

Abstract
Ion-ion reactions are valuable tools in mass-spectrometry-based peptide and protein sequencing. To boost the generation of sequence-informative fragment ions from low charge-density precursors, supplemental activation methods, via vibrational and photoactivation have become widely adopted. However, long-lived radical peptide cations undergo intramolecular hydrogen atom transfer from c-type ions to z•-type ions. Here we investigate the degree of hydrogen transfer for thousands of unique peptide cations where electron transfer dissociation (ETD) was performed and was followed by beam-type collisional activation (EThcD), resonant collisional activation (ETcaD), or concurrent infrared photoirradiation (AI-ETD). We report on precursor charge density and local amino acid environment surrounding bond cleavage to illustrate the effects of intramolecular hydrogen atom transfer for various precursor ions. Over 30% of fragments from EThcD spectra comprise distorted isotopic distributions, whereas over 20% of fragments from ETcaD have distorted distributions and less than 15% of fragments derived from ETD and AI-ETD reveal distorted isotopic distributions. Both ETcaD and EThcD give a relatively high degree of hydrogen migration, especially when D, G, N, S, and T residues were directly C-terminal to the cleavage site. Whereas all post-activation methods boost the number of c- and z•-type fragment ions detected, the collision-based approaches produce higher rates of hydrogen migration, yielding fewer spectral identifications when only c- and z•-type ions are considered. Understanding hydrogen rearrangement between c- and z•-type ions will facilitate better spectral interpretation.
Graphical Abstract:
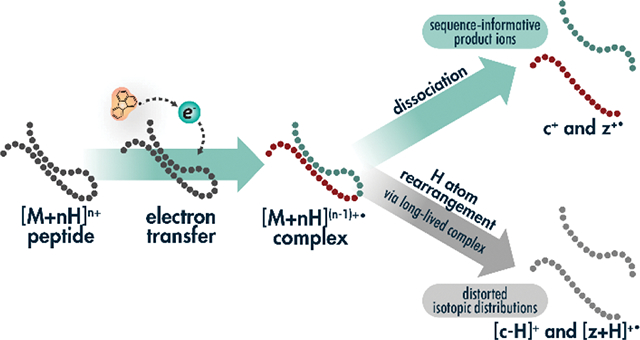
INTRODUCTION
Tandem mass spectrometry (MS/MS) continues to be a cornerstone in protein and peptide analyses. Multiply charged peptide and protein cations may dissociate along various pathways, producing mass spectra rich in sequence information. The source of ion activation can be diverse, although collisions with gases are most common[1]. These collision-based activation methods typically cleave amide backbone bonds to produce b- and y-type ion products[2–5].
Electron-driven activation, emerging over the past two decades as a viable alternative, can be achieved via free electron capture, as in electron capture dissociation (ECD)[6], or electron transfer from small molecule radical anions, as in electron transfer dissociation (ETD)[7]. In contrast to collision-based approaches, ECD and ETD typically cleave N-Cα backbone bonds to form c- and z•-type product ions[6–8]. While the specific dissociation mechanisms for formation of c- and z•-type product ions by ECD and ETD have been debated, electron-based methods are widely recognized to preserve labile covalent bonds and non-covalent interactions concomitant to peptide backbone dissociation[9, 10]. As a result, ECD and ETD are particularly useful in analyses of labile post-translational modifications (PTMs) and intact proteins[7, 11–14]. Proposed mechanisms include the Cornell mechanism in which electron capture or transfer to a protonated nitrogen atom and subsequent hydrogen rearrangement to a proximal carbonyl oxygen leads to nonergodic backbone cleavage[7, 15, 16]; the Utah-Washington mechanism describes electron capture or transfer at an amide moiety prompting intramolecular proton abstraction by the highly basic amide moiety and causing peptide backbone fragmentation[17–19]; and a radical cascade mechanism contends that electron capture at a protonated nitrogen causes initial hydrogen atom migration and iterative neutral loss production[20–23].
Electron-driven dissociation methods are inherently dependent on precursor ion charge density, where low charge density ions have less intramolecular Coulombic repulsion and more non-covalent intramolecular interactions. Precursor ions of high mass-to-charge (m/z) have lower charge density, which leads to a higher proclivity for a phenomenon known as non-dissociative electron capture/transfer (ECnoD/ETnoD)[24–27]. Here, electron capture or transfer will occur to generate peptide backbone cleavage, but more compact precursor ion secondary structures lead to non-covalent intramolecular interactions that hold product ions together in an ECnoD/ETnoD complex. Instead of forming sequence-informative product ions desired of the fragmentation methods, these species appear simply as charge-reduced, intact precursors within a mass spectrum[24].
Efforts to explore this non-dissociative behavior in ECD led to a number of ion activation methods to improve fragment ion generation, including supplemental activation with collisions, increased temperatures, and photons[12, 28–35]. The advent of ETD garnered similar investigations for mitigating ETnoD. Whereas ECD is routinely performed within the low-pressure magnetic resonance chamber of a FT-ICR instrument, ETD typically occurs within RF ion trapping devices that have several orders of magnitude higher pressures. Ultimately, three major strategies emerged for supplemental activation in ETD reactions. ETcaD uses resonant excitation of ETnoD products after the ETD reaction, imparting enough vibrational energy via collisional activation to release fragment ions[36, 37]. This approach generally performs the activation within the same ion trapping device where the ETD reaction occurred. Several strategies explored beam-type collisional activation of all or some of ETD products after the ion-ion reaction (including unreacted precursor ions and ETnoD species)[38–42]. A widely used version is called EThcD, where broadband beam-type collisional dissociation in a separate reaction cell is used to activate all ions present following ETD reactions[43, 44]. Both EThcD and ETcaD tend to increase production of sequence-informative ions, although they can complicate spectra as well (vide infra).
A third alternative called activated-ion ETD (AI-ETD) relies on irradiation of the trapping volume of the ETD reaction cell with infrared photons simultaneous to the ion-ion reaction[45, 46]. Vibrational activation from photon absorption disrupts the precursor ion secondary structure, reducing production of ETnoD species. Importantly, the concomitant nature of AI-ETD means it adds no additional activation time to the scan sequence and minimizes the lifetime of ETnoD complexes. The benefits of AI-ETD have been demonstrated for numerous biomolecules, including tryptic[45, 47] and post-translationally modified peptides[48–52], intact proteins[53–56], and oligonucleotides[57].
The initial implementation of AI-ETD revealed limited intramolecular hydrogen atom rearrangement, an undesired side reaction especially common for precursors of low charge density that is common with collision-based supplemental activation strategies like ETcaD and EThcD[46]. This phenomenon, during which a hydrogen atom migrates from a c-type ion to a z•-type ion, distorts fragments’ isotopic distributions and can challenge downstream fragment mapping. The side reaction creates “[c+H]●” ions and “[z+2H]” ions. Note, there have been several efforts to establish nomenclature for peptide fragment ions that can lead to confusion[58–62]. The explicit nomenclature proposed by Chu et al. accounts for all hydrogens and protons, making discussion of various fragment ion types straightforward[58]. The typical even-electron c-type ions and odd-electron z•-type ions in ETD spectra are referred to as [c+2H]1+ and [z+H]●1+ ions, respectively. Thus, it becomes clear that hydrogen rearrangements occur to form odd-electron [c+H]●1+ and even-electron [z+2H]1+ due to hydrogen loss and gain, respectively. Because we feel this is more intuitive than comparing c/ z•-type ions to (c-1)●/ (z+1)-type ions, we use the explicit Chu et al. nomenclature throughout the rest of this work. We also note that all fragment ions discussed here are singly charged, so we have omitted charge state values. The odd electron [c+H]● ion shifts the monoisotopic mass one Dalton lower, which reduces the intensity of what was the original/expected [c+2H] monoisotopic peak intensity, distorting the overall isotopic distribution, and widening the tolerance necessary for confident fragment matching. In contrast, the even electron [z+2H] ion overlaps with the fragment’s first C13-containing peak, in turn reducing monoisotopic peak intensity and distorting the overall isotopic distribution, leading to potential misassignment of the monoisotopic peak.
Hydrogen atom rearrangement has been extensively studied in ECD spectra, where precursor charge, amino acid composition, and local environment where found to play prominent roles[63–70]. Hydrogen atom migration in ETD spectra has also been studied[71–73], but hydrogen migrations in the various supplemental activation methods remains largely uncharacterized. Beyond aiding our fundamental understanding of each method, surveying hydrogen migration behavior in various ETD approaches provides practical knowledge for both manual spectral interpretation and performance of search algorithms[74–80]. Additionally, new implementations of electron-driven activation methods continue to emerge[81–85], highlighting the need for detailed understanding of fragment ions generated by various activation regimes. Covalent bond formation during gas-phase ion-ion reactions may be a useful probe of gas-phase protein and peptide structure[86]. Herein, we leverage thousands of doubly-charged precursor ions to investigate hydrogen atom migration across a diverse population of tryptic peptides derived from HAP1 human cell lysate. We compare effects on c- and z•-type product ion formation and isotopic distributions from ETD, ETcaD, EThcD, and AI-ETD to ascertain the importance of activation method, charge-density and proximal environment, on the distortion of isotopic distributions via intramolecular hydrogen atom migration.
