

Abstract
Aging is a complex biological process that is accompanied by changes in gene expression and mutational load. In many species, including humans, older fathers pass on more paternally-derived de novo mutations; however, the cellular basis and cell types driving this pattern are still unclear. To explore the root causes of this phenomenon, we performed single-cell RNA-sequencing (scRNA-seq) on testes from young and old male Drosophila, as well as genomic sequencing (DNA-seq) on somatic tissues from the same flies. We found that early germ cells from old and young flies enter spermatogenesis with similar mutational loads, but older flies are less able to remove mutations during spermatogenesis. Mutations in old cells may also increase during spermatogenesis. Our data reveal that old and young flies have distinct mutational biases. Many classes of genes show increased post-meiotic expression in the germlines of older flies. Late spermatogenesis-biased genes have higher dN/dS than early spermatogenesis-biased genes, supporting the hypothesis that late spermatogenesis is a source of evolutionary innovation. Surprisingly, genes biased in young germ cells show higher dN/dS than genes biased in old germ cells. Our results provide novel insights into the role of the germline in de novo mutation.
Introduction
Aging is a process that is accompanied by phenotypic changes in animals. These phenotypic changes include both observable traits and intermediate traits, such as gene expression. Aging can impact not only the health of offspring, but also evolution. When reproductive organs age, they may pass a higher amount of de novo mutations to the offspring. Most novel mutations are inherited from the paternal germline, and the number of mutations inherited increases with paternal age 1–3. Some studies have attributed excess paternal mutations to the increased number of cell divisions that cycling spermatogonial stem cells undergo throughout the life of the male 4–6. Conversely, other reports have found that the excess cell divisions do not track with the ratio of maternal to paternal mutations during aging 1,7, which suggests instead that lifestyle, chemical and environmental factors may cause this discrepancy 8,9. Previous studies of the effect of age on paternally inherited mutations have inferred de novo mutations through sequencing of parents and offspring 3. These methods are highly useful, but they only capture de novo mutations that have evaded repair mechanisms, ended up inside a viable gamete, fertilized an egg, and created a viable embryo. Much less is known about the dynamics of mutation and repair inside the male germline. One study found that mutations arise least frequently in human spermatogonia 10. In our previous work 11, however, we instead found that mutational load is highest in the earliest stages of spermatogenesis. Taken together, these results imply that most mutations occur prior to germline stem cell (GSC) differentiation and are removed during spermatogenesis. Are these mutations replicative in origin? If so, we would expect germline stem cells from older flies to be more mutated than those from younger flies.
In addition to these mutational effects, aging is known to cause other germline phenotypes such as lower numbers of germ cells and reduced germline stem cell proliferative capacity 12. The GSC microenvironment also undergoes chemical changes associated with reductions in fecundity 13. These phenotypic consequences, in fact, could be linked to mutation, as the germline mutational rate in young adults correlates with longevity 14. As such, the germline mutation rate has consequences for both an organism and its descendants.
In our previous study 11, we used single-cell RNA-sequencing (scRNA-seq) to follow germline mutations throughout Drosophila spermatogenesis and found evidence that germline mutations decline in abundance throughout spermatogenesis. We also found evidence that some germline genome maintenance genes are more highly expressed in GSCs and early spermatogonia, which are the earliest male germ cells. Our results were in line with the idea that active DNA repair plays a role in the male germline 15, but since age is an important factor for mutational load, in this study we directly compare patterns in young and old testis.
To study transcriptional and mutational signatures in the aging germline, we generated scRNA-seq data from Drosophila melanogaster testes 48 hours and 25 days after eclosion (“Young” and “Old” respectively). We also sequenced correlated genomic DNA from each sample to confirm that each detected mutation was a real de novo germline mutation. Our results support our previous observation that the proportion of mutated cells declines throughout spermatogenesis for young flies. For old flies, however, the proportion of mutated cells begins high and remains high throughout spermatogenesis. We found that on a molecular level, each class of older germ cell has a higher mutational burden than comparable cells from young flies. Additionally, older flies carry a higher proportion of C>G and C>A mutations. Our results also indicate that the old germline is more highly mutated and transcriptionally dysregulated. We did not find evidence of a profound shift in the expression of genome maintenance genes; however, a number of these genes were highly expressed in young and old flies. We also find that patterns of global gene expression differ between young and old testes, including increased post-meiotic expression of de novo genes, TEs, and canonical genes. We found that early spermatogenesis-biased genes have lower dN/dS than late spermatogenesis-biased genes, and that genes biased in older germ cells have lower dN/dS than genes biased in younger germ cells. These findings provide a deeper insight into the process of spermatogenesis as a key source of de novo mutations.
Results
A cell atlas of the aged male germline
We aimed to capture representative cell types from the major somatic and germline cell types of the testis (Fig. 1A). We generated testes scRNA-seq data from male flies 48 hours (young) and 25 days (old) after eclosion to facilitate the identification of de novo mutations (Fig. 1B, Supplementary Table 1). Each of the six libraries was made with approximately 30 pairs of fly testes. We used Cellranger 16 to align these libraries against the FlyBase 17 D. melanogaster genome (version R6.32). We used previously established marker genes 18 to annotate cell types for the young and old flies separately. Dot plots showing the expression of key marker genes are shown in Extended Data Fig. 1. To confirm that mutations observed are from the germline, we prepared and sequenced somatic genomic DNA libraries from the carcasses of the same flies used for scRNA-seq and used these samples as control.
Fig. 1: Overview of experimental design and visualization of old and young datasets.
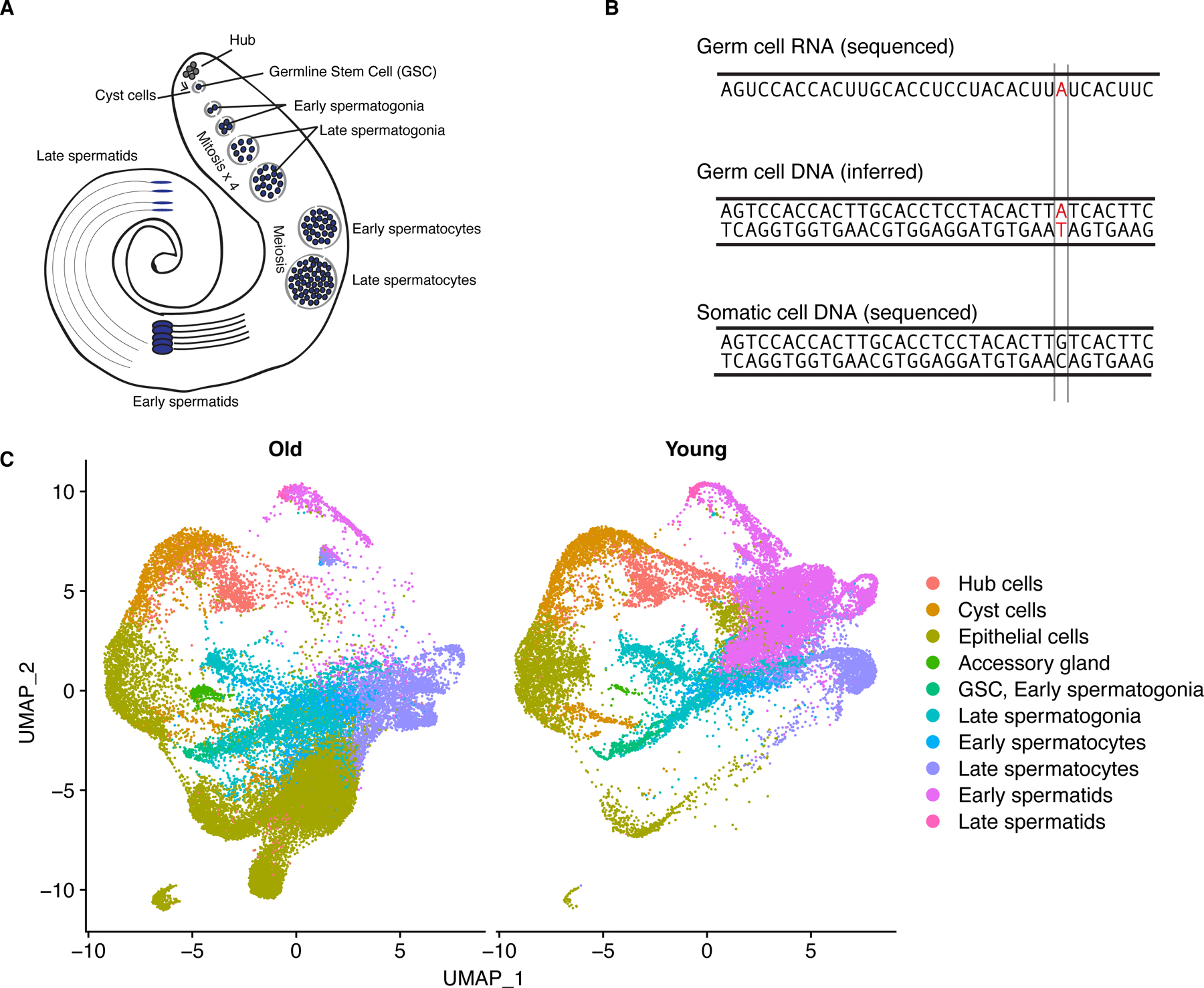
A) Diagram of major cell types in Drosophila testis 11. B) experimental rationale: we infer mutated genomic sites in germ cells using scRNA-seq data. If the same locus is unmutated in somatic cell DNA, we call the SNP in red a de novo mutation. We can detect a mutation if it is present on both strands or only the template strand. C) dimensional reduction showing the cell-type assignments of scRNA-seq data from young and old flies.
