

Abstract
Brain iron homeostasis is maintained through the normal function of blood–brain barrier and iron regulation at the systemic and cellular levels, which is fundamental to normal brain function. Excess iron can catalyze the generation of free radicals through Fenton reactions due to its dual redox state, thus causing oxidative stress. Numerous evidence has indicated brain diseases, especially stroke and neurodegenerative diseases, are closely related to the mechanism of iron homeostasis imbalance in the brain. For one thing, brain diseases promote brain iron accumulation. For another, iron accumulation amplifies damage to the nervous system and exacerbates patients’ outcomes. In addition, iron accumulation triggers ferroptosis, a newly discovered iron‐dependent type of programmed cell death, which is closely related to neurodegeneration and has received wide attention in recent years. In this context, we outline the mechanism of a normal brain iron metabolism and focus on the current mechanism of the iron homeostasis imbalance in stroke, Alzheimer's disease, and Parkinson's disease. Meanwhile, we also discuss the mechanism of ferroptosis and simultaneously enumerate the newly discovered drugs for iron chelators and ferroptosis inhibitors.
Keywords: ferroptosis, iron chelators, iron homeostasis, neurodegenerative diseases, stroke
As neurological diseases that seriously affect the survival and prognosis of patients, stroke and neurodegenerative diseases cause excessive accumulation of brain iron by disturbing the normal mechanism of brain iron absorption and export, accompanied by the outbreak of oxidative stress, which leads to programmed cell death called ferroptosis, and eventually leads to cognitive and motor dysfunction of patients.
1. INTRODUCTION
Iron is a redox‐active element of life. Iron can accept or donate electrons to facilitate other redox reactions in the conversion between ferric (Fe3+) and ferrous (Fe2+) forms. Due to its redox ability, iron is widely utilized in nearly every biochemical pathway in the form of iron–sulfur (Fe–S) clusters and heme, ranging from oxygen transport as key a component for hemoglobin synthesis, to DNA metabolism as a cofactor for multiple enzymes including DNA repair enzymes and ribonucleotide reductase, to oxidative phosphorylation being an indispensable substance for complex I–IV of the mitochondrial respiratory chain, to myelin and neurotransmitter synthesis (e.g., as a cofactor for tyrosine hydroxylase). 1 , 2 , 3 , 4 , 5 Therefore, balanced iron metabolism plays a critical role in maintaining the body's metabolism normal neurological function.
Iron accumulation in the brain has been reported to be closely related to a variety of neurological conditions in middle‐aged and elderly people, such as stroke, Alzheimer's disease (AD), and Parkinson's disease (PD). Local or extensive brain iron accumulation was found to affect the progression of these diseases, impairing the cognitive and motor ability of patients. 6 In addition, iron accumulation triggers ferroptosis, which is a newly discovered iron‐dependent regulated cell death and different from autophagy, apoptosis, necroptosis, and other forms of cell death. The most essential biochemical attributes of ferroptosis are iron overload and the lethal accumulation of ROS and lipid peroxides accompanied by glutathione (GSH) depletion, glutathione peroxidase (GPX) 4 inactivation within the cell, leading to fatal damage and disorganization of the cell membrane at last. 7 As a consequence of iron accumulation, ferroptosis is an important cause of neurodegeneration in brain diseases. 8 Thus, elucidating how the central nervous system incorporates and manages iron under physiological and pathological conditions and how iron homeostasis imbalance cause ferroptosis is helpful to provide potential targets for the treatment of stroke and neurodegenerative diseases and provide clues to solve the puzzles in the field of imbalance of brain iron homeostasis.
In this review, we focus on (i) the mechanism of normal iron regulation in the brain involves iron absorption, storage, and export; (ii) iron accumulation in brain diseases, mostly focusing on stroke, AD, and PD; (iii) the mechanism of ferroptosis; (iv) the latest therapeutic agents for treating iron accumulation and ferroptosis, including iron chelators and ferroptosis inhibitors.
2. IRON HOMEOSTASIS IN THE BRAIN
2.1. Brain iron absorption
Most (90%) of the daily iron consumed by a healthy adult derives from the recycling of iron within senescent erythrocytes by reticuloendothelial macrophages in the spleen and liver, with the remaining 10% from dietary iron absorption by enterocytes. 9 Iron recycled from erythrocytes and absorbed from diet enters the circulation as Fe3+. 10 Fe3+ is more stable compared with Fe2+, which can generate ROS via the Fenton reaction, a type of reaction between Fe2+ and hydrogen peroxide (H2O2), with the ability to oxidize a wide range of organic substrates. 11 Fe3+ that has lost three electrons cannot participate in the oxidation reaction again, making the transportation of Fe3+ in circulation relatively safe.
Iron is transported to the brain mainly in two forms: the iron–transferrin complex (TF–Fe3+) and the nontransferrin‐bound iron (NTBI). The regulatory mechanism of iron absorption in the brain is highly strict. The blood–brain barrier (BBB) is the first barrier for iron to enter the brain cells. The BBB consists of endothelial cells, astrocytic endfeet, pericytes, and basement membranes that collaborate to control the entry of substances or cells in circulation into the brain to maintain a stable environment for normal brain function. 12 , 13 And brain microvascular endothelial cells (BMVECs) are the main components of the BBB and also the target cells recognized by TF–Fe3+ complex. 14 The uptake and recycling of TF–Fe3+ complex include several steps (Figure 1): (i) due to the tight junctions (TJs) between BMVECs, TF–Fe3+ complex can enter cells only in the transcellular pathway (endocytosis) by binding to transferrin receptor 1 (TfR1) on the membrane of BMVECs, which is clathrin dependent 15 ; (ii) the internalized Tf/TfR1 complex is coated with membranes to form vesicles that are transported to the endosome and fuse with it; (iii) Fe3+ is released from Tf inside the endosomal compartment of BMVECs in an acidic pH (around 5.5) achieved by the activity of proton pumps; (iv) the reduction of Fe3+ to Fe2+ in the presence of ferric reductases, such as STEAP316; (v) Fe2+ then crosses the endosomal membrane into the labile iron pool in the cytoplasm via divalent metal transporter 1 (DMT1) 17 , 18 ; (vi) return of non‐iron‐bound Tf (apo‐Tf) with TfR1 to the membrane where apo‐Tf is released into the blood. 19 The vasculature of the brain is almost ensheathed by astrocytic end‐feet, which are specialized units that serve the function to maintain the ionic and osmotic homeostasis of the brain. 20 , 21 Once iron is released from the BMVECs, it may be taken up by the end‐feet of astrocytes. As the most abundant glial cells in the brain, the primary role of astrocytes is to transport iron ions to other glial cells and neurons rather than to store them. 22 Before being expelled out of cells, Fe2+ is oxidized to Fe3+ by multicopper ferroxidases, including ceruloplasmin (Cp) and hephaestin (Heph). The major type of ferroxidase in astrocytes is Cp, while Heph is found in neurons, oligodendrocytes, and microglia. 23 , 24 , 25 , 26 The ferroxidase activity of Cp can facilitate iron export coupled to ferroportin 1 (Fpn1). After entering into interstitial fluid or cerebrospinal fluid in ventricles, Fe3+ binds with extracellular transferrin (apo‐Tf) readily and supplies iron to the cells expressing TfR1 within the central nervous system. 15 , 27 , 28
FIGURE 1.
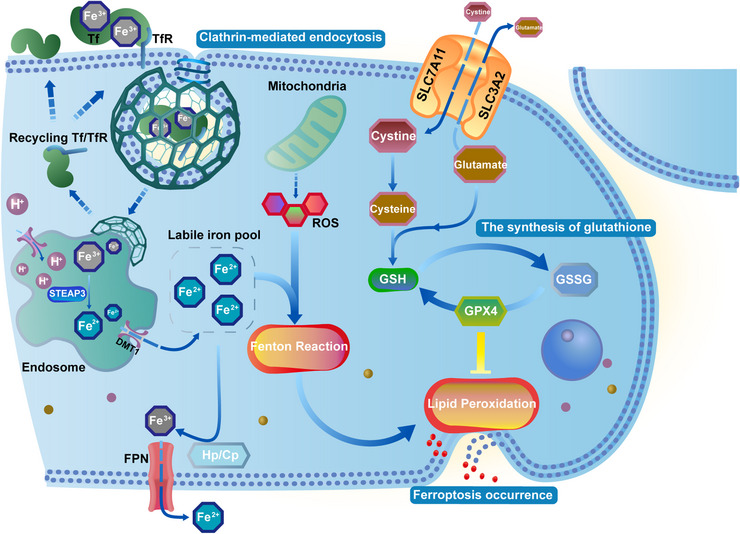
Metabolic processes of iron transport by transcellular pathway (endocytosis), lipid peroxidation related to the occurrence of ferroptosis, and the body's representative antioxidant system. The Fenton reaction stimulated by liable iron induces lipid peroxidation, while intracellular GSH is insufficient to resist it due to the rapid rise of liable iron so ferroptosis occurs due to the rupture of the unstable membrane. Tf, transferrin; TfR, transferrin receptor; DMT1, divalent metal transporter 1; FPN, ferroportin; Cp, ceruloplasmin; Hp, hephaestin; ROS, reactive oxygen species; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GSSG, oxidized glutathione; GPX4, glutathione peroxidase 4.
NTBI is potentially toxic in the plasma, so it may still exist but can be barely detectable under physiological conditions. 29 One transferrin molecule can bind 2 atoms of iron, and normally, there are left with ∼70% of the iron binding sites in the plasma transferrin pool are unoccupied at any time, which means transferrin saturation≈30%, thereby providing a considerable buffering capacity against the generation of NTBI. 11 But TFs are over‐saturated under conditions of iron overload, resulting in NTBI redundancy. Due to its high redox activity, NTBI can react with biomolecules such as sugars, lipids, and proteins to cause peroxidative tissue damage by producing harmful ROS in cells. 30 NTBI transport relies on DMT1 and ferric reductase in the traditional model. Fe2+, from the reduction of Fe3+ mediated by reductases, cotranslocates with H+ across the cell membrane, which is mediated by DMT1. 31 Moreover, the translocation of NTBI by DMT1 is pH dependent. The increase of H+ in the local brain promotes the transport of NTBI, leading to the accumulation of Fe2+ in the impaired nervous system. Indeed, astrocytes, oligodendrocytes, and microglia take up iron in the form of NTBI in a certain proportion even under physiological conditions, given that their low expression of TF cannot satisfy physiological needs. However, NTBI uptake will be proportionately increased especially in cases of iron overload, which might aggravate intracellular oxidative stress. 32 A recent study reports that ferritin (FT) heavy chain as a transport protein helps NTBI to cross the BBB by binding with T‐cell immunoglobulin and mucin domain‐containing 1 receptor 1 in human endothelial cells. 33 In addition, prion protein, a ubiquitously expressed plasma membrane glycoprotein that is most abundant in neuronal cells, can promote the transport of NTBI mediated by DMT1. 17
2.2. Brain iron storage and export
FTs, which serve as the major cellular iron storage protein, are both nontoxic and bioavailable forms in the sequestration of free iron (Fe2+). 34 , 35 Excess iron is mainly stored in the cytoplasmic FT or partly exported to the extracellular fluid to keep the intracellular labile iron pool relatively stable. In addition, as the major intracellular iron‐utilizing organelles where the Fe–S cluster is synthesized to keep the normal function of the respiratory chain complexes I–IV and provide a steady energy output, mitochondria also have their exclusive FTs, called mitochondrial FT (FtMt), to prevent the detrimental effect of oxidative damage, such as the release of cytochrome c and the inhibition of Fe–S cluster enzymes. 36 , 37 , 38 Iron over the reserve is transferred out of the cell, which relies on Fpn1, the only‐known iron export protein on the cell membrane. 39
Because of the essential function of Fpn1 in cellular and systemic iron homeostasis, the expression and activity of Fpn1 are regulated at multiple levels. At the cellular level, Fpn1 is regulated by the iron regulatory protein (IRP)/iron responsive element (IRE) system that maintains cellular iron homeostasis by regulating iron transport and storage. 40 It controls the translation of proteins responsible for iron uptake (DMT1 and TfR1), storage (FT), and export (Fpn1). Under cellular iron deficiency, IRPs bind to the IRE, a stem‐loop structure in the 5′‐untranslated region of Fpn1 messenger RNA, to inhibit the translation of Fpn1, thereby limiting iron export. 41 Furthermore, the IRP/IRE system can downregulate FT assembly and translation, and upregulate the expression of TfR1 and DMT1, so finally enhancing the absorption and reducing the storage and export of iron. 42 , 43 , 44 , 45 Under iron redundancy, the IRP/IRE system downregulates the translation of TfR1 and upregulates the translation of FT and Fpn1, so limiting the absorption while promoting the storage and export of iron. 46 At the systemic level, Fpn1 activity is regulated by hepcidin, a 25 amino‐acid peptide hormone mainly expressed by the liver. 47 The expression of hepcidin is influenced by several physiological conditions, such as systemic iron levels, inflammation, hypoxia, anemia, erythropoiesis, and infection. By binding Fpn1 on the cytoplasmic membrane, hepcidin is able to induce the ubiquitination of Fpn1 mediated by the E3 ubiquitin ligase RNF217. 48 Subsequently, nonfunctional Fpn1 is under internalization and degradation, thereby inhibiting iron export to circulation. 49 Moreover, it is reported that hepcidin functions by downregulating TfR1 and DMT1 in BMVECs, astrocytes, and neurons, which suggests that hepcidin can regulate iron import and export in the brain simultaneously. 50 , 51 , 52 , 53
Although the mechanisms by which the IRP/IRE system and hepcidin function differ, they reach the same goal with mutual dependence, and both are affected by intracellular iron levels. 45 IRP1 and IRP2, which are the two forms of IRP protein, are the main regulators of cellular iron in humans. The regulation of Fpn1 by hepcidin is mainly dependent on IRP2. Animal studies have shown that IRP2 dominates the mouse brain, and IRP2 knockdown significantly affected the downregulation of Fpn1 regulated by hepcidin. 54 Therefore, the regulatory pathway of hepcidin and IRP/IRE system partially overlap.
From what has been discussed above, we found that iron transport is chemically modified by a variety of enzymes and combined with different proteins. Through this sophisticated transport mode, tissues are effectively protected from iron‐mediated oxidative damage. With the normal function of BBB, the regulation of the IRP/IRE system, and hepcidin, brain iron metabolism can reach an equilibrium state. And it was reported to be largely unaffected even when peripheral iron homeostasis was dysregulated. 55
3. IRON METABOLISM IN BRAIN DISEASES
Under physiological conditions, brain iron content is in a dynamic balance of uptake, storage, and exportation to maintain the normal metabolism and function of neurocytes. Brain iron homeostasis largely depends on the integrity of the BBB and the accurate regulation of related proteins at the systemic and cellular levels, including TF, TfR1, DMT1, FT, Fpn1, and so on.
3.1. Iron metabolism in stroke
Stroke is the second leading cause of death and disability in the adult population in modern society, including ischemic stroke (IS) caused by the obstruction of the artery, accounting for about 87% of all stroke cases, and hemorrhagic stroke (HS) mainly caused by arterial rupture, accounting for about 13%. 56 The causal association between stroke and brain iron accumulation has long been confirmed by a large number of clinical data, including neuroimaging and basic medical research. 57 , 58 , 59 , 60 As the gatekeeper of substance homeostasis in the brain, BBB plays an important role in controlling the import of brain iron. 60 Once the BBB is breached, the homeostasis of iron in the brain will be disrupted, along with the altered expression of iron‐related proteins. Though both IS and HS can trigger brain iron homeostasis imbalance, the pathways differ between the two types of strokes. Clarifying the differences between the two will be favorable for precision treatment in the future.
3.1.1. Ischemic stroke
BBB has extremely low paracellular permeability and high electrical resistance due to the existence of TJs between adjacent BMVECs. 61 , 62 In IS, the paracellular permeability of the BBB increases greatly for the disruption of TJs, so free iron ions and FT in the circulation penetrate the brain parenchyma through weak barriers, resulting in iron overload in the brain. Recombinant tissue plasminogen activator (rt‐PA) is the preferred treatment for acute IS within 4.5 h of the onset of symptoms and has been proven effective in thrombolysis and recanalization of blood vessels. 63 , 64 However, the administration of rt‐PA increases the extent of BBB leakage, which may result in hemorrhagic transformation related to high mortality. 65 Therefore, patients treated with rt‐PA are prone to severe iron overload, which will further aggravate neurological dysfunction caused by ferroptosis if combined with hemorrhagic transformation, even the risk of death. In addition, influenced by ischemia/excitotoxicity, the endocytosis of the Tf–TfR1 complex and expression of DMT1 in the ipsilateral ischemic region is increased without changing neuronal TfR1 levels, implying the circulation rate of TfR1 increased rapidly in response to iron overload. 66 , 67 Mass Tf–TfR1 complexes containing iron enter the endosome, releasing Fe2+ under an acidic medium, which finally overflows the labile iron pool.
