

Abstract
Disruption of the transcriptional activity of the Hippo pathway members YAP1 and TAZ has become a major target for cancer treatment. However, detailed analysis of the effectiveness and networks affected by YAP1/TAZ transcriptional targeting is limited. In this study, we utilize TEAD inhibitor, an inhibitor of the binding of YAP1 and TAZ with their main transcriptional target TEAD in a mouse model of basal cell carcinoma, to unveil the consequences of YAP1/TAZ transcriptional blockage in cancer cells. Both TEAD inhibitor and YAP1/TAZ knockdown lead to reduced proliferation and increased differentiation of mouse basal cell carcinoma driven by oncogenic hedgehog-smoothened (SmoM2) activity. Although TEAD-transcriptional networks were essential to inactivate differentiation, this inactivation was found to be indirect and potentially mediated through the repression of KLF4 by SNAI2. By comparing the transcriptional effects of TEAD inhibition with those caused by YAP1/TAZ depletion, we determined YAP1/TAZ–TEAD–independent effects in cancer cells that impact STAT3 and NF-κB. Our results reveal the gene networks affected by targeting YAP1/TAZ–TEAD in basal cell carcinoma tumors and expose the potential pitfalls for targeting TEAD transcription in cancer.
INTRODUCTION
The Hippo pathway is one of the main signaling networks altered in human cancer (Sanchez-Vega et al., 2018), with genomic and signaling changes in Hippo components leading to activation and nuclear translocation of YAP1 and TAZ (WWTR1). YAP1 and TAZ are paralog proteins that interact with a wide range of cytoplasmic and nuclear transcriptional effectors, ultimately modulating the proliferative and differentiation status of cells in response to chemical and mechanical microenvironmental cues (Yu et al., 2015; Zanconato et al., 2016).
In skin, YAP1 and TAZ are activated in basal cell carcinoma (BCC) and squamous cell carcinoma (Zhang et al., 2011), and YAP1 and TAZ knockout reduces or prevents tumor formation in BCC and squamous cell carcinoma mouse models (Debaugnies et al., 2018; Maglic et al., 2018). This evidence indicates that disruption of YAP1 and TAZ signaling could have therapeutic benefits for BCC and squamous cell carcinoma treatment. One common YAP1 and TAZ downstream pathway involved in their oncogenic effects is the activation of TEAD, and efforts are underway to develop YAP1/TAZ–TEAD interaction inhibitors that could be used for cancer and other hyperproliferative diseases. However, a major challenge in studying the effectiveness of this approach is the lack of preclinical models to characterize the consequences of TEAD inhibition.
To circumvent some of the limitations to study TEAD inhibition, our group developed TEAD inhibitor (TEADi), a genetically encoded fluorescently traceable dominant negative protein that blocks nuclear interaction of TEAD with YAP1 and TAZ (Yuan et al., 2020). TEADi presents several advantages, including rapid inhibition of TEAD transcription and concomitant blockage of both YAP1 and TAZ without altering the structural or cytoplasmic functions of these proteins. TEADi can be used to dissect in more detail the consequences of TEAD blockage and could serve as a resource to differentiate TEAD-dependent and -independent effects, providing additional clues to suppress YAP1/TAZ activity in cancer.
In this study, we utilize TEADi in a mouse model of BCC driven by oncogenic hedgehog-smoothened activity to analyze the transcriptional and cell fate consequences of TEAD blockage in skin cancer. We find that TEAD inhibition in BCC triggers rapid activation of differentiation programs, both in cell culture and in mouse skin. The regulation of differentiation by TEAD is indirect and is dependent on the repression of KLF4. YAP1/TAZ–TEAD transcription regulates the expression of SNAI2, a known repressor of KLF4 that could mediate the effects of TEAD in differentiation. We also analyze TEAD-dependent and -independent gene networks in BCC cells. Overall, our results indicate that repression of KLF4 transcriptional networks by YAP1/TAZ–TEAD is essential for maintaining basal cell identity and block differentiation downstream of oncogenic hedgehog-smoothened activity.
RESULTS
YAP1/TAZ–TEAD regulates differentiation gene networks in BCC
For our studies, we utilized a mouse model of BCC driven by constitutively active SmoM2 oncogene. Mice that conditionally express SmoM2 under the ROSA26 promoter (abbreviated lox-stop-lox [LSL]-SmoM2 mice) (Mao et al., 2006) were crossed with mice carrying a tamoxifeninducible cre-recombinase under control of the cytokeratin 14 promoter (K14CreERT) (Vasioukhin et al., 1999) to target basal cells in the skin (named keratin 14 [K14]-SmoM2 in the remaining part of this paper) (Figure 1a). Tamoxifen treatment in K14-SmoM2 mice led to a rapid development of BCC, with lesions appearing 2 weeks after tamoxifen primarily in ears and tail, as it has been reported in other BCC mouse models (Iglesias-Bartolome et al., 2015; Pasca di Magliano and Hebrok, 2003; Youssef et al., 2010). These lesions are characterized by expansion of basal keratinocytes (KCs) expressing K14 and show nuclear staining for p63 (Figure 1b) as well as increase nuclear staining of YAP1 when compared with that in wild type mice (Figure 1c).
Figure 1. TEAD-transcriptional networks regulate differentiation in BCC.
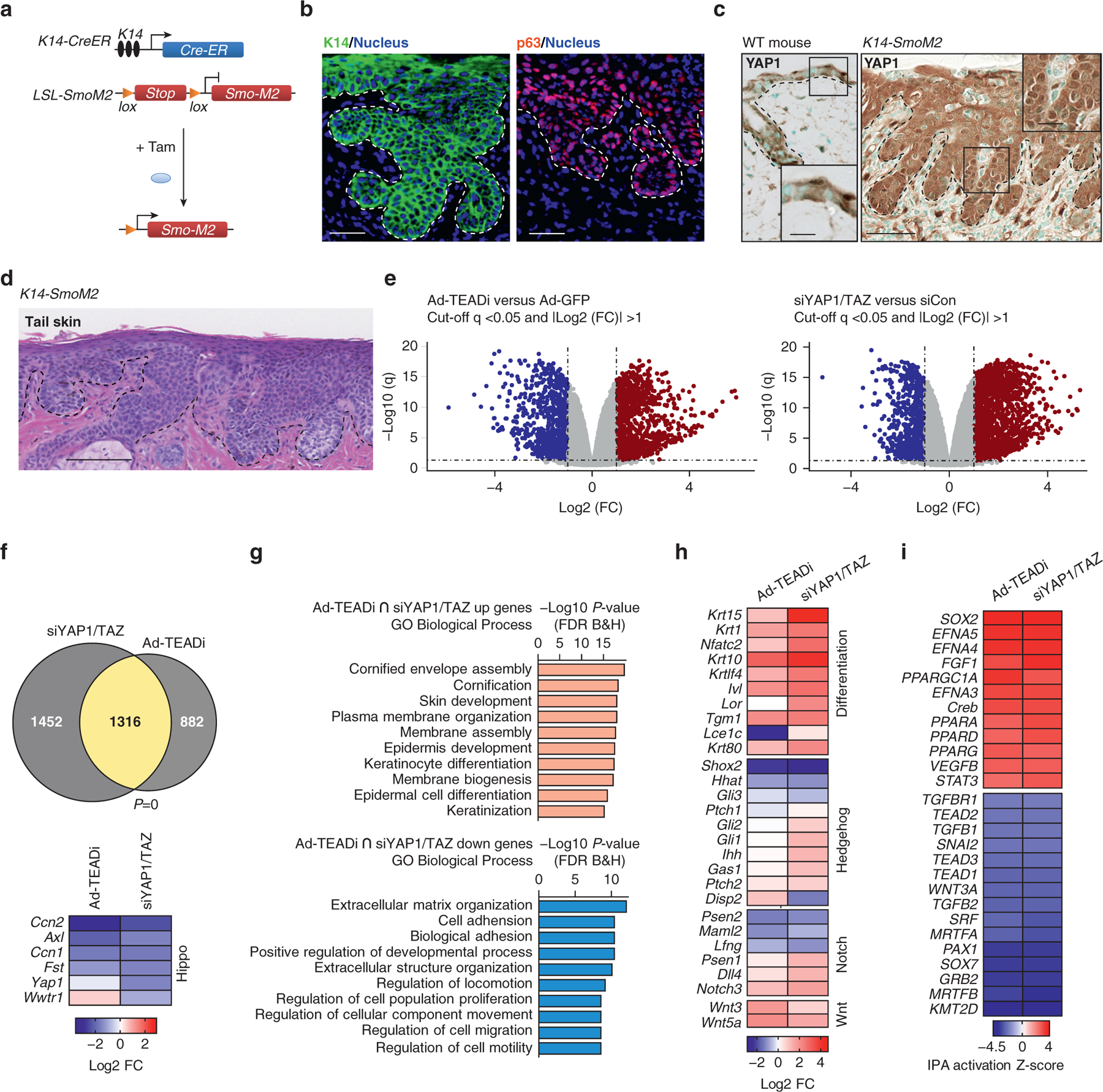
