

INTRODUCTION
With advances in cystic fibrosis (CF) care, endocrine comorbidities, including diabetes (CFRD) and bone disease (CFBD), have become increasingly important medical considerations in people with CF. Although the underlying pathophysiology remains elusive, the medical community is leveraging novel technologies and therapeutics to manage these conditions while addressing patient-reported concerns. Previously recognized conditions in CF such as pubertal delay and short stature have been tempered whereas others such as hypogonadism are increasingly recognized. Enter the success of highly effective modulator therapies (HEMT) for many patients, and the established phenotypes for several of these conditions are now less clearly defined. Given the rapidly changing landscape, this review will discuss the more recent literature surrounding endocrine comorbidities and introduce non–CF-specific innovations that are being extended to address these co-occurring conditions in CF.
CYSTIC FIBROSIS-RELATED DIABETES
Introduction
The relevance of diabetes in CF cannot be understated. CFRD is common, traditionally associated with worse CF outcomes, adds additional patient burden,1 and, like other forms of diabetes, associates with microvascular sequelae.
Cystic Fibrosis-Related Diabetes Screening Guidelines
As CFRD does not generally present with classic diabetes symptoms (polyuria, polydipsia) but can negatively impact pulmonary function and nutritional status despite its generally “indolent” nature, the Cystic Fibrosis Foundation (CFF) recommends annual screening with oral glucose tolerance testing (OGTT) starting by age 10 years,2 Table 1. Unlike in type 2 diabetes (T2D) in which fasting glucose and hemoglobin A1c (HbA1c) are useful screening tools, fasting hyperglycemia (glucose >125 mg/dL) tends to be a late finding in CF although HbA1C is frequently normal (HbA1c <6.5%).
Table 1.
Definitions of glucose tolerance
| Plasma Glucose (PG) Mg/dL | |||
|---|---|---|---|
| Fasting | OGTT | ||
| 1-h | 2-h | ||
| Normal | <100 | <140 | <140 |
| Impaired fasting glucose | 100–125 | ||
| Fasting hyperglycemia | ≥126 | ||
| Early Glucose Intolerance a | ≥155 and < 200 | <140 | |
| Indeterminate Glucose Intolerance (IND) | ≥200 | <140 | |
| Impaired glucose tolerance (IGT) | 140–199 | ||
| CFRD without fasting hyperglycemia | <126 | ≥200 | |
| CFRD | Random glucose ≥200 mg/dL with classic symptoms (polyuria, polydipsia) HbA1c ≥ 6.5% |
||
Not included in 2010 CFF guidelines.
Cystic Fibrosis-Related Diabetes Screening Research
Recent clinical research has also attempted to find alternatives to OGTT for screening for CFRD. This work was recently briefly reviewed by Chan.3 Lowering of the HbA1C threshold from the traditional threshold of 6.5% to 5.8% does not overcome the issue of lack of sensitivity of HbA1c, and further lowering renders the test completely nonspecific.4,5 Clinically, HbA1c is frequently obtained in patients with CF because (1) an elevated HbA1c is consistent with CFRD and (2) high-normal values heighten awareness that CFRD or abnormal glucose tolerance (AGT) may be operative. Indeed, HbA1c has been proposed as a prescreen to identify the subset of individuals requiring OGTT for formal CFRD screening; patients with HbA1c greater than or equal to 6.5% do not require OGTT as they meet the diagnostic criteria and those with HbA1c less than 5.5% have a low likelihood of having CFRD and might delay OGTT.6 This approach has not been rigorously tested beyond the cited study and is not a currently recommended approach for CFRD screening. Similarly, the nonfasting, single sample, 50-g glucose challenge test has been proposed as a prescreen to identify the subset of individuals who should undergo OGTT, but the limited number of patients with CFRD in that group of 27 adolescents and young adults with pancreatic insufficient CF (PI-CF) made drawing conclusions surrounding sensitivity challenging.7 Continuous glucose monitoring (CGM) has received increasing attention as a potential screening tool for CFRD, and although CGM identifies glycemic variability and frequent hyperglycemia under free-living conditions, its agreement with the OGTT diagnosis of CFRD is inconsistent.8–12 Investigators and clinicians are now focusing on the CGM thresholds that associate with worse outcomes even among those with normal glucose tolerance8,13–15 and at which treatment imparts improvements.
Mechanisms of Cystic Fibrosis-Related Diabetes Pathogenesis
CFRD shares features with but is distinct from type 1 diabetes (T1D) and T2D, and arises largely from insulin secretion defects (Fig. 1) with a contribution of reduced insulin sensitivity that has been best characterized in adults.16 As a pancreatogenic form of diabetes, pancreatic exocrine inflammation extending to pancreatic islet tissue where fatty infiltration and fibrosis contribute to reductions in islet mass has traditionally been considered responsible for impaired insulin secretion. Indeed, pancreatic islets from individuals with CFRD also show disrupted architecture, abnormal aggregation, and variable encasement in fatty or fibrotic tissue as well as immune cell infiltration.17 More recently, however, a 50% relative reduction in beta-cell number per islet, smaller insulin-positive area, and lower markers of beta-cell proliferation and ductal cells were found in pancreata from children with CF aged <4 years; these reductions were independent of the extent of pancreatic exocrine damage.18 These data suggest beta-cell loss is not simply a by-product of exocrine tissue damage extending to islets but may reflect reduced survival of beta-cell progenitors, reduced beta-cell proliferation, or given the relative abundance of other islet cells, reduced beta-cell fate specification of progenitors.18 Interestingly, neonatal CF transmembrane regulator (CFTR) knockout ferret islets also demonstrate 30% to 50% reductions in insulin content but upregulate insulin secretion.19
Fig. 1.
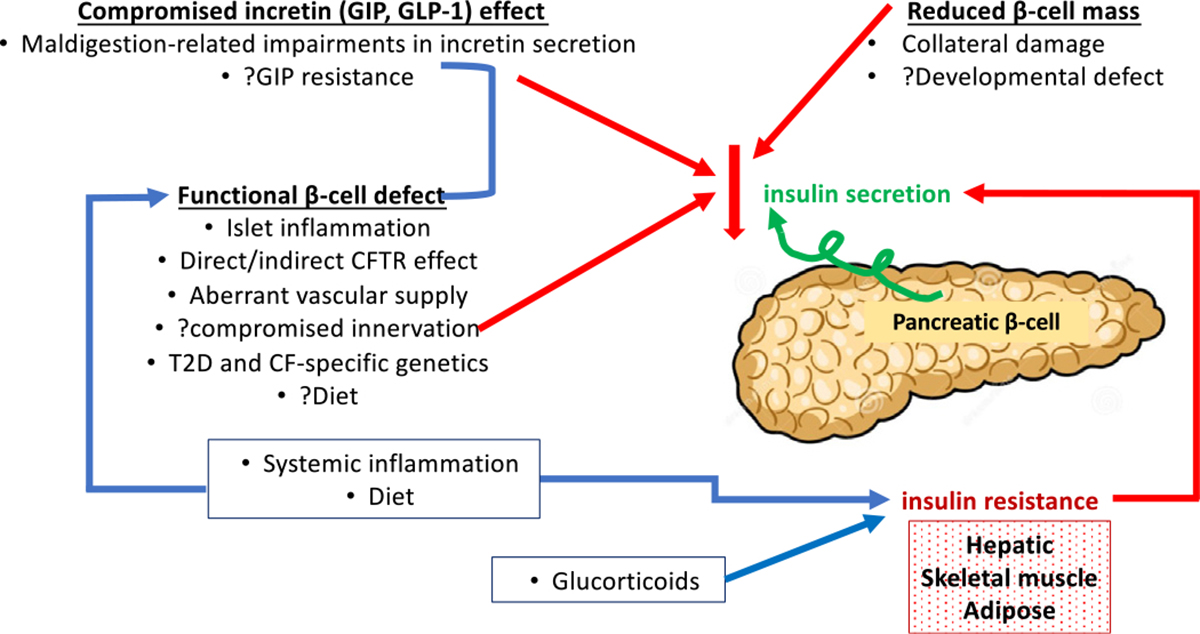
Pathogenesis of CFRD arises largely from insulin secretion defects.
Research into Mechanisms of Cystic Fibrosis-Related Diabetes Pathogenesis
With the goal of delineating the mechanisms underlying insulin secretion defects in CF to develop interventions that delay, prevent, and treat CFRD, much attention has focused on the endocrine pancreas and its pancreatic exocrine neighborhood.
A primary role for defective beta-cell function in the insulin secretion defects of CF is receiving increasing attention. Insulin secretion defects in the pig model, in which islet mass is generally preserved, are unlikely fully explained by the modest reductions in overall insulin content of the pancreas.20 Also suggesting that the islet “neighborhood” may be relevant, insulin secretion studies of islets isolated from pancreata of individuals with CFRD documented preserved insulin and glucagon secretion.17 CF pigs demonstrate intact islet vasculature but reduction in neural fibers20; the contribution of aberrant innervation to islet function is not yet known albeit actively being studied. Supporting a role for inflammation in islet dysfunction are (1) immune cell infiltration of islets from people with CFRD,17 (2) increased secretion of interleukin-6 by CFTR knockout ferret islets, and (3) the recapitulation of the neonatal CFTR knockout ferret model’s reduced insulin content but upregulation of insulin secretion following interleukin-6 application to the wild-type ferret.19 Further highlighting the inflammatory milieu’s potential negative impact upon beta-cell insulin secretion, increased interleukin-1 beta accompanied relatively preserved beta-cell area and higher alpha-cell area in pancreata from patients with CF with and without CFRD including in youth.21
The direct role of CFTR in islet-cell function has been debated. CFTR RNA expression is very low in beta cells,17,22–24 and in extensive studies by Hart and colleagues, immunocytochemistry did not identify CFTR protein coexpression with insulin-positive, glucagon-positive, or somatostatin-positive cells in human islets whereas CFTR modulators and inhibitors did not appear to impact in vitro insulin secretion even at high glucose concentrations.17 These studies contrast with other in vitro murine and human islet studies identifying impaired GLP-1-augmented and forskolin-augmented glucose-potentiated insulin secretion with CFTR inhibition—findings that were attributed to interfere with cyclic AMP-mediated exocytosis of insulin secretory granules.25 By way of explanation, CFTR inhibition similarly reduced insulin secretion from isolated human, wild-type ferret, and CF ferret islets19–findings that suggest these CFTR inhibitors may be operating nonspecifically.