Using Seurat 4 19, we classified somatic cells into four broad types: hub cells, cyst cells, accessory gland, and epithelial cells. We split germ cells into six types, listed from earliest to latest: germline stem cells/early spermatogonia, late spermatogonia, early spermatocytes, late spermatocytes, early spermatids, and late spermatids. In total, we characterized 23489 cells from young flies and 28861 cells from old flies (Fig. 1C, Supplementary Table 1). We found that for each age group and cell type, the 3 replicates from each age group largely corroborate each other, with Pearson’s r values over 0.91 between replicates and cells of the same age (Extended Data Fig. 2). After cell type assignments, we used Seurat 4 to perform downstream analyses on the integrated dataset.
Old flies show impaired mutational repair in spermatogenesis
We identified germline SNPs in each sample and matched them to every cell with reads corroborating a given SNP (Fig. 2). To assess the mutational burden between young and old flies, we compared, for each cell type, the proportion of cells where at least one mutation was detected. We also compared, for every cell type, the number of detected SNPs per unique molecular index (UMI) to account for differences in coverage between libraries. We did not find any SNPs in more than one replicate, indicating that recurrent age-related transcriptional errors or RNA editing events did not bias our results.
Fig. 2: The proportion of mutated cells and mutation load across cell types for young and old flies.
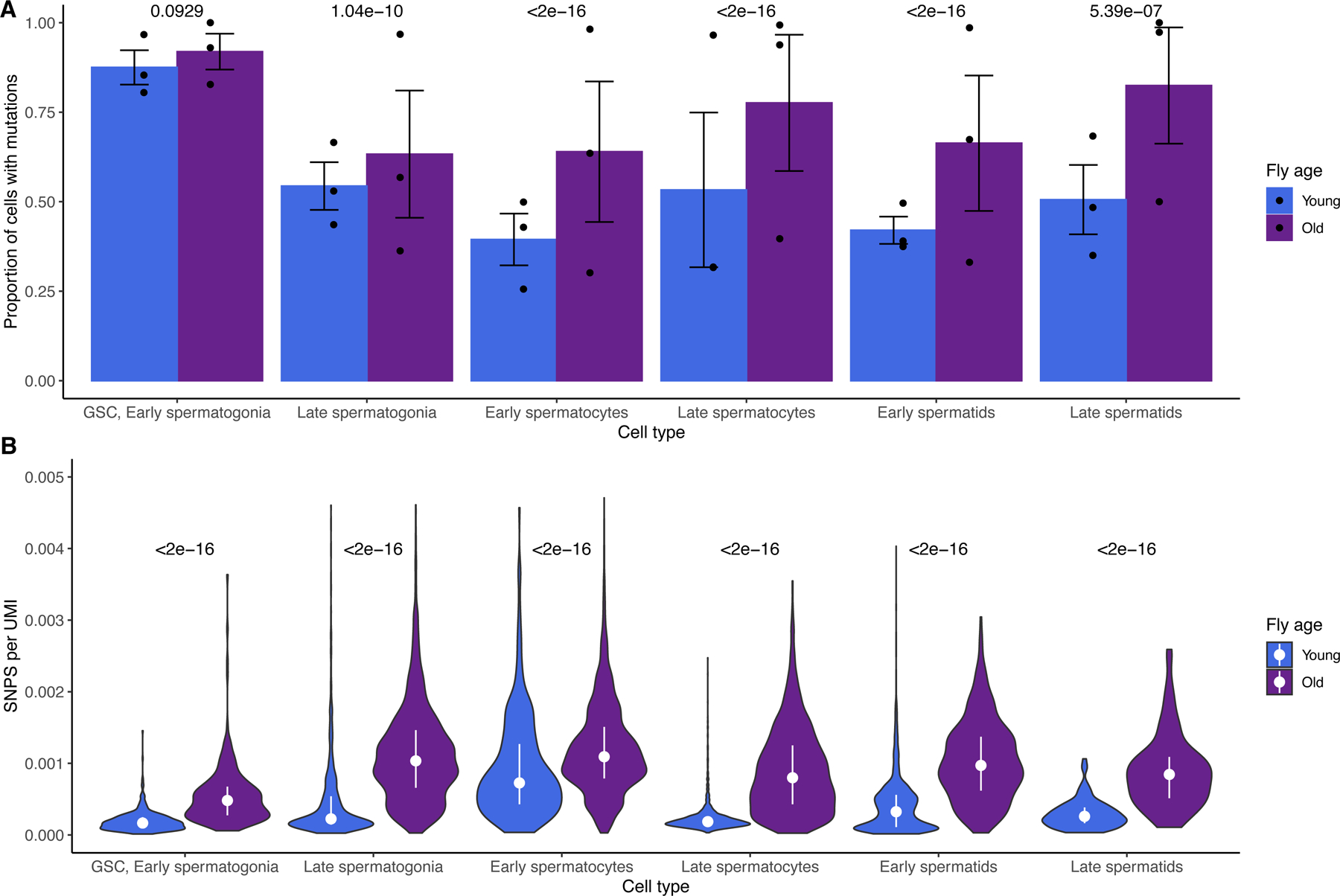
A) For old and young flies and every cell type, shown are the proportions of cells of each type carrying at least one mutation. Error bars are +/− 1 standard error, centered at the mean proportion. P values are Bonferroni-corrected from a two-sided chi-square test of mean proportions between young and old cells of a type. The proportion of mutated cells declines more for young flies than not old flies, during spermatogenesis. Raw p values, from left to right: 0.093, 3.5e-11, 1.9e-32, 1.5e-70, 1.8e-44, and 2.7e-07. B) for each cell, the number of SNPs divided by the number of Unique Molecular Indices (UMIs) detected, a proxy for read depth. P values are Bonferroni-corrected from a Wilcoxon test comparing young and old cells of a type. Bars represent the interquartile range, and the dot represents the median SNPs/UMI across biological replicates for each cell type. Statistical analyses were performed using full datasets. Every class of older germ cells has more mutations per RNA molecule detected. This indicates that the higher mutational load of older flies precedes spermatogenesis and may increase during spermatogenesis. Raw p values, from left to right: 5.36e-199, 7.22e-17, 1.97e-257, 0.00e+00, 2.79e-56, and 4.38e-20.
In young flies, the proportion of mutated cells declines drastically during spermatogenesis, indicating that lesions are either repaired or that mutated cells are removed from the population. Old flies begin spermatogenesis with a similar proportion of mutated GSC/early spermatogonia, but their mutational burden remains high throughout spermatogenesis. Proportions of mutated cells are statistically similar for young and old flies in GSC/Early spermatogonia but begin to diverge in later cell types. By the end of spermatogenesis, young flies achieve a lower proportion of mutated cells compared to older flies, whose proportion of mutated cells remains high throughout.
Old spermatocytes and spermatids have significantly higher proportions of mutated cells (Fig. 2A). To confirm that this trend was not confounded by different read depths across replicates, we counted the number of detected SNPs per Unique Molecular Index (UMI) for every cell type and found that RNA molecules from old cells are consistently more likely to carry mutations than young cells of the same type (Fig. 2B). This suggests that much of the elevated mutational load of the older germline occurs before spermatogenesis, accumulated within cycling germ cells. However, we also observe that later germ cells from old flies have more mutations per RNA molecule than older GSC/early spermatogonia. This observation would be expected if some germline mutations arose during spermatogenesis.
Old and young testes show distinct mutational biases
We compared the relative proportions of the six major classes of mutation between young and old flies. Young flies and old flies have distinct mutational signatures. In young flies, we found that C>T and T>G mutations are enriched compared to old flies (Fig. 3). Using a chi-square test of proportions, we found that old flies were significantly enriched for T>C and C>G mutations. This suggests an age-related mutational or repair bias during spermatogenesis. We asked whether these mutational signatures were due to differential activity of genome maintenance genes 11,20 and found that, as a group, genome maintenance genes are similarly expressed in most cell types as other annotated genes (Extended Data Fig. 3). As such, the mutational load of older flies cannot be purely attributed to age-related global downregulation of genomic maintenance genes, although it is likely that the regulation of genome maintenance genes at the protein level differs between young and old testis.
Fig. 3: Age-related trends in mutational signatures.
