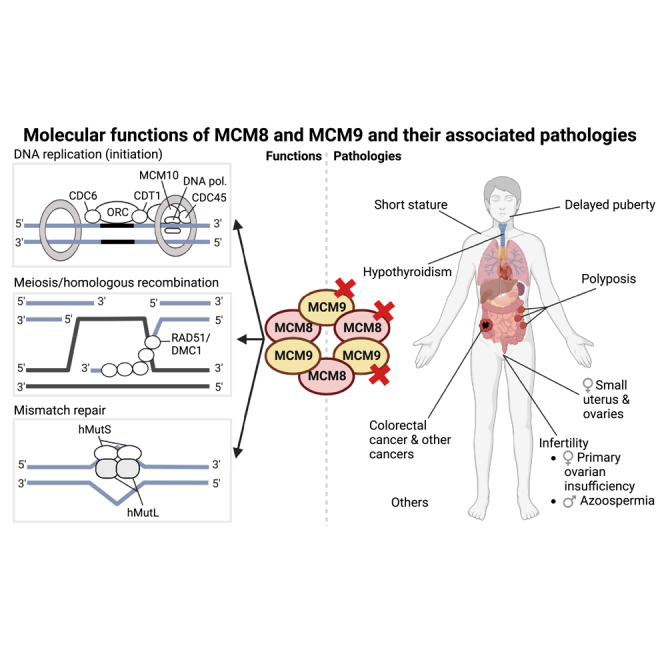
Following the identification of MCM8 and MCM9, which may also interact to form a hexameric ring complex,6 a wide variety of experimental approaches were used to explore their molecular functions. As with other MCM components, MCM8 and MCM9 have been implicated in initiation of DNA replication,7,8,9,10 as well as in meiosis,9,11,12,13,14 homologous recombination (HR)15,16,17,18,19,20,21 and mismatch repair (MMR).20,22,23 Correspondingly, the number of studies describing the possible involvement of MCM8 and MCM9 in pathologies has increased enormously, with disrupting variants of the MCM8/MCM9 genes that follow an autosomal recessive inheritance pattern linked to infertility in both males and females,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44 as well as recently highlighted roles for the MCM8/MCM9 genes in polyposis,23,37,38 (early onset) colorectal cancer (OMIM 114500)23,37,38 and multiple other cancer types.23,34,37,38
Despite the increasing number of studies focusing on MCM8 and MCM9, comprehensive literature reviews are scarce and those published have generally focused on a subset of molecular functions or pathologies.6,37 Taking a broader view, we aim to provide an overview of all associated molecular functions of both proteins, whereas also covering current evidence of their roles in distinct pathologies. We will explore potential clinical implications of MCM8/MCM9 variant carriership and highlight important future directions of MCM8 and MCM9 research.
Results
Functions of MCM8 and MCM9
DNA replication (initiation)
Since their discovery, MCM8 and MCM9 have been associated with multiple DNA-related processes. MCM8 was the first to be associated with the initiation of DNA replication, which is strictly controlled and regulated by the cell cycle, requiring the assembly of a pre-replicative complex in G1 phase.45 In short, initiation of DNA replication in eukaryotic cells begins with the mobilization of a six-subunit origin recognition complex (ORC) at an origin of DNA replication (Figure 1A). Subsequently, ORC recruits CDC6 (OMIM 602627) and CDT1 (OMIM 605525), which stabilize the ORC and load the MCM2-7 protein complex onto chromatin.46 The MCM2-7 complex possesses DNA helicase activity, which is activated through phosphorylation of the complex by CDK2/CYCLIN E (OMIM 116953; OMIM 123837, respectively) and CDC7/DBF4 kinases (OMIM 603311; OMIM 604281, respectively) and by the assembly of the CMG complex, consisting of MCM2-7, GINS (OMIM 610608) and CDC45 (OMIM 603465).10,47,48,49 The recruitment of CDC45 to the MCM2-7 complex is facilitated by MCM10, which also recruits other replication factors.10,49 The CMG complex is also required for the recruitment of DNA polymerases, allowing DNA replication to commence.
Figure 1.

Simplified overview of the proposed molecular functions of MCM8/MCM9
(A) In eukaryotic cells, DNA replication initiation starts with the mobilization of ORC at each origin. MCM8 and MCM9 may subsequently support ORC in recruiting CDC6 and CDT1, respectively, which stabilize ORC and help load the MCM2-7 protein complex on chromatin. The helicase activity of the MCM2-7 complex is then activated through phosphorylation by CDK2/Cyclin E and CDC7/DBF4 kinases and by assembly of the CMG complex (GINS-CDC45-MCM2-7), thereby opening the DNA double helix. The CMG complex is also required for the recruitment of DNA polymerases, allowing DNA replication to begin. The recruitment of CDC45 to the MCM2-7 complex is facilitated by MCM10, which also attaches to the MCM2-7 complex and additional recruits other replication factors. During DNA replication, MCM8-9 complexes can, under some circumstances, drive fork progression, for instance in case of MCM2-7 dysfunction. By recruiting BRCA1 and RAD51, the MCM8-9 complexes may also prevent fork degradation when a transient or persistent block is encountered.
(B) [1] The formation of COs is essential for genetic diversity. This process is initiated by the formation of a DSB on one chromatid, which is then resected by the MRE11-RAD50-NBS1 complex to generate 3′ single-stranded overhangs. MCM8-9 complexes may be responsible for the function of MRE11-RAD50-NBS1 nuclease activity and the recruitment of MRE11 to foci of DNA damage. Next, one of the 3′ single-stranded overhangs invades the homologous, non-sister chromatid. The latter depends on RAD51, the recruitment of which may be facilitated by MCM8-9 complexes. MCM8-9 complexes themselves may be recruited by HROB, which localizes on damaged chromatin by interacting with RPA1 and/or single-stranded DNA. Following the formation of several intermediate DNA products, resolution of DSBs could eventually lead to COs or NCOs, with MCM8-9 complexes potentially shifting the balance in favor of CO formation. [2] During the repair of crosslinks, MCM8-9 may exert similar functions as during the formation of COs.
(C) Base mismatches and insertion/deletion variants are recognized by hMutS, consisting of MSH2 and MSH6 (hMutSα) or MSH2 and MSH3 (hMutSβ), respectively. hMutS may then recruit MCM8-9 complexes to the site of the mutation, which through DNA helicase activity may open the DNA double helix. This, in turn, could trigger the recruitment of hMutL, consisting of MLH1 and PMS2 (hMutLɑ) or MLH1 and MLH3 (hMutLɣ), which initiates the degradation of the mismatch-containing strand by exonucleases and the synthesis of a new strand by DNA polymerases and ligases. NOTE: This figure visualizes simplified versions of complex processes. In each of the three processes, a multitude of components are involved that are not depicted or mentioned. Adapted from “DNA Replication Process”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates. CO, crossover; DSB, double-strand break; HR, homologous recombination; MMR, mismatch repair; NCO, non-crossover; ORC, origin recognition complex.
In 2005, Volkening et al.10 used immunoprecipitation assays to show that although MCM8 accumulated on chromatin throughout the cell cycle, this peaked in early G1 phase and involved interactions with ORCs and CDC6. As downregulation of MCM8 led to a reduction of CDC6 and MCM2-7 loading, Volkening et al.10 hypothesized that MCM8 is responsible for the recruitment of CDC6 to ORCs, possibly in cooperation with CDT1, and is therefore required for MCM2-7 loading and the assembly of the pre-replicative complex.10 The latter conclusion was supported by their finding that endogenous depletion of MCM8 by RNA interference reduced DNA replication by delaying entry into S phase.
Similarly, Maiorano et al.50 suggested that MCM8 is directly involved in DNA replication, rather than simply the initiation of DNA replication. In contrast to Volkening et al.,10 Maiorano et al.50 were unable to identify chromatin loading of MCM8 during the formation of pre-replicative complexes whilst studying a Xenopus homolog of MCM8. Instead, they detected maximal chromatin binding of MCM8 during processive DNA synthesis and showed that MCM8, like the MCM2-7 complex, displays DNA helicase activity, as determined by displacement of 40 base-labeled oligonucleotides annealed to single-stranded DNA.50 This postulated DNA helicase activity suggests that MCM8 may be able to unwind DNA during DNA synthesis, an inference supported by the fact that cellular MCM8 depletion led to a reduction of chromatin-bound RPA34, which specifically recognizes single-stranded DNA and recruits DNA polymerase-α (OMIM 312040) at replication forks.50
The contrasting findings of Volkening et al.10 and Maiorano et al.50 highlight the complexity of DNA replication (initiation) and the potential role(s) for MCM8 in these processes. The discrepancies could be the result of differences in analysis methods, as well as the use of different models and MCM8 homologs. Of interest, Kinoshita et al.51 found that MCM8 colocalizes with proteins involved in both the initiation of DNA replication (e.g., CDC6, CDK2) and the DNA replication process (RPA70; OMIM 179835), implying that both of the proposed roles for MCM8 may exist in parallel.
In 2008, Lutzmann et al.7,8 described a possible role for MCM9 in pre-replicative complexes, showing that MCM9 forms a stable complex with CDT1 and like MCM8 harbors a helicase domain (Figure 2) which opens up the double strand during G1 phase to allow loading of MCM2-7 complexes that facilitate replication fork movement later in S phase. This introduced the hypothesis that MCM8 and MCM9 may exert similar functions in DNA replication (initiation), a proposition which gained support from later findings showing that MCM8 and MCM9 may form a complex, resembling the MCM2-7 complex in both size (600kD) and structure (hexamer).18 By forming this complex, MCM8 and MCM9 may be able to stabilize each other, because both MCM8 and MCM9 silencing reduces the protein concentration of the other partner.16 Although future studies should define the precise stoichiometry of the MCM8-9 complex, MCM8 and MCM9 subunits have been proposed to form a hetero-hexameric ring, which includes a central channel that may be used to accommodate DNA.6 The N-terminal domains of this ring were found to be more stable than the C-terminal domains, with the relative positions of the N- and C-terminal domains being able to change during functional state conversion.52 Considering the structural resemblance to and sequence homology with the MCM2-7 complex, an analogous role of MCM8-9 in DNA replication (initiation) could be envisioned.
Figure 2.

MCM8 and MCM9 human protein domains
(A) Protein domains of MCM8 are similarly organized as those of MCM2-7 and MCM9, containing an N-terminal and C-terminal domain.6,52 The N-terminal domain contains a zinc-finger (ZF) motif and is able to bind DNA. The N terminus consists of a structurally disorder region (amino acids 1–60). The C-terminal domain is composed of an AAA+ (ATPase) domain and a winged-helix (WH) domain, with the latter also being able to bind DNA.53 The AAA+ domain contains the highly conserved Walker A (WA) and Walker B (WB) motifs, which are required for ATP hydrolysis and helicase activity, and an arginine-finger (RF) domain.18 The N-terminal domain and C-terminal domain are linked by the N-C linker domain.
(B) Protein domains of MCM9 are similarly organized as those of MCM2-7 and MCM8, containing an N-terminal and C-terminal domain.6,52 The N-terminal domain contains a ZF motif. The C-terminal domain is composed of an AAA+ domain and a WH domain. The AAA+ domain contains the highly conserved WA and WB motifs and an RF domain.18 At its C terminus, MCM9 contains a unique long tail (amino acids 685–1143), which may mediate interactions with other proteins.6 The N-terminal domain and C-terminal domain are linked by the N-C linker domain. RF, arginine finger; WA, Walker A; WB, Walker B; WH, winged-helix; ZF, zinc-finger.