EXPERIMENTAL SECTION
Materials
Human HAP1 cells were lysed in a buffer of 6 M guanidine followed by probe sonication. Protein precipitation was performed with 90% methanol. Tryptic digestion was performed similarly as described previously[87–89]. The supernatant was decanted, and proteinaceous pellets were suspended in 8 M urea, 100 mM TRIS, pH 8.0, and 40 mM chloroacetamide with lysyl endopeptidase (LysC) (1:50 enzyme/protein, Wako Chemicals). After a four-hour room temperature incubation, digested samples were diluted to 1.5 M urea with 100 mM TRIS, pH 8.0 and a second digestion was performed with trypsin (1:50 enzyme/protein, Promega, Madison, WI) for 12 hours. Quenching of trypsin activity was performed by adding trifluoroacetic acid (TFA) to a final pH less than 2.0. Peptides were desalted using Strata-X columns (Phenomenex Strata-X 33 μm Polymeric Reverse Phase, 10 mg/mL, Torrance, CA), which were equilibrated using one column volume of 100% acetonitrile (ACN) followed by one column volume of 0.1% TFA. Acidified peptides were added to the column, rinsed with three column volumes of 0.1% TFA and eluted with 500 μL of 80% ACN with 0.1% TFA. Protein concentration was determined using a Quantitative Colorimetric Peptide Assay (Thermo Pierce, Rockford, IL).
LC-MS/MS
A reverse-phase column was made in-house using 75 μm inner diameter, 360 μm outer diameter bare fused silica capillary and packed with 1.7 μm diameter, 130 Å pore size ethylene bridged hybrid C18 particles (Waters) to a length of 30 cm. A high-pressure packing station, capable of reaching pressures of 30,000 psi was used, as described previously[90].
All MS and MS/MS experiments were performed on a quadrupole-Orbitrap-quadrupole linear ion trap (q-OT-QLT) MS system (Orbitrap Fusion Lumos, Thermo Fisher Scientific, San Jose, CA, USA) that was modified to include a Firestar T-100 Synrad 60 Watt (W) CO2 continuous wave laser (Mukilteo, WA) to allow for the excitation of precursor ions within the QLT during ion-ion reactions[47, 53]. A multi-mode hollow-core fiber capable of transmitting mid-IR wavelength photons was obtained (Opto-Knowledge Systems, Torrance, CA) and coupled between the laser and hybrid mass spectrometer, as described previously[53].
The reverse-phase column was installed on a Dionex Ultimate 3000 UPLC system and heated to 55 °C. Solvent A consisted of 0.2% formic acid (FA) and solvent B was 70% acetonitrile (ACN) with 0.2% FA in water. 2.5 μg of tryptic HAP1 peptides were injected onto the column and a gradient elution was performed at 350 nL/min. The gradient ramped from 0% B to 5% B over 5 minutes, followed by an increase to 40% B at 70 minutes, a wash at 100% B for 2 minutes, and a re-equilibration of the column at 0% B for 11 minutes, for a total of 90 minutes.
Precursors were ionized using electrospray ionization at 2.3 kV with respect to ground. The inlet capillary was held at 275 °C and the ion funnel RF was held at 30%. All MS1 survey spectra were collected at a resolving power of 60,000 in the Orbitrap with a scan range of 200 – 2000 m/z, an AGC target of 1,000,000 charges and a maximum injection time of 54 ms. Monoisotopic precursor selection was enabled for peptide isotopic distributions. Dynamic exclusion was set to exclude resampling of precursors within 20 seconds. MS2 scans were conducted on precursors of charge state z = 2 for a one second cycle time. Scans were conducted in the Orbitrap at a resolving power of 120,000 at 200 m/z and an AGC target of 30,000 with a maximum injection time of 54 ms. Precursors were isolated with a wide isolation width of 3 Th using the quadrupole to ensure precursor isotopic distributions were present in MS2 scans. For increased peak quality, MS2 spectra are the result of 5 averaged scans (i.e., five microscans) for ETD, AI-ETD, EThcD, and ETcaD experiments.
For ETD experiments, precursor cations were isolated by the quadrupole and accumulated in the middle section of the high-pressure trap of the QLT, followed by accumulation of fluoranthene reagent anion (202 m/z) within the front section of the trap, and subsequent charge sign independent trapping for the ETD reaction[91]. Calibrated charge-dependent ETD parameters were enabled to determine ETD reagent ion AGC and ETD reaction times[92]. Normalized collision energies (nce) of 30 and 25 were set for ETcaD and EThcD experiments, respectively, whereas the IR laser power for AI-ETD was 30% maximal power, as indicated in the text. The supplier indicates a maximum laser output of 60 W.
Data Analysis
Mass spectra and chromatography information were viewed in the vendor’s post-acquisition software (XCalibur Qual Browser, version 4.0). The OMSSA algorithm and COMPASS software suite were used for searching and processing data[93, 94]. The .raw files were converted to dta text files and scored against a human reference proteome database, downloaded from Uniprot. Prior to the search, spectra were processed, with charge-reduced product ions and neutral losses removed within the window from 60 Da below to 5 Da above the charge-reduced precursor[79, 80]. For all analyses, c- and z•-type product ions were searched. Separate database searches were carried out for which b-, c-, y-, and z•-type ions were included in searches for all methods, as indicated in the text. Enzymatic cleavage was set to trypsin with up to three missed cleavages allowed and no cleavage on proline residues. The maximum expectation value was cut off at 10 and cysteine carbamidomethylation was set as a fixed modification, whereas methionine oxidation was specified as a variable modification. Peptides were searched with a 25-ppm tolerance around the monoisotopic precursor and a 10-ppm tolerance on fragment ion masses. Search results were filtered to a 1% unique peptide FDR based on expectation value (E-value) and ppm error using COMPASS[93, 95, 96].
Note, a C# script using the C# Mass Spectrometry Library (CSMSL, https://github.com/dbaileychess/CSMSL) was written to extract precursor and fragment ions and their intensities as well as perform in silico fragmentation of peptides. All fragment matches were made within a 10-ppm tolerance. Fragment matches without a monoisotopic, +1 C13, and +2 C13 peak were discarded. See the Supporting Information for more detailed methods.
RESULTS AND DISCUSSION
To elucidate the prevalence and effects of hydrogen atom migration on large-scale shotgun proteomics analyses with ETD, AI-ETD, ETcaD, and EThcD, we performed back-to-back-to-back-to-back MS2 workflows to ensure that every precursor was isolated and activated with each of the four separate methods once it was selected for tandem MS. As precursors of low charge density have previously shown the most noticeable hydrogen migration rates[46] and are the most common species within tryptic digests[37], we specifically targeted doubly-charged precursors in our analyses. Figure 1, which exemplifies trends in the entire dataset, shows the isotopic distributions of a c-type ion and z•-type ion derived from ETD, AI-ETD, ETcaD, and EThcD. For the c17+ ion of the tryptic peptide having the sequence GAEAANVTGPGGVPVQGSK (m/z = 848.44), hydrogen migration presents itself in the form of a (c17-1)+ peak, one hydrogen atom lighter than the monoisotopic peak. Due to fragment ions’ varied intensities and differences between c- and z•-type ion chemistries, displaying hydrogen migration rates as relative [c+H]●/[c+2H] intensity ratios and [z+2H]/[z+H]• intensity ratios, respectively, allows for global comparison across fragments, fragment types, and activation methods. As the theoretical intensity of any c-1 peak is zero, the expected [c+H]●/[c+2H] ratio will also be zero, meaning any ratio greater than zero represents a distorted spectrum. Note, expected isotope intensities are denoted by solid circles in each panel. For this example, ETD gave a [c+H]●/[c+2H] ratio for the c17+ fragment of 0.82, indicating that hydrogen migration occurs even in the absence of supplemental activation. AI-ETD reduced the [c+H]●/[c+2H] ratio to 0.70, while ETcaD increased the [c+H]●/[c+2H] ratio to 0.91 and EThcD increased it to 1.07. For the EThcD spectrum, the c-1 peak, with an ideal signal of zero, overtook the monoisotopic peak to further convolute spectral interpretation.
Figure 1. Hydrogen migration creates distorted isotopic distributions for c- and z•-type product ions.