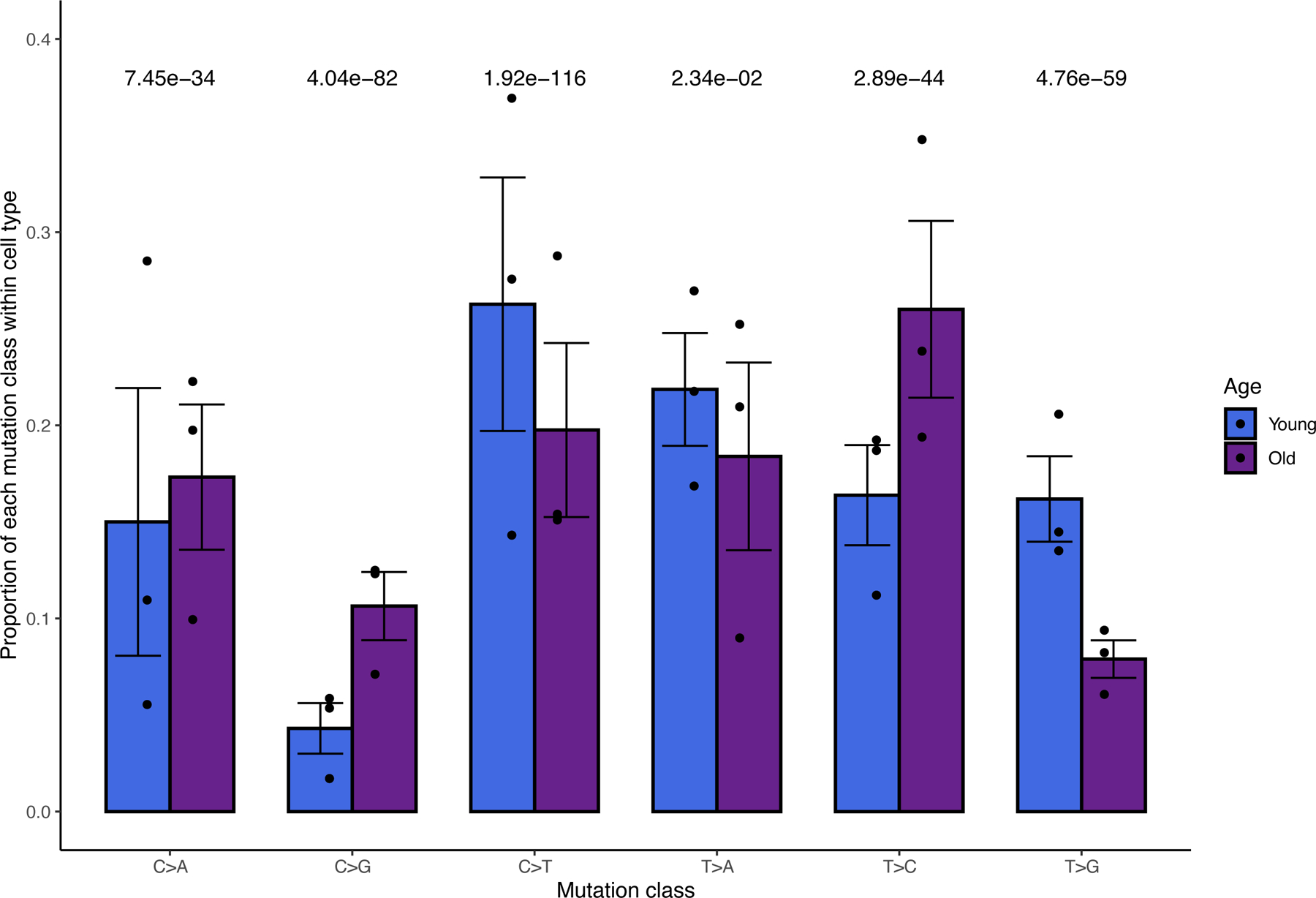
For young and old flies, shown are the relative proportions of the 6 types of mutations (each class is equivalent to a complementary mutation, for example, T>G also represents A>C). Error bars are +/− 1 standard error, centered at the mean proportion. Bonferroni-corrected P values are from a two-sided chi square test of proportions comparing the mean proportions between young and old flies (n=2 mean proportions for each signature, one from 3 young libraries and one from 3 old libraries). T>C and C>G mutations are enriched in old flies, while C>T and T>G mutations are underrepresented. Statistical analyses were performed using full datasets. Raw p values, from left to right: 1.9e-34, 4e-83, 1.6e-117, 0.012, 4.8e-45, and 5.9e-60.
Interestingly, although our method detects novel SNPs that only occur on the template strand during spermatogenesis, we observed that C>T mutations occur frequently. C>T mutations are the most common mutational class for young flies and the second most common class for old testis. This enrichment is in line with comparative genomic analysis from previous work 21, although our work and previous published works detected mutations at different timescales.
Many genome maintenance genes show age-biased expression
We performed differential expression testing between every cell type (Table 1) and focused on a list of 211 genes related to DNA damage repair compiled from our previous work 11,20. We found that in GSC/early spermatogonia, 7 genome maintenance genes were more highly expressed in young flies and 14 more highly expressed in old flies (Extended Data Fig. 3, Supplementary Table 2). In spermatocytes and spermatids, comparatively few genes are differentially expressed between young and old testes. Depleted expression of genome maintenance genes in the earliest germ cells could impact the efficiency of germline DNA repair throughout spermatogenesis.
Table 1: Numbers of age-biased genes per cell type.
Arranged from top (early) to bottom (latest). For young flies, the cell type with the most biased genes is GSC/Early spermatogonia, the earliest germ cell class. Late spermatids are the most mutated class of cell in older flies. In every class of cells except GSC, early and late spermatogonia, old flies have more biased genes than young flies.
| Cell type | # genes biased in young flies | # genes biased in old flies |
|---|---|---|
| GSC, Early spermatogonia | 273 | 251 |
| Late spermatogonia | 144 | 337 |
| Early spermatocytes | 40 | 367 |
| Late spermatocytes | 80 | 281 |
| Early spermatids | 167 | 277 |
| Late spermatids | 52 | 68 |
Transcription-related genes, Rbp8 (FBgn0037121) and RpII15 (FBgn0004855) are more highly expressed in young GSC/Early spermatogonia than in old flies. Rbp8, also known as B52, is essential for DNA topoisomerase I recruitment to chromatin during transcription 22. DNApol-iota (FBgn0037554), which is a gene involved in translesion synthesis 23 and may be important UV damage response 20, was highly expressed in young GSCs. This suggests that our observed mutational signatures of older flies might be caused by defects in transcription-coupled repair or DNA damage related repair, although the mechanisms of germline genomic surveillance are yet to be fully understood. In old flies, lower expression of RpII15, also known as RNA Polymerase II, subunit I, might further explain reduced transcription in these cell types from old flies. The gene 14–3-3epsilon (FBgn0020238) is highly expressed in old GSC and spermatogonia, which may play a role in cell division and apoptosis in early germ cells for old testis 24. The cellular-level mutational signature (Extended Data Fig. 4) is in line with previous work suggesting that highly expressed genes evolve more slowly 25,26.
Increased post-meiotic expression for de novo genes
In our previous work we found that de novo genes are highly enriched in meiotic cells 11. We asked whether transcriptional dysregulation of the aging germline could impact the expression of genetic novelties such as de novo genes and transposable elements (TEs). We performed a parallel analysis of our scRNA-seq data with a custom reference containing 267 testis-expressed de novo genes identified from our 2019 study, as well as 239 TEs 27. We then scaled expression of every gene, centered at zero to compare the expression patterns of groups of genes (Fig. 4).
Fig. 4: Global expression patterns of de novo genes and transposable elements changes with age in each cell type.