(a) Schematic representation of the K14-SmoM2 Tam inducible BCC mouse model. (b) Staining in the ear sections with the indicated markers 5 weeks after Tam induction. The basal layer of the epidermis is indicated with a dotted line (Bar = 20μm). (c) IHC staining with YAP1 in WT and K14-SmoM2 ear sections 5 weeks after Tam induction (Bar = 50 μm). The basal layer of the epidermis is indicated with a dotted line. Box indicates a magnified area. (d) H&E staining of tail skin from K14-SmoM2 mice showing BCC tumors covering the entire epidermis (Bar = 100 μm). (e) Volcano plots from RNA-seq of BCC cells treated with Ad-TEADi or siYAP1/TAZ; n = 3 for each condition; dotted lines indicate significant genes (respective logarithmic value corresponding to q < 0.05 and absolute FC ≥ 2). Blue indicates significantly downregulated genes, and red indicates significantly upregulated genes. (f) Venn diagram showing the overlap between differentially regulated genes from Figure 1e, and heatmap indicating FC (log2 FC) of YAP1/TAZ and target genes; P-value of the overlap is indicated; Fisher’s exact test. (g) Top GO biological process terms for genes enriched in upregulated (orange, q < 0.05 and FC ≥ 2) or downregulated (light blue, q < 0.05 and FC ≤ −2) genes indicated in yellow in Figure 1f. (h) Heatmap showing the FC of selected genes related to differentiation, Hedgehog, Notch, and Wnt signaling pathways. (i) Heatmap showing activation Z-score for upstream transcriptional regulators calculated using IPA for genes differentially regulated by both siYAP1/TAZ and TEADi. Ad-TEADi, adenoviruses expressing a GFP-tagged TEAD inhibitor; BCC, basal cell carcinoma; FC, fold change; FDR, false discovery rate; GO, gene ontology; IHC, immunohistochemistry; IPA, ingenuity pathway analysis; K, keratin; LSL, lox-stop-lox; RNA-seq, RNA sequencing; siYAP1/TAZ, small interfering RNAs targeting YAP1 and TAZ; Tam, tamoxifen; TEADi, TEAD inhibitor; WT, wild Type
Although the role of YAP1 and TAZ in the development and progression of BCC in K14-SmoM2 mice has been described before (Debaugnies et al., 2018; Maglic et al., 2018), the precise effects of TEAD inhibition in BCC are not clear. To assess TEAD role in BCC, we utilized adeno-viruses expressing a GFP-tagged TEADi (Ad-TEADi) or adenoviruses expressing a GFP as a control and pooled small interfering RNAs targeting YAP1 and TAZ (siYAP1/TAZ) or a random sequence (small interfering RNA targeting control) in isolated skin KCs from the tail of mice with BCC 5 weeks after tamoxifen induction. At this time point, BCC lesions covered the entire tail epidermis (Figure 1d). RNA sequencing of BCC KCs with Ad-TEADi or siYAP1/TAZ revealed dysregulated expression of numerous transcripts (Figure 1e and Supplementary Table S1). A significant overlap between differentially regulated genes in both conditions was observed (Figure 1f), indicating common gene networks. Both TEADi and siYAP1/TAZ led to signif-cant downregulation of the known TEAD targets Ccn1 and Ccn2 (also known as Cyr61 and Ctgf, respectively) and Axl and Fst (Figure 1f). siYAP1/TAZ led to a significant reduction in Yap1 and Taz (Wwtr1) expression, whereas TEADi did not significantly alter these genes (Figure 1f). Gene ontology analysis of genes regulated by both TEADi and siYAP1/TAZ indicated an upregulation of processes related to skin and KC differentiation, whereas down-regulated gene networks wereenriched for terms associated with extracellular matrix organization, cell adhesion, and cell migration (Figure 1g).
Although BCC is characterized by dysregulation of hedgehog signaling, we did not find significant alterations on Gli or Ptch expression or other hedgehog targets by TEADi (Figure 1h). Multiple alterations were present in genes central for KC differentiation, including Klf4, Notch3, Ivl, and K10, among others (Figure 1h). A broader analysis of differentiation genes revealed that both TEADi and siYAP1/TAZ led to the activation of differentiation markers related to every differentiation stage (Supplementary Figure S1a). These results indicate that the activation of YAP1/TAZ–TEAD transcriptional networks is essential for hedgehog-mediated blockage of differentiation in BCC. TEADi- and siYAP1/TAZ-downregulated genes showed enrichment for extracellular matrix and cell adhesion family members (Figure 1g). Components of the basal membrane, including laminins and collagens, were downregulated by siYAP1/TAZ and TEADi, whereas several metalloproteases were upregulated (Supplementary Figure S1b), suggesting that TEAD transcription participates in regulating the integrity of the basal membrane and cell adhesion. Adhesion to the basal membrane is closely related to cell renewal in KCs, and downregulation of these gene networks could be contributing to the observed differentiation in BCC TEADi cells.
Ingenuity pathway analysis (IPA) of upstream transcriptional regulators affected by siYAP1/TAZ and TEADi confirmed the downregulation of networks related to TEAD transcription factors (Figure 1i). In addition, genes related to SRF/MRTF were downregulated (Figure 1i). SRF and MRTF have been linked to therapeutic-resistant BCC (Whitson et al., 2018). In contrast, networks related to EFN and peroxisome proliferator–activated receptors were activated (Figure 1i). TAZ can regulate peroxisome proliferator–activated receptor-γ (Hong and Yaffe, 2006), and peroxisome proliferator–activated receptors activation has been implicated in KC differentiation (Sertznig et al., 2008). EFN has also been involved in reducing KC proliferation and increased differentiation (Lin et al., 2012). Interestingly, IPA analysis revealed a similarity between the transcriptional networks activated by YAP1/TAZ knockdown and TEADi to those activated by cAMP and protein kinase A signaling (Supplementary Figure S1c). This suggests that transcriptional regulation downstream of protein kinase A is partially mediated by YAP1/TAZ and TEAD blockage.
Overall, our results show that YAP1- and TAZ–TEAD–dependent transcriptional networks are involved in the maintenance of the undifferentiated state of BCC cells and that TEAD inhibition leads to rapid activation of differentiation genes.
KLF4 transcriptional networks are activated by YAP1/TAZ and TEAD inhibition in BCC
To better understand the differentiation pathways triggered by TEAD blockage in BCC, we performed a transcription factor binding site enrichment analysis in proximal promoters of genes regulated by both TEADi and siYAP1/TAZ. Over-represented transcription factor binding sites in downregulated genes included TEAD and SRF (Figure 2a), supporting a direct role of TEAD in regulating the expression of these transcripts. Binding sites in genes upregulated by YAP1/TAZ–TEAD inhibition showed a clear enrichment for KLF4 (Figure 2a). KLF4 is a central regulator of KC differentiation (Li et al., 2012; Segre et al., 1999), and our group has previously shown that YAP1/TAZ–TEAD regulates the activity of this transcription factor in normal KCs (Yuan et al., 2020).
Figure 2. YAP1/TAZ and TEAD inhibition in BCC activate KLF4 transcription.
