

Abstract
Indole is a significant structural moiety and functionalization of the C–H bond in indole-containing molecules expands their chemical space, and modifies their properties and/or activities. Indole prenyltransferases (IPTs) catalyze the direct regiospecific installation of prenyl, C5 carbon units, on indole-derived compounds. IPTs have shown relaxed substrate flexibility enabling them to be used as tools for indole functionalization. However, the mechanism by which certain IPTs target a specific carbon position is not fully understood. Herein, we use structure-guided site-directed mutagenesis, in vitro enzymatic reactions, kinetics and structural-elucidation of analogs to verify the key catalytic residues that control the regiospecificity of all characterized regiospecific C6 IPTs. Our results also demonstrate that substitution of PriB_His312 to Tyr leads to the synthesis of analogs prenylated at different positions than C6. This work contributes to understanding of how certain IPTs can access a challenging position in indole-derived compounds.
Keywords: biocatalysis, dimethylallyl tryptophan synthase, enzyme catalysis, histidine, protein engineering
Graphical Abstract
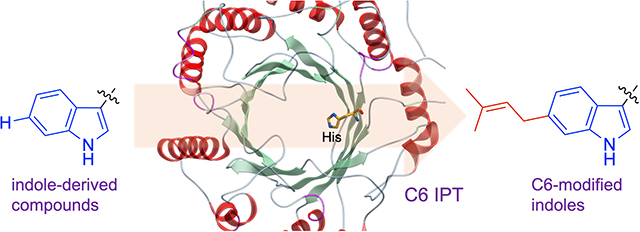
The functionalization of indole-derived compounds modifies their physicochemical properties and/or pharmacological activities. Activation of the indole C6 position is challenging. Yet, certain indole prenyltransferases (IPTs) catalyze this reaction efficiently, regiospecifically and under physiological conditions. Through enzyme engineering, in-vitro enzymatic reactions and NMR experiments, guided by protein structure analyses, we verify the catalytic mechanism by which C6 IPTs catalyze their reactions.
Introduction
Functionalization of C–H bonds is a powerful strategy to generate new carbon-carbon or carbon-heteroatom bonds. This can reduce the number of steps involved in a synthetic route for a compound or improve the starting material availability, which opens up avenues for drug discovery and development. Within this context, the indole scaffold is encountered in numerous biological active compounds.[1,2] Indole-containing molecules can bind to various receptors with high affinity. Thus, it is not surprising that indole is among the most prevalent heteroaromatic groups in approved drugs or those in clinical trials.[1] This has attracted efforts to functionalize the C–H bonds of indole-derived compounds as a means of modifying their structures and enhance their biological properties.[3] The C2 and C3 carbons are more activated towards attack by electrophiles than the C4–C7 benzene ones.[4] Noteworthy, selective activation of the indole C6 position via synthetic methods has proven to be particularly challenging due to its lower nucleophilicity when compared to other C–H benzenoid sites.[5,6] Nevertheless, several groups have accessed the C6 indole position (Figure 1A).[6–11] These methods have been met with challenges such as the requirement of transition metals or directing or blocking groups and/or suffer from the formation of multiple products.
Figure 1.
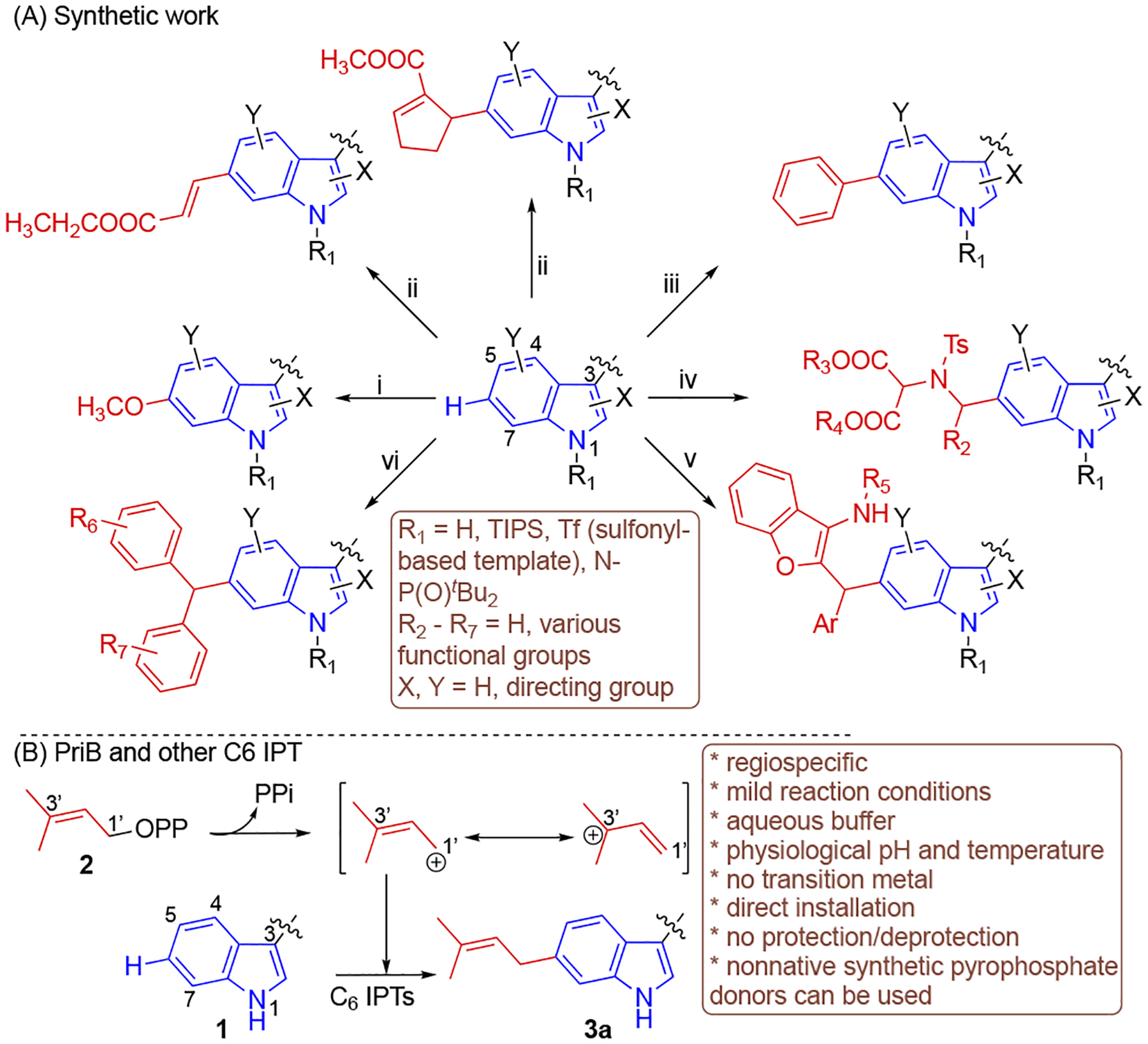
Functionalization of C6 position in Trp and/or indole-containing compounds using (A) synthetic efforts and (B) PriB and other C6 indole prenyltransferase. i,[6] LiHMDS, TIPS-Cl, Ir[(cod)OMe]2, B2Pin2, 80 °C, Cu(OAc)2, MeOH; ii,[7] CH2=CHCOOCH2CH3 or C5H5COOCH3, Pd(OAc)2, Ac-Gly-OH, AgOAc, HFIP, 55 – 90 °C; iii,[8] (C6H5)2IOTf, CuO, 80 °C; iv,[9] Sc(OTf)3, N–Ts aziridines, rt; v,[10] azadiene, (R)-phosphoric acid, 40 °C; vi,[11] (aza-)quinonemethides, Brønsted acid, rt. OPP, pyrophosphate; PPi, inorganic pyrophosphate.
