

Abstract
Constructing a comprehensive understanding of macromolecular behavior from a set of correlated small angle scattering (SAS) data is aided by tools that analyze all scattering curves together. SAS experiments on biological systems can be performed on specimens that are more easily prepared, modified, and formatted relative to those of most other techniques. An X-ray SAS measurement (SAXS) can be performed in less than a milli-second in-line with treatment steps such as purification or exposure to modifiers. These capabilities are valuable since biological macromolecules (proteins, polynucleotides, lipids, and carbohydrates) change conformation or assembly under specific conditions that often define their biological role. Furthermore, mutation or post-translational modification change their behavior and provides an avenue to tailor their mechanics. Here, we describe tools to combine multiple correlated SAS measurements for analysis and review their application to biological systems. The SAXS Similarity Map (SSM) compares a set of scattering curves and quantifies the similarity between them for display as a color on a grid. Visualizing an entire correlated data set with SSMs helps identify patterns that reveal biological functions. The SSM analysis is available as a web-based tool at https://sibyls.als.lbl.gov/saxs-similarity/. To make data available and promote tool development, we have also deployed a repository of correlated SAS data sets called Simple Scattering (available at https://simplescattering.com). The correlated data sets used to demonstrate the SSM are available on the Simple Scattering website. We expect increased utilization of correlated SAS measurements to characterize the tightly controlled mechanistic properties of biological systems and fine-tune engineered macromolecules for nanotechnology-based applications.
1. Introduction
Biology fundamentally relies on precisely orchestrated chemical reactions. To coordinate the mechanism and timing that select which atomic bonds will be made or broken, macromolecular architectures have evolved to identify molecules and, through conformational changes, direct them into controlled environments. These environments concentrate reactants, promote specific rotamer orientations or orbital overlaps, and prevent reversion to initial states or unwanted side products. The macromolecules are also precisely tuned to function in context-specific ways based on solvent conditions and chemical inputs from other macromolecules. These functionalities remain unmatched by synthetic systems because the complexity and speed of macromolecular mechanisms are difficult to decipher experimentally or computationally. While recent advances in cryo-electron microscopy (cryo-EM) and continued innovation in macromolecular crystallography (MX) and nuclear magnetic resonance (NMR) provide atomic resolution information, these techniques host several shortcomings related to capturing the aforementioned macromolecular complexity of biological systems. SAS is well positioned to fill the gaps in our knowledge of macromolecular function, provide additional information that is unobtainable through other means, and enhance the value of every determined atomic resolution structure.
SAS analysis has proven to provide valuable, and often definitive, constraints on macromolecular conformational change and dynamics at the nanoscale. The number of constraints SAS generates on macromolecular structure has been estimated using a Shannon sampling formalism (Konarev & Svergun, 2015). Applying this formalism is necessary as SAS information content is not equivalent to the number of points in a SAS curve. The sampling of momentum transfer (q) as a function of scattered intensity usually far exceeds the number of points required to reconstruct the SAS curve. According to Shannon analysis, a single SAS measurement on a solution of macromolecules typically has 10–40 unique pieces of information (Konarev & Svergun, 2015). While alone, this information is insufficient to define the relative positions of each atom within a macromolecule and how they evolve over time, but when the information is used to constrain conformation based on a model generated by other methods, the resolution is high, can be generated in high-throughput, and, importantly, reflects the solution state. When multiple SAS measurements that probe multiple states are collected and analyzed together, the information content can be higher than the sum of Shannon channels and uniquely complementary to tools that resolve static structures.
The malleability of SAS measurements is an important asset during their application to providing insights on top of those gleaned from static structures. Due to low-throughput constraints needed to make measurements, cryo-EM, MX, and NMR often produce a set of atomic coordinates representing a single structure inhabiting a limited physiological context. Further, SAS profiles can be accurately calculated from an atomic model (Schneidman-Duhovny, Hammel, & Sali, 2010; Shin et al., 2009; Svergun, Barberato, & Koch, 1995). If a poor match is found between the measured and calculated profiles, the atomic model can be modified and a new SAS profile can be calculated and compared to generate new conformations that more closely match the measured data (Fig. 1). Therefore, a single atomic resolution structure, generated by a lower throughput technique, can be leveraged by SAS to define nanoscale changes that occur in other contexts. Integrating information from multiple contextualized SAS measurements will be central to applications of SAS over the next decade.
Fig. 1.
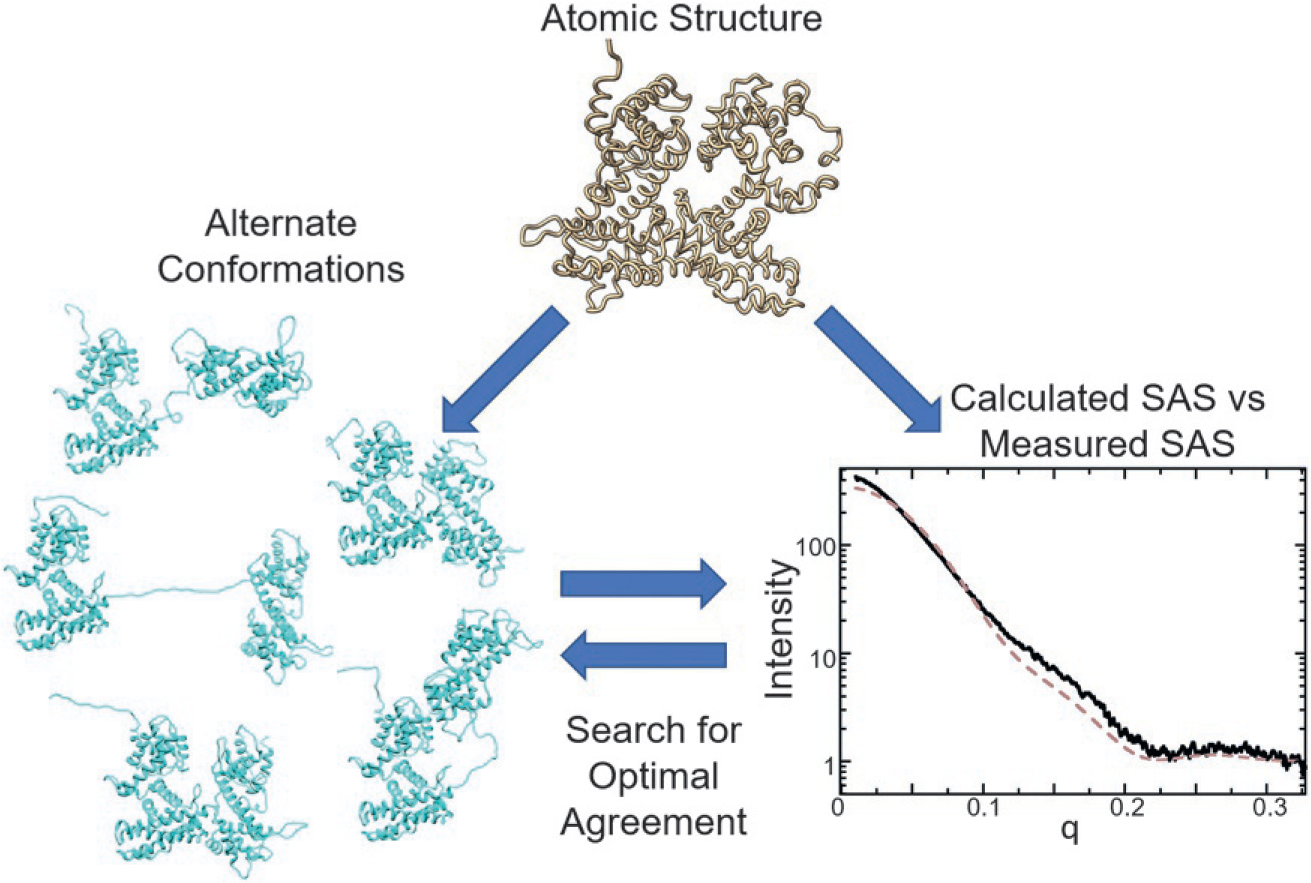
Integrating SAS with atomic resolution measurements. A SAS signal can be accurately calculated from atomic coordinates determined either by a high-resolution technique or through structure prediction. Disagreement indicates a different solution state than that provided from the high-resolution model. Alternate conformations can be generated based on the atomic coordinates to search for models or ensembles of models that best fit the SAS data.
To facilitate the integration of information from multiple contextualized SAS measurements, the SAS community needs both access to correlated data sets and tools that analyze those data together. In this chapter, we describe our efforts to address both of these needs in two principle sections. In Section 2, we describe SAXS Similarity Maps (SSMs), which is a tool designed to visualize tens of SAXS measurements collected from a macromolecule or related macromolecules under a variety of contexts. We provide examples and also demonstrate the application of SSMs to derive insights in several cases. In Section 3, we describe a bespoke repository, called Simple Scattering, for organizing and holding this kind of data. Further, Simple Scattering can also be used to hold other types of data beyond those usefully analyzed by SSMs. Simple Scattering holds SAXS data generated during size-exclusion coupled SAXS (SEC-SAXS) and can also hold high-throughput (HT-SAXS), time-resolved, micro-fluidic-associated, and neutron-based SAS (SANS) data. These resources will be useful as the structural biology community seeks to leverage success in resolving static structures and produce dynamic and solution state perspectives.
SAXS is particularly well suited to collect structural information on multiple states or conditions of a macromolecule, can be conducted at a higher throughput than other structural techniques, and data acquisition times are often less than a second at synchrotron user facilities. (Yang et al., 2021). Many SAXS beamlines have automated sample loading schemes that allow hundreds of samples to be collected in only a few hours (Grant et al., 2011; Hura et al., 2009; Nielsen, Moller, & Gillilan, 2012; Round et al., 2015; Yang et al., 2021). Sample preparation is also straightforward, requiring microliter volumes of microgram quantities, and measurements can be performed in almost any biologically relevant condition. Additionally, sample preparation for SAXS data collection can also be automated with liquid handling robotics. The throughput and ease of sample preparation and data collection lends itself to conducting many experiments.