These questions surrounding the role of CFTR in islet cell function are not arcane. Case reports and series documenting improvements in insulin secretion and glucose excursion/tolerance with CFTR modulator therapy with ivacaftor26,27 and the trend toward lower CFRD rates in the US and UK registries over the 4 to 5 years following ivacaftor treatment28 underscore their relevance. Unfortunately, these data are unable to differentiate direct potentiation of insulin secretion by beta-cell CFTR modulation from indirect enhancements in insulin secretion related to improvements in systemic or peri-islet inflammation. In a multi-center French study of OGTT in individuals aged 12 years or older with IGT (n = 31) or CFRD (n = 9) at baseline, glucose tolerance improved following 1 year of lumacaftor/ivacaftor29; the enthusiasm was tempered with the reminder of the well-recognized variability in OGTT.30 In contrast, the overall lack of improvement in glucose tolerance and insulin secretion in the US-based PROSPECT with lumacaftor/ivacaftor was disheartening but not unexpected given the modest impact of the modulator upon CF outcomes.31 More recent CGM data from people with CFRD identified improvements in glucose excursion with elexacaftor/tezacaftor/ivacaftor (ETI).32 The CFF-funded PROMISE endocrine sub-study was organized to test the impact of 24 to 30 months of ETI on various aspects of glucose excursion as well as islet hormone and incretin secretion using multisample oral glucose tolerance tests performed at baseline, 12 to 18, and 24 to 30 months.33 PROMISE would be unable to discern the direct and indirect impacts of ETI on beta-cell function, but analyses of the relationships of changes in OGTT outcomes to changes in BMI, body composition, pulmonary function, and, potentially, systemic inflammation, liver stiffness, and gastrointestinal health are planned. Worthy of consideration, aging and the emergence of overweight/obesity may unmask the residual compromised beta-cell function.
Enrichment in genome-wide associated (GWAS) T2D insulin secretion-affecting variants confers increased CFRD risk.34 Candidate gene-based studies identified increased CFRD risk, specifically with T2D-related variants in TCF7L2, CDKAL1, CDKN2A/B, and IGF2BP235. Variants in the gene encoding SLC26A9, a widely expressed anion transporter that has not been documented to associate with T2D, are associated with CFRD onset34,35 and with meconium ileus in CF.36 Providing further compelling evidence for a modifying role of SLC26A9, this protein has recently been demonstrated to be coexpressed with CFTR in a subset of pancreatic ductal cells where low-risk variants confer enhanced SLC26A9 expression.22 A second CFRD-specific variant on chromosome 2 near PTMA, encoding prothymosin-α, has also been identified,34 but its role remains undefined. Based upon these variants, a personalized CFRD risk assessment tool based on genetic and clinical measures at birth is now available online.37
Additional patient-oriented studies are attempting to distill the mechanisms underlying progressive insulin secretion defects and development of CFRD. Loss of early-phase insulin secretion (secretion within first 30 minutes of meal or glucose consumption) is one of the earliest markers of insulin secretion defects in CF, is accompanied by augmented second-phase insulin secretion that occurs 60 to 90 minutes after ingestion, and manifests clinically as an isolated OGTT 1-hour glucose equal to or greater than 155 mg/dL,38,39 a threshold that is increasingly being recognized as abnormal in adults who will later advance to T2D,40,41 Fig. 2. Insulin secretion is likely abnormal even prior to this “early glucose intolerance” (EGI) threshold as individuals with PI-CF and 1-hour glucose less than 155 mg/dL (1) have glucose concentrations that are higher at 1 hour than in otherwise healthy individuals without CF and (2) demonstrate augmented second-phase insulin secretion.38 The mechanisms underlying the gradual loss of early-phase insulin secretion, worsening of glucose tolerance, loss of second-phase insulin secretion as glucose tolerance worsens,38 and, in a subset of patients, emergence of fasting hyperglycemia are not known. The animal models described above will hopefully provide clarity on some of these questions.
Fig. 2.
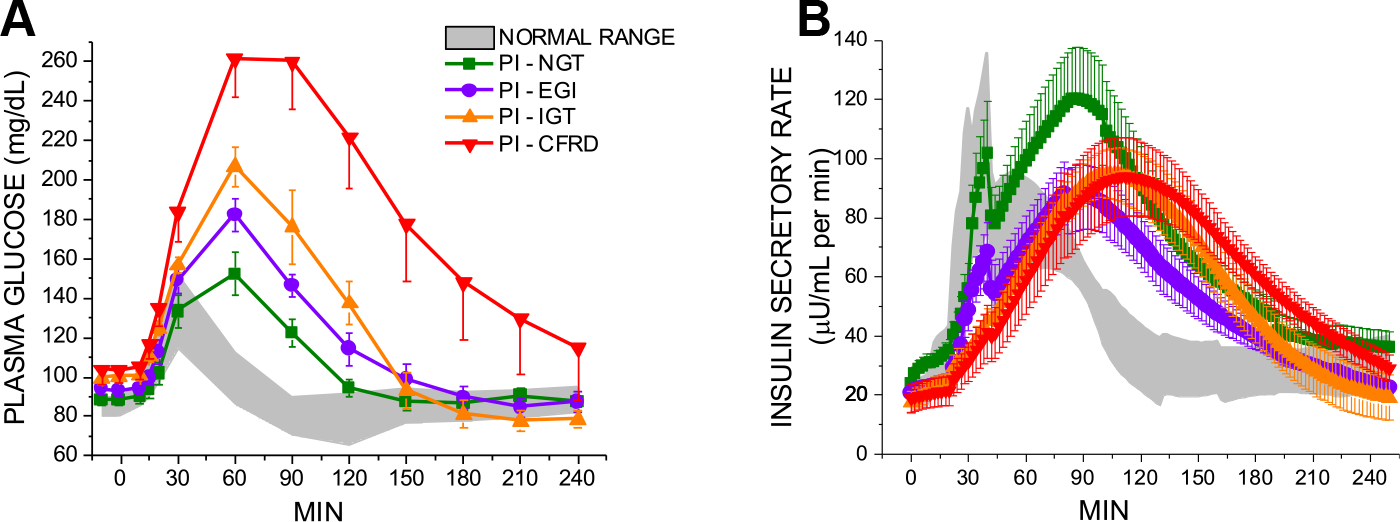
Plasma glucose (A) and insulin secretory rates (B) in response to the mixed-meal tolerance test (MMTT) in subjects with pancreatic insufficient CF (PI-CF). Individuals were categorized based on a preceding oral glucose tolerance test (NGT, normal glucose tolerance; EGI, early glucose intolerance (plasma glucose at 1 hour > 155 mg/dL and plasma glucose at 2 hours < 140 mg/dL; IGT, impaired glucose tolerance; CFRD, CF-related diabetes). Significant decline in beta-cell secretory capacity is evident in PI-EGI. Reprinted under STM Permissions Guidelines from “Beta-cell secretory defects are present in pancreatic insufficient cystic fibrosis with 1-h oral glucose tolerance test glucose >/ = 155 mg/dL”. (From Nyirjesy SC, Sheikh S, Hadjiliadis D, De Leon DD, Peleckis AJ, Eiel JN, Kubrak C, Stefanovski D, Rubenstein RC, Rickels MR, Kelly A. β-Cell secretory defects are present in pancreatic insufficient cystic fibrosis with 1-hour oral glucose tolerance test glucose ≥155 mg/dL. Pediatr Diabetes. 2018 Nov;19(7):1173–1182.)
Insulin resistance is recognized but has not been considered a major contributor to CFRD. Basing glucose tolerance strictly on OGTT 2-hour glucose in adults with pancreatic exocrine sufficient (PS) and pancreatic exocrine insufficient (PI) CF, Boudreau and colleagues identified baseline differences in OGTT insulin area under the curve (AUC) from 0 to 30 min and 30 to 120 min in the stable, improved, and worsened glucose tolerance status groups over approximately 21 months.42 Worsening of these insulin AUC phases was not apparent in the group whose glucose tolerance worsened. Instead, lower insulin sensitivity was found.42 Increasing rates of overweight and obesity in CF may propel a greater role for insulin resistance in CFRD development. At the University of Minnesota (2015–2017), overweight was present in approximately 25%, obesity in 6.6%, and underweight in only 5.2% of adults with CF.43 In 2012, the University of Pittsburgh reported 15% overweight and 8% obesity in youth aged 2 to 18 years with CF.44 With increasing use of highly effective modulator therapy and improvements in body mass index (BMI) that accompany that therapy,45 undernutrition may become even less common.
Treatment of Cystic Fibrosis-Related Diabetes
Current CFRD clinical care guidelines recommend insulin therapy as the mainstay of CFRD treatment2,46 These guidelines are based on associations of CFRD with worsening nutritional status and pulmonary function47,48 that have been ascribed to reduced insulin secretion49; the former improved with a 12-month intervention with rapid-acting insulin. Many insulin formulations are available and the approach to CFRD management is highly individualized to meet the lifestyle and nutritional needs of patients. Current guidelines do not differentiate the management of CFRD among those with insulin deficiency versus insulin resistance; however, research exploring the use of T2D agents is ongoing. Compromised early-phase insulin secretion (robust insulin secretion occurring within the first 30 minutes following a meal) leads to early hyperglycemia and a compensatory, albeit delayed, insulin secretion. This excessive insulin secretion with a potential contribution of inadequate glucagon response appear to contribute to postprandial hypoglycemia in CF.50,51 No studies have systematically addressed interventions to mitigate hypoglycemia development, but clinically (1) avoidance of quick acting sugars, (2) pairing quick-acting sugars with complex carbohydrates, proteins, and fats, (3) administering rapid-acting insulin 15 to 20 minutes before meals, and (4) switching premeal rapid-acting insulin to ultrafast-acting insulin are all trialed. The emerging technologies presented below may also help combat this nuisance.