A selective autophagy pathway known as ferritinophagy, a process in which iron‐loaded FT is transmitted to the lysosome for degradation along with the free iron release, occupies a critical place in systemic iron homeostasis. 34 , 68 The concentration of labile iron increased significantly along with reduced content of FT 6 h after middle cerebral artery occlusion (MCAO), suggesting that the course of ferritinophagy in cortical neurons is highly activated by stroke. 69 In addition, Fe2+ can be released even after bounding to FT, which will be potentiated especially when the intracellular fluid gets acidic and in the presence of superoxide radicals. 70 , 71
The limited iron excretion is related to Fpn1 dysfunction. Recently, evidence shows that the microtubule‐associated protein tau, which regulates the stability and dynamics of microtubules in neurons, is closely related to the function of Fpn1. 72 In addition to being the major component of neurofibrillary tangles in AD and frontotemporal dementia, tau is prone to progressive hyperphosphorylation in neural disease, which makes it easy to aggregate/deposit and interferes with normal cellular functions. 73 Therefore, tau seems to contribute to the development of neurodegeneration. Patients were found to face more severe neurological damage following MCAO, including dilated motor function deficit, high frequency of epileptiform discharge activity, and markedly larger cerebral infarction, in the presence of tau. 74 , 75 The existence of tau connects iron overload with ipsilateral hemispheric stroke to some extent. 76 Tau facilitates iron export by trafficking amyloid precursor protein possessed with ferroxidase activity to stabilize Fpn1, while this process is suppressed by ischemia‐reperfusion injury, resulting in intracellular iron retention as a new mechanism of iron accumulation after stroke. 76 , 77 Indeed, compared with wild‐type neurons, the iron export rate and half‐maximal lethal dose for Fe2+ toxicity were significantly lower in tau‐knockout neurons, which implies getting rid of excessive iron is anticytotoxic. 78
Compared with iron accumulation, poststroke iron deficiency in the brain has rarely been reported. However, in recent years, there has been evidence linking IS to serum FT deficiency, or called anemia in stroke recovery. 79 Anemia after IS was found to be associated with lower muscle strength and cognitive impairment in patients, so early identification of poor iron status might help to identify patients with poor functional outcomes after rehabilitation. 80 However, the pathomechanism linking serum iron deficiency to IS remains unknown. Moreover, brain iron homeostasis is not vulnerable to circulating iron omeostasis imbalance due to the protection of BBB, so it remains to be unclear whether brain experiences a state of iron deficiency under the condition of serum iron deficiency after AIS and whether serum iron deficiency is associated with brain iron accumulation after stroke. Addressing these issues will provide new insights into improving iron chelation therapy.
3.1.2. Hemorrhagic stroke
The excess iron in HS comes mainly from the intracerebral. The most common cause of HS is aneurysm rupture caused by hypertension. Mass blood‐borne cells, chemicals, and fluid from blood vessels invade local brain parenchyma across the ruptured vessel walls. 81 Hematomas composed of numerous red blood cells are formed surrounded by the brain parenchyma. During the absorption of the hematoma, the red blood cells disintegrate and shed hemoglobin. The phagocytosis of erythrocytes by microglia is mediated by CD36, a well‐recognized integral cell membrane protein. 82 On the one hand, microglia are activated to express mass heme oxygenases (HOs), which engulfs and decomposes heme within hemoglobin, on the other hand, they highly express FT to chelate iron. 83 , 84 Excess iron that cannot be chelated will either be transferred into neurons via the Tf–TfR1 system or released in the cytoplasm of microglia, both of which exert neurotoxicity. 85 , 86
In addition, It is reported that voltage‐gated calcium channels act as a facile conduit for the entry of free ferrous iron into neurons under iron overload conditions. 87 L‐type calcium channel antagonists represented by nimodipine are used in the treatment of secondary arterial vasospasm after aneurysmal subarachnoid hemorrhage and intracerebral hemorrhage. Studies found that nimodipine may serve as a protective agent for cells highly susceptible to iron toxicity but with limited neurological function improvement. 88 , 89 Therefore, L‐type calcium channels seem to aggravate iron‐induced neurotoxicity, however, the fraction of free iron being imported through this pathway under iron overload conditions has not been elucidated. Whether the inhibition of this channel can achieve the desired effect of iron clearance without affecting ion transport in other cells (e.g., cardiomyocytes) is still unknown. Further studies are still needed to confirm the protective role of L‐type calcium channel inhibitors in preventing neurons from iron‐induced neurotoxicity after HS.
3.1.3. Neurogliocytes in stroke
Neurogliocytes are the major regulators of the peri‐infarct/hematoma environment in the central nervous system and have been implicated in poststroke immune response and iron regulation. 90 , 91 The response of neurogliocytes in vivo would reflect the development of neuroinflammation after stroke to some extent. Therefore, tracking the response patterns of neurogliocytes could provide insight into finding targeted treatments for intralesional iron accumulation and neurological injury.
Microglia, as the resident immune cells in the brain, play a fundamental role in mediating poststroke neuroinflammation and iron absorption, usually react faster than astrocytes to pathological stimuli, thereby regulating and activating nearby astrocytes and microglia. 92 After stroke, microglia is capable to switch phenotype from proinflammatory (M1) to anti‐inflammatory (M2) phenotype so facilitating immune response to damage and repair in the nervous system, and the two phenotypes coexist but predominate at different stages. 93 In murine models of IS, the M1 phenotype predominates during the first two weeks of injury, which corresponds to the upregulated expression of FT heavy chain and FT light chain, suggesting that the phagocytosis of microglia is highly active to iron in the early IS. In contrast, the M2 phenotype was found low at the early stage and gradually increases and predominates over two weeks, which corresponds to the increased main proteins in the myelin sheath, suggesting that the remyelination of damaged axons and neurologic repairment by using iron is under process in the later period. 94 Compared with IS, the M1–M2 phenotype switch occurs earlier in HS (Figure 2). 95 , 96 , 97 , 98 Augmented M2 activation promotes vascularization in the injured zone, thus contributing to improvements in both tissue reorganization and functional recovery. A recent study shows that the interferon regulatory factor (IRF) 5–IRF4 regulatory axis functions to switch the polarization of microglia in stroke brains and change the outcome of stroke by tilting the axis. 99
FIGURE 2.
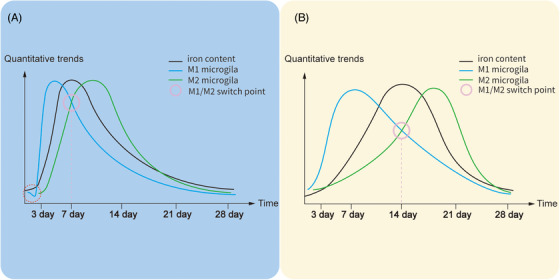
Timeline of microglia and iron after hemorrhage stroke (A) and ischemic stroke (B). These timelines are based on the data from the male rat model of AIS and HS. 95 , 96 , 97 , 98 Iron deposition in hemorrhage stroke is more rapid than in ischemic stroke due to the release of iron by the dissolution of mass red blood cells. The microglia numbers decrease (the red cycle) in the peri‐hematoma area shortly after HS, which might ascribe to necroptosis, and apoptosis caused by iron release from the hematoma, and significantly increase and peak at days 3−7. In contrast, microglia numbers in the core infarct area reach the peak during days 7−14 after IS and return to normal after day 28. Therefore, the reaction speed and proliferation ability of microglia cells were stronger in hemorrhage stroke, leading to an earlier peaking time of microglia proliferation as well as the earlier switch of the microglial phenotype (M1–M2).
Astrocytes and oligodendrocytes play a crucial role in injury and repair response after stroke. Astrocytes are the most abundant glial cells in the brain, playing an important role in the blood–brain barrier maintenance, neuroinflammatory response, and the absorption and transport of iron. 29 , 100 , 101 , 102 , 103 , 104 , 105 As a buffer pool of iron, astrocytes possess a high‐speed capacity for iron transport mainly due to their high expression of TfR1, DMT1, Fpn1, and Cp, which facilitates taking up iron from the abluminal side of BMECs and mobilizing it into the brain interstitial fluid, precisely regulating the iron concentration in neurons. 23 , 29 In response to stress, NADPH oxidase (NOX) 4 inside astrocytes was quickly activated to produce ROS, which further induces NF‐κB signaling and upregulates inflammation and lipocalin‐2 (LCN2) gene expression. LCN2 is capable to promote iron accumulation in neurons and glial cells by acting as an iron transporter, 106 , 107 , 108 , 109 and triggers the shift of astrocytes from an anti‐inflammatory towards a proinflammatory phenotype and contributes to BBB breakdown. 110 , 111 Therefore, astrocytic response serves an important role in oxidative stress and brain injury following stroke. Oligodendrocytes possess the highest iron levels in the brain due to their metabolic needs associated with the process of myelin formation, accurate wrapping, and repair in various brain diseases. Microglia and astrocytes are capable of releasing Fe3+ contained in FT to support oligodendrocytes for myelination or remyelination. 112 However, as iron over accumulates after stroke, oxidative stress can damage oligodendrocytes and myelination, thus amplifying tissue damage. 113 Compared with mature oligodendrocytes, oligodendrocyte progenitor cells (OPCs) are more susceptible to excitotoxic injury. 114 Combined with recent research that hemoglobin causes damage to mitochondrial function in primary cultured OPCs, inhibits OPCs proliferation, and even cell death, it should be emphasized the importance of protecting OPCs against cell death after stroke to reduce brain injury and promote recovery. 115 , 116
Therefore, controlling the response of neurogliocytes, like the phenotype switch of microglia, the expression of inflammatory mediators like LCN2, and protecting OPCs might provide attractive therapeutic targets for stroke. However, it also should be noted that though M1 microglia might increase inflammation levels in the brain, the long‐term and improper inhibition of the M1 microglia might cause an adverse effect on the phagocytosis to iron and other fragments, thus leading to the deterioration of neurological function. Similarly, irrational or overused administration of iron chelators might interfere with the remyelination of damaged axons by oligodendrocytes during the recovery, thus causing poor outcomes though iron chelators have been proven effective in treating iron overload.
3.1.4. Delayed iron deposition after stroke
Iron is an important component of the redox balance in the nervous system, and the regulation of brain iron homeostasis is of great significance to the recovery of neurological function after stroke. Despite breakthroughs in treatment of acute stroke in recent years, which have greatly improved the survival rate of stroke patients, how to reduce the long‐term functional impairment for stroke patients is still a matter of concern. The disruption of brain iron homeostasis after stroke will not only affect the ischemic areas but also impose a long‐lasting effect (the delayed iron deposition) on the remote but functionally or anatomically connected tissues, which get disconnected for primary ischemic injury. The delayed iron deposition would not be observed shortly after stroke but is gradually emerging over time. With a progressive increase in locally deposited iron, neurodegeneration of corresponding brain areas is obvious progressively, which eventually leads to poor outcomes for patients. 117 As early as 1991, A Tamura et al. 118 found that ipsilateral thalamic atrophy 1 year later, as well as alterations of the thalamic MRI signal, follows distant infarcts caused by MCAO, which is initial evidence for stroke‐related remote change. In 2017, Kuchcinski et al. 119 found in a longitudinal study that thalamic R2* signal remains unchanged in the subacute phase of stroke (24–72 h after symptom onset) but was elevated 1 year after the cerebrovascular acute event and confirmed that infarcts in different regions of the brain lead to delayed iron deposition in the corresponding nuclei of the thalamus and ultimately worsen cognitive function in patients, which provides important evidence for the link between delayed iron deposition and neurodegeneration. In 2019, Linck et al. 120 found infarcts of the striatum caused chronic iron deposition in the ipsilateral SN. Further, the asymmetry of bilateral iron deposition signal captured by MRI R2* is positively correlated with the poorer motor function of the nondominant arm. In other words, chronic iron deposition in the ipsilateral SN after stroke aggravates the defective motor ability of patients. Therefore, the evidence of neuroimaging above shows that stroke not only disrupts the brain iron homeostasis at the early stage of the disease but also changes the distribution of brain iron even after recovery, thus affecting the motor and cognitive functions of patients in the long term. Furthermore, histological studies demonstrate this association by verifying that the neurodegeneration of SN and thalamus induced by striatal lesions or corticothalamic disconnection respectively are related to the increased number of iron‐containing macrophages and/or microglia and extracellular iron deposition. 121 The foundation of these collaborative changes lies in a large array of functional connections between different brain regions. This provides us with a new perspective for understanding the different patterns of iron deposition in the brain responding to the different stroke regions.
3.2. Iron metabolism in AD
Alzheimer's disease is a progressive and irreversible disease of neurodegeneration by far, which is typically characterized by an abnormal deposition of β‐amyloid (Aβ) plaques in extracellular plaques and neurofibrillary tangles formed by phosphorylated tau in neurons. 122 Studies have found that iron content in the AD brain is significantly increased, which promotes the cognitive deficits of AD brains. 123 , 124 , 125 , 126 The association of iron accumulation with AD pathology is demonstrated in quantitative susceptibility mapping (QSM), which is highly sensitive to tissue iron, further confirming the reliability of iron as a biomarker for AD progression. 127 , 128 , 129 Therefore, elucidating the mechanism of iron metabolism disorder in AD is crucial for its future treatment. Although the exact mechanisms of iron accumulation in AD remain unclear, it was thought to be involved in several following mechanisms: (i) Aβ plaques aggregation; (ii) phosphorylated tau deposition; (iii) neuroinflammation; (iv) dysfunction of BBB.
3.2.1. Aβ and iron accumulation: a vicious cycle
Aβ generates from the amyloidogenic processing of Aβ precursor protein (APP), a ubiquitously expressed type 1 transmembrane protein in cells. 130 The amyloidogenic way starts from the endocytic internalization of APP on the cell membrane. Internalized APP is trafficked to endosomes via the endocytic trafficking regulator ADP ribosylation factor 6 (ARF6), where APP is cleaved by β‐secretase (mostly BACE1 in the brain) and γ‐secretase, along with Aβ releasing. 131 , 132 , 133 In contrast, the nonamyloidogenic processing of APP, as the predominant way, generates neurotrophic sAPPα with the help of α‐secretase cleavage at the cell membrane.
Previous studies suggested that APP promotes neuronal iron export by stabilizing Fpn1, the only iron export protein of cells. 134 , 135 This means that the cleaving of APP would decrease iron export, either in the amyloidogenic or nonamyloidogenic way. However, sAPPα, instead of APP, was found to work on iron export by stabilizing Fpn1 in the latest research, which further proves the amyloidogenic processing of APP is a harmful way to iron retention. 136 , 137 , 138 , 139 Aβ is able to activate microglia and promote the production of lactoferrin by binding to the NLR family pyrin domain containing 3. The interaction between the iron‐bound form of lactoferrin and APP diverted neuronal APP endocytosis from the clathrin‐dependent pathway (amyloidogenic process) to the nonamyloidogenic process mediated by ARF6, further aggravating iron retention. 140 Therefore, rational inhibition of the amyloidogenic process appears to be a viable approach to treating AD. However, the functional abnormalities reduction mediated by BACEi inhibition in AD brains is found unstable either in animal models or clinical trials in humans. 141 , 142 This may attribute to differences in patient populations, mouse models, or methods used, and whether compensatory pathways for the amyloidogenic process exist remains to be explored.
The interaction between iron retention with Aβ plaque within neurons appears to form a vicious cycle that promotes cytotoxicity. First, Fe2+ and Fe3+ play key roles in Aβs folding and aggregation. Iron ions, especially Fe3+, can promote β‐sheet formations on the Aβs C‐terminal, which is favorable to reducing negative surface charges of Aβs. 143 When electrostatic repulsion between the negatively charged domains of Aβs reduces, the Aβs aggregation becomes energetically favorable. Second, excess intracellular iron prevents IRP from binding to IREs in the 5′ untranslated regions of APP mRNA, leading to elevated APP generation, which could contribute to Aβ pathology partly. 144 In addition, Fe3+ can be reduced to Fe2+ by binding plaque Aβ along with the generation of H2O2, thus propagating a continued source of ROS, and contributing to lipid peroxidation levels. 145 , 146 , 147 This seems to explain how AD, as a progressive disease, promotes the production of its key pathogenic substances continuously by using the imbalance of iron homeostasis as a mediator, leading to a high redox state in the nervous system, neuronal vulnerability to ROS, and eventually develops into a neurodegenerative disease.