SAXS from many conditions that inform on macromolecules can be generated in several ways. Many complex macromolecular systems interact with multiple metabolites for their function. A common example are proteins that couple energy from ATP hydrolysis to other complex reactions (Krukenberg, Street, Lavery, & Agard, 2011; Rosenberg, Deindl, Sung, Nairn, & Kuriyan, 2005; Williams et al., 2011). Because these systems recognize both ATP and other proteins or metabolites and also need to release hydrolyzed ATP and the product of the reaction, they often cycle through a range of conformations. Multiple correlated SAXS measurements have been successfully used to identify which combinations of chemical signals activate different mechanisms. SAXS measurements can screen solution conditions or small molecules that trigger conformational changes. Using a mutation scanning approach on proteins, SAXS can be used to identify amino acid residues essential for facilitating substrate and partner binding and overall conformational change. Amino acids create specific and often nonintuitive interacting networks of hydrogen bonds that enable long distance communication between one region of a protein and another. Mutations are often critical to engineering macromolecules for specific structural and dynamic outcomes, which are readily identified by SAS.
Correlated SAXS data on macromolecules can also be obtained by collecting SAXS data during purification, exposure to light, or mixing. The flux of synchrotron X-ray sources and detector efficiencies ensures SAXS can be monitored as a function of time. The use of component mixing, light exposure, or temperature and pressure jumps can initiate reactions that induce conformational changes over time. For example, microfluidic technologies enable controlled mixing and modifications to a flow of macromolecules which can be examined at different points of the fluid flow (Malaby et al., 2015). Additionally, many macromolecular systems are transient in assembly and conformation. The equilibrium solution state of these transient systems is a mixture that can often be dissected during purification. SEC-SAXS provides access to transient structures from within a larger pool of mixed states and macromolecular species (Malaby et al., 2015; Mathew, Mirza, & Menhart, 2004).
SAS results do not always fit in a similar category as results from cryo-EM, NMR, and MX. At the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB, https://www.rcsb.org/), each entry consists of the atomic coordinates of a single macromolecular conformation. A database with a similar purpose has been developed for SAS data, the Small Angle Scattering Biological Data Bank (SASBDB) (Kikhney, Borges, Molodenskiy, Jeffries, & Svergun, 2020). SASBDB is a valuable SAS community resource, as was its predecessor BioISIS (Hura et al., 2009). However, these databases have generally been created for a single type of SAS experimental configuration and result.
To compliment structural biology tools that provide perspectives on static structures, like the SASBDB, we have implemented Simple Scattering to store correlated data sets and deployed SSMs for the analysis of an important subset of these experiments. We have made the tools we describe in detail, below, accessible through a web browser. The SSMs are accessed at https://sibyls.als.lbl.gov/saxs-similarity/. The SAS data sets are stored at https://simplescattering.com/.
2. Visualization of correlated SAXS data
Integrating information from correlated SAXS data is not trivial. Ideally this integration would occur when atomic models have been determined from each independent measurement, as outlined in Fig. 1. However, as an averaging solution-based technique, many phenomena may be occurring at the same time. In complex macromolecular systems, for example, heterogeneity in conformation and assembly are common. Changes in solution conditions and the presence of substrates or products may push populations of conformations or assemblies toward one form or another. To diagnose these phenomena and extract meaningful biological insights, analysis of a data set as a whole is useful.
2.1. SAXS Similarity Map (SSM) test and demonstration
We have implemented a SAXS Similarity Map (SSM) or heat map approach to visualize correlated SAXS data sets (Hura et al., 2013). These maps are information rich and often provide mechanistic details. The order in which data are presented also often improves analysis. Before we present experimental examples, we will describe how this approach has been implemented and demonstrate its utility on calculated data.
An SSM displays data in a heat map form commonly employed with other types of data. SSMs are essentially symmetric matrices where each entry quantifies the difference between two SAXS curves. The score is represented as a color along a gradient from high to low. The central diagonal of the matrix compares a curve against itself (perfect agreement) and is blacked out to provide greater contrast. Every contributing curve is compared against all other curves in non-diagonal elements. Element 1,2 of the matrix compares the first curve against the second and 2,1 compares the second curve against the first. With most metrics developed to compare SAXS curves, these elements should have the same value.
Several metrics have been developed to compare SAXS curves. The most frequently used is as defined in Eq. 1, where is momentum transfer, and are the intensities, and the intensity means, and and the errors for separate SAXS measurements. We have developed our own metric called Volatility of Ratio which is defined in Eq. 2, where is the ratio of intensities at (Hura et al., 2013). The mathematical measure of volatility is borrowed from the world of finance (Roll, 1984) and has been useful to describe how much variation there is over a stable trend. is not without shortcomings. For instance, using as a fitness measure for optimization in other applications is not as straightforward as with . Also, a describes an optimal fit, given the errors in the data, whereas has not been implemented with the error of measurement considered. However, has several valuable characteristics relative to for comparison of SAXS data that prove its utility and are demonstrated below.
| (1) |
| (2) |
The first reason we favor is that it operates on the ratio of two SAXS curves rather than the difference of two curves, as is the case with . SAXS data decay exponentially as a function of . Therefore, differences in small , which have exponentially higher values, can vastly outweigh differences in high . The rate of exponential decay varies from structure to structure. However, with correlated SAXS measurements of the type typically analyzed in the SSMs—that is, a given macromolecular system in varying experimental conditions—the structures are often similar as is their rate of exponential decay. Thus, a ratio reduces the overwhelming impact of the exponential decay and provides more equal weighting to all regions of . This can also be over-factored in that a range of covers a Bragg spacing of while a range of covers . The former range is frequently more important than the latter in terms of SAXS data. Therefore, our implementation of by default only uses a range of . A second reason we use is that it bins the data to approximate Shannon sampling. Binning has the effect of increasing signal to noise since most SAXS profiles are over-sampled. The spacing between bins is linear in momentum transfer. Non-linear binning may have advantages that have vet to be explored.
We have calculated SAXS data from each step in a linear geometric morph of a designed protein cage (Lai, Cascio, & Yeates, 2012; Lai, Tsai, Sawaya, Asturias, & Yeates, 2013; Padilla, Colovos, & Yeates, 2001) starting from a compact state and gradually morphing to an expanded and open state (Fig. 2A and B). The cage resembles a closed triangular pyramid. The 11 curves calculated from models along the trajectory have been randomly ordered in an SSM (Fig. 2C, left). The first row in the map coincidently is the first step in the trajectory. Scanning the row of the curve that scored most similar (most red), the second step in the trajectory can be identified. By applying a simple clustering algorithm, the randomized curves align into a meaningful heatmap array that correlates with the closeness of atomic models along the morph trajectory (Fig. 2C, right). The clustering algorithm first finds the two curves with best agreement and pairs them at the start of the matrix list. It then finds the curves with the most disagreement relative to the pair with greatest agreement and places them at the end of the list. Finally, it works from both ends of the list finding the next nearest agreements and adds them to the list from both bottom and top.
Fig. 2.
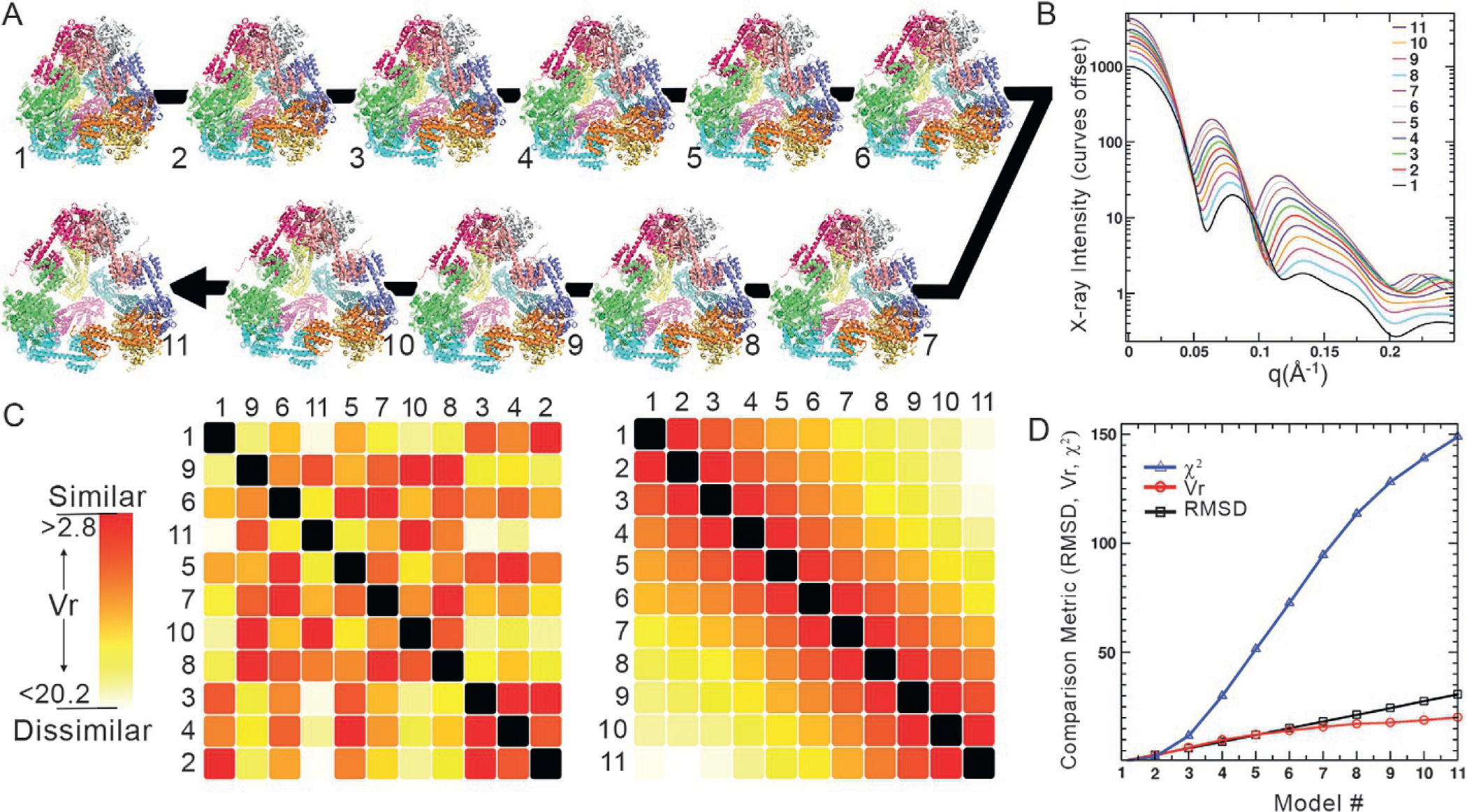
Similarity of SAXS curves correlates directly to similarity in structure. (A) A designed tetrahedral protein cage was crystallized in a collapsed and compact state (conformation 1) relative to its idealized expanded design (conformation 11). A linear morph was created between the two (intervening conformations). (B) SAXS profiles were calculated from each step in the morph. The curves are offset in X-ray intensity for clarity. (C) These SAXS profiles were used to create a SAXS Similarity Map (SSM) where each cell is a comparison of two profiles (compared curves from each step in the morph are identified left and top of SSM). The similarity, numerically quantified by the metric , is depicted as a color on a red to white gradient with the most similar curves (lowest score) colored red and most dissimilar are white. On the SSM, (C, left) the curves were input in a random order. An automated clustering algorithm was applied to organize the SSM (C, right). (D) The root mean square deviation (RMSD) between the most compact structure (1) and later steps in the morph increases linearly (black squares), in agreement with the algorithm that created the morph. In contrast, the agreement between SAXS curves exponentially increases (blue triangles). This extreme deviation complicates the interpretation of how different two structures are based on SAXS profile. The agreement (red circles) follows the linear RMSD behavior more closely between morph steps and provides more correlated guidance on similar structures.