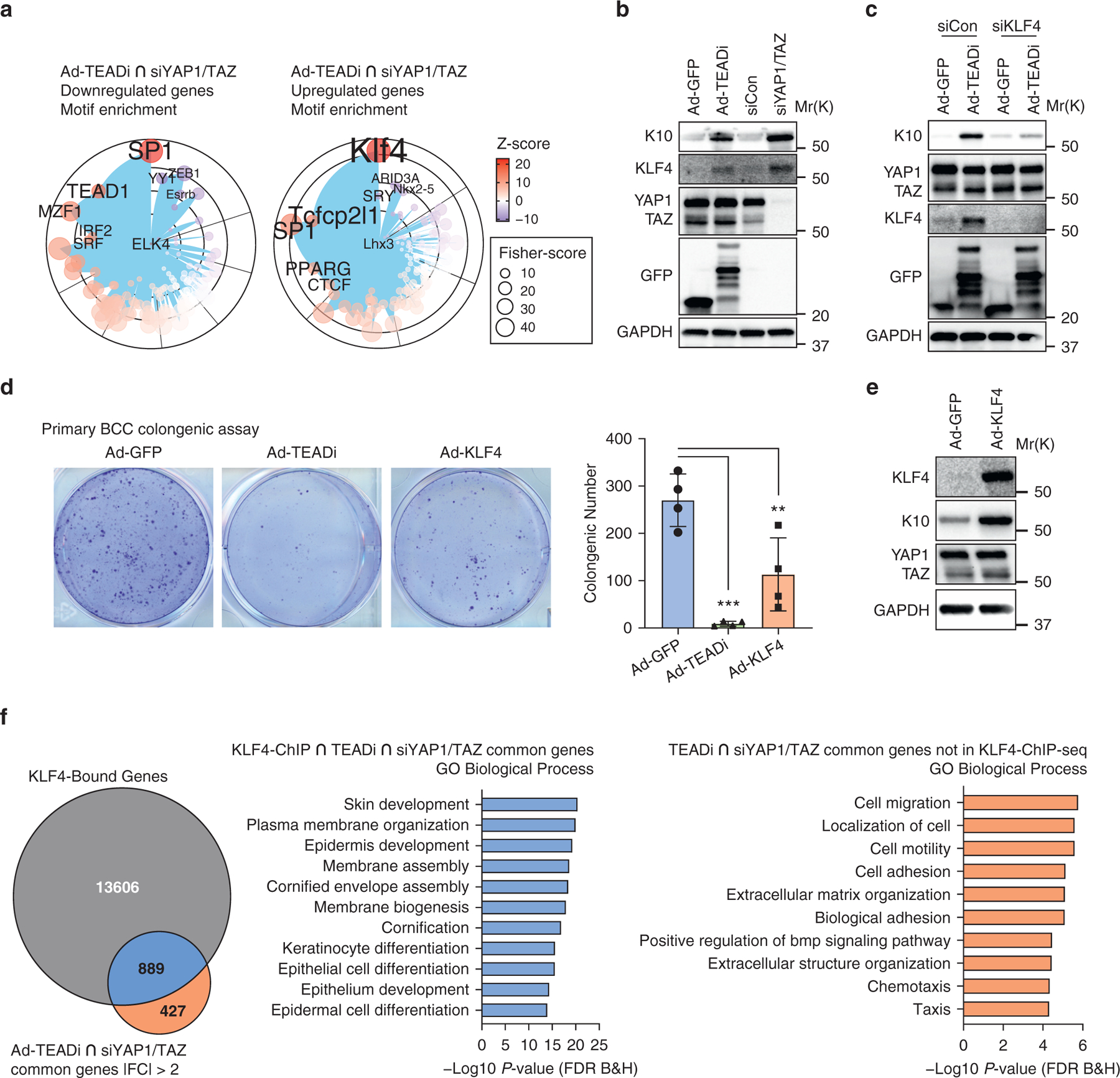
(a) Graph showing the transcription factor binding site enrichment analysis using genes differentially regulated by both siYAP1/TAZ and TEADi (indicated in yellow in Figure 1f); only the top enriched transcription factors are highlighted; color represents Z-score, and size represents Fisher score. (b) Western blot analysis of the indicated markers 48 hours after transduction of cultured BCC keratinocytes with Ad-TEADi (GFP) and control Ad-GFP or with pooled siYAP1/TAZ or control siCon. (c) Western blot analysis of the indicated markers in BCC cultured keratinocytes infected with Ad-TEADi (GFP) or control Ad-GFP, transduced 12 hours later with siRNAs targeting KLF4 or negative siCon and harvested after 48 hours. (d) Clonogenic assay of cultured BCC keratinocytes transduced with control Ad-GFP, Ad-TEADi, or Ad-KLF4. Representative picture and colony numbers from four replicates. Two-way ANOVA with Dunnett’s multiple comparisons test was used (**P < 0.01, ***P < 0.001). (e) Western blot of the indicated markers in BCC-cultured keratinocytes transduced with Ad-KLF4 or control Ad-GFP for 48 hours. (f) Venn diagram indicating the overlap between genes with KLF4 binding (KLF4 ChIP-seq) and genes commonly regulated by TEADi and siYAP1/TAZ and top GO biological process terms from genes in the overlap area (indicated in blue) or exclusively in the TEADi–siYAP1/TAZ dataset (indicated in orange). Ad-GFP, adenoviruses expressing a GFP; Ad-KLF4, adenoviruses expressing KLF4; Ad-TEADi, adenoviruses expressing a GFP-tagged TEAD inhibitor; BCC, basal cell carcinoma; ChIP, chromatin immunoprecipitation; ChIP-seq, chromatin immunoprecipitation sequencing; FC, fold change; FDR, false discovery rate; GO, gene ontology; K, keratin; siCon, small interfering RNAs targeting control; siKLF4, small interfering RNAs targeting KLF4; siRNA, small interfering RNA; siYAP1/TAZ, small interfering RNAs targeting YAP1 and TAZ; TEADi, TEAD inhibitor
The remarkable activation of KLF4 and differentiation in BCC cells triggered by TEAD inhibition without alterations in hedgehog signaling led us to explore this pathway in more detail. BCC KCs transduced with TEADi or siYAP1/TAZ showed an upregulation of KLF4 expression and the differentiation marker K10 (Figure 2b), which could be prevented by the downregulation of KLF4 by pooled small interfering RNAs (Figure 2c). Furthermore, BCC KCs transduced with adenoviruses expressing TEADi or KLF4 presented reduced clonogenic capacity (Figure 2d), and KLF4 overexpression was sufficient to induce the expression of K10 (Figure 2e). These results validate the necessity of TEAD activation and KLF4 inhibition for cell growth in BCC cells.
To confirm the core function of KLF4 in BCC KC differentiation, we utilized available information on KLF4 chromatin immunoprecipitation and sequencing (chromatin immunoprecipitation sequencing [ChIP-seq]) from mouse epidermis (Szigety et al., 2020). Integration of KLF4 ChIP-seq and our RNA sequencing data by binding and expression target analysis (Wang et al., 2013) showed significant activation of transcriptional function in TEADi KCs (Supplementary Figure S2a). Predicted KLF4-activated genes were enriched for differentiation gene ontology terms (Supplementary Figure S2a). Furthermore, by cross-referencing KLF4-bound genes with genes activated by both TEADi and siYAP1/TAZ, we were able to show that KLF4-bound genes were related to skin development and KCs differentiation, whereas the set of genes without KLF4 binding did not show enrichment for differentiation terms (Figure 2f). Our results indicate that repression of KLF4 transcriptional networks by YAP1/TAZ–TEAD is essential for maintaining basal cell identity and block differentiation in BCC.
YAP1/TAZ–TEAD directly regulate SNAI2 expression in KCs
Although the activation of differentiation networks in KCs downstream of TEAD-transcriptional inhibition is clear, it is not known how YAP1/TAZ–TEAD modulates KLF4 activity to regulate differentiation. One possibility is that direct binding of YAP1/TAZ to TEAD in differentiation genes leads to the observed transcriptional changes. To explore this possibility, we analyzed genes bound by YAP1 by ChIP-seq in N/TERT2G human KCs, which recapitulate the effects of TEAD and YAP1/TAZ inhibition in BCC (Yuan et al., 2020). YAP1 ChIP-seq peaks showed a strong enrichment for TEAD and activator protein-1 transcription factors (Figure 3a). By cross-referencing genes bound by YAP1 with those altered by TEADi and siYAP1/TAZ in N/TERT2G cells (Yuan et al., 2020), we were able to identify the core factors regulated by YAP1 binding in KCs. Remarkably, these genes did not show significant enrichment for genes that regulate KC differentiation (Figure 3b), suggesting that regulation of differentiation networks by YAP1/TAZ–TEAD is indirect. Furthermore, we found minimal binding of YAP1 in the KLF4 promoter region (Figure 3c), the main regulator of differentiation downstream of Hippo signaling in KCs (Yuan et al., 2020).
Figure 3. YAP1/TAZ–TEAD target SNAI2 in keratinocytes.