3.2.2. Phosphorylated tau: decreased iron export
Tau, as the major component of neurofibrillary tangles, functions to mediate iron export from cells by trafficking sAPP to stabilize Fpn1. 122 Loss of soluble tau protein due to deposition in neurofibrillary tangles may lead to iron retention. It was reported that the sites of iron deposition were found spatially colocalized with areas of tau burden, which further confirms the association between iron homeostasis disorder and tau pathology. 148
As described in AD, Aβ deposition can cause iron homeostatic imbalance in the AD brain. The formation of neurofibrillary tangles follows after that of Aβ plaque in AD. Iron accumulation is able to enhance the hyperphosphorylation and aggregation of tau by stimulating the activity of the cyclin‐dependent kinase 5 complex and glycogen synthase kinase‐3β kinase. 149 , 150 In addition, HO‐1 protein can be detected in the neurofibrillary tangles in AD brains. 85 On the one hand, elevated HO‐1 reduces the expression of memory‐related synaptic proteins, aggravates iron accumulation, and enhances tau phosphorylation in HO‐1 transgenic mice. 151 On the other hand, Mass generation of HO‐1 in AD may lead to the metabolism of heme from damaged mitochondria, thus releasing Fe2+, and finally increasing oxidative stress level and neuronal damage. 152 Slight cognitive impairment in AD patients has been shown to correlate with the increased concentration of HO‐1 in the frontal cortex and hippocampus. 153
3.2.3. Neuroinflammation
Neuroinflammation, resulting from microglia activation, is another hallmark of AD. On the one hand, the deposition of Aβs attracts chemotaxis of microglia and drives neuroinflammation by activating NLRP3 inflammasomes in microglia. 154 , 155 Meanwhile, NLRP3 inflammasome activation is also amplified by the interaction between Aβs and the adaptor protein apoptosis‐associated speck‐like protein containing a CARD (ASC), thus further recruiting and activating procaspase‐1 and contributing to pyroptotic neuron death. 156 On the other hand, disruption in iron homeostasis triggers microglial polarization to the proinflammatory (M1) phenotype via ROS, resulting in a higher secretion of proinflammatory cytokines in microglia, such as TNF‐α and IL‐1β, and further aggravating neuroinflammation. 157 And IL‐1β and TNF‐α function to promote iron retention in neurons by upregulating TfR1 and DMT1 and downregulating Fpn1. 158 In addition, despite a strong resistance to iron accumulation of reactive astrocytes, astrocytes can still be activated by Aβs in AD, then releasing proinflammatory mediators and cytotoxic molecules, leading to neuroinflammation, and AD pathology exacerbation. 22 , 159
3.2.4. Dysfunction of BBB
The ε4 allele of the apolipoprotein E (APOE) gene is validated as the strongest genetic risk factor for late‐onset AD and greatly affects the progression of Aβ pathology. 160 APOE‐ε4 can reduce the expression of aquaporin‐4 (AQP4), a set of perivascular water channels, contributing to aberrant Aβ plaque accumulation. 161 Notably, the BBB dysfunction caused by APOE‐ε4 in the capillaries is similar to the pathomechanism of cerebral small vessel disease, where iron leakage occurs in the brain due to BBB dysfunction. 162 APOE‐ε4 and mild cognitive impairment were associated with higher cortical iron in a neuroimaging study using a 7.0T QSM, which may suggest iron leakage by impaired BBB in an APOE‐ε4 carrier. 163 However, the association between APOE gene and iron accumulation seems to be less than strong in iron heritability by APOE. 148 , 164 In addition, vascular permeability factors secreted by activated astrocytes, such as vascular endothelial growth factors, can participate in the dysfunction of BBB and may affect the association between APOE and iron status. 165 Therefore, the relation between APOE genotype and brain iron status remains elusive and to be researched.
3.3. Iron metabolism and PD
Parkinson's disease is the second most common disorder after AD, with a global prevalence of more than 6 million individuals. 166 The major pathological hallmarks of PD include (i) the progressive iron deposition along with loss of DA neurons in the substantia nigra pars compacta (SNpc); (ii) the misfolding and aggregated α‐synuclein (α‐syn) result in the formation of Lewy bodies. 167 The correlations between iron deposition and loss of DA neurons in the SNpc of PD brains have been proven in neuropathological and imaging studies, indicating that the imbalance of iron homeostasis participates in the pathology of PD progression. 168 , 169
3.3.1. α‐Synuclein and iron accumulation: mutual reinforcement
α‐Syn is a water‐soluble protein consisting of 140 amino acids and highly enriched in the presynaptic membrane. 170 Although the physiological function of α‐syn is remain unclear yet, it may play a role in maintaining the synaptic function, regulating the storage and release of synaptic neurotransmitters. 171 In PD, α‐syn goes from the physiological monomer form to the pathological oligomers, protofibrils form, and aggregates to insoluble clumps after stepwise modification. Finally, α‐syn aggregates and interact with membranous organelles to produce Lewy bodies. 172 Postmortem analysis of patients with PD shows that iron and α‐syn coexist in the Lewy bodies in the SNpc, suggesting an interaction between iron deposition and α‐syn pathology. 173 Indeed, α‐syn aggregation plays a vital role in disrupting iron homeostasis. Guo et al. 174 have reported intranasal injection of exogenous α‐syn preformed fibrils can cause iron deposition in the substantia nigra–striatum system of monkeys. On the one hand, as an iron‐reductase and iron‐binding protein, α‐syn is able to reduce Fe3+ to Fe2+, increasing the intracellular ferrous iron content, thereby increasing the generation of ROS and oxidative toxicity. 175 On the other hand, many studies show the expression of α‐syn is associated with the Tf–TfR1 pathway of iron uptake. It has been reported that α‐syn enhanced clathrin‐mediated endocytosis in neuronal cultured cells. 176 The hidden mechanism of this interaction was uncovered by recent evidence that α‐syn accelerates the rate of synaptic vesicle endocytosis alongside reducing the fraction of released synaptic vesicles through its activity to enrich the plasma membrane with phosphatidylinositol 4,5‐bisphosphate and phosphatidylinositol 3,4‐bisphosphate. 177 Additionally, decreasing TfR1 and increasing FT were found in α‐syn‐knockout cells in culture, indicating that α‐syn functions to promote iron import and inhibit its storage, which is consistent with previous research. 178 , 179 The research above further describes evidence for a pathogenic role for α‐syn in dysregulating iron in PD.
At the same time, a lot of research has studied the role of iron in α‐syn aggregation and the regulation of posttranslational modification of α‐syn. It evidenced that α‐syn oligomerization and iron accumulation is a mutually reinforcing process. On the one hand, iron promotes α‐syn aggregation by binding to α‐syn and inducing its conformational changes. On the other hand, increased iron grabs IRP from the α‐syn mRNA, thus promoting the translation of the α‐syn mRNA. 144 However, the link between iron and α‐syn goes deeper than this. A recent study reported that iron enrichment occurring in particular areas of PD brains affected the spreading pathology of α‐syn in mice models. 180 Striatal iron retention alters the connectivity of the brain connectome, leading to a redistribution of α‐syn aggregates in the brain, or rather the restriction of phosphorylated α‐syn spread. Previous research with PD patients has reinforced this conclusion, suggesting iron modulates the degree of cross‐talk between the striatum/substantia nigra and cerebral cortex. 181 , 182 In addition, iron levels in the brain are known to increase with age, which seems to provide a reasonable explanation for the recent research that task functional connectivity in brains of the elder was found less strongly correlated as compared with the younger. 183 Therefore, the rise in brain iron levels seems to play a protective role in PD to a certain extent; however, whether the protective effect of brain iron is related to its content or the different stages of PD remains to be explored.
3.3.2. Neuromelanin: storage of iron and spark of neuroinflammation
Another typical neuropathological feature of PD is the depigmentation of dopaminergic neurons containing neuromelanin (NM) in SNpc along with the iron deposition throughout PD. 184 Neuromelanin is the main iron chelator in SN neurons due to the low expression of FT, which plays its role mainly in the glia. 185 Similar to FT, neuromelanin also sequesters iron by binding it in a redox‐stabilized ferric form and forming iron oxide clusters. 184 , 186 Zecca et al. 187 examined metal contents in neuromelanin and showed that per gram of neuromelanin from SN contained 10.9 μg iron, which confirms the ability of neuromelanin to chelate iron. As an excellent chelator of iron, neuromelanin protects neurons from toxic compounds under physiological conditions.
However, excessive accumulation of NM in the course of PD increases the vulnerability of dopaminergic neurons and induces their death to a certain extent. Intraneuronal neuromelanin was found to increase the content of major histocompatibility complex class I (MHC‐I) in neuromelanin‐containing neurons in SN. 188 MHC‐I functions to bind antigenic peptides and present them on neuronal membranes so that cytotoxic CD8+ lymphocytes could target neurons inducing their death. 188 , 189 Furthermore, recent neuroimaging studies also confirm the neuronal loss in dopaminergic neuromelanin‐pigmented neurons of the SNpc by providing evidence that dopaminergic function in striatum and substantia nigra negatively relates to NM‐sensitive signal and lateral SN R2* (on behalf of iron content). 190 , 191 After neuron death and collapse, neuromelanin is released into the extraneuronal space and slowly phagocytosed by microglial cells. The degradation of neuromelanin can not only activate microglial cells leading to the release of inflammatory and toxic mediators including TNF‐α and IL‐6 but also discharge toxic compounds and excess iron ions. 192 In addition, IL‐6, via its trans‐signaling pathway, induces changes in the neuronal iron transcriptome that promote ferrous iron uptake and decrease cellular iron export via the cellular iron sequestration response. 193 The increased inflammatory factors and iron cause the generation of free hydroxyl radicals in the cytoplasm, exacerbating the peroxidation of lipids and aggregation of α‐syn, resulting in PD progression and neuronal loss.
With the rapid development of neuroimaging research in recent years, the traces of iron and neuromelanin in PD have been gradually uncovered. 7T‐MRI brings us clear images in comparison of substantia‐nigra and R2* signal intensity between PD patients and the normal, intuitively showing elevated iron deposition and decreased neuromelanin in PD patients. 194 The temporal changes of PD progression show that PD begins with striatal dopaminergic dysfunction, followed by SNpc iron accumulation preceding SNpc neuromelanin loss and, finally, dopaminergic cell loss, which suggests the interdependent changes between dopaminergic neurons, neuromelanin, and iron; moreover, brain iron deposition is more likely a consequence rather than a primary driver of neurodegeneration. 168 The ability to detect neuromelanin with NM‐MRI and iron changes with R2∗ or QSM in the SNpc is valuable in helping the early diagnosis and monitoring progression of PD. More importantly, a comprehensive set of imaging biomarkers derived from neuromelanin and iron content in SNpc for PD diagnosis gives a better result than the detection of either alone and potentially, could be used to diagnose PD clinically. 195
3.3.3. Mitochondrial dysfunction and defective mitophagy
Mitochondria are the main organelles in the cells that produce ATP. Therefore, the functional stability of mitochondria is essential for cell survival, especially in neurons. Because brain is the most energy‐intensive organ in the body and consumes 20% of body oxygen for energy production, which implies that any disturbances of mitochondria function might cause oxidative stress and even neuronal death, for instance, iron overload. Iron plays an important role in the mitochondria and is mainly used in three pathways: heme synthesis, Fe–S cluster generation, and iron storage in the FtMt. Multiple evidence indicates that oxidative stress induced by mitochondrial dysfunction mainly due to decreased activity of respiratory chain complex I is critical to PD pathogenesis. 196 , 197 Treatment with complex I inhibitor rotenone was able to reproduce features of the PD in animal models, including SN dopaminergic degeneration, Lewy bodies generation, and oxidative stress. 198 Recent research reported intracellular neuromelanin increased mitochondrial oxidative stress and exacerbated mitochondrial bioenergetic deficits mediated by heterocyclic aromatic amines, a substance that causes dose‐dependent cell death specifically to dopaminergic neurons potentially through inhibition of specific complexes of the electron transport chain. 199 , 200 In addition, it is also suggested that the accumulation of α‐syn in PD inhibits mitochondrial function by affecting the level of mitochondrial sirtuin 3 protein, an NAD+‐dependent deacetylase that regulates the acetylation/deacetylation level of complex I. 201 The sirtuin 3 in mitochondria has been proven to prevent dopaminergic neuron loss in a rodent model of PD. 202 This indicates that dysfunction of complex I plays an important role in mediating PD pathology. In addition, complex II–IV are also inhibited to varying degrees in PD, suggesting impaired mitochondrial oxidative respiration in PD. 203 And this lessened capacity of mitochondrial electron transport chain is able to elevate mitochondrial iron uptake and ROS production. 204
Mitophagy is a fundamental mechanism that regulates mitochondrial quality by a selective clearance of dysfunctional mitochondria in dependence on lysosomes. 205 In this way, neurons can protect themselves from ROS damage and maintain physiological activity. It has been known to us that defective mitophagy due to Pink1/Prkn mutations leads to iron accumulation during a neurodegenerative process, such as PD. 206 , 207 , 208 Recently, subcortical brain iron deposition was found highly predictive of mitochondrial impairment in patients with PD in vivo, which suggests a synergistic role between iron accumulation and mitochondrial damage in neurodegenerative diseases. 209 Impaired mitochondria and defective mitophagy may disrupt iron homeostasis, and altered iron metabolism may, in turn, cause mitochondrial damage, forming a detrimental vicious cycle. Until now, it is still unclear whether mitochondria damage and defective mitophagy are causes or consequences of iron accumulation, but there is no doubt that both of them can accelerate PD progress and concur with the pathology of age‐associated neurodegeneration.
3.4. Other neurological diseases associated with iron metabolism
Huntington's disease (HD) is a progressive neurodegenerative disorder induced by the expansion of a CAG repeat in the huntingtin gene on chromosome 4, which leads to mutant huntingtin protein misfolding in neurons and microglial cells. 210 Characteristic iron accumulation in HD patients occurs mainly in the striatum and cerebral cortex. The change of iron homeostasis in the HD brain may be associated with the mutation HTT. On the one hand, It was demonstrated that mutation of HTT upregulates the protein related to iron import, such as IRP1, Tf, and TfR, leading to iron accumulation in the striatum and cortex of HD mice. 211 On the other hand, HTT mutation can disrupt mitochondria energy metabolism including inhibition of mitochondrial respiratory complexes II–IV, mitochondrial trafficking abnormality, and the impairment of mitochondrial, thus leading to abnormal ATP generation and neuronal death. 212 , 213 , 214 , 215 In addition, previous studies have evidence that microglial can promote the progression of HD under the activation of iron accumulation, however, the related mechanism is unclear. It was recently reported that iron contributes to HD progression and neurodegeneration by activating the microglial enzyme indoleamine‐2,3‐dioxygenase in the kynurenine pathway to generate increased amounts of neurotoxic intermediates 3‐hydroxykynurenine and quinolinic acid. 216 This further supports a causal relation between iron accumulation, microglial activation, and neurodegeneration in HD and provides a potential target for the treatment of HD in microglia‐related mechanisms.
In addition, some of the rare neurodegenerative diseases with iron homeostasis imbalance involve amyotrophic lateral sclerosis, multiple sclerosis, Wilson's disease, and a disease group called neurodegeneration with brain iron accumulation (NBIA), which is a clinical syndrome comprised of a genetically heterogeneous group of disorders. NBIA is characterized by iron accumulation in different regions of the brain, mainly within the basal ganglia, with the symptoms of movement disorders (dystonia–parkinsonism, cerebellar ataxia), pyramidal dysfunction, dysgnosia, neuropsychiatric abnormalities, and early death. The diagnosis of NBIA relies upon iron accumulation detected by brain MRI combined with clinical features and is finally confirmed by gene analyses. It includes but is not limited to Friedreich's ataxia, aceruloplasminemia, neuroferritinopathy, and pantothenate kinase‐associated neurodegeneration. The genetic basis of the majority of NBIA disorders has been elucidated so further detail is not discussed here. 217
4. FERROPTOSIS: THE ENDING OF IRON OVERLOAD
Ferroptosis is a newly identified iron‐dependent form of regulated cell death, morphologically characterized by a normal nucleus, but shrinking mitochondria with condensed membrane density, a reduction of mitochondrial cristae, and ruptured outer membrane compared with programmed cell death such as apoptosis and autophagy. 218 In recent years, the mechanism of the iron homeostasis imbalance represented by ferroptosis in the progression of neurodegenerative diseases has been supported by increasing amounts of evidence. Ferroptosis, characterized by altered brain iron homeostasis, dysregulation of the antioxidant system, and oxidative damage, plays an important role in the progression of neurodegenerative diseases such as stroke, AD, PD, and so on. 116 , 219 , 220 , 221 , 222 , 223 , 224 , 225 , 226 Therefore, elucidating the mechanisms involved in ferroptosis is crucial for the treatment of neurodegeneration. Taking the classical mechanism of ferroptosis as an example, starting from the accumulation of intracellular iron, ferroptosis follows closely behind the trilogy: (i) mass production of ROS; (ii) massive peroxidation of phospholipids (PLs) containing polyunsaturated fatty acids (PUFA‐PL) in the plasma membrane; (iii) collapse of the antioxidant system caused by reduced GSH (Figure 1).