The most challenging aspect in functionalization is to enable the precise activation of the desired C–H bond over another. Nevertheless, enzymes have served as a valuable tool to achieve selective functionalization.[12–15] Enzymes are highly selective and can catalyze their reactions under mild and physiological conditions without the need of protection/deprotection or directing groups. In particular, indole prenyltransferase (IPT) enzymes catalyze the installation of prenyl moieties using pyrophosphate donors such as dimethylallyl, geranyl and farnesyl pyrophosphates (DMAPP, GPP, FPP) onto indole-containing molecules.[16,17] Release of the diphosphate moiety from the prenyl donor leads to the formation of an allylic carbocation that is stabilized by electron delocalization. This creates electron deficiency on the primary carbon, C1’ and the tertiary carbon C3’ facilitating interaction with the electron rich indole and leading to subsequent normal or reverse prenylation, respectively (Figure 1B). IPTs have relaxed acceptor and donor specificities allowing them to be used as a valuable tool for indole late-stage functionalization and labelling.[18–20] IPTs have shown to facilitate the synthesis of a complex natural product alkaloid[21] as well as the semipreparative synthesis of regiospecific l- and d-indole derivatives.[22]
Different IPTs have been functionally characterized that catalyze the C–H bond activation at each of the non-fusion carbons including C6 via Friedel-Crafts alkylation.[23,24] In this regard, PriB is a promiscuous C6 IPT that accommodates various indole-derived compounds and achieves a C1’ normal prenylation reaction (Figure 1B, 3a).[25] PriB is also able to install an array of native and synthetic pyrophosphate donors onto indoles. Structural and enzymatic studies have revealed insights into the mechanistics of C6 IPTs and key residues that govern catalysis. His residues positioned close to the indole moiety were suggested[25] and shown[26] to be responsible for C6 prenylation. Yet, it is unclear if the involvement of His is prevalent among C6 IPTs. In this study, we confirmed the catalytic base for PriB regiospecificity using protein structure analysis, site directed mutagenesis and in vitro enzymatic reactions and kinetics. We generated a PriB mutant capable of prenylating indole at different positions other than C6 as evidenced by extensive NMR analyses of the purified analogs. We expand our studies to other C6 IPTs and reveal that the catalytic residue is universal for all regiospecific C6 IPTs currently characterized.
Results and Discussion
His312 plays a major role in the catalytic efficiency of PriB.
Similar to other IPTs, the overall structure of PriB (PDB ID 5INJ) is the ABBA barrel fold[27] which contains antiparallel beta strands forming the core of the protein surrounded by alpha helices (Figure S1).[25] The native acceptor and donor substrates, l-Trp (1) and DMAPP (2)-homolog, occupy the center of the tunnel inside the ABBA barrel. To investigate the catalysis of C6 prenylation, the PriB binding pocket was carefully examined to identify residues that make close contacts with 1. The binding of l-Trp is facilitated by the close interaction of several aromatic and non-polar amino acids (Figure 2A). The 1-indole ring is held in place by π-stacking and hydrophobic interactions. The Trp and DMAPP-homolog are docked closely together, where the dimethylallyl moiety is positioned under the benzene ring of l-Trp. The closest indole carbons from the DMAPP PriB C1’ carbon are C6 and C7 which are located at 3.6 Å (Figure 2B, Figure S2A). Infact, PriB was found to prenylate the 1-C7 position when the C6 is blocked.[25] Furthermore, the indole carbon is positioned at less than 4 Å from only two residues, His312 and Tyr364 (Figure 2A). Noteworthy, the indole C6 carbon in PriB is located at 3.7 Å from the His312 basic nitrogen, closer than any other Trp carbon (Figure 2B, Figure S2B). The Tyr364 hydroxyl group is too far to propose a role in the catalysis of C6 (Figure 2B). Structural alignment of PriB (C6) with selected non-C6 IPTs [DMATS1 (PDB ID 8DB0, N-1)[28], AmbP3 (PDB ID 5Y7C, C2), FtmPT1 (PDB ID 3O2K, C3 rearranges to C-2)[29,30], CdpNPT (PDB ID 4E0U, C3 rearranges to N1)[31,32], AnaPT (PDB ID 4LD7, C3)[33], FgaPT2 (PDB ID 3I4X, C4)[27], 5DMATSsc (PDB ID 6ZRZ, C5)[26], MpnD (PDB ID 4YLA, C7)[34]] reveal that His312 in PriB replaces a conserved Tyr residue that is present in non C6 IPTs (Figure 2C, Figures S3–S4). Recently, a His residue was determined as the key catalytic residue for 6DMATSmo.[26] Thus, we hypothesized that His312 plays a major role in controlling PriB regiospecificity by acting as a catalytic base and abstracting the 1 H6. To test our hypothesis, saturation mutagenesis of His312 was carried out (Table S1) and mutations were sequence verified. The plasmids with successful mutations were transformed in E. coli BL21 (DE3) cells and the corresponding N-His6 recombinant proteins were overproduced and purified to homogeneity via metal chelation with yields ranging from 5–11 mg L−1. SDS-PAGE for all proteins revealed the presence of products with the expected molecular weights (Figure S5). Circular dichroism confirmed proper folding of the purified PriB mutants compared to the wild-type (Figure S6). In addition, we synthesized dimethylallyl pyrophosphate by reacting dimethylallyl chloride with Tris(tetrabutylammonium) hydrogen pyrophosphate (TBAPP) at room temperature[35] with overall yield of 40% and confirmed its structure using HR-MS and 1H and 31P NMR (Supporting Mass and NMR Spectra). Analytical scale reactions containing 0.5 mM l-Trp (1), 1 mM DMAPP (2) and 8.3 μM wild-type or engineered protein in Tris 50 mM (pH 8.0) were prepared and incubated at 37 °C for 16 h (Figure 3A). HPLC-MS analyses of quenched reactions revealed that most His312 mutants showed absence or reduced prenylated Trp 3a formation (Figure 3B, Figure S7). This agrees with the proposed role of His312. To obtain insights on the catalytic efficiency, Michaelis-Menten kinetics of PriB compared to the four mutants with the highest conversions (H312E, H312G, H312K, H312Q) were determined using GraphPad Prism 9.5.0 with a non-linear regression (Figure S8). These mutants exhibit Km values in the range of 26.8 × 10−2 − 94.7 × 10−2 mM (Table 1). In addition, the PriB mutants possessed turnover numbers kcat in the range of 5.0 × 10−2 − 39.3 × 10−2 min−1, 50 – 400-fold lower than that of the wild-type. The catalytic efficiencies of the mutants ranged from 5.5 × 10−2 − 41.5 × 10−2 mM−1 min−1 100 – 900-fold lower than that of PriB wild-type (Table 1). The ability of these four mutants to cause partial conversion could be attributed to their ability to be involved in acid/base catalysis directly or mediated by water.[26,27,36] His312 mutation to Asp or Asn resulted in almost complete loss of activity compared to that of Glu and Gln and could be related to enzyme structural changes caused by size differences. These data are consistent with our hypothesis that highlights the significance of His312 for the efficient catalysis of PriB.
Figure 2.
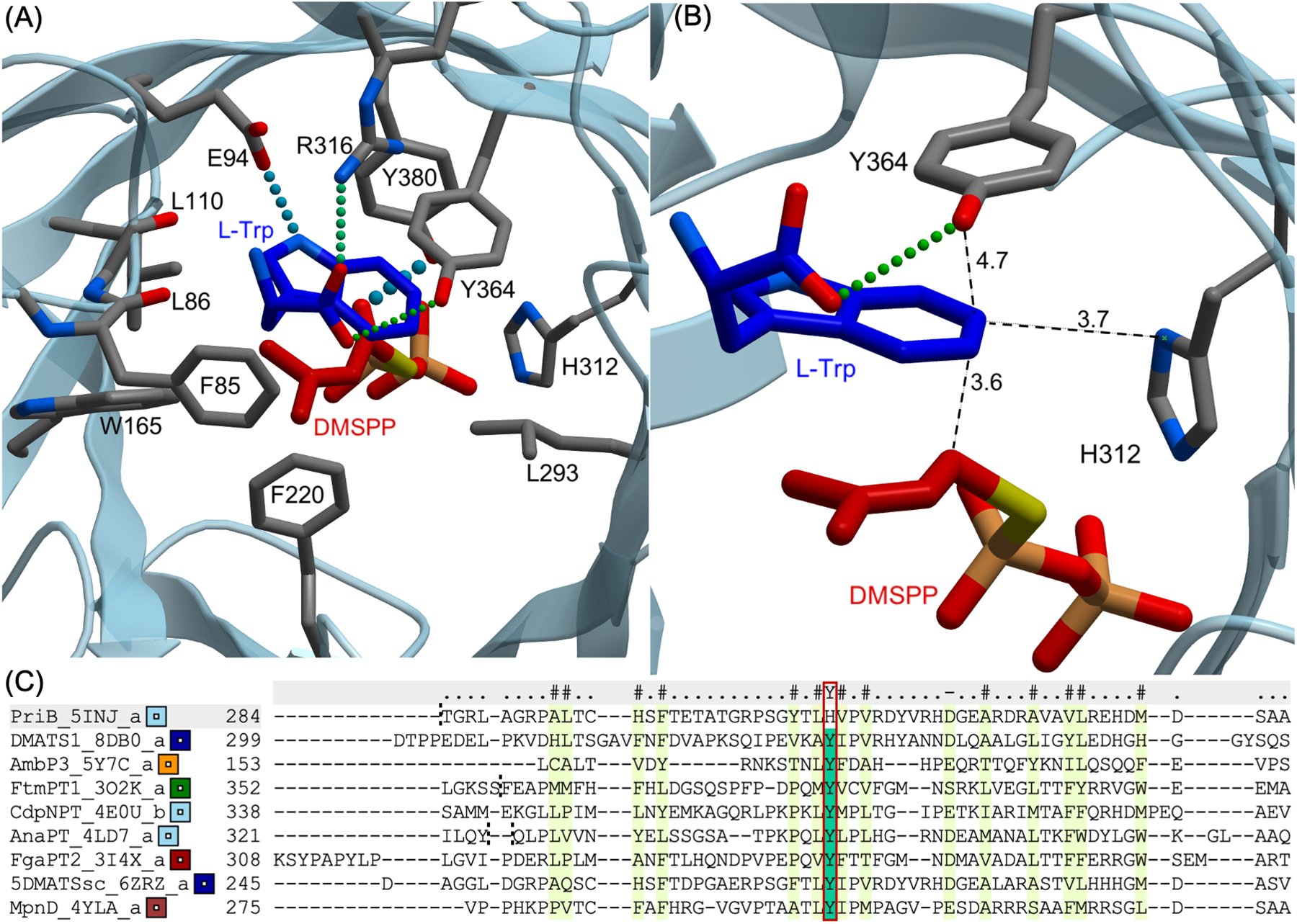
Significance of His312 in PriB catalysis. (A) Active site of ligand-bound PriB showing residues that are within 4 Å from l-Trp (1). (B) Close-up view of PriB active site showing residues that are within 4 Å from Trp C6; (C) Structure-based alignment of PriB with selected IPTs. Red square highlighting proposed catalytic residue. Names of protein and PDB ID are stated. The ligands are illustrated as ball-and-stick models with the following color code: carbon, dark blue (l-Trp) and red (DMAPP-homolog, DMSPP, dimethylallyl S-thiolopyrophosphate); oxygen, light red; nitrogen, light blue; phosphorous, orange; sulfur, yellow. Hydrogen bonds are shown in thick and thin spheres to indicate strong and weak bonds, respectively. Distances are given in Å. DMSPP is a DMAPP-homolog where the cleave site oxygen atom has been replaced with a sulfur resulting in a less reactive substrate suitable for the three-dimensional structure studies of IPTs.
