

1. Introduction:
The development of therapeutics for Alzheimer’s disease (AD) has been hindered by the difficulty of obtaining timely and accurate diagnosis, which contributes up to 80% screen failure rates in AD clinical trials [1]. Despite the availability of amyloid-beta (Aβ) positron emission tomography (PET) and cerebrospinal fluid (CSF) biomarker assays for Aβ isoforms (Aβ42, Aβ40), tau, and other neuroproteins (A/T/N classifiers) used to identify brain amyloid pathology, there remains an unmet need for an accessible, radiation-free, minimally invasive, cost-effective, rapid, and analytically validated diagnostic method to facilitate clinical trial enrollment and aid in clinical AD diagnosis [2].
One AD risk factor, the apolipoprotein E (APOE) gene, exists as three polymorphic alleles—APOE2, APOE3 and APOE4 – which give rise to six common genotypes, APOE2/2, APOE2/3, APOE2/4, APOE3/3, APOE3/4, and APOE4/4 [3]. The APOE protein is vital for peripheral and central nervous system (CNS) lipid metabolism [4,5], and studies show that APOE genotypes strongly affect Aβ clearance, deposition, and senile plaque formation in AD brains [6]. Aβ deposition in the form of senile plaques is more abundant in APOE4 carriers compared with noncarriers [7–10]. Individuals with one copy of APOE4 (heterozygotes) have a three-fold higher risk, while those who inherit two copies of APOE4 (homozygotes) have an eight to 12-fold greater risk of developing AD [4,11]. Conversely, one or two APOE2 alleles appear to protect against AD [12].
Studies have demonstrated that a lower plasma Aβ42/Aβ40 ratio, in combination with the well-established risk factors of APOE4 status and age, correlate with brain amyloidosis as measured using amyloid PET imaging [13,14].
In response to this need C2N Diagnostics has developed two liquid chromatography tandem mass spectrometry (LC-MS/MS) based assays that simultaneously quantify Aβ isoforms in human plasma (Precivity-Aβ™) and determine common APOE proteotype (Precivity-APOE™). C2N previously reported that the plasma Aβ42/40 ratio accurately identifies the presence of brain amyloidosis [15], and more recently, we improved the accuracy for identifying brain amyloidosis by combining plasma Aβ42/40 ratio with APOE proteotype and age into an Amyloid Probability Score (APS, PrecivityAD™) [16]. Based on the clinical evidence available at the time of this manuscript, the test is currently intended for use as a prescription only test in individuals 60 years of age or older with early cognitive impairment or dementia undergoing evaluation for Alzheimer’s disease. Here, we report the analytical validation parameters and metrics for these novel LC-MS/MS based plasma biomarkers.
2. Materials and Methods
2.1. Specimen Collection.
Blood was collected in K2 EDTA tubes (Becton Dickinson, Franklin Lakes, NJ), placed on ice, and centrifuged at 1800 – 2300 x g to obtain plasma. Plasma was aliquoted into 2.0 mL polypropylene vials, frozen on dry ice, and stored at −65 to −80°C. Specimens were shipped overnight on dry ice to C2N Diagnostics.
2.2. Aβ40 and Aβ42 Quantification
On the day of use, frozen calibrators, quality control (QC) materials, plasma specimens, and uniformly labeled 15N full length Aβ40 and Aβ42 internal standards (IS) (rPeptide, Watkinsville, GA) were thawed. Once thawed, 450 μL of plasma, 9 μL of 2.5% (w/v) Tween-20, 23 μL of PBS, 45 μL of 5M guanidine, and 10 μL of protease inhibitors were added to each well of a 2 mL 96-well plate (Thermo Fisher Scientific, Waltham, MA). Final concentrations of full-length 15N IS proteins were 200 pg/mL and 30 pg/mL for Aβ40 and Aβ42, respectively. Aβ was extracted from plasma by immunoprecipitation using a monoclonal antibody, HJ5.1 (Aβ amino acid residues 13–28; C2N owns the proprietary HJ 5.1 monoclonal antibody that is manufactured at Rockland Immunochemical, Inc), conjugated to magnetic beads (DynaBeads M-270 Epoxy, Thermo Fisher Scientific). After 90 minutes of immunocapture, the Aβ bound magnetic beads were removed and washed with PBS and triethylammonium bicarbonate (TEABC) to reduce non-specific binding contaminants prior to sample digestion. After washing, the bound amyloid protein containing beads were placed in a TEABC at 37°C containing 0.94 ng/μL of the metalloendopeptidase Lys-N (Thermo Fisher Scientific), which digests Aβ species into peptides Aβ28–40 (amino acid sequence KGAIIGLMVGGVV) and Aβ28–42 (amino acid sequence KGAIIGLMVGGVVIA). After 120 minutes, the digestion was quenched by the addition of 2% formic acid. The Aβ peptide digests were further purified using reverse-phase solid phase extraction (Waters Corporation, Milford, MA), washed with 20% MeOH/1% formic acid to remove contaminating substances, and eluted using 55% ACN/1% formic acid onto a 96-well collection plate (Waters Corp.). Eluted Aβ peptides were dried under vacuum and reconstituted in 16 μL of 10% ACN/10% formic acid for injection onto the LC-MS/MS system.