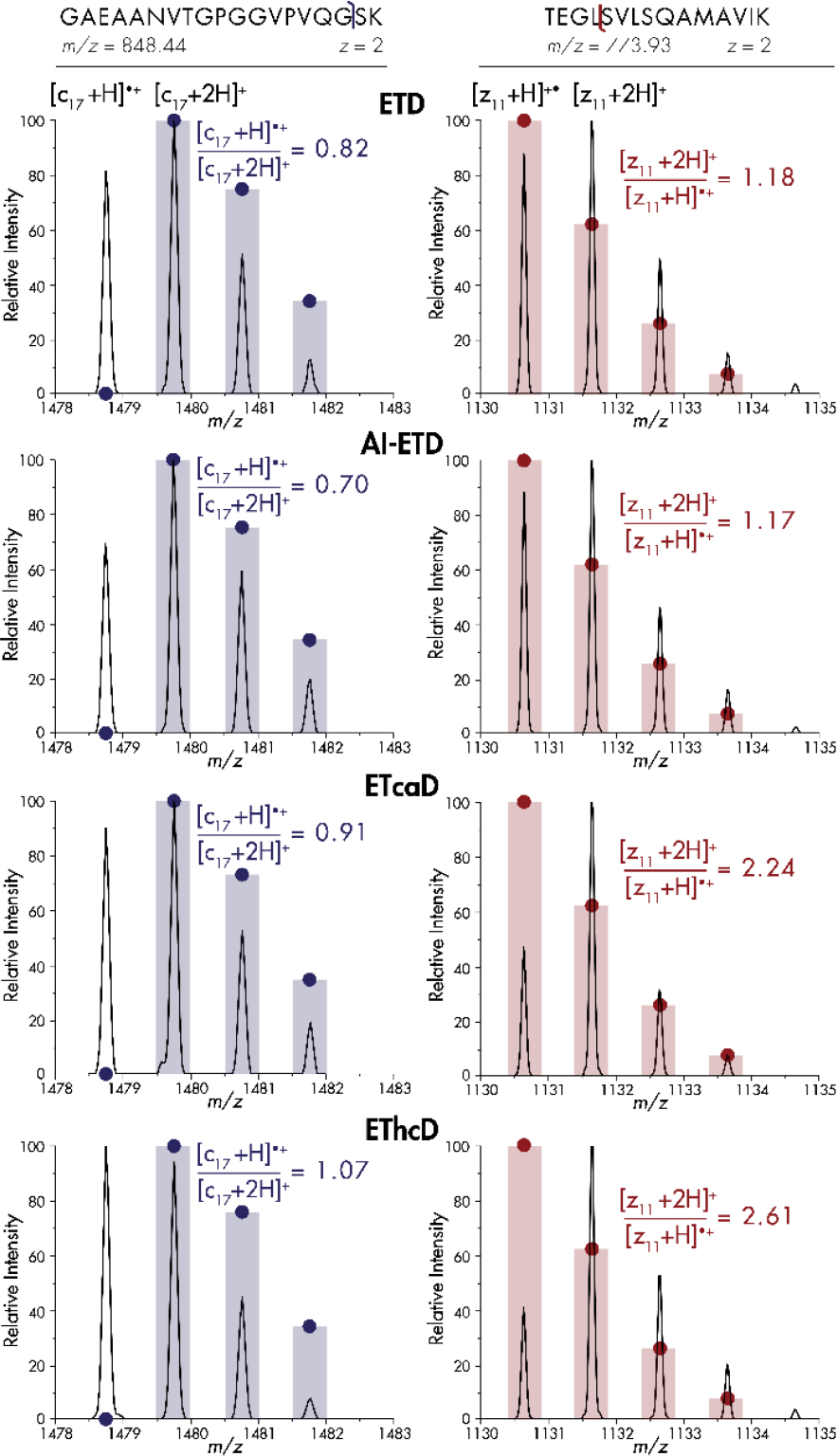
Spectral comparisons for ETD, AI-ETD, ETcaD, and EThcD for the c17+ and z11+• fragment ions of doubly-charged tryptic peptides GAEAANVTGPGGVPVQGSK and TEGLSVLSQAMAVIK, respectively. Intensities are normalized to the highest peak for that method. Bars and solid circles represent theoretical isotopic distributions.
C-terminus containing z•-type fragment isotopic distributions may also appear distorted by hydrogen atom migration. A [z+2H]/[z+H]• ratio can portray the relative amount of hydrogen rearrangement for a given fragment’s isotopic distribution (Figure 1). As the introduced hydrogen atom and an additional neutron are very similar in mass, the [z+2H] peak overlaps with the fragment’s first C13-containing peak. For the z11+• fragment of the tryptic peptide having the sequence TEGLSVLSQAMAVIK (m/z 773.93), hydrogen atom migration causes the [z+2H] peak to overtake the monoisotopic peak intensity for all four methods, even though the theoretical [z+2H] peak is ~60% of the monoisotopic peak (solid circles). ETD generates a [z+2H]/[z+H]• ratio of 1.18, and AI-ETD provides virtually the same ratio slightly at 1.17. Conversely, ETcaD increases the prevalence of hydrogen atom migration such that [z+2H]/[z+H]• is 2.24 and for EThcD the ratio is again highest at 2.61.
In a more global assessment on the prevalence of hydrogen atom migration across a wide variety of tryptic peptides, we examined all fragments produced from ETD, AI-ETD, ETcaD, and EThcD vs. mass-to-charge (Figure 2). These results demonstrate a strong correlation between hydrogen atom migration and precursor mass-to-charge. For both c- and z•-type fragment ions, distorted isotopic distributions are present at higher m/z values. [c+H]●/[c+2H] and [z+2H]/[z+H]• ratios are nearly always higher with collision based supplemental methods, especially at high m/z values. AI-ETD produces [c+H]●/[c+2H] and [z+2H]/[z+H]• values equal to or slightly less than that of ETD alone. Interestingly, both ETD and AI-ETD produce much lower [c+H]●/[c+2H] rates than ETcaD and EThcD across precursor m/z values but produce only slightly lower [z+2H]/[z+H]• values than the collision-based methods. Increasing peptide mass alters the isotopic distribution. The higher values with z•-type ions, caused by overlapping of the [z+2H] peak with the first C13-containing peak, may mask differences between methods. The increase in hydrogen migration with precursor m/z can likely be attributed to lowered Coulombic repulsion and more densely folded secondary structure, both of which contribute to a longer-lived radical ETnoD species with ample time and reduced distance for intramolecular hydrogen atom migration [86, 97]. Whereas AI-ETD demands no additional scan time after the ETD reaction, ETcaD resonant excitation requires activation on the order of tens of milliseconds and EThcD shuttles all ETD products to the ion routing multipole (IRM) for beam-type collisional activation. Additional vibrational energy is imparted on peptides via IR photons, as with AI-ETD, or collisions with background gases, as with ETcaD and EThcD. The degrees of freedom of the molecule coupled with local vibrational energy accumulation may induce backbone bond cleavage or hydrogen rearrangement. However, rather than vibrational activation itself solely causing rearrangements, these results suggest that the additional time and movement of ETD products during ETcaD and EThcD give the long-lived radical ETnoD complexes enough time for hydrogen atom transfer to occur at increased rates.
Figure 2: Dependence of intramolecular hydrogen atom migration on precursor m/z for c- and z•-type ion populations.
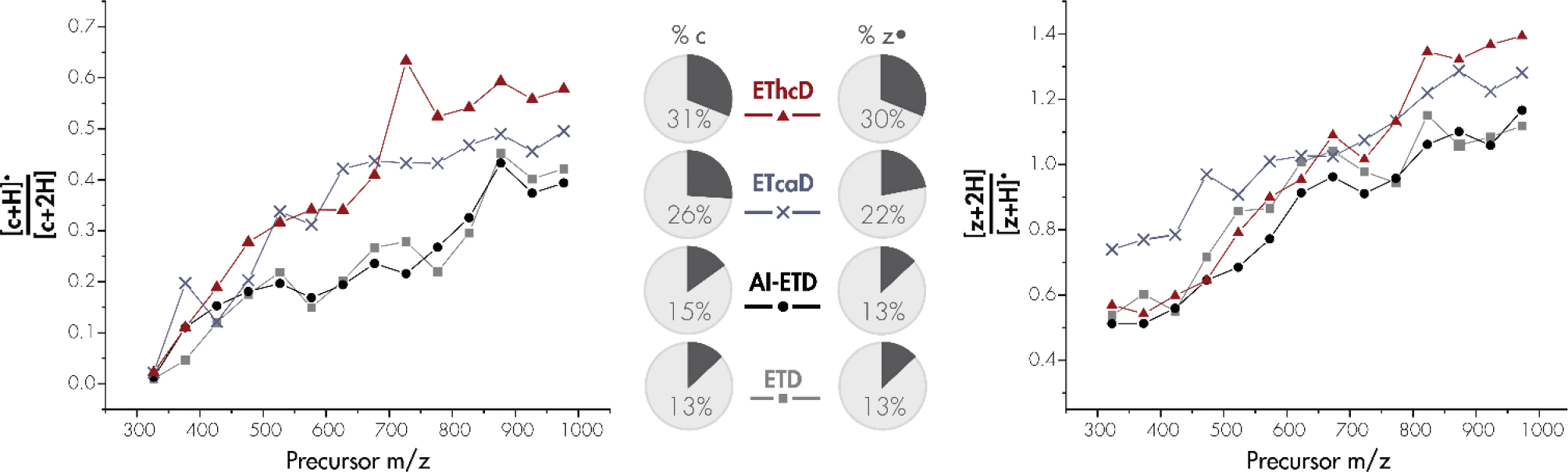
Hydrogen migration is shown as the average peak area ratios, with 50 Th bins, for [c+H]•/[c+2H] (left) and [z+2H]/[z+H]• (right) across all doubly charged fragment ions. EThcD (red triangle), ETcaD (pink cross), AI-ETD (green circle), and ETD without supplemental activation (blue square) are shown for each fragment ion type. Pie charts display the percentage of c-and z•-type isotopic distributions which display hydrogen migration.
In all, ETD produces hydrogen atom migration in 13% of all c- and z•-type ion distributions. For the same precursors, AI-ETD produces spectra with c- and z•-type ions with hydrogen rearrangement detected 15% and 13% of the time, respectively, as a fraction of all fragment ions weighted equally. ETcaD produces spectra with c- and z•-type ions with hydrogen rearrangement detected 26% and 22% of the time, respectively, and EThcD gives c- and z•-type fragment ions with isotopic distributions impacted by hydrogen migration 31% and 30% of the time, respectively.