The remarkable transformation of the randomized matrix to an ordered one shows that organizing SAXS data by their similarity correlates with organizing similar structures in solution. Additionally, using to quantify SAXS, and therefore structural similarity, has advantages over . Fig. 2D plots the various comparison metrics (RMSD, ) vs. models generated from the morph. better captures the linearity of the morph RMSD relative to a calculation on the same set of data. changes exponentially and, as a result, loses its capacity to identify similarity with even modest changes in the Guinier region of experimental data since that region is weighted so heavily. performs more as a binary metric; identifying data sets as either very similar or very different.
2.2. Data requirements and web-based application of SAXS similarity maps
As mentioned above, SSMs can be generated via a web-based interface at https://sibyls.als.lbl.gov/saxs-similarity/. The inputs are ASCI tabulated SAS data that take the form of three columns. The first column lists , the second X-ray intensity, and the third error in measured intensity. The columns can be tab or space delimited.
Public web-based platforms are targets for hacking and security imperatives change frequently. Currently, pound (“#”) symbol headers are tolerated. However, the closer the SAS files are to a simple three column format, the greater the chance they pass evolving security filtering process. Similarly, for security reasons, only files with “.dat” extensions are currently tolerated.
To be useful, more than two SAS profiles are required as the tool is purposed to compare different SAS profiles. Multiple SAS profiles can be selected and dropped into an active area in the web application or a folder can be dropped in the same way. The data do not need to have the same spacing as an interpolation script is applied to all data sets. Errors in loading specific files are noted in a visual display. Once the simple file requirements are fulfilled and the files are dropped, an SSM is displayed.
The off-diagonal scores are used to adjust the range of the gradient. The two profiles that are most similar set one limit of the gradient while the profiles that score least similar set the other. The gradient can be adjusted with three options: red-yellow-white, blue-white-red and black-gray-white. The current implementation does not allow for further contrast adjustment. However, the SSM can be saved as an image and further manipulated by image processing software to highlight features that may not be visible by the default contrast settings.
The SSM can be manipulated on the web interface in several ways. The order of the files can be adjusted by dragging file names up or down on the list. Once a file is dragged to a different position, the entire SSM is reorganized as indicted with a visible effect. Tabs allow for the automated clustering or reversal of the order of data in the SSM.
More information about the difference between any two files is provided by clicking on the cell that compares the two. Once clicked, the two profiles are plotted below the SSM. The names of the two profiles are listed in the plot along with their calculated radii of gyration. The clicked cell in the SSM also lists the difference in radius of gyration, , calculated from the two profiles. All can be made visible by using a tab option.
An important capability is the adjustment of range used for comparison. This capability makes possible the analysis of data with varying amounts of aggregation, other dependent anomalies or when a specific region of contains the most discrimination. For example, in drug screening, some drugs may be more insoluble than others. These may create large aggregates that solely affect the small region. Adjusting the range so that the small region is ignored improves analysis. To adjust the range, a cell must be clicked and, once the two profiles are plotted as described above, a range selector is displayed for manipulation.
Four different metrics of comparison are available for the SSM, including . The default is . As has yet to consider relative signal to noise, the SSM may appear asymmetric for low signal data. is ratio-dependent and low signal data will be near zero, thus affecting its scoring. For low signal data, the score using Profile 1 divided by Profile 2 may not be the same as the score using Profile 2 divided by Profile 1. To adjust this asymmetry, the range must be adjusted so that only the higher signal data is utilized. The matrix of numbers that quantifies the similarity can be viewed using a tab feature.
Tutorials, instructions, and test data are also available at the web site. Users of the web site that have trouble dragging and dropping their files are recommended to download the test data sets. Once downloaded, these data can be uploaded to the web site to generate an SSM, testing the web site’s functionality. Once functionality is demonstrated, the test files can be inspected and mimicked in the data sets of interest.
2.3. Similarity Map of substrate binding
Many DNA repair proteins are significant in size and carefully orchestrate events to preserve genome integrity. Simply keeping DNA together is insufficient since several cellular malfunctions can occur with just one nucleotide base slip. Because of this complexity, DNA repair proteins are some of the most complicated macromolecular machines. The original motivating application for developing SSMs stemmed from mechanistic studies of a DNA repair protein MutS that binds multiple substrates and metabolites (Hura et al., 2013). MutS is greater than 100kDa and forms homo- or heterodimers. In most higher organisms, MutS is known as MSH2 and binds the homologous MSH3 or MSH6 to shift specificity for recognizing damaged DNA. MSH2/MSH3, also referred to as MutSβ (Lang et al., 2011), identifies unpaired loops or hairpins while MSH2/MSH6, also referred to as MutSα (Graham, Putnam, & Kolodner, 2018), identifies mismatches. Identification of damaged DNA is followed by a repair cascade with multiple binding partners that derive energy from an ATPase domain. Outstanding questions include how specificity is achieved, the mechanistic role of ATP hydrolysis, specific actions on DNA, and binding with partner proteins during the repair cascade. Knowledge of this system is key for cancer and antibody fields, where DNA mismatches are important for maintaining genomic integrity and generating new antibodies. Achieving a comprehensive understanding of MutS mechanisms with cryo-EM or MX alone has proven very challenging because many structures are necessary but have thus far been challenging to obtain. Correlated SAXS data sets were used to enhance overall understanding of these complex systems.
SAXS data was collected from a ten-member correlated SAXS data set: MutSβ either alone or in the presence of loop DNA with either ADP, ATP, ATPγS, or AMPpMp (Fig. 3). The latter two metabolites are non-hydrolysable analogues of ATP. The first row of the SSM compares the apo MutSβ against the other conditions. The most similar condition to apo MutSβ, along the first row, is in the presence of the substrate it is designed to recognize, a DNA loop. This is surprising as the protein surrounds the DNA in a clamp like manner and must involve a conformational change, though of an apparent lesser degree than the other conditions. For example, a greater dissimilarity in underlying conformation occurs in the presence of ADP and ATP.
Fig. 3.
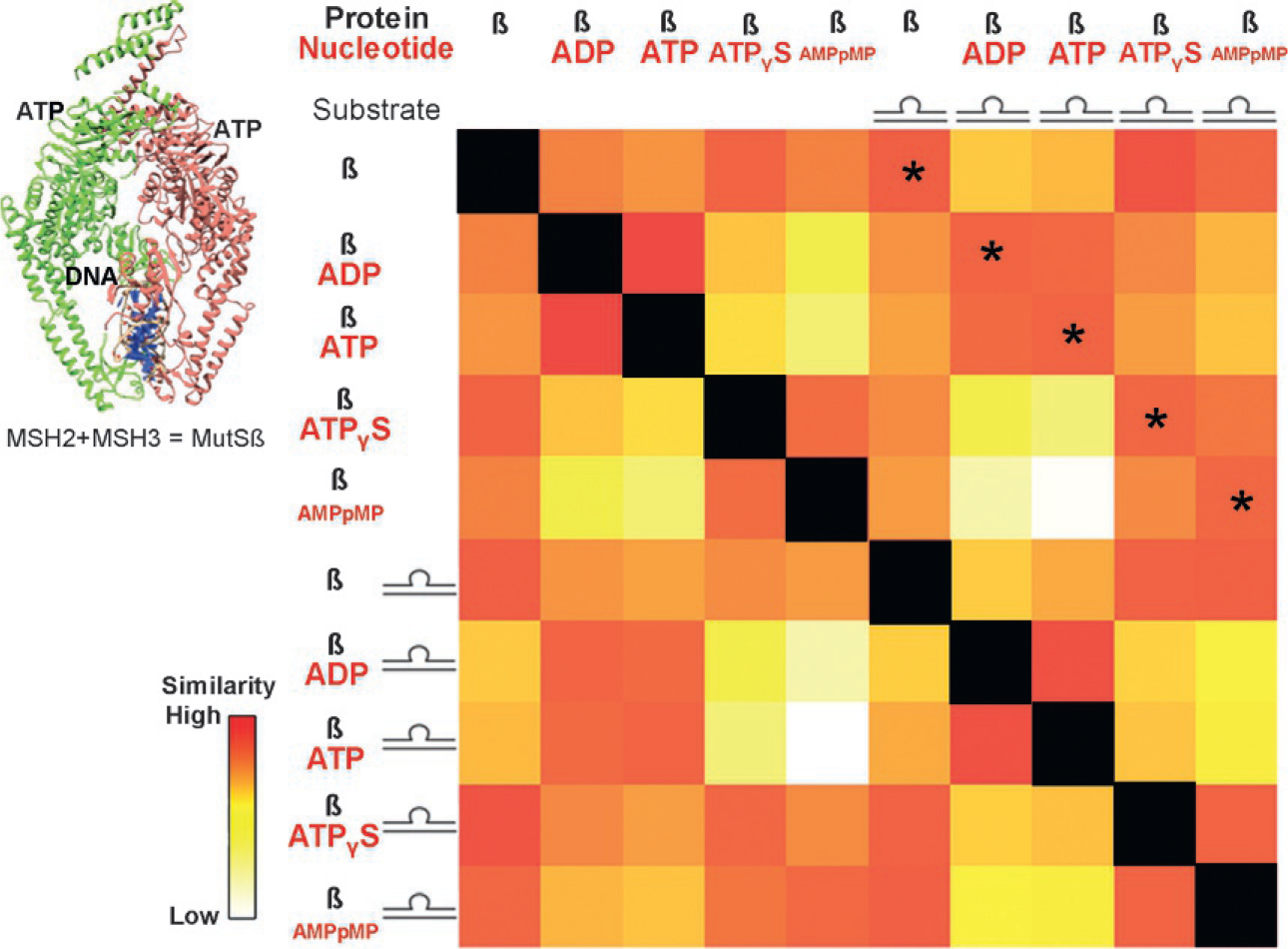
Conformational changes of MutSβ due to substrate binding. MutSβ aids in repairing DNA loops and is powered by ATP hydrolysis. SAXS collected on MutSβ with and without DNA and with nucleotides ADP, ATP, and non-hydrolysable analogues AMPpMP and ATPγS were compared against one another in an SSM. The asterisked diagonal compares measurements with and without DNA in the presence of the same nucleotide. The indicated similarity across this diagonal highlights that, mechanistically, the nucleotide state determines the conformation of the protein, not DNA binding.