Figure 3.
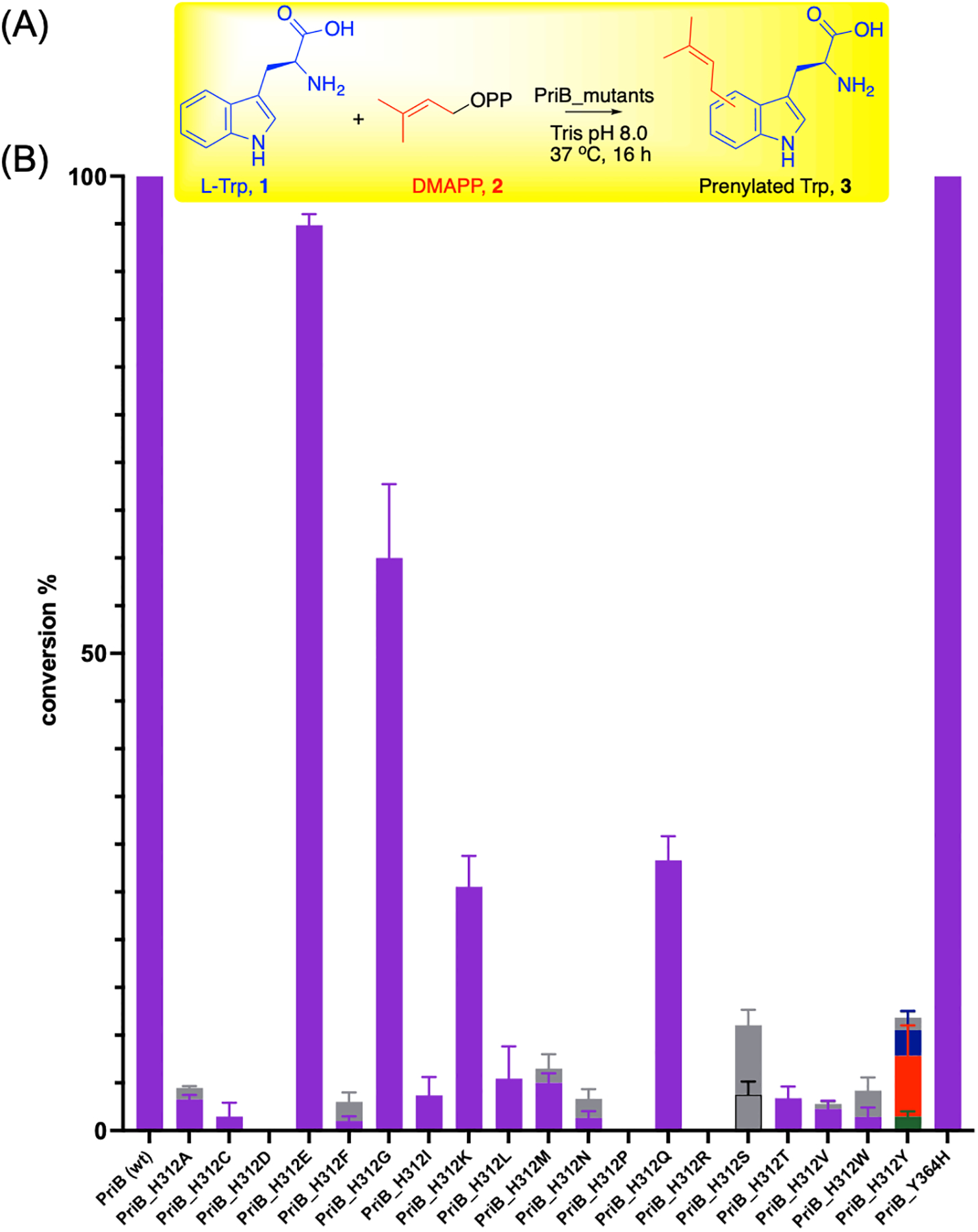
Role of PriB His312 in the C6 catalysis. (A) General reaction scheme of PriB mutants screening with 0.5 mM l-Trp (1) and 1 mM DMAPP (2) and 8.3 μM enzyme in Tris 50 mM pH (8.0) incubated at 37 °C for 16 h. (B) Product yields of the conversion of PriB variants monitored via HPLC A280. All reactions were carried out in triplicates and positive reactions were confirmed by high-resolution mass spectrometry (HR-MS). Peaks were assigned based on HPLC retention times represented in Figure S7. Purple, 3a; green, 3b; orange, 3c; blue, 3d; gray, monoprenylated tryptophans that had different retention times or could not be unambiguiously assigned.
Table 1.
Kinetic parameters of selected wild-type and mutant C6 IPTs using varying concentrations (5 × 10−2 − 5 mM) of l-Trp (1) and 2 mM DMAPP (2).
| l-Trp (1) | |||
|---|---|---|---|
| Enzyme | Km (mM) | kcat (min−1) | kcat/Km (mM−1 min−1) |
| PriB_wt | (42.9 ± 6.9) × 10−2 | 19.8 ± 0.8 | 46.2 |
| PriB_H312E | (94.7 ± 14.9) × 10−2 | (39.3 ± 3.3) × 10−2 | 41.5 × 10−2 |
| PriB_H312G | (26.8 ± 2.2) × 10−2 | (5.0 ± 0.1) × 10−2 | 18.7 × 10−2 |
| PriB_H312K | (91.2 ± 8.4) × 10−2 | (5.0 ± 0.2) × 10−2 | 5.5 × 10−2 |
| PriB_H312Q | (58.7 ± 4.8) × 10−2 | (9.0 ± 0.2) × 10−2 | 15.3 × 10−2 |
| PriB_H312Y | (26.4 ± 3.2) × 10−2 | (2.8 ± 0.8) × 10−2 | 10.6 × 10−2 |
| PriB_Y364H | (65.4 ± 6.5) × 10−2 | 9.4 ± 0.3 | 14.4 |
| 6DMATSmo_wt | (33.1 ± 2.5) × 10−2 | 5.5 ± 0.1 | 16.6 |
| 6DMATSmo_Y277H | (38.2 ± 0.1) × 10−2 | (6.5 ± 0.2) × 10−2 | 17.0 × 10−2 |
| 6DMATSmo_H329Y | (26.7 ± 1.6) × 10−2 | (4.7 ± 0.07) × 10−2 | 17.6 × 10−2 |
| 6DMATSmo_Y277H_H329Y | 1.0 ± 0.1 | (10.9 ± 0.2) × 10−2 | 10.9 × 10−2 |
Mutagenesis-based alteration of the PriB prenylation regiospecificity.