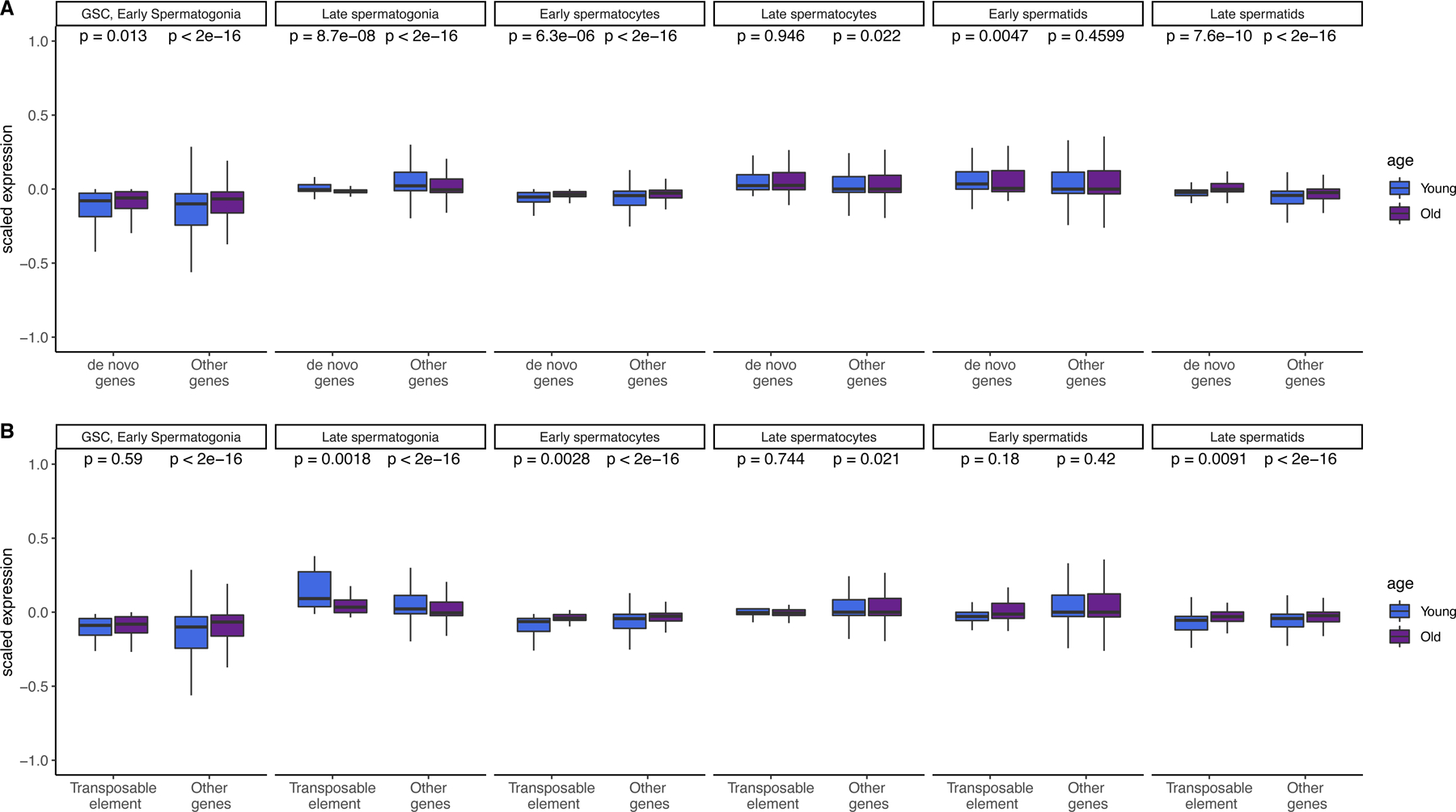
A.) Scaled expression of de novo genes and other genes (not including TEs) across cell types. Expression of both gene types is enriched in the late spermatids of older flies as compared with a two-sided Wilcoxon test (raw p values shown, n = 150 de novo genes, 15479 other genes) B.) Scaled expression of TEs and other genes (not including de novo genes) across cell types. Transposable element expression is highly enriched in late spermatogonia of young flies, and enriched in the late spermatids of older flies (n = 239 TEs, 15479 other genes. Both panels show raw p values by 2-sided Wilcoxon rank sum tests. Boxes represent the 75th to 25th percentiles, the top whisker represents the largest value within 1.5 times the interquartile range, and the bottom whisker represents the smallest value within 1.5 times the interquartile range of the 25th percentile.
Overall, the expression patterns for de novo genes resemble those of other genes, and both de novo genes and other genes are more highly expressed in the late spermatids of older flies (Fig. 4A, p = 7. 6e-10, p < 2e-16, Supplementary Table 3). This suggests that de novo genes show similar expression regulation related to aging compared to old genes and that the regulatory environment acts similarly to conserved and young genes.
We were also interested in the expression of TEs, since TE suppression is important for germ cell development 28. In other tissues, such as the aging brain, the amount of certain types of TEs change with age 29. We found TE expression is also globally enriched in older late spermatids (Fig. 4B, Supplementary Table 4); however, this pattern is not more extreme compared to annotated genes. Since gene expression is supposed to stop after meiosis, it is possible that elevated gene expression after meiosis is a consequence of reduced post-meiotic transcriptional suppression. Although all three classes of genes were also more highly expressed in the late spermatogonia of young flies, this increase was strikingly large for transposable elements (p = 0.0018). This result corroborates earlier work which found a similar burst of transposon activity in early spermatogenesis, likely when spermatogonia transit to spermatocytes 27. As such, the reduced early TE expression in older germlines may reflect a dysregulated transcriptional environment.
Early spermatogenesis-biased genes have lower dN/dS
We asked whether functional constraint varies for age-biased or cell-type biased genes. We defined “age-biased” genes as genes differentially expressed between old and young flies in the same cell type. We defined “cell type-biased” genes as genes differentially expressed between cell types of the same age. To find cell type-biased genes, we split our dataset into “old” and “young” cells and then performed Seurat’s FindMarkers function between GSC/early spermatogonia (early germline) and late spermatids (late germline) for each age group. Using dN/dS data from flyDIVaS 30, we compared dN/dS values for early-stage-biased and late-stage-biased genes. In both young and old flies, the genes with expression bias toward later stages exhibit higher dN/dS than genes biased in GSC/early spermatogonia (Fig. 5A), which is in line with the idea that genes expressed in late spermatogenesis may evolve rapidly. Note that spermatocytes and spermatids are also hotspots for the expression of novel genes including de novo originated genes. Our findings are also similar to a murine study which found that genes expressed in early spermatogenesis are under more evolutionary constraint than genes expressed late germ cells 31.
Fig. 5: dN/dS trends of cell type-biased and age-biased genes.
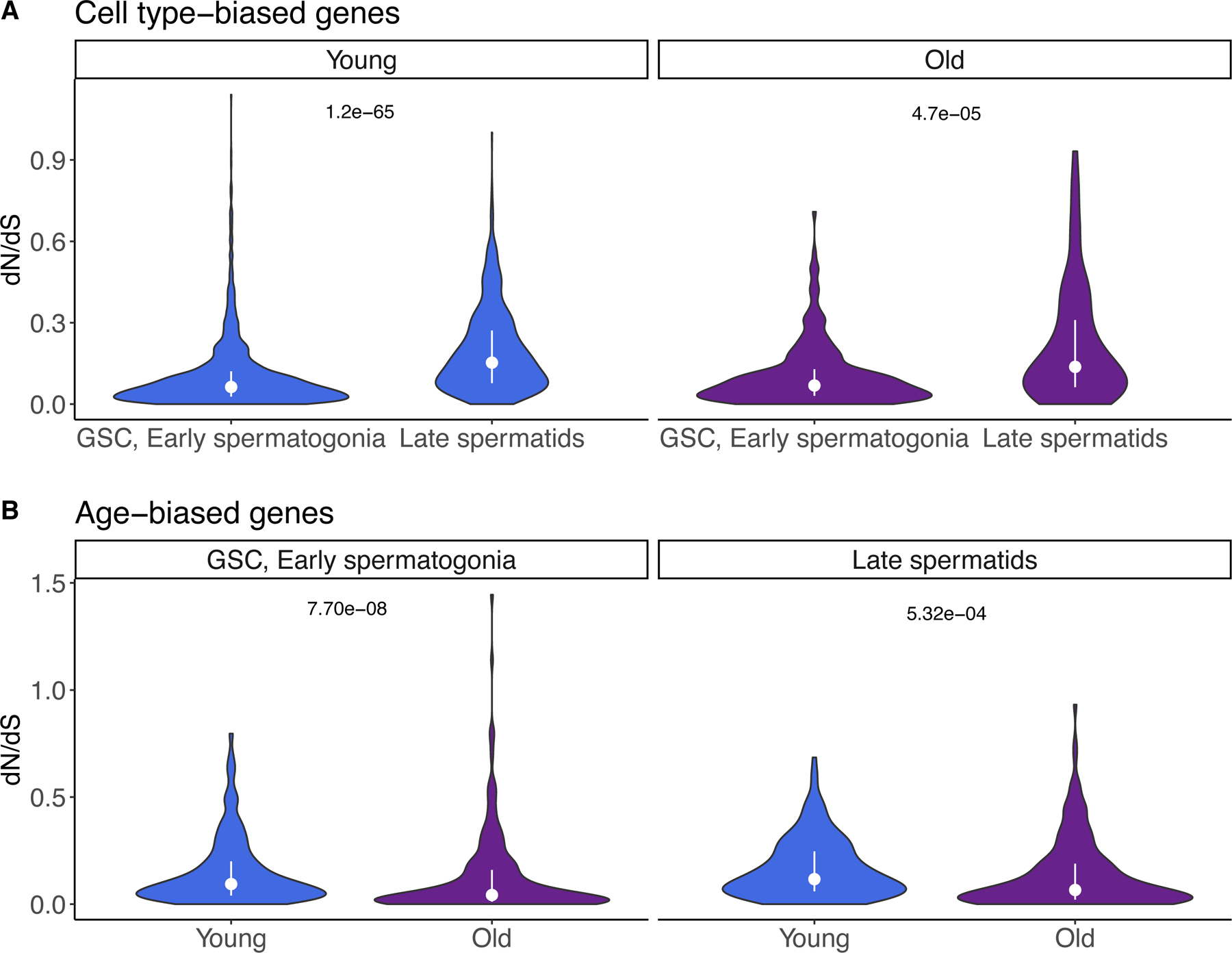
We calculated gene expression bias in two different ways. First, we calculated gene expression bias between GSC/early spermatogonia and late spermatids separately for young and old flies. Then, we identified genes biased in young and old cells within a cell type. A) Cell type-biased genes: in both old and young flies, genes biased in late spermatids have higher dN/dS than genes biased in GSC/early spermatogonia (two-sided Wilcoxon rank sum test, raw p values 1.2e-65, 4.7e-5, respectively). B) Age-biased genes: In GSC/early spermatogonia, but not late spermatids, young-biased genes have higher dN/dS than old-biased genes (two-sided Wilcoxon rank sum test, raw p values 7.70e-08 and 5.32e-04, respectively). dN/dS values are from flyDIVaS. For all panels, bars represent interquartile range and the dot represents the median.