4.1. Mass generation of ROS
ROS generation is an important prerequisite for lipid peroxidation and ferroptosis. Under physiological conditions, intracellular ROS mainly comes from the respiratory chain in mitochondria, where the redox reaction is approaching equilibrium. Generally, incompletely reduced forms of dioxygen, such as superoxide anion (O2 •−) and H2O2, are produced at considerably high rates (about 1−4% dioxygen undergoes a reduction reaction) in the process of reducing dioxygen to water completely. And such reduced dioxygen in vivo can be generated from other enzymes, such as oxygenases, oxidases, and peroxidases. For example, H2O2 produced by xanthine oxidase is able to significantly aggravate ischemic/reperfusion injury. 227 , 228 Reduced dioxygen can not only cause or promote oxidative stress directly, but also lead to the mass generation of secondary products with oxidative activity and destructiveness by reacting with other specific substances, such as hydroxyl radical (OH•) which is extremely toxic and produced by the Fenton reaction.
OH• is able to react with multifarious biological molecules, such as DNA, proteins, and unsaturated lipids, thus damaging cell functions. 229
4.2. Massive peroxidation of PUFA‐PL
Peroxidation of PUFA‐PL is the key to executing ferroptosis. When peroxidation exceeds the buffer capacity provided by the antioxidant system, it will lead to the fatal accumulation of lipid peroxides on the cell membrane and the subsequent collapse and rupture of the cell membrane, then ferroptosis happens as a result. Acyl‐CoA synthetase long‐chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are acknowledged as the key regulatory factors for PUFA‐PL synthesis. ACSL4 catalyzes the addition of CoA to the long‐chain polyunsaturated bonds of PUFA, thereby promoting the formation of coenzyme A‐activated polyunsaturated fatty acid (PUFA‐CoA), followed by the esterification of PUFA‐CoA to PLs mediated by LPCAT3. 230 , 231 Lipid peroxidation is initiated in two ways: enzymatic reaction meditated by lipoxygenases (LOXs) and nonenzymatic reaction, which is iron dependent. The formation of lipid radicals (R‐OOH) can be directly catalyzed by LOXs, a family of enzymes that contains nonheme iron for their activity and is critical for multiple signaling pathways, such as arachidonic acid LOXs. 232 Multiple enzymes have been proven to drive lipid peroxidation, for instance, 15‐LOX, by binding to phosphatidylethanolamine binding protein 1, is able to switch the substrate specificity of the enzyme from free PUFAs to PUFA tails of PLs thus promoting the peroxidation of PUFA‐PL, 12‐LOX is crucial to ferroptosis mediated by p53, and cytochrome P450 oxidoreductase is involved in lipid peroxidation in ferroptosis. 233 , 234 , 235 Moreover, these enzymes are all iron dependent.
Lipid peroxidation occurs mainly through nonenzymatic pathways, which, as ROS dependent, can be divided into three phases: initiation, propagation, and termination (Figure 3). In the initiation phase, labile oxidants (OH•) grab hydrogen atoms from the bisallylic methylene of a membrane PL‐PUFA, leaving a phospholipid radical (PL’•) centered on carbon. The PL’• cannot keep self‐stable and reacts with oxygen producing another substrate with oxidative capacity—phospholipid peroxy radical (PLOO•), which is able to extract hydrogen atoms from surrounding PL to generate a phospholipid hydroperoxide (PLOOH) along with a new PL’•. PLOOH is an essential free radical precursor that initiates the next peroxidation chain cycle(progression) in which the Fenton reaction is involved. As a circulating substrate in the phospholipid peroxidation, if the generation of PLOOH in neurons exceeds the capacity of the peroxide scavenging system, it can propagate peroxidation to adjacent PUFA‐PLs on the PL membrane in the presence of labile iron, thereby disrupting the permeability and fluidity of the membrane and ultimately causing ferroptosis. 236 Antioxidants, like GSH, can terminate radical propagation by donating electrons to radical compounds without being a radical, which is a primary mechanism of defense against lipid peroxidation and other oxidative damage.
FIGURE 3.
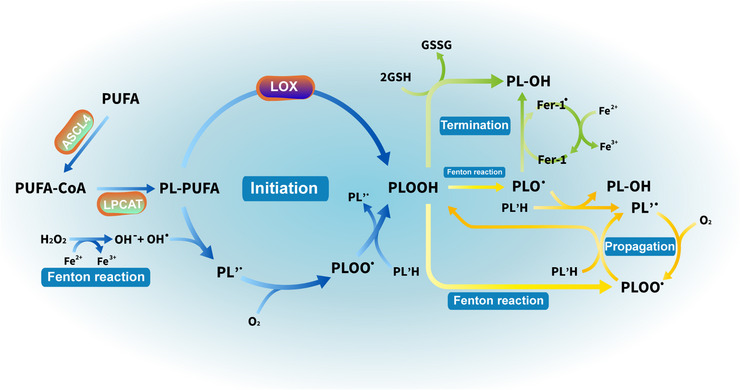
The three phases of lipid peroxidation. (i) Initiation is the process that generates radical compounds (PLOOH/PLOO•) from nonradical molecules (PL‐PUFA). (ii) Propagation, starting from lipid peroxides (PLOOH), is a peroxidative chain reaction giving rise to new radicals while the number of radicals remains constant. (iii) Termination is the process in which the peroxidative chain is broken by donating electrons to radical compounds so that transferring the active product (radical compounds) to a stable one (PL‐OH). It should be noted that the iron‐involved Fenton reaction, which participates in the process of initiation and propagation, is a free electron provider. PUFA, polyunsaturated fatty; ACSL4, acyl‐CoA synthetase long‐chain family member 4; PUFA‐CoA, coenzyme A‐activated polyunsaturated fatty acid; LPCAT3, lysophosphatidylcholine acyltransferase 3; PL‐PUFA/PL/PL'H, phospholipids containing polyunsaturated fatty acids; LOX, lipoxygenase; PLOO•, phospholipid peroxy radical; PLOOH, phospholipid hydroperoxide; PLO•, the phospholipid alkoxyl radical; GSH, glutathione; GSSG, oxidized glutathione; Fer‐1, ferrostatin‐1; PL‐OH, phospholipid hydroxides.
4.3. Reduction of GSH: the collapse of antioxidant system
As the major intercellular antioxidant against oxidative stress, GSH plays a critical role in protecting cells from ROS attack. 237 By binding to intracellular Fe2+, GSH is able to effectively reduce the labile iron pool, which in turn reduces the generation of lipid peroxides and ROS mediated by Fe2+. The import of raw materials for GSH synthesis is dependent on the system xc− and the excitatory amino acid transporter (EAAT). The system xc− on the cell membrane, as a heterodimeric protein complex, is composed of solute carrier family 3 member 2 (SLC3A2) and solute carrier family 7 member 11 (SLC7A11) and serves its function by importing the extracellular cystine along with exporting the intracellular glutamate by a ratio of 1:1. 238 The cystine transferred into the cell is decomposed into cysteine for further reaction. Neuronal glutamate transporters, mainly EAAT3 in postsynaptic neurons, exert functions by clearing extracellular excess glutamate to maintain a normal concentration gradient of intra‐to‐extra cellular glutamate for normal functioning of the system xc−. In addition, EAAT3 also functions as a unidirectional cysteine import channel that facilitates cysteine entry into the cell. 239 Under the catalysis of γ‐glutamylcysteine synthetase, cysteine combines with glutamate to form γ‐glutamylcysteine, which will next combine with glycine to form GSH. 240 , 241
Under physiological conditions, most glutamates are rapidly cleared by EAAT3 at the postsynaptic membrane after exerting its physiological role at the neuronal synapse, leaving extracellular glutamate at a low level, which is critical to the normal function of synapses and neural circuits. 242 However, pathological conditions, such as ischemia after stroke, cause the dysfunction of EAAT3, resulting in excess accumulation of extracellular glutamate, which inhibits the normal function of the system xc−.246 This is because the glutamate/cystine exchange proceeding depends on the gradient of glutamate toward the cell, rather than consuming adenosine triphosphate actively. With the excess accumulation of extracellular glutamate, cystine turnover is inhibited, ultimately leading to a rapid decline in GSH synthesis. Therefore, normal GSH content is essential for the maintenance of the cellular antioxidant system. The lower baseline plasma GSH levels were reported to be associated with an increased risk of developing neurodegenerative diseases, such as AD and PD. 244 , 245
In addition, the body's antioxidant system also faces the loss of enzyme activity in GSH synthesis, which is crucial to the antioxidant response. As an important antioxidant enzyme, GPX4 conducts its biological activity by converting two molecules of GSH to oxidized glutathione (GSSG) along with reducing phospholipid hydroperoxides (PL‐OOH) to phospholipid hydroxides (PL‐OH), to prevent the accumulation of lipid peroxides, which is toxic. 246 The reserve of GSH is consumed by constant Fenton reaction induced by mass labile iron during stroke and neurodegenerative diseases, leading to a decline in the concentration of GSH as enzymatic reactants, which in turn inhibits GPX4 activity. 247 Moreover, evidence shows that promoting GPX4 activity and supplementation of GSH in neurodegenerative diseases and stroke both can mitigate oxidative stress and inflammatory pathology related to ferroptosis, thereby contributing to better preservation of executive functioning longitudinally. 244 , 248 , 249 , 250 , 251 , 252
Therefore, GSH supplementation appears to be a promising target for stroke and neurodegenerative diseases. However, due to the isolation of BBB and the omnipresent γ‐glutamyl transpeptidase, which is responsible for GSH cleavage, GSH supplementation in vitro fails to achieve the ideal effect in neurological protection. 253 , 254 It was reported in recent years that some stable GSH analogs, prodrugs, and GSH‐coupled nanocarriers, such as ψ‐GSH, l‐cysteine, and γ‐glutamylcysteine, can overcome the disadvantages of GSH administration to achieve the aim of elevating GSH levels in the brain. 249 , 255 , 256 , 257 , 258 , 259 Therefore, drug development in GSH remains to be explored and will focus on the delivery and functional properties of drugs. In addition, although GPX4 plays the central part in ferroptosis inhibition, researchers have found four other systems capable of suppressing ferroptosis act independently of GPX4 in the past few years, including ferroptosis suppressor protein 1 (FSP1)/CoQ10 pathway, GTP cyclohydrolase 1 (GCH1)/tetrahydrobiopterin (BH4) pathway, dihydroorotate dehydrogenase (DHODH) /CoQ Pathway, and p62/Keap1/Nrf2 Pathway. 260 , 261 , 262 , 263 , 264 , 265 Understanding new mechanisms will contribute to developing new targeted drugs and clinical trials.
5. THERAPEUTIC AGENTS FOR TREATING IRON ACCUMULATION AND FERROPTOSIS: IRON CHELATORS AND FERROPTOSIS INHIBITORS
The accumulation of labile iron is an initiating trigger of ferroptosis, which is followed by the peroxidation of membrane lipids by the mass production of ROS, ultimately leading to neuronal death and impairment of neurological function. Therefore, keeping labile iron content in control or inhibiting the occurrence of ferroptosis both work for neurological protection.
5.1. Iron chelators
Deferoxamine (DFO), the most widely used iron chelator approved by the Food and Drug Administration in America, has been used clinically for a long time as a first‐line therapeutic agent to scavenge excess iron in diseases such as thalassemia, and its effectiveness as a neuroprotectant has been demonstrated in experimental stroke models. 266 , 267 The rationale that iron chelators exert neuroprotective effects is mainly based on chelating and scavenging free iron in the blood and enhancing iron excretion by the urine. When Fe3+ comes into contact with DFO, the linear chains of DFO perssad will wrap around Fe3+, forming a stable complex containing iron, blocking the entry of Fe3+ into the Haber Weiss reaction and interrupt the toxic redox cycle, ultimately reducing the generation of ROS and the accumulation of lipid peroxides. 268 The iron chelators reported in recent years and their related mechanisms are summarized in Table 1.
TABLE 1.
Iron chelators for neurological diseases
| Name | Mechanism/effect | Target disease | References |
|---|---|---|---|
| Deferiprone |
|
PD, AD, ALS, NBIA | 269 , 270 , 271 , 272 , 273 , 274 , 275 |
| Deferasirox |
↓The intracellular Fe2+ accumulation, ROS, and lipid peroxidation Maintain BBB permeability ↓Neuroinflammation |
HS, IS | 266 , 276 |
|---|---|---|---|
| TPP‐DFO |
|
NBIA | 277 |
| Quercetin |
Chelate iron ions inside and outside the cell Shuttle intracellular free iron ions to extracellular space through GLUTs (when quercetin's concentrations≦ 1 μM) ↑Hepcidin expression by Nrf2 upregulation |
Stroke, AD, PD | 278 , 279 , 280 |
|---|---|---|---|
| M30 | ↑ HIF‐1 protein, growth factors, and antioxidant enzymes | AD, PD, ALS | 281 |
| PBPT |
Iron chelation ↓Copper‐mediated amyloid‐β aggregation ↓Oxidative stress ↓AChE activity ↑Autophagy |
AD | 284 |
|---|---|---|---|
| Naringin |
|
AD | 285 |
| NBMI | Iron chelation preferential to labile Fe2+ | – | 286 |
|---|---|---|---|
| YL‐939 |
|
– | 287 |
Note: ↑, activate/upregulate; ↓, inhibit/suppress.
Abbreviations: PD, Parkinson's disease; AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; NBIA, neurodegeneration with brain iron accumulation; ROS, reactive oxygen species; BBB, blood–brain barrier; HS, hemorrhagic stroke; IS, ischemic stroke; TPP, triphenylphosphonium; Aβ, β‐amyloid; Nrf2, nuclear factor erythroid 2‐related factor 2; HIF‐1, hypoxia‐inducible factor‐1; SLC7A11, solute carrier family 7 member 11; GPX4, glutathione peroxidase 4; AchE, acetylcholinesterase; NBMI, N, N’‐bis(2‐mercaptoethyl)isophthalamide; NCOA4, nuclear receptor coactivator 4.
Interestingly, it was reported that two new forms of DFO (PEGylated DFO and liposome‐DFO) seem to achieve results at opposite poles in the treatment of stroke, with the former a greater half‐life, lower cytotoxicity, and better iron chelation than traditional DFO, while the latter aggravating nerve damage and delaying neurological function recovery, which suggests that iron chelators might be a promising therapy for treating iron‐overload conditions in the clinic but still need to be further verified by lots of experiments. 94 , 288 In addition, though the safety of DFO has also been further validated in clinical trials, the starting and during time, and dosage of iron chelators should be individualized and disease‐specific, which are worthy of further exploration. 289 And as we have discussed above, the irrational use of iron chelators may be counterproductive.
5.2. Ferroptosis inhibitors
In contrast, ferroptosis inhibitors mainly function to break the autoxidation of chain‐propagating ROS and prevent excess ROS from lipid peroxidation. Ferrostatin‐1 (Fer‐1) has been regarded as the most potential ferroptosis inhibitor and a standard drug in experimental studies to judge the effects of other inhibitors. 218 , 290 As a member of the lipophilic radical‐trapping antioxidant (RTA) family, the mechanism of Fer‐1 in inhibiting ferroptosis has been widely explored in recent years. The latest researches suggest that Fer‐1 can bind the 15‐LOX/PEBP1 complex and disrupt its allosteric movement required for catalysis, thus inhibiting the catalytic activity of 15‐LOX for lipid peroxidation and further blocking ferroptosis caused by GPX4 insufficiency. 218 , 291 And Fer‐1 is capable to alleviate angiotensin II‐induced inflammation and ferroptosis by suppressing the ROS levels and activating the Nrf2/HO‐1 signaling pathway. 292 , 293 Moreover, Fer‐1 is associated with the downregulated expression of ferroptosis‐related genes and products (ireb2 and PTGS2), leading to the clearance of iron and ROS. 294 In addition, Liprostatin, another member of the RTAs, has similar mechanisms to Fer‐1 while better absorption, distribution, and effects with lower doses in the body. 295 Moreover, several other enzymes, like ACSL4, LPCAT3, and LOX, can promote lipid peroxidation during ferroptosis, therefore, drugs that inhibit these enzymes antagonize the onset of ferroptosis. The ferroptosis inhibitors reported in recent years and their related mechanisms are summarized in Table 2.