As mentioned before, the His312 replaces the conserved Tyr present in other non-C6 IPTs. In contrast to other His312 mutants, analysis of the HPLC-MS chromatograms of the reaction containing PriB_H312Y show the presence of multiple monoprenylated products different than that of wild-type (Figure 3B, Figure S7). Michaelis-Menten kinetics of PriB_H312Y revealed Km to be 26.4 × 10−2 mM and kcat to be 2.8 × 10−2 min−1 with catalytic efficiency > 400-fold lower than that of the wild-type (Table 1, Figure S8). To investigate the structures of the PriB_H312Y-prenylated products, a large-scale reaction containing 0.8 mM l-Trp, 1.6 mM DMAPP, 24 μM PriB_H312Y in Tris 50 mM pH (8.0) was incubated at 37 °C for 16 h. Preparative HPLC led to the purification of three monoprenylated products 3b–3d as indicated by the HPLC-HRMS (Table S2). The signals of 1H NMR spectra for all three compounds showed δH = 1.51–1.81 with HMBC correlations to a quaternary carbon δC = 131.9 – 134.9 and was indicative of a gem dimethyl. A methine carbon δC = 119.7–122.2 attached to a H with δH = 5.04 – 5.42 shows a COSY correlation to methylene hydrogens at C1’. This was consistent with the attachment of a dimethylallyl group indicating a normal prenylation (C1’) reaction. Based on the cumulative 1-and 2D NMR spectroscopic data, 3b–3d were confirmed to be prenylated at the C3, C7 and N1 positions, respectively (Figures S9–S11, Tables S3–S4, Supporting Mass and NMR Spectral data). The C3 prenylated Trp 3b, formed a bond between C2 and N10 and a subsequent hexahydropyrrolo-[2,3-b]indole structure (Figure 4B) typical of other C3 prenylated indoles.[19,37] The 2S/3R relative configuration of 3b was established by1H-1H NOESY correlation between H2 and H1’ (Figure S9).
Figure 4.
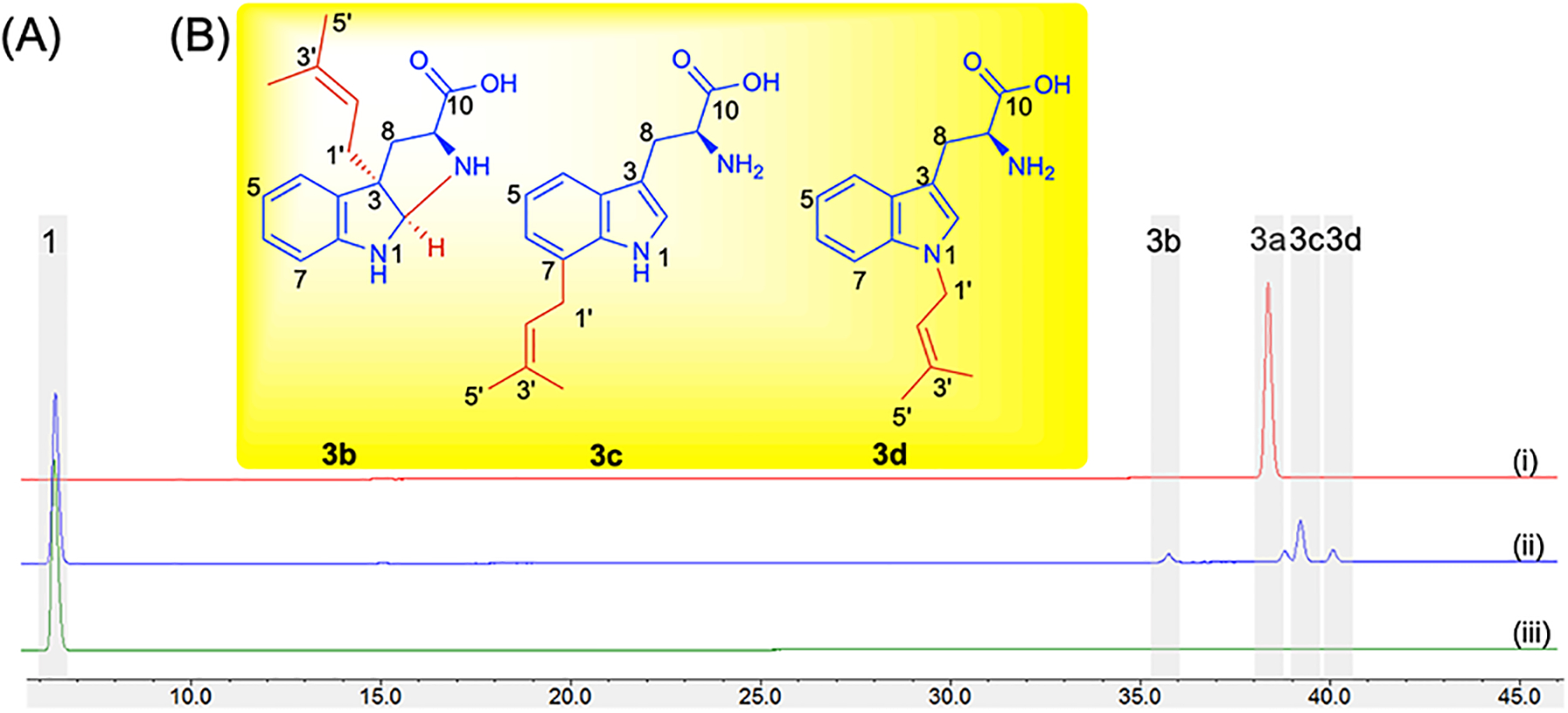
Effect of mutation of PriB His312 to Tyr. (A) HPLC chromatograms of analytical enzymatic reactions containing 0.5 mM l-Trp (1) and 1 mM DMAPP (2) and 8.3 μM wild-type PriB (i), PriB_H312Y (ii) and no enzyme (iii) in Tris 50 mM pH (8.0). Reactions were incubated at 37 °C for 16 h. (B) Structures of prenylated tryptophans 3b, 3c and 3d synthesized by PriB_H312Y. 1- and 2D data correlations are provided in Figures S9–S11, Tables S3–S4 and Supporting Mass and NMR spectral data. One peak was not elucidated due to presence of a mixture of insufficient quantity.
Role of His residue in other C6 IPTs.
To determine whether His plays the similar catalytic role in other C6 IPTs, we analyzed the three-dimensional structures of other regiospecific C6 IPT enzymes functionally characterized so far. We compared the X-ray crystal structures of IptA[38] (PDB ID 7W8V) and 6DMATSmo[26] (PDB ID 6ZRX) as well as the homology models of 6DMATSsa[39] and 6DMATSsv[39] with the crystal structure of PriB. Structural alignment of all five C6 IPTs enzymes reveals a conserved His at the same position (His294, IptA; His284, 6DMATSsa; His287, 6DMATSsv) except for 6DMATSmo[26] which was replaced with Tyr277 (Figure 5, Figures S12–S13). Interestingly, a different residue, His329 is located at less than 4 Å away was close to the Trp C6 position (Figures S13E–S14) and was shown to act as the catalytic residue for 6DMATSmo.[26] The docking position of 1 in 6DMATSmo in the enzyme active site is shifted compared to that in other C6 IPTs (Figure S2, Figure S14) consistent with the reported data.[26] This suggests that the His residue in all C6 IPTs currently characterized is responsible for the regiospecificity at the C6 position. To confirm our hypothesis, the coding nucleic acid sequences for all four C6 IPT enzymes were synthesized (Figures S15–S18), cloned and transformed into E. coli. The His residue in the four enzymes were mutated to Tyr using site-directed mutagenesis. In addition, we created the 6DMATSmo_Y277H as well as the double mutant 6DMATSmo_Y277H_H329Y which mimics the environment surrounding 1 present in the active site of other C6 IPTs. The corresponding wild-type and mutant enzymes were overproduced and purified and proper folding was confirmed by circular dichroism (Figures S19–S20). Enzymatic reactions were carried out in Tris 50 mM (pH 8.0) containing 0.5 mM Trp, 1 mM DMAPP and 18 μM protein and incubated at 37 °C for 16 h. HPLC-MS of the quenched reactions showed complete/partial loss of activity and regiospecificity with little or no C6 derivative (Figure 6A). IptA_H294Y and 6DMATSsa_H284Y lost almost complete activity, while 6DMATSsv_H287Y and 6DMATSmo_H329Y lost 65–70% of activities compared to that of their corresponding wild-types (Figure 6B). Most importantly, mutation of His to Tyr in 6DMATSsv and 6DMATSmo seem to have lost regiospecificity as evidenced by the formation of multiple products similar to the case with PriB_H312Y (Figure 6A). The 6DMATSmo_H329Y result is consistent with previously reported loss of ~80% activity.[26]. The two mutants, 6DMATSmo_Y277H and 6DMATSmo_Y277H_H329Y lost 30–40% of the activity compared to that of the wild-type. Michalis-Menten kinetics for 6DMATSmo mutants showed that the catalytic efficiency of each of Tyr277 and His329 mutants was reduced by ~ 100-fold (Table 1, Figure S21). The catalytic efficiency of the 6DMATSmo_Y277H_H329Y, which mimics the key catalytic residues of other C6 IPTs, was 150-fold lower than that of the wild-type. Even though the 6DMATSmo double mutant simulates the key catalytic residues of other C6 IPTs, the decrease in activity and loss of regiospecificity is most probably caused by the shift in the 1 position in 6DMATSmo active site.[26] The residue that aligns with 6DMATSmo_His329 in PriB is Tyr364 (Figure 4A, Figure S13, Table S5). To determine the significance of PriB_Tyr364, PriB_Y364H was generated and the corresponding protein was overproduced and proper folding was confirmed by circular dichroism (Figure S5, Figure S6D, Table S1). The purified PriB_Y364H showed near-quantitative conversion (Figure 3B, Figure S7) and its catalytic efficiency was three-fold lower than that of wild-type PriB (Table 1, Figure S8G). This suggests that Tyr364 binds to the indole ring by π-stacking and the activity can be partially retained when replaced with another aromatic amino acid residue.