The dependence of intramolecular hydrogen atom migration on the amino acid environment surrounding N-Cα peptide bond cleavage is illustrated in Figure 3. “Position n-1” denotes the amino acid residue toward the N-terminus, while “position n” corresponds to the amino acid residue toward the C-terminus of the peptide, relative to peptide bond cleavage. The average [c+H]●/[c+2H] ratios from all c-type ion distributions in which the respective amino acid was neighboring bond breakage are plotted in purple. When backbone cleavage occurred at sites where the residue at position n contained a polar side chain, a high degree of hydrogen rearrangement occurred, on average. Specifically, aspartic acid (D), asparagine (N), glutamine (Q), serine (S), and threonine (T), as well as glycine (G) with its two primary hydrogen atoms, gave a high degree of intramolecular hydrogen rearrangement when at position n. For nearly all residues, hydrogen atom transfer proceeded at higher rates in EThcD and ETcaD than AI-ETD and ETD. The amino acid residue at position n-1, i.e., the residue N-terminal to the cleavage site, appears to have less of an impact on hydrogen atom migration, although N, Q and G increase the rate of hydrogen migration, also seen when these amino acids are C-terminal to bond breakage. Polar hydrogen atoms – those bonded to electronegative atoms like oxygen and nitrogen – near the N-Cα bond cleavage on proximal side chains may be easier to abstract from c-type ions to z•-type ions. ETcaD gave relatively high rates of hydrogen atom transfer when bond cleavage occurred near a tryptophan (W) residue. Glycine residues allow for increased degrees of freedom to the peptide structure, as well as an increase in the number of hydrogen atoms neighboring the bond breakage site available for rearrangement. Interestingly, ETD produces. higher rates of intramolecular hydrogen rearrangement than AI-ETD for c-type ions when bond cleavage neighbors a glycine at position n, indicating that by unfolding the peptide and potentially removing intramolecular interactions, a hydrogen atom transfer is less likely.
Figure 3: Dependence of intramolecular hydrogen atom migration on proximal amino acids.
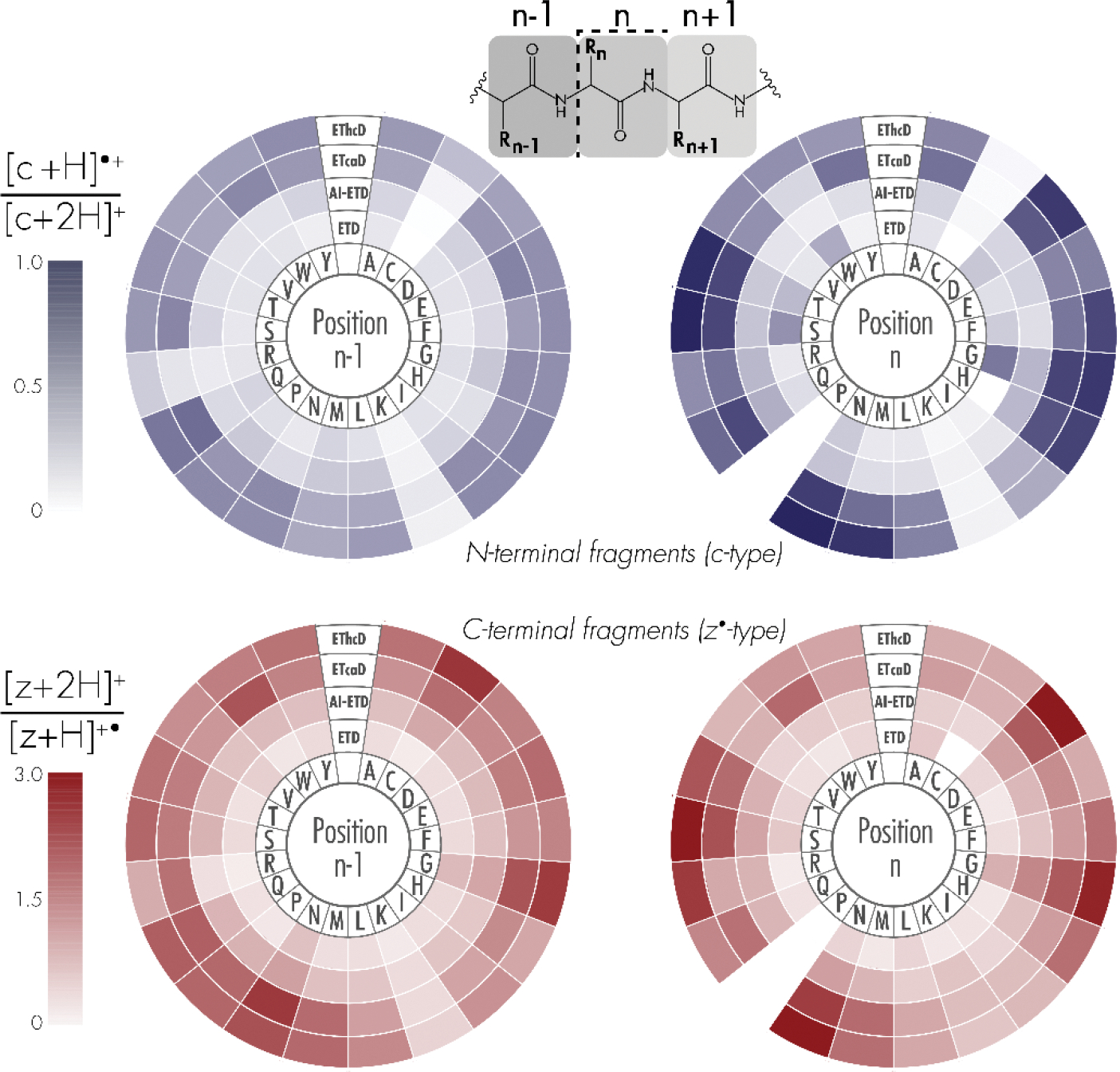
Shading corresponds to the average peak area ratios of [c+H]•/[c+2H] (top) and [z+2H]/[z+H]• (bottom) for each of the twenty amino acid residues. Position “n-1” denotes the N-terminal amino acid relative to N-Cα peptide bond cleavage, whereas position “n” denotes the C-terminal amino acid upon N-Cα bond cleavage. Note, fragments N-terminal to proline residues were not considered.
To illustrate the impact of hydrogen migration on the distortion of isotopic distributions after gas phase ion-ion reactions, relative intensities of all precursor and c- and z•-type ion peaks were plotted vs. precursor mass (Figure S1). Peak intensities within precursor isotopic distributions were normalized to the total peak intensity of the monoisotopic and isotopic peaks and include the c-1 peak intensity for c-type ion distributions. The increased probability of heavy C13 incorporation with increasing molecular mass causes a decrease in the relative abundance of the monoisotopic peak intensity[98]. Figure 4a shows the dot product of fragments’ theoretical monoisotopic peak intensity and observed monoisotopic peaks intensities. Intensities of all peaks in the isotopic distribution were normalized to the total intensity within the distribution. Any hydrogen migration should decrease the observed monoisotopic peak intensity for both c- and z•-type product ions, causing the dot product to decrease. Therefore, higher dot products generally indicate more similarity with the theoretical isotopic distribution, whereas scores closer to zero indicate dissimilar spectra as a result of hydrogen migration, akin to cosine similarity scores[99]. While a distribution in observed vs. theoretical isotopic patterns is revealed by these data, AI-ETD spectra more closely resemble ETD spectra, while ETcaD and EThcD are more distorted and have lower values. AI-ETD gives slightly higher values than ETD without supplemental activation for both c- and z•-type fragment dot products. Theoretical distributions were calculated based on amino acid sequence and chemical composition of each peptide fragment[98, 100, 101]. Note, for implementations of ETD following proton transfer reaction (PTR), a c-type product ion may have the same nominal mass as a [c+H]● product ion. As the experiments and analyses described herein involve only doubly-charged tryptic peptides and fluoranthene heavily favors electron transfer over PTR, this side-product should not convolute the data.
Figure 4: Comparison of observed and theoretical isotopic distributions reveals more distortion of isotopic distributions in ETcaD and EThcD spectra.
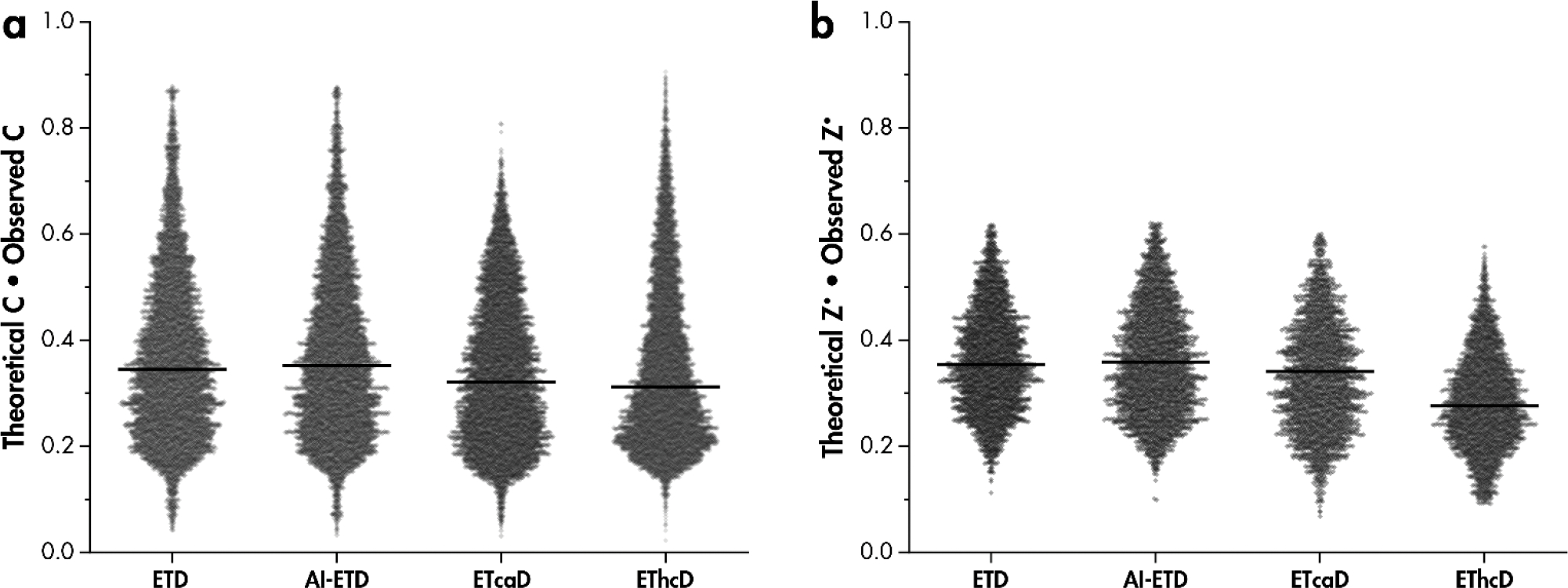
Dot product of the theoretical monoisotopic peak intensity and observed monoisotopic peak intensity for a) c-type and b) z•-type product isotopic distributions. Both theoretical and observed monoisotopic peak intensities are normalized to the total intensity of the first three isotopic peaks for z•-type fragments and include the [c+H]• peak for c-type fragments. Black bars are median values.