TABLE 2.
Ferroptosis inhibitors for neurological diseases
| Name | Mechanism/effect | Target disease | References |
|---|---|---|---|
| N‐acetylcysteine | ↓Toxic arachidonic acid products of nuclear ALOX5 | HS | 296 |
| ADA‐409‐052 |
↓TBHP‐induced lipid peroxidation ↓Activation of BV2 microglia Maintain mitochondrial morphology |
IS | 299 |
|---|---|---|---|
| Thiazolidinediones | ↓ACSL4 activity selectively | IS | 300 |
| Baicalin | ↑The mRNA expression of GPX4 and SLC7A11 in the peri‐hematoma brain tissues | HS | 303 |
|---|---|---|---|
| Dauricine | ↑GPX4 and glutathione reductase coexpression | HS | 304 |
| Melatonin | ↓5‐LOX level by circPtpn14/miR‐351‐5p signaling | TBI | 305 |
|---|---|---|---|
| DHI | ↑SATB1/SLC7A11/HO‐1 pathway | IS | 306 |
| β‐Caryophyllene | ↑Nrf2/HO‐1 signaling pathway | IS | 307 |
|---|---|---|---|
| Eriodictyol |
|
AD | 308 |
| α‐Lipoic acid |
↓p‐tau Iron redistribution mediated by TfR and Fpn1 |
PD | 309 |
|---|---|---|---|
| TS, TP | Inhibit 15‐LOX by competing for the substrate binding site of PUFA | _ | 310 |
Note: ↑, activate/upregulate;↓, inhibit/suppress.
Abbreviations: HS, hemorrhagic stroke; SSAT1, spermidine/spermine N1‐acetyltransferase 1; ALOX5, arachidonate 5‐lipoxygenase; IS, ischemic stroke; TfR1, transferrin receptor 1; DMT1, divalent metal transporter 1; SLC7A11, solute carrier family 7 member 11; GPX4, glutathione peroxidase 4; GSH, glutathione; TBHP, tert‐Butyl hydroperoxide; ACSL4, Acyl‐CoA synthetase long‐chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; 5‐LOX, 5‐lipoxygenase; TBI, traumatic brain injury; DHI, danhong injection; SATB1, special AT‐rich sequence‐binding protein 1; SLC7A11, solute carrier family 7 member 11; HO‐1, heme oxygenase‐1; Nrf2, nuclear factor erythroid 2‐related factor 2; Aβ, β‐amyloid; VDR, vitamin D receptor; p‐tau, phosphorylated tau; TfR, transferrin receptor; Fpn1, ferroportin 1; TS, α‐Tocopherol succinate; TP, α‐Tocopherol phosphate; 15‐LOX, 15‐lipoxygenase; PUFA, polyunsaturated fatty acids.
Apart from pharmacological inhibitors, there are two essential fat‐soluble vitamins (vitamin E and vitamin K) in the human body that can act as antioxidant molecules and exert antiferroptosis effects. Vitamin E terminates the propagation reactions by providing hydrogen atoms to PLOO∙ along with the formation of vitamin E free radicals (TOC∙). Finally, nonradical products are generated from the reaction of TOC∙ with another PLOO∙, representing an interruption of the oxidative transport chain. 311 Vitamin E can also regulate ferroptosis by LOX inhibition. 312 Besides, vitamin K was shown to confer robust antiferroptotic activity via its reduced form (VKH2), which is a potent RTA and PL peroxidation inhibitor. 313 Ferroptosis suppressor protein 1 (FSP1), a NAD(P)H‐ubiquinone reductase, plays a crucial role in reducing vitamin K to VKH2. Given the inhibitory role of vitamin E and vitamin K in ferroptosis, there may be nutritional approaches to the treatment of poststroke and neurodegenerative diseases combined with ferroptotic inhibitors
6. CONCLUSION AND PROSPECTS
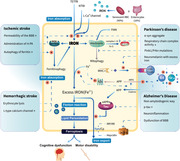
Iron is an important micronutrient for life maintenance, and it is closely related to the physiological metabolism of the human body. The pathogenesis of iron metabolism imbalance in brain diseases has been the focus of researchers in recent years. The regulation of iron homeostasis in the brain is very strict and involves the blood–brain barrier, the IRP/IRE system, the regulation of hepcidin, and the expression of many transporter proteins and enzymes. However, acute onset represented by stroke and chronic neurodegeneration represented by AD and PD interfere with the maintenance of brain iron homeostasis through their unique pathological mechanisms, leading to excessive accumulation of iron in the brain (Figure 4), which is evidenced by neuroimaging, pathology, and animal experiments. Excessive accumulation of iron in the brain triggers ferroptosis, a specific pattern of apoptosis, which significantly affects the prognosis of patients. Since its discovery and identification in 2012, many advances have been made to understand the mechanisms of ferroptosis. Many pharmacological agents like iron chelators and ferroptosis inhibitors have been used to bring iron metabolism imbalance back on track and modulate the ferroptotic response to attenuate morbidity and mortality. In contrast, many unresolved problems in iron metabolism and ferroptosis still need to be tackled in the future.
FIGURE 4.
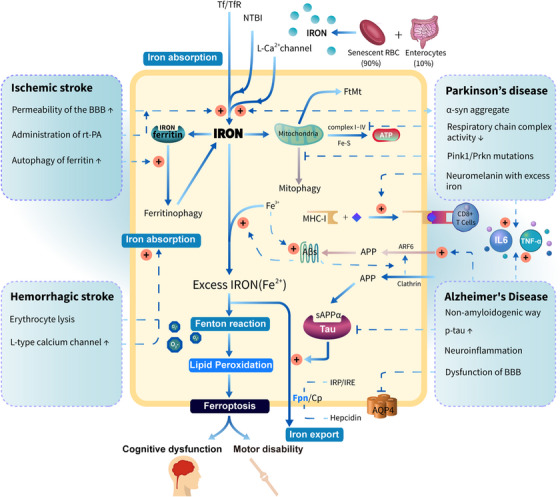
A generalization of the pathogenesis of brain iron accumulation in ischemic stroke, hemorrhagic stroke, Alzheimer's disease, and Parkinson's disease. Stroke greatly increases the import of brain iron, resulting in iron accumulation. In contrast, the progression of neurodegeneration and iron accumulation are mutually reinforcing. Mass labile iron, as a central link in stroke and neurodegenerative diseases, involves in the Fenton reaction and triggers the imbalance between the oxidation reaction and the antioxidant system in the neurological system, which leads to ferroptosis and then poor outcomes for patients. Tf, transferrin; TfR, transferrin receptor; NTBI, nontransferrin‐bound iron; Fpn, ferroportin; Cp, ceruloplasmin; FtMt, mitochondrial ferritin; MHC‐I, major histocompatibility complex class I; APP, amyloid precursor protein; ARF6, ADP ribosylation factor 6; p‐tau, phosphorylated tau; BBB, the blood–brain barrier; AQP4, aquaporin‐4. Note: "+ in the red cycle” means to promote.
In terms of pathological mechanisms, although the link between brain iron accumulation and neurodegeneration is increasingly being demonstrated, it is not clear yet whether iron deposition plays a cause or a consequence of these diseases. In addition, whether iron accumulation in the early stages of the disease is an initial reaction for self‐protection of the nervous system in facing noxious stimuli remains unclear, if so, then what are the thresholds for iron accumulation to be neuroprotective or ferroptosis‐inductive? On top of these, we still face a conundrum: who is the executor of ferroptosis? In terms of clinical treatments, although iron chelators have been proven beneficial in diseases related to iron metabolism imbalance, the dose and timing pattern of their administration in stroke patients remains to be determined. And the efficacy of clinical trials of ferroptosis inhibitors is still unknown. Developing key molecular markers to identify disease progression in order to facilitate the rational use of clinical drugs will be valuable. At last, the effects of comorbidities on iron metabolism and iron chelation must be taken into consideration in the future. Integrating the pathogenetic background of patients with iron chelation therapy to achieve the purpose of personalized and precise treatment in the future is worth looking forward to.
In summary, our understanding of the mechanisms involved in the regulation of iron homeostasis and ferroptosis has advanced enormously in the past several decades. Elucidating the mechanisms by which iron accumulation and ferroptosis occur not only helps to gain insight into the process of iron homeostasis imbalance and neurodegeneration but also facilitates the discovery of new therapeutic targets for related brain diseases. Nevertheless, many unknown aspects of iron metabolism and ferroptosis in brain disease remain to be unfolded, which requires a huge effort in basic animal research and clinical drug research.
AUTHOR CONTRIBUTION
Z. J. G. proposed the topic; Z. J. G., W. L. M., and Z. W. S. analyzed the structure and intellectual content of this review and made critical revisions. L. H. N. prepared, revised the manuscript, and drew the figures. All the authors agreed to publish this review.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81901845) and Shanghai Key Clinical Specialty (N0. shslczdzk03203). All Figures were created by Adobe Illustrator 2022 by using icons from Reactome (reactome.org).
Long H, Zhu W, Wei L, Zhao J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm. 2023;4:e298. 10.1002/mco2.298
Contributor Information
Liming Wei, Email: weilimingnj@126.com.
Jungong Zhao, Email: zhaojungongradio@hotmail.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Shi R, Hou W, Wang ZQ, Xu X. Biogenesis of iron‐sulfur clusters and their role in DNA metabolism. Front Cell Dev Biol. 2021;9:735678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gell DA. Structure and function of haemoglobins. Blood Cells Mol Dis. 2018;70:13‐42. [DOI] [PubMed] [Google Scholar]
- 3. Sukhbaatar N, Weichhart T. Iron regulation: macrophages in control. Pharmaceuticals (Basel). 2018;11(4):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Read AD, Bentley RE, Archer SL, Dunham‐Snary KJ. Mitochondrial iron‐sulfur clusters: structure, function, and an emerging role in vascular biology. Redox Biol. 2021;47:102164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fitzpatrick PF. The aromatic amino acid hydroxylases: structures, catalysis, and regulation of phenylalanine hydroxylase, tyrosine hydroxylase, and tryptophan hydroxylase. Arch Biochem Biophys. 2023;735:109518. [DOI] [PubMed] [Google Scholar]
- 6. Jopowicz A, Wiśniowska J, Tarnacka B. Cognitive and physical intervention in metals' dysfunction and neurodegeneration. Brain Sci. 2022;12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carbonell T, Rama R. Iron, oxidative stress and early neurological deterioration in ischemic stroke. Curr Med Chem. 2007;14(8):857‐874. [DOI] [PubMed] [Google Scholar]
- 9. Zhang DL, Ghosh MC, Ollivierre H, Li Y, Rouault TA. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood. 2018;132(19):2078‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chifman J, Laubenbacher RC, Torti SV. A systems biology approach to iron metabolism. Adv Exp Med Biol. 2014;844:201‐225. JAiem, biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaasch JA, Lockman PR, Geldenhuys WJ, Allen DD, Van der Schyf CJ. Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem Res. 2007;32(7):1196‐1208. [DOI] [PubMed] [Google Scholar]
- 12. Jiang X, Andjelkovic AV, Zhu L, et al. Blood‐brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018;163‐164:144‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim K‐A, Kim D, Kim J‐H, et al. Autophagy‐mediated occludin degradation contributes to blood‐brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS. 2020;17(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCarthy RC, Kosman DJ. Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood‐brain barrier. Front Mol Neurosci. 2015;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCarthy RC, Kosman DJ. Iron transport across the blood‐brain barrier: development, neurovascular regulation and cerebral amyloid angiopathy. Cell Mol Life Sci. 2015;72(4):709‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burkhart A, Skjørringe T, Johnsen KB, et al. Expression of iron‐related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood‐brain barrier. Mol Neurobiol. 2016;53(10):7237‐7253. [DOI] [PubMed] [Google Scholar]
- 17. Haldar S, Tripathi A, Qian J, et al. Prion protein promotes kidney iron uptake via its ferrireductase activity. J Biol Chem. 2015;290(9):5512‐5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55‐63. [DOI] [PubMed] [Google Scholar]
- 19. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10(9):597‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kadry H, Noorani B, Cucullo L. A blood‐brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. 2020;17(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng H, Wang N, Ma X, et al. Spatial‐temporal changes of iron deposition and iron metabolism after traumatic brain injury in mice. Front Mol Neurosci. 2022;15:949573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen Z, Jiang R, Chen M, et al. Multi‐copper ferroxidase deficiency leads to iron accumulation and oxidative damage in astrocytes and oligodendrocytes. Sci Rep. 2019;9(1):9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ji C, Steimle BL, Bailey DK, Kosman DJ. The ferroxidase hephaestin but not amyloid precursor protein is required for ferroportin‐supported iron efflux in primary hippocampal neurons. Cell Mol Neurobiol. 2018;38(4):941‐954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schulz K, Vulpe CD, Harris LZ, David S. Iron efflux from oligodendrocytes is differentially regulated in gray and white matter. J Neurosci. 2011;31(37):13301‐13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zarruk JG, Berard JL, Passos dos Santos R, et al. Expression of iron homeostasis proteins in the spinal cord in experimental autoimmune encephalomyelitis and their implications for iron accumulation. Neurobiol Dis. 2015;81:93‐107. [DOI] [PubMed] [Google Scholar]
- 27. Helman SL, Zhou J, Fuqua BK, et al. The biology of mammalian multi‐copper ferroxidases. Biometals. 2022;36:1‐19. doi: 10.1007/s10534-022-00370-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang Q, Liu W, Zhang S, Liu S. The cardinal roles of ferroportin and its partners in controlling cellular iron in and out. Life Sci. 2020;258:118135. [DOI] [PubMed] [Google Scholar]
- 29. Knutson MD. Non‐transferrin‐bound iron transporters. Free Radic Biol Med. 2019;133:101‐111. [DOI] [PubMed] [Google Scholar]
- 30. Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood. 2003;102(7):2670‐2677. [DOI] [PubMed] [Google Scholar]
- 31. Morré DJ, Morré DM. Plasma membrane electron transport. a metabolic process deserving of renewed interest. Biofactors. 2004;20(4):183‐187. [DOI] [PubMed] [Google Scholar]
- 32. Moos T. Immunohistochemical localization of intraneuronal transferrin receptor immunoreactivity in the adult mouse central nervous system. J Comp Neurol. 1996;375(4):675‐692. [DOI] [PubMed] [Google Scholar]
- 33. Chiou B, Neal EH, Bowman AB, Lippmann ES, Simpson IA, Connor JR. Endothelial cells are critical regulators of iron transport in a model of the human blood‐brain barrier. J Cereb Blood Flow Metab. 2019;39(11):2117‐2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plays M, Müller S, Rodriguez R. Chemistry and biology of ferritin. Metallomics. 2021;13(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Przybyla‐Toscano J, Christ L, Keech O, Rouhier N. Iron‐sulfur proteins in plant mitochondria: roles and maturation. J Exp Bot. 2021;72(6):2014‐2044. [DOI] [PubMed] [Google Scholar]
- 37. Pedroletti L, Moseler A, Meyer AJ. Assembly, transfer and fate of mitochondrial iron‐sulfur clusters. J Exp Bot. 2023:erad062. [DOI] [PubMed] [Google Scholar]
- 38. Levi S, Ripamonti M, Dardi M, Cozzi A, Santambrogio P. Mitochondrial ferritin: its role in physiological and pathological conditions. Cells. 2021;10(8):1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qian ZM, Shen X. Brain iron transport and neurodegeneration. Trends Mol Med. 2001;7(3):103‐108. [DOI] [PubMed] [Google Scholar]
- 40. Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823(9):1468‐1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang DL, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis ‐ an update. Front Pharmacol. 2014;5:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sammarco MC, Ditch S, Banerjee A, Grabczyk E. Ferritin L and H subunits are differentially regulated on a post‐transcriptional level. J Biol Chem. 2008;283(8):4578‐4587. [DOI] [PubMed] [Google Scholar]
- 43. Casey JL, Koeller DM, Ramin VC, Klausner RD, Harford JB. Iron regulation of transferrin receptor mRNA levels requires iron‐responsive elements and a rapid turnover determinant in the 3' untranslated region of the mRNA. EMBO J. 1989;8(12):3693‐3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gunshin H, Allerson CR, Polycarpou‐Schwarz M, et al. Iron‐dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509(2):309‐316. [DOI] [PubMed] [Google Scholar]
- 45. Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2(8):406‐414. [DOI] [PubMed] [Google Scholar]
- 46. Silva B, Faustino P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta. 2015;1852(7):1347‐1359. [DOI] [PubMed] [Google Scholar]
- 47. Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem. 2002;277(40):37597‐37603. [DOI] [PubMed] [Google Scholar]
- 48. Jiang L, Wang J, Wang K, et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood. 2021;138(8):689‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090‐2093. [DOI] [PubMed] [Google Scholar]
- 50. Du F, Qian C, Qian ZM, et al. Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP‐protein kinase A pathway. Glia. 2011;59(6):936‐945. [DOI] [PubMed] [Google Scholar]
- 51. Du F, Qian ZM, Luo Q, Yung WH, Ke Y. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron‐overloaded rats. Mol Neurobiol. 2015;52(1):101‐114. [DOI] [PubMed] [Google Scholar]