Further evidence of a role for MCM8-9 in DNA replication came from more recent studies by Natsume et al.17 and Griffin et al.,54 both of whom linked MCM8-9 complex functioning to the progression of replication forks. First, Natsume et al.17 showed that MCM2-depleted cells maintained some DNA synthesis, which required the helicase activity of MCM8-9 complexes, hypothesizing that these MCM8-9 complexes could be alternative drivers of fork progression in case of MCM2-7 dysfunction. Correspondingly, Griffin et al.54 demonstrated that replication fork progression and the overall replication rate were reduced in MCM8KO or MCM9KO cells. Moreover, they showed that MCM8-9 complexes were involved in fork protection during replication stress via recruitment of downstream proteins such as BRCA1 (OMIM 113705) and RAD51 (OMIM 179617), which stabilize the replication fork and prevent fork degradation.54
In summary, current evidence defines multifunctional roles of MCM8 and MCM9 in the initiation of DNA replication and in DNA replication itself: MCM8 and MCM9 may assist the assembly of pre-replicative complexes, may alternatively drive replication fork progression using their DNA helicase activity and they may be able to protect replication forks during replication stress. Although a subset of these functions was initially described for each protein individually, they conceivably acquire MCM8 and MCM9 to function in complex. Future studies exploring molecular architecture of the MCM8-9 complex, as well as the timing of MCM8-9 functioning and the specific consequences of MCM8/MCM9 silencing on DNA replication (initiation) are necessary for a better understanding of these complex processes.
Meiosis and HR
In addition to the aforementioned association with DNA replication (initiation), a number of studies have implicated MCM8 and MCM9 in meiosis. Meiosis is responsible for the generation of haploid gametes (containing a single set of chromosomes), such as sperm or egg cells, from diploid precursors (containing two sets of chromosomes).55 In short, each chromosome must recognize and align with its homologous pairing partner during early prophase, after which the aligned chromosomes are held together by the assembly of a synaptonemal complex between the chromosomes. Next, the formation of DNA double-strand breaks (DSBs) induces crossover (CO) recombination events between the DNA of the aligned and synapsed homologs.55 These COs form physical linkages between chromosome pairs and are essential for the introduction of genetic diversity.11 During late prophase, the connected homologs orient away from each other, resulting in bipolar attachment of homologs to the meiosis I spindle and segregation of homologous chromosomes at anaphase I. A similar separation of sister chromatids during meiosis II completes the meiotic program.55
The first evidence of a potential role for MCM8 in meiosis originated from a study on recombination defective (rec), the Drosophila ortholog of MCM8, which found that rec mutants exhibited a defect in meiotic recombination, as indicated by high levels of chromosome nondisjunction and reduced fertility.56 It was hypothesized that rec is involved in the generation of COs, because the rec mutants showed a severe reduction in CO formation but were able to pair homologs normally.11,56 Briefly, CO formation is initiated with the formation of a DSB on one chromatid, which is then resected to generate 3′ single-stranded overhangs, one of which invades the homologous, non-sister chromatid and primes DNA repair synthesis (Figure 1B).11 Following the formation of several intermediate DNA products, resolution of the DSBs may eventually lead to COs, in which genetic material is exchanged between the two homologous chromosomes' non-sister chromatids. Alternatively, non-crossover (NCO) products may be produced that do not result in exchange of genetic material.11
Of interest, Blanton et al.11 demonstrated that rec mutant females had about twice the number of NCOs compared to wildtype females, suggesting that rather than the initiation of recombination itself, repair of DSBs as COs is impaired in rec mutants. The hypothesis that rec facilitates DSB repair during meiotic recombination was also supported by studies in Arabidopsis thaliana, a frequently used plant model because of its relatively simple genome, because three Atmcm8 mutants showed a limited level of chromosome fragmentation at meiosis, a process that normally depends on DSB repair.12 Moreover, a study using Drosophila melanogaster models showed that rec participates in multiple protein complexes that show differential rec-dependent ATP-binding and ATP-hydrolyzing requirements and, in part, regulate CO formation.14
One of the mechanisms responsible for the repair of DSBs into COs during meiosis is HR, which additionally has multiple other functions in DNA maintenance and repair that involve a multiplex of pathways and mediators. Of interest, considerable evidence implicates MCM8 but also MCM9 in HR. The first association between MCM8-9 and HR was reported by Nishimura et al.,18 who demonstrated that MCM8-9 is involved in DNA interstrand crosslink repair, because MCM8KO and MCM9KO cells were both found to be highly sensitive to DNA crosslinking agents, showing more chromosomal aberrations following mitomycin C treatment as compared to wildtype cells. Interstrand crosslink repair is orchestrated by the Fanconi anemia proteins and depends on nucleotide excision repair, translesion synthesis and HR. Lutzmann et al.16 confirmed these findings, showing that HR was impaired in both MCM8KO and MCM9KO mice during gametogenesis and that MCM8-9 deficient cells were hypersensitive to DSBs and replication stress. Of interest, in the latter study, the potential involvement of MCM8-9-mediated HR in meiosis was highlighted by the fact that female MCM8KO mice were sterile, showed atrophied ovaries with dysplastic primary follicles, and were prone to development of colon adenomas and sex cord stromal tumors. Similarly, male MCM8KO mice were sterile, showed testis with atrophic seminiferous tubules, elevated numbers of apoptotic cells and no post-meiotic cells.16 Female MCM9KO mice were also sterile, with ovaries completely devoid of oocytes, while male MCM9KO mice were fertile but with testes that showed severe early proliferation defects of germ cells, leading to an abundance of atrophied seminiferous tubules.16 Correspondingly, Hartford et al.9 showed that MCM9-mutant mice underwent P53-independent embryogenic germ-cell depletion in both sexes, with males also exhibiting defective spermatogonial stem-cell renewal.
Remarkably, although the studies of Nishimura et al.18 and Lutzmann et al.16 both supported a role for MCM8-9 in HR, they conflicted with regards to the exact timing of MCM8-9 functioning in HR. To distinguish early events in HR, such as end resection, from late events, including strand invasion and DSB resolution, genetic studies regularly rely on RAD51 appearance (or DMC1, its meiotic homolog; OMIM 602721).57 Of interest, though Nishimura et al.18 hypothesized that complexes of MCM8 and MCM9 act at some point after RAD51 loading and are therefore only involved in the late events of HR, Lutzmann et al.16 reported that MCM8-9 complexes are required before RAD51 loading. Furthermore, Park et al.19 and McKinzey et al.20 found that MCM8-9 complexes directly promote RAD51 recruitment, whereas Lee et al.15 demonstrated that MCM8-9 complexes are required for DNA resection by the MRE11-RAD50-NBS1 complex (OMIM 600814; OMIM 604040; OMIM 602667, respectively) at DSBs to generate 3′ single-stranded overhangs, with an essential role for the ATPase activity of MCM9 in the function of the MRE11-RAD50-NBS1 nuclease and the recruitment of MRE11 to foci of DNA damage.
Several recent studies additionally demonstrated that the OB-fold containing protein HROB (also referred to as C17orf53/MCM8IP; OMIM 618611) is able to support MCM8-9 functioning during HR.58,59 It is hypothesized that HROB may interact with RPA1 (OMIM 179835) and/or directly bind to single-stranded DNA.58,59 Once present on damaged chromatic, HROB was found to increase the affinity of MCM8-9 for single-stranded DNA and to remarkably stimulate (∼6-fold increase) MCM8-9 helicase activity.58 With HROB-deficient cells showing severely impaired HR and increased sensitivity to DNA crosslinking agents, the interactions of HROB and MCM8-9 appear to facilitate HR and protect against crosslinking agents by promoting replication fork progression and cellular viability.58,59 Future studies are needed to test the biochemical basis of these interactions and should for example explore whether HROB may drive MCM8-9 conformational changes that underlie the increased affinity of MCM8-9 for single-stranded DNA and the increased DNA helicase activity of MCM8-9. Moreover, future studies are necessary to define whether the interaction of HROB and MCM8-9 is also required for MCM8-9 activity in other DNA-related processes, including DNA replication (initiation).
Collectively, these findings provide convincing evidence for the role of MCM8-9 in meiosis and HR, yet future functional studies are vital to better understand the precise functioning of MCM8-9 in these processes. Although MCM8 and MCM9 may also have distinct functionalities, indicated for example by modest differences in the phenotypes of MCM8KO/MCM9KO mice, most of the functions in (HR-mediated) meiosis appear dependent on MCM8-9 complexes. As was the case for MCM8-9 in DNA replication (initiation), the MCM8-9 complex is most likely involved in more than one step of these complex reactions.57
MMR
The most recently suggested function of MCM8-9 involves MMR. The MMR system serves as a post-replicative proofreading and editing system60 responsible for the repair of variants caused by slippage of DNA polymerases,61,62 as well as for the repair of diverse types of endo- and exogenous DNA damage.63,64 Briefly, base mismatches and short insertion/deletion variants are initially recognized by hMutSα, consisting of MSH2 (OMIM 609309) and MSH6 (OMIM 600678) dimers, whereas longer insertion-deletion loops are detected by hMutSβ, consisting of MSH2 and MSH3 (OMIM 600887; Figure 1C). Upon detection of a mismatch, hMutS recruits and forms a complex with hMutL, which subsequently coordinates the degradation of the mismatch-containing strand and the synthesis of a new strand. The hMutL complex either involves hMutLɑ or hMutLɣ, consisting of MLH1 (OMIM 120436) and PMS2 (OMIM 600259) or MLH1 and MLH3 (OMIM 604395), respectively.60
As MCM9 co-immunoprecipitates with multiple MMR initiation proteins, including MSH2, MSH3, MLH1 and PMS1 (OMIM 600258), it was hypothesized that MCM9 plays a role in MMR.21,65 This hypothesis gained traction when Traver et al.21 reported that MCM9KO cells display MMR deficiency and MSI. Of interest, although MMR activity was restored in MCM9KO cells following transfection with wildtype MCM9 protein, transfection with helicase-dead MCM9 did not restore MMR activity in MCM9KO cells, suggesting that MCM9 helicase activity is required for efficient MMR. Moreover, whereas MSH2 was found to be essential for MCM9 loading on chromatin, MCM9 was responsible for the recruitment of MLH1.21 The latter finding was supported by Liu et al.,22 who showed that aberrantly expressed HORMAD1, which binds to MCM9 and prevents the efficient nuclear localization of MCM8-9 complexes, led to compromised MMR via reduced MLH1 loading.
A possible role for MCM8 in MMR was first proposed by Golubicki et al.,23 who demonstrated that MCM8KO cells were microsatellite instable (MSI), with their DNA reflecting the single-base signature 20, which is associated with concurrent POLD1 (OMIM 174761) pathogenic variants and MMR deficiency.23,66 Of interest, Golubicki et al.23 also found higher frequencies of insertions and deletions larger than 5 base pairs in MCM8KO cells, and in a comet assay these cells were less able to repair DNA damage caused by oxaliplatin compared with MCM8WT cells. The investigators hypothesized that MCM8 deficiencies can concurrently impair both MMR and other DNA repair pathways, such as HR.
Collectively, these findings suggest the following role for MCM9 (and MCM8) in MMR, which should be tested and evaluated in future studies: once a mismatch is detected, MSH2 recruits MCM9, perhaps in a complex with MCM8. The DNA helicase activity of MCM9 and/or the MCM8-9 complex subsequently opens the DNA double helix at the site of the mismatch, triggering the recruitment of MLH1 to the mismatch site, which then forms a complex with PMS2 or MLH3 to repair the mismatch.