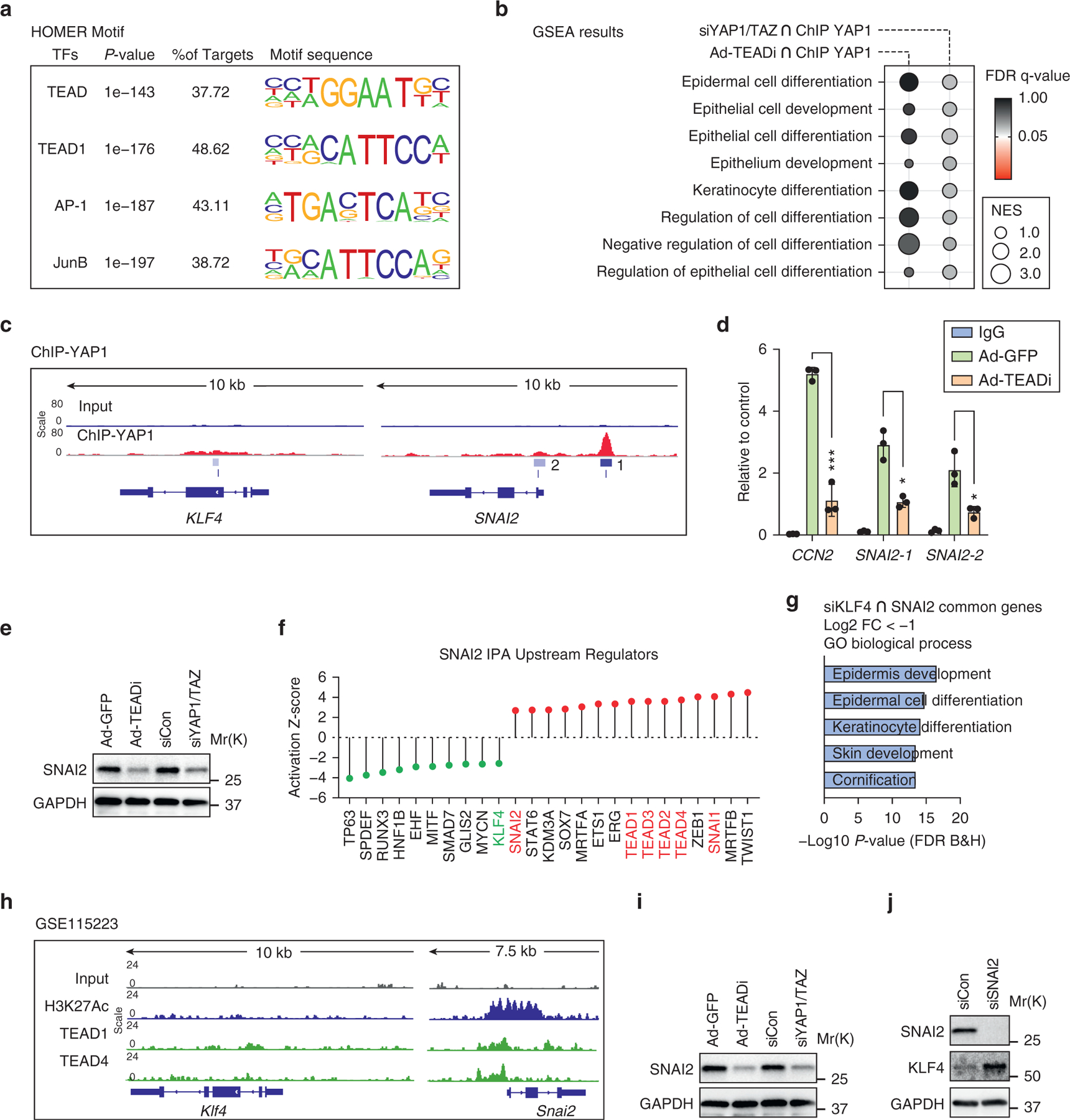
(a) Top enriched TF motif sequences analyzed by Homer in ChIP-sequencing data of YAP1 in NTERT-2G human keratinocytes. (b) GSEA terms related to differentiation present in gene sets differentially regulated by TEADi or siYAP1/TAZ and bound by YAP1 in NTERT-2G cells; color represents FDR q-value, and size represents NES. (c) Gene tracks showing YAP1 occupancy in regions surrounding the transcriptional start site of KLF4 and SNAI2 in NTERT-2G cells. The scale represents the normalized read density of reads per million. 1 and 2 indicate YAP1 binding peaks in SNAI2. (d) ChIP qPCR (by CUT&RUN) in NTERT-2G cells showing binding intensities of YAP1 to the target gene promoter sites in CCN2 and SNAI2. SNAI-1 and −2 indicate YAP1-binding sites in Figure 3c. Unpaired t-test (*P < 0.05, ***P < 0.001). (e)Western blot of SNAI2 in NTERT-2G cells transduced for 48 hours with Ad-TEADi or siYAP1/TAZ and a control treatment with Ad-GFP or nontargeting siRNA (siCon). (f) Bar plot showing the activation Z-score for upstream transcriptional regulators calculated using IPA for genes in SNAI2 over-expression dataset. (g) Diagram showing the top GO biological process terms from common genes downregulated by siKLF4 and SNAI2 overexpression in keratinocytes. (h) Gene tracks showing TEAD1/4 occupancy in regions surrounding the transcriptional start site of Klf4 and Snai2 in BCC keratinocytes. The scale represents the normalized read density of reads per million. (i) Western blot of SNAI2 in BCC keratinocytes transduced as indicated for 48 hours. (j) Western blot of BCC keratinocytes transfected with siRNA targeting SNAI2 for 48 hours. Ad-GFP, adenoviruses expressing a GFP; Ad-TEADi, adenoviruses expressing a GFP tagged TEAD inhibitor; AP-1, activator protein-1; BCC, basal cell carcinoma; ChIP, chromatin immunoprecipitation; FC, fold change; FDR, false discovery rate; GO, gene ontology; GSEA, Gene Set Enrichment Analysis; IPA, ingenuity pathway analysis; Kb, kilobase; NES, normalized enrichment score; siCon, small interfering RNAs targeting control; siKLF4, small interfering RNAs targeting KLF4; siRNA, small interfering RNA; siSNAI2, small interfering RNAs targeting SNAI2; siYAP1/TAZ, small interfering RNAs targeting YAP1 and TAZ; TEADi, TEAD inhibitor; TF, transcription factor.
Because our data indicate an indirect regulation of KLF4 by YAP1/TAZ–TEAD, we examined known KLF4 regulators that are changed in TEADi and siYAP1/TAZ datasets and found that SNAI2 gene networks are downregulated in both conditions (Figure 1i). SNAI2, also known as SNAIL2 or SLUG, is a well-known transcriptional repressor that directly blocks KLF4 expression and differentiation in KCs and other cells (Liu et al., 2012; Mistry et al., 2014). Supporting the role of YAP1/TAZ–TEAD in regulating SNAI2, we found significant binding of YAP1 to the SNAI2 promoter in human KCs (Figure 3c). Expression of TEADi but not of control GFP resulted in reduced binding of YAP1 to promoters of the known YAP1 target CCN2 and to SNAI2 (Figure 3d). In addition, both TEADi and siYAP1/TAZ were able to reduce SNAI2 protein expression in human KCs (Figure 3e). IPA analysis of a publicly available dataset from SNAI2-overexpressing KCs (Mistry et al., 2014) revealed that gene expression changes induced by SNAI2 are enriched for networks related to TEAD transcription factors, whereas gene networks related to KLF4 are depleted (Figure 3f). Moreover, genes commonly downregulated by small interfering RNA depletion of KLF4 (Yuan et al., 2020) and SNAI2 over-expression (Mistry et al., 2014) showed enrichment for KC differentiation gene ontology terms (Figure 3g), indicating common gene networks under both conditions.
To corroborate that our findings translate to the mouse, we analyzed available TEAD ChIP-seq from mouse BCC KCs and found that TEAD1 and TEAD4 show binding to mouse Snai2 but not to Klf4 (Figure 3h). Treatment of BCC KCs with TEADi and siYAP1/TAZ resulted in reduced SNAI2 protein expression, and pooled small interfering RNAs targeting Snai2 were able to upregulate KLF4 (Figure 3i and j), recapitulating the effect of TEADi and siYAP1/TAZ in BCC KCs. Therefore, our data support the notion that although TEAD-transcriptional networks are essential to inactivate differentiation in normal and BCC KCs, this inactivation is indirect and is potentially mediated by repression of KLF4 by SNAI2.