- 52. McDonald EA, Gundogan F, Olveda RM, Bartnikas TB, Kurtis JD, Friedman JF. Iron transport across the human placenta is regulated by hepcidin. Pediatr Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang H, Ostrowski R, Jiang D, et al. Hepcidin promoted ferroptosis through iron metabolism which is associated with DMT1 signaling activation in early brain injury following subarachnoid hemorrhage. Oxid Med Cell Longev. 2021;2021:9800794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang L, Liu X, You LH, et al. Hepcidin and iron regulatory proteins coordinately regulate ferroportin 1 expression in the brain of mice. J Cell Physiol. 2019;234(5):7600‐7607. [DOI] [PubMed] [Google Scholar]
- 55. Rutgers MP, Pielen A, Gille M. Chronic cerebellar ataxia and hereditary hemochromatosis: causal or coincidental association? J Neurol. 2007;254(9):1296‐1297. [DOI] [PubMed] [Google Scholar]
- 56. Saini V, Guada L, Yavagal DR. Global epidemiology of stroke and access to acute ischemic stroke interventions. Neurology. 2021;97(2):S6‐S16. 20 Suppl. [DOI] [PubMed] [Google Scholar]
- 57. Righy C, Turon R, Freitas G, et al. Hemoglobin metabolism by‐products are associated with an inflammatory response in patients with hemorrhagic stroke. Rev Bras Ter Intensiva. 2018;30(1):21‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dávalos A, Castillo J, Marrugat J, et al. Body iron stores and early neurologic deterioration in acute cerebral infarction. Neurology. 2000;54(8):1568‐15674. [DOI] [PubMed] [Google Scholar]
- 59. van Etten ES, van der Grond J, Dumas EM, van den Bogaard SJ, van Buchem MA, Wermer MJ. MRI susceptibility changes suggestive of iron deposition in the thalamus after ischemic stroke. Cerebrovasc Dis. 2015;40(1‐2):67‐72. [DOI] [PubMed] [Google Scholar]
- 60. Wiegertjes K, Chan KS, Telgte AT, et al. Assessing cortical cerebral microinfarcts on iron‐sensitive MRI in cerebral small vessel disease. J Cereb Blood Flow Metab. 2021;41(12):3391‐3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Amruta N, Bix G. ATN‐161 ameliorates ischemia/reperfusion‐induced oxidative stress, fibro‐inflammation, mitochondrial damage, and apoptosis‐mediated tight junction disruption in bEnd.3 Cells. Inflammation. 2021;44(6):2377‐2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sasson E, Anzi S, Bell B, et al. Nano‐scale architecture of blood‐brain barrier tight‐junctions. eLife. 2021;10:e63253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Berge E, Whiteley W, Audebert H, et al. European Stroke Organisation (ESO) guidelines on intravenous thrombolysis for acute ischaemic stroke. Eur Stroke J. 2021;6(1):I‐LXII. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344‐e418. [DOI] [PubMed] [Google Scholar]
- 65. Jaffer H, Adjei IM, Labhasetwar V. Optical imaging to map blood‐brain barrier leakage. Sci Rep. 2013;3:3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DeGregorio‐Rocasolano N, Martí‐Sistac O, Ponce J, et al. Iron‐loaded transferrin (Tf) is detrimental whereas iron‐free Tf confers protection against brain ischemia by modifying blood Tf saturation and subsequent neuronal damage. Redox Biol. 2018;15:143‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vaslin A, Puyal J, Borsello T, Clarke PG. Excitotoxicity‐related endocytosis in cortical neurons. J Neurochem. 2007;102(3):789‐800. [DOI] [PubMed] [Google Scholar]
- 68. Mancias JD, Pontano Vaites L, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2‐mediated proteolysis. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li C, Sun G, Chen B, et al. Nuclear receptor coactivator 4‐mediated ferritinophagy contributes to cerebral ischemia‐induced ferroptosis in ischemic stroke. Pharmacol Res. 2021;174:105933. [DOI] [PubMed] [Google Scholar]
- 70. Ho KJ, Liu TK, Huang TS, Lu FJ. Humic acid mediates iron release from ferritin and promotes lipid peroxidation in vitro: a possible mechanism for humic acid‐induced cytotoxicity. Arch Toxicol. 2003;77(2):100‐109. [DOI] [PubMed] [Google Scholar]
- 71. Daher R, Ducrot N, Lefebvre T, et al. Crosstalk between acidosis and iron metabolism: data from in vivo studies. Metabolites. 2022;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ballatore C, Lee VM, Trojanowski JQ. Tau‐mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8(9):663‐672. [DOI] [PubMed] [Google Scholar]
- 73. Götz J, Ittner A, Ittner LM. Tau‐targeted treatment strategies in Alzheimer's disease. Br J Pharmacol. 2012;165(5):1246‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bi M, Gladbach A, van Eersel J, et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun. 2017;8(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dong Y, Yu H, Li X, et al. Hyperphosphorylated tau mediates neuronal death by inducing necroptosis and inflammation in Alzheimer's disease. J Neuroinflammation. 2022;19(1):205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tuo QZ, Lei P, Jackman KA, et al. Tau‐mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22(11):1520‐1530. [DOI] [PubMed] [Google Scholar]
- 77. Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA. β‐Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One. 2014;9(12):e114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lei P, Ayton S, Finkelstein DI, et al. Tau deficiency induces parkinsonism with dementia by impairing APP‐mediated iron export. Nat Med. 2012;18(2):291‐295. [DOI] [PubMed] [Google Scholar]
- 79. Doehner W, Scherbakov N, Schellenberg T, et al. Iron deficiency is related to low functional outcome in patients at early rehabilitation after acute stroke. J Cachexia Sarcopenia Muscle. 2022;13(2):1036‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Scherbakov N, Sandek A, Valentova M, et al. Iron deficiency and reduced muscle strength in patients with acute and chronic ischemic stroke. J Clin Med. 2022;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Keaney J, Campbell M. The dynamic blood‐brain barrier. FEBS J. 2015;282(21):4067‐4079. [DOI] [PubMed] [Google Scholar]
- 82. Li Q, Lan X, Han X, et al. Microglia‐derived interleukin‐10 accelerates post‐intracerebral hemorrhage hematoma clearance by regulating CD36. Brain Behav Immun. 2021;94:437‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang Z, Song Y, Zhang Z, et al. Distinct role of heme oxygenase‐1 in early‐ and late‐stage intracerebral hemorrhage in 12‐month‐old mice. J Cereb Blood Flow Metab. 2017;37(1):25‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9‐17. [DOI] [PubMed] [Google Scholar]
- 85. Schipper HM, Song W, Tavitian A, Cressatti M. The sinister face of heme oxygenase‐1 in brain aging and disease. Prog Neurobiol. 2019;172:40‐70. [DOI] [PubMed] [Google Scholar]
- 86. Kim S, Lee W, Jo H, et al. The antioxidant enzyme peroxiredoxin‐1 controls stroke‐associated microglia against acute ischemic stroke. Redox Biol. 2022;54:102347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gaasch JA, Geldenhuys WJ, Lockman PR, Allen DD, Van der Schyf CJ. Voltage‐gated calcium channels provide an alternate route for iron uptake in neuronal cell cultures. Neurochem Res. 2007;32(10):1686‐1693. [DOI] [PubMed] [Google Scholar]
- 88. Qin Y, Li G, Sun Z, Xu X, Gu J, Gao F. Comparison of the effects of nimodipine and deferoxamine on brain injury in rat with subarachnoid hemorrhage. Behav Brain Res. 2019;367:194‐200. [DOI] [PubMed] [Google Scholar]
- 89. Lockman JA, Geldenhuys WJ, Bohn KA, Desilva SF, Allen DD, Van der Schyf CJ. Differential effect of nimodipine in attenuating iron‐induced toxicity in brain‐ and blood‐brain barrier‐associated cell types. Neurochem Res. 2012;37(1):134‐142. [DOI] [PubMed] [Google Scholar]
- 90. von Bartheld CS, Bahney J, Herculano‐Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. 2016;524(18):3865‐3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xu S, Lu J, Shao A, Zhang JH, Zhang J. Glial cells: role of the immune response in ischemic stroke. Front Immunol. 2020;11:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu LR, Liu JC, Bao JS, Bai QQ, Wang GQ. Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol. 2020;11:1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hu X, Leak RK, Shi Y, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. 2015;11(1):56‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Guo X, Jin X, Han K, et al. Iron promotes neurological function recovery in mice with ischemic stroke through endogenous repair mechanisms. Free Radic Biol Med. 2022;182:59‐72. [DOI] [PubMed] [Google Scholar]
- 95. Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood‐brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation. 2015;12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sillerud LO, Yang Y, Yang LY, Duval KB, Thompson J, Yang Y. Longitudinal monitoring of microglial/macrophage activation in ischemic rat brain using Iba‐1‐specific nanoparticle‐enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2020;40(1):S117‐S133. _suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rawlinson C, Jenkins S, Thei L, Dallas ML, Chen R. Post‐ischaemic immunological response in the brain: targeting microglia in ischaemic stroke therapy. Brain Sci. 2020;10(3):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ye F, Yang J, Hua Y, Keep RF, Xi G. Novel approach to visualize microglia death and proliferation after intracerebral hemorrhage in mice. Stroke. 2022;53(11):e472‐e476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Al Mamun A, Chauhan A, Qi S, et al. Microglial IRF5‐IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proc Natl Acad Sci USA. 2020;117(3):1742‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Preininger MK, Kaufer D. Blood‐brain barrier dysfunction and astrocyte senescence as reciprocal drivers of neuropathology in aging. Int J Mol Sci. 2022;23(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Daneman R, Prat A. The blood‐brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci. 2021;24(10):1475‐1487. [DOI] [PubMed] [Google Scholar]
- 103. Lanfranco MF, Sepulveda J, Kopetsky G, Rebeck GW. Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia. 2021;69(6):1478‐1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li H, Zhang N, Lin HY, et al. Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis‐induced ischemia in adult mice. BMC Neurosci. 2014;15:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. You L, Yu PP, Dong T, et al. Astrocyte‐derived hepcidin controls iron traffic at the blood‐brain‐barrier via regulating ferroportin 1 of microvascular endothelial cells. Cell Death Dis. 2022;13(8):667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ranjbar Taklimie F, Gasterich N, Scheld M, et al. Hypoxia induces astrocyte‐derived lipocalin‐2 in ischemic stroke. Int J Mol Sci. 2019;20(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Makhezer N, Ben Khemis M, Liu D, et al. NOX1‐derived ROS drive the expression of lipocalin‐2 in colonic epithelial cells in inflammatory conditions. Mucosal Immunol. 2019;12(1):117‐131. [DOI] [PubMed] [Google Scholar]
- 108. Liu R, Wang J, Chen Y, et al. NOX activation in reactive astrocytes regulates astrocytic LCN2 expression and neurodegeneration. Cell Death Dis. 2022;13(4):371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917‐921. [DOI] [PubMed] [Google Scholar]
- 110. Zhao N, Xu X, Jiang Y, et al. Lipocalin‐2 may produce damaging effect after cerebral ischemia by inducing astrocytes classical activation. J Neuroinflammation. 2019;16(1):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pan P, Zhao H, Zhang X, et al. Cyclophilin a signaling induces pericyte‐associated blood‐brain barrier disruption after subarachnoid hemorrhage. J Neuroinflammation. 2020;17(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cheli VT, Santiago González DA, Wan Q, et al. H‐ferritin expression in astrocytes is necessary for proper oligodendrocyte development and myelination. Glia. 2021;69(12):2981‐2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cheli VT, Correale J, Paez PM, Pasquini JM. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. ASN Neuro. 2020;12:1759091420962681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20(24):9235‐9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pandya CD, Vekaria H, Joseph B, et al. Hemoglobin induces oxidative stress and mitochondrial dysfunction in oligodendrocyte progenitor cells. Transl Res. 2021;231:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Shen D, Wu W, Liu J, et al. Ferroptosis in oligodendrocyte progenitor cells mediates white matter injury after hemorrhagic stroke. Cell Death Dis. 2022;13(3):259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhang J, Zhang Y, Xing S, Liang Z, Zeng J. Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke. 2012;43(6):1700‐1705. [DOI] [PubMed] [Google Scholar]
- 118. Tamura A, Tahira Y, Nagashima H, et al. Thalamic atrophy following cerebral infarction in the territory of the middle cerebral artery. Stroke. 1991;22(5):615‐618. [DOI] [PubMed] [Google Scholar]
- 119. Kuchcinski G, Munsch F, Lopes R, et al. Thalamic alterations remote to infarct appear as focal iron accumulation and impact clinical outcome. Brain. 2017;140(7):1932‐1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Linck PA, Kuchcinski G, Munsch F, et al. Neurodegeneration of the substantia nigra after ipsilateral infarct: mRI R2* mapping and relationship to clinical outcome. Radiology. 2019;291(2):438‐448. [DOI] [PubMed] [Google Scholar]
- 121. Justicia C, Ramos‐Cabrer P, Hoehn M. MRI detection of secondary damage after stroke: chronic iron accumulation in the thalamus of the rat brain. Stroke. 2008;39(5):1541‐1547. [DOI] [PubMed] [Google Scholar]
- 122. Kent SA, Spires‐Jones TL, Durrant CS. The physiological roles of tau and Aβ: implications for Alzheimer's disease pathology and therapeutics. Acta Neuropathol. 2020;140(4):417‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Guan X, Guo T, Zhou C, et al. Altered brain iron depositions from aging to Parkinson's disease and Alzheimer's disease: a quantitative susceptibility mapping study. Neuroimage. 2022;264:119683. [DOI] [PubMed] [Google Scholar]
- 124. Derry PJ, Hegde ML, Jackson GR, et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer's disease from a ferroptosis perspective. Prog Neurobiol. 2020;184:101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dusek P, Hofer T, Alexander J, Roos PM, Aaseth JO. Cerebral iron deposition in neurodegeneration. Biomolecules. 2022;12(5):714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pan R, Luo S, Huang Q, et al. The associations of cerebrospinal fluid ferritin with neurodegeneration and neuroinflammation along the Alzheimer's disease continuum. J Alzheimers Dis. 2022;88(3):1115‐1125. [DOI] [PubMed] [Google Scholar]
- 127. Gong N‐J, Dibb R, Bulk M, van der Weerd L, Liu C. Imaging beta amyloid aggregation and iron accumulation in Alzheimer's disease using quantitative susceptibility mapping MRI. Neuroimage. 2019;191:176‐185. [DOI] [PubMed] [Google Scholar]
- 128. Ayton S, Fazlollahi A, Bourgeat P, et al. Cerebral quantitative susceptibility mapping predicts amyloid‐β‐related cognitive decline. Brain. 2017;140(8):2112‐2119. [DOI] [PubMed] [Google Scholar]
- 129. Chen L, Soldan A, Oishi K, et al. Quantitative susceptibility mapping of brain iron and β‐Amyloid in MRI and PET relating to cognitive performance in cognitively normal older adults. Radiology. 2021;298(2):353‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhang Y‐W, Thompson R, Zhang H, Xu H. APP processing in Alzheimer's disease. Mol Brain. 2011;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tang W, Tam JHK, Seah C, et al. ATF6 controls beta‐amyloid production by regulating macropinocytosis of the amyloid precursor protein to lysosomes. Mol Brain. 2015;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cai H, Wang Y, McCarthy D, et al. BACE1 is the major beta‐secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4(3):233‐234. [DOI] [PubMed] [Google Scholar]
- 133. Hur J‐Y. γ‐Secretase in Alzheimer's disease. Exp Mol Med. 2022;54(4):433‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Dlouhy AC, Bailey DK, Steimle BL, Parker HV, Kosman DJ. Fluorescence resonance energy transfer links membrane ferroportin, hep haestin but not ferroportin, amyloid precursor protein complex with iron efflux. J Biol Chem. 2019;294(11):4202‐4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. McCarthy RC, Kosman DJ. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell iron efflux. J Biol Chem. 2013;288(24):17932‐17940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. McCarthy RC, Park Y‐H, Kosman DJ. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO reports. 2014;15(7):809‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Dlouhy AC, Bailey DK, Steimle BL, Parker HV, Kosman DJ. Fluorescence resonance energy transfer links membrane ferroportin, hep haestin but not ferroportin, amyloid precursor protein complex with iron efflux. J Biol Chem. 2019;294(11):4202‐4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tsatsanis A, Wong BX, Gunn AP, et al. Amyloidogenic processing of Alzheimer's disease β‐amyloid precursor protein induces cellular iron retention. Mol Psychiatry. 2020;25(9):1958‐1966. [DOI] [PubMed] [Google Scholar]