MCM8 and MCM9 in pathology
Infertility
MCM8
Following the first reports suggesting a role for MCM8 in meiosis and the observation that both male and female MCM8KO mice were sterile, a potential link between MCM8 and (in)fertility was hypothesized. This link was further explored by multiple genome-wide association studies in women, which identified an association between single nucleotide polymorphisms in the MCM8 gene and age at natural menopause.67,68,69,70,71 Validation of these associations in several cohorts of women undergoing early menopause showed that single nucleotide polymorphisms in MCM8 significantly increase the odds for early menopause and are additionally associated with a decreased length of reproductive lifespan and number of ovarian follicles.72,73,74
Early menopause, a decreased reproductive lifespan and a decreased number of ovarian follicles are all hallmarks of primary ovarian insufficiency (POI), also referred to as premature ovarian failure (POF; OMIM 311360).75 In POI-affected females, ovaries cease to produce mature oocytes before the age of 40 years, leading to secondary amenorrhea, infertility, hypoestrogenism and elevated serum levels of follicle-stimulating hormone, among other effects.75
Additional evidence for an association between MCM8 and POI originated from a study by AlAsiri et al.,24 who were the first to describe a consanguineous family with three homozygous MCM8 variant [p.(Pro149Arg)] carriers affected by POI. The heterozygous carriers in this family were all healthy. Similarly, Tenenbaum-Rakover et al.25 reported two consanguineous families in whichMCM8 variant carriers were affected by POI. In the first family, a brother and sister, both homozygous carriers of the MCM8 variant (c.1954-1G>A), were affected by azoospermia at the age of 17 years and POI at the age of 15 years, respectively. In the second family, three sisters, all homozygous MCM8 variant (c.1469-1470insTA) carriers, were each affected by POI, whereas two of their first-degree cousins, also homozygous carriers, were diagnosed with primary hypergonadotropic hypogonadism. Of interest, the heterozygous carriers in both families were healthy, albeit that some showed delayed puberty.25 Moreover, the fibroblasts or lymphocytes of homozygous carriers from both studies showed significantly increased chromosomal breakage following exposure to mitomycin C (DNA crosslinker) as compared to cells of unaffected family members, suggesting impaired HR because of the absence of functional MCM8.24,25
Numerous other studies have since reported MCM8 variant carriers with POI26,27,28,29,30,31,32,33 and/or (non-obstructive) azoospermia (Table 1).76 Of interest, Tucket et al.33 described a POI patient with variants of HROB, which is believed to interact with and support MCM8-9 functioning, suggesting that variants of genes with functions related to MCM8-9 may cause similar phenotypes.33,59
Table 1.
Pathologies associated with MCM8/MCM9
| Pathology | MCM8 | MCM9 |
|---|---|---|
| Abnormal uterine bleeding | Shen et al.77 | |
| Alzheimer disease | Ratnakumar et al.78 | |
| Birth of child with Down syndrome | Pal et al.79 | |
| Infertility, female | AlAsiri et al., Tenenbaum-Rakover et al., Dou et al., Desai et al., Bouali et al., Zhang et al., Heddar et al., Wang et al., Jin et al., Tucker et al.24,25,26,27,28,29,30,31,32,33 | Desai et al., Alvarez-Mora et al., Fauchereau et al., França et al., Goldberg et al., Guo et al., Liu et al., Shen et al., Turkyilmaz et al., Wood-Trageser et al., Yang et al., and Jolly et al.27,34,35,36,38,39,40,41,42,43,44,80 |
| Born small for gestational age | Tenenbaum-Rakover et al., Heddar et al.25,30 | |
| Delayed puberty | AlAsiri et al., Tenenbaum-Rakover et al., Bouali et al., Heddar et al.24,25,28,30 | Fauchereau et al., Turkyilmaz et al., Wood-Trageser et al., Yang et al.35,42,43,44 |
| Facial naevi | Wang et al31 | |
| Hearing loss | Tenenbaum-Rakover et al.25 | |
| Hypothyroidism | AlAsiri et al and Heddar et al.24,30 | |
| Infantile uterus | AlAsiri et al., Tenenbaum-Rakover et al., Zhang et al.24,25,29 | Fauchereau et al.,Guo et al., Shen et al., Turkyilmaz et al., Wood-Trageser et al., Yang et al.35,39,41,42,43,44 |
| Invisible/small ovaries | AlAsiri et al., Tenenbaum-Rakover et al., Bouali et al., Zhang et al., Wang et al., Tucker et al.24,25,28,29,31,33 | Fauchereau et al., Turkyilmaz et al., Wood-Trageser et al., Yang et al.35,42,43,44 |
| Kidney agenesis | Tenenbaum-Rakover et al.25 | |
| Mental retardation | Tenenbaum-Rakover et al.25 | |
| Osteoporosis/delayed bone age | Heddar et al and Wang et al.30,31 | Fauchereau et al., Wood-Trageser et al., Yang et al.35,43,44 |
| Peltate chest | Wang et al.31 | |
| Pilomatricomas | Heddar et al.30 | |
| Short stature | Wang et al.31 | França et al.,Guo et al., Turkyilmaz et al., Wood-Trageser et al.36,39,42,43 |
| Temporal epilepsy | Tenenbaum-Rakover et al.25 | |
| Infertility, male | Tenenbaum-Rakover et al and Kherraf et al.25,76 | Goldberg et al and Chen et al.37,81 |
| Born small for gestational age | Tenenbaum-Rakover et al.25 | |
| Delayed puberty | Tenenbaum-Rakover et al.25 | |
| Tremor | Bally et al.82 |
| Cancer | Tumor suppressive role |
Oncogenic role |
Tumor suppressive function(s) |
Oncogenic role |
||
|---|---|---|---|---|---|---|
| (Loss of function) variants/deletions | Increased expression | Gain of copy number | (Loss of function) variants/deletions | Increased expression | Gain of copy number | |
| Acute myeloid leukemia | He et al.83 | |||||
| Adrenocortical carcinoma | He et al.83 | |||||
| Bladder cancer | Zhu et al.84 | He et al.83 | Lee et al.15 | |||
| Breast cancer | Golubicki et al., Verdiesen et al, and Michailidou et al.23,85,86 | He et al.83 | He et al.83 | Lee et al.15 | ||
| (HPV18+) Cervical cancer | Goldberg et al.37 | Sample.87 | ||||
| Cholangiocarcinoma | Hao et al.88 | |||||
| Chronic myelogenous leukemia | Cai et al.89 | |||||
| Colorectal cancer | Golubicki et al.23 | He et al.83 | Golubicki et al., Goldberg et al and Goldberg et al.23,37,38 | |||
| Esophageal (adeno)carcinoma | He et al., Li and Xu83,90 | |||||
| Gastric cancer | Huang et al.91 | |||||
| Germ cell tumor | Alvarez-Mora et al.34 | |||||
| Glioblastoma (multiforme) | He et al.83 | He et al.83 | Lee et al.15 | |||
| Glioma | He et al and Wang et al83,92 | |||||
| Head and neck squamous cell carcinoma | He et al.83 | Lee et al.15 | ||||
| Hepatocellular carcinoma | He et al., Liu et al., Wan et al., Wen et al, and Xiong et al.83,93,94,95,96 | |||||
| Kidney renal clear cell carcinoma | Lee et al.15 | |||||
| Liver cancer | He et al.83 | |||||
| Lung cancer | Li et al and Liu et al.97,98 | |||||
| Non-small cell lung cancer | He et al and Xie et al.83,99 | He et al.83 | ||||
| Lymphoma | Braggio et al and Sung et al.100,101 | |||||
| Medulloblastoma | He et al.83 | |||||
| Melanoma | Lee et al.15 | |||||
| Nasopharyngeal carcinoma | He et al.83 | |||||
| Osteosarcoma | Ren et al.102 | |||||
| Ovarian cancer | He et al.83 | Lee et al.15 | ||||
| Pancreatic cancer | Peng et al.103 | He et al.83 | ||||
| Polyposis | Golubicki et al and Soares de Lima et al.23,104 | |||||
| Primary salivary adenoid cystic sarcoma | Feng et al.105 | |||||
| Primitive neuroectodermal tumor | He et al.83 | |||||
| Prostate cancer | He et al.83 | He et al.83 | Lee et al and Kim et al.15,106 | |||
| Rhabdoid tumor | He et al.83 | |||||
| Sarcoma | He et al.83 | |||||
| Serous ovarian cancer | Li and Xu.90 | |||||
| T cell acute lymphoblastic leukemia | He et al.83 | |||||
| Thyroid carcinoma | He et al.83 | |||||
| Uterine corpus endometrial carcinoma | He et al83 | |||||
| Uterine leiomyomas | Rafnar et al107 | |||||
HPV18, Human papillomavirus 18.
Strikingly, the phenotype of infertile MCM8 variant carriers is additionally characterized by several other clinical features (Table 1). For instance, a majority of POI-affected MCM8 variant carriers were found to have infantile uteri24,25,29 and/or invisible or small ovaries on ultrasound,24,25,28,29,31,33 indicating a potential need to include the MCM8 gene in screening panels for unexplained gonadal dysgenesis. Moreover, multiple POI-affected cases were diagnosed with hypothyroidism, suggesting that MCM8 dysfunction may concurrently affect endocrine homeostasis.24,30 Several other observed characteristics are typical for patients with POI and/or azoospermia, including delayed puberty,24,25,28,30 osteoporosis/delayed bone age30,31 and a short stature,31 whereas other associated pathologies seem less directly related, such as kidney agenesis,25 temporal epilepsy,25 mental retardation,25 hearing loss,25 pilomatricomas,30 facial naevi,31 a peltate chest31 and being small for gestational age at birth.25,30
MCM9
Considering the significant overlap in their cellular functions, the pathologies associated with MCM8 and MCM9 also show commonalties, with MCM9 variants being similarly linked to fertility problems (Table 1). Wood-Trageser et al.43 were the first to describe MCM9 variants in patients with POI, detecting homozygous MCM9 variants in two unrelated, consanguineous families. In the first family, two daughters of a union between first-degree cousins were found to be homozygous for the MCM9 c.1732 + 2T>C variant and presented with a Turner-like phenotype of primary amenorrhea, short stature and low weight. In the second family, a 16-year-old girl with homozygous MCM9 variants [p.(Arg132∗)] similarly presented with primary amenorrhea, short stature and low weight, as well as additionally showing a lack of breast development. In all affected females in both families, ovaries were invisible on ultrasound and the uteri were infantile. All unaffected (fe)males of both families were healthy, except for a younger sister of the two affected daughters from the first family, who also showed developmental delay and short stature. Repair of chromosome breaks was impaired in lymphocytes from affected, but not unaffected, females in both families, consistent with an MCM9 function in HR.43 Moreover, in a cohort of 109 women with idiopathic POI, Wood-Trageser et al.43 detected one additional heterozygous carrier for a likely damaging MCM9 variant p.(Val229Gly).
Numerous other female MCM9 carriers were subsequently reported with POI.27,34,35,36,38,39,40,41,42,44,80 Of interest, as was the case for MCM8 variant carriers with POI, the POI-affected MCM9 variant carriers typically presented with short stature,36,39,42,43 delayed puberty,35,42,43,44 delayed bone age,35,43,44 infantile uteri35,39,41,42,43,44 and/or small or invisible ovaries.35,42,43,44 The other pathologies found in MCM8 variant carriers with POI, including hypothyroidism amongst others, have not been reported in POI-affected MCM9 variant carriers.
MCM9 variants have further been linked to a predisposition for infertility in male carriers. For instance, in a cohort of 314 unrelated male patients with non-obstructive azoospermia or severe oligospermia, a homozygous MCM9 variant [p.(Gln434Pro)] was identified in a carrier with oligozoospermia, arrest of male meiosis and abnormal germ cell apoptosis.81 Like-wise, Goldberg et al.37 described a carrier of a heterozygous MCM9 variant [p.(Glu495∗)] diagnosed with severe oligoteratoasthenozoospermia (OMIM 619379) at the age of 35 years.