YAP1 and TAZ regulate inflammatory-related gene networks independently of TEAD
We next focused on TEAD-independent functions of YAP1/TAZ in BCC by evaluating the pathways preferentially regulated by siYAP1/TAZ but not by TEADi. YAP1 and TAZ knockdown led to a significant upregulation of Gli1, the hedgehog regulating gene Gas1, and Ihh (Figure 1h), indicating the possibility of a TEAD-independent cross-talk between hedgehog and Hippo signaling. IPA analysis for YAP1/TAZ- and TEADi-regulated genes indicated preferential activation by YAP1/TAZ knockdown of several networks related to NF-κB signaling (Figure 4a). YAP1 has been described to reduce NF-κB activation and the concomitant expression of proinflammatory cytokines IL-6, TNF-α, and IL-1β (Lv et al., 2018), and our IPA analysis showed that siYAP1/TAZ but not TEADi increases IL6, TNF, and IL1 gene networks (Figure 4a). Other immune-modulatory pathways showed increased Z-score IPA activation in YAP1 and TAZ knockdown compared with that in TEADi, including IFN-γ and STAT3 signaling (Figure 4a). Transduction of BCC KCs with siYAP1/TAZ and TEADi confirmed differential levels of activated phosphorylated STAT3 (Figure 4b).
Figure 4. YAP1 and TAZ regulate inflammatory signals through TEAD-independent gene networks.
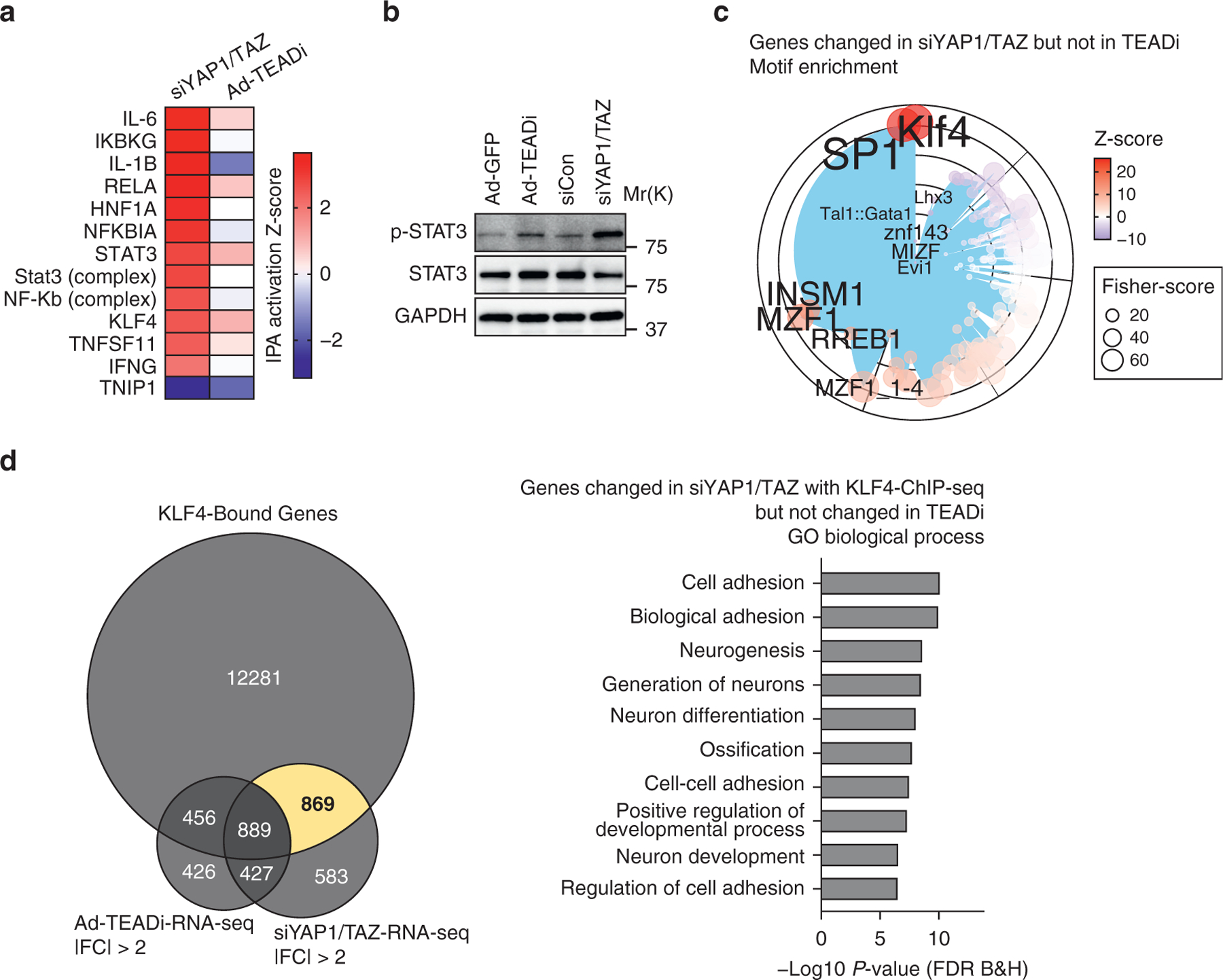
(a) Heatmap showing the activation Z-score for selected IPA upstream transcriptional regulators for genes differentially regulated in siYAP1/TAZ or TEADi datasets. (b) Western blot showing the expression level of p- and total STAT3 in BCC cells treated with the indicated conditions. (c) Graph showing the transcription factor binding site enrichment analysis in genes exclusively regulated by siYAP1/TAZ; only the top enriched transcription factors are highlighted; color represents Z-score, and size represents Fisher score. (d) Venn diagram indicating the overlap between KLF4 ChIP-seq-target genes and genes differentially regulated by siYAP1/TAZ and TEADi and selected GO biological process terms from the area highlighted in yellow. Ad-GFP, adenoviruses expressing GFP; Ad-TEADi, adenoviruses expressing a GFP-tagged TEAD inhibitor; BCC, basal cell carcinoma; ChIP-seq, chromatin immunoprecipitation sequencing; FC, fold change; FDR, false discovery rate; GO, gene ontology; IPA, ingenuity pathway analysis; p-, phosphorylated; RNA-seq, RNA sequencing; siCon, small interfering RNAs targeting control; siYAP1/TAZ, small interfering RNAs targeting YAP1 and TAZ; TEADi, TEAD inhibitor
Interestingly, IPA analysis also indicated a stronger activation of KLF4 gene networks by YAP1/TAZ knockdown (Figure 4a). Examination of over-represented transcription factor binding sites in the genes differentially regulated by siYAP1/TAZ but not by TEADi also showed enrichment for KLF4 (Figure 4c). It is worth noting that this TEADi-independent set of genes did not present enrichment for TEAD binding sites (Figure 4c). Cross-reference of TEADi and siYAP1/TAZ differentially regulated genes with KLF4-ChIP-seq data indicated that KLF4 networks were activated in both conditions (Figure 4d). Genes exclusively regulated by YAP1/TAZ that present KLF4 binding were not related to epithelial differentiation but to cell adhesion (Figure 4d). These results suggest a TEAD dependency for activation of KLF4 differentiation networks resulting from YAP1/TAZ inhibition.
TEADi leads to rapid elimination of tumor cells in BCC lesions
To study the effect of TEADi in BCC tumors, we crossed K14-SmoM2 mice with mice carrying LSL-rtTA (Belteki et al., 2005) and tetracycline-inducible TEADi (TRE-TEADi) (Yuan et al., 2020) (Figure 5a). The resulting mouse model allowed us to trigger BCC formation with tamoxifen while controlling the expression of TEADi by feeding mice doxycycline chow at different points during tumor development.
Figure 5. TEAD inhibition leads to the rapid elimination of tumor cells in BCC lesions.