Proceeding to the second row, where MutSβ with ADP is compared against all other conditions, a relative similarity is found with the solution containing ATP and a relative dissimilarity is found with non-hydrolysable ATP analogues. This suggested, and was later corroborated by other methods, that MutSβ hydrolyzes ATP immediately, regardless of whether DNA is present (Hura et al., 2013). However, prior to hydrolysis, the ATP-bound state is in a different conformation that is more similar to the apo-state.
The above conclusions and a primary mechanistic insight about the system are further summarized in the diagonal of similarity, highlighted by the asterisks in Fig. 3. This diagonal compares ATP hydrolyzation states both on and off DNA. The SAXS curves from each ATP analogue, regardless of whether DNA is bound, are similar. Having verified that DNA is indeed bound using gel electrophoresis, the implication is that conformation is fixed by the ATP hydrolysis state and that its recognition process relies on finding DNA that is able to conform to this structure. This can be rationalized by understanding that DNA possessing a loop is more malleable than fully complementary DNA. SSM analysis requires ordering the conditions in a specific way and a careful examination for patterns. The benefit is that each measurement is represented and can be placed in the context of all other measurements for a comprehensive analysis.
2.4. Studies of conformation and assembly based on buffer conditions
Solution conditions influence macromolecular assembly and conformation. With designed proteins, understanding these factors can help improve or control the system (Lai et al., 2016). The protein cage system shown in Fig. 2 and Section 2.1, was designed, expressed and purified for further experimental analysis using correlated SAXS. A monomer in the dodecameric cage assembly is a fusion of a protein that forms trimers (the vertices of the pyramid) with a protein that forms dimers (the edges of the pyramid) (Padilla et al., 2001) (Lai et al., 2012, 2013). Successful formation of the pyramid was first demonstrated by cryo-stained EM and later by crystallography. In the crystallographic studies several crystal forms were found. However, none of the crystal structures matched the idealized pyramid. All of them were collapsed to varying extents, with compressed edges and a smaller internal cavity than expected from the initial design.
To determine whether salt or pH in the varying crystal forms affected the conformation of the cage we performed an extensive salt and pH screen. We modified the NaCl concentration using 10, 100, 300 and 500mM. We also analyzed each of these salt concentrations in single pH steps from pH of 4–11. This resulted in a total of 28 conditions. An SSM of these conditions is shown in Fig. 4.
Fig. 4.
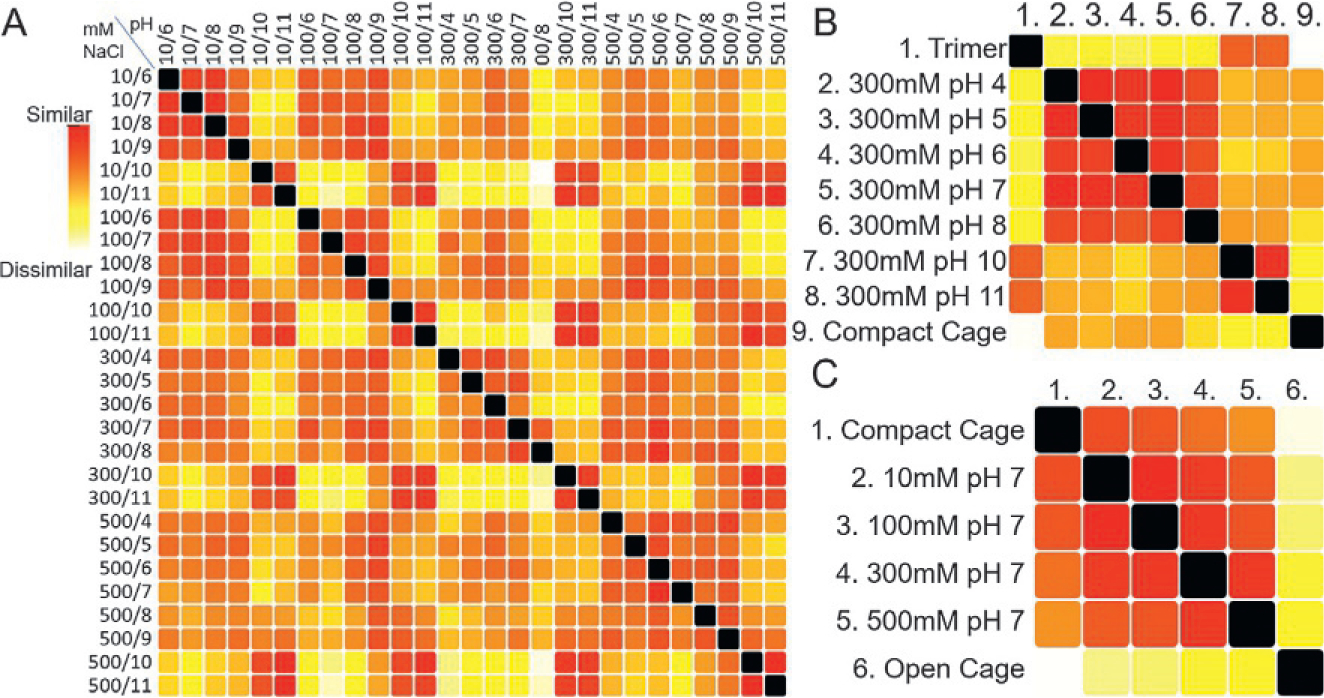
Protein cage assembly can be controlled by NaCl concentration and pH. (A) SAXS data was collected on a dodecameric protein cage (atomic model shown in Fig. 2) as a function NaCl concentration and pH. These are identified at the top and left of the SSM using a mM NaCl/pH format. The striking grid like patterns in the SSM can be further understood looking at subsets of the data (B) pH can be used to control assembly. Looking at an SSM of a subset that varies pH but holds salt concentration constant (300mM) shows that at the high pH values (10 and 11) the SAXS curves are most similar to that calculated from a trimeric disassembly product rather than the full cage. (C) Salt concentration modifies conformation of the cage. Comparing solutions that held a pH of 7 but varying salt concentration shows that the structures in solution are most similar to a compact state. However, increasing salt moves the overall structure toward the designed open state.
The most obvious feature from the overall SSM is a grid like pattern. The vertical and horizontal strips of yellow and white occur at pH values of 10 and 11. Applying the clustering algorithm, all curves with pH of 10 and 11 cluster together (not shown).
Interpretation of SSMs is aided by including SAXS calculated from models expected in solution. In Fig. 4B we show an SSM of the 300mM NaCl pH series including SAXS calculated from a model of the disassembled trimeric vertices of the cage and a crystal structure of the compact cage. At high pH the agreement to the model of the trimer is high, indicating that pH weakens the interaction between the dimer forming part of the fusion. Therefore, increasing pH can be used to disassemble the system. An SSM representing pH 7 with varying salt concentration (Fig. 4C) reveals that higher salt concentration increases the openness of the cage. Interpretation is aided by including theoretical SAXS curves calculated from the compact cage and the idealized open cage. A gradient in similarity toward the open conformation is revealed as salt concentration is increased. Changing the salt concentration is therefore a means to control cage expansion. While the designed conformation was not observed with this construct in any of the solution conditions tested, the results suggest that modifying the charges on the inside of the cage may help form the more open structure. With the current construct, salt and pH allow for significant control of assembly and conformation.
2.5. Structural impact of point mutations
Screening mutations in a protein has many applications in biology. In some cases, a single point mutation can inactivate or unfold a protein. Mutations can also affect affinity, allosteric ability to undergo structural rearrangement or multimerization. SAXS is sensitive to these types of changes.
The structural impact of mutations upon the protein Apoptosis-inducing factor (AIF) was assayed by SAXS (Brosey et al., 2016). A mitochondrial oxidoreductase, AIF facilitates metabolism by supporting import of OXPHOS components into mitochondria (Hangen et al., 2015). However, during DNA damage and PARP-1 hyperactivation, AIF is released from mitochondria and participates in a cell death pathway (Fatokun, Dawson, & Dawson, 2014; Morales et al., 2014). AIF functional regulation is believed to derive from architectural switching stimulated by binding of the metabolite NADH. NADH allosterically triggers AIF dimerization and release of a 50-residue surface loop (C-loop) from the C-terminal domain. Because the NADH binding site is distant from AIF’s dimerization interface and C-loop (Fig. 5A), the “information” of NADH binding is communicated via intervening amino-acid cascades. Identifying amino acids along these cascades opens additional avenues for regulating AIF switching and functional responses. To define the amino acid cascade and controlling mechanisms of NADH responses, HT-SAXS was performed on a panel of AIF mutants with and without NADH.