Cancer
MCM8
In view of the proposed involvement of MCM8 in (multiple) DNA repair mechanisms, including HR and MMR, a potential role in cancer as a tumor suppressor gene is plausible. This was confirmed by the in vivo studies of Lutzmann et al.,108 who found that MCM8KO mice show increased bone marrow DNA damage, leading to the development of myeloid tumors. The first evidence for a role in human cancer originated from a study by Golubicki et al.,23 who described a male patient with fertility issues and Lynch-like syndrome (LLS) presenting as early onset colorectal cancer without detectable germline MMR variants and MLH1 hypermethylation. This patient was found to be a compound heterozygous carrier of two possibly pathogenic MCM8 variants, p.(Lys118Glufs∗5) and p.(Ile138Met) (Table 1). Moreover, in a cohort of 131 Dutch unaffiliated familial cancer cases, Golubicki et al.23 identified a compound heterozygous MCM8 variant [p.(Ile231Lys), p.(Thr332Ala)] carrier with breast cancer (OMIM 114480), as well as five heterozygous MCM8 variant [p.(Ile717Val), p.(Ile138Met), p.(Asn629Ser), p.(Arg278Cys), p.(Ala737Thr)] carriers with either colonic polyposis or MMR-proficient familial CRC. The link with polyposis was further strengthened by findings of Soares de Lima et al.,104 who detected a likely pathogenic deletion in MCM8 (c.876-1delG) in a family with serrated polyposis syndrome (OMIM 617108), whilst other studies have linked the MCM8 variant p.(Glu341Lys) to an increased risk for breast cancer85,86 and uterine leiomyomas (OMIM 150699).107
Intriguingly, high expression levels of MCM8 have also been observed in a wide variety of cancer types and are associated with aggressive tumor behavior102 and poorer clinical outcomes (Table 1).91,97,98,103 In line with this, multiple cancer types show MCM8 gains of copy number, as identified by analyses of cancer datasets from The Cancer Genome Atlas.83,90 These findings suggest a potential oncogenic role for MCM8 in cancer development, in contrast to the previously discussed tumor suppressive functions. He et al.83 hypothesized that overexpression of MCM8 facilitates and exacerbates chromosomal rearrangement in malignant cells, which through the rearrangement of genes involved in cell survival, growth and migration may contribute to cancer progression and metastasis.
One model potentially reconciling the conflicting roles of MCM8 in cancer development requires MCM8 expression levels to remain within a specific reference range for normal functioning, with decreased expression because of loss of function variants leading to impaired DNA repair, whereas increased expression (e.g., through amplification) leads to uncontrolled DNA rearrangements (Figure 3). Further studies are needed to test the plausibility of this model and to gain a better understanding of MCM8 (dys)function during cancer development and progression. These studies would likely benefit from analyses of the mutational signatures of DNA from MCM8-deficient and MCM8-enhanced tumors, which has previously proven effective in identifying underlying mutational mechanisms caused by novel cancer-predisposing genes.109
Figure 3.

Proposed model for the role for MCM8 in cancer
If the expression of MCM8 must remain within a certain reference range for normal functioning, decreased expression through loss of function variants may lead to impaired DNA repair, promoting tumor development. Increased expression levels, through amplifications for instance, may lead to uncontrolled DNA rearrangements, which could similarly promote tumorigenesis. Of note, although MCM8 shows increased expression levels in a wide variety of cancer types, increased expression of MCM9 is only reported infrequently. Created with BioRender.com (2022).
MCM9
Similarly to MCM8, the proposed molecular functions of MCM9 suggest a tumor suppressive role in cancer development. This is supported by in vivo studies showing that MCM9-mutant mice are predisposed to cancer, including sex-specific cancers and hepatocellular carcinomas,9,108 as well as by the fact that human cancers frequently lose the MCM9 allele(s) via homo- and heterozygous deletions or translocations on 6q22.31, the genomic region containing the MCM9 gene (Table 1).15,100,101,106 In 2015, Goldberg et al.38 linked MCM9 variants [p.Glu225Lysfs∗4] to a predisposition to hereditary mixed polyposis and CRC, describing two sisters from consanguineous parents exhibiting primary amenorrhea, multiple types of colorectal polyps and early onset CRC. Of interest, the CRCs from both sisters were MMR-proficient, contradicting previous studies which implied that loss of MCM9 always leads to MMR deficiency.
However, this proposed role for germline MCM9 variants as predisposing factors for polyposis and/or cancer was not confirmed by numerous later studies. For instance, Liu et al.110 tested the presence of MCM9 variants in 109 patients with LLS, finding 15 variants [p.(Ser191Ser), p.(Gly242Gly), p.(Asn304Ser), p.(Arg424Arg), p.(Glu507Asp), p.(Ile531Thr), p.(Cys558Ser), p.(Gln658His), p.(Arg666Trp), p.(Thr758Ala), p.(Met887Arg), p.(Ser898Phe), p.(Asp963Asp), p.(Pro967Pro), p.(Met1096Val)], none of which were predicted to be (possibly) pathogenic. Similarly, Belhadj et al.111 found no enrichment of MCM9 variants in a cohort of 473 familial/early onset CRC cases compared to controls, whereas Terradas et al.112 were unable to detect homozygous or compound heterozygous MCM9 variant carriers in a cohort of 177 unrelated patients with nonaffiliated polyposis.
More recent findings, however, do support a role for MCM9 as a predisposing gene for (early onset) CRC and polyposis. In a cohort of 131 Dutch unaffiliated familial cancer cases, Golubicki et al.23 identified a compound heterozygous MCM9 variant [p.(Arg548Trp), p.(Asn51Ile)] carrier with LLS and POI, as well as a compound heterozygous MCM9 variant [p.(Lys1142Arg), p.(Leu547Pro)] carrier with MMR-proficient CRC. Moreover, Golubicki et al.23 identified 12 heterozygous carriers of nine distinct MCM9 variants [p.(Met1096Val), p.(Glu610∗), p.(Glu1012Gln), p.(Glu507Asp), p.(Asp715Val), p.(Lys1142Arg), p.(Ser663fs), p.(Leu639Val), p.(Asn304Ser)], of which five were affected by LLS, six by MMR-proficient familial cancer and one by familial CRC of unknown MMR status. Moreover, Goldberg et al.37 described another consanguineous family carrying an MCM9 variant [p.(Glu495∗)], with two homozygous sisters both affected by POI. One of these sisters was diagnosed with polyposis and CRC at the age of 31 years, whereas the other sister was diagnosed with a clear cell carcinoma of the cervix at age 37 years. Both parents were heterozygous carriers and were diagnosed with three polyps at the ages of 66–68 years, whereas the heterozygous brother of the two sisters was diagnosed with microsatellite stable CRC at age 35 years, as well as polyps and severe oligoteratoasthenozoospermia.
Taken together, the studies of Goldberg et al.37,38support a strong association between germline MCM9 variants and the development of (early onset) CRC and/or polyposis, whereas the studies by Liu et al.,110 Belhadj et al.111 and Terradas et al.112 suggest that the prevalence of pathogenic MCM9 variants in cohorts of patients with unexplained familial CRC, early onset CRC and polyposis is limited. Of interest, Alvarez-Mora et al.34 described a homozygous MCM9 variant [p.(Thr492Tyrfs∗4)] carrier with POI and a germ cell tumor, suggesting that germline MCM9 variants may potentially predispose carriers to other types of cancer as well.
Studies reporting high expression levels of MCM9 in tumors are scarce compared to similar studies of MCM8 (Table 1). Although higher expressions levels of MCM9 have been associated with poorer outcomes for human papillomavirus 18 positive cervical cancer (OMIM 603956) patients,87 they were also associated with better overall survival in patients with non-small cell lung cancer.113 Moreover, lower expression levels of MCM9 have been associated with resistance to radiotherapy in patients with nasopharyngeal carcinomas (OMIM 607107),114 as well as with a favorable prognosis in patients with breast cancer.115
Alzheimer disease (AD)
MCM8
Ratnakumar et al.78 found damaging MCM8 variants in five out of 1208 female AD cases but none in 2162 female controls, suggesting a link to the development of AD in females (Table 1). These authors hypothesized that estrogen loss at menopause confers increased vulnerability to AD in women, based on the fact that estrogen upregulates synapse genes78 and correlates with hippocampal size throughout the menstrual cycle.116,117 Moreover, surgical menopause was found to increase the lifetime risk for dementia, cognitive decline and AD.118,119 As early menopause is one of the hallmarks of POI, which is frequently diagnosed in MCM8 variant carriers, this provides a hypothetical underlying mechanism explaining the predisposition of MCM8 variant carriers to develop AD. The function of MCM8 does, however, substantially contrast with the functions of other genes that have been associated with AD, which for instance are involved in amyloid precursor protein metabolism (e.g., PSEN2 (OMIM 600759), APP (OMIM 104760), PSEN1 (OMIM 104311), etc.), cholesterol metabolism (e.g., APOE4 (OMIM 107741)), immune response (e.g., TREM2 (OMIM 605086)) and endocytosis (e.g., SORL1 (OMIM 602005), MEF2C (OMIM 600662)).120 Whether or not MCM8 variants may also contribute to AD through low estrogen levels therefore remains highly speculative, and should be tested in future studies.
Other conditions
MCM8
Two coding variants of MCM8 (rs3761873, rs16991617) were associated with abnormal uterine bleeding following use of copper intrauterine devices. However, because both variants are synonymous and no underlying mechanism has been proposed, the level of evidence for this association appears minimal.77
MCM9
MCM9 variants have also been associated with other diseases and/or clinical features, albeit to a limited extent (Table 1). For example, Pal et al.79 identified a multitude of MCM9 polymorphisms in the genomes of women with a Down syndrome child (OMIM 190685), hypothesizing that the variants may increase the chance of a child with Down syndrome because of their association with the recombination and nondisjunction of chromosome 21 at meiosis I stage of oogenesis in a maternal age-independent manner. Moreover, Bally et al.82 detected an MCM9 variant [p.(Arg247Trp)] in a family with hearing loss, balance issues and tremor. Although the balance and hearing loss were most likely explained by a COCH (OMIM 603196) variant [p.(Pro51Ser)], the tremor was linked to an MCM9 variant [p.(Arg247Trp)], because it was present in five out of five affected carriers but absent in all five unaffected carriers. The molecular mechanism(s) underlying the association between MCM9 variants and tremor remain to be clarified.
Discussion
Clinical implications and future recommendations
Following two decades of research, the (patho)physiological functions of MCM8 and MCM9 have become increasingly apparent. Considerable evidence supports a role for MCM8 and MCM9 in DNA replication (initiation), meiosis, HR and MMR, with potential involvement in more than one step of these complex reactions. Future studies are vital to define the exact functionalities of the MCM8-9 complex in each of the implicated DNA-related processes, but should also evaluate potential individual functionalities of MCM8 and MCM9, which are implied based on modest differences in associated pathologies of both proteins.
Consistent with the molecular functions, MCM8 and MCM9 have been associated with a variety of pathologies, including infertility and cancer. In the event of solid confirmation of the predisposing roles of germline MCM8/MCM9 variants to infertility and cancer, the clinical implications are numerous. With regards to infertility, for instance, current evidence suggests a need for universal screening of the MCM8/MCM9 genes in patients with unexplained (fe)male infertility and/or gonadal dysgenesis. This has already been implemented by a minority of laboratories121 and may result in the identification of additional MCM8/MCM9 variant carriers, allowing personalized genetic counseling of these otherwise unexplained infertility cases. The phenotype of infertile MCM8/MCM9 variant carriers, characterized by short stature, gonadal dysgenesis and hypothyroidism, among other features, should raise suspicion among clinicians regarding potential MCM8/MCM9 variant carriership. Of interest, several other genomic instability disorders, including Bloom syndrome (OMIM 210900),122 Nijmegen breakage syndrome (OMIM 251260),123 Ataxia telangiectasia (OMIM 208900)124 and Fanconi anemia (OMIM 227650),125 have similarly been associated with short stature, hypogonadism and/or endocrine dysfunctions, suggesting comparable etiologies.43
Despite the abundance of evidence supporting the causative role for MCM8/MCM9 variants in infertility, several important questions remain before the relevance of these variants to disease can be clearly defined. For example, it is currently uncertain whether heterozygous MCM8/MCM9 variant carriers face an increased risk for infertility as compared to the general population, and whether the type or location of variants within the MCM8/MCM9 genes influences the degree of risk. The latter is of special interest as at least half of reported MCM8/MCM9 variants are missense variants, the effect of which on MCM8-9 function generally remains unknown. Of interest, a previous review from Griffin et al.6 demonstrated a clear bias in the number of POI- or cancer-associated variants within the ATPase domain (61%) of MCM8, with the remaining variants located in the DNA binding domain of MCM8. For MCM9, on the other hand, the DNA binding domain was most commonly (43%) affected, followed by its ATPase (30%) and C-terminal domains (27%). Consequently, Griffin et al.6 hypothesized that missense variants within the conserved DNA binding and ATPase domains of MCM8/MCM9 may negatively alter their respective activities, and further proposed that missense variants within the extended C-terminal domain of MCM9 might affect protein-protein interactions and in this way may perturb the function of the MCM8-9 complex. Additional studies are needed to explore these proposed genotype-phenotype correlations.