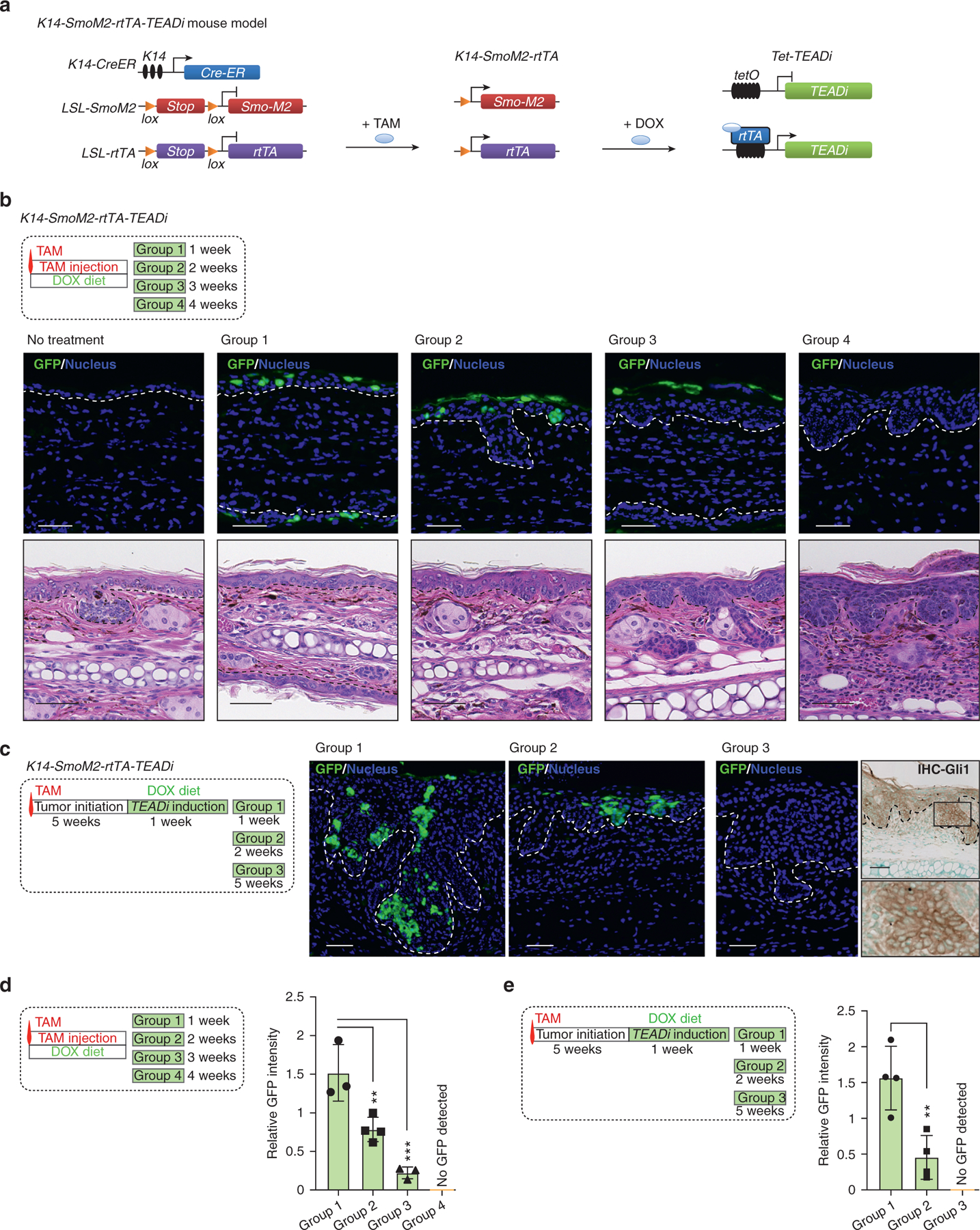
(a) Schematic diagram presenting the mouse model used to trigger TEADi expression in BCC tumors. (b) Timeline and histological analysis of ear tissue from mice induced with one dose of TAM and concomitantly given DOX to activate TEADi expression; GFP staining shows TEADi-positive cells at the indicated time points (Bar = 20 μm); representative pictures from mice are analyzed (Bar = 50 μm); n = 3 for no treatment group and groups 1 and 3; n = 4 for groups 2 and 4. The basal layer of the epidermis is indicated with a dotted line. (c) Timeline and TEADi expression (GFP) in ear tissue from mice induced with one dose of TAM and given DOX 5 weeks later to activate TEADi expression after tumor formation; representative pictures are from n = 4 mice analyzed for groups 1−3 (Bar = 50 μm). The last right panel indicates IHC staining for GLI1 in group 3 ear section, and magnification of the area is indicated with a box. The basal layer of the epidermis is indicated with a dotted line. (d, e) Quantifications of GFP expression level from each time point as indicated in Figure 5b and c; one-way ANOVA with Dunnett’s multiple comparisons test (**P < 0.01, ***P < 0.001). BCC, basal cell carcinoma; DOX, doxycycline; IHC, immunohistochemistry; K14, keratin 14; TAM, tamoxifen; TEADi, TEAD inhibitor
When mice were induced with a single dose of tamoxifen and concomitantly fed doxycycline chow, we observed scattered cells in the ear epidermis positive for TEADi (GFP, Figure 5b). Over time, TEADi-expressing cells were eliminated from the epidermis and were localized to upper and differentiated layers of the skin by week 3 (Figure 5b and d and Supplementary Figure S3a). Although BCC lesions still arose in these mice, resulting tumors were smaller and TEADi negative (Figure 5b and Supplementary Figure S3b and c), indicating that cells in which TEAD was blocked did not proceed to form tumors.
In another set of experiments, mice were treated with tamoxifen, and TEADi expression was induced 5 weeks later. Although in this case, BCC lesions were already developed before the expression of the inhibitor, we observed a similar trend in which all TEADi-positive cells were eliminated from ear BCC tumors over time (Figure 5c and e and Supplementary Figure S3a). No difference was observed in overall tumor growth in these mice (Supplementary Figure S3d), which could be explained by the low percentage of TEADi-expressing cells in this condition (Supplementary Figure S3a). The appearance of BCC lesions negative for GFP in our model could be due to incomplete recombination of LSL-rtTA in a subset of cells that express SmoM2, allowing TEADi-negative cells to proceed with tumor formation. Indeed, lesions remaining in mice after TEADi cell depletion were positive for the hedgehog-smoothened signaling marker GLI1 (Figure 5c), indicating activity of SmoM2. A similar scenario was reported in BCC studies with YAP1/TAZ-knockout animals (Debaugnies et al., 2018; Maglic et al., 2018), in which BCC tumors arise in YAP1/TAZ- and YAP1-knockout mice that are positive for YAP1 owing to incomplete recombination.
The low penetrance of TEADi expression in our model allowed us to test the activation of proliferation and differentiation pathways in TEADi-positive versus that in TEADi-negative cells in the same tumor. Intratumoral TEADi cells in the ear presented a significant increase in expression of the differentiation markers K10 and KLF4 (Figure 6a and b) and showed reduced labeling for the PCNA when compared with surrounding tumor cells (Figure 6c). Our in vivo data show that inhibition of TEAD during or after BCC tumor formation leads to rapid activation of differentiation pathways and elimination of cells from tumor lesions and validate our conclusion that activation of TEAD is necessary for preventing differentiation downstream from hedgehog signaling in BCC.
Figure 6. TEAD inhibition induces differentiation and reduces proliferation in BCC cells.