Figure 5.
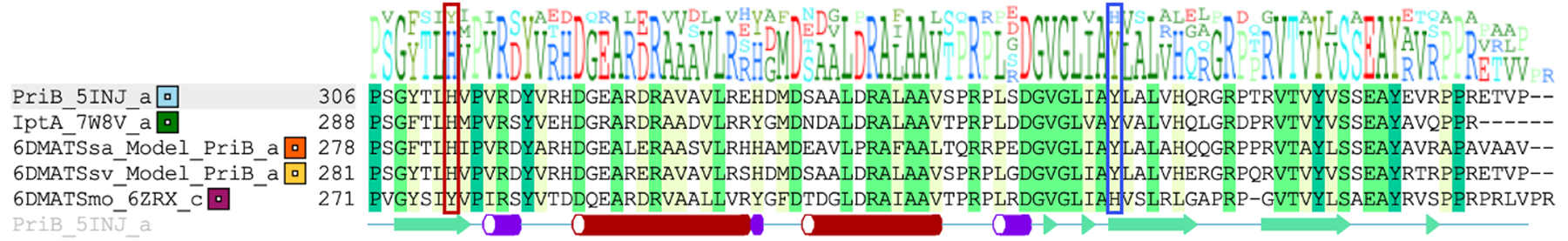
Structure-based alignment of PriB with other C6 IPTs. Red square highlighting residues that align with PriB His312 while blue square highlights residues that align with Tyr364. Names of protein and PDB ID are stated.
Figure 6.
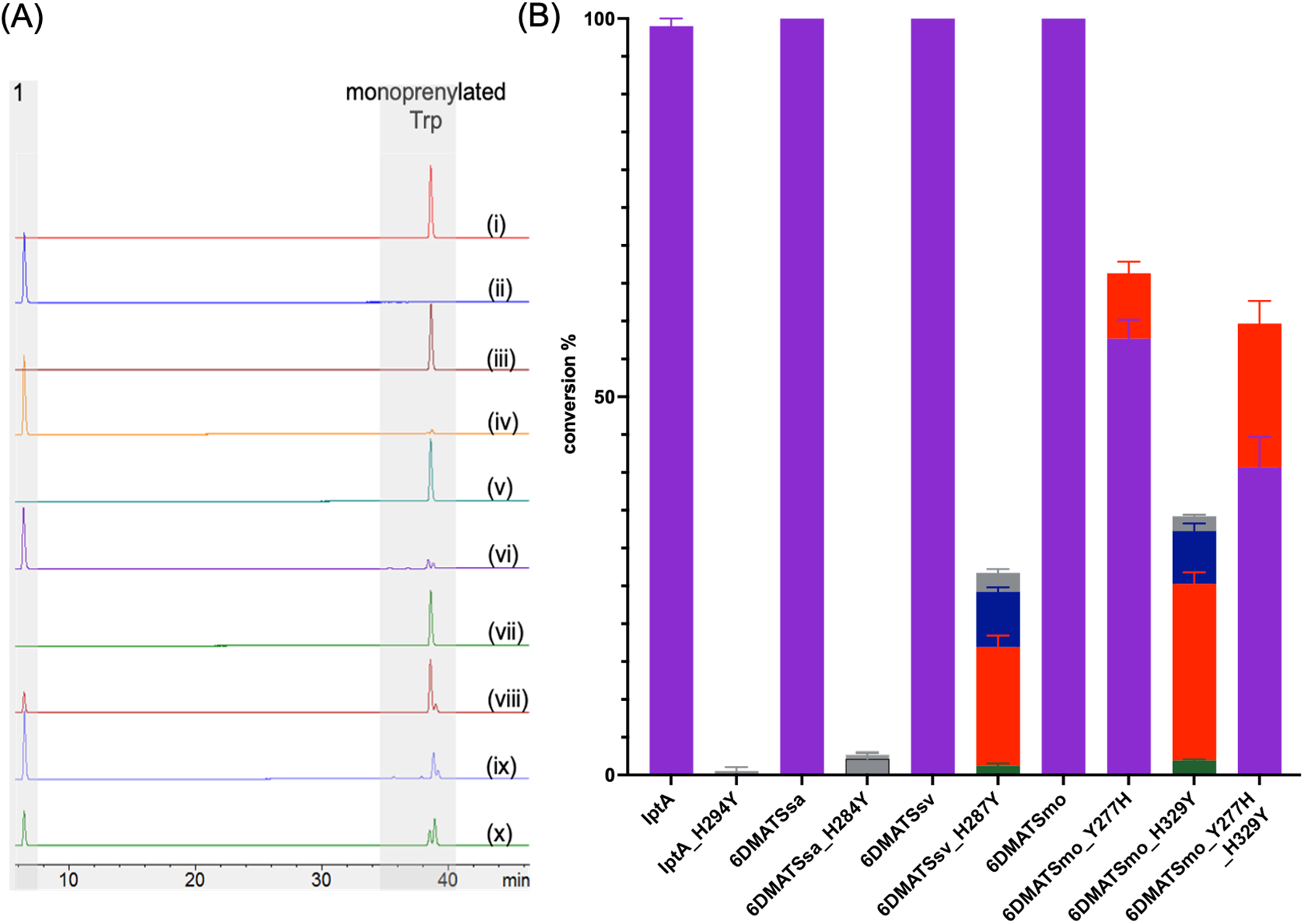
The significance of His residue in the catalysis of C6 PTs. (B) HPLC chromatograms of enzymatic reactions containing 0.5 mM l-Trp (1) and 1 mM DMAPP (2) 18 μM wild-type IptA (i), IptA_H294Y (ii), wild-type 6DMATSsa (iii), 6DMATSsa_H284Y (iv), wild-type 6DMATSsv (v), 6DMATSsv_H287Y (vi), wild-type 6DMATSmo (vii), 6DMATSmo_Y277H (viii), 6DMATSmo_H329Y (ix), 6DMATSmo_Y277H_H329Y (x). (C) Product yields of the conversion of wild-type and mutant C6 IPTs monitored via HPLC at A280. Peaks were assigned based on HPLC retention times. Purple, 3a; green, 3b; orange, 3c; blue, 3d; gray, monoprenylated tryptophans that had different retention times or could not be unambiguiously assigned. All reactions were carried out in triplicates and positive reactions were confirmed by high-resolution mass spectrometry (HRMS).
Several residues make close contacts with the indole moiety in IPTs active site. Of particular interest, are the two residues that correspond to His312 (position 1) and Tyr364 (position 2) in PriB (Figure 2B). These positions are occupied by aromatic amino acids in different wild-type enzymes (Table S5). The conserved Tyr in position 2 seems to play a role in binding the acceptor, and mutation to Ala led to a four-fold reduction in DMATS1 activity.[28] Only in IPTs that catalyze C6 prenylation, there is a strict need for one of those two residues to be His to act as a catalytic base. A His in C6 IPTs needs to be perfectly positioned close to the indole C6 carbon. The residue pair His312/Tyr364 are conserved in C6 IPTs except for 6DMATSmo where they are switched (Tyr277/His329, Table S5). In addition to the above mentioned regiospecific C6 IPTs, other IPTs were reported to catalyze the prenylation at multiple indole positions including C6. AtmD was found to synthesize mono- and diprenyled indoles at C4, C5 and/or C6 positions.[40] PaxD[41] and JanD[42,43] catalyze the diprenylation of paxilline and the janthitremane shearinine alkaloids, respectively at both C5 and C6 indole positions. Interestingly, the three enzymes also have His in one the two positions (Figure S22 and Table S5) consistent with the significance of the presence of His for the efficient indole C6 catalysis. His occupies position 1 in PriB, IptA, 6DMATSsa and 6DMATSsv and position 2 in 6DMATSmo as well as the non-regiospecific IPTs, AtmD, JanD and PaxD (Table S5). The use of His in different positions for homologous IPT enzymes to achieve the same C6 regiospecificity could be attributed to the change of the indole orientation in the active site or participation of the active site waters.