Emerging Technologies
Rapid advances in diabetes technologies over the last decade have improved the care of people with CFRD. CGM monitors are wearable technologies that measure and report interstitial glucose values every 5 to 15 minutes with optional alerts for both hypoglycemia and hyperglycemia.52 Since its development in 1999, CGM has become increasingly user friendly such that devices are factory calibrated, do not require calibration, and allow for diabetes treatment decisions to be made without a confirmatory fingerstick glucose value. Differences between the gold-standard Yellow Springs Instrument measurements of plasma glucose concentrations and CGM measurements are less than 10% among commercially available systems. The accuracy of CGM among people with CF is comparable to that among individuals without CF.53 Most CGM sensors are worn for 7 to 14 consecutive days before being changed at home by the user with simple applicator devices. Glucose values may be sent to the user’s smartphone or to a dedicated receiver that can display the glucose values (see example in Fig. 3).
Fig. 3.
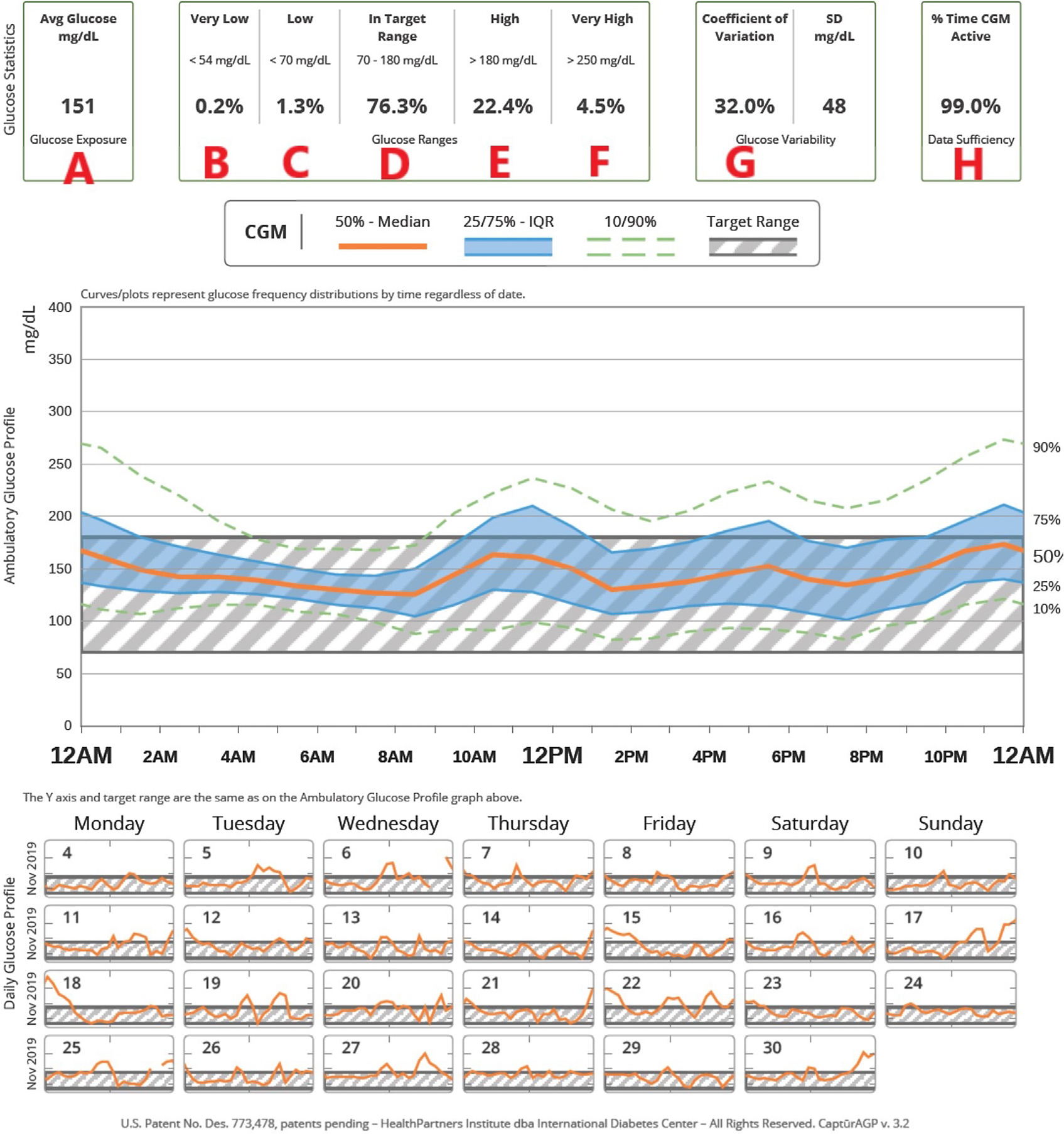
Example of continuous glucose monitoring (CGM) usage. (From Marks BE, Wolfsdorf JI. Monitoring of Pediatric Type 1 Diabetes. Front Endocrinol (Lausanne). 2020 Mar 17;11:128.)
CGM-defined glycemic targets have not yet been established for individuals with CFRD. However, International Consensus Guidelines for most individuals with T1D and T2D recommend targeting at least 70% of sensor glucose values between 70 to 180 mg/dL (time in range) and no more than 5% of time below 70 mg/dL (time below range).54 These targets are derived from data demonstrating a strong correlation between 70% time in range and HbA1c less than 7%.55 Given the HbA1c goal of less than or equal to 7% in CFRD, applying these targets for individuals with CFRD seems reasonable. Rates of CGM use in routine clinical care were 75% among a selected group of individuals with CFRD.56 Despite positive perceptions of this technology among the CF community, including those with and without CFRD, and many perceived benefits of CGM use, 19% discontinued CGM mostly commonly due to cost and increased worry about glycemia. Greater evidence to support the benefits of CGM use among people with CFRD and better insurance coverage of this technology coupled with improved education about CGM may increase uptake and sustained use of this technology.
The last decade has also witnessed tremendous advances in insulin delivery systems. Insulin pumps provide a continuous subcutaneous infusion of rapid-acting insulin that allows for greater customization of insulin delivery than with injection-based therapy. Data in this area are extremely limited, but continuous insulin infusion may optimize glycemic control for those with common gastrointestinal comorbidities of CF including exocrine pancreatic insufficiency, gastroparesis with alterations in intestinal transit, and periods of insulin resistance during illness. Improvements in lean body mass and glycemic control with insulin pump therapy have been demonstrated in CFRD.57 Despite its potential benefits, insulin pump use is not sustained among a subset of people with CFRD and is less highly regarded than CGM among the CF community.56
Automated insulin delivery (AID), which uses CGM glucose data to guide pump delivery of insulin, is the most promising development in diabetes technology to date. AID systems increase insulin delivery in response to hyperglycemia and can also decrease or suspend insulin delivery in response to predicated or impending hypoglycemia. Among individuals with T1D, AID use improved glycemic control while simultaneously decreasing patient burden.58,59 A pilot study (n = 3) of a dual hormone AID system employing insulin and glucagon showed nonsignificant improvements in mean CGM sensor glucose and patient reported outcomes with decreased treatment burden.60 However, AID systems and insulin delivery algorithms were developed to meet the needs of individuals with T1D, who in contrast to people with CFRD, have near complete insulin deficiency. Further studies of AID efficacy and patient-reported outcomes in individuals with the unique physiology of CFRD are needed.
Research into Therapeutic Approaches for Cystic Fibrosis-Related Diabetes
Associations of early glucose abnormalities with worse CF outcomes have prompted interventions with insulin in the prediabetes state.61–65 Results of various insulin regimens (long-acting, rapid) in prediabetes have been variable and may be more effective with worse baseline health.65 Many of the recent clinical studies in CF report fairly preserved nutritional status in people with CFRD (average body mass index z-score (BMIZ): −0.6 to −0.8),66 adults with abnormal glucose tolerance (average BMIZ: −0.05)50 or abnormal glucose tolerance/CFRD without fasting hyperglycemia (FH),67 and youth (NGT: −0.18; AGT: 0.14, CFRD: −0.65).39 As a result, the focus of management may shift from nutritional status (and perhaps pulmonary function) to traditional diabetes-related microvascular outcomes, though less likely in CFRD than in T1D and T2D.
With the changing CF landscape and the goal of less encumbered CFRD care, several case series and acute, short-term, and longer-term trials are examining the role of T2D interventions in people with CF and abnormal glucose tolerance. Following 12 weeks of combined aerobic and resistance exercise program in 8 sedentary adults with abnormal glucose tolerance, glucose excursion improved while remaining unchanged in the control group.68 Similarly, a randomized trial examining the impact of aerobic interval training upon glucose tolerance is planned for youth with CF.69
T2D incretin-based therapies are predicated on the ability of the gut derived hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), to augment insulin secretion in response to a meal or oral glucose load. In T2D, GLP-1 agonist treatment has the added advantage of weight loss, whereas dipeptidyl peptidase 4 inhibitors (DPP-4) like sitagliptin interfere with incretin catabolism but tend to be weight neutral. Incretin-based therapies are attractive in CF because data demonstrate improved meal-related incretin secretion and glucose excursion with pancreatic enzyme replacement in individuals with pancreatic insufficient CF.70,71 Moreover, in the CF ferret, hyperglycemia is accompanied by dampened insulin, GLP-1, and GIP responses.72 In a randomized, cross-over study of a single dose of either the short-acting GLP-1 agonist, exenatide, or placebo in 6 individuals aged 11 to 24 years with IGT, exenatide was associated with lower glucose and insulin following a high carbohydrate (50 g) meal (Table 2).73 In a 6-month, randomized, placebo-controlled study, the oral DDP-4 inhibitor, sitagliptin, was well tolerated, associated with increased GLP-1 and GIP and improved early-phase insulin (first 30 minutes) secretion in response to a meal, but no improvement in glucose tolerance/excursion in adults with pancreatic insufficient CF and abnormal glucose tolerance; BMI remained unchanged.67 A case series of 3 patients with CFRD treated with sitagliptin showed short-term improvements in glucose excursion as defined by CGM and continued CFRD control beyond 5 years in 2 in whom BMI was maintained or improved; the third participant had worsening obesity (BMI 32–30 kg/m2) and required insulin therapy beyond the 5 years.74 These preliminary data suggest incretin-based therapies may have a role in a subset of people with CFRD in whom under-nutrition is not a concern. A 6-week, cross-over, proof-of-concept study will test the impact of weekly dulaglutide on early phase insulin secretion during a meal and will collect important data on gastrointestinal side effects and BMI (NCT04731272).