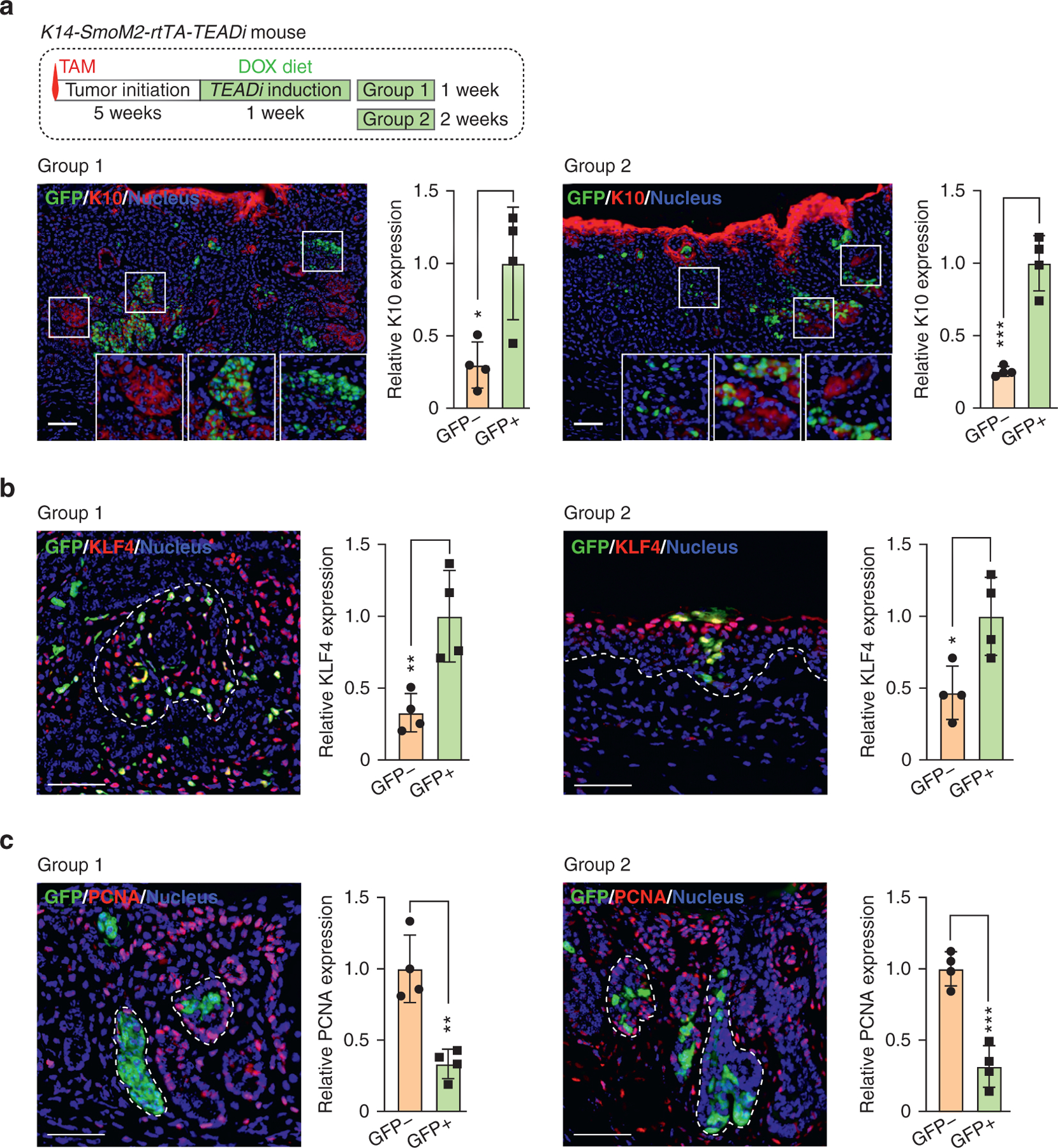
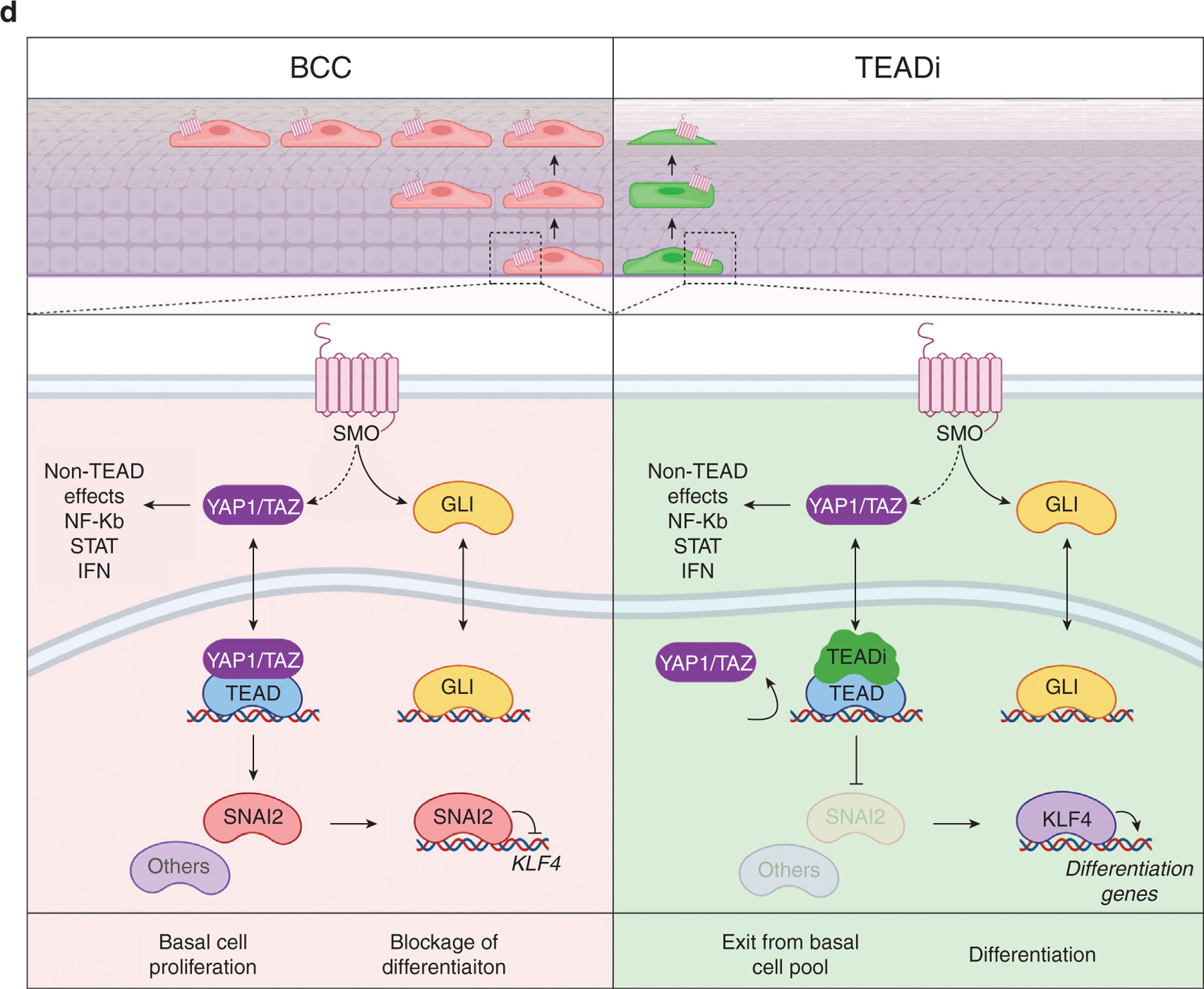
(a−c) Timeline and immunofluorescence analysis of indicated markers in ear tissue from mice induced with one dose of TAM and given DOX 5 weeks later to activate TEADi expression after tumor formation. Graphs show the relative expression of the indicated markers quantified in TEADi-positive cells (GFP+) and surrounding control cells (GFP−); each value represents the average expression in sections from one mouse, n = 4 mice; Bar = 20 μm; unpaired t-test (*P < 0.05, **P < 0.01, ***P < 0.001). The basal layer of the epidermis is indicated with a dotted line. (d) Simplified model summarizing the results from our study. See Discussion for details. BCC, basal cell carcinoma; DOX, doxycycline; K10, keratin 10; SMO, hedgehog-smoothened; TAM, tamoxifen; TEADi, TEAD inhibitor.
DISCUSSION
In this study, we use knockdown strategies and specific transcriptional inhibition to discern TEAD-dependent and -independent effects of YAP1/TAZ in cancer. We observe that blockage of TEAD leads to rapid differentiation of BCC cells by activating KLF4. One surprising finding is that although SmoM2 requires YAP1/TAZ–TEAD to block differentiation, activation of differentiation pathways downstream of TEAD inhibition is indirect and is mediated by repression of KLF4. TEAD directly regulates the transcriptional repressor SNAI2 in KCs, a known inhibitor of KLF4 and differentiation (Mistry et al., 2014). This evidence suggests that inhibition of differentiation by TEAD might be mediated by SNAI2, although other factors could participate in this repressive effect (Figure 6d).
The central role of KLF4 in regulating differentiation downstream of oncogenic activation in KCs highlights its potential role as a cancer target. Genomic alterations in KLF4 are not common in skin cancer, but KLF4 is down-regulated in BCC and squamous cell carcinoma tumors, and KLF4-knockout mice present increased sensitivity to tumor formation (Li et al., 2012). KLF4 is also responsible for reduced self-renewal and increased differentiation of skin cancer-initiating cells in mouse models (Sastre-Perona et al., 2019). Because KLF4 expression can also result in concomitant inhibition of TEAD (Yuan et al., 2020) and its inactivation is mainly mediated by transcriptional inhibition, finding avenues to regain or increase KLF4 activity in BCC could prove effective in therapeutic strategies for these tumors.
Our results show that although YAP1/TAZ knockdown recapitulates TEAD inhibition, it results in additional effects in cancer cells. siYAP1/TAZ but not TEADi leads to activation of IFNγ, STAT, and NF-κB gene networks as well as increases in levels of some hedgehog pathway members. Reduced IFN and STAT signaling has been attributed to cytoplasmic effects mediated by YAP1 and TAZ (Fang et al., 2019; Wang et al., 2017). YAP1 has also been shown to dampen NF-kB activation by interacting directly with TRAF6 (Lv et al., 2018) and TAK1 (Deng et al., 2018). Together, these data indicate that TEAD-independent effects of YAP1 and TAZ might be mediated mainly through cytoplasmic rather than nuclear interactions with other proteins, although the precise mechanisms regulating these pathways require further investigation.