Experimentally observed c- and z•-type fragment isotopic distributions were plotted across fragment mass, revealing similar patterns to the theoretical spectra, with noticeably more distortion (Figure S1). For c-type fragments, ETD and AI-ETD more closely resemble theoretical spectra than ETcaD and EThcD which generally have monoisotopic peaks lower in intensity and c-1 peaks higher in intensity. For z•-type fragments, ETD and AI-ETD again closely resemble theoretical spectra, although some isotopic distortion is present. Both collision-based supplemental methods give generally more distorted fragment isotopic distributions, with the monoisotopic peak intensity decreased and [z+2H]1+ increased relative to theoretical spectra. In many cases of excessive hydrogen atom transfer, the monoisotopic peak is no longer the most abundant peak in the distribution, even for these relatively low molecular weight molecules. This can cause dispersal of signal across multiple peaks and even monoisotopic peak misassignment. Differences between theoretical and experimental values for c- and z•-type ions across activation methods are shown in Figure S2, illustrating the degree of spectral distortion detected in many ETcaD and EThcD spectra.
To assess the potential detriment of hydrogen migration on peptide spectral identifications, we carried out ETD, AI-ETD, ETcaD, and EThcD analyses of tryptic HAP1 peptides. Fragment identifications were made when the theoretical isotopic distribution contained an intensity value greater than one for the monoisotopic peak as well as the first two isotopic peaks (Figure 5). For the same doubly-charged peptide precursors, ETD gave 62,831 c- and z•-type fragment ion distributions detected, while AI-ETD gave 135,052 at 18W laser power. These correspond to a boost in unique fragment generation of 115% over ETD. ETcaD gave 95,499, and EThcD gave 94,670 c- and z•-type fragment ion distributions detected. These values correspond to boosts of 52% and 51%, respectively, over ETD alone. Interestingly, upon a human database search analyzing only c- and z•-type ions, ETD gave 4,192 unique peptide spectral matches (PSMs), while AI-ETD gave 8,924, ETcaD gave 5,443, and EThcD gave 3,500 PSMs. Therefore, while additional c- and z•-type fragments detected from AI-ETD translate directly into additional PSMs, with 113% boost in PSMs over ETD, the additional fragments from ETcaD and EThcD do not. Hydrogen migration affects EThcD spectra to the extent that 17% fewer PSMs are made from c- and z•-type ions compared to ETD without any supplemental activation.
Figure 5. Hydrogen migration affects spectral interpretation.
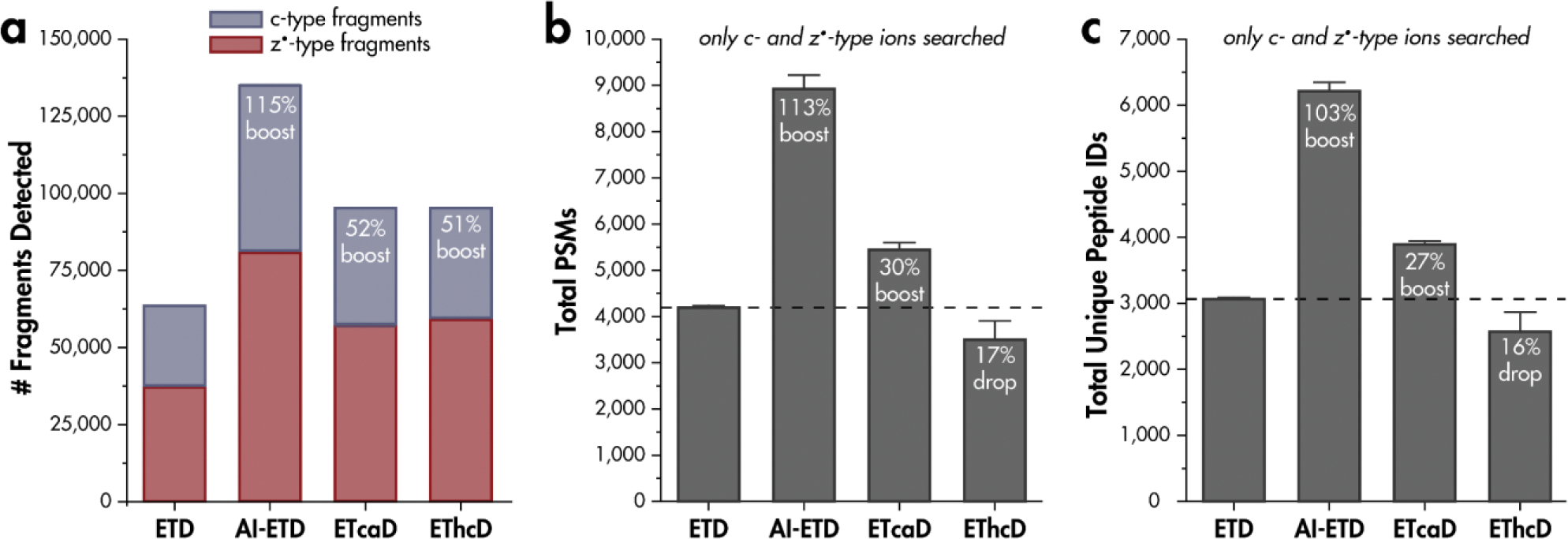
a) Number of c- and z•-type fragment isotopic distributions generated from ETD, AI-ETD, ETcaD, and EThcD shotgun proteomics experiments. Tryptic, doubly-charged precursors were considered. b) Unique peptide spectral matches (PSMs) obtained when including only c- and z•-type fragment ions in searching parameters and c) corresponding unique peptide identifications when using only c- and z•-type ions. Percentages are relative to ETD without any supplemental activation.
Relating unique peptide identifications for each activation method, overall boosts in identification over ETD of 103% for AI-ETD at 18W and 60% for ETcaD are obtained, while a 16% drop in identification for EThcD are calculated. While additional fragment ions detected in AI-ETD spectra relative to ETD spectra translate into additional peptide identifications, the additional c- and z•-type ions in ETcaD and EThcD do not translate into additional identifications and hydrogen migration hampers algorithmic spectral interpretation and peptide identification by diminishing the monoisotopic peak intensity and widening the tolerance needed for identification. Note, EThcD, and AI-ETD are capable of generating b- and y-type fragment ions, which greatly benefit spectral searches over ETD alone, when b-, c-, y-, and z•-type ions are considered (Figure S3)[47]. While EThcD enables additional peptide identifications through the generation of b- and y-type ions, the benefits of ETD for analyses of post-transcriptional modifications and intact proteins often necessitate maximal c- and z•-type ion formation and minimal hydrogen migration, realized through gentler activation.
CONCLUSION
Intramolecular hydrogen migration events are known to happen in electron-driven activation of peptide cations, although the degree of migration can depend on a number of variables. Especially important in understanding migration proclivity are the amino acids surrounding the cleavage site and the time even- and odd- electron product ions are held in close proximity. In this work we compared hydrogen migration for four different activation modes: ETD, ETcaD, EThcD, and AI-ETD. The three latter methods all involve supplemental activation of ETD products, although the timing, energy, and specificities of which ions are activated differ. ETcaD and EThcD both involve vibrational activation of product ions after the ETD reaction concludes, although ETcaD uses resonant excitation of only ETnoD products while EThcD uses broadband beam-type activation of all ETD product ions. Both of these post-activation methods yielded a relatively high degree of hydrogen migration, especially when D, G, N, S, and T residues were directly C-terminal to the cleavage site. In contrast, AI-ETD, which uses concurrent vibrational activation of the entire ion-ion reaction volume via infrared photons, showed relatively less hydrogen migration relative to ETcaD and EThcD. AI-ETD and ETD generally appeared more similar in their hydrogen migration profiles.