Table 2.
Trials of type 2 diabetes medications in CFRD
| Authors. Year | Study Design | Sample Size | Inclusion Criteria | Treatment | Outcomes |
|---|---|---|---|---|---|
| Geyer, et al,74 2018 | Double-blinded, Randomized, placebo-controlled, cross-over | N = 6 | Ages 11–24 IGT |
Single-dose Exenatide 2.5 mg vs Placebo | During MMTT, AUC over 240 min for blood glucose, insulin, and GLP-1 was lower with exenatide |
| Kelly, et al,68 2021 | Randomized, placebo-controlled | N = 26 | Adults AGT (1 h >155 and 2h <140), IGT, or CFRD |
6-mo double-blind Sitagliptin 100 mg daily vs Placebo | During MMTT GLP-1, GIP insulin secretion rates improved while glucagon suppression increased without changes in postprandial glycemia |
| Olatunbosun,75 2021 | Case series | N = 3 | CFRD (44yo M, 38yo M, 19yo F) | 5–10 y of Sitagliptin 100 mg daily | Improved BMI, HbA1c <6%; insulin required after 7 and 10 y in 2 patients |
| Ballmann, et al,30 2017 | Open-label, randomized | N = 67 | >10 y Newly diagnosed CFRD |
24 mo Repaglinide vs Insulin | No significant difference in HbA1c over time between the two groups |
Ballmann and colleagues completed a 24-month, multicenter study of 3 times per day, premeal, oral repaglinide (n = 34; 14 of whom discontinued before 24 months) to 3 times per day, premeal, regular insulin (n = 41; 16 of whom discontinued before 24 months) in individuals aged 10 years or older with newly diagnosed CFRD. No between-group differences in HbA1c, FEV1%-predicted, or BMI were found at 24 months.66 An additional ongoing study in people with CFRD is the cross-over MIRE study of 3-month of preprandial aspart insulin versus post-prandial faster-acting aspart insulin which will compare time in range by CGM (NCT04381429).
With individuals with CF overall healthier, a number of the traditional paradigms are being challenged. On a cautionary note, this recent literature many not be generalizable to the subsets of individuals around the world with limited access to care and highly effective modulator therapies, with CFTR mutations that are not amenable to highly effective modulator therapies or who do not tolerate highly effective modulator therapy, or with such profound insulin secretion defects that the prospect of using T2D therapies is unlikely to impact hyperglycemia.
CYSTIC FIBROSIS-RELATED BONE DISEASE
Introduction
Cystic fibrosis-related bone disease (CFBD) was first described by Mischler and colleagues in 1979, as an association of low bone density in CF. In their study, they found that 44% of CF individuals had reduced bone mineral content (BMC).75 An interesting bit of foreshadowing for future research studies, they noted that these findings were confounded by short stature, delayed bone age and low body weight. In the past 40 years since that first report, numerous studies have documented bone disease in CF. These studies demonstrated the multitude of factors that contribute to bone disease. Probably the greatest presentation of bone disease in CF is found in the individuals referred for lung transplant, in whom 57% had osteoporosis,76 translating to a 100-fold greater risk of vertebral compression fractures (a potential disqualifier for transplantation). In contrast, lower bone density and content does not necessarily translate to an increase fracture risk in youth. Rovner and colleagues, in a study of 186 CF children and young adults with mild to moderate lung disease, reported fracture rates that were comparable to those of healthy children.77 Underscoring the potential implications of bone fragility in youth are case reports of significant fracture, including the spontaneous sternal fracture in a 16-year-old woman that caused severe respiratory distress.78
Screening Guidelines
The 2019 Cystic Fibrosis Foundation Patient Registry (CFFPR) reports a history fracture, osteopenia, and osteoporosis as 0.2%, 1.1%, and 0.3% (respectively) for children less than 18 years and 0.2%, 17.9%, and 7.0% (respectively) for adults older than 18 years.79 In much need of revision, the CF guidelines for bone health screening in CF currently recommend baseline dual-energy x-ray absorptiometry (DXA) for all individuals aged 18 years or older, and for children aged 8 years or older with risk factors.80 The guidelines also recommend DXA results guide timing of subsequent DXA. The CFFPR documents that only 59.3% of individuals aged 18 years or older underwent DXA during 2015 to 2019.79 Ongoing studies aim to improve both patient and CF Center compliance with the guidelines.
Research into Mechanisms of Cystic Fibrosis-Related Bone Disease
Briefly stated, CFBD is an example of a multifactorial disorder, with contributions from vitamin D deficiency, malnutrition, hypogonadism, increased inflammatory cytokines, and glucocorticoid therapies.80–91 Additionally, male gender, advanced lung disease, malnutrition, and low fat-free body mass are established additional risk factors for CFBD.80,92–94 Furthermore, emerging data suggest a direct genetic component to the development of low bone density. The F508del-CFTR mutation is the most common mutation resulting in CF in the United States, with almost 85.3% of CF individuals having at least 1 copy and 44.4% homozygous for this genotype.95 More severe mutations, and especially the F508del-CFTR genotype, are more commonly associated with reduced bone density.96
Although many factors associated with CFBD are established, other disease states provide insights into additional contributors. Bone health is maintained through a dynamic bone turnover process. In CF, uncoupled bone turnover (a state of decreased bone formation in the presence of increased osteoclast bone resorption) is present.
A well-described contributor to CFBD in the literature, vitamin D deficiency with resultant reduced calcium gut absorption, continues to be confounding bone health today. Guidelines can assist with vitamin D absorption difficulties in CF patients, proposing target concentrations of 25-hydroxy vitamin D and approaches to meet these. Reduced 25-hydroxy vitamin D and calcium concentrations induce release of parathyroid hormone (PTH) and consequently osteoclastic bone turnover to restore circulating calcium and the body’s immediate needs.
Corticosteroid therapy has long been appreciated as a contributor to CFBD, especially in the posttransplant population. Corticosteroids impact bone health through multiple mechanisms: (1) reduced gastrointestinal calcium absorption, (2) increased urinary calcium excretion, and (3) increased osteoclast-driven bone resorption (via production of receptor activator of nuclear factor-kappa B ligand or RANKL).84 Through similar mechanisms, increased and chronic inflammation (IL-8 and others) upregulates the same pathway, and correlates with BMC.85–87,96,97
The chronic insulin deficient state found in CF and CFRD is a less-recognized contribution to CFBD. Insulin (an anabolic hormone) is a potent stimulator of osteoblast proliferation and function. Rana and colleagues compared DXA outcomes by CFRD status in 81 youth aged 18 years or younger, demonstrating an association with reduced bone density.92 Other proanabolic hormones, such as insulin-like growth factor 1 (IGF-1) and sex hormones (testosterone and estrogen) directly affect bone health. Although reduced IGF-1 concentrations are repeatedly documented in CF individuals and animal models of CF, serum IGF-1 correlates with bone density in CF. The contributions of testosterone and estrogen are more theoretic in CF, based on their contributions in healthy individuals. One retrospective study by Wu and colleagues demonstrated higher BMD in women who had been treated with supplemental estrogen before age 21.98 Ongoing studies are examining the influence of estrogen supplementation upon CF bone health.
Studies also identify a direct contribution of CFTR to CFBD. Multiple animal studies in CF deficient mice (CFTR knockout and the F508del-CFTR models) demonstrate reduced bone density, and a study of CF rats found reduced bone content. Notably, the rodent model does not develop overt lung manifestations (prior to these findings in the CF rat).99–102 Reduced bone mineral content was found in the newborn CF piglets compared with wild-type littermates (and simultaneously lower IGF-1 concentrations).103 At the cellular level, Cftr mRNA expression and immunohistochemistry staining of CFTR have been documented in murine osteoblasts, but not murine osteoclasts. Reduced bone formation and fewer osteoblasts but increased osteoclasts were also found in CFTR-deficient murine cell cultures. Osteoblast–osteoclast cell signaling in dynamic cell cultures revealed reduced expression of the RANKL competitive inhibitor, osteoprotegerin (OPG) and an increased Rankl to OPG ratio that drives osteoclastogenesis.