- 139. McCarthy RC, Park YH, Kosman DJ. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. 2014;15(7):809‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tsatsanis A, McCorkindale AN, Wong BX, et al. The acute phase protein lactoferrin is a key feature of Alzheimer's disease and predictor of Aβ burden through induction of APP amyloidogenic processing. Mol Psychiatry. 2021;26(10):5516‐5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Egan MF, Kost J, Tariot PN, et al. Randomized trial of verubecestat for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2018;378(18):1691‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Johnson ECB, Ho K, Yu G‐Q, et al. Behavioral and neural network abnormalities in human APP transgenic mice resemble those of APP knock‐in mice and are modulated by familial Alzheimer's disease mutations but not by inhibition of BACE1. Mol Neurodegener. 2020;15(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Vahed M, Sweeney A, Shirasawa H, Vahed M. The initial stage of structural transformation of Aβ(42) peptides from the human and mole rat in the presence of Fe(2+) and Fe(3+): related to Alzheimer's disease. Comput Biol Chem. 2019;83:107128. [DOI] [PubMed] [Google Scholar]
- 144. Zhou ZD, Tan EK. Iron regulatory protein (IRP)‐iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol Neurodegener. 2017;12(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rottkamp CA, Raina AK, Zhu X, et al. Redox‐active iron mediates amyloid‐beta toxicity. Free Radic Biol Med. 2001;30(4):447‐450. [DOI] [PubMed] [Google Scholar]
- 146. Everett J, Céspedes E, Shelford LR, et al. Ferrous iron formation following the co‐aggregation of ferric iron and the Alzheimer's disease peptide β‐amyloid (1‐42). J R Soc Interface. 2014;11(95):20140165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Yan N, Zhang J. Iron metabolism, ferroptosis, and the links with Alzheimer's disease. Front Neurosci. 2019;13:1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ayton S, Wang Y, Diouf I, et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry. 2020;25(11):2932‐2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Xie L, Zheng W, Xin N, Xie JW, Wang T, Wang ZY. Ebselen inhibits iron‐induced tau phosphorylation by attenuating DMT1 up‐regulation and cellular iron uptake. Neurochem Int. 2012;61(3):334‐340. [DOI] [PubMed] [Google Scholar]
- 150. Wu TY, Zhao LX, Zhang YH, Fan YG. Activation of vitamin D receptor inhibits Tau phosphorylation is associated with reduction of iron accumulation in APP/PS1 transgenic mice. Neurochem Int. 2022;153:105260. [DOI] [PubMed] [Google Scholar]
- 151. Wang D, Hui Y, Peng Y, et al. Overexpression of heme oxygenase 1 causes cognitive decline and affects pathways for tauopathy in mice. J Alzheimers Dis. 2015;43(2):519‐534. [DOI] [PubMed] [Google Scholar]
- 152. Consoli V, Sorrenti V, Grosso S, Vanella L. Heme Oxygenase‐1 signaling and redox homeostasis in physiopathological conditions. Biomolecules. 2021;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Cheignon C, Tomas M, Bonnefont‐Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sebastian Monasor L, Müller SA, Colombo AV, et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xiao S, Zhou D, Luan P, et al. Graphene quantum dots conjugated neuroprotective peptide improve learning and memory capability. Biomaterials. 2016;106:98‐110. [DOI] [PubMed] [Google Scholar]
- 156. Friker LL, Scheiblich H, Hochheiser IV, et al. β‐Amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep. 2020;30(11):3743‐3754. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Meng FX, Hou JM, Sun TS. In vivo evaluation of microglia activation by intracranial iron overload in central pain after spinal cord injury. J Orthop Surg Res. 2017;12(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wang J, Song N, Jiang H, Wang J, Xie J. Pro‐inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim Biophys Acta. 2013;1832(5):618‐625. [DOI] [PubMed] [Google Scholar]
- 159. Lopez‐Rodriguez AB, Hennessy E, Murray CL, et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer's disease: iL‐1β drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimers Dement. 2021;17(10):1735‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Raulin AC, Doss SV, Trottier ZA, Ikezu TC, Bu G, Liu CC. ApoE in Alzheimer's disease: pathophysiology and therapeutic strategies. Mol Neurodegener. 2022;17(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Uchida Y, Kan H, Sakurai K, et al. APOE ɛ4 dose associates with increased brain iron and β‐amyloid via blood‐brain barrier dysfunction. J Neurol Neurosurg Psychiatry. 2022. [DOI] [PubMed] [Google Scholar]
- 162. Uchida Y, Kan H, Sakurai K, et al. Iron leakage owing to blood‐brain barrier disruption in small vessel disease CADASIL. Neurology. 2020;95(9):e1188‐e1198. [DOI] [PubMed] [Google Scholar]
- 163. van Bergen JM, Li X, Hua J, et al. Colocalization of cerebral iron with amyloid beta in mild cognitive impairment. Sci Rep. 2016;6:35514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hofer E, Pirpamer L, Langkammer C, et al. Heritability of R2* iron in the basal ganglia and cortex. Aging (Albany N Y). 2022;14(16):6415‐6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Matsuno H, Tsuchimine S, O'Hashi K, et al. Association between vascular endothelial growth factor‐mediated blood‐brain barrier dysfunction and stress‐induced depression. Mol Psychiatry. 2022;27(9):3822‐3832. [DOI] [PubMed] [Google Scholar]
- 166. Tolosa E, Garrido A, Scholz SW, Poewe W. Challenges in the diagnosis of Parkinson's disease. Lancet Neurol. 2021;20(5):385‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. 2021;397(10291):2284‐2303. [DOI] [PubMed] [Google Scholar]
- 168. Biondetti E, Santin MD, Valabrègue R, et al. The spatiotemporal changes in dopamine, neuromelanin and iron characterizing Parkinson's disease. Brain. 2021;144(10):3114‐3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Khedher L, Bonny JM, Marques A, et al. Intrasubject subcortical quantitative referencing to boost MRI sensitivity to Parkinson's disease. Neuroimage Clin. 2022;36:103231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α‐synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Liu C, Zhao Y, Xi H, Jiang J, Yu Y, Dong W. The membrane interaction of alpha‐synuclein. Front Cell Neurosci. 2021;15:633727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Mahul‐Mellier AL, Burtscher J, Maharjan N, et al. The process of Lewy body formation, rather than simply α‐synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci USA. 2020;117(9):4971‐4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Castellani RJ, Siedlak SL, Perry G, Smith MA. Sequestration of iron by Lewy bodies in Parkinson's disease. Acta Neuropathol. 2000;100(2):111‐114. [DOI] [PubMed] [Google Scholar]
- 174. Guo JJ, Yue F, Song DY, et al. Intranasal administration of α‐synuclein preformed fibrils triggers microglial iron deposition in the substantia nigra of Macaca fascicularis. Cell Death Dis. 2021;12(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Sian‐Hulsmann J, Riederer P. The role of alpha‐synuclein as ferrireductase in neurodegeneration associated with Parkinson's disease. J Neural Transm (Vienna). 2020;127(5):749‐754. [DOI] [PubMed] [Google Scholar]
- 176. Ben Gedalya T, Loeb V, Israeli E, Altschuler Y, Selkoe DJ, Sharon R. Alpha‐synuclein and polyunsaturated fatty acids promote clathrin‐mediated endocytosis and synaptic vesicle recycling. Traffic. 2009;10(2):218‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schechter M, Atias M, Abd Elhadi S, Davidi D, Gitler D, Sharon R. α‐Synuclein facilitates endocytosis by elevating the steady‐state levels of phosphatidylinositol 4,5‐bisphosphate. J Biol Chem. 2020;295(52):18076‐18090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Shekoohi S, Rajasekaran S, Patel D, et al. Knocking out alpha‐synuclein in melanoma cells dysregulates cellular iron metabolism and suppresses tumor growth. Sci Rep. 2021;11(1):5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Baksi S, Tripathi AK, Singh N. Alpha‐synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin‐bound iron: implications for visual manifestations of Parkinson's disease. Free Radic Biol Med. 2016;97:292‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Dauer Née Joppe K, Tatenhorst L, Caldi Gomes L, et al. Brain iron enrichment attenuates α‐synuclein spreading after injection of preformed fibrils. J Neurochem. 2021;159(3):554‐573. [DOI] [PubMed] [Google Scholar]
- 181. Guan X, Zhang Y, Wei H, et al. Iron‐related nigral degeneration influences functional topology mediated by striatal dysfunction in Parkinson's disease. Neurobiol Aging. 2019;75:83‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Salami A, Avelar‐Pereira B, Garzón B, Sitnikov R, Kalpouzos G. Functional coherence of striatal resting‐state networks is modulated by striatal iron content. Neuroimage. 2018;183:495‐503. [DOI] [PubMed] [Google Scholar]
- 183. Hughes C, Faskowitz J, Cassidy BS, Sporns O, Krendl AC. Aging relates to a disproportionately weaker functional architecture of brain networks during rest and task states. Neuroimage. 2020;209:116521. [DOI] [PubMed] [Google Scholar]
- 184. Zucca FA, Segura‐Aguilar J, Ferrari E, et al. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog Neurobiol. 2017;155:96‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5(11):863‐873. [DOI] [PubMed] [Google Scholar]
- 186. Bjørklund G, Hofer T, Nurchi VM, Aaseth J. Iron and other metals in the pathogenesis of Parkinson's disease: toxic effects and possible detoxification. J Inorg Biochem. 2019;199:110717. [DOI] [PubMed] [Google Scholar]
- 187. Zecca L, Stroppolo A, Gatti A, et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A. 2004;101(26):9843‐9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Cebrián C, Zucca FA, Mauri P, et al. MHC‐I expression renders catecholaminergic neurons susceptible to T‐cell‐mediated degeneration. Nat Commun. 2014;5:3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Wang BY, Ye YY, Qian C, et al. Stress increases MHC‐I expression in dopaminergic neurons and induces autoimmune activation in Parkinson's disease. Neural Regen Res. 2021;16(12):2521‐2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Depierreux F, Parmentier E, Mackels L, et al. Parkinson's disease multimodal imaging: f‐DOPA PET, neuromelanin‐sensitive and quantitative iron‐sensitive MRI. NPJ Parkinsons Dis. 2021;7(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Gaurav R, Yahia‐Cherif L, Pyatigorskaya N, et al. Longitudinal changes in neuromelanin MRI signal in Parkinson's disease: a progression marker. Mov Disord. 2021;36(7):1592‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. García‐Revilla J, Herrera AJ, de Pablos RM, Venero JL. Inflammatory animal models of Parkinson's disease. J Parkinsons Dis. 2022;12(s1):S165‐S182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sterling JK, Kam TI, Guttha S, et al. Interleukin‐6 triggers toxic neuronal iron sequestration in response to pathological α‐synuclein. Cell Rep. 2022;38(7):110358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Lakhani DA, Middlebrooks EH. 7‐T Neuromelanin and R2* MRI in Parkinson disease. Radiology. 2022;305(2):296. [DOI] [PubMed] [Google Scholar]
- 195. Jokar M, Jin Z, Huang P, et al. Diagnosing Parkinson's disease by combining neuromelanin and iron imaging features using an automated midbrain template approach. Neuroimage. 2023;266:119814. [DOI] [PubMed] [Google Scholar]
- 196. Gao XY, Yang T, Gu Y, Sun XH. Mitochondrial dysfunction in Parkinson's disease: from mechanistic insights to therapy. Front Aging Neurosci. 2022;14:885500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Anandhan A, Jacome MS, Lei S, et al. Metabolic dysfunction in Parkinson's disease: bioenergetics, redox homeostasis and central carbon metabolism. Brain Res Bull. 2017;133:12‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Liang T, Qian ZM, Mu MD, Yung WH, Ke Y. Brain hepcidin suppresses major pathologies in experimental parkinsonism. iScience. 2020;23(7):101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Lawana V, Um SY, Foguth RM, Cannon JR. Neuromelanin formation exacerbates HAA‐induced mitochondrial toxicity and mitophagy impairments. NeuroToxicology. 2020;81:147‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Cruz‐Hernandez A, Agim ZS, Montenegro P, McCabe GP, Rochet J‐C, Cannon JR. Selective dopaminergic neurotoxicity of three heterocyclic amine subclasses in primary rat midbrain neurons. Neurotoxicology. 2018;65:68‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Park JH, Burgess JD, Faroqi AH, et al. Alpha‐synuclein‐induced mitochondrial dysfunction is mediated via a sirtuin 3‐dependent pathway. Mol Neurodegener. 2020;15(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Jiang DQ, Zang QM, Jiang LL, Lu CS, Zhao SH, Xu LC. SIRT3 expression alleviates microglia activation‑induced dopaminergic neuron injury through the mitochondrial pathway. Exp Ther Med. 2022;24(5):662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. He L, Wang J, Yang Y, Li J, Tu H. Mitochondrial sirtuins in Parkinson's disease. Neurochem Res. 2022;47(6):1491‐1502. [DOI] [PubMed] [Google Scholar]
- 204. Urrutia PJ, Mena NP, Núñez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 2014;5:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40(3):e104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Valente EM, Abou‐Sleiman PM, Caputo V, et al. Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158‐1160. [DOI] [PubMed] [Google Scholar]
- 207. Gan ZY, Callegari S, Cobbold SA, et al. Activation mechanism of PINK1. Nature. 2022;602(7896):328‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Burbulla LF, Song P, Mazzulli JR, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. 2017;357(6357):1255‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Prasuhn J, Göttlich M, Gerkan F, et al. Relationship between brain iron deposition and mitochondrial dysfunction in idiopathic Parkinson's disease. Mol Med. 2022;28(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Jansen AH, van Hal M, Op den Kelder IC, et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell‐type‐specific. Glia. 2017;65(1):50‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Niu L, Ye C, Sun Y, et al. Mutant huntingtin induces iron overload via up‐regulating IRP1 in Huntington's disease. Cell Biosci. 2018;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Napoli E, Wong S, Hung C, Ross‐Inta C, Bomdica P, Giulivi C. Defective mitochondrial disulfide relay system, altered mitochondrial morphology and function in Huntington's disease. Hum Mol Genet. 2013;22(5):989‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Okada N, Yako T, Nakamura S, Shimazawa M, Hara H. Reduced mitochondrial complex II activity enhances cell death via intracellular reactive oxygen species in STHdhQ111 striatal neurons with mutant huntingtin. J Pharmacol Sci. 2021;147(4):367‐375. [DOI] [PubMed] [Google Scholar]
- 214. Franco‐Iborra S, Plaza‐Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez‐Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 2021;17(3):672‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Li XJ, Orr AL, Li S. Impaired mitochondrial trafficking in Huntington's disease. Biochim Biophys Acta. 2010;1802(1):62‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Donley DW, Realing M, Gigley JP, Fox JH. Iron activates microglia and directly stimulates indoleamine‐2,3‐dioxygenase activity in the N171‐82Q mouse model of Huntington's disease. PLoS One. 2021;16(5):e0250606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Dusek P, Hofer T, Alexander J, Roos PM, Aaseth JO. Cerebral iron deposition in neurodegeneration. Biomolecules. 2022;12(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Zhang Y, Lu X, Tai B, Li W, Li T. Ferroptosis and its multifaceted roles in cerebral stroke. Front Cell Neurosci. 2021;15:615372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Guo J, Tuo QZ, Lei P. Iron, ferroptosis, and ischemic stroke. J Neurochem. 2023. [DOI] [PubMed] [Google Scholar]
- 221. Lane DJR, Metselaar B, Greenough M, Bush AI, Ayton SJ. Ferroptosis and NRF2: an emerging battlefield in the neurodegeneration of Alzheimer's disease. Essays Biochem. 2021;65(7):925‐940. [DOI] [PubMed] [Google Scholar]