The carrier frequency and lifetime prevalence of infertility among MCM8/MCM9 variant carriers also remains to be determined. To date, 26 MCM8 and 27 MCM9 variants have been reported in the ClinVar database,126 of which six and seven variants, respectively, are predicted to result in loss of protein function. Likewise, 1354 MCM8 and 1155 MCM9 variants are described in the gnomAD database.127 Of these, respectively 83 and 55 variants are predicted to result in loss of protein function and 498 and 562 variants, respectively, are missense variants, in-frame insertions or deletions. The estimated carrier frequency, calculated based on data from the gnomAD database,127 is 2.04 and 2.41 for any MCM8 or MCM9 variant, respectively. The carrier frequency of predicted loss of function variants is estimated to be 1.4e−3 for MCM8 and 2.5e−3 for MCM9, whereas the estimated carrier frequency of missense variants, in-frame insertions or deletions is 0.46 for MCM8 and 1.17 for MCM9. These estimates suggest that the carrier frequency of MCM8/MCM9 variants is relatively low, although future population-based studies are needed to confirm this suspicion.
The questions discussed above also apply to the potential role of MCM8/MCM9 variants in cancer. Currently, the association of homozygous MCM8/MCM9 variants with CRC and/or polyposis is supported by the strongest evidence, having been described in multiple unrelated families.23,37,38 This association is of considerable interest, because a sizable proportion of familial CRC aggregation currently remains unexplained: 16–35% of CRC cases are thought to have a hereditary origin, but variants in any of the high-penetrance CRC genes explain only 4–8% of cases.37,128,129 This suggests that variants of potential novel cancer-predisposing genes, such as MCM8/MCM9, may be responsible for at least part of the unexplained familial CRC aggregation, arguing for the inclusion of the MCM8/MCM9 genes in diagnostic testing for these cases, especially when infertility is also diagnosed.
Similarly, the MCM8/MCM9 genes should be included in diagnostic testing for LLS, because germline MMR variants and MLH1 hypermethylation are undetectable in about 30% of MSI CRCs.130,131 The presence of MSI in these cases may be explained by double somatic hits in the MMR genes, undetected germline variants in MMR genes or by a germline variant of other genes that are potentially involved in MMR, including MCM8/MCM9.23 Moreover, patients with double somatic hits in the MMR genes, explaining their MSI, could also be evaluated for germline predisposition beyond the canonical genes. Although the exact role for MCM8-9 in MMR remains to be clarified, illustrated by the fact that multiple MCM8/MCM9 variant carriers show MMR-proficient/microsatellite stable CRCs, inclusion of the MCM8/MCM9 genes in diagnostic testing for LLS cases may potentially resolve a proportion of these cases and at the same time yield a better understanding of the role of MCM8/MCM9 in MMR.23,37,38 In the latter case, one could argue for the inclusion of fertility evaluation in the diagnostic algorithm of LLS to identify patients at risk for germline MCM8/MCM9 variants.
A possible explanation for the lack of MMR-deficiency/MSI in MCM9-deficient tumors may hypothetically involve the assessment method of MSI and the type of frameshift variants caused by MCM9 deficiency, with MCM9 deficiency possibly resulting in tri- or tetranucleotide frameshifts rather than mono- or dinucleotide frameshifts. Such elevated microsatellite instability at selected tetranucleotide repeats (EMAST) is for example observed in MSH3-deficient tumors and is generally not part of the conventional MSI detection methods, which focus on mono- and dinucleotide markers.132 Although highly speculative and confirmation in future studies is needed, this would indicate the use of inappropriate MSI tests rather than the absence of MMR deficiency/MSI in MCM9-deficient tumors. In addition to MSI, MCM8/MCM9-deficienct tumors may also be characterized by homologous recombination deficiency (HRD), considering their implicated roles in HR.
A predisposing role for germline MCM8/MCM9 variants in CRC development and polyposis suggests that MCM8/MCM9 variant carriers may benefit from early surveillance using colonoscopy protocols. This has already been recommended for all Lynch syndrome carriers (OMIM 120435) as a preventive measure regarding CRC development, and has proven effective in reducing both CRC incidence and mortality.133,134,135,136,137
Besides germline variants, high expression levels of MCM8 have been detected in a wide variety of tumor types, suggesting a Janus-like role for MCM8 in cancer. Although the postulated requirement for MCM8 to remain within a certain reference range, to maintain control of DNA repair and rearrangements, may explain the dual role of MCM8 in cancer, this model remains hypothetical and needs to be evaluated in future studies. This dual role appears less prominent in the case of MCM9, and high expression levels of MCM9 are far less commonly reported.
The therapeutic implications of MCM8-9 functioning in cancer are an active subject of discussion, because multiple studies have reported that depletion or loss of function of MCM8/MCM9 hypersensitizes cancer cells to interstrand crosslinking agents (e.g., cisplatin, oxaliplatin)15,19,23,127 and poly(ADP-ribose) polymerase inhibitor-based chemotherapy (e.g., olaparib).138 This most likely depends on the role of MCM8-9 in HR, which is responsible for the repair of DNA modifications induced by both types of therapy. If confirmed in future studies this could have important clinical implications. Firstly, it suggests that MCM8/MCM9 variant carriers affected by cancer may especially benefit from these types of therapies. The latter prediction is clinically supported by the complete response to interstrand crosslinking agents observed in two homozygous MCM9 variant [p.(E225Kfs∗4)] carriers affected by CRC.38 Secondly, it highlights the potential clinical use of MCM8/MCM9 inhibitors as interstrand crosslinking agent/poly(ADP-ribose) polymerase inhibitor-based chemotherapy sensitizers. The latter approach may be applicable to multiple types of cancer and should therefore be evaluated in future studies.
For most other pathology associations, for example those linking MCM8 to abnormal uterine bleeding and Alzheimer disease or MCM9 to tremor and birth of a child with Down syndrome, the level of evidence remains limited, because it is predominantly based on single studies and relatively small numbers of cases. As such, these associations must be viewed with caution during genetic counseling, and future studies are essential to further solidify these putative associations.
Conclusions
Although MCM8 and MCM9 are both involved in DNA-related processes, further studies are needed to clarify their precise molecular functions and to improve our understanding of the mechanisms underlying associated pathologies. Ultimately, our goal should be to provide a complete picture of MCM8-9 (patho)physiological functioning and the MCM8/MCM9 variant carrier phenotype, allowing optimization of MCM8/MCM9 variant carrier management and the potential exploitation of MCM8 and MCM9 in other facets of scientific research and medical care.
Acknowledgments
The authors gratefully acknowledge the editing assistance of MedicalEditing.nl. MG and MA were supported by Foundation Nelia et Amadeo Barletta and the Argentinian National Cancer Institute. L.B. and S.C.B. were supported by Fondo de Investigación Sanitaria/FEDER (20/00113), Fundació La Marató de TV3 (2019-202008-10), Fundación Científica de la Asociación Española Contra el Cáncer (PRYGN211085CAST), CERCA Program (Generalitat de Catalunya), and Agència de Gestió d'Ajuts Universitaris i de Recerca, Generalitat de Catalunya, GRPRE2017SGR21). CIBEREHD is funded by the Instituto deSalud Carlos III. The work was carried out (in part) at the Esther Koplowitz Center, Barcelona. The funders had no role in the study design, data acquisition and analysis, decision to publish, or preparation of the manuscript.
Author contributions
N.C.H.: Conceptualization, Writing – Original Draft, Writing – Review and Editing, Visualization; D.T.: Writing – Review and Editing; L.B.: Writing – Review and Editing; M.G.: Writing – Review and Editing; M.A.: Writing – Review and Editing; H.M.: Writing – Review and Editing; T.v.W.: Writing – Review and Editing; S.C-B.: Writing – Review and Editing; Y.G.: Writing – Review and Editing; M.N.: Conceptualization, Writing – Original Draft, Writing – Review and Editing.
Declaration of interests
None.
References
- 1.Gozuacik D., Chami M., Lagorce D., Faivre J., Murakami Y., Poch O., Biermann E., Knippers R., Bréchot C., Paterlini-Bréchot P. Identification and functional characterization of a new member of the human Mcm protein family: hMcm8. Nucleic Acids Res. 2003;31:570–579. doi: 10.1093/nar/gkg136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson E.M., Kinoshita Y., Daniel D.C. A new member of the MCM protein family encoded by the human MCM8 gene, located contrapodal to GCD10 at chromosome band 20p12.3-13. Nucleic Acids Res. 2003;31:2915–2925. doi: 10.1093/nar/gkg395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida K. Identification of a novel cell-cycle-induced MCM family protein MCM9. Biochem. Biophys. Res. Commun. Jun 3 2005;331:669–674. doi: 10.1016/j.bbrc.2005.03.222. [DOI] [PubMed] [Google Scholar]
- 4.Maiorano D., Lutzmann M., Méchali M. MCM proteins and DNA replication. Curr. Opin. Cell Biol. 2006;18:130–136. doi: 10.1016/j.ceb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Tye B.K. MCM proteins in DNA replication. Annu. Rev. Biochem. 1999;68:649–686. doi: 10.1146/annurev.biochem.68.1.649. [DOI] [PubMed] [Google Scholar]
- 6.Griffin W.C., Trakselis M.A. The MCM8/9 complex: a recent recruit to the roster of helicases involved in genome maintenance. DNA Repair. 2019;76:1–10. doi: 10.1016/j.dnarep.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lutzmann M., Méchali M. MCM9 binds Cdt1 and is required for the assembly of prereplication complexes. Mol. Cell. 2008;31:190–200. doi: 10.1016/j.molcel.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Lutzmann M., Méchali M. How to load a replicative helicase onto chromatin: a more and more complex matter during evolution. Cell Cycle. 2009;8:1309–1313. doi: 10.4161/cc.8.9.8216. [DOI] [PubMed] [Google Scholar]