Fig. 5.
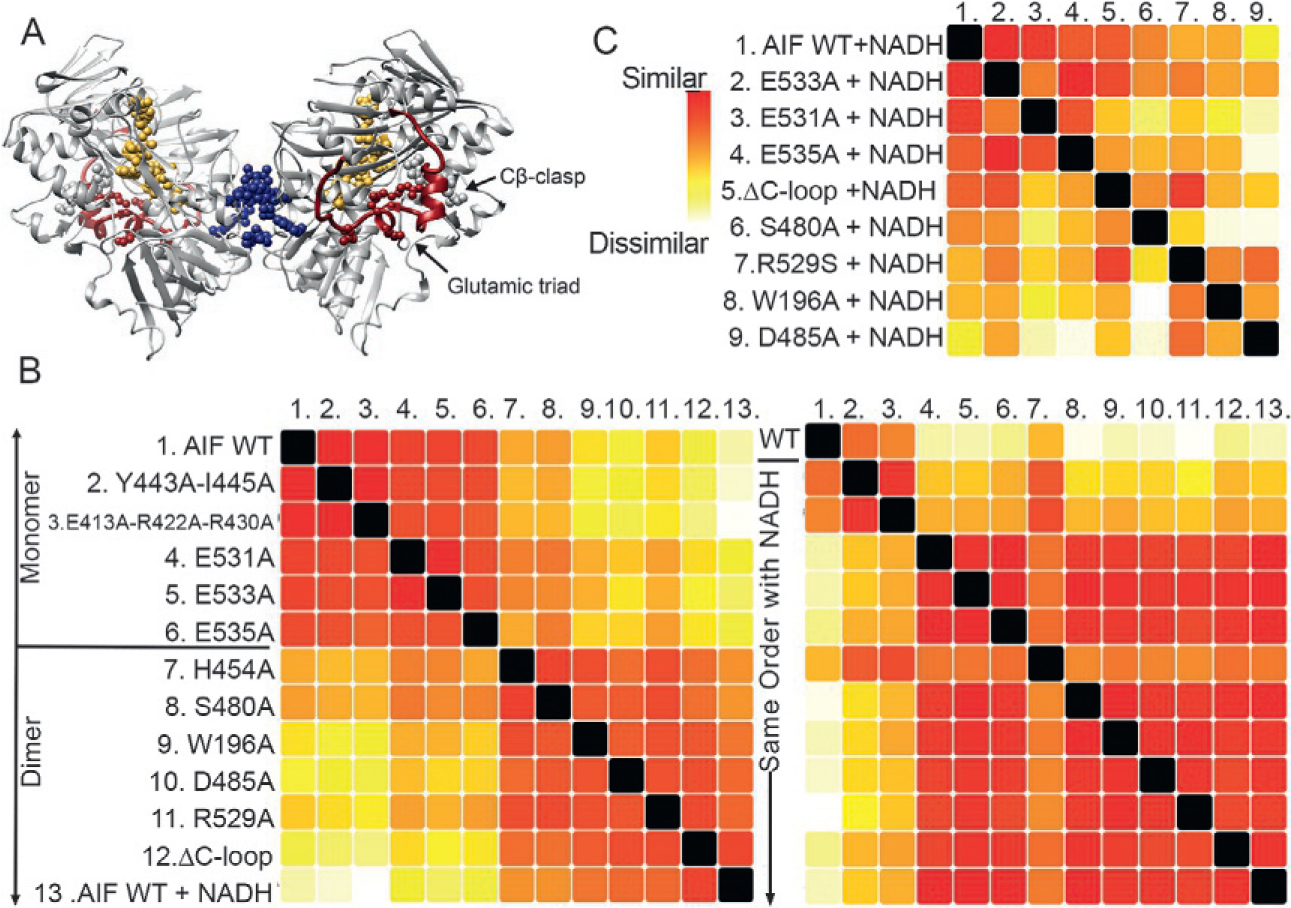
SAXS uncovers allosteric communication between a metabolite binding site and dimer interface. (A) The protein AIF binds the metabolite NADH at its central active site (gold), triggering dimerization at a distal surface (blue) and release of a surface C-loop (red). Allosteric residues targeted for mutation are shown as spheres. Dimeric AIF supports OXPHOS biogenesis, a target of interest for cancer treatment. (B) SSM analysis of AIF mutants relative to wild-type (WT) monomeric AIF without NADH (left panel) distinguishes AIF mutants consistent with the AIF monomer (top) from AIF mutants that dimerize without NADH (bottom). Complementary SSM analysis of NADH-saturated AIF mutants (right panel) suggest high similarity to the wild-type AIF dimer except for Y443A-I445A and E413A-R422A-R430A (defective for dimerization) and H454A (defective for C-loop release). (C) Extended SSM analysis of NADH-saturated AIF mutants relative to wild-type AIF dimer conformationally partitions mutants of AIF’s glutamic acid triad and Cβ-clasp.
An SSM helps characterize function-breaking mutations (Fig. 5B and C). In the absence of NADH, wild-type AIF is monomeric. However, mutants which remove the C-loop (ΔC-loop) or target residues S480, H454, W196, D485, and R529 resemble the dimeric NADH-bound wild-type state, pointing to their role in allosteric communication (Fig. 5B, left). In contrast, mutations at the dimer interface, E413A-R422A-R430A and Y443A-I445A, prevent dimer formation, regardless of the presence of NADH (Fig. 5B, right). This result identified residues Y443 and I445 as novel contributors to the AIF dimerization interface. Notably, the H454A mutant exhibits intermediate similarity to wild-type AIF monomer and dimer. Complementary biochemistry experiments revealed that this mutant obligately dimerizes but does not release its C-loop in response to NADH (Brosey et al., 2016), uncovering its unique role in linking the active site and dimerization interface. Thus, SSM analysis also highlights mutants that present unusual and informative phenotypes.
To extract further detail, SSMs can be recalculated by systematic removal of outliers (here Y443A-I445A, E413A-R422A-R430A, and H454A). In Fig. 5C, NADH-saturated AIF mutants that successfully form dimers and release the C-loop can be compared directly to NADH-saturated wild-type AIF dimer to distinguish subtle impacts from individual mutations. As before, SSM analysis partitions different classes of allosteric mutants. The glutamic acid triad (E531, E533, E535) forms a network of exterior salt bridges securing the C-loop to AIF’s surface, while residues of the Cβ-clasp (R529A, W196, D485) form a complex allosteric hydrogen bond network that is disrupted to release the C-loop upon NADH binding. Here, the SSM analysis for the wild-type dimer ranks collective similarity of the dimeric glutamic acid mutants higher than the dimeric Cβ-clasp mutants, suggesting a difference in conformation or monomer-dimer equilibria. As such, the SSM results frame and prioritize mutant classes for follow-up analyses, whether by detailed examination of the underlying SAXS curves or complementary structural and biochemical approaches.
2.6. Validating protein engineering designs with SSMs
A major motivator for establishing HT-SAXS is to confirm macromolecular designs from computationally driven macromolecular engineering. Recent machine learning computational programs have been successful in predicting fold based on sequence (Alexander et al., 2021; Baek et al., 2021; Jumper et al., 2021). Tackling the converse problem, desired structure to predicted sequence, has also been successful in several cases. Many engineered proteins express well, making them highly amenable to protein expression pipelines. The Baker lab and other macromolecular engineers have employed SAXS for experimental validation of their designs (Boyken et al., 2016; Chen et al., 2020; Langan et al., 2019).
In a foundational study on the simple alpha helix-turn-helix motif, the Baker lab has investigated the possible topologies that could be created (Brunette et al., 2015). Surprisingly many structures not yet observed in natural peptides seemed computationally feasible. Several of the topologies are shown in Fig. 6A. To test whether these computationally designed structures could be stabilized, sequences for over 40 topologies were expressed, purified, and verified by SAXS. Several were also verified from crystal structures.
Fig. 6.