When PriB_His312 was replaced with Tyr, activation of the indole C6 was shifted to the more activated indole positions, C3, N1, as well as C7. The presence of Tyr residue in place of His enables binding of the indole moiety via π-stacking, yet does not act as a base in the reaction. In the absence of a catalytic base, prenylation is shifted to the C3 and N1 and the formation of 3b and 3d, respectively. These indole positions are more activated towards electrophilic attack due to their higher electron density.[4] In the three C6 IPTs that have been structurally characterized, the distances between each of 2-C1’ as well as His-basic nitrogen is at a distance less than 4 Å from the indole C6 or C7 (Figures S2, S23). Thus, the absence of a basic residue in PriB_H312Y enables the activation of the indole C7 and formation of 3c. This is consistent with earlier studies that showed that C7 is the second favorable position for C6 wild-type IPTs.[25,44] It is also possible that PriB_H312Y catalyzes its reaction via active-site water similar to the Gln residue and 5DMATSsc.[26] Based on the previous data, we propose a general acid-base mechanism that relies on His side chain participation for C6 indole functionalization (Figure 7). The release of the pyrophosphate moiety from 2 generates an allylic carbocation 2a. A strictly conserved glutamate residue[24,27,28] in IPTs (Glu94 in PriB) positioned at close proximity to N1 forms a hydrogen bond with the indole N–H. This hydrogen bond increases the indole ring reactivity towards electrophilic substitution by 2a at the 1 C6 position.[27] The σ-complex intermediate 1a is stabilized via charge complementation. A His, (His312 in PriB) strategically positioned at close proximity to the indole substrate, abstracts H6 from the σ-complex 1a and subsequently regenerates aromaticity (3a). Indeed, previous work has shown IPTs catalysis proceeds via a dissociative SN1 mechanism where the reaction is initiated by the ionization of the prenyl donor to form a prenyl/diphosphate ion pair.[45] This is followed by a C–C bond formation between the indole carbon and the prenyl carbocation resulting in an arenium ion intermediate. One would not preclude an associative SN2 mechanism for the mutants. This would proceed via a nucleophilic attack by the Trp indole onto the donor with subsequent release of the diphosphate moiety and formation of the arenium ion intermediate. In both mechanisms, the arenium ion intermediate is deprotonated and rearomatized to form the indole ring.
Figure 7.
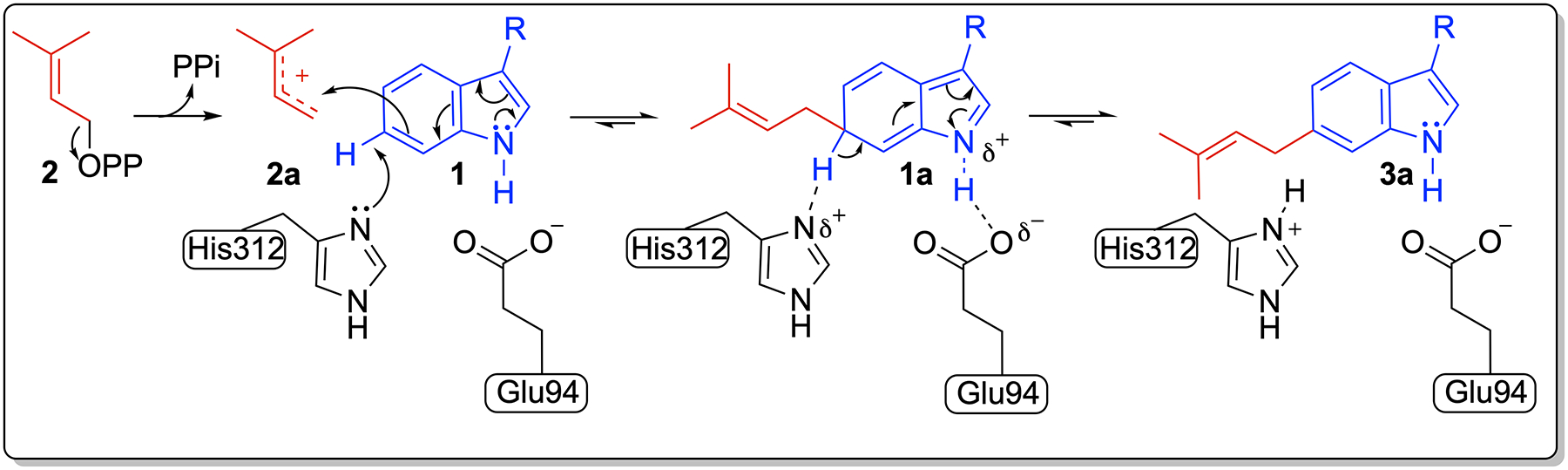
Proposed mechanism for the formation of PriB-mediated C-6 prenylation. Similar mechanism is proposed for other C6-indole prenyltransferases using conserved His and Glu residues.
Conclusion
In this work, we verified the catalytic role of PriB His312 in the C6 indole regiospecificity which was previously proposed.[25] We show that His is crucial in other C6 IPTs as well (IptA, 6DMATSsa and 6DMATSsv). The data reported here confirms the key residue that catalyzes the activation of indole C6 position in several C6 IPTs. We show that His for all characterized regiospecific C6 IPTs is necessary for the efficient functionalization of a difficult-to-access indole position in align with what was reported earlier with 6DMATSmo.[26] Our work shows that structure-guided mutagenesis of His in this region alters the regiospecificity of C6 enzymes. The regiospecificity of IPTs appears to be determined by various factors; i) presence of a catalytic residue at an appropriate distance from the targeted indole position as in the case with Glu and N1[28] and C2,[29] Lys and C4,[27] Gln and C5[26] and His in C6 as characterized in this study and others.[26] The function of this residue is to abstract proton, restore aromaticity and reduce activation energy; ii) distance of the catalytic residue from the position to be prenylated; iii) relative orientation and proximity between C1’ or C3’ of the donor and the targeted carbon position at the acceptor within the IPT binding pocket; iv) potential rearrangement as in the case of CdpNPT and FtmPT1;[30,32] v) nature and size of the substrates that can cause steric hindrance and lead to subsequent shift in the active site[19,37]; vi) potential participation of active site waters as proposed with Gln and 5DMATSsc.[26] This work suggests that analysis of the active site of an uncharacterized IPT and presence/absence of His at these two positions can determine its ability to catalyze C6 indole prenylation. Due to their relaxed substrate flexibility, IPTs have been used to achieve late-stage functionalization showing strong potential in the development of new compounds.[18–20] The current work contributes to previous engineering efforts towards changing the IPTs donor,[34,38,46,47] acceptor[48] and regiospecificities[26,29] and is another example of how structure-guided mutagenesis can lead functional characterization and reveal catalytic mechanisms of enzymes.[49] This study broadens the current knowledge of IPTs regiospecificity and promiscuity and provide valuable guidance for the engineering of efficient biocatalysts for the functionalization of C–H bond in indoles and the development of key compounds through rationale design.
Experimental Section
Parental plasmid, protein overproduction and purification
PriB plasmid was graciously provided by Professor Jon Thorson at the University of Kentucky.[25] The genes encoding IptA, 6-DMATSsa, 6-DMATSsv, and 6-DMATSmo were obtained from synthetic nucleic acids (Twist Biosciences). Synthetic genes were digested with NdeI and HindIII restriction enzymes (New England Biolab) and the corresponding fragments were cloned pET28a (Novagen), transformed into E. coli DH5α chemically competent cells. Plasmids were purified using Monarch® Plasmid Miniprep kit (New England Biolabs) and sequence-verified. The validated expression plasmids were subsequently transformed into E. coli BL21 (DE3) competent cells. Production strains for mutants were constructed in a similar fashion. All studies employed the corresponding N-terminal-His6 fusion proteins. DNA quality and quantity were assessed using gel electrophoresis and Nanodrop (Thermo Scientific NanoDrop 2000 Spectrophotometer).