The use of novel CFTR modulators has added to the evidence of a direct CFTR contribution (or at least a systemic one). Recently, improvements in cortical microarchitecture, as measured by high-resolution peripheral quantitative computed tomography, were found in adults with the G551D-CFTR genotype and treated with ivacaftor, but no differences in children were found.104
Research Studies of Cystic Fibrosis-Related Bone Disease Treatment
The CFRD treatment paradigm for CFBD has largely focused on disease prevention or interrupting disease progression. The prevention arm focuses on vitamin D, calcium, and vitamin K supplementation although newer studies focus on factors like insulin (in CFRD) and sex hormone replacement. The CFF guidelines suggest the following for vitamin D supplementation: 400 to 500 IU/day for children 12 months and younger, 800 to 1000 IU/day for 1 to 10 years of age, and 800 to 2000 IU/day for 11 years and older. Additionally, a serum 25-hydroxyvitamin D concentration at least 30 ng/mL is recommended.105 CFBD treatment is limited to bisphosphonate medications in adults with DXA BMD Z-scores less than 2. These medications increase BMD in CFBD but the impact upon fracture risk is not known.106 Newer therapies, such as teriparatide (which stimulates osteoblast activity) could address the uncoupled nature of bone disease. Growth hormone may also improve bone content in CF,107 but studies have focused more on the linear growth potential. IGF-1 therapy and oxandrolone, which also stimulate osteoblast activity, have yet to be studied in CFBD. Finally, other antiresorptive agents such as denosumab (monoclonal antibody against RANKL) are attractive for targeting the increased inflammatory contribution to CFBD.108
GROWTH RESTRICTION IN CYSTIC FIBROSIS
Introduction
Linear bone growth and bone health are intimately related. Growth, as a function of weight and BMI, has long been associated with improved health and lung function in CF. Issues of weight gain and the direct contribution of nutrition are beyond the scope of this review; therefore we focus here on the issue of linear growth itself. However, it is notable that a limited focus on height, and more focus on weight and BMI, may have led to an underrecognized linear growth problem in CF. Recently, investigators have discovered that despite improvements in weight and BMI to that of the 50th% or better, 20% of individuals had a height less than the 10th%.109 More alarming, impaired linear growth can be an early indicator of pulmonary disease, even before spirometry can be reliably obtained.110 In a study by Sanders and colleagues, maintaining a height for age above the 50th percentile was a predictor for pulmonary function despite BMI.111 The more dramatic depiction of stunting, defined as height below the 5th percentile, is a predictor of mortality in CF.112 In our brief discussion, we will review the potential role of CFTR itself in linear growth, and the expected benefits of a new era of therapies.
Mechanisms of Linear Growth Impairment
Much like bone health, linear growth is multifactorial with many positive and negative contributors. In CF, the weight of these confounders is unbalanced to a less productive endpoint. Unfortunately some clinical treatments that are used in CF may impair growth further (such as corticosteroids).113 The GH hormone and IGF-1 axis contributing to linear growth has been studied clinically and in animal models.103,114,115 Controversies over GH secretion exist, but consistently IGF-1 concentrations are low, believed to reflect the impact of chronic inflammation on this axis.116,117 This factor lowers the anabolic drive of the growing child. Other nonhormonal factors that negatively impact growth such as chronic inflammation, inflammatory cytokines, and corticosteroid usage can directly interfere with the growth plate itself.113
However, poor growth in CF begins very early in life and may be linked to the degree of CFTR deficiency (or severity of the genotype). In the BONUS study, an MCT of infants with CF diagnosed by newborn screen, weight for age and length for age had already declined by 1 to 3 months of age (Fig. 4).118 While weight for age recovers by 8 to 10 months of age with the general population, length for age does not by 1 year in CF. In that particular study, the investigators discovered that the presence of pancreatic insufficiency was a predictor of whether infants would meet their expected length for age by 1 year.
Fig. 4.
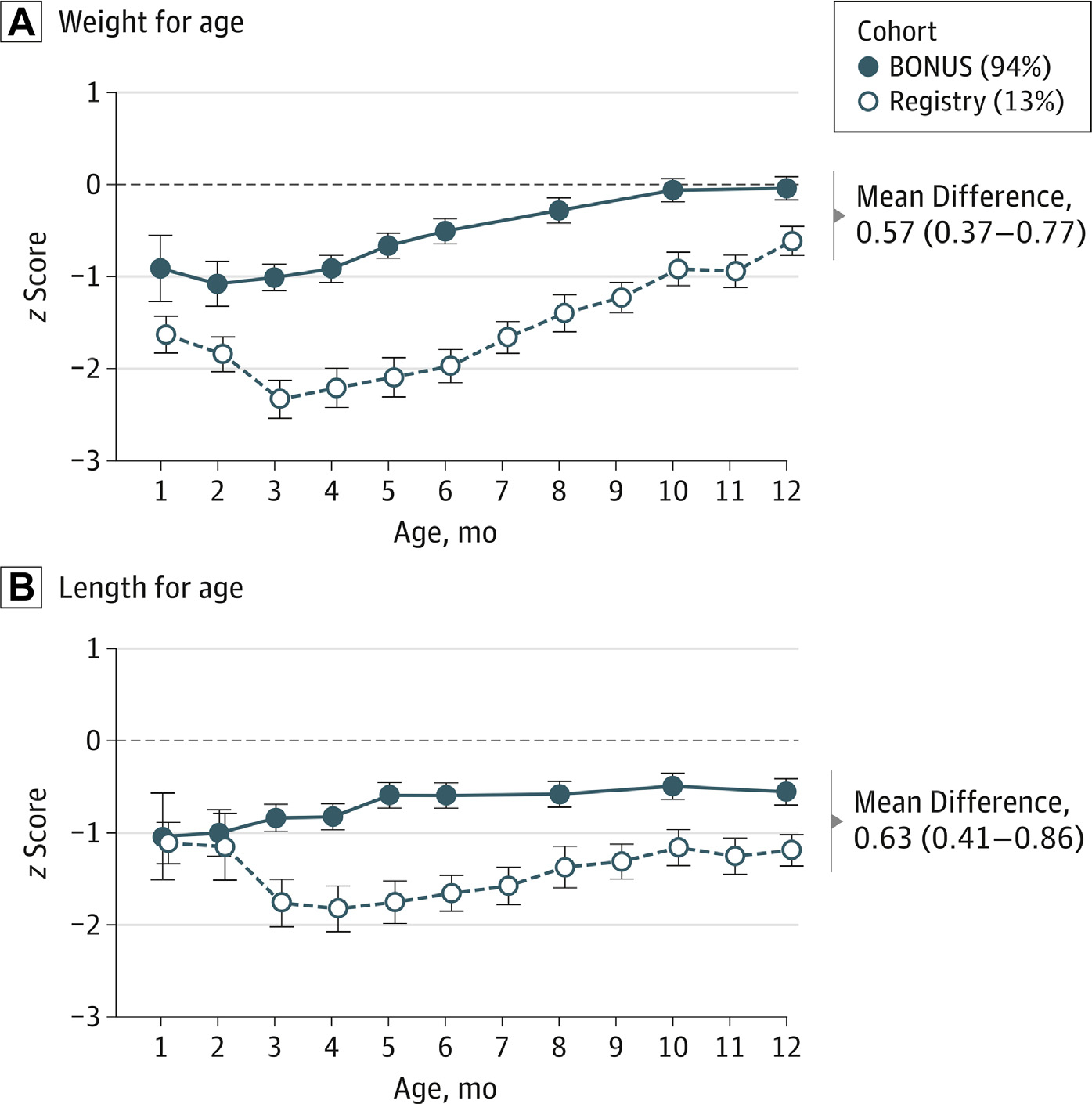
Baby Observational and Nutrional Study (BONUS) Cohort and Historic Infant Cohort z Scores for Growth During the First Year of Life. (A) Weight for age, ( B) Length for age. (From Leung DH, Heltshe SL, Borowitz D, Gelfond D, Kloster M, Heubi JE, Stalvey M, Ramsey BW; Baby Observational and Nutrition Study (BONUS) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Effects of Diagnosis by Newborn Screening for Cystic Fibrosis on Weight and Length in the First Year of Life. JAMA Pediatr. 2017 Jun 1;171(6):546–554.)
Additionally, the animal models discussed above in bone health and CFBD demonstrate growth restriction despite a lack of pulmonary manifestations and/or pancreatic disease. In an early paper by Rosenberg and colleagues, the authors studied growth in the CFTR-deficient mouse. CFTR-deficient mice had reduced weight and length compared with their wild-type counterparts. Additionally, the CF mice had reduced IGF-1 concentrations.114 A similar finding was detected by Rogan and colleagues in the CF newborn piglets, in which even at birth they had shorter humeral lengths and IGF-1 levels than non-CF pigs.103 Lastly, the CF rat model was found to have analogous findings of reduced femoral length, weight, and IGF-1.115 These discoveries led to some of the early evidence that CFTR may directly contribute to linear growth. However, a better understanding of the contribution to growth by CFTR is needed, and whether it occurs as a systemic or direct effect.
Clinical evidence that linear growth is potentially impacted directly by CFTR dysfunction in childhood was first demonstrated by improvements in growth following ivacaftor treatment in pre-pubertal CF children with at least one copy of the G551D-CFTR genotype.119 In that report, children (ages 6–11 years) in the multi-center observational study GOAL and the placebo-controlled study ENVISION, demonstrated an improvement in height Z-scores as well as annualized growth velocities following treatment with ivacaftor. Ongoing and future studies are attempting to evaluate the changes in growth following the initiation of newer HEMT in both infants and toddlers, as well as prepubertal and pubertal children with CF. These newer modulators under investigation target the F508del genotype which will encompass a much broader clinical base and may have a larger effect on the CF population.
Treatment Approaches to Linear Growth Impairment
Unfortunately, despite the advances to date, interventions to optimize growth have been limited, that is outside of the nutritional recommendations. Multiple studies through the past couple of decades have examined the use of human growth hormone (hGH) as a supplemental therapy. In a randomized MCT, treatment with hGH in prepubertal CF children demonstrated a 0.5 SDS improvement in height over 1 year, and mean height velocity of 8.2 cm/y compared to 5.3 cm/y in the observation group.120 However, growth hormone treatment-related improvements in linear growth, bone mineral density, weight, and lean body mass did not translate into dramatic improvements in pulmonary function. Additionally, growth hormone carries the theoretic risk of worsening or precipitating CFRD, however data from the above MC trial did not support that concern.