The 96-well plate containing the resolubilized Aβ peptides was placed in the temperature-controlled LC autosampler (Waters Acquity LC), and 3 μL from each well were injected onto a monolithic divinylbenzene column (Thermo Fisher Scientific) where Aβ28–40 and Aβ28–42 were separated, identified, and quantified using LC-MS/MS (Acquity UPLC M-Class liquid chromatography unit (Waters Corp) interfaced to a Thermo Fisher Scientific Fusion Lumos Mass Spectrometer (Waltham, MA)). Following chromatographic separation, the Aβ peptides were introduced into the MS inlet via electrospray ionization, where precursor ions were filtered by quadrupole isolation of 1.6 m/z and detected within the orbitrap at a mass resolution of 30,000. Automatic gain control (AGC) targets for Aβ40 and Aβ42 were set at 1.0e5 and 5.0e5, respectively. Aβ40 and Aβ42 concentrations were calculated by the summation of peak areas from three product ions that result after fragmentation of their respective precursor ion.
After summation, the total peak area for the endogenous 14N Aβ peptides were divided by the total peak area for the exogenously added, uniform labeled 15N Aβ peptide internal standards to obtain a peak area ratio (PAR). The PAR for each Aβ peptide was compared to a PAR vs. peptide concentration of an external eight-point standard curve that spans the analytical measurement range, and Aβ peptide concentrations (pg/mL) were determined from the standard curve. The measured concentrations are expressed as a Aβ42/40 ratio. These data were assembled and assessed by TraceFinder 4.1 General Quan software (Thermo Fisher Scientific).
2.3. Aβ Calibration and Quality Control
Each 96-well plate included eight levels of calibrators, three QC samples and test specimens. The three QC samples were prepared from human plasma pools; the high QC was spiked with additional recombinant Aβ42 and Aβ40. Calibrators were prepared in a surrogate matrix composed of 2% recombinant human serum albumin. Aβ calibrator concentrations were assigned by amino acid analysis (AAA) quantification using a method traceable to the United States Pharmacopeia (USP) harmonized standard. AAA quantification was performed on both 14N and uniformly labeled 15N versions of Aβ40 and Aβ42. Acceptance criteria for assay performance were based on the defined total allowable error (TAE). TAE for Aβ40 was defined as 20% or 10 pg/mL, whichever is greater, while TAE for Aβ42 was defined as the greater of 20% or 4 pg/mL.
2.4. Aβ Performance Validation
2.4.1. Precision
A nested multifactor analysis-of-variance design was used to evaluate within-run precision (repeatability) and within-laboratory precision. The study implemented a 10 × 2 × 2 design where five samples were analyzed over ten days, with two runs per day and two replicates of each test sample per run, using a single lot of reagents and a single instrument. Each plate/run included calibrators; two independent calibrator lots were used, one lot for each 5-day period. Mean, standard deviation (SD) and coefficient of variation (CV) were calculated, and total and within-run precision were calculated by analysis of variance (ANOVA) according to CLSI guidelines [17]. The five samples included:
Human plasma sample, near the lower limit detection for Aβ40 and Aβ42.
Human plasma sample, near the medical decision point for the Aβ42/Aβ40 ratio (~0.096).