The practical effects of hydrogen migration events were shown in a database search of LC-MS/MS human tryptic peptide data that only accounts for expected [c+2H]1+ and [z+H]●1+. All three supplemental activation methods generated more total [c+2H]1+ and [z+H]●1+ fragment ions than ETD alone; however, the benefit of more peptide identifications was only realized for AI-ETD. This is because product ion distributions were altered in ETcaD and EThcD spectra because of hydrogen rearrangements while product ions remained more similar to traditional ETD fragments that are expected by the search algorithm. This indicates that manual spectral interpretation and database searching must properly account for product ions specific to different ETD-based fragmentation methods, and that AI-ETD is more suited to algorithms already specifically built with expected ETD [c+2H]1+ and [z+H]●1+ in mind. This work provides a holistic view of hydrogen migration patterns in traditional shotgun proteomic data. Additionally, new implementations of electron-driven activation methods continue to emerge[81–85], highlighting the need for detailed understanding of fragment ions generated by various supplemental activation schemes. Our work here provides a framework to investigate such data at proteome scale.
Supplementary Material
ACKNOWLEDGMENT
We thank John E. P. Syka, Graeme McAlister, Christopher Mullen and Dain Brademan for helpful discussions. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (Grant P41GM108538 to J.J.C.) and the National Human Genome Research Institution through a training grant to the Genomic Science Training Program (Grant T32HG002760 to T.M.P.C.).
Footnotes
J.J.C. is a consultant for Thermo Fisher Scientific. Raw data files are available online on Chorus (Project ID 1734).
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Expanded materials and methods descriptions. Figure S1, Expected and observed isotopic distributions for thousands of c- and z•-type fragments generated by ETD, AI-ETD, ETcaD, and EThcD; Figure S2, Differences between theoretical and isotopic fragment distributions are more pronounced in ETD with supplemental collision-based activation; Figure S3, Peptide identification rates when b-, c-, y-, and z•-type ions are searched; Figure S4, Tandem mass spectra for the peptide TEGLSVLSQAMAVIK; Figure S5, Tandem mass spectra for the peptide GAEAANVTGPGGVPVQGSK; Figure S6, Distribution of precursors sampled for ETD, AI-ETD, ETcaD, and EThcD in triplicate.
REFERENCES
- 1.Brodbelt JS: Ion activation methods for peptides and proteins. Anal. Chem. 88, 30–51 (2016). 10.1021/acs.analchem.5b04563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooks RG: Collision Spectroscopy. Anal. Chem. 51, 1979 (1979) [Google Scholar]
- 3.McLuckey SA: Principles of collisional activation in analytical mass spectrometry. J. Am. Soc. Mass Spectrom. 3, 599–614 (1992) [DOI] [PubMed] [Google Scholar]
- 4.Shukla AK, Futrell JH: Tandem mass spectrometry: Dissociation of ions by collisional activation. J. Mass Spectrom. 35, 1069–1090 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Madsen JA, Brodbelt JS: Comparison of Infrared Multiphoton Dissociation and Collision-Induced Dissociation of Supercharged Peptides in Ion Traps. J. Am. Soc. Mass Spectrom. 20, 349–358 (2009). 10.1016/j.jasms.2008.10.018 [DOI] [PubMed] [Google Scholar]
- 6.Zubarev RA, Kelleher NL, McLafferty FW, October RV: Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120, 3265–3266 (1998). 10.1021/ja973478k [DOI] [Google Scholar]
- 7.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF: Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 101, 9528–9533 (2004). 10.1073/pnas.0402700101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW: Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal. Chem. 72, 563–573 (2000). 10.1021/ac990811p [DOI] [PubMed] [Google Scholar]
- 9.Zhurov KO, Fornelli L, Wodrich MD, Tsybin YO: Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev. 42, 5014–5030 (2013). 10.1039/c3cs35477f [DOI] [PubMed] [Google Scholar]
- 10.Lermyte F, Valkenborg D, Loo JA, Sobott F: Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom. Rev. 37, 750–771 (2018). 10.1002/mas.21560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith LM, Kelleher NL: Proteoform: a single term describing protein complexity. Nat. Methods. 10, 186–187 (2013). 10.1038/nmeth.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW: Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. J. Am. Chem. Soc. 124, 672–678 (2002). 10.1021/ja011335z [DOI] [PubMed] [Google Scholar]
- 13.Toby TK, Fornelli L, Kelleher NL: Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 9, 499–519 (2016). 10.1146/annurev-anchem-071015-041550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riley NM, Coon JJ: The Role of electron transfer dissociation in modern proteomics. Anal. Chem. 90, 40–64 (2018). 10.1021/acs.analchem.7b04810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breuker K, Oh H, Lin C, Carpenter BK, McLafferty FW: Nonergodic and conformational control of the electron capture dissociation of protein cations. Proc. Natl. Acad. Sci. U. S. A. 101, 14011–14016 (2004). 10.1073/pnas.0406095101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW: Detailed Unfolding and Folding of Gaseous Ubiquitin Ions Characterized by Electron Capture Dissociation. J. Am. Chem. Soc. 124, 6407–6420 (2002). 10.1021/ja012267j [DOI] [PubMed] [Google Scholar]
- 17.Syrstad EA, Tureček F: Toward a general mechanism of electron capture dissociation. J. Am. Soc. Mass Spectrom. 16, 208–224 (2005). 10.1016/j.jasms.2004.11.001 [DOI] [PubMed] [Google Scholar]
- 18.Tureček F, Chen X, Hao C: Where does the electron go? Electron distribution and reactivity of peptide cation radicals formed by electron transfer in the gas phase. J. Am. Chem. Soc. 130, 8818–8833 (2008). 10.1021/ja8019005 [DOI] [PubMed] [Google Scholar]
- 19.Sawicka A, Skurski P, Hudgins RR, Simons J: Model calculations relevant to disulfide bond cleavage via electron capture influenced by positively charged groups. J. Phys. Chem. B. 107, 13505–13511 (2003). 10.1021/jp035675d [DOI] [Google Scholar]
- 20.Leymarie N, Costello CE, O’Connor PB: Electron capture dissociation initiates a free radical reaction cascade. J. Am. Chem. Soc. 125, 8949–8958 (2003). 10.1021/ja028831n [DOI] [PubMed] [Google Scholar]
- 21.O’Connor PB, Lin C, Cournoyer JJ, Pittman JL, Belyayev M, Budnik BA: Long-Lived Electron Capture Dissociation Product Ions Experience Radical Migration via Hydrogen Abstraction. (2006). 10.1016/j.jasms.2005.12.015 [DOI] [PubMed]
- 22.Lin C, O’Connor PB, Cournoyer JJ: Use of a Double Resonance Electron Capture Dissociation Experiment to Probe Fragment Intermediate Lifetimes. J. Am. Soc. Mass Spectrom. 17, 1605–1615 (2006). 10.1016/j.jasms.2006.07.007 [DOI] [PubMed] [Google Scholar]
- 23.Prell JS, O’Brien JT, Holm AIS, Leib RD, Donald WA, Williams ER: Electron capture by a hydrated gaseous peptide: Effects of water on fragmentation and molecular survival. J. Am. Chem. Soc. 130, 12680–12689 (2008). 10.1021/ja8022434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Mcluckey SA: Electron transfer dissociation: Effects of cation charge state on product partitioning in ion/ion electron transfer to multiply protonated polypeptides. Int. J. Mass Spectrom. 332, 174–181 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitteri SJ, Chrisman PA, McLuckey SA: Electron-transfer ion/ion reactions of doubly protonated peptides: Effect of elevated bath gas temperature. Anal. Chem. 77, 5662–5669 (2005). 10.1021/ac050666h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA: Electron transfer ion/ion reactions in a three-dimensional quadrupole ion trap: Reactions of doubly and triply protonated peptides with SO2•−. Anal. Chem. 77, 1831–1839 (2005). 10.1021/ac0483872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Y, Gunawardena HP, Erickson DE, McLuckey SA: Effects of cation charge-site identity and position on electron-transfer dissociation of polypeptide cations. J. Am. Chem. Soc. 129, 12232–12243 (2007). 10.1021/ja0736764 [DOI] [PubMed] [Google Scholar]
- 28.Horn DM, Ge Y, McLafferty FW: Activated Ion Electron Capture Dissociation for Mass Spectral Sequencing of Larger (42 kDa) Proteins. Anal. Chem. 72, 4778–4784 (2000). 10.1021/ac000494i [DOI] [PubMed] [Google Scholar]
- 29.Horn DM, Breuker K, Frank AJ, McLafferty FW, C.U. V, York N, August R V: Kinetic Intermediates in the Folding of Gaseous Protein Ions Characterized by Electron Capture Dissociation Mass Spectrometry. J. Am. Chem. Soc. 123, 9792–9799 (2001). 10.1021/ja003143u [DOI] [PubMed] [Google Scholar]
- 30.Tsybin YO, Witt M, Baykut G, Kjeldsen F, Håkansson P: Combined infrared multiphoton dissociation and electron capture dissociation with a hollow electron beam in Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 17, 1759–1768 (2003). 10.1002/rcm.1118 [DOI] [PubMed] [Google Scholar]