Research Efforts to Treat Linear Growth Impairment
A study by Zhang in 2013 reviewed peak height velocity and adult height in CF,121 the authors found about a 6-month delay and 15% reduction in peak height velocity. This delay in peak height velocity may represent a second-hit following the early restriction seen in infants. As pubertal delays in CF improve in the HEMT era, these delay may be further attenuated in the future. In a more recent paper by Zysman-Colman, peak height velocity already shows a more consistent timing with the general pediatric population, although remaining still shorter than their peers.122
Current data from the CFF Patient Registry Annual Report documents the average length of individuals under the age of 24 months to be 30th percentile, and those between the ages of 2 and 19 years to be 38.4th percentile.79 Regardless, we have seen improvements over the past 15 years, with height less than 5th percentile decreasing from 15% of individuals to 10%. As growth restriction affects individuals with severe genotypes and those who are pancreatic insufficient to a greater extent,118 we could predict that if HEMT does improve growth, we would see much improvement in the years to come.
SUMMARY
As we celebrate highly effective modulator therapy and longevity, endocrine comorbidities are becoming increasingly important considerations in the care of people with CF. Insights into the pathophysiology of CF-related endocrinopathies from the PROMISE study and improvements in the overall health among people using highly effective modulator therapy may lead to radical changes in the prevalence of and management options for CFRD, CFBD, and linear growth.
CLINICS CARE POINTS.
Progressive insulin secretion defects underlie abnormal glucose tolerance in cystic fibrosis (CF).
Leveraging diabetes technologies and type 2 diabetes therapeutics may advance care in youth and adults with CF-related abnormal glucose tolerance.
Advances in care and the introduction of highly effective modulator therapies may challenge traditional paradigms that focus on the nutritional compromise that accompanies insulin secretion defects in CF.
The complexity of clinical care in CF confounds and may worsen bone health, thus close supervision is warranted to exacerbate an existing problem.
When possible, prevention of bone disease is the best course.
Growth restriction in CF can be a prognostic indicator and should not be overlooked for the sake of focusing weight and BMI.
KEY POINTS.
Progressive insulin secretion defects underlie abnormal glucose tolerance in cystic fibrosis (CF).
Leveraging diabetes technologies and type 2 diabetes therapeutics may advance care in youth and adults with CF-related abnormal glucose tolerance.
Advances in care and the introduction of highly effective modulator therapies may challenge the traditional paradigms that focus on the nutritional compromise that accompanies insulin secretion defects in CF.
Footnotes
DISCLOSURE
A. Kelly has nothing to disclose. B. Marks has received investigator initiated research support from Tandem Diabetes Care and Dexcom. M. Stalvey has nothing to disclose.
REFERENCES
- 1.Kwong E, Desai S, Chong L, et al. The impact of cystic fibrosis-related diabetes on health-related quality of life. J Cystic Fibrosis 2019;7–9. [DOI] [PubMed] [Google Scholar]
- 2.Moran A, Brunzell C, Cohen RC, et al. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care 2010;33(12):2697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan CL. Cystic fibrosis related diabetes: Revisiting the OGTT and alternate screening tests. J Cyst Fibros 2020;19(5):671–2. [DOI] [PubMed] [Google Scholar]
- 4.Burgess JC, Bridges N, Banya W, et al. HbA1c as a screening tool for cystic fibrosis related diabetes. J Cyst Fibros 2016;15(2):251–7. [DOI] [PubMed] [Google Scholar]
- 5.Boudreau V, Coriati A, Desjardins K, et al. Glycated hemoglobin cannot yet be proposed as a screening tool for cystic fibrosis related diabetes. J Cyst Fibros 2016;15(2):258–60. [DOI] [PubMed] [Google Scholar]
- 6.Gilmour JA, Sykes J, Etchells E, et al. Cystic fibrosis-related diabetes screening in adults: a Gap analysis and evaluation of accuracy of Glycated hemoglobin levels. Can J Diabetes 2019;43(1):13–8. [DOI] [PubMed] [Google Scholar]
- 7.Sheikh S, Localio AR, Kelly A, et al. Abnormal glucose tolerance and the 50-gram glucose challenge test in Cystic fibrosis. J Cyst Fibros 2020;19(5):696–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor-Cousar JL, Janssen JS, Wilson A, et al. Glucose >200 mg/dL during continuous glucose monitoring identifies adult patients at risk for development of cystic fibrosis related diabetes. J Diabetes Res 2016;2016:1527932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CL, Pyle L, Vigers T, Zeitler PS, Nadeau KJ.J Clin Endocrinol Metab. The Relationship Between Continuous Glucose Monitoring and OGTT in Youth and Young Adults With Cystic Fibrosis. 2022. Jan 18;107(2):e548–e560. doi: 10.1210/clinem/dgab692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemente Leon M, Bilbao Gasso L, Moreno-Galdo A, et al. Oral glucose tolerance test and continuous glucose monitoring to assess diabetes development in cystic fibrosis patients. Endocrinol Diabetes Nutr (Engl Ed 2018;65(1):45–51. [DOI] [PubMed] [Google Scholar]
- 11.Elidottir H, Diemer S, Eklund E, et al. Abnormal glucose tolerance and lung function in children with cystic fibrosis. Comparing oral glucose tolerance test and continuous glucose monitoring. J Cyst Fibros 2021;20(5):779–84. [DOI] [PubMed] [Google Scholar]
- 12.Gojsina B, Minic P, Todorovic S, et al. Continuous glucose monitoring as a valuable tool in the early detection of diabetes related to cystic fibrosis. Front Pediatr 2021;9:659728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan CL, Vigers T, Pyle L, et al. Continuous glucose monitoring abnormalities in cystic fibrosis youth correlate with pulmonary function decline. J Cyst Fibros 2018;17(6):783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inman TB, Proudfoot JA, Lim M, et al. Continuous glucose monitoring in a cystic fibrosis patient to predict pulmonary exacerbation? J Cyst Fibros 2017;16(5):628–30. [DOI] [PubMed] [Google Scholar]
- 15.Leclercq A, Gauthier B, Rosner V, et al. Early assessment of glucose abnormalities during continuous glucose monitoring associated with lung function impairment in cystic fibrosis patients. J Cyst Fibros 2014;13(4):478–84. [DOI] [PubMed] [Google Scholar]
- 16.Colomba J, Boudreau V, Lehoux-Dubois C, et al. The main mechanism associated with progression of glucose intolerance in older patients with cystic fibrosis is insulin resistance and not reduced insulin secretion capacity. J Cyst Fibros 2019;18(4):551–6. [DOI] [PubMed] [Google Scholar]
- 17.Hart NJ, Aramandla R, Poffenberger G, et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight 2018;3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogdani M, Blackman SM, Ridaura C, et al. Structural abnormalities in islets from very young children with cystic fibrosis may contribute to cystic fibrosis-related diabetes. Sci Rep 2017;7(1):17231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X, Yi Y, Xie W, et al. CFTR influences beta cell function and insulin secretion through non-cell Autonomous exocrine-derived factors. Endocrinology 2017;158(10):3325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uc A, Olivier AK, Griffin MA, et al. Glycaemic regulation and insulin secretion are abnormal in cystic fibrosis pigs despite sparing of islet cell mass. Clin Sci (Lond) 2015;128(2):131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hull RL, Gibson RL, McNamara S, et al. Islet interleukin-1beta Immunoreactivity is an early feature of cystic fibrosis that may contribute to beta-cell Failure. Diabetes Care 2018;41(4):823–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam AN, Aksit MA, Vecchio-Pagan B, et al. Increased expression of anion transporter SLC26A9 delays diabetes onset in cystic fibrosis. J Clin Invest 2020;130(1):272–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Segerstolpe A, Palasantza A, Eliasson P, et al. Single-cell Transcriptome Profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 2016;24(4):593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baron M, Veres A, Wolock SL, et al. A single-cell Transcriptomic Map of the human and mouse pancreas Reveals inter- and Intra-cell population Structure. Cell Syst 2016;3(4):346–360 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edlund A, Esguerra JL, Wendt A, et al. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med 2014;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellin MD, Laguna T, Leschyshyn J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes 2013;14(6):417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly A, De Leon DD, Sheikh S, et al. Islet hormone and incretin secretion in cystic fibrosis after Four Months of ivacaftor therapy. Am J Respir Crit Care Med 2019;199(3):342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volkova N, Moy K, Evans J, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: data from national US and UK registries. J Cyst Fibros 2020;19(1):68–79. [DOI] [PubMed] [Google Scholar]
- 29.Misgault B, Chatron E, Reynaud Q, et al. Effect of one-year lumacaftor-ivacaftor treatment on glucose tolerance abnormalities in cystic fibrosis patients. J Cyst Fibros 2020;19(5):712–6. [DOI] [PubMed] [Google Scholar]
- 30.Ballmann M, Prinz N, Glass A, et al. Comment on “Effect of one-year lumacaftor-ivacaftor treatment on glucose tolerance abnormalities in cystic fibrosis patients. J Cyst Fibros 2020;19(5):839. [DOI] [PubMed] [Google Scholar]