For protein production, 1L of LB broth (Becton, Dickinson and Company) supplemented with kanamycin (80 μg mL−1) was inoculated with 0.1% (v/v) of overnight corresponding plasmids in E. coli BL21 (DE3) seed culture and grown at 37 °C with shaking (250 rpm). Cultures were induced at OD600 of ~0.6–0.8 with isopropyl-β-D-thiogalactopyranoside (0.5 mM final concentration) and allowed to grow for an additional 16 h at 21 °C. Cells were harvested by centrifugation and stored in lysis buffer (10 mM imidazole, 50 mM sodium monobasic phosphate, and 300 mM NaCl, pH 8.0) at −80 °C until used. All subsequent steps were carried out on ice. Cells were allowed to thaw and were subsequently lysed by sonication (Virtis VirSonic 475 with a microtip, 100W, 10 × 10 sec pulses, 20 sec between pulses). Insoluble debris was removed by centrifugation at 20,000 g for 1 h. The supernatant was collected and filtered using 0.22 μm filters, and the desired N-His6 fusion proteins were purified via HiTrap nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography using standard protocols and Bio-Rad NGS chromatography system. Buffer exchange of each sample was performed using a PD-10 column (GE Healthcare) eluted with 50 mM Tris and 100 mM NaCl, pH 8.0, to yield 5 – 11 mg L−1 of proteins. Fractions were collected and concentrated using Amicon Ultra Centrifuge filters 30,000 MWCO (EMD Millipore) and stored in 50 mM Tris, 100 mM NaCl and 10% glycerol (pH 8.0) at −80 °C. Protein concentrations were determined by Bradford assay (Bio-Rad) using bovine serum albumin as a standard. Purity and presence of proteins were confirmed by SDS-PAGE gel (Blot™ 4–12% Bis-Tris Plus, Invitrogen) electrophoresis. The N-His6 fusion proteins were used for all studies. Production of prenyltransferase mutants for the overall study followed the same protocol.
Procedure for the synthesis of dimethylallyl pyrophosphate (DMAPP, 2)
In a 25 mL round-bottomed flask equipped with a magnetic bar containing dimethylallyl chloride 0.104 g (1.0 mmol, 1.0 eq) dissolved in dry acetonitrile 1.178 g (2 mL) and Tris(tetrabutylammonium) hydrogen pyrophosphate (TBAPP) (1.3 mmol, 1.3 eq) were allowed to stir at rt for 2–3 h. The reaction was monitored by TLC in isopropanol/NH4OH/H2O in 7:2:1 with phosphomolybdic acid stain solution. After reaction completion, the solvent was removed under reduced pressure and the crude residue was purified on silica gel (60–120 mesh) using isopropanol/NH4OH/H2O in 7:2:1 ratio as the mobile phase. The combined fractions containing the Tris(tetrabutylammonium) (TBA) salt of desired products were concentrated under reduced pressure, passed through Dowex-50WX8 ion exchange column (1 × 8 cm) pre-equilibrated with concentrated NH4OH:H2O (3:1) and flushed with buffer (0.025 M NH4HCO3) until the pH was 8.0. The ammonium salt of 2 (DMAPP.3NH4+) was eluted with two column volumes of 0.025 M NH4HCO3 buffer and lyophilized until dryness.
white solid (120 mg, 40% yield). 1H NMR (400 MHz, D2O) δ 5.33 (s, 1H), 4.32 (t, J = 6.4 Hz, 2H), 1.64 (s, 3H), 1.60 (s, 3H). 31P NMR (162 MHz, D2O) δ −6.85 (d, J = 21.3 Hz), −10.39 (d, J = 21.7 Hz). HRMS (TOF) m/z [M – H]− Calcd for C5H11O7P2 244.9996 found 244.9941.
Enzymatic analytical assays
Standard in vitro assays were conducted in 1.5 mL tubes in a volume of 100 μL of Tris 50 mM (pH 8.0) containing a final concentration of 0.5 mM l-Trp and 1 mM DMAPP. After preincubation of the reaction mixture at 37 °C for 10 min, the reactions were initiated with the addition of 8.3 μM enzyme and allowed to proceed for 16 h (unless otherwise noted) at 37 °C. Positive and negative reactions with and without the wild-type PriB, respectively were performed under the same conditions. Reactions were quenched by the addition of 100 μL of MeOH and mixing followed by centrifugation (22 000 g, 15 min, 4 °C) to remove precipitated proteins. The supernatants were analyzed by high-performance liquid chromatography (HPLC) using method A (see Supporting Methods and Materials) to calculate the conversion rate based on the area of the substrate and the prenylated product peaks.
Construction of mutants and purification of corresponding proteins
Point mutations were generated by PCR and the Q5 Site Directed Mutagenesis Kit (New England Biolabs) using the appropriate primer pair (Table S1, engineered codons are underlined) and plasmids carrying sequence of wild-type enzymes as the template. PCR for each reaction included 20 ng template DNA, 1× of Q5 Hot Start High Fidelity master mix, and 0.5 μM of each primer. The PCR program included an initial hold at 98 °C for 30 s followed by 23 cycles of 98 °C for 10 s, annealing temperature ranging from 60 to 72 °C for 20 s, 72 °C for 3 min 20 s, and 1 cycle of 72 °C for 5 min. All amplicons were confirmed and quantified using gel electrophoresis and a ND-1000 Spectrophotometer (Thermo). The PCR reaction was treated with kinase, ligase & DpnI (KLD) according to the manufacturer’s protocol. The mutated plasmids were transformed to E. coli DH5α chemically competent cells (New England Biolabs) and plated on LB medium containing agar supplemented with kanamycin 80 μg mL−1 overnight at 37 °C. Single colonies were used to inoculate 3 mL cultures of LB medium supplemented with kanamycin and incubated for 16 h at 37 °C and 225 rpm. Plasmid DNA was isolated from each culture using the Monarch® Plasmid Miniprep kit according to the manufacturer’s instructions. The isolated plasmid DNA for each desired mutant was confirmed by sequencing and subsequently transformed into chemically competent E. coli BL21 (DE3) according to the manufacturer’s instructions, plated on LB medium containing agar supplemented with kanamycin, and incubated at 37 °C. Isolated single colonies were used to inoculate LB medium supplemented with kanamycin and incubated for 16 h at 37 °C and 225 rpm. A glycerol stock [with a final concentration of 15% glycerol (v/v)] of each culture was prepared and frozen at −80 °C. Protein overproduction and purification followed the same protocol as that for wild-type proteins (described in the previous section). To confirm proper folding of the purified proteins, the CD spectra of all wild-type and mutant enzymes used in this study in their substrate-free state were measured using a Jasco J1500 spectropolarimeter at 25 °C using quartz cells (Alpha Nanotech) with a pathlength of 0.1 cm. The enzymes were dissolved at a concentration of approximately 200 μg mL−1 in 10 mM sodium phosphate buffer and 100 mM sodium fluoride. Low noise CD spectra were measured by averaging 5 scans, and the final spectra were corrected by subtracting the corresponding baseline with the buffer.
Determination of kinetics parameters
Reaction mixtures consisted of 50 mM Tris (pH 8.0) containing almost saturating DMAPP (2 mM) with variable l-Trp (5 × 10−2 − 5 mM). Reactions were pre-heated to 37 °C in the absence of substrate and initiated by substrate addition. The reactions were performed at 37 °C with 184.6 × 10−3 − 37 μM enzyme for 240 min and analyzed under initial velocity conditions. Products formation were determined using HPLC and method B (see Supporting Material and Methods). Each data point represents a minimum of three replicate end-point assays; kinetic constants were obtained by nonlinear regression fit to the Michaelis-Menten equation using GraphPad Prism version 9.5.0.
Synthesis, isolation and purification of modified Trp analogs 3b–3d
Reactions containing 80 mL of Tris 50mM (pH 8.0) supplemented with l-Trp 1 (0.8 mM), DMAPP 2 (1.6 mM) and 24 μM PriB_H312Y were prepared. Reactions were incubated at 37 °C for 16 h followed by freezing and lyophilization. To calculate the conversion percentage, a 100 μL sample from the reaction was taken and quenched by adding equivalent amount of methanol and centrifuged at 20,000 × g for 15 min and analyzed using methods A and B and comparing the integrations of the HPLC peaks corresponding to product and starting material 1. Crude reaction mixtures were subsequently redissolved in DMSO and purified via semi-preparative HPLC method C followed by an additional purification method using HPLC method D. The fractions corresponding to the product were combined and dried under reduced pressure. Purity was confirmed via HPLC using methods B and HR-ESI-MS was determined using HPLC method E. Finally, the compounds were structurally elucidated by 1- and 2D NMR and HR-ESI-MS (Supporting MS and NMR data).
(2S)-3a-(3-methylbut-2-en-1-yl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole-2-carboxylic acid 3b. white solid (3 mg, 9% yield). 1H NMR and 13C NMR see Table S3. HRMS (TOF) m/z [M + H]+ Calcd for C16H21N2O2 273.1598 found 273.1589.
(S)-2-amino-3-(7-(3-methylbut-2-en-1-yl)-1H-indol-3-yl)propanoic acid 3c. white solid (9 mg, 30% yield). 1H NMR and 13C NMR see Table S4. HRMS (TOF) m/z [M + H]+ Calcd for C16H21N2O2 273.1598 found 273.1589.