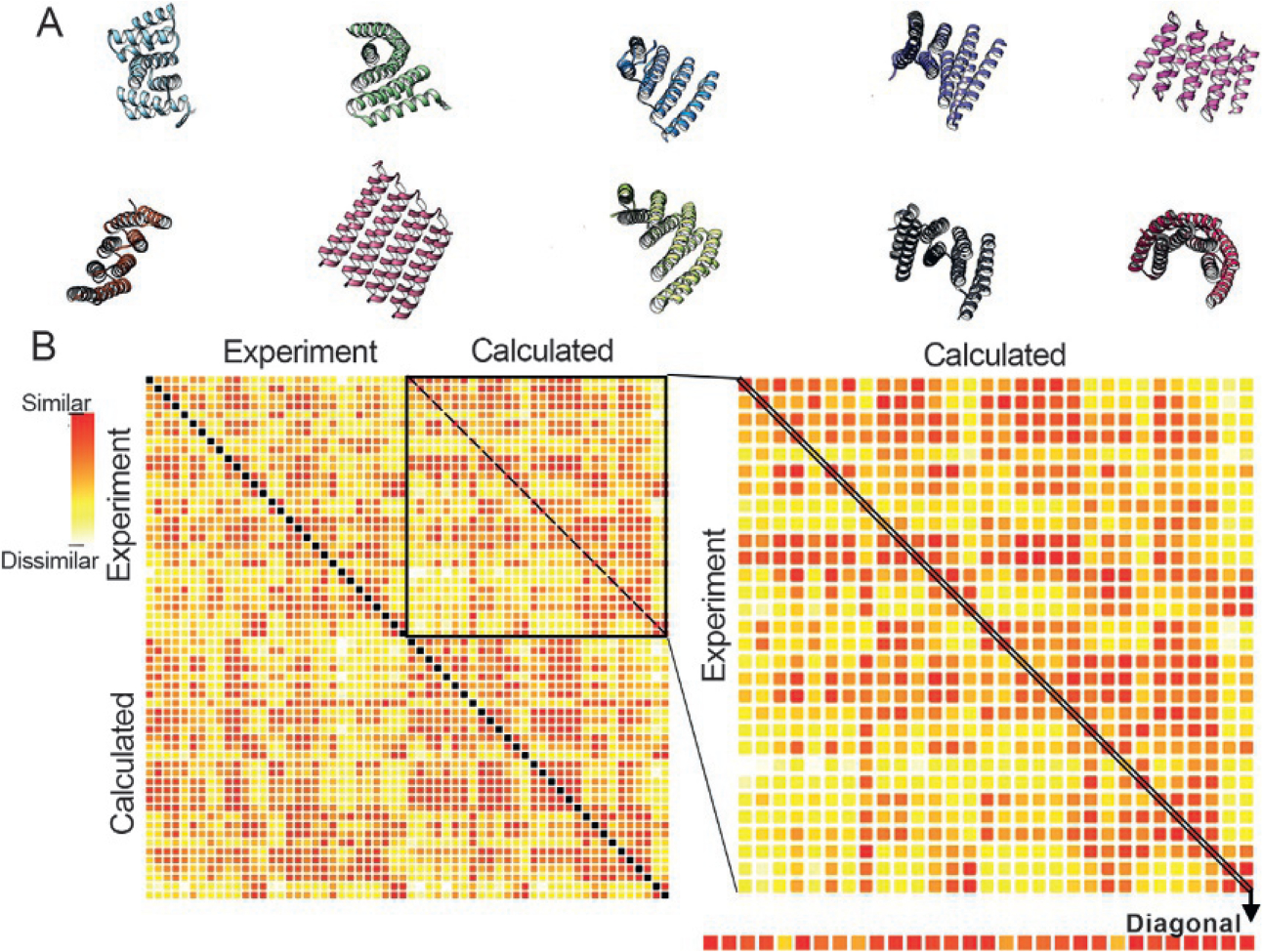
Validating protein engineering principles with SSMs. Using a repeat helix-turn-helix design, over 80 protein topologies were computationally created. The designed topologies varied stacking, torsions, and angles between helices. Examples of topologies are shown in (A). Sequences from designed topologies were expressed, purified, and were measured by HT-SAXS. An SSM of 30 measured SAXS profiles and the corresponding calculated SAXS profiles is shown in (B). The SSM is ordered with all measured SAXS profiles in the first half followed by the calculated. The calculated profiles are in the same order (top to bottom) as the measured. Success could be assessed by analyzing the diagonal of the top right-hand quadrant of and SSM from all profiles, which is blown up on the right. The mostly red diagonal in this quadrant, which is further extracted in a linear form at the bottom, indicates success in many designs. Off-diagonal red squares indicate either poor SAXS discrimination between the topologies or sequences that favor other folds, as diagnosed by other quadrants.
An SSM was used to rapidly analyze the success of the computational designs (Fig. 6B). All experimental profiles are listed first followed by the calculated profiles in the same order as the experimental profiles. The SSM can be analyzed in quadrants; top left, top right, bottom left, and bottom right. The bottom right compares all the calculated scattering curves against one another. Given that constructs are approximately the same size and the overall shapes are mostly flat and elongated, the degree of variation in similarity in the SAXS curves confirms SAXS sensitivity to high resolution information. Some of the topologies create more unique SAXS profiles than others.
Experimental evidence for a match to the computational designed topologies can be observed in several ways from the SSM. The matching of patterns in the SSM of the top left quadrant (experiments against experiments) vs. the bottom right (computation against computation) is one perspective. A more direct approach is examining the top right quadrant (Fig. 6B, blowup right) where experimental data is compared to computation data. Since the topologies are listed in the same order, the agreement can be assessed by the degree to which there is a diagonal of similarity. The diagonal can be extracted to identify topologies that failed (Fig. 6B, bottom). These can be reviewed in detail for improvements in sequence or to help identify constraints on topology that have yet to be understood. The SSM therefore provides both a global perspective on the ability to form novel helix-turn-helix motifs and highlights specific instances for improvement in design.
3. Simple Scattering data set repository
The applications of SSMs described above is a necessary first step in a deeper analysis of those data sets. The drag and drop option of SSMs on a web browser make their application straightforward and potentially provide non-experts a tool to highlight outstanding conditions or constructs from a SAXS perspective. However, many other tools have been developed and could be applied in their analysis. Moreover, it is our sincere hope that new tools will continue to develop to extract further information from these rich data sets. To promote further analysis, we have deposited the data sets described above into the Simple Scattering web repository. The repository is available to store and access SAS data collected by others and we describe how SAS data can be deposited in Simple Scattering below.
Correlated SAS data can be generated in many ways, as described in the introduction. Besides the SSM examples described above that were collected by high-throughput means, Simple Scattering is also designed to hold correlated SAS data generated by other methods. Currently, Simple Scattering is also designed to hold SEC-SAS, time-resolved SAS (TR-SAS), and SAS from microfluidics. As a developing resource, to accommodate these various forms of data, data entry has significant flexibility with many optional fields while also accepting meta-data in various formats. This reduces the time to deposit a data set which is valuable for a technique that generates data quickly. The Simple Scattering client interface has been designed to allow automated deposition from particular types of correlated experiments directly from instruments, as described below. The repository was designed with a singular purpose to hold data rather than perform analysis on the data itself. From its inception Simple Scattering has also been designed with the goal of achieving FAIR (Findable, Accessible, Interoperable, and Reusable) principles of data management (Wilkinson et al., 2016).
Simple Scattering’s purpose is distinct from the existing SAS repository SASBDB. The SASBDB (Kikhney et al., 2020) at https://www.sasbdb.org/ and its predecessor BioISIS (Hura et al., 2009) hold individual SAS curves that are analyzed to as full an extent as possible including atomic models derived from the SAS curves. The SASBDB and BioISIS resources are modeled after the PDB. As SASBDB and PDB entries are partially curated, database entry typically requires significant time. These databases were not specifically designed to store multiple data sets that characterize multiple correlated states. Simple Scattering, on the other hand, has been expressly designed to hold tens to hundreds of SAS curves from a correlated set of experiments collected from samples in varying solution conditions, related but slightly different constructs, or representing different sample preparation regimes (as exemplified in Section 2).
Simple Scattering provides several advantages over general purpose data repositories like Zenodo (https://zenodo.org/). As a niche site, such as SASBDB and the PDB, Simple Scattering provides a publicly available repository for researchers to discover large correlated SAS data sets. Simple Scattering provides control over how data is deposited, uniformity over how data may be displayed and the ability to further fine tune the site for future access by other tools. As noted above, we anticipate increased generation of correlated SAS data sets with a corresponding need for improved analysis. By building the database, we provide access and draw further attention to data set types that require the development of unique tools to complete their analysis.
Once deposition is complete an entry is paired with a unique identifier similar to those generated by the PDB and SASBDB. Simple Scattering identifiers start with the letters “XS” followed by six unique alphanumeric ASCII characters. A search tool is available to find an entry by the identifier among other key words described in proceeding sections.
3.1. Simple Scattering user storyboard
The current workflow for a user depositing correlated scattering data for public access has four main steps. (1) A data depositor sets up an account that is verified by the maintenance staff, (2) a single or correlated set of one-dimensional SAS data is uploaded by the depositor through a typical web-based graphical user interface or by an automated process from an instrument and is assigned a unique identifier, (3) the depositor adds descriptions and relevant meta-data, and (4) maintenance staff validate the deposition and release the entry for public access.
For the public, the latest entries may be found on the Simple Scattering “Dashboard” page, along with site statistics and blog posts. Entries for all available data are listed on the Simple Scattering “Open Datasets” pages. If the unique identifier code for a particular set of data is known, it can be appended to the site’s URL after a slash (e.g., https://simplescattering.com/open_dataset/XSYY8DN2) and the page will be displayed. Alternatively, the code can be entered in the search bar atop each page to retrieve the entry. Data can also be identified by searching keywords associated with the deposition. The SAS data and any associated supplementary data can then be downloaded by clicking a link.
Data presented in Simple Scattering is intended as a supplement to a publication. In its current form, SAS data deposited in Simple Scattering is not extensively reviewed. Automated validation and analysis tools have yet to be developed and linked to the deposition process. Therefore, details associated with the Simple Scattering entry, such as data collection method and the file nomenclature used to delineate each SAS profile is the responsibility of the depositor. Ideally, the data set is associated with a reviewed publication and provides readers and reviewers access to the data described in the publication. To this end, depositors can update their deposition at any time so long as their Simple Scattering account is valid.
3.2. Deposition of data to Simple Scattering
Deposition of data into Simple Scattering can be approached in two ways. The first is by manual entry online through a web graphical user interface (Fig. 7A); the second is through an application programming interface (API) (Fig. 7B). The latter method lends itself to automation, which matches the pace of data collection at modern facilities wielding high-throughput configurations. The API is currently implemented at the SIBYLS beamline for its SEC-SAXS users. We will describe both processes below.
Fig. 7.