- 31.Moheet A, Beisang D, Zhang L, et al. Lumacaftor/ivacaftor therapy fails to increase insulin secretion in F508del/F508del CF patients. J Cyst Fibros 2021;20(2):333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scully KJ, Marchetti P, Sawicki GS, Uluer A, Cernadas M, Cagnina RE, Kennedy JC, The effect of elexacaftor/tezacaftor/ivacaftor (ETI) on glycemia in adults with cystic fibrosis. Putman MS.J Cyst Fibros. 2022. Mar;21(2):258–263. doi: 10.1016/j.jcf.2021.09.001. Epub 2021 Sep 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nichols DP, Donaldson SH, Frederick CA, et al. PROMISE: Working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J Cyst Fibros 2021;20(2):205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aksit MA, Pace RG, Vecchio-Pagan B, et al. Genetic modifiers of cystic fibrosis-related diabetes have extensive Overlap with type 2 diabetes and related Traits. J Clin Endocrinol Metab 2020;105(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blackman SM, Commander CW, Watson C, et al. Genetic modifiers of cystic fibrosis-related diabetes. Diabetes 2013;62(10):3627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun L, Rommens JM, Corvol H, et al. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat Genet 2012;44(5):562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin YC, Keenan K, Gong J, et al. Cystic fibrosis-related diabetes onset can be predicted using biomarkers measured at birth. Genet Med 2021;23(5):927–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nyirjesy SC, Sheikh S, Hadjiliadis D, et al. beta-Cell secretory defects are present in pancreatic insufficient cystic fibrosis with 1-hour oral glucose tolerance test glucose >/=155 mg/dL. Pediatr Diabetes 2018;19(7):1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tommerdahl KL, Brinton JT, Vigers T, et al. Delayed glucose peak and elevated 1-hour glucose on the oral glucose tolerance test identify youth with cystic fibrosis with lower oral disposition index. J Cyst Fibros 2021;20(2):339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tschritter O, Fritsche A, Shirkavand F, et al. Assessing the shape of the glucose curve during an oral glucose tolerance test. Diabetes Care 2003;26(4):1026–33. [DOI] [PubMed] [Google Scholar]
- 41.Abdul-Ghani MA, Abdul-Ghani T, Ali N, et al. One-hour plasma glucose concentration and the metabolic syndrome identify subjects at high risk for future type 2 diabetes. Diabetes Care 2008;31(8):1650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boudreau V, Coriati A, Hammana I, et al. Variation of glucose tolerance in adult patients with cystic fibrosis: what is the potential contribution of insulin sensitivity? J Cyst Fibros 2016;15(6):839–45. [DOI] [PubMed] [Google Scholar]
- 43.Harindhanavudhi T, Wang Q, Dunitz J, et al. Prevalence and factors associated with overweight and obesity in adults with cystic fibrosis: a single-center analysis. J Cyst Fibros 2020;19(1):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanna RM, Weiner DJ. Overweight and obesity in patients with cystic fibrosis: a center-based analysis. Pediatr Pulmonol 2015;50(1):35–41. [DOI] [PubMed] [Google Scholar]
- 45.Middleton PG, Mall MA, Drevinek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019;381(19):1809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moran A, Pillay K, Becker D, et al. ISPAD Clinical Practice Consensus Guidelines 2018: management of cystic fibrosis-related diabetes in children and adolescents. Pediatr Diabetes 2018;19(Suppl 27):64–74. [DOI] [PubMed] [Google Scholar]
- 47.Milla CE, Warwick WJ, Moran A. Trends in pulmonary function in patients with cystic fibrosis correlate with the degree of glucose intolerance at baseline. Am J Respir Crit Care Med 2000;162(3 Pt 1):891–5. [DOI] [PubMed] [Google Scholar]
- 48.Lanng S, Thorsteinsson B, Nerup J, et al. Influence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. Eur J Pediatr 1992;151(9):684–7. [DOI] [PubMed] [Google Scholar]
- 49.Peraldo M, Fasulo A, Chiappini E, et al. Evaluation of glucose tolerance and insulin secretion in cystic fibrosis patients. Horm Res 1998;49(2):65–71. [DOI] [PubMed] [Google Scholar]
- 50.Kilberg MJ, Harris C, Sheikh S, Stefanovski D, Cuchel M, Kubrak C, Hadjiliadis D, Rubenstein RC, Rickels MR, Kelly A. Hypoglycemia and Islet Dysfunction Following Oral Glucose Tolerance Testing in Pancreatic-Insufficient Cystic Fibrosis. J Clin Endocrinol Metab. 2020. Oct 1;105(10):3179–89. doi: 10.1210/clinem/dgaa448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kilberg MJ, Sheikh S, Stefanovski D, et al. Dysregulated insulin in pancreatic insufficient cystic fibrosis with post-prandial hypoglycemia. J Cyst Fibros 2020;19(2):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marks BE, Wolfsdorf JI. Monitoring of pediatric type 1 diabetes. Front Endocrinol (Lausanne) 2020;11:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Riordan SM, Hindmarsh P, Hill NR, et al. Validation of continuous glucose monitoring in children and adolescents with cystic fibrosis: a prospective cohort study. Diabetes Care 2009;32(6):1020–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Battelino T, Danne T, Bergenstal RM, et al. Clinical targets for continuous glucose monitoring data Interpretation: recommendations from the International Consensus on time in range. Diabetes Care 2019;42(8):1593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beck RW, Bergenstal RM, Cheng P, et al. The relationships between time in range, hyperglycemia Metrics, and HbA1c. J Diabetes Sci Technol 2019;13(4):614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marks BE, Kilberg MJ, Aliaj E, et al. Perceptions of diabetes technology Use in cystic fibrosis-related diabetes management. Diabetes Technol Ther 2021;23(11):753–9. [DOI] [PubMed] [Google Scholar]
- 57.Hardin DS, Rice J, Rice M, et al. Use of the insulin pump in treat cystic fibrosis related diabetes. J Cyst Fibros 2009;8(3):174–8. [DOI] [PubMed] [Google Scholar]
- 58.Pinsker JE, Deshpande S, McCrady-Spitzer S, et al. Use of the Interoperable Artificial pancreas system for type 1 diabetes management during Psychological stress. J Diabetes Sci Technol 2021;15(1):184–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown SA, Kovatchev BP, Raghinaru D, et al. Six-month randomized, Multicenter trial of Closed-Loop control in type 1 diabetes. N Engl J Med 2019;381(18):1707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherwood JS, Jafri RZ, Balliro CA, et al. Automated glycemic control with the bionic pancreas in cystic fibrosis-related diabetes: a pilot study. J Cyst Fibros 2020;19(1):159–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dobson L, Hattersley AT, Tiley S, et al. Clinical improvement in cystic fibrosis with early insulin treatment. Arch Dis Child 2002;87(5):430–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bizzarri C, Lucidi V, Ciampalini P, et al. Clinical effects of early treatment with insulin glargine in patients with cystic fibrosis and impaired glucose tolerance. J Endocrinol Invest 2006;29(3):RC1–4. [DOI] [PubMed] [Google Scholar]
- 63.Mozzillo E, Franzese A, Valerio G, et al. One-year glargine treatment can improve the course of lung disease in children and adolescents with cystic fibrosis and early glucose derangements. Pediatr Diabetes 2009;10(3):162–7. [DOI] [PubMed] [Google Scholar]
- 64.Moran A, Pekow P, Grover P, et al. Insulin therapy to improve BMI in cystic fibrosis-related diabetes without fasting hyperglycemia: results of the cystic fibrosis related diabetes therapy trial. Diabetes Care 2009;32(10):1783–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minicucci L, Haupt M, Casciaro R, et al. Slow-release insulin in cystic fibrosis patients with glucose intolerance: a randomized clinical trial. Pediatr Diabetes 2012;13(2):197–202. [DOI] [PubMed] [Google Scholar]
- 66.Ballmann M, Hubert D, Assael BM, et al. Repaglinide versus insulin for newly diagnosed diabetes in patients with cystic fibrosis: a multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol 2018;6(2):114–21. [DOI] [PubMed] [Google Scholar]
- 67.Kelly A, Sheikh S, Stefanovski D, Peleckis AJ, Nyirjesy SC, Eiel JN, Sidhaye A, Localio R, Gallop R, De Leon DD, Hadjiliadis D, Rubenstein RC, Effect of Sitagliptin on Islet Function in Pancreatic Insufficient Cystic Fibrosis With Abnormal Glucose Tolerance. Rickels MR.J Clin Endocrinol Metab. 2021. Aug 18;106(9):2617–2634. doi: 10.1210/clinem/dgab365. Epub 2021 May 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beaudoin N, Bouvet GF, Coriati A, et al. Combined exercise training improves glycemic control in adult with cystic fibrosis. Med Sci Sports Exerc 2017;49(2):231–7. [DOI] [PubMed] [Google Scholar]
- 69.Monteiro KS, Azevedo MP, Jales LM, et al. Effects of aerobic interval training on glucose tolerance in children and adolescents with cystic fibrosis: a randomized trial protocol. Trials 2019;20(1):768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuo P, Stevens JE, Russo A, et al. Gastric emptying, incretin hormone secretion, and post-prandial glycemia in cystic fibrosis–effects of pancreatic enzyme supplementation. J Clin Endocrinol Metab 2011;96(5):E851–5. [DOI] [PubMed] [Google Scholar]
- 71.Perano SJ, Couper JJ, Horowitz M, et al. Pancreatic enzyme supplementation improves the incretin hormone response and attenuates postprandial glycemia in adolescents with cystic fibrosis: a randomized crossover trial. J Clin Endocrinol Metab 2014;99(7):2486–93. [DOI] [PubMed] [Google Scholar]
- 72.Sun X, Yi Y, Liang B, et al. Incretin dysfunction and hyperglycemia in cystic fibrosis: role of acyl-ghrelin. J Cyst Fibros 2019;18(4):557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geyer MC, Sullivan T, Tai A, et al. Exenatide corrects postprandial hyperglycaemia in young people with cystic fibrosis and impaired glucose tolerance: a randomized crossover trial. Diabetes Obes Metab 2019;21(3):700–4. [DOI] [PubMed] [Google Scholar]