1-(3-methylbut-2-en-1-yl)-l-tryptophan 3d. white solid (3 mg, 9% yield). 1H NMR and 13C NMR see Table S4. HRMS (TOF) m/z [M + H]+ Calcd for C16H21N2O2 273.1598 found 273.1591.
Supplementary Material
Acknowledgements
This work was supported by NIH grant R03AI168826 (S.I.E) and Chapman University School of Pharmacy (CUSP). We thank Dr. Innokentiy Maslennikov and the CUSP Nuclear Magnetic Resonance facility for NMR measurements, Basir Syed for technical assistance and the CUSP core lab facility.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Norwood VM, Huigens RW, Chembiochem 2019, 20, 2273–2297. [DOI] [PubMed] [Google Scholar]
- [2].Li T, Xu H, Mini Rev. Med. Chem 2022, 22, 2702–2725. [DOI] [PubMed] [Google Scholar]
- [3].Wan Y, Li Y, Yan C, Yan M, Tang Z, Eur. J. Med. Chem 2019, 183, 111691. [DOI] [PubMed] [Google Scholar]
- [4].Hu J-J, He P-Y, Li Y-M, J. Pept. Sci 2021, 27, e3286. [DOI] [PubMed] [Google Scholar]
- [5].Leitch JA, Bhonoah Y, Frost CG, ACS Catal. 2017, 7, 5618–5627. [Google Scholar]
- [6].Feng Y, Holte D, Zoller J, Umemiya S, Simke LR, Baran PS, J. Am. Chem. Soc 2015, 137, 10160–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang G, Lindovska P, Zhu D, Kim J, Wang P, Tang R-Y, Movassaghi M, Yu J-Q, J. Am. Chem. Soc 2014, 136, 10807–10813. [DOI] [PubMed] [Google Scholar]
- [8].Yang Y, Li R, Zhao Y, Zhao D, Shi Z, J. Am. Chem. Soc 2016, 138, 8734–8737. [DOI] [PubMed] [Google Scholar]
- [9].Liu H, Zheng C, You S-L, J. Org. Chem 2014, 79, 1047–1054. [DOI] [PubMed] [Google Scholar]
- [10].Huang W-J, Ma Y-Y, Liu L-X, Wu B, Jiang G-F, Zhou Y-G, Org. Lett 2021, 23, 2393–2398. [DOI] [PubMed] [Google Scholar]
- [11].Yan J, Zhang Z, Chen M, Lin Z, Sun J, ChemCatChem 2020, 12, 5053–5057. [Google Scholar]
- [12].Devine PN, Howard RM, Kumar R, Thompson MP, Truppo MD, Turner NJ, Nat. Rev. Chem 2018, 2, 409–421. [Google Scholar]
- [13].Hanefeld U, Hollmann F, Paul CE, Chem. Soc. Rev 2022, 51, 594–627. [DOI] [PubMed] [Google Scholar]
- [14].Miller DC, Athavale SV, Arnold FH, Nat. Synth 2022, 1, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Winkler CK, Schrittwieser JH, Kroutil W, ACS Cent. Sci 2021, 7, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Walsh CT, ACS Chem. Biol 2014, 9, 2718–2728. [DOI] [PubMed] [Google Scholar]
- [17].Mori T, J. Nat. Med 2020, 74, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mupparapu N, Lin Y-HC, Kim TH, Elshahawi SI, Chem. Eur. J 2021, 27, 4176–4182. [DOI] [PubMed] [Google Scholar]
- [19].Mupparapu N, Brewster L, Ostrom KF, Elshahawi SI, Chem. Eur. J 2022, 28, e202104614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Colombano A, Dalponte L, Dall’Angelo S, Clemente C, Idress M, Ghazal A, Houssen WE, Angew. Chem. Int. Ed Engl 2023, 62, e202215979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kelly SP, Shende VV, Flynn AR, Dan Q, Ye Y, Smith JL, Tsukamoto S, Sigman MS, Sherman DH, J. Am. Chem. Soc 2022, 144, 19326–19336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eggbauer B, Schrittwieser JH, Kerschbaumer B, Macheroux P, Kroutil W, Chembiochem 2022, 23, e202200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leveson-Gower RB, Roelfes G, ChemCatChem 2022, 14, e202200636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tanner ME, Nat. Prod. Rep 2015, 32, 88–101. [DOI] [PubMed] [Google Scholar]
- [25].Elshahawi SI, Cao H, Shaaban KA, Ponomareva LV, Subramanian T, Farman ML, Spielmann HP, Phillips GN, Thorson JS, Singh S, Nat. Chem. Biol 2017, 13, 366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ostertag E, Zheng L, Broger K, Stehle T, Li S-M, Zocher G, J. Mol. Biol 2021, 433, 166726. [DOI] [PubMed] [Google Scholar]
- [27].Metzger U, Schall C, Zocher G, Unsöld I, Stec E, Li S-M, Heide L, Stehle T, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eaton SA, Ronnebaum TA, Roose BW, Christianson DW, Biochemistry 2022, 61, 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jost M, Zocher G, Tarcz S, Matuschek M, Xie X, Li S-M, Stehle T, J. Am. Chem. Soc 2010, 132, 17849–17858. [DOI] [PubMed] [Google Scholar]
- [30].Mahmoodi N, Tanner ME, Chembiochem 2013, 14, 2029–2037. [DOI] [PubMed] [Google Scholar]
- [31].Chen J, Morita H, Kato R, Noguchi H, Sugio S, Abe I, Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun 2012, 68, 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ruan H-L, Yin W-B, Wu J-Z, Li S-M, Chembiochem 2008, 9, 1044–1047. [DOI] [PubMed] [Google Scholar]
- [33].Yu X, Zocher G, Xie X, Liebhold M, Schütz S, Stehle T, Li S-M, Chem. Biol 2013, 20, 1492–1501. [DOI] [PubMed] [Google Scholar]
- [34].Mori T, Zhang L, Awakawa T, Hoshino S, Okada M, Morita H, Abe I, Nat. Commun 2016, 7, 10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD, J. Org. Chem 1986, 51, 4768–4779. [Google Scholar]
- [36].Ribeiro AJM, Tyzack JD, Borkakoti N, Holliday GL, Thornton JM, J. Biol. Chem 2020, 295, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liebhold M, Xie X, Li S-M, Org. Lett 2013, 15, 3062–3065. [DOI] [PubMed] [Google Scholar]
- [38].Suemune H, Nishimura D, Mizutani K, Sato Y, Hino T, Takagi H, Shiozaki-Sato Y, Takahashi S, Nagano S, Biochem. Biophys. Res. Commun 2022, 593, 144–150. [DOI] [PubMed] [Google Scholar]
- [39].Winkelblech J, Li S-M, Chembiochem 2014, 15, 1030–1039. [DOI] [PubMed] [Google Scholar]
- [40].Liu C, Minami A, Noike M, Toshima H, Oikawa H, Dairi T, Appl. Environ. Microbiol 2013, 79, 7298–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu C, Noike M, Minami A, Oikawa H, Dairi T, Appl. Microbiol. Biotechnol 2014, 98, 199–206. [DOI] [PubMed] [Google Scholar]
- [42].Nicholson MJ, Eaton CJ, Stärkel C, Tapper BA, Cox MP, Scott B, Toxins 2015, 7, 2701–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu C, Minami A, Dairi T, Gomi K, Scott B, Oikawa H, Org. Lett 2016, 18, 5026–5029. [DOI] [PubMed] [Google Scholar]
- [44].Winkelblech J, Xie X, Li S-M, Org. Biomol. Chem 2016, 14, 9883–9895. [DOI] [PubMed] [Google Scholar]
- [45].Luk LYP, Tanner ME, J. Am. Chem. Soc 2009, 131, 13932–13933. [DOI] [PubMed] [Google Scholar]
- [46].Shen Y, Zhang L, Yang M, Shi T, Li Y, Li L, Yu Y, Deng H, Lin H-W, Zhou Y, ACS Chem. Biol 2023, 18, 123–133. [DOI] [PubMed] [Google Scholar]
- [47].Liao G, Mai P, Fan J, Zocher G, Stehle T, Li S-M, Org. Lett 2018, 20, 7201–7205. [DOI] [PubMed] [Google Scholar]
- [48].Fan A, Zocher G, Stec E, Stehle T, Li S-M, J. Biol. Chem 2015, 290, 1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Elshahawi SI, Ramelot TA, Seetharaman J, Chen J, Singh S, Yang Y, Pederson K, Kharel MK, Xiao R, Lew S, Yennamalli RM, Miller MD, Wang F, Tong L, Montelione GT, Kennedy MA, Bingman CA, Zhu H, Phillips GN, Thorson JS, ACS Chem. Biol 2014, 9, 2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.