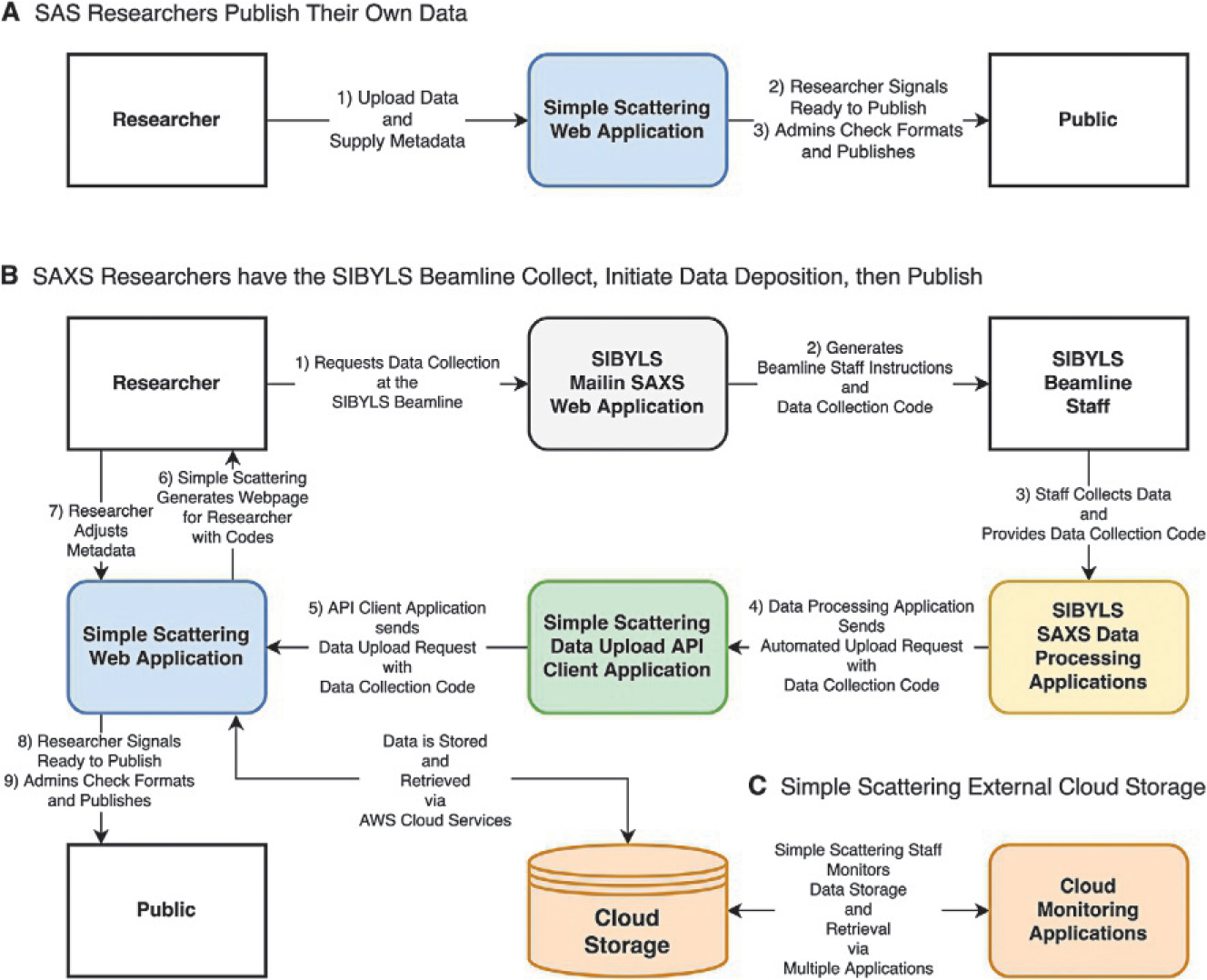
Workflow for upload of data to Simple Scattering for SIBYLS beamline. (A) Upload and release of data from the Simple Scattering web application. (B) In a more automated pipeline, beamline users first request beamtime through our MailinSAXS site (https://bl1231.als.lbl.gov/htsaxs), which generates instructions and a unique data collection code. Beamline staff then collects data and converts image readouts to individual files that contain the scattering angle, intensities, and errors using in-house data processing applications. These applications can then initiate a data upload request to Simple Scattering on behalf of the user through a data upload API client application. Simple Scattering then generates its unique code for the data set and makes a request to store the data on Amazon Web Services. Upon success, Simple Scattering generates a draft web page for the beamline user, who then adds additional metadata and supplementary files. Upon publishing the data, the web page for the particular data set is made accessible to site visitors, and data can be retrieved from storage and sent to the visitor by clicking a link. (C) Staff monitors cloud storage through AWS cloud monitoring applications.
To manually deposit scattering data the user must register for an account. Registration requires the user’s full name and email address, followed by the user’s project leader, institute, and the country that the researcher belongs to. To deposit data, the account requires vetting of the user by the Simple Scattering system administration.
Once registered, the user uploads data sets by filling out a minimal set of required fields: Title of experiment, Data collection technique, Experimental description, File description. A user may also add additional metadata about the macromolecule, or in the case of complexes, macromolecules, including but not limited to: the sample type (protein, DNA, lipid, etc.), the sample name, the abbreviated name, and its polymer sequence (amino acid or nucleotide sequence) or chemical formula. Lastly, at minimum, a compressed (zip) file containing the data is uploaded. Other supporting files may also be uploaded.
The compressed file containing the data should consist of individual plain text files, one for each data collection experiment (if a single experiment was performed, the file should still be compressed). Each file within the compressed zip file should contain plain text, with three columns representing the momentum transfer, intensities and errors, in that order. Or alternatively, with two columns representing the momentum transfer and intensities. Headers or footers may be included in the individual data files provided each line is preceded by a hashtag or pound (“#”) symbol. Each file within the compressed zip file should have the extension “.dat” or “.txt.” To note, currently there is a memory limit of 20MB for the compressed file. The raw data collection image files should not be included in the compressed file or any other files. These raw files could by uploaded to a service like Zendo (see above) and the unique identifier could be noted in the deposition.
In addition to the compressed file containing the data, researcher’s may upload additional files. First, up to 100 individual experimental files contained within the compressed file may be uploaded. The purpose for this is so that visitors to the site may download one or a few files to inspect prior to downloading the larger data set. Second, researchers may add additional files that support the data set. This may include atomic coordinates generated by modeling or experimental methods such as MX, NMR, cryo-EM (.pdb or .cif files), or other data collected while running a size-exclusion column such as multi-angle light scattering (MALS) or quasi-elastic light scattering (QELS). Additionally, plain text or portable document format (.pdf) files with additional explanations or insights for the experiment may be included. MALS or QELS may be submitted in the form of portable document format (.pdf) files, or comma separated value (.csv) files. The Experimental description field is free text and serves a similar purpose as the methods and materials section in journals. Sample preparation information, the conditions tested, and the parameters that remained constant throughout the experiment are valuable for future analysis. Details of where and how the data was collected should also be included.
The file description field is intended to allow researchers to describe the file naming convention of their data files within the main compressed file. This does not preclude investigators adding pound tagged headers or footers inside the files themselves. If non-descriptive file names or headers/footers are used, the file description field will need to contain a file by file description of the SAS filenames. For a simple SEC-SAXS experiment, noting that the files are sequentially numbered according to the elution is sufficient. For more complex experiments, the investigators downloading data sets may need indicators that distinguish one file from another in a correlated data set. Often file base names provide a sufficient number of characters to be descriptive and are a useful way to distinguish one file from another. For example, in the case of the metabolite screen described in Section 2.2, the file named MutSBeta_DNA_ATP.dat is the SAXS profile collected from protein MutSβ mixed with hairpin DNA and ATP in contrast to the file named MutSBeta_ADP.dat which is the SAXS profile of MutSβ mixed with ADP. For the salt and pH screen conducted on the cage system described in Section 2.3, the files have names in the form of 10mM_pH7_PCTrip. dat. The mutation analysis (Section 2.4) contains the one-letter code mutations in the name.
Once all required information is input, the depositor can upload the data, and signal the Simple Scattering system administrators that a data set is either still in draft mode or ready for release to the public. System administrators can approve a data set for release or contact the user if there are issues with the submission. Depositors can continue to edit the information contained in their submission as necessary.
Simple Scattering also includes a private API that allows beamline scientists collecting data for their users to upload their data on their behalf to ease the deposition process. The API is implemented at the SIBYLS beamline at the Advanced Light Source to deposit SEC-SAXS data through an automated deposition system (Fig. 7B). When the SIBYLS beamline is operating, SEC-SAXS data are collected at a rate of approximately 20 samples per week. The deposition of an SEC-SAXS data set is accomplished by first using data processing software. This is followed by using a Python-based data upload API client application that sends requests to the currently private Simple Scattering API. The private API upload requires an email address and the location of the SAXS data files. The data is transferred off the local beamline file system to cloud storage, mediated by the Simple Scattering database. A unique identification code is generated for the data set and data availability is communicated to the user. At this stage the data remain private. However, the user is able to enter the other required information (meta data) described above. Once the user has filled out all required fields, the data can be submitted for approval by Simple Scattering system administration and publicly released.
3.3. Simple Scattering infrastructure
Simple Scattering is hosted on the Heroku cloud platform and offered to the public by the SIBYLS beamline. The frontend servers are provided via Heroku and as such the details of administering, maintaining and upgrading the physical frontend servers is left to them. By using an enterprise class cloud service provider such as Heroku, we are able to offload much of the nitty gritty details of server configuration, database maintenance, and service scalability to Heroku. If the demands on server capacity or database size/speed increase, Heroku provides the ability to seamlessly scale Simple Scattering should demand necessitate.
Simple Scattering is a Ruby on Rails application whose Object-Relational Mapping system managed by Active Record is coupled to a PostgreSQL relational database management system. Ruby on Rails is a pre-packaged server-side (backend) framework that utilizes Ruby to support the model-view-controller design pattern that is frequently used in web applications. It also includes the tooling for the HTML, CSS, and JavaScript, a high-level asynchronous programming language, required for client-side (frontend) programming to generate the web pages that outside users interact with. All metadata is stored in the PostgreSQL database provided as part of the Heroku service and is protected by both a local snapshot disaster recovery backup called “Postgres Continuous Protection” and a longer-term offsite backup solution called “PG Backups.” All data files uploaded to Simple Scattering are stored in Amazon’s Web Service (AWS) high durability, high availability S3 standard buckets which provide 99.999999999% durability and 99.99% availability over a given year (https://aws.amazon.com/s3/storage-classes/). We additionally monitor data flow to the cloud servers though Amazon’s CloudWatch and CloudTrail services, and maintain separate backups of both the database and data. To connect to the API, a set of Python programs are used.
As the minimal core functionality of the Simple Scattering site has been completed, there are many avenues for improvements and new features. Future directions and features will be user focused, and we hope to include integrations with other beamlines. Suggestions include a public API for deposit, searching records and to download files. The ability to automate fetching data may allow other applications such as SAXS Similarity to access data from the unique Simple Scattering identification codes.