- 74.Olatunbosun ST. Chronic incretin-based therapy in cystic fibrosis-related diabetes: A tale of 3 patients treated with sitagliptin for over 5 years. J Cyst Fibros. 2021. Nov;20(6):e124–e128. doi: 10.1016/j.jcf.2021.02.005. Epub 2021 Mar 2. [DOI] [PubMed] [Google Scholar]
- 75.Mischler EH, Chesney PJ, Chesney RW, et al. Demineralization in cystic fibrosis detected by direct photon absorptiometry. Am J Dis Child 1979;133(6):632–5. [DOI] [PubMed] [Google Scholar]
- 76.Aris RM, Renner JB, Winders AD, et al. Increased rate of fractures and severe kyphosis: sequelae of living into adulthood with cystic fibrosis. Ann Intern Med 1998;128(3):186–93. [DOI] [PubMed] [Google Scholar]
- 77.Rovner AJ, Zemel BS, Leonard MB, et al. Mild to moderate cystic fibrosis is not associated with increased fracture risk in children and adolescents. J Pediatr 2005;147(3):327–31. [DOI] [PubMed] [Google Scholar]
- 78.Latzin P, Griese M, Hermanns V, et al. Sternal fracture with fatal outcome in cystic fibrosis. Thorax 2005;60(7):616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foundation CF. Cystic Fibrosis Foundation Patient Registry 2019 Annual Data Report. Bethesda (MD): 2020. [Google Scholar]
- 80.Aris RM, Merkel PA, Bachrach LK, et al. Guide to bone health and disease in cystic fibrosis. J Clin Endocrinol Metab 2005;90(3):1888–96. [DOI] [PubMed] [Google Scholar]
- 81.Aris RM, Ontjes DA, Buell HE, et al. Abnormal bone turnover in cystic fibrosis adults. Osteoporos Int 2002;13(2):151–7. [DOI] [PubMed] [Google Scholar]
- 82.Aris RM, Lester GE, Dingman S, et al. Altered calcium homeostasis in adults with cystic fibrosis. Osteoporos Int 1999;10(2):102–8. [DOI] [PubMed] [Google Scholar]
- 83.Hall WB, Sparks AA, Aris RM. Vitamin d deficiency in cystic fibrosis. Int J Endocrinol 2010;2010:218691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hofbauer LC, Gori F, Riggs BL, et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 1999;140(10):4382–9. [DOI] [PubMed] [Google Scholar]
- 85.DiMango E, Ratner AJ, Bryan R, et al. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest 1998;101(11):2598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ionescu AA, Nixon LS, Evans WD, et al. Bone density, body composition, and inflammatory status in cystic fibrosis. Am J Respir Crit Care Med 2000;162(3 Pt 1):789–94. [DOI] [PubMed] [Google Scholar]
- 87.Haworth CS, Selby PL, Webb AK, et al. Inflammatory related changes in bone mineral content in adults with cystic fibrosis. Thorax 2004;59(7):613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shead EF, Haworth CS, Barker H, et al. Osteoclast function, bone turnover and inflammatory cytokines during infective exacerbations of cystic fibrosis. J Cyst Fibros 2010;9(2):93–8. [DOI] [PubMed] [Google Scholar]
- 89.Shead EF, Haworth CS, Gunn E, et al. Osteoclastogenesis during infective exacerbations in patients with cystic fibrosis. Am J Respir Crit Care Med 2006;174(3):306–11. [DOI] [PubMed] [Google Scholar]
- 90.Stead RJ, Hodson ME, Batten JC, et al. Amenorrhoea in cystic fibrosis. Clin Endocrinol (Oxf) 1987;26(2):187–95. [DOI] [PubMed] [Google Scholar]
- 91.Leifke E, Friemert M, Heilmann M, et al. Sex steroids and body composition in men with cystic fibrosis. Eur J Endocrinol 2003;148(5):551–7. [DOI] [PubMed] [Google Scholar]
- 92.Rana M, Munns CF, Selvadurai H, et al. The impact of dysglycaemia on bone mineral accrual in young people with cystic fibrosis. Clin Endocrinol (Oxf) 2013;78(1):36–42. [DOI] [PubMed] [Google Scholar]
- 93.Gordon CM, Binello E, LeBoff MS, et al. Relationship between insulin-like growth factor I, dehydroepiandrosterone sulfate and proresorptive cytokines and bone density in cystic fibrosis. Osteoporos Int 2006;17(5):783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rossini M, Del Marco A, Dal Santo F, et al. Prevalence and correlates of vertebral fractures in adults with cystic fibrosis. Bone 2004;35(3):771–6. [DOI] [PubMed] [Google Scholar]
- 95.Foundation CF. Cystic fibrosis Foundation patient Registry, 2019. Bethesda, MD: Annual Data Report; 2020. [Google Scholar]
- 96.King SJ, Topliss DJ, Kotsimbos T, et al. Reduced bone density in cystic fibrosis: DeltaF508 mutation is an independent risk factor. Eur Respir J 2005;25(1):54–61. [DOI] [PubMed] [Google Scholar]
- 97.Lam GY, Desai S, Fu J, et al. IL-8 correlates with reduced baseline femoral neck bone mineral density in adults with cystic fibrosis: a single center retrospective study. Sci Rep 2021;11(1):15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu M, Bettermann EL, Arora N, et al. Relationship between estrogen treatment and Skeletal health in women with cystic fibrosis. Am J Med Sci 2020;360(5):581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dif F, Marty C, Baudoin C, et al. Severe osteopenia in CFTR-null mice. Bone 2004;35(3):595–603. [DOI] [PubMed] [Google Scholar]
- 100.Haston CK, Li W, Li A, et al. Persistent osteopenia in adult cystic fibrosis transmembrane conductance regulator-deficient mice. Am J Respir Crit Care Med 2008;177(3):309–15. [DOI] [PubMed] [Google Scholar]
- 101.Pashuck TD, Franz SE, Altman MK, et al. Murine model for cystic fibrosis bone disease demonstrates osteopenia and sex-related differences in bone formation. Pediatr Res 2009;65(3):311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Le Henaff C, Gimenez A, Hay E, et al. The F508del mutation in cystic fibrosis transmembrane conductance regulator gene impacts bone formation. Am J Pathol 2012;180(5):2068–75. [DOI] [PubMed] [Google Scholar]
- 103.Rogan MP, Reznikov LR, Pezzulo AA, et al. Pigs and humans with cystic fibrosis have reduced insulin-like growth factor 1 (IGF1) levels at birth. Proc Natl Acad Sci U S A 2010;107(47):20571–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Putman MS, Greenblatt LB, Bruce M, et al. The effects of ivacaftor on bone density and microarchitecture in children and adults with cystic fibrosis. J Clin Endocrinol Metab 2021;106(3):e1248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tangpricha V, Kelly A, Stephenson A, et al. An update on the screening, diagnosis, management, and treatment of vitamin D deficiency in individuals with cystic fibrosis: evidence-based recommendations from the Cystic Fibrosis Foundation. J Clin Endocrinol Metab 2012;97(4):1082–93. [DOI] [PubMed] [Google Scholar]
- 106.Conwell LS, Chang AB. Bisphosphonates for osteoporosis in people with cystic fibrosis. Cochrane Database Syst Rev 2012;4:CD002010. [DOI] [PubMed] [Google Scholar]
- 107.Hardin DS, Ahn C, Prestidge C, et al. Growth hormone improves bone mineral content in children with cystic fibrosis. J Pediatr Endocrinol Metab 2005;18(6):589–95. [DOI] [PubMed] [Google Scholar]
- 108.Putman MS, Anabtawi A, Le T, et al. Cystic fibrosis bone disease treatment: Current knowledge and future directions. J Cyst Fibros 2019;18(Suppl 2):S56–65. [DOI] [PubMed] [Google Scholar]
- 109.Konstan MW, Pasta DJ, Wagener JS, et al. BMI fails to identify poor nutritional status in stunted children with CF. J Cyst Fibros 2017;16(1):158–60. [DOI] [PubMed] [Google Scholar]
- 110.Assael BM, Casazza G, Iansa P, et al. Growth and long-term lung function in cystic fibrosis: a longitudinal study of patients diagnosed by neonatal screening. Pediatr Pulmonol 2009;44(3):209–15. [DOI] [PubMed] [Google Scholar]
- 111.Sanders DB, Slaven JE, Maguiness K, et al. Early-life height Attainment in cystic fibrosis is associated with pulmonary function at age 6 Years. Ann Am Thorac Soc 2021;18(8):1335–42. [DOI] [PubMed] [Google Scholar]
- 112.Vieni G, Faraci S, Collura M, et al. Stunting is an independent predictor of mortality in patients with cystic fibrosis. Clin Nutr 2013;32(3):382–5. [DOI] [PubMed] [Google Scholar]
- 113.Wong SC, Dobie R, Altowati MA, et al. Growth and the growth hormone-insulin like growth factor 1 Axis in children with chronic inflammation: Current evidence, Gaps in knowledge, and future directions. Endocr Rev 2016;37(1):62–110. [DOI] [PubMed] [Google Scholar]
- 114.Rosenberg LA, Schluchter MD, Parlow AF, et al. Mouse as a model of growth retardation in cystic fibrosis. Pediatr Res 2006;59(2):191–5. [DOI] [PubMed] [Google Scholar]
- 115.Stalvey MS, Havasi V, Tuggle KL, et al. Reduced bone length, growth plate thickness, bone content, and IGF-I as a model for poor growth in the CFTR-deficient rat. PLoS One 2017;12(11):e0188497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pascucci C, De Biase RV, Savi D, et al. Deregulation of the growth hormone/insulin-like growth factor-1 axis in adults with cystic fibrosis. J Endocrinol Invest 2018;41(5):591–6. [DOI] [PubMed] [Google Scholar]
- 117.Gifford AH, Nymon AB, Ashare A. Serum insulin-like growth factor-1 (IGF-1) during CF pulmonary exacerbation: trends and biomarker correlations. Pediatr Pulmonol 2014;49(4):335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leung DH, Heltshe SL, Borowitz D, et al. Effects of diagnosis by newborn screening for cystic fibrosis on weight and length in the first Year of life. JAMA Pediatr 2017;171(6):546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stalvey MS, Pace J, Niknian M, et al. Growth in pre-pubertal children with cystic fibrosis treated with ivacaftor. Pediatrics 2017;139(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stalvey MS, Anbar RD, Konstan MW, et al. A multi-center controlled trial of growth hormone treatment in children with cystic fibrosis. Pediatr Pulmonol 2012;47(3):252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Z, Lindstrom MJ, Lai HJ. Pubertal height velocity and associations with prepubertal and adult heights in cystic fibrosis. J Pediatr 2013;163(2):376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zysman-Colman ZN, Kilberg MJ, Harrison VS, et al. Genetic potential and height velocity during childhood and adolescence do not fully account for shorter stature in cystic fibrosis. Pediatr Res 2021;89(3):653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]