- 9.Hartford S.A., Luo Y., Southard T.L., Min I.M., Lis J.T., Schimenti J.C. Minichromosome maintenance helicase paralog MCM9 is dispensible for DNA replication but functions in germ-line stem cells and tumor suppression. Proc. Natl. Acad. Sci. USA. 2011;108:17702–17707. doi: 10.1073/pnas.1113524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volkening M., Hoffmann I. Involvement of human MCM8 in prereplication complex assembly by recruiting hcdc6 to chromatin. Mol. Cell Biol. 2005;25:1560–1568. doi: 10.1128/MCB.25.4.1560-1568.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanton H.L., Radford S.J., McMahan S., Kearney H.M., Ibrahim J.G., Sekelsky J. REC, Drosophila MCM8, drives formation of meiotic crossovers. PLoS Genet. 2005;1:e40. doi: 10.1371/journal.pgen.0010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crismani W., Portemer V., Froger N., Chelysheva L., Horlow C., Vrielynck N., Mercier R. MCM8 is required for a pathway of meiotic double-strand break repair independent of DMC1 in Arabidopsis thaliana. PLoS Genet. 2013;9:e1003165. doi: 10.1371/journal.pgen.1003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohl K.P., Jones C.D., Sekelsky J. Evolution of an MCM complex in flies that promotes meiotic crossovers by blocking BLM helicase. Science. 2012;338:1363–1365. doi: 10.1126/science.1228190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann M., Kohl K.P., Sekelsky J., Hatkevich T. Meiotic MCM proteins promote and inhibit crossovers during meiotic recombination. Genetics. 2019;212:461–468. doi: 10.1534/genetics.119.302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K.Y., Im J.S., Shibata E., Park J., Handa N., Kowalczykowski S.C., Dutta A. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat. Commun. 2015;6:7744. doi: 10.1038/ncomms8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lutzmann M., Grey C., Traver S., Ganier O., Maya-Mendoza A., Ranisavljevic N., Bernex F., Nishiyama A., Montel N., Gavois E., et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell. 2012;47:523–534. doi: 10.1016/j.molcel.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 17.Natsume T., Nishimura K., Minocherhomji S., Bhowmick R., Hickson I.D., Kanemaki M.T. Acute inactivation of the replicative helicase in human cells triggers MCM8-9-dependent DNA synthesis. Genes Dev. 2017;31:816–829. doi: 10.1101/gad.297663.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimura K., Ishiai M., Horikawa K., Fukagawa T., Takata M., Takisawa H., Kanemaki M.T. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell. 2012;47:511–522. doi: 10.1016/j.molcel.2012.05.047. [DOI] [PubMed] [Google Scholar]
- 19.Park J., Long D.T., Lee K.Y., Abbas T., Shibata E., Negishi M., Luo Y., Schimenti J.C., Gambus A., Walter J.C., Dutta A. The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol. Cell Biol. 2013;33:1632–1644. doi: 10.1128/MCB.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinzey D.R., Gomathinayagam S., Griffin W.C., Klinzing K.N., Jeffries E.P., Rajkovic A., Trakselis M.A. Motifs of the C-terminal domain of MCM9 direct localization to sites of mitomycin-C damage for RAD51 recruitment. J. Biol. Chem. 2021;296:100355. doi: 10.1016/j.jbc.2021.100355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traver S., Coulombe P., Peiffer I., Hutchins J.R.A., Kitzmann M., Latreille D., Méchali M. MCM9 is required for mammalian DNA mismatch repair. Mol. Cell. 2015;59:831–839. doi: 10.1016/j.molcel.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Liu K., Wang Y., Zhu Q., Li P., Chen J., Tang Z., Shen Y., Cheng X., Lu L.Y., Liu Y. Aberrantly expressed HORMAD1 disrupts nuclear localization of MCM8-MCM9 complex and compromises DNA mismatch repair in cancer cells. Cell Death Dis. 2020;11:519. doi: 10.1038/s41419-020-2736-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golubicki M., Bonjoch L., Acuna-Ochoa J.G., Díaz-Gay M., Muñoz J., Cuatrecasas M., Ocaña T., Iseas S., Mendez G., Cisterna D., et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight. 2020;5 doi: 10.1172/jci.insight.140698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.AlAsiri S., Basit S., Wood-Trageser M.A., Yatsenko S.A., Jeffries E.P., Surti U., Ketterer D.M., Afzal S., Ramzan K., Faiyaz-Ul Haque M., et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J. Clin. Invest. 2015;125:258–262. doi: 10.1172/JCI78473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tenenbaum-Rakover Y., Weinberg-Shukron A., Renbaum P., Lobel O., Eideh H., Gulsuner S., Dahary D., Abu-Rayyan A., Kanaan M., Levy-Lahad E., et al. Minichromosome maintenance complex component 8 (MCM8) gene mutations result in primary gonadal failure. J. Med. Genet. 2015;52:391–399. doi: 10.1136/jmedgenet-2014-102921. [DOI] [PubMed] [Google Scholar]
- 26.Dou X., Guo T., Li G., Zhou L., Qin Y., Chen Z.J. Minichromosome maintenance complex component 8 mutations cause primary ovarian insufficiency. Fertil. Steril. 2016;106:1485–1489.e2. doi: 10.1016/j.fertnstert.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Desai S., Wood-Trageser M., Matic J., Chipkin J., Jiang H., Bachelot A., Dulon J., Sala C., Barbieri C., Cocca M., et al. MCM8 and MCM9 nucleotide variants in women with primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 2017;102:576–582. doi: 10.1210/jc.2016-2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouali N., Francou B., Bouligand J., Imanci D., Dimassi S., Tosca L., Zaouali M., Mougou S., Young J., Saad A., Guiochon-Mantel A. New MCM8 mutation associated with premature ovarian insufficiency and chromosomal instability in a highly consanguineous Tunisian family. Fertil. Steril. 2017;108:694–702. doi: 10.1016/j.fertnstert.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y.X., He W.B., Xiao W.J., Meng L.L., Tan C., Du J., Lu G.X., Lin G., Tan Y.Q. Novel loss-of-function mutation in MCM8 causes premature ovarian insufficiency. Mol. Genet. Genomic Med. 2020;8:e1165. doi: 10.1002/mgg3.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heddar A., Beckers D., Fouquet B., Roland D., Misrahi M. A novel phenotype combining primary ovarian insufficiency growth retardation and pilomatricomas with MCM8 mutation. J. Clin. Endocrinol. Metab. 2020;105:dgaa155. doi: 10.1210/clinem/dgaa155. [DOI] [PubMed] [Google Scholar]
- 31.Wang F., Guo S., Li P. Two novel mutations in the MCM8 gene shared by two Chinese siblings with primary ovarian insufficiency and short stature. Mol. Genet. Genomic Med. 2020;8:e1396. doi: 10.1002/mgg3.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin H., Ahn J., Park Y., Sim J., Park H.S., Ryu C.S., Kim N.K., Kwack K. Identification of potential causal variants for premature ovarian failure by whole exome sequencing. BMC Med. Genom. 2020;13:159. doi: 10.1186/s12920-020-00813-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tucker E.J., Bell K.M., Robevska G., van den Bergen J., Ayers K.L., Listyasari N., Faradz S.M., Dulon J., Bakhshalizadeh S., Sreenivasan R., et al. Meiotic genes in premature ovarian insufficiency: variants in HROB and REC8 as likely genetic causes. Eur. J. Hum. Genet. 2022;30:219–228. doi: 10.1038/s41431-021-00977-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarez-Mora M.I., Todeschini A.L., Caburet S., Perets L.P., Mila M., Younis J.S., Shalev S., Veitia R.A. An exome-wide exploration of cases of primary ovarian insufficiency uncovers novel sequence variants and candidate genes. Clin. Genet. 2020;98:293–298. doi: 10.1111/cge.13803. [DOI] [PubMed] [Google Scholar]
- 35.Fauchereau F., Shalev S., Chervinsky E., Beck-Fruchter R., Legois B., Fellous M., Caburet S., Veitia R.A. A non-sense MCM9 mutation in a familial case of primary ovarian insufficiency. Clin. Genet. 2016;89:603–607. doi: 10.1111/cge.12736. [DOI] [PubMed] [Google Scholar]
- 36.França M.M., Funari M.F.A., Lerario A.M., Santos M.G., Nishi M.Y., Domenice S., Moraes D.R., Costalonga E.F., Maciel G.A.R., Maciel-Guerra A.T., et al. Screening of targeted panel genes in Brazilian patients with primary ovarian insufficiency. PLoS One. 2020;15:e0240795. doi: 10.1371/journal.pone.0240795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldberg Y., Aleme O., Peled-Perets L., Castellvi-Bel S., Nielsen M., Shalev S.A. MCM9 is associated with germline predisposition to early-onset cancer-clinical evidence. NPJ Genom. Med. 2021;6:78. doi: 10.1038/s41525-021-00242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldberg Y., Halpern N., Hubert A., Adler S.N., Cohen S., Plesser-Duvdevani M., Pappo O., Shaag A., Meiner V. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015;208:621–624. doi: 10.1016/j.cancergen.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Guo T., Zheng Y., Li G., Zhao S., Ma J., Qin Y. Novel pathogenic mutations in minichromosome maintenance complex component 9 (MCM9) responsible for premature ovarian insufficiency. Fertil. Steril. 2020;113:845–852. doi: 10.1016/j.fertnstert.2019.11.015. [DOI] [PubMed] [Google Scholar]
- 40.Liu H., Wei X., Sha Y., Liu W., Gao H., Lin J., Li Y., Tang Y., Wang Y., Wang Y., Su Z. Whole-exome sequencing in patients with premature ovarian insufficiency: early detection and early intervention. J. Ovarian Res. 2020;13:114. doi: 10.1186/s13048-020-00716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen J., Qu D., Gao Y., Sun F., Xie J., Sun X., Wang D., Ma X., Cui Y., Liu J., Diao F. Genetic etiologic analysis in 74 Chinese Han women with idiopathic premature ovarian insufficiency by combined molecular genetic testing. J. Assist. Reprod. Genet. 2021;38:965–978. doi: 10.1007/s10815-021-02083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turkyilmaz A., Cayir A., Yarali O., Kurnaz E., Kartal Baykan E., Arslan Ates E., Demirbilek H. Clinical characteristics and molecular genetic analysis of a cohort with idiopathic congenital hypogonadism. J. Pediatr. Endocrinol. Metab. 2021;34:771–780. doi: 10.1515/jpem-2020-0590. [DOI] [PubMed] [Google Scholar]
- 43.Wood-Trageser M.A., Gurbuz F., Yatsenko S.A., Jeffries E.P., Kotan L.D., Surti U., Ketterer D.M., Matic J., Chipkin J., Jiang H., et al. MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am. J. Hum. Genet. 2014;95:754–762. doi: 10.1016/j.ajhg.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X., Touraine P., Desai S., Humphreys G., Jiang H., Yatsenko A., Rajkovic A. Gene variants identified by whole-exome sequencing in 33 French women with premature ovarian insufficiency. J. Assist. Reprod. Genet. 2019;36:39–45. doi: 10.1007/s10815-018-1349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Méndez J., Stillman B. Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. Bioessays. 2003;25:1158–1167. doi: 10.1002/bies.10370. [DOI] [PubMed] [Google Scholar]
- 46.Mizushima T., Takahashi N., Stillman B. Cdc6p modulates the structure and DNA binding activity of the origin recognition complex in vitro. Genes Dev. 2000;14:1631–1641. [PMC free article] [PubMed] [Google Scholar]
- 47.Im J.S., Ki S.H., Farina A., Jung D.S., Hurwitz J., Lee J.K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. USA. 2009;106:15628–15632. doi: 10.1073/pnas.0908039106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moyer S.E., Lewis P.W., Botchan M.R. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mimura S., Masuda T., Matsui T., Takisawa H. Central role for cdc45 in establishing an initiation complex of DNA replication in Xenopus egg extracts. Gene Cell. 2000;5:439–452. doi: 10.1046/j.1365-2443.2000.00340.x. [DOI] [PubMed] [Google Scholar]
- 50.Maiorano D., Cuvier O., Danis E., Méchali M. MCM8 is an MCM2-7-related protein that functions as a DNA helicase during replication elongation and not initiation. Cell. 2005;120:315–328. doi: 10.1016/j.cell.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Kinoshita Y., Johnson E.M., Gordon R.E., Negri-Bell H., Evans M.T., Coolbaugh J., Rosario-Peralta Y., Samet J., Slusser E., Birkenbach M.P., Daniel D.C. Colocalization of MCM8 and MCM7 with proteins involved in distinct aspects of DNA replication. Microsc. Res. Tech. 2008;71:288–297. doi: 10.1002/jemt.20553. [DOI] [PubMed] [Google Scholar]
- 52.Li J., Yu D., Liu L., Liang H., Ouyang Q., Liu Y. Structural study of the N-terminal domain of human MCM8/9 complex. Structure. 2021;29:1171–1181.e4. doi: 10.1016/j.str.2021.05.006. [DOI] [PubMed] [Google Scholar]
- 53.Zeng H., Li J., Xu H., Li H., Liu Y. Crystal structure of the winged-helix domain of MCM8. Biochem. Biophys. Res. Commun. 2020;526:993–998. doi: 10.1016/j.bbrc.2020.03.150. [DOI] [PubMed] [Google Scholar]