- 31.Zabrouskov V, Whitelegge JP: Increased coverage in the transmembrane domain with activated-ion electron capture dissociation for top-down fourier-transform mass spectrometry of integral membrane proteins. J. Proteome Res. 6, 2205–2210 (2007). 10.1021/pr0607031 [DOI] [PubMed] [Google Scholar]
- 32.Håkansson K, Chalmers MJ, Quinn JP, McFarland MA, Hendrickson CL, Marshall AG: Combined electron capture and infrared multiphoton dissociation for multistage MS/MS in a Fourier transform ion cyclotron resonance mass spectrometer. Anal. Chem. 75, 3256–3262 (2003). 10.1021/ac030015q [DOI] [PubMed] [Google Scholar]
- 33.Cooper HJ, Heath JK, Jaffray E, Hay RT, Lam TKT, Marshall AG: Identification of sites of ubiquitination in proteins: A fourier transform ion cyclotron resonance mass spectrometry approach. Anal. Chem. 76, 6982–6988 (2005). 10.1021/ac0401063 [DOI] [PubMed] [Google Scholar]
- 34.Oh H, McLafferty FW: A variety of activation methods employed in “Activated-Ion” electron capture dissociation mass spectrometry: A test against Bovine Ubiquitin 7+ ions. Bull. Korean. Chem. Soc. 27, 389–394 (2006) [Google Scholar]
- 35.Mikhailov VA, Cooper HJ: Activated Ion Electron Capture Dissociation (AI ECD) of Proteins: Synchronization of Infrared and Electron Irradiation with Ion Magnetron Motion. J. Am. Soc. Mass Spectrom. 20, 763–771 (2009). 10.1016/j.jasms.2008.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JEP, Coon JJ: Supplemental Activation Method for High-Efficiency Electron-Transfer Dissociation of Doubly Protonated Peptide Precursors. Anal. Chem. 79, 477–485 (2007). 10.1021/ac061457f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swaney DL, McAlister GC, Coon JJ: Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods. 5, 959–964 (2008). 10.1038/NMETH.1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell JL, Hager JW, Le Blanc JCY: On Performing Simultaneous Electron Transfer Dissociation and Collision-Induced Dissociation on Multiply Protonated Peptides in a Linear Ion Trap. J. Am. Soc. Mass Spectrom. 20, 1672–1683 (2009). 10.1016/j.jasms.2009.05.009 [DOI] [PubMed] [Google Scholar]
- 39.Xia Y, Han H, Mcluckey SA: Activation of Intact Electron-Transfer Products of Polypeptides and Proteins in Cation Transmission Mode Ion/Ion Reactions. Anal. Chem. 80, 1111–1117 (2008). 10.1021/ac702188q [DOI] [PubMed] [Google Scholar]
- 40.Han H, Xia Y, McLuckey SA: Beam-type collisional activation of polypeptide cations that survive ion/ion electron transfer. Rapid Commun. Mass Spectrom. 21, 1567–1573 (2007). 10.1002/rcm.2994 [DOI] [PubMed] [Google Scholar]
- 41.Han H, Xia Y, McLuckey SA: Ion trap collisional activation of c and z• ions formed via gas-phase ion/ion electron-transfer dissociation. J. Proteome Res. 6, 3062–3069 (2007). 10.1021/pr070177t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu CW, Lai CC: Effects of electron-transfer coupled with collision-induced dissociation (ET/CID) on doubly charged peptides and phosphopeptides. J. Am. Soc. Mass Spectrom. 22, 57–66 (2011). 10.1007/s13361-010-0020-9 [DOI] [PubMed] [Google Scholar]
- 43.Frese CK, Altelaar AFM, Van Den Toorn H, Nolting D, Griep-Raming J, Heck AJR, Mohammed S: Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 84, 9668–9673 (2012). 10.1021/ac3025366 [DOI] [PubMed] [Google Scholar]
- 44.Frese CK, Zhou H, Taus T, Altelaar AFM, Mechtler K, Heck AJR, Mohammed S: Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 12, 1520–1525 (2013). 10.1021/pr301130k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ledvina AR, Beauchene NA, McAlister GC, Syka JEP, Schwartz JC, Griep-Raming J, Westphall MS, Coon JJ: Activated-ion electron transfer dissociation improves the ability of electron transfer dissociation to identify peptides in a complex mixture. Anal. Chem. 82, 10068–10074 (2010). 10.1021/ac1020358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ledvina AR, McAlister GC, Gardner MW, Smith SI, Madsen JA, Schwartz JC, Stafford GC, Syka JEP, Brodbelt JS, Coon JJ: Infrared photoactivation reduces peptide folding and hydrogen- atom migration following ETD tandem mass spectrometry. Angew. Chemie Int. Ed. 48, 8526–8528 (2009). 10.1002/anie.200903557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riley NM, Westphall MS, Hebert AS, Coon JJ: Implementation of activated ion electron transfer dissociation on a quadrupole-Orbitrap-linear ion trap hybrid mass spectrometer. Anal. Chem. 89, 6358–6366 (2017). 10.1021/acs.analchem.7b00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riley NM, Hebert AS, Westphall MS, Coon JJ: Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun. 10, 1311 (2019). 10.1038/s41467-019-09222-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riley NM, Hebert AS, Dü G, Stanek F, Mechtler K, Westphall MS, Coon JJ: Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal. Chem. 89, 6367–6376 (2017). 10.1021/acs.anal [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buch-Larsen SC, Hendriks IA, Lodge JM, Rykaer M, Furtwängler B, Shishkova E, Westphall MS, Coon JJ, Nielsen ML: Mapping physiological ADP-ribosylation using Activated Ion Electron Transfer Dissociation (AI-ETD). bioRxiv. (2020). 10.1101/2020.01.27.921650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ledvina AR, Rose CM, McAlister GC, Syka JEP, Westphall MS, Griep-Raming J, Schwartz JC, Coon JJ: Activated ion ETD performed in a modified collision cell on a hybrid QLT-Oribtrap mass spectrometer. J. Am. Soc. Mass Spectrom. 24, 1623–1633 (2013). 10.1007/s13361-013-0621-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leung KK, Wilson GM, Kirkemo LL, Riley NM, Coon JJ, Wells JA: Broad and thematic remodeling of the surfaceome and glycoproteome on isogenic cells transformed with driving proliferative oncogenes. Proc. Natl. Acad. Sci. U. S. A. 117, 7764–7775 (2020). 10.1073/pnas.1917947117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peters-Clarke TM, Schauer KL, Riley NM, Lodge JM, Westphall MS, Coon JJ: Optical Fiber-Enabled Photoactivation of Peptides and Proteins. Anal. Chem. 92, 12363–12370 (2020). 10.1021/acs.analchem.0c02087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riley NM, Westphall MS, Coon JJ: Activated ion electron transfer dissociation for improved fragmentation of intact proteins. Anal. Chem. 87, 7109–7116 (2015). 10.1021/acs.analchem.5b00881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riley NM, Westphall MS, Coon JJ: Activated ion-electron transfer dissociation enables comprehensive top-down protein fragmentation. J. Proteome Res. 16, 2653–2659 (2017). 10.1021/acs.jproteome.7b00249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riley NM, Westphall MS, Coon JJ: Sequencing larger intact proteins (30–70 kDa) with activated ion electron transfer dissociation. J. Am. Soc. Mass Spectrom. 29, 140–149 (2018). 10.1007/s13361-017-1808-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peters-Clarke TM, Quan Q, Brademan DR, Hebert AS, Westphall MS, Coon JJ: Ribonucleic Acid Sequence Characterization by Negative Electron Transfer Dissociation Mass Spectrometry. Anal. Chem. 92, 4436–4444 (2020). 10.1021/acs.analchem.9b05388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chu IK, Siu C-K, Kai-Chi Lau J, Kit Tang W, Mu X, Kuen Lai C, Guo X, Wang X, Li N, Xia Y, Kong X, Bin Oh H, Ryzhov V, Tureček F, Hopkinson AC, Michael Siu K: Proposed nomenclature for peptide ion fragmentation. Int. J. Mass Spectrom. 390, 24–27 (2015). 10.1016/j.ijms.2015.07.021 [DOI] [Google Scholar]
- 59.Tureček F, Syrstad EA: Mechanism and energetics of intramolecular hydrogen transfer in amide and peptide radicals and cation-radicals. J. Am. Chem. Soc. 125, 3353–3369 (2003). 10.1021/ja021162t [DOI] [PubMed] [Google Scholar]
- 60.Palzs B, Suhal S: Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 (2005). 10.1002/mas.20024 [DOI] [PubMed] [Google Scholar]
- 61.Tureček F, Julian RR: Peptide Radicals and Cation Radicals in the Gas Phase. Chem. Rev. 113, 6691–6733 (2013). 10.1021/cr400043s [DOI] [PubMed] [Google Scholar]
- 62.Medzihradszky KF, Chalkley RJ: Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom. Rev. 34, 43–63 (2015). 10.1002/mas.21406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Savitski MM, Kjeldsen F, Nielsen ML, Zubarev RA: Hydrogen rearrangement to and from radical z Fragments in electron capture dissociation of peptides. J. Am. Soc. Mass Spectrom. 18, 113–120 (2007). 10.1016/j.jasms.2006.09.008 [DOI] [PubMed] [Google Scholar]
- 64.Rand KD, Adams CM, Zubarev RA, Jørgensen TJD: Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens. J. Am. Chem. Soc. 130, 1341–1349 (2008). 10.1021/ja076448i [DOI] [PubMed] [Google Scholar]