Pooled human plasma with concentrations of Aβ40 and Aβ42 between the limit of detection and midpoint of the analytical measurement range (AMR).
Spiked human plasma sample with concentrations of Aβ40 and Aβ42 between midpoint of the AMR and the highest calibrator (Aβ40: 1558 pg/mL; Aβ42: 235 pg/mL).
Spiked human plasma sample with concentrations of Aβ40 and Aβ42 near the high calibrator.
The two modified plasma samples were spiked with known amounts of Aβ40 and Aβ42 USP-traceable recombinant protein standards. Acceptable total (within-laboratory) and within-run (repeatability) imprecision was defined as ½ TAE limits.
2.4.2. Analytical Measurement Range (AMR)
Linearity, or AMR, was assessed using five samples prepared by intermixing high and low plasma pools as recommended by CLSI-EP07 [18]. The low concentration pool (Level 1) was prepared with 2% recombinant human albumin solution. The high concentration pool was created by spiking 2% recombinant human serum albumin with recombinant Aβ40 or Aβ42 to achieve a concentration within 5–10% of the highest calibrator (Level 5). Three admixtures that incorporate different amounts of the Level 1 and Level 5 pools were prepared to create levels 2,3, and 4. These samples were analyzed randomly in triplicate using one lot of reagents on one instrument. Concentration (Y-axis) vs. level (X-axis) was plotted and linear regression was used to calculate expected concentrations. Bias was calculated as the observed-expected value for each level. Polynomial regression analysis was conducted, as recommended by CLSI-EP07, to see if nonlinear “fit” was statistically better than linear fit. A two-tailed t-test was calculated to test whether the non-linear coefficients were statistically significant (p<0.05), indicating that the assay was statistically nonlinear [18]. Linearity was acceptable if deviation from linearity for each level did not exceed 1/2 TAE even if nonlinear coefficients were statistically significant.
2.4.3. Accuracy
There is no certified reference material (CRM) or reference measurement procedure (RMP) for Aβ40 and Aβ42 in a human plasma matrix. Therefore, trueness for plasma Aβ40 and Aβ42 was assessed by recovery experiments using native plasma samples spiked with commercially available full length recombinant Aβ40 and Aβ42 protein standards that were value-assigned by a USP-traceable amino acid analysis. Full length Aβ40 and Aβ42 recombinant standards were spiked together into two separate lots of biological matrix, K2EDTA plasma, at the following three final concentration levels:
| Level | Aβ40 (pg/mL) | Aβ42 (pg/mL) |
|---|---|---|
| 1 | 50 | 7.5 |
| 2 | 200 | 30 |
| 3 | 1000 | 150 |
Spiked biological matrix samples, along with corresponding unspiked biological matrix samples, were analyzed in the same run with three replicates at each concentration. This experiment was performed on two different instruments using two different reagent lots. Percentage recovery was calculated by taking the ratio of the recovery concentration to the expected concentration. Recovery less than or equal to ½ TAE was considered acceptable.
2.4.4. Carryover
Carryover was assessed with two sets of plasma samples: one approximately five-fold higher in Aβ40 and Aβ42 concentrations than the highest calibrator (Aβ40: 1558 pg/mL; Aβ42: 235 pg/mL) and one near the limit of quantitation. The concentrations selected for the high sample were much greater than the highest physiological concentration likely to be measured in plasma. The high concentration sample was created by spiking recombinant Aβ40 and Aβ42 into native plasma, and the low concentration sample was created by diluting native plasma with 2% recombinant human serum albumin to obtain final Aβ concentrations near the detection limit of the assays.
Testing was performed in a single run in the following sequence: five replicates of the low concentration samples followed by two replicates of the high concentration sample followed by one replicate of the low concentration sample. This “high, high, low” sequence was repeated five times. The initial set of five low concentration samples were used as the protected low concentration samples, whereas the five low concentration samples following the high concentration repeating set were used as the unprotected low concentration sample. Unpaired, one-tailed t-test was performed to compare the Aβ concentrations in protected low samples to unprotected low samples. Percentage carryover was calculated for each individual sample for each of the five unprotected low samples as follows:
% Carryover = (concentration of unprotected low sample - average concentration of protected low samples)/(average concentration of high concentration samples) × 100%.