- 222. Bao WD, Pang P, Zhou XT, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell Death Differ. 2021;28(5):1548‐1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Plascencia‐Villa G, Perry G. Preventive and therapeutic strategies in Alzheimer's disease: focus on oxidative stress, redox metals, and ferroptosis. Antioxidants & redox signaling. 2021;34(8):591‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Mahoney‐Sánchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson's Disease. Prog Neurobiol. 2021;196:101890. [DOI] [PubMed] [Google Scholar]
- 225. Wang Z‐L, Yuan L, Li W, Li J‐Y. Ferroptosis in Parkinson's disease: glia‐neuron crosstalk. Trends Mol Med. 2022;28(4):258‐269. [DOI] [PubMed] [Google Scholar]
- 226. Angelova PR, Choi ML, Berezhnov AV, et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020;27(10):2781‐2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Letourneau M, Wang K, Mailloux RJ. Protein S‐glutathionylation decreases superoxide/hydrogen peroxide production xanthine oxidoreductase. Free Radic Biol Med. 2021;175:184‐192. [DOI] [PubMed] [Google Scholar]
- 228. Maciejczyk M, Nesterowicz M, Zalewska A, et al. Salivary xanthine oxidase as a potential biomarker in stroke diagnostics. Front Immunol. 2022;13:897413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Salvador GA. Iron in neuronal function and dysfunction. Biofactors. 2010;36(2):103‐110. [DOI] [PubMed] [Google Scholar]
- 230. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604‐1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta. 2015;1851(4):308‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628‐641. e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Chu B, Kon N, Chen D, et al. ALOX12 is required for p53‐mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81(2):355‐369. e10. [DOI] [PubMed] [Google Scholar]
- 236. Catalá A, Díaz M. Editorial: impact of lipid peroxidation on the physiology and pathophysiology of cell membranes. Front Physiol. 2016;7:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Sun Y, Zheng Y, Wang C, Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018;9(7):753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation‐dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143‐170. [DOI] [PubMed] [Google Scholar]
- 239. Shanker G, Allen JW, Mutkus LA, Aschner M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001;902(2):156‐163. [DOI] [PubMed] [Google Scholar]
- 240. Lin W, Wang C, Liu G, et al. SLC7A11/xCT in cancer: biological functions and therapeutic implications. Am J Cancer Res. 2020;10(10):3106‐3126. [PMC free article] [PubMed] [Google Scholar]
- 241. Chen L, Na R, Danae McLane K, et al. Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci Rep. 2021;11(1):12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Nieoullon A, Canolle B, Masmejean F, Guillet B, Pisano P, Lortet S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a major actor at the brain excitatory synapse? J Neurochem. 2006;98(4):1007‐1018. [DOI] [PubMed] [Google Scholar]
- 243. Banjac A, Perisic T, Sato H, et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27(11):1618‐1628. [DOI] [PubMed] [Google Scholar]
- 244. Charisis S, Ntanasi E, Yannakoulia M, et al. Plasma GSH levels and Alzheimer's disease. a prospective approach.: results from the HELIAD study. Free Radic Biol Med. 2021;162:274‐282. [DOI] [PubMed] [Google Scholar]
- 245. Charisis S, Ntanasi E, Stamelou M, et al. Plasma glutathione and prodromal Parkinson's disease probability. Mov Disord. 2022;37(1):200‐205. [DOI] [PubMed] [Google Scholar]
- 246. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175‐185. [DOI] [PubMed] [Google Scholar]
- 247. Latunde‐Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861(8):1893‐1900. [DOI] [PubMed] [Google Scholar]
- 248. Qu XX, He JH, Cui ZQ, Yang T, Sun XH. PPAR‐α agonist GW7647 protects against oxidative stress and iron deposit via GPx4 in a transgenic mouse model of Alzheimer's diseases. ACS Chem Neurosci. 2022;13(2):207‐216. [DOI] [PubMed] [Google Scholar]
- 249. Raza A, Xie W, Kim KH, et al. Dipeptide of ψ‐GSH inhibits oxidative stress and neuroinflammation in an Alzheimer's disease mouse model. Antioxidants (Basel). 2022;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Fu RH, Tsai CW, Liu SP, et al. Neuroprotective capability of narcissoside in 6‐OHDA‐exposed Parkinson's disease models through enhancing the MiR200a/Nrf‐2/GSH axis and mediating MAPK/Akt associated signaling pathway. Antioxidants (Basel). 2022;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Bai L, Yan F, Deng R, Gu R, Zhang X, Bai J. Thioredoxin‐1 rescues MPP(+)/MPTP‐Induced ferroptosis by Increasing glutathione peroxidase 4. Mol Neurobiol. 2021;58(7):3187‐3197. [DOI] [PubMed] [Google Scholar]
- 252. Guan X, Li X, Yang X, et al. The neuroprotective effects of carvacrol on ischemia/reperfusion‐induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 2019;235:116795. [DOI] [PubMed] [Google Scholar]
- 253. Muraoka M, Yoshida S, Ohno M, et al. Reactivity of γ‐glutamyl‐cysteine with intracellular and extracellular glutathione metabolic enzymes. FEBS Lett. 2022;596(2):180‐188. [DOI] [PubMed] [Google Scholar]
- 254. Hanigan MH. Gamma‐glutamyl transpeptidase: redox regulation and drug resistance. Adv Cancer Res. 2014;122:103‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Christopher Kwon YI, Xie W, Zhu H, et al. γ‐glutamyl‐transpeptidase‐resistant glutathione analog attenuates progression of Alzheimer's disease‐like pathology and neurodegeneration in a mouse model. Antioxidants (Basel). 2021;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. More SS, Vince R. Potential of a γ‐glutamyl‐transpeptidase‐stable glutathione analogue against amyloid‐β toxicity. ACS Chem Neurosci. 2012;3(3):204‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Garg G, Singh S, Singh AK, Rizvi SI. N‐acetyl‐l‐cysteine attenuates oxidative damage and neurodegeneration in rat brain during aging. Can J Physiol Pharmacol. 2018;96(12):1189‐1196. [DOI] [PubMed] [Google Scholar]
- 258. Ni G, Hu Z, Wang Z, et al. Cysteine donor‐based brain‐targeting prodrug: opportunities and challenges. Oxid Med Cell Longev. 2022;2022:4834117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Braidy N, Zarka M, Jugder BE, et al. The precursor to glutathione (GSH), γ‐glutamylcysteine (GGC), can ameliorate oxidative damage and neuroinflammation induced by Aβ(40) oligomers in human astrocytes. Front Aging Neurosci. 2019;11:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione‐independent ferroptosis suppressor. Nature. 2019;575(7784):693‐698. [DOI] [PubMed] [Google Scholar]
- 261. Yusuf RZ, Saez B, Sharda A, et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood;136(11):1303‐1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS central science;6(1):41‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Wang L, Liu C, Wang L, Tang B. Astragaloside IV mitigates cerebral ischaemia‐reperfusion injury via inhibition of P62/Keap1/Nrf2 pathway‐mediated ferroptosis. Eur J Pharmacol. 2023;944:175516. [DOI] [PubMed] [Google Scholar]
- 264. Wang L, Zhang X, Xiong X, et al. Nrf2 regulates oxidative stress and its role in cerebral ischemic stroke. Antioxidants (Basel). 2022;11(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Fan Z, Wirth AK, Chen D, et al. Nrf2‐Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6(8):e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Abdul Y, Li W, Ward R, et al. Deferoxamine treatment prevents post‐stroke vasoregression and neurovascular unit remodeling leading to improved functional outcomes in type 2 male diabetic rats: role of endothelial ferroptosis. Transl Stroke Res. 2021;12(4):615‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Guo X, Qi X, Li H, et al. Deferoxamine alleviates iron overload and brain injury in a rat model of brainstem hemorrhage. World Neurosurg. 2019;128:e895‐e904. [DOI] [PubMed] [Google Scholar]
- 268. Keberle H. The biochemistry of desferrioxamine and its relation to iron metabolism. Ann N Y Acad Sci. 1964;119:758‐768. [DOI] [PubMed] [Google Scholar]
- 269. Martin‐Bastida A, Ward RJ, Newbould R, et al. Brain iron chelation by deferiprone in a phase 2 randomised double‐blinded placebo controlled clinical trial in Parkinson's disease. Sci Rep. 2017;7(1):1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Rao SS, Lago L, Volitakis I, et al. Deferiprone treatment in aged transgenic tau mice improves Y‐Maze performance and alters tau pathology. Neurotherapeutics. 2021;18(2):1081‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 271. Rao SS, Portbury SD, Lago L, et al. The iron chelator deferiprone improves the phenotype in a mouse model of tauopathy. J Alzheimers Dis. 2020;77(2):753‐771. [DOI] [PubMed] [Google Scholar]
- 272. Fawzi SF, Menze ET, Tadros MG. Deferiprone ameliorates memory impairment in Scopolamine‐treated rats: the impact of its iron‐chelating effect on β‐amyloid disposition. Behav Brain Res. 2020;378:112314. [DOI] [PubMed] [Google Scholar]
- 273. Moreau C, Danel V, Devedjian JC, et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid Redox Signal. 2018;29(8):742‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Romano N, Baiardi G, Pinto VM, et al. Long‐term neuroradiological and clinical evaluation of NBIA patients treated with a deferiprone based iron‐chelation therapy. J Clin Med. 2022;11(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Marchand F, Moreau C, Kuchcinski G, Huin V, Defebvre L, Devos D. Conservative iron chelation for neuroferritinopathy. Mov Disord. 2022;37(9):1948‐1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Imai T, Tsuji S, Matsubara H, et al. Deferasirox, a trivalent iron chelator, ameliorates neuronal damage in hemorrhagic stroke models. Naunyn Schmiedebergs Arch Pharmacol. 2021;394(1):73‐84. [DOI] [PubMed] [Google Scholar]
- 277. Alta RYP, Vitorino HA, Goswami D, Terêsa Machini M, Espósito BP. Triphenylphosphonium‐desferrioxamine as a candidate mitochondrial iron chelator. Biometals. 2017;30(5):709‐718. [DOI] [PubMed] [Google Scholar]
- 278. Vlachodimitropoulou E, Sharp PA, Naftalin RJ. Quercetin‐iron chelates are transported via glucose transporters. Free Radic Biol Med. 2011;50(8):934‐944. [DOI] [PubMed] [Google Scholar]
- 279. Lesjak M, Balesaria S, Skinner V, Debnam ES, Srai SKS. Quercetin inhibits intestinal non‐haem iron absorption by regulating iron metabolism genes in the tissues. Eur J Nutr. 2019;58(2):743‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Horniblow RD, Henesy D, Iqbal TH, Tselepis C. Modulation of iron transport, metabolism and reactive oxygen status by quercetin‐iron complexes in vitro. Mol Nutr Food Res. 2017;61(3). [DOI] [PubMed] [Google Scholar]
- 281. Youdim MBH. Monoamine oxidase inhibitors, and iron chelators in depressive illness and neurodegenerative diseases. J Neural Transm (Vienna). 2018;125(11):1719‐1733. [DOI] [PubMed] [Google Scholar]
- 282. Lakey‐Beitia J, Burillo AM, La Penna G, Hegde ML, Rao KS. Polyphenols as potential metal chelation compounds against Alzheimer's disease. J Alzheimers Dis. 2021;82(s1):S335‐S357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Yuan Y, Zhai Y, Chen J, Xu X, Wang H. Kaempferol ameliorates oxygen‐glucose deprivation/reoxygenation‐induced neuronal ferroptosis by activating Nrf2/SLC7A11/GPX4 axis. Biomolecules. 2021;11(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Palanimuthu D, Poon R, Sahni S, et al. A novel class of thiosemicarbazones show multi‐functional activity for the treatment of Alzheimer's disease. Eur J Med Chem. 2017;139:612‐632. [DOI] [PubMed] [Google Scholar]
- 285. Jahanshahi M, Khalili M, Margedari A. Naringin chelates excessive iron and prevents the formation of amyloid‐beta plaques in the hippocampus of iron‐overloaded mice. Front Pharmacol. 2021;12:651156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Cheng R, Gadde R, Fan Y, et al. Reversal of genetic brain iron accumulation by N,N'‐bis(2‐mercaptoethyl)isophthalamide, a lipophilic metal chelator, in mice. Arch Toxicol. 2022;96(7):1951‐1962. [DOI] [PubMed] [Google Scholar]
- 287. Yang W, Mu B, You J, et al. Non‐classical ferroptosis inhibition by a small molecule targeting PHB2. Nat Commun. 2022;13(1):7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Xu J, Sun T, Zhong R, You C, Tian M. PEGylation of deferoxamine for improving the stability, cytotoxicity, and iron‐overload in an experimental stroke model in rats. Front Bioeng Biotechnol. 2020;8:592294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Millán M, DeGregorio‐Rocasolano N, Pérez de la Ossa N, et al. Targeting pro‐oxidant iron with deferoxamine as a treatment for ischemic stroke: safety and optimal dose selection in a randomized clinical trial. Antioxidants (Basel). 2021;10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Anthonymuthu TS, Tyurina YY, Sun WY, et al. Resolving the paradox of ferroptotic cell death: ferrostatin‐1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE‐PE, and protects against ferroptosis. Redox Biol. 2021;38:101744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Liu G, Hosomi N, Hitomi H, et al. Angiotensin II induces human astrocyte senescence through reactive oxygen species production. Hypertens Res. 2011;34(4):479‐483. [DOI] [PubMed] [Google Scholar]
- 293. Li S, Zhou C, Zhu Y, et al. Ferrostatin‐1 alleviates angiotensin II (Ang II)‐induced inflammation and ferroptosis in astrocytes. Int Immunopharmacol. 2021;90:107179. [DOI] [PubMed] [Google Scholar]
- 294. Ge H, Xue X, Xian J, et al. Ferrostatin‐1 alleviates white matter injury via decreasing ferroptosis following spinal cord injury. Mol Neurobiol. 2022;59(1):161‐176. [DOI] [PubMed] [Google Scholar]
- 295. Cao Y, Li Y, He C, et al. Selective ferroptosis inhibitor liproxstatin‐1 attenuates neurological deficits and neuroinflammation after subarachnoid hemorrhage. Neurosci Bull;37(4):535‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Karuppagounder SS, Alin L, Chen Y, et al. N‐acetylcysteine targets 5 lipoxygenase‐derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84(6):854‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Zhao J, Wu Y, Liang S, Piao X. Activation of SSAT1/ALOX15 axis aggravates cerebral ischemia/reperfusion injury via triggering neuronal ferroptosis. Neuroscience. 2022;485:78‐90. [DOI] [PubMed] [Google Scholar]
- 298. Lan B, Ge J‐W, Cheng S‐W, et al. Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J Integr Med. 2020;18(4):344‐350. [DOI] [PubMed] [Google Scholar]
- 299. Keuters MH, Keksa‐Goldsteine V, Dhungana H, et al. An arylthiazyne derivative is a potent inhibitor of lipid peroxidation and ferroptosis providing neuroprotection in vitro and in vivo. Sci Rep. 2021;11(1):3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol;13(1):91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Sayan‐Ozacmak H, Ozacmak VH, Barut F, Jakubowska‐Dogru E. Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion. Neurochem Int;61(3):287‐290. [DOI] [PubMed] [Google Scholar]
- 302. Reed A, Ichu TA, Milosevich N, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol. 2022;17(6):1607‐1618. [DOI] [PubMed] [Google Scholar]
- 303. Duan L, Zhang Y, Yang Y, et al. Baicalin inhibits ferroptosis in intracerebral hemorrhage. Front Pharmacol. 2021;12:629379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Peng C, Fu X, Wang K, et al. Dauricine alleviated secondary brain injury after intracerebral hemorrhage by upregulating GPX4 expression and inhibiting ferroptosis of nerve cells. Eur J Pharmacol. 2022;914:174461. [DOI] [PubMed] [Google Scholar]
- 305. Wu C, Du M, Yu R, et al. A novel mechanism linking ferroptosis and endoplasmic reticulum stress via the circPtpn14/miR‐351‐5p/5‐LOX signaling in melatonin‐mediated treatment of traumatic brain injury. Free Radic Biol Med. 2022;178:271‐294. [DOI] [PubMed] [Google Scholar]
- 306. Zhan S, Liang J, Lin H, et al. SATB1/SLC7A11/HO‐1 axis ameliorates ferroptosis in neuron cells after ischemic stroke by danhong injection. Mol Neurobiol. 2023;60(1):413‐427. [DOI] [PubMed] [Google Scholar]
- 307. Hu Q, Zuo T, Deng L, et al. β‐Caryophyllene suppresses ferroptosis induced by cerebral ischemia reperfusion via activation of the NRF2/HO‐1 signaling pathway in MCAO/R rats. Phytomedicine. 2022;102:154112. [DOI] [PubMed] [Google Scholar]
- 308. Li L, Li WJ, Zheng XR, et al. Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor‐mediated Nrf2 activation. Mol Med. 2022;28(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Zhang YH, Wang DW, Xu SF, et al. α‐lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S tau transgenic mice. Redox Biol. 2018;14:535‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054‐2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16(5):e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Mishima E, Ito J, Wu Z, et al. A non‐canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.