Genes biased in young cells have higher dN/dS
To identify age-biased genes, we divided the dataset by cell type and identified genes biased in old or young cells of the same type. We found that in both GSC/early spermatogonia and late spermatids, genes biased in young cells have higher dN/dS than genes biased in old cells (Fig. 5B). This result show that many early-germ-cell-biased genes in young flies evolve rapidly, that is, a higher proportion of rapidly evolving genes are biased in young-age germline stem cells and spermatogonia than in old ones. This suggests that, if antagonistic pleiotropy in aging plays an important role in the shift of gene expression 32,33, rapidly evolving genes are often expressed and function in young animals and are subject to antagonistic pleiotropy.
Discussion
Mutational load is an equilibrium between mutation and repair. In old flies, this equilibrium may shift away from repair. In this work, we show that the germline of older flies is less able to remove de novo mutations compared to the germline of young flies. Throughout spermatogenesis, we observed that germ cells from older flies have more mutations per RNA molecule than comparable cells from younger flies. This finding adds a new explanation for the still-controversial mechanism behind the increased age-dependent mutational load of the male germline. Our finding of increased mutational load in older GSC/early spermatogonia suggests that much of the mutational load of older flies accumulates prior to spermatogenesis. However, later germ cells of older flies have more SNPs per UMI, suggesting that some mutations may also accumulate during spermatogenesis. Our work corroborates previous work that found that the huge excess of male germline divisions is too large to explain the much smaller ratio of male/female-inherited mutations during parental aging 1. Our finding that early germ cells from young and old flies are similarly mutated supports the notion that many age-related germline mutations are not due to replicative processes. Some of our conclusions are in line with recent findings that germline stem cells have the lowest mutation rate of any human cell type 10.
In addition to being highly mutated, the older germline shows distinct mutational signatures compared to the younger germline. For example, we found a statistical overrepresentation of C>A and C>G mutations in old flies and an underrepresentation of C>T mutations, although C>T still ranks the second most abundant mutation class in old testis. These altered ratios of single nucleotide polymorphisms could be caused by differential activity of DNA repair pathways in the old germline. We did not find strong evidence of global downregulation of genome maintenance gene expression but found lower expression of a few key transcriptional genes in GSC of older flies. Altered types and numbers of de novo mutations would likely have implications for a population, affecting the type and frequency of genetic novelties that emerge 34. In the future, it would be interesting to understand the molecular mechanisms contributing to mutational bias in germ cells. Additionally, these methods should be reproduced with mated flies to examine if germline mutational bias can be affected by a male’s reproductive activity.
We observed that scaled expression of all genes is generally down in GSC/early spermatogonia but up in late spermatids. The latter result is intriguing because transcription largely ceases after meiosis in the male Drosophila germline 35. While the downregulation of transcription in early germ cells could have implications for germline DNA repair, the potential effects of increased post-meiotic transcription are less clear. It could have no effect, or it could affect spermatid maturation or sperm competition, potentially affecting fertility. Indeed, increased male age associates with reduced fertility in humans 36.
Other studies have proposed that the testis uses ubiquitous gene expression to detect genomic lesions and repair them with transcription-coupled repair 15,37. Due to our bias towards detecting mutations in expressed genes, our data is not ideal to test this hypothesis. We noted, however, that genes with many detectable SNPs tend to be lowly expressed across replicates (Extended Data Fig. 4), a finding that appears to be consistent with the transcriptional scanning model. Broadly, this is in line with previous work that reported a pattern of lower dN/dS in highly expressed genes and the pattern is driven by various types of selective pressures 25,26. Our results surprisingly found this pattern in mutational bias, even before natural selection acts on the genes. This result also supports the notion that it is very unlikely that the germline mutations we observed are directly from transcription errors, as that would lead to more mutations in highly expressed genes. Impaired mutational repair of the older male germline may be partially due to deficiencies in transcription-coupled repair 38.
The global post-meiotic upregulation of transcription extends beyond conserved genes. We observed that many transposable elements are highly enriched in the early germline of young flies. Beyond implications for transposable element mobilization, this finding could also indicate heritable changes in chromatin structure, signaling, or gene expression 39,40.
The global deregulation of gene expression during aging also has interesting implications for evolution. Consistent with prior work 31, we found that genes biased in late germ cells have higher dN/dS than genes biased in early germ cells for both young and old flies. This result suggests that spermatocytes and spermatids are sources of rapid evolution or positive selection. Considering that spermatocytes and spermatids are also the stages where de novo gene are the most abundant and where they likely function 11 our results highlight the importance of late spermatogenesis in transcriptional and functional innovation.
Unexpectedly, we also found that, within analogous cell types, genes biased in cells from young flies have higher dN/dS than genes in old flies. There are two possible explanations. First, rapidly evolving genes or genes biased with more adaptive changes may provide a greater evolutionary advantage to young flies than old flies. Second, since we used the scaled expression, old-biased genes may reflect a specific set of genes that are essential for aging animals, which may more likely be house-keeping genes. Our results are interesting in the light of recent work which found that genes expressed later in life tend to fix nonsynonymous mutations more frequently 41. One should note that the methodology from Cheng and Kirkpatrick41 is different from ours: their age-biased genes were identified from whole-body data, whereas ours were calculated just from male germ cells. Gene expression in the testis is often an outlier compared to other tissues 42, so the results of these two studies are not necessarily in conflict. Nevertheless, the consistent pattern between this study and that of Cheng and Kirkpatrick 41 is that that genes biased in late spermatids have a higher dN/dS than those biased in early germ cells. In this sense, at both whole-organism development and germline development level, late-stage biased genes tend to evolve more rapidly.
Our study design limits the detection of mutations in expressed transcripts. While we have strict criteria for the identification of novel SNPs, the abundance of false negatives could vary between cell types due to cell-specific variation in transcriptional activity. We are reassured because the most commonly mutated cell type in our datasets is GSC/early spermatogonia, consistent with our previous observations using different datasets and slightly different analytical pipelines. If transcriptional activity biased our inference of mutational load, we would expect spermatocytes, the most transcriptionally active cell type, to appear the most mutated instead. These potential confounding effects from using transcriptional activity would be resolved if a method became available to simultaneously perform scRNA-seq and whole-genome sequencing on thousands of single cells simultaneously. Current methods may not capture enough cells to comprehensively profile rarer cell types, but we expect this technical challenge to improve with future technological advances.
One important note is that the mutations and mutational patterns observed in this study may not be analogous to published work21,43. Previous work characterized mutations observed in a species or a population, and the patterns are a combination of mutational bias, selection, and drift. Here, we identified de novo mutations during spermatogenesis before passing them to the next generation and compared the patterns between young and old germline, revealing different patterns associated with aging. Our patterns may be indirectly compared with published work, and the differences may be partly attributed to the differences caused by natural selection and drift. Additionally, our method is biased towards mutations on the template strands of expressed genes. Rather than a comprehensive catalog of germline mutations, the primary utility of our method is to compare mutational load between cell types within the germline.
Another open question is whether patterns found in one replicate would be observed in another biological replicate. If mutations in spermatogenesis are strongly affected by a few stereotypical mechanisms, then we should expect the patterns to be consistently similar in different replicates. On the other hand, if mutations are overall random or created by many distinctive mechanisms acting independently, then we may not see highly repeatable patterns in terms of mutational signatures in different replicates. The results we have seen sit in between the two extremes. In the future, it will be important to dissect mechanisms contributing to de novo mutations in aging germline.
A potential confounder is that aging might create the appearance of germline SNPs through reduced transcriptional fidelity44. We do not think this is a significant source of error, since our SNPs are verified by multiple independent reads and are not allowed to be present in more than one dataset, in fact, we did not observe mutations occur in multiple datasets after filtering, suggesting that hidden common transcription errors or RNA editing do not impact our work. To understand the role that age-related transcriptional fidelity significantly plays in our results, this topic would benefit from high throughput combined scRNA/scDNA sequencing from the same cells from isolated cells through spermatogenesis. Another future direction is to trace mutations through trio-studies, specifically sequencing a large number of zygotes and scRNA/scDNA sequencing from the correlated testis and ovary. The technology allowing us to trace de novo mutations throughout the germline is still very new, and we look forward to technological advancements in this exciting field.
Methods
ScRNA-seq library preparation and sequencing
In this study, we used young and old RAL517 for experiments. Briefly, flies were reared in the standard corn syrup medium at room temperature with synchronized 12:12 hour light:dark cycle. Both age groups of flies were kept as virgins after eclosion. Young and old samples were collected 48 hours and 25 days after eclosion, respectively. In detail, virgin males were collected within one hour after eclosion and were transferred to new vials. After 48 hours, we dissected 30 pairs of young testes for single-cell suspension and scRNA-seq and kept the carcasses for DNA sequencing. After 25 days, we dissected 30 pairs of old testes for single-cell suspension and scRNA-seq and kept the carcasses for DNA sequencing. We generated three biological replicates for young and old flies, respectively. Flies were all dissected in the morning (ZT1-ZT3 in our lab environment) to reduce expression fluctuation due to circadian rhythm. Single-cell testis suspensions were prepared as described in our previous work 11. Libraries were prepared with 10X Chromium 3’ V3 kit and sequenced with Illumina Hiseq 4000.