- 54.Griffin W.C., McKinzey D.R., Klinzing K.N., Baratam R., Eliyapura A., Trakselis M.A. A multi-functional role for the MCM8/9 helicase complex in maintaining fork integrity during replication stress. Nat. Commun. 2022;13:5090. doi: 10.1038/s41467-022-32583-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hillers K.J., Jantsch V., Martinez-Perez E., Yanowitz J.L. WormBook. Vol. 2017. 2017. Meiosis; pp. 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grell R.F. Time of recombination in the DROSOPHILA MELANOGASTER oocyte. III. Selection and characterization of temperature-sensitive and -insensitive, recombination-deficient alleles in Drosophila. Genetics. 1984;108:425–443. doi: 10.1093/genetics/108.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guilbaud G., Sale J.E. Unwinding to recombine. Mol. Cell. 2012;47:493–494. doi: 10.1016/j.molcel.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 58.Huang J.W., Acharya A., Taglialatela A., Nambiar T.S., Cuella-Martin R., Leuzzi G., Hayward S.B., Joseph S.A., Brunette G.J., Anand R., et al. MCM8IP activates the MCM8-9 helicase to promote DNA synthesis and homologous recombination upon DNA damage. Nat. Commun. 2020;11:2948. doi: 10.1038/s41467-020-16718-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hustedt N., Saito Y., Zimmermann M., Álvarez-Quilón A., Setiaputra D., Adam S., McEwan A., Yuan J.Y., Olivieri M., Zhao Y., et al. Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes Dev. 2019;33:1397–1415. doi: 10.1101/gad.329508.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Helderman N.C., Bajwa-Ten Broeke S.W., Morreau H., Suerink M., Terlouw D., van der Werf-' T Lam A.S., van Wezel T., Nielsen M. The diverse molecular profiles of lynch syndrome-associated colorectal cancers are (highly) dependent on underlying germline mismatch repair mutations. Crit. Rev. Oncol. Hematol. 2021;163:103338. doi: 10.1016/j.critrevonc.2021.103338. [DOI] [PubMed] [Google Scholar]
- 61.Peltomäki P. Update on Lynch syndrome genomics. Fam. Cancer. 2016;15:385–393. doi: 10.1007/s10689-016-9882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamura K., Kaneda M., Futagawa M., Takeshita M., Kim S., Nakama M., Kawashita N., Tatsumi-Miyajima J. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int. J. Clin. Oncol. 2019;24:999–1011. doi: 10.1007/s10147-019-01494-y. [DOI] [PubMed] [Google Scholar]
- 63.Bridge G., Rashid S., Martin S.A. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers. 2014;6:1597–1614. doi: 10.3390/cancers6031597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stojic L., Brun R., Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair. 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 65.Hutchins J.R.A., Traver S., Coulombe P., Peiffer I., Kitzmann M., Latreille D., Méchali M. Proteomic data on the nuclear interactome of human MCM9. Data Brief. 2016;6:410–415. doi: 10.1016/j.dib.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He C., Kraft P., Chen C., Buring J.E., Paré G., Hankinson S.E., Chanock S.J., Ridker P.M., Hunter D.J., Chasman D.I. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat. Genet. 2009;41:724–728. doi: 10.1038/ng.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carty C.L., Spencer K.L., Setiawan V.W., Fernandez-Rhodes L., Malinowski J., Buyske S., Young A., Jorgensen N.W., Cheng I., Carlson C.S., et al. Replication of genetic loci for ages at menarche and menopause in the multi-ethnic Population Architecture using Genomics and Epidemiology (PAGE) study. Hum. Reprod. 2013;28:1695–1706. doi: 10.1093/humrep/det071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen C.T.L., Liu C.T., Chen G.K., Andrews J.S., Arnold A.M., Dreyfus J., Franceschini N., Garcia M.E., Kerr K.F., Li G., et al. Meta-analysis of loci associated with age at natural menopause in African-American women. Hum. Mol. Genet. 2014;23:3327–3342. doi: 10.1093/hmg/ddu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spencer K.L., Malinowski J., Carty C.L., Franceschini N., Fernández-Rhodes L., Young A., Cheng I., Ritchie M.D., Haiman C.A., Wilkens L., et al. Genetic variation and reproductive timing: african American women from the population architecture using genomics and epidemiology (PAGE) study. PLoS One. 2013;8:e55258. doi: 10.1371/journal.pone.0055258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coignet M.V., Zirpoli G.R., Roberts M.R., Khoury T., Bandera E.V., Zhu Q., Yao S. Genetic variations, reproductive aging, and breast cancer risk in African American and European American women: the Women's Circle of Health Study. PLoS One. 2017;12:e0187205. doi: 10.1371/journal.pone.0187205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murray A., Bennett C.E., Perry J.R.B., Weedon M.N., Jacobs P.A., Morris D.H., Orr N., Schoemaker M.J., Jones M., Ashworth A., et al. Common genetic variants are significant risk factors for early menopause: results from the Breakthrough Generations Study. Hum. Mol. Genet. 2011;20:186–192. doi: 10.1093/hmg/ddq417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen C.T.L., Fernández-Rhodes L., Brzyski R.G., Carlson C.S., Chen Z., Heiss G., North K.E., Woods N.F., Rajkovic A., Kooperberg C., Franceschini N. Replication of loci influencing ages at menarche and menopause in Hispanic women: the Women's Health Initiative SHARe Study. Hum. Mol. Genet. 2012;21:1419–1432. doi: 10.1093/hmg/ddr570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schuh-Huerta S.M., Johnson N.A., Rosen M.P., Sternfeld B., Cedars M.I., Reijo Pera R.A. Genetic markers of ovarian follicle number and menopause in women of multiple ethnicities. Hum. Genet. 2012;131:1709–1724. doi: 10.1007/s00439-012-1184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nelson L.M. Clinical practice. Primary ovarian insufficiency. N. Engl. J. Med. 2009;360:606–614. doi: 10.1056/NEJMcp0808697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kherraf Z.E., Cazin C., Bouker A., Fourati Ben Mustapha S., Hennebicq S., Septier A., Coutton C., Raymond L., Nouchy M., Thierry-Mieg N., et al. Whole-exome sequencing improves the diagnosis and care of men with non-obstructive azoospermia. Am. J. Hum. Genet. 2022;109:508–517. doi: 10.1016/j.ajhg.2022.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen Y., Xu L., Zhu W., Zhang Z., Liu J., Jiang L., Liu X., Mao Y., Xu J., Yan X., et al. Associations of MCM8 rs3761873 and rs16991617 variants with abnormal uterine bleeding induced by copper intrauterine device. J. Obstet. Gynaecol. Res. 2022;48:440–447. doi: 10.1111/jog.15101. [DOI] [PubMed] [Google Scholar]
- 78.Ratnakumar A., Zimmerman S.E., Jordan B.A., Mar J.C. Estrogen activates Alzheimer's disease genes. Alzheimers Dement (N Y). 2019;5:906–917. doi: 10.1016/j.trci.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pal U., Halder P., Ray A., Sarkar S., Datta S., Ghosh P., Ghosh S. The etiology of Down syndrome: maternal MCM9 polymorphisms increase risk of reduced recombination and nondisjunction of chromosome 21 during meiosis I within oocyte. PLoS Genet. 2021;17:e1009462. doi: 10.1371/journal.pgen.1009462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jolly A., Bayram Y., Turan S., Aycan Z., Tos T., Abali Z.Y., Hacihamdioglu B., Coban Akdemir Z.H., Hijazi H., Bas S., et al. Exome sequencing of a primary ovarian insufficiency cohort reveals common molecular etiologies for a spectrum of disease. J. Clin. Endocrinol. Metab. 2019;104:3049–3067. doi: 10.1210/jc.2019-00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen S., Wang G., Zheng X., Ge S., Dai Y., Ping P., Chen X., Liu G., Zhang J., Yang Y., et al. Whole-exome sequencing of a large Chinese azoospermia and severe oligospermia cohort identifies novel infertility causative variants and genes. Hum. Mol. Genet. 2020;29:2451–2459. doi: 10.1093/hmg/ddaa101. [DOI] [PubMed] [Google Scholar]
- 82.Bally J.F., Zhang M., Dwosh E., Sato C., Rutka J., Lang A.E., Rogaeva E. Genomic study of a large family with complex neurological phenotype including hearing loss, imbalance and action tremor. Neurobiol. Aging. 2022;113:137–142. doi: 10.1016/j.neurobiolaging.2021.12.004. [DOI] [PubMed] [Google Scholar]
- 83.He D.M., Ren B.G., Liu S., Tan L.Z., Cieply K., Tseng G., Yu Y.P., Luo J.H. Oncogenic activity of amplified miniature chromosome maintenance 8 in human malignancies. Oncogene. 2017;36:3629–3639. doi: 10.1038/onc.2017.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu W., Gao F., Zhou H., Jin K., Shao J., Xu Z. Knockdown of MCM8 inhibits development and progression of bladder cancer in vitro and in vivo. Cancer Cell Int. 2021;21:242. doi: 10.1186/s12935-021-01948-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Verdiesen R.M., van der Schouw Y.T., van Gils C.H., Verschuren W.M., Broekmans F.J., Borges M.C., Soares A.L., Lawlor D.A., Eliassen A.H., Kraft P., et al. Genome-wide association study meta-analysis identifies three novel loci for circulating anti-Mullerian hormone levels in women. medRxiv. 2020 doi: 10.1101/2020.10.29.20221390. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Michailidou K., Lindström S., Dennis J., Beesley J., Hui S., Kar S., Lemaçon A., Soucy P., Glubb D., Rostamianfar A., et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–94. doi: 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sample K.M. DNA repair gene expression is associated with differential prognosis between HPV16 and HPV18 positive cervical cancer patients following radiation therapy. Sci. Rep. 2020;10:2774. doi: 10.1038/s41598-020-59383-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hao J., Deng H., Yang Y., Chen L., Wu Q., Yao P., Li J., Li B., Jin X., Wang H., Duan H. Downregulation of MCM8 expression restrains the malignant progression of cholangiocarcinoma. Oncol. Rep. 2021;46:235. doi: 10.3892/or.2021.8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cai L., Zhao K., Yuan X. Expression of minichromosome maintenance 8 in chronic myelogenous leukemia. Int. J. Clin. Exp. Pathol. 2015;8:14180–14188. [PMC free article] [PubMed] [Google Scholar]
- 90.Li Z., Xu X. Post-translational modifications of the mini-chromosome maintenance proteins in DNA replication. Genes. 2019;10:331. doi: 10.3390/genes10050331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang B., Lin M., Lu L., Chen W., Tan J., Zhao J., Cao Z., Zhu X., Lin J. Identification of mini-chromosome maintenance 8 as a potential prognostic marker and its effects on proliferation and apoptosis in gastric cancer. J. Cell Mol. Med. 2020;24:14415–14425. doi: 10.1111/jcmm.16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X., Zhang L., Song Y., Jiang Y., Zhang D., Wang R., Hu T., Han S. MCM8 is regulated by EGFR signaling and promotes the growth of glioma stem cells through its interaction with DNA-replication-initiating factors. Oncogene. 2021;40:4615–4624. doi: 10.1038/s41388-021-01888-1. [DOI] [PubMed] [Google Scholar]