3.4. Current statistics from the Simple Scattering database
If Simple Scattering were to remain static with the current deposits, it is the largest public repository of correlated SAXS and SEC-SAXS data sets thus far. For developers of software tools, Simple Scattering is therefore a unique source of experimental data. As the site is just now being advertised to researchers, there are ~40 users. Thus far, over 180 SEC-SAXS data sets (each containing over 600 scattering profiles) have been deposited with nearly 40 released to the public. Three of the correlated SAXS data sets, reported above in Section 2, are also available. The Simple Scattering unique identifiers for the MutSβ, PCtrip, and AIF studies are XSYY8DN2, XSSQ87OY, and XSUTRZQL, respectively. Site statistics reveal that others from around the world are already accessing the sites, as anyone can download data without registration.
As noted above, there is a dashboard, https://simplescattering.com/dashboard, that shows the most recent uploads, the latest news and statistics about the data contained within the database. Currently, project titles, macromolecular names, and data set codes are searchable through the search field in the site’s navbar. More advanced search features and page views will be available in the near future.
4. Summary and outlook
Macromolecular structural biology has made tremendous gains in recent years, including advances in cryo-EM and machine learning structure prediction such as AlphaFold2 (Jumper et al., 2021) and RoseTTAFold (Humphreys et al., 2021). These technologies, in conjunction with MX and NMR, are capable of providing near atomic resolution models of most soluble macromolecules. The remaining open challenges in characterizing macromolecules has shifted in light of these capabilities. One of the challenges receiving renewed focus is to derive an understanding of mechanisms given an atomic structure.
SAXS is well positioned to help address this challenge. Not only do SAXS data provide a means to confirm an atomic resolution model in solution (Fig. 1), but SAXS data can also be collected from macromolecules in hundreds of contexts. Above, we showed SAXS data collected from macromolecules as a function of multiple substrates (Section 2.3), pH and salt concentration (Section 2.3), and mutations (Section 2.5). When the data from multiple SAXS measurements are integrated together and displayed as an SSM, the conditions that maintain a structure or induce changes therein are readily apparent. In some cases, the SAXS calculated from atomic models are helpful as a reference. Conditions that deviate from this reference can be identified and further investigated. The available atomic model is modified to better agree with the SAXS data and provides insights into the factors necessary to change conformation or assembly. These approaches are powerful and can be applied to the engineering of designed macromolecules where the effect of many sequence modifications can to be tested for desired outcomes.
To provide access to correlated SAS data sets for further verification and analysis we have introduced the data depository Simple Scattering. Many of the data sets can be analyzed by SSMs or other algorithms designed for this purpose. SEC-SAXS has emerged as a powerful approach and analysis tools continue to develop and improve. We hope other synchrotron beamlines will also join in the effort in depositing SEC-SAXS data using the API. Providing access to many SEC-SAXS, HT-SAXS, SANS, microfluidic SAXS, and TR-SAXS data sets is expected to spur development of computational tools and drive sophisticated SAS analysis to advance our understanding of macromolecules and their mechanisms.
Acknowledgments
The acquisition of data at the SIBYLS beamline and the development of the web-based applications were largely funded by the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by the DOE Office of Biological and Environmental Research under contract DE-AC02-05CH11231. Additional funding was provided by the National Cancer Institute (NCI) grants for Structural Biology of DNA Repair (SBDR) PO1 CA092584, CA220430, and the National Institute of Health project, ALS-ENABLE (P30 GM124169).
References
- Alexander LT, Lepore R, Kryshtafovych A, Adamopoulos A, Alahuhta M, Arvin AM, et al. (2021). Target highlights in CASP14: Analysis of models by structure providers. Proteins: Structure, Function, and Bioinformatics, 89(12), 1647–1672. 10.1002/prot.26247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science, 373(6557), 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyken SE, Chen Z, Groves B, Langan RA, Oberdorfer G, Ford A, et al. (2016). De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science, 352(6286), 680–687. 10.1126/science.aad8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosey CA, Ho C, Long WZ, Singh S, Burnett K, Hura GL, et al. (2016). Defining NADH-driven Allostery regulating apoptosis-inducing factor. Structure, 24(12), 2067–2079. 10.1016/j.str.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunette TJ, Parmeggiani F, Huang PS, Bhabha G, Ekiert DC, Tsutakawa SE, et al. (2015). Exploring the repeat protein universe through computational protein design. Nature, 528(7583), 580–584. 10.1038/nature16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Kibler RD, Hunt A, Busch F, Pearl J, Jia M, et al. (2020). De novo design of protein logic gates. Science, 368(6486), 78–84. 10.1126/science.aay2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, & Dawson TM (2014). Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. British Journal of Pharmacology, 171(8), 2000–2016. 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham WJT, Putnam CD, & Kolodner RD (2018). The properties of Msh 2-Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair. Journal of Biological Chemistry, 293(47), 18055–18070. 10.1074/jbc.RA118.005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant TD, Luft JR, Wolfley JR, Tsuruta H, Martel A, Montelione GT, et al. (2011). Small angle X-ray scattering as a complementary tool for high-throughput structural studies. Biopolymers, 95(8), 517–530. 10.1002/bip.21630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM, et al. (2015). Interaction between AIF and CHCHD4 regulates respiratory chain biogenesis. Molecular Cell, 58(6), 1001–1014. 10.1016/j.molcel.2015.04.020. [DOI] [PubMed] [Google Scholar]
- Humphreys IR, Pei JM, Baek M, Krishnakumar A, Anishchenko I, Ovchinnikov S, et al. (2021). Computed structures of core eukaryotic protein complexes. Science, 374(6573), 1340. 10.1126/science.abm4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, et al. (2013). Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nature Methods, 10(6), 453–454. 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, Tsutakawa SE, et al. (2009). Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nature Methods, 6(8), 606–612. 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikhney AG, Borges CR, Molodenskiy DS, Jeffries CM, & Svergun DI (2020). SASBDB: Towards automatically curated and validated repository for biological scattering data. Protein Science, 29(1), 66–75. 10.1002/pro.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, & Svergun DI (2015). A posteriori determination of the useful data range for small-angle scattering experiments on dilute monodisperse systems. IUCrJ, 2(Pt 3), 352–360. 10.1107/S2052252515005163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukenberg KA, Street TO, Lavery LA, & Agard DA (2011). Conformational dynamics of the molecular chaperone Hsp90. Quarterly Reviews of Biophysics, 44(2), 229–255. 10.1017/s0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YT, Cascio D, & Yeates TO (2012). Structure of a 16-nm cage designed by using protein oligomers. Science, 336(6085), 1129. 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- Lai YT, Hura GL, Dyer KN, Tang HY, Tainer JA, & Yeates TO (2016). Designing and defining dynamic protein cage nanoassemblies in solution. Science Advances, 2(12), e1501855. 10.1126/sciadv.1501855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YT, Tsai KL, Sawaya MR, Asturias FJ, & Yeates TO (2013). Structure and flexibility of nanoscale protein cages designed by symmetric self-assembly. Journal of the American Chemical Society, 135(20), 7738–7743. 10.1021/ja402277f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang WH, Coats JE, Majka J, Hura GL, Lin YY, Rasnik I, et al. (2011). Conformational trapping of mismatch recognition complex MSH2/MSH3 on repair-resistant DNA loops. Proceedings of the National Academy of Sciences of the United States of America, 108(42), E837–E844. 10.1073/pnas.1105461108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan RA, Boyken SE, Ng AH, Samson JA, Dods G, Westbrook AM, et al. (2019). De novo design of bioactive protein switches. Nature, 572(7768), 205–210. 10.1038/s41586-019-1432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaby AW, Chakravarthy S, Irving TC, Kathuria SV, Bilsel O, & Lambright DG (2015). Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering. Journal of Applied Crystallography, 48, 1102–1113. 10.1107/s1600576715010420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew E, Mirza A, & Menhart N (2004). Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. Journal of Synchrotron Radiation, 11, 314–318. 10.1107/s0909049504014086. [DOI] [PubMed] [Google Scholar]
- Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. (2014). Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Critical Reviews in Eukaryotic Gene Expression, 24(1), 15–28. 10.1615/critreveukaryotgeneexpr.2013006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SS, Moller M, & Gillilan RE (2012). High-throughput biological small-angle X-ray scattering with a robotically loaded capillary cell. Journal of Applied Crystallography, 45, 213–223. 10.1107/s0021889812000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla JE, Colovos C, & Yeates TO (2001). Nanohedra: Using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2217–2221. 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll R (1984). A simple implicit measure of the effective bid-ask spread in an efficient market. The Journal of Finance, 39(4), 1127–1139. 10.1111/j.1540-6261.1984.tb03897.x. [DOI] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, & Kuriyan J (2005). Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell, 123(5), 849–860. 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Round A, Felisaz F, Fodinger L, Gobbo A, Huet J, Villard C, et al. (2015). BioSAXS sample changer: A robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallographica Section D-Structural Biology, 71, 67–75. 10.1107/s1399004714026959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, & Sali A (2010). FoXS: A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Research, 38(Web Server issue), W540–W544. 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, DiDonato M, Barondeau DP, Hura GL, Hitomi C, Berglund JA, et al. (2009). Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: Structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. Journal of Molecular Biology, 385(5), 1534–1555. 10.1016/j.jmb.2008.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D, Barberato C, & Koch MHJ (1995).CRYSOL—A programto evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography, 28, 768–773. 10.1107/S0021889895007047. [DOI] [Google Scholar]
- Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, et al. (2016). The FAIR guiding principles for scientific data management and stewardship. Scientific Data, 3, 160018. 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, et al. (2011). ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nature Structural & Molecular Biology, 18(4), 423–U454. 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lazo E, Byrnes J, Chodankar S, Antonelli S, & Rakitin M (2021). Tools for supporting solution scattering during the COVID-19 pandemic. Journal of Synchrotron Radiation, 28, 1237–1244. 10.1107/s160057752100521x. [DOI] [PMC free article] [PubMed] [Google Scholar]