Genomic DNA preparation
Fly carcasses were frozen at −80°C, then ground in 200 µL 100 mM Tris-HCl, pH 7.5, 100 mM EDTA, 100mM NACl, 0.5% SDS. The mixtures were incubated at 65°C for 40 minutes. Then, 160 µl KAc and 240 µl 6M LiCl were added, tubes were inverted 10 times and placed on ice for 30 minutes. Samples were then centrifuged at 18000g at 4°C for 15 minutes. The supernatant was transferred into a new tube and an equivalent volume of isopropanol was added and mixed by inversion. Samples were spun for 15 minutes at 18000g, and the supernatant was discarded. Pellets were washed with 800 µL 70% ethanol and samples were spun at 18000g for 5 minutes, and supernatant discarded. Pellet was air dried for 5 minutes and resuspended in 100 µL nuclease-free water. DNA was then sent for Illumina library preparation and sequencing by Novogene.
ScRNA-seq data processing
ScRNA-seq data were aligned with Cellranger Count and further processed with Seurat. To assign cell types with Seurat, we used marker genes described in Witt et al. 2021 18. We normalized the replicates with SCTransform in Seurat and integrated them into a combined Seurat object. We performed clustering and annotation together on all replicates, using normalized counts from the “SC T” slot. Cells were annotated based on the expression patterns of marker genes. Clusters enriched in bam and aub are GSC/early spermatogonia11,18,45, and adjacent clusters with less bam/aub and less His2Av are late spermatogonia. Clusters enriched in fzo and twe are early and late spermatocytes, respectively 46,47. Clusters with enriched soti, but not p-cup are early spermatids 35 and clusters enriched in p-cup are late spermatids 35. Clusters enriched in MtnA are somatic cells, dlg1 defines cyst cells 48 and Fas3 defines hub cells. Epithelial cells are enriched in MtnA but not Rab11 or Fas3 18.
SNPs were called with bcftools 49 separately for each single-cell library and each gDNA library. Per-base coverage was calculated for every gDNA sample with Samtools 50. For each young and old scRNA-seq library, bcftools isec was used to extract mutations only present in the SC data and not the somatic gDNA. Using Samtools, we identified every cell barcode in the scRNA-seq data that corroborated every SNP (details in accompanying code). For each mutated position, we then verified that the corresponding locus in the gDNA file had at least 10 reads supporting the reference allele, and 0 reads supporting the alternative allele. We also required that every SNP be present only in a single scRNA-seq dataset, to reduce the chance that RNA editing events or transcription errors caused us to infer a SNP incorrectly. We also required every SNP to have >=2 reads corroborating it to reduce the potential impact of sequencing errors.
Comparisons using scaled expression
To compare gene expression for groups of genes across replicates, we scaled expression using the ScaleData Seurat function separately on each replicate. Expression is scaled such that 0 represents a gene’s median expression across all cells; 1 represents 1 standard deviation above that gene’s mean expression; and −1 represents 1 standard deviation below. Within a cell type, each gene’s scaled expression was averaged between cells. Groups of genes were compared using a two-sample Wilcoxon rank sum test, and p values were adjusted with Bonferroni’s correction.
Differential expression testing
For each germ cell type, we made a subset Seurat object containing just that cell type with old and young flies, assigning “age” as the cell identifier. We then used Seurat’s FindMarkers function with ident.1 as “Young” and ident.2 as “Old”. We classified genes with a Bonferroni-adjusted p value < 0.05 and Log2 fold change > 0 as enriched in young, and <0 as enriched in old. We then constructed volcano plots with the EnhancedVolcano package, while including differentially expressed genes from our list of 211 genome maintenance genes from our previous paper 11.
De novo gene and TE analysis
De novo genes from our previous paper 11 were added to a reference GTF containing transposable elements from another study 27. This alternate reference was used to align reads from all libraries with Cellranger. Cell-type annotations were copied from the annotations made for the main Seurat object. Enriched de novo genes and TEs were detected with the FindMarkers function in Seurat.
Extended Data
Extended Data Fig. 1:
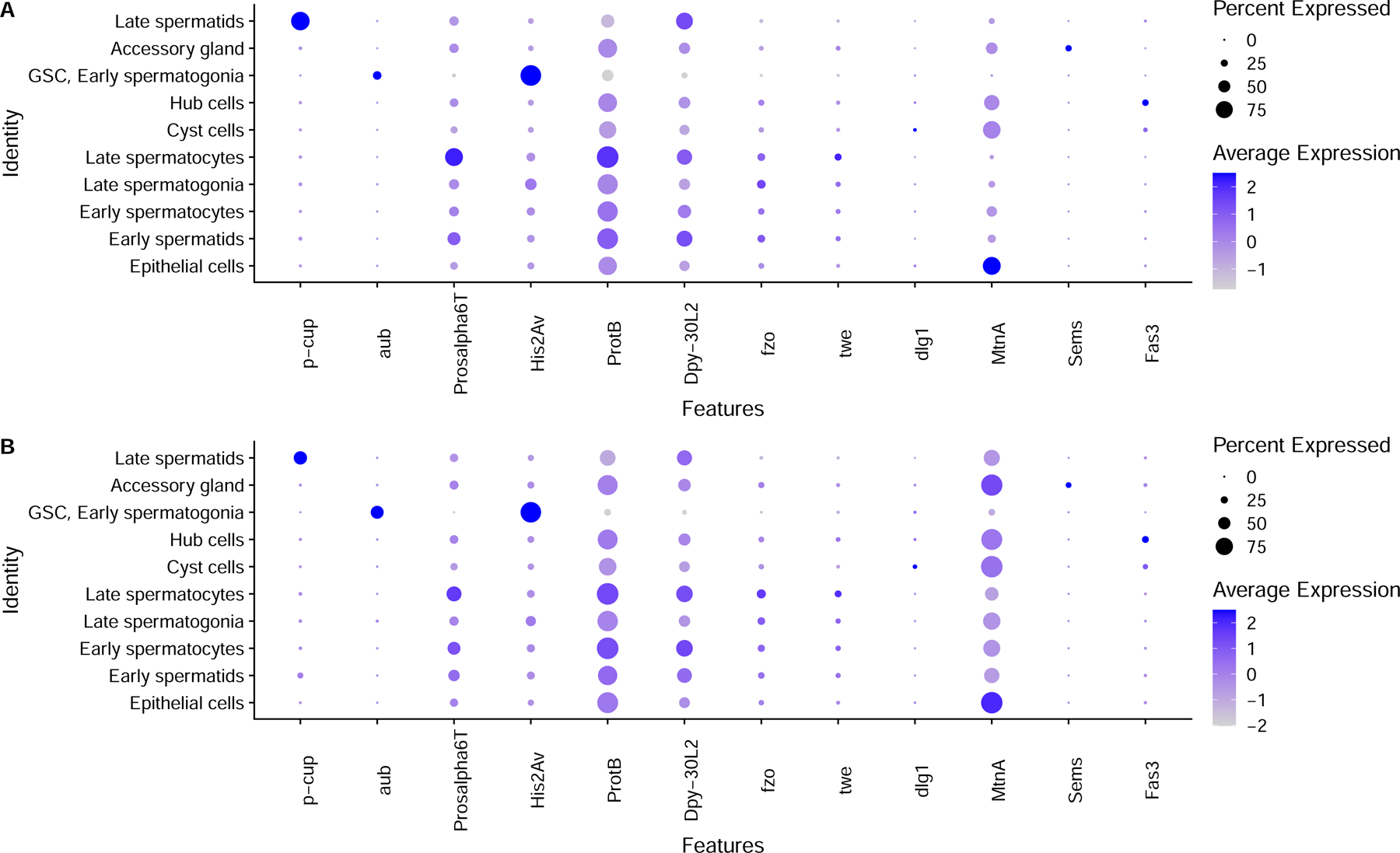
Dot plots of key marker genes in old and young fly testes.