- 93.Liu Z., Li J., Chen J., Shan Q., Dai H., Xie H., Zhou L., Xu X., Zheng S. MCM family in HCC: MCM6 indicates adverse tumor features and poor outcomes and promotes S/G2 cell cycle progression. BMC Cancer. 2018;18:200. doi: 10.1186/s12885-018-4056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wan W., Shen Y., Li Q. MCM10 acts as a potential prognostic biomarker and promotes cell proliferation in hepatocellular carcinoma: integrated bioinformatics analysis and experimental validation. Cancer Manag. Res. 2020;12:9609–9619. doi: 10.2147/CMAR.S267493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wen D.Y., Huang J.C., Wang J.Y., Pan W.Y., Zeng J.H., Pang Y.Y., Yang H. Potential clinical value and putative biological function of miR-122-5p in hepatocellular carcinoma: a comprehensive study using microarray and RNA sequencing data. Oncol. Lett. 2018;16:6918–6929. doi: 10.3892/ol.2018.9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiong D.D., Feng Z.B., Lai Z.F., Qin Y., Liu L.M., Fu H.X., He R.Q., Wu H.Y., Dang Y.W., Chen G., Luo D.Z. High throughput circRNA sequencing analysis reveals novel insights into the mechanism of nitidine chloride against hepatocellular carcinoma. Cell Death Dis. 2019;10:658. doi: 10.1038/s41419-019-1890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li S., Jiang Z., Li Y., Xu Y. Prognostic significance of minichromosome maintenance mRNA expression in human lung adenocarcinoma. Oncol. Rep. 2019;42:2279–2292. doi: 10.3892/or.2019.7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu K., Kang M., Liao X., Wang R. Genome-wide investigation of the clinical significance and prospective molecular mechanism of minichromosome maintenance protein family genes in patients with Lung Adenocarcinoma. PLoS One. 2019;14:e0219467. doi: 10.1371/journal.pone.0219467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xie G., Li Y., Jiang Y., Ye X., Tang J., Chen J. Silencing HIPPI suppresses tumor progression in non-small-cell lung cancer by inhibiting DNA replication. OncoTargets Ther. 2021;14:3467–3480. doi: 10.2147/OTT.S305388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Braggio E., Dogan A., Keats J.J., Chng W.J., Huang G., Matthews J.M., Maurer M.J., Law M.E., Bosler D.S., Barrett M., et al. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Mod. Pathol. 2012;25:651–660. doi: 10.1038/modpathol.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sung C.O., Kim S.C., Karnan S., Karube K., Shin H.J., Nam D.H., Suh Y.L., Kim S.H., Kim J.Y., Kim S.J., et al. Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood. 2011;117:1291–1300. doi: 10.1182/blood-2010-07-297861. [DOI] [PubMed] [Google Scholar]
- 102.Ren Z., Li J., Zhao S., Qiao Q., Li R. Knockdown of MCM8 functions as a strategy to inhibit the development and progression of osteosarcoma through regulating CTGF. Cell Death Dis. 2021;12:376. doi: 10.1038/s41419-021-03621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peng Y.P., Zhu Y., Yin L.D., Zhang J.J., Guo S., Fu Y., Miao Y., Wei J.S. The expression and prognostic roles of MCMs in pancreatic cancer. PLoS One. 2016;11:e0164150. doi: 10.1371/journal.pone.0164150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Soares de Lima Y., Arnau-Collell C., Diaz-Gay M., Bonjoch L., Franch-Expósito S., Muñoz J., Moreira L., Ocaña T., Cuatrecasas M., Herrera-Pariente C., et al. Germline and somatic whole-exome sequencing identifies new candidate genes involved in familial predisposition to serrated polyposis syndrome. Cancers. 2021;13 doi: 10.3390/cancers13040929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Feng X., Matsuo K., Zhang T., Hu Y., Mays A.C., Browne J.D., Zhou X., Sullivan C.A. MicroRNA profiling and target genes related to metastasis of salivary adenoid cystic carcinoma. Anticancer Res. 2017;37:3473–3481. doi: 10.21873/anticanres.11715. [DOI] [PubMed] [Google Scholar]
- 106.Kim J.H., Dhanasekaran S.M., Mehra R., Tomlins S.A., Gu W., Yu J., Kumar-Sinha C., Cao X., Dash A., Wang L., et al. Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res. 2007;67:8229–8239. doi: 10.1158/0008-5472.CAN-07-1297. [DOI] [PubMed] [Google Scholar]
- 107.Rafnar T., Gunnarsson B., Stefansson O.A., Sulem P., Ingason A., Frigge M.L., Stefansdottir L., Sigurdsson J.K., Tragante V., Steinthorsdottir V., et al. Variants associating with uterine leiomyoma highlight genetic background shared by various cancers and hormone-related traits. Nat. Commun. 2018;9:3636. doi: 10.1038/s41467-018-05428-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lutzmann M., Bernex F., da Costa de Jesus C., Hodroj D., Marty C., Plo I., Vainchenker W., Tosolini M., Forichon L., Bret C., et al. MCM8- and MCM9 deficiencies cause lifelong increased hematopoietic DNA damage driving p53-dependent myeloid tumors. Cell Rep. 2019;28:2851–2865.e4. doi: 10.1016/j.celrep.2019.07.095. [DOI] [PubMed] [Google Scholar]
- 109.Grolleman J.E., de Voer R.M., Elsayed F.A., Nielsen M., Weren R.D.A., Palles C., Ligtenberg M.J.L., Vos J.R., Ten Broeke S.W., de Miranda N.F., et al. Mutational signature analysis reveals NTHL1 deficiency to cause a multi-tumor phenotype. Cancer Cell. 2019;35:256–266.e5. doi: 10.1016/j.ccell.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 110.Liu Q., Hesson L.B., Nunez A.C., Packham D., Hawkins N.J., Ward R.L., Sloane M.A. Pathogenic germline MCM9 variants are rare in Australian Lynch-like syndrome patients. Cancer Genet. 2016;209:497–500. doi: 10.1016/j.cancergen.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 111.Belhadj S., Terradas M., Munoz-Torres P.M., Aiza G., Navarro M., Capellá G., Valle L. Candidate genes for hereditary colorectal cancer: mutational screening and systematic review. Hum. Mutat. 2020;41:1563–1576. doi: 10.1002/humu.24057. [DOI] [PubMed] [Google Scholar]
- 112.Terradas M., Munoz-Torres P.M., Belhadj S., Aiza G., Navarro M., Brunet J., Capellá G., Valle L. Contribution to colonic polyposis of recently proposed predisposing genes and assessment of the prevalence of NTHL1- and MSH3-associated polyposes. Hum. Mutat. 2019;40:1910–1923. doi: 10.1002/humu.23853. [DOI] [PubMed] [Google Scholar]
- 113.Huang C., Lei C., Pan B., Fang S., Chen Y., Cao W., Liu L. Potential prospective biomarkers for non-small cell lung cancer: mini-chromosome maintenance proteins. Front. Genet. 2021;12:587017. doi: 10.3389/fgene.2021.587017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun Z., Wang X., Wang J., Wang J., Liu X., Huang R., Chen C., Deng M., Wang H., Han F. Key radioresistance regulation models and marker genes identified by integrated transcriptome analysis in nasopharyngeal carcinoma. Cancer Med. 2021;10:7404–7417. doi: 10.1002/cam4.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu X., Liu Y., Wang Q., Song S., Feng L., Shi C. The alterations and potential roles of MCMs in breast cancer. JAMA Oncol. 2021;2021:7928937. doi: 10.1155/2021/7928937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barth C., Steele C.J., Mueller K., Rekkas V.P., Arélin K., Pampel A., Burmann I., Kratzsch J., Villringer A., Sacher J. In-vivo dynamics of the human hippocampus across the menstrual cycle. Sci. Rep. 2016;6:32833. doi: 10.1038/srep32833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Protopopescu X., Butler T., Pan H., Root J., Altemus M., Polanecsky M., McEwen B., Silbersweig D., Stern E. Hippocampal structural changes across the menstrual cycle. Hippocampus. 2008;18:985–988. doi: 10.1002/hipo.20468. [DOI] [PubMed] [Google Scholar]
- 118.Bove R., Secor E., Chibnik L.B., Barnes L.L., Schneider J.A., Bennett D.A., De Jager P.L. Age at surgical menopause influences cognitive decline and Alzheimer pathology in older women. Neurology. 2014;82:222–229. doi: 10.1212/WNL.0000000000000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rocca W.A., Bower J.H., Maraganore D.M., Ahlskog J.E., Grossardt B.R., de Andrade M., Melton L.J., 3rd Increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause. Neurology. 2007;69:1074–1083. doi: 10.1212/01.wnl.0000276984.19542.e6. [DOI] [PubMed] [Google Scholar]
- 120.Konig T., Stogmann E. Genetics of Alzheimer's disease. Wien Med. Wochenschr. 2021;171:249–256. doi: 10.1007/s10354-021-00819-9. Genetik der Alzheimer-Demenz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.The portal for rare diseases and orphan drugs. Orphanet Encyclopedia. 2022. https://www.orpha.net/consor/cgi-bin/index.php?lng=EN
- 122.German J. Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine. 1993;72:393–406. [PubMed] [Google Scholar]
- 123.Maraschio P., Peretti D., Lambiase S., Lo Curto F., Caufin D., Gargantini L., Minoli L., Zuffardi O. A new chromosome instability disorder. Clin. Genet. 1986;30:353–365. doi: 10.1111/j.1399-0004.1986.tb01892.x. [DOI] [PubMed] [Google Scholar]
- 124.Barlow C., Hirotsune S., Paylor R., Liyanage M., Eckhaus M., Collins F., Shiloh Y., Crawley J.N., Ried T., Tagle D., Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 125.Wong J.C.Y., Alon N., McKerlie C., Huang J.R., Meyn M.S., Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum. Mol. Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- 126.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.You Y.N., Borras E., Chang K., Price B.A., Mork M., Chang G.J., Rodriguez-Bigas M.A., Bednarski B.K., Meric-Bernstam F., Vilar E. Detection of pathogenic germline variants among patients with advanced colorectal cancer undergoing tumor genomic profiling for precision medicine. Dis. Colon Rectum. 2019;62:429–437. doi: 10.1097/DCR.0000000000001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jiao S., Peters U., Berndt S., Brenner H., Butterbach K., Caan B.J., Carlson C.S., Chan A.T., Chang-Claude J., Chanock S., et al. Estimating the heritability of colorectal cancer. Hum. Mol. Genet. 2014;23:3898–3905. doi: 10.1093/hmg/ddu087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Antelo M., Golubicki M., Roca E., Mendez G., Carballido M., Iseas S., Cuatrecasas M., Moreira L., Sanchez A., Carballal S., et al. Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int. J. Cancer. 2019;145:705–713. doi: 10.1002/ijc.32160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eikenboom E.L., van der Werf-'t Lam A.S., Rodríguez-Girondo M., Van Asperen C.J., Dinjens W.N.M., Hofstra R.M.W., Van Leerdam M.E., Morreau H., Spaander M.C.W., Wagner A., Nielsen M. Universal immunohistochemistry for lynch syndrome: a systematic review and meta-analysis of 58,580 colorectal carcinomas. Clin. Gastroenterol. Hepatol. 2022;20:e496–e507. doi: 10.1016/j.cgh.2021.04.021. [DOI] [PubMed] [Google Scholar]
- 132.Carethers J.M. Microsatellite instability pathway and EMAST in colorectal cancer. Curr. Colorectal Cancer Rep. 2017;13:73–80. doi: 10.1007/s11888-017-0352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brenner H., Chang-Claude J., Jansen L., Knebel P., Stock C., Hoffmeister M. Reduced risk of colorectal cancer up to 10 years after screening, surveillance, or diagnostic colonoscopy. Gastroenterology. 2014;146:709–717. doi: 10.1053/j.gastro.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 134.Järvinen H.J., Aarnio M., Mustonen H., Aktan-Collan K., Aaltonen L.A., Peltomäki P., De La Chapelle A., Mecklin J.P. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 135.Järvinen H.J., Renkonen-Sinisalo L., Aktán-Collán K., Peltomäki P., Aaltonen L.A., Mecklin J.P. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J. Clin. Oncol. 2009;27:4793–4797. doi: 10.1200/JCO.2009.23.7784. [DOI] [PubMed] [Google Scholar]
- 136.Newton K., Green K., Lalloo F., Evans D.G., Hill J. Colonoscopy screening compliance and outcomes in patients with Lynch syndrome. Colorectal Dis. 2015;17:38–46. doi: 10.1111/codi.12778. [DOI] [PubMed] [Google Scholar]
- 137.Renkonen-Sinisalo L., Aarnio M., Mecklin J.P., Järvinen H.J. Surveillance improves survival of colorectal cancer in patients with hereditary nonpolyposis colorectal cancer. Cancer Detect. Prev. 2000;24:137–142. [PubMed] [Google Scholar]
- 138.Morii I., Iwabuchi Y., Mori S., Suekuni M., Natsume T., Yoshida K., Sugimoto N., Kanemaki M.T., Fujita M. Inhibiting the MCM8-9 complex selectively sensitizes cancer cells to cisplatin and olaparib. Cancer Sci. 2019;110:1044–1053. doi: 10.1111/cas.13941. [DOI] [PMC free article] [PubMed] [Google Scholar]