- 65.Ly T, Julian RR: Tracking Radical Migration in Large Hydrogen Deficient Peptides with Covalent Labels: Facile Movement does not Equal Indiscriminate Fragmentation. J. Am. Soc. Mass Spectrom. 20, 1148–1158 (2009). 10.1016/j.jasms.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 66.Nielsen ML, Budnik BA, Haselmann KF, Olsen JV, Zubarev RA: Intramolecular hydrogen atom transfer in hydrogen-deficient polypeptide radical cations. Chem. Phys. Lett. 330, 558–562 (2000) [Google Scholar]
- 67.Ledvina AR, Coon JJ, Tureček F: Competitive hydrogen atom migrations accompanying cascade dissociations of peptide cation-radicals of the z+• type. Int. J. Mass Spectrom. 377, 44–53 (2015). 10.1016/j.ijms.2014.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riffet V, Jacquemin D, Frison G: H-atom loss and migration in hydrogen-rich peptide cation radicals: The role of chemical environment. Int. J. Mass Spectrom. 390, 28–38 (2015). 10.1016/j.ijms.2015.08.001 [DOI] [Google Scholar]
- 69.Bythell BJ: To Jump or Not To Jump? Cα Hydrogen Atom Transfer in Post-cleavage Radical-Cation Complexes. J. Phys. Chem. 117, 1189–1196 (2013). 10.1021/jp305277v [DOI] [PubMed] [Google Scholar]
- 70.Siu C, Ke Y, Orlova G, Hopkinson AC, Siu KWM: Dissociation of the N – C Bond and Competitive Formation of the [ z n – H ] •+ and [cn + 2H]+ Ions Containing Tyrosine and Tryptophan : The Influence of Proton Affinities on Product Formation. J. Am. Soc. Mass Spectrom. 19, 1799–1807 (2008). 10.1016/j.jasms.2008.09.026 [DOI] [PubMed] [Google Scholar]
- 71.Sun R-X, Dong M-Q, Song C-Q, Chi H, Yang B, Xiu L-Y, Tao L, Jing Z-Y, Liu C, Wang L-H, Fu Y, He S-M: Improved Peptide Identification for Proteomic Analysis Based on Comprehensive Characterization of Electron Transfer Dissociation Spectra. J. Proteome Res. 9, 6354–6367 (2010). 10.1021/pr100648r [DOI] [PubMed] [Google Scholar]
- 72.Baker PR, Medzihradszky KF, Chalkley RJ: Improving software performance for peptide electron transfer dissociation data analysis by implementation of charge state- and sequence-dependent scoring. Mol. Cell. Proteomics. 9, 1795–1803 (2010). 10.1074/mcp.M110.000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chalkley RJ, Medzihradszky KF, Lynn AJ, Baker PR, Burlingame AL: Statistical analysis of peptide electron transfer dissociation fragmentation mass spectrometry. Anal. Chem. 82, 579–584 (2010). 10.1021/ac9018582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Good DM, Wirtala M, McAlister GC, Coon JJ: Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics. 6, 1942–1951 (2007). 10.1074/mcp.M700073-MCP200 [DOI] [PubMed] [Google Scholar]
- 75.Sadygov RG, Good DM, Swaney DL, Coon JJ: A new probabilistic database search algorithm for ETD spectra. J. Proteome Res. 8, 3198–3205 (2009). 10.1021/pr900153b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu X, Shan B, Xin L, Ma B: Better score function for peptide identification with ETD MS/MS spectra. BMC Bioinformatics. 11, 1–8 (2010). 10.1186/1471-2105-11-S1-S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li W, Song C, Bailey DJ, Tseng GC, Coon JJ, Wysocki VH: Statistical analysis of electron transfer dissociation pairwise fragmentation patterns. Anal. Chem. 83, 9540–9545 (2011). 10.1021/ac202327r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sridhara V, Bai DL, Chi A, Shabanowitz J, Hunt DF, Bryant SH, Geer LY: Increasing peptide identifications and decreasing search times for ETD spectra by pre-processing and calculation of parent precursor charge. Proteome Sci. 10, 1–10 (2012). 10.1186/1477-5956-10-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Good DM, Wenger CD, McAlister GC, Bai DL, Hunt DF, Coon JJ: Post-Acquisition ETD Spectral Processing for Increased Peptide Identifications. J. Am. Soc. Mass Spectrom. 20, 1435–1440 (2009). 10.1016/j.jasms.2009.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Good DM, Wenger CD, Coon JJ: The effect of interfering ions on search algorithm performance for electron-transfer dissociation data. Proteomics. 10, 164–167 (2010). 10.1002/pmic.200900570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cannon JR, Holden DD, Brodbelt JS: Hybridizing ultraviolet photodissociation with electron transfer dissociation for intact protein characterization. Anal. Chem. 86, 10970–10977 (2014). 10.1021/ac5036082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shaw JB, Malhan N, Vasil’Ev YV, Lopez NI, Makarov A, Beckman JS, Voinov VG: Sequencing Grade Tandem Mass Spectrometry for Top-Down Proteomics Using Hybrid Electron Capture Dissociation Methods in a Benchtop Orbitrap Mass Spectrometer. Anal. Chem. 90, 10819–10827 (2018). 10.1021/acs.analchem.8b01901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zubarev RA, Yang H: Multiple soft ionization of gas-phase proteins and swift backbone dissociation in collisions with ≤ 99 eV electrons. Angew. Chemie - Int. Ed. 49, 1439–1441 (2010). 10.1002/anie.200905977 [DOI] [PubMed] [Google Scholar]
- 84.Baba T, Ryumin P, Duchoslav E, Chen K, Chelur A, Loyd B, Chernushevich I: Dissociation of Biomolecules by an Intense Low-Energy Electron Beam in a High Sensitivity Time-of-Flight Mass Spectrometer. J. Am. Soc. Mass Spectrom. (2021). 10.1021/jasms.0c00425 [DOI] [PubMed] [Google Scholar]
- 85.Li H, Sheng Y, McGee W, Cammarata M, Holden D, Loo JA: Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal. Chem. 89, 2731–2738 (2017). 10.1021/acs.analchem.6b02377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Webb IK, Kit MCS, Shepherd SO, Prell JS: Experimental determination of activation energies for covalent bond formation via ion/ion reactions and competing processes. J. Am. Soc. Mass Spectrom. (2021). 10.1021/jasms.1c00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ: The One Hour Yeast Proteome. Mol. Cell. Proteomics. 13, 339–347 (2014). 10.1074/mcp.M113.034769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Richards AL, Hebert AS, Ulbrich A, Bailey DJ, Coughlin EE, Westphall MS, Coon JJ: One-hour proteome analysis in yeast. Nat. Protoc. 10, 701–714 (2015). 10.1038/nprot.2015.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stefely JA, Kwiecien NW, Freiberger EC, Richards AL, Jochem A, Rush MJP, Ulbrich A, Robinson KP, Hutchins PD, Veling MT, Guo X, Kemmerer ZA, Connors KJ, Trujillo EA, Sokol J, Marx H, Westphall MS, Hebert AS, Pagliarini DJ, Coon JJ: Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol. 34, 1191–1197 (2016). 10.1038/nbt.3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shishkova E, Hebert AS, Westphall MS, Coon JJ: Ultra-High Pressure (>30,000 psi) Packing of Capillary Columns Enhancing Depth of Shotgun Proteomic Analyses. Anal. Chem. 90, 11503–11508 (2018). 10.1021/acs.analchem.8b02766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Riley NM, Mullen C, Weisbrod CR, Sharma S, Senko MW, Zabrouskov V, Westphall MS, Syka JEP, Coon JJ: Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 27, 520–531 (2016). 10.1007/s13361-015-1306-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rose CM, Rush MJP, Riley NM, Merrill AE, Kwiecien NW, Holden DD, Mullen C, Westphall MS, Coon JJ: A Calibration Routine for Efficient ETD in Large-Scale Proteomics. J. Am. Soc. Mass Spectrom. 26, 1848–1857 (2015). 10.1007/s13361-015-1183-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ: COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. (2011). 10.1002/pmic.201000616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH: Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 (2004). 10.1021/pr0499491 [DOI] [PubMed] [Google Scholar]
- 95.Elias JE, Gygi SP: Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 4, 207–214 (2007). 10.1038/nmeth1019 [DOI] [PubMed] [Google Scholar]
- 96.Nesvizhskii AI, Aebersold R: Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteomics. 4, 1419–1440 (2005). 10.1074/mcp.R500012-MCP200 [DOI] [PubMed] [Google Scholar]
- 97.Lermyte F, Łacki MK, Valkenborg D, Baggerman G, Gambin A, Sobott F: Understanding reaction pathways in top-down ETD by dissecting isotope distributions: A mammoth task. Int. J. Mass Spectrom. 390, 146–154 (2015). 10.1016/j.ijms.2015.08.008 [DOI] [Google Scholar]
- 98.Senko MW, Beu SC, McLafferty FW: Determination of Monoisotopic Masses and Ion Populations for Large Bio... (Averagine algorithm).pdf. (1994) [DOI] [PubMed] [Google Scholar]
- 99.Tabb DL, MacCoss MJ, Wu CC, Anderson SD, Yates JR: Similarity among tandem mass spectra from proteomic experiments: Detection, significance, and utility. Anal. Chem. 75, 2470–2477 (2003). 10.1021/ac026424o [DOI] [PubMed] [Google Scholar]
- 100.Valkenborg D, Jansen I, Burzykowski T: A Model-Based Method for the Prediction of the Isotopic Distribution of Peptides. J. Am. Soc. Mass Spectrom. 19, 703–712 (2008). 10.1016/j.jasms.2008.01.009 [DOI] [PubMed] [Google Scholar]
- 101.Yergey JA: A general approach to calculating isotopic distributions for mass spectrometry. Int. J. Mass Spectrom. Ion Phys. 52, 337–349 (1983). 10.1016/0020-7381(83)85053-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.