The activation of KLF4 and differentiation pathways resulting from TEAD inhibition in BCC cells closely recapitulates our findings in normal human KCs and mouse skin (Yuan et al., 2020), raising potential limitations with this tumor model. Although oncogene-driven models such as SmoM2 provide invaluable information on cancer initiation and progression, they do not completely reflect the complex mutational landscape of tumors. Carcinomas accumulate numerous genomic and epigenomic modifications that render them incapable of differentiation, compared with single oncogene models. Although our data suggest that TEAD inhibition in normal and mutant Smo cancer cells would trigger their elimination by activating differentiation, more research is needed to understand the networks resulting from TEAD inhibition in more genetically complex cancer cells.
MATERIALS AND METHODS
Detailed methodology is available in the Supplementary Materials and Methods.
Cell culture
BCC tumor cells were isolated from tail skin of K14-SmoM2 mice 5 weeks after tamoxifen induction. N/TERT2G human KCs, transfections, and adenoviral infections were described before (Yuan et al., 2020). To assess colony-forming efficiency, an equal number of KCs were infected and plated in triplicate and grown for 7 days. Plates were fixed using 4% paraformaldehyde for 15 minutes and stained with crystal violet (0.5%, Sigma-Aldrich, St. Louis, MO). Immunoblot analysis on cell lysates was performed as previously described (Yuan et al., 2020).
Mice
All mouse studies were carried out according to approved protocols from the National Institute of Health Intramural Animal Care and Use Committee of the National Cancer Institute (Bethesda, MD). TRE-TEADi mice were described before (Yuan et al., 2020). Other mouse lines were obtained from the Jackson Laboratory (Bar Harbor, ME): LSL-SmoM2 (stock 005130), LSL-rtTA (stock 005670), and K14CreERT (stock 005107). Immunofluorescence and immunohistochemistry were performed on ear tissue sections embedded in paraffin as previously described (Iglesias-Bartolome et al., 2015; Yuan et al., 2020). Both male and female mice were used for analysis.
Statistics
All analyses were performed in triplicate or greater, and the means obtained were used for ANOVA or independent t-tests. Statistical analyses were carried out using Prism 7 statistical analysis program (GraphPad Software, La Jolla, CA). Statistical analysis of intersections in Venn diagrams was performed by hypergeometric test (one-tailed Fisher’s exact test). Asterisks denote statistical significance.
Data availability statement
RNA sequencing and chromatin immunoprecipitation sequencing primary and processed data generated in this manuscript are available from Gene Expression Omnibus databases GSE156913 and GSE172323, respectively. Processed RNA sequencing data are provided in Supplementary Table S1. RNA sequencing data from human keratinocytes with TEAD inhibitor, small interfering RNAs targeting YAP1 and TAZ, and KLF4 are from GSE136876, GSE137531, and GSE137410, respectively (Yuan et al., 2020). KLF4 chromatin immunoprecipitation sequencing data are from GSE137232 (Szigety et al., 2020). Data from keratinocytes with SNAI2 overexpression are from GSE55269 (Mistry et al., 2014). TEAD1 and TEAD4 chromatin immunoprecipitation sequencing data are from GSE115223 (Maglic et al., 2018).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (ZIA BC 011764 and ZIA BC 011763). This work used the computational resources of the National Institute of Health High-Performance Computing Biowulf Cluster. Analysis and management of H&E images were supported by the National Cancer Institute HALO Image Analysis Resource. We thank Jennifer E. Dwyer for assistance with slide scanning and members of the Center for Cancer Research Sequencing Facility at Frederick National Laboratory for Cancer Research for their help during sample preparation, sequencing, and data processing.
Abbreviations:
- Ad-TEADi
adenoviruses expressing a GFP-tagged TEAD inhibitor
- BCC
basal cell carcinoma
- ChIP-seq
chromatin immunoprecipitation sequencing
- IPA
ingenuity pathway analysis
- K
keratin
- KC
keratinocyte
- LSL
lox-stop-lox
- siYAP1/TAZ
small interfering RNAs targeting YAP1 and TAZ
- TEADi
TEAD inhibitor
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2021.06.020.
REFERENCES
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, et al. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res 2005;33:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debaugnies M, Sánchez-Danés A, Rorive S, Raphaël M, Liagre M, Parent MA, et al. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Lu J, Li W, Wu A, Zhang X, Tong W, et al. Reciprocal inhibition of YAP/TAZ and NF-kB regulates osteoarthritic cartilage degradation. Nat Commun 2018;9:4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, Li J, Qi S, Lei Y, Zeng Y, Yu P, et al. An alternatively transcribed TAZ variant negatively regulates JAK-STAT signaling. EMBO Rep 2019;20:e47227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Yaffe MB. TAZ: a beta-catenin-like molecule that regulates mesenchymal stem cell differentiation. Cell Cycle 2006;5:176–9. [DOI] [PubMed] [Google Scholar]
- Iglesias-Bartolome R, Torres D, Marone R, Feng X, Martin D, Simaan M, et al. Inactivation of a Gα(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat Cell Biol 2015;17: 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zheng H, Yu F, Yu T, Liu C, Huang S, et al. Deficiency of the Kruppel-like factor KLF4 correlates with increased cell proliferation and enhanced skin tumorigenesis. Carcinogenesis 2012;33:1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Wang B, Getsios S. Eph/ephrin signaling in epidermal differentiation and disease. Semin Cell Dev Biol 2012;23:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor beta-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol 2012;32:941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Y, Kim K, Sheng Y, Cho J, Qian Z, Zhao YY, et al. YAP controls endothelial activation and vascular inflammation through TRAF6. Circ Res 2018;123: 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglic D, Schlegelmilch K, Dost AF, Panero R, Dill MT, Calogero RA, et al. YAP-TEAD signaling promotes basal cell carcinoma development via a c-Jun/AP1 axis. EMBO J 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res 2006;66:10171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry DS, Chen Y, Wang Y, Zhang K, Sen GL. SNAI2 controls the undifferentiated state of human epidermal progenitor cells. Stem Cells 2014;32: 3209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 2003;3:903–11. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173:321–337 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre-Perona A, Hoang-Phou S, Leitner MC, Okuniewska M, Meehan S, Schober M. De novo PITX1 expression controls bi-stable transcriptional circuits to govern self-renewal and differentiation in squamous cell carcinoma. Cell Stem Cell 2019;24:390–404.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet 1999;22:356–60. [DOI] [PubMed] [Google Scholar]
- Sertznig P, Seifert M, Tilgen W, Reichrath J. Peroxisome proliferator-activated receptors (PPARs) and the human skin: importance of PPARs in skin physiology and dermatologic diseases. Am J Clin Dermatol 2008;9:15–31. [DOI] [PubMed] [Google Scholar]
- Szigety KM, Liu F, Yuan CY, Moran DJ, Horrell J, Gochnauer HR, et al. HDAC3 ensures stepwise epidermal stratification via NCoR/SMRT-reliant mechanisms independent of its histone deacetylase activity. Genes Dev 2020;34:973–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA 1999;96:8551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 2013;8:2502–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKϵ-mediated phosphorylation. Nat Immunol 2017;18:733–43. [DOI] [PubMed] [Google Scholar]
- Whitson RJ, Lee A, Urman NM, Mirza A, Yao CY, Brown AS, et al. Noncanonical hedgehog pathway activation through SRFeMKL1 promotes drug resistance in basal cell carcinomas. Nat Med 2018;24:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef KK, Van Keymeulen A, Lapouge G, Beck B, Michaux C, Achouri Y, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol 2010;12:299–305. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015;163:811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Park J, Feng A, Awasthi P, Wang Z, Chen Q, et al. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun 2020;11:1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016;29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA 2011;108:2270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing and chromatin immunoprecipitation sequencing primary and processed data generated in this manuscript are available from Gene Expression Omnibus databases GSE156913 and GSE172323, respectively. Processed RNA sequencing data are provided in Supplementary Table S1. RNA sequencing data from human keratinocytes with TEAD inhibitor, small interfering RNAs targeting YAP1 and TAZ, and KLF4 are from GSE136876, GSE137531, and GSE137410, respectively (Yuan et al., 2020). KLF4 chromatin immunoprecipitation sequencing data are from GSE137232 (Szigety et al., 2020). Data from keratinocytes with SNAI2 overexpression are from GSE55269 (Mistry et al., 2014). TEAD1 and TEAD4 chromatin immunoprecipitation sequencing data are from GSE115223 (Maglic et al., 2018).