Extended Data Fig. 2:
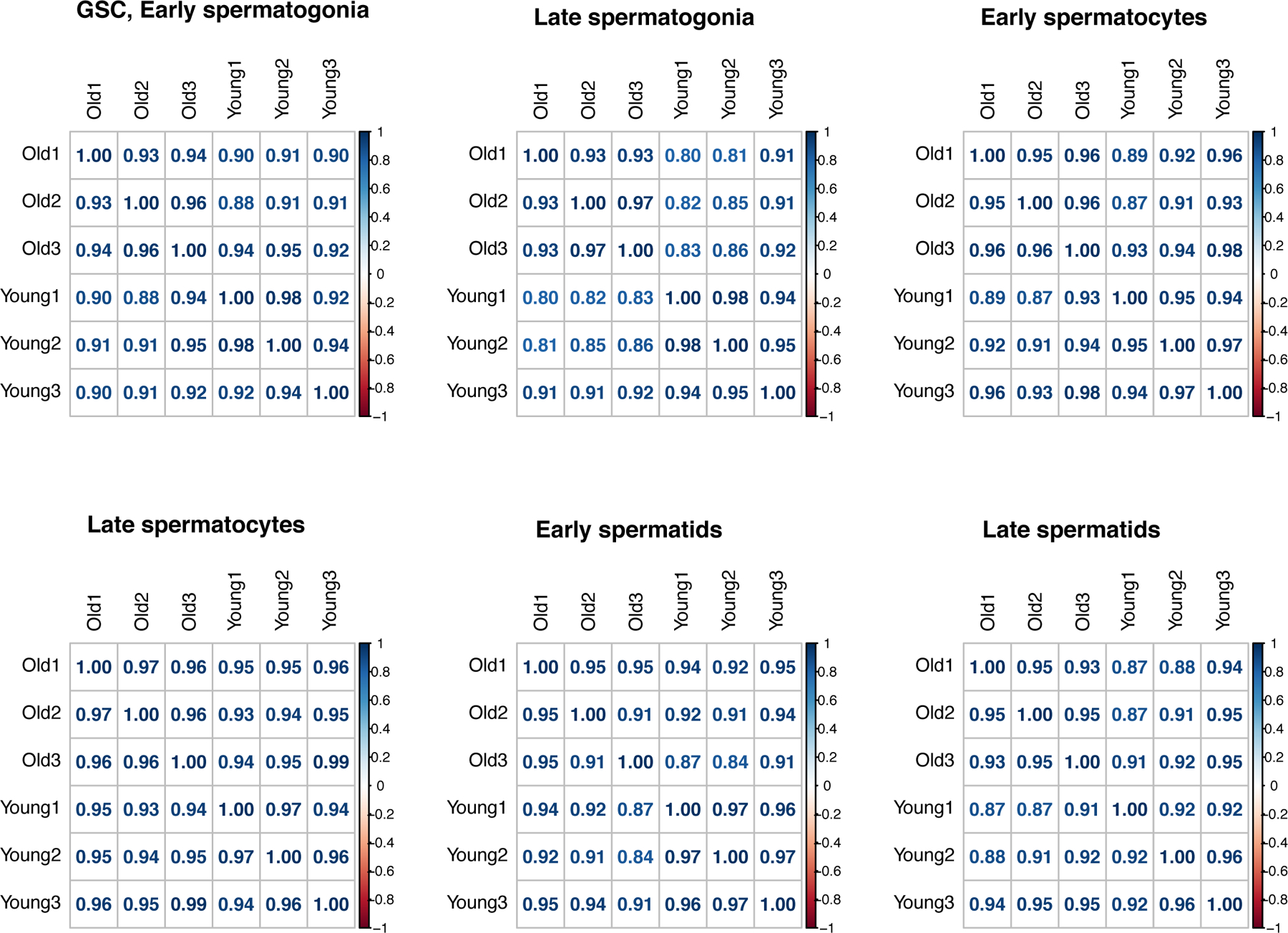
Correlograms of germ cells between scRNA-seq replicates.
Extended Data Fig. 3:
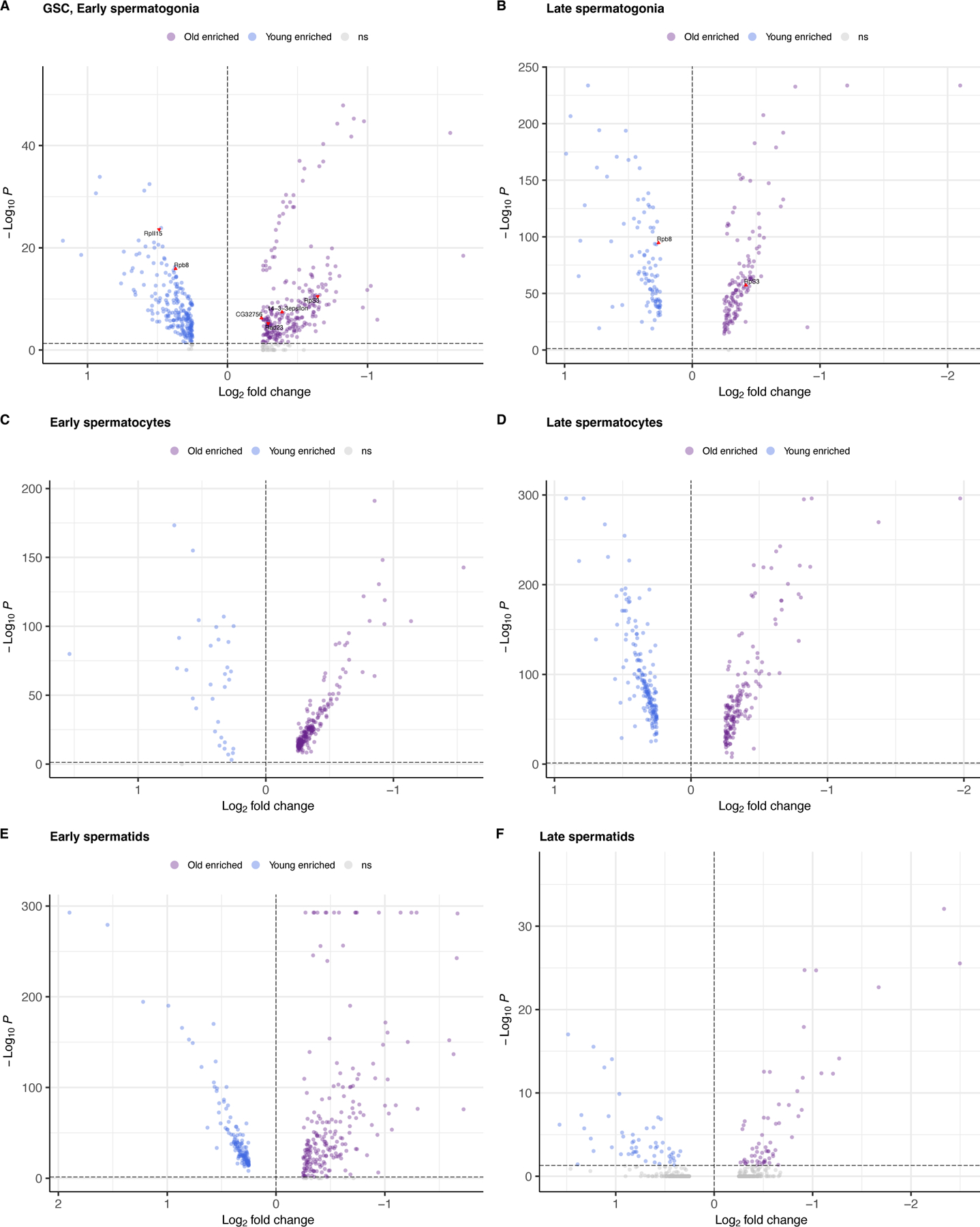
Age-related differential expression of genes, including genome maintenance genes.
Extended Data Fig. 4:
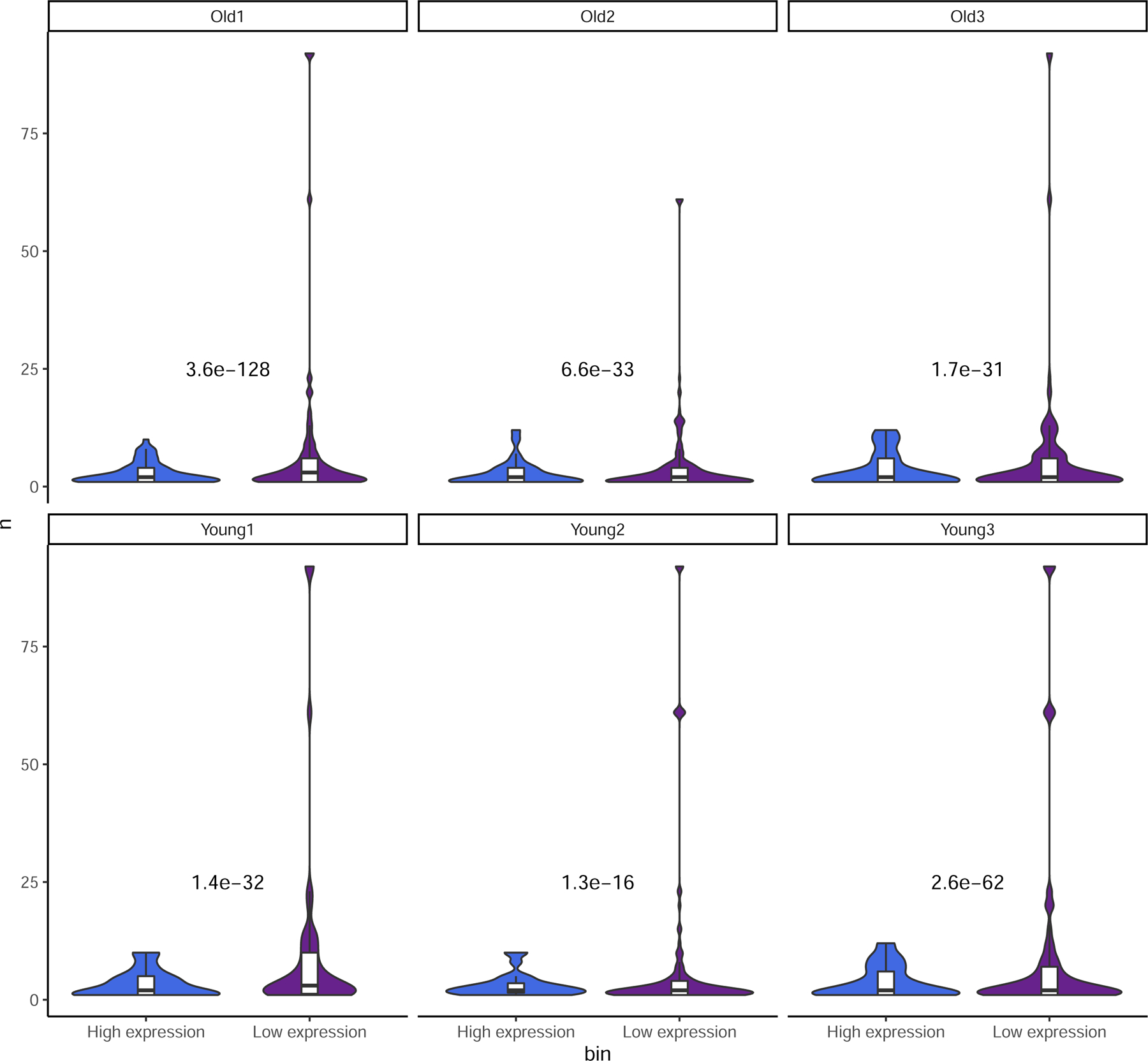
Expression vs. number of SNPs detected.
Supplementary Material
Acknowledgements
We thank Hong Duan and Connie Zhao at Genomics Resource Center of Rockefeller University for their help with the scRNA-seq libraries, and members of Zhao lab for their helpful comments and suggestions. We thank Dr. Ziyue Gao from UPenn for the suggestions on interpreting mutational signatures.
Funding
The work was supported by NIH MIRA R35GM133780, the Robertson Foundation, a Monique Weill-Caulier Career Scientist Award, a Rita Allen Foundation Scholar Program, and a Vallee Scholar Program (VS-2020-35), and an Alfred P. Sloan Research Fellowship (FG-2018-10627) to L. Z.
Footnotes
Declaration of interests
The authors declare no competing interests.
Data availability
Raw sequence data has been deposited to NCBI BioProject PRJNA777411.
Data availability
Code used for processing of data is deposited at https://github.com/LiZhaoLab/Mutation_project. This repository also include permanent links to large data files including a Seurat RDS and mutation database.
Reference
- 1.Gao Z et al. Overlooked roles of DNA damage and maternal age in generating human germline mutations. Proc. Natl. Acad. Sci 116, 9491 LP – 9500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crow JF The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet 1, 40–47 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Gao Z, Wyman MJ, Sella G & Przeworski M Interpreting the dependence of mutation rates on age and time. PLoS Biol 14, 1–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drost JB & Lee WR Biological basis of germline mutation: Comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ. Mol. Mutagen 25, 48–64 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Gao J-J et al. Highly variable recessive lethal or nearly lethal mutation rates during germ-line development of male Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A 108, 15914–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li WH, Ellsworth DL, Krushkal J, Chang BH & Hewett-Emmett D Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis. Mol. Phylogenet. Evol 5, 182–187 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Huttley GA, Jakobsen IB, Wilson SR & Easteal S How important is DNA replication for mutagenesis? Mol. Biol. Evol 17, 929–937 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Irigaray P et al. Lifestyle-related factors and environmental agents causing cancer: An overview. Biomed. Pharmacother 61, 640–658 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Parkin DM, Boyd L & Walker LC 16. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010. Br. J. Cancer 105, S77–S81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore L et al. The mutational landscape of human somatic and germline cells. Nature 597, 381–386 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Witt E, Benjamin S, Svetec N & Zhao L Testis single-cell RNA-seq reveals the dynamics of de novo gene transcription and germline mutational bias in Drosophila. Elife 8, e47138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee M-H, Luo H-R, Bae SH & San-Miguel A Genetic and Chemical Effects on Somatic and Germline Aging. Oxid. Med. Cell. Longev 2020, 4684890 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones DL Aging and the germ line: where mortality and immortality meet. Stem Cell Rev 3, 192–200 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Cawthon RM et al. Germline mutation rates in young adults predict longevity and reproductive lifespan. Sci. Rep 10, 10001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia B et al. Widespread transcriptional scanning in testes modulates gene evolution rates. Cell 248–262 (2020). [DOI] [PMC free article] [PubMed]
- 16.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thurmond J et al. FlyBase 2.0: the next generation. Nucleic Acids Res 47, D759–D765 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witt E, Shao Z, Hu C, Krause HM & Zhao L Single-cell RNA-sequencing reveals pre-meiotic X-chromosome dosage compensation in Drosophila testis. PLOS Genet 17, e1009728 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svetec N, Cridland JM, Zhao L & Begun DJ The Adaptive Significance of Natural Genetic Variation in the DNA Damage Response of Drosophila melanogaster. PLoS Genet 12, e1005869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh ND, Bauer DuMont VL, Hubisz MJ, Nielsen R & Aquadro CF Patterns of mutation and selection at synonymous sites in Drosophila. Mol. Biol. Evol 24, 2687–97 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Juge F, Fernando C, Fic W & Tazi J The SR Protein B52/SRp55 Is Required for DNA Topoisomerase I Recruitment to Chromatin, mRNA Release and Transcription Shutdown. PLOS Genet 6, e1001124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishikawa T et al. Mutagenic and Nonmutagenic Bypass of DNA Lesions byDrosophila DNA Polymerases dpolη and dpolι. J. Biol. Chem 276, 15155–15163 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Su TT et al. Cell cycle roles for two 14–3-3 proteins during Drosophila development. J. Cell Sci 114, 3445–54 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Good JM & Nachman MW Rates of protein evolution are positively correlated with developmental timing of expression during mouse spermatogenesis. Mol. Biol. Evol 22, 1044–1052 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Drummond DA, Bloom JD, Adami C, Wilke CO & Arnold FH Why highly expressed proteins evolve slowly. Proc. Natl. Acad. Sci. U. S. A 102, 14338–43 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawlor MA, Cao W & Ellison CE A transposon expression burst accompanies the activation of Y-chromosome fertility genes during Drosophila spermatogenesis. Nat. Commun 12, 6854 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee YCG & Langley CH Transposable elements in natural populations of Drosophila melanogaster. Philos. Trans. R. Soc. B Biol. Sci 365, 1219–1228 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W et al. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci 16, 529–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stanley CEJ & Kulathinal RJ flyDIVaS: A Comparative Genomics Resource for Drosophila Divergence and Selection. G3 (Bethesda) 6, 2355–2363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schumacher J & Herlyn H Correlates of evolutionary rates in the murine sperm proteome. BMC Evol. Biol 18, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Austad SN & Hoffman JM Is antagonistic pleiotropy ubiquitous in aging biology? Evol. Med. Public Heal 2018, 287–294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams GC Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution (N. Y) 11, 398 (1957). [Google Scholar]
- 34.Loewe L & Hill WG The population genetics of mutations: good, bad and indifferent. Philos. Trans. R. Soc. B Biol. Sci 365, 1153–1167 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barreau C, Benson E, Gudmannsdottir E, Newton F & White-Cooper H Post-meiotic transcription in Drosophila testes. Development 135, 1897–1902 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Harris ID, Fronczak C, Roth L & Meacham RB Fertility and the aging male. Rev. Urol 13, e184–e190 (2011). [PMC free article] [PubMed] [Google Scholar]
- 37.Xia B & Yanai I Gene expression levels modulate germline mutation rates through the compound effects of transcription-coupled repair and damage. Hum. Genet 141, 1211–1222 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Deger N, Yang Y, Lindsey-Boltz LA, Sancar A & Selby CP Drosophila, which lacks canonical transcription-coupled repair proteins, performs transcription-coupled repair. J. Biol. Chem 294, 18092–18098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanciano S & Cristofari G Measuring and interpreting transposable element expression. Nat. Rev. Genet 21, 721–736 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Chuong EB, Elde NC & Feschotte C Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet 18, 71–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng C & Kirkpatrick M Molecular evolution and the decline of purifying selection with age. Nat. Commun 12, 2657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witt E, Svetec N, Benjamin S & Zhao L Transcription Factors Drive Opposite Relationships between Gene Age and Tissue Specificity in Male and Female Drosophila Gonads. Mol. Biol. Evol 38, 2104–2115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharp NP & Agrawal AF Low Genetic Quality Alters Key Dimensions of the Mutational Spectrum. PLoS Biol 14, e1002419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verheijen BM & van Leeuwen FW Commentary: The landscape of transcription errors in eukaryotic cells. Front. Genet 8, 219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawase E Gbb/Bmp signaling is essential for maintaining germline stem cells and for repressing bam transcription in the Drosophila testis. Development 131, 1365–1375 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Hwa JJ, Hiller MA, Fuller MT & Santel A Differential expression of the Drosophila mitofusin genes fuzzy onions (fzo) and dmfn. Mech. Dev 116, 213–216 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Courtot C, Fankhauser C, Simanis V & Lehner CF The Drosophila cdc25 homolog twine is required for meiosis. Development 116, 405–16 (1992). [DOI] [PubMed] [Google Scholar]
- 48.Papagiannouli F & Mechler BM discs large regulates somatic cyst cell survival and expansion in Drosophila testis. Cell Res 19, 1139–1149 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Narasimhan V et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data has been deposited to NCBI BioProject PRJNA777411.
Code used for processing of data is deposited at https://github.com/LiZhaoLab/Mutation_project. This repository also include permanent links to large data files including a Seurat RDS and mutation database.