Carryover was considered clinically significant if p ≤ 0.05 and/or percentage carryover > 2%.
2.4.5. Sensitivity
The limit of blank (LoB) is defined as the highest expected value in a series of sample replicates that contain no analyte. This experiment utilized a single blank sample analyzed on two instruments over three days with two runs per day with two different reagent lots (one lot paired with one instrument). The sample was tested three times per run for a total number of 36 measurements. The blank sample was created using 2% recombinant human serum albumin, because native plasma, even if immunodepleted, contains trace amounts of Aβ peptides as previously stated. The following equation from CLSI EP17 was used to calculate LoB [19]:
where meanBlank is the mean value of replicates; Cβ is a correction for the biased estimate of the population standard deviation (SD) due to sample size (Cβ = 1.645/(1 – (1/(4 * df)); df = degrees of freedom (n-1); and n= number of samples.
The limit of detection (LoD) experiment followed a similar design as the LoB experiment with three samples analyzed on two instruments over three days with three replicates per run using two reagent lots (one for each instrument). Test samples were prepared by spiking 2% recombinant human serum albumin with recombinant Aβ standards to a concentration near the lowest calibrator. The following equation from CLSI EP17 was used to calculate LoD [19]:
where Cβ is defined as above; df = n-k-1; k=number of samples (different concentrations); and n= number of replicates per sample.
The limit of quantitation (LoQ) is defined as the lowest actual concentration at which analyte is reliably detected and at which the uncertainty of the observed test result is less than or equal to the total allowable error goal set by the lab (20%). If the %CV for the samples used in the LoD experiment are ≤20% the LoQ is equal to the LoD.
2.4.6. Clinically Reportable Range
The clinically reportable range (CRR) is the range of analyte values that can be reported, allowing for specimen dilution, concentration, or other pretreatment used to extend the direct analytical measurement range (AMR). There will be times when patient sample Aβ40 and Aβ42 concentrations exceed the previously defined AMR of this assay and will need to be diluted to bring that patient’s Aβ concentrations within the AMR. This study was conducted using one lot of reagents and one instrument. Due to the lack of native plasma samples with endogenously high concentrations of Aβ40 and Aβ42, 2% recombinant human serum albumin was spiked with full length recombinant Aβ40 and Aβ42 at concentrations approximately three-fold higher than the highest calibrator. The Aβ40 and Aβ42 recombinant protein standards were value-assigned by a USP-traceable amino acid analysis, as described previously.
Briefly, four dilutions, 1/4, 1/8, and 1/16 were each measured in duplicate along with undiluted spiked plasma. The duplicates were averaged and divided against the target value. Dilution was considered acceptable if recovery for each dilution was within ½TAE.
2.4.7. Interference
Potential interferents, at test concentrations recommended in CLSI EP37 [20,21], were spiked into native plasma samples with high or low Aβ40 and Aβ42 concentrations. Stock solutions of concentrated interferents, approximately 20-fold higher than the test concentration, were prepared in suitable solvents or were obtained from Sun Diagnostics (New Gloucester, ME). Each of the two Aβ concentrations were tested in triplicate: low Aβ40 and Aβ42/high interferent, high Aβ40 and Aβ42/high interferent, and compared to control samples spiked with solvent instead of interferent. Potential endogenous and exogenous interferents tested are listed in Table 1. Test specimens were created by spiking human plasma pools with a specific amount of interferent stock solution to yield desired final concentrations of respective interferent. Control samples were also created by adding appropriate solvent in a volume equal to the interferent volume used in the test sample. Both test samples and controls were analyzed on one day, using one instrument, one reagent lot, with five replicates each. If an interference was found during this screening experiment, a dose-response experiment was conducted based on CLSI EP07-A3 to determine the degree of interference as a function of interference concentration [20].
Table 1:
List of potential interfering substances and maximum interferent concentrations tested.
| Interferent | Concentration Tested | Maximum Interferent Aβ42/40 | Maximum Interferent APOE |
|---|---|---|---|
| Proteins (albumin and gamma-globulins; 50:50 mix) | 12.5 g/dL | 12.5 g/dL | 12.5 g/dL |
| Bilirubin | 40 mg/dL | 2 mg/dL | 40 mg/dL |
| Hemolysate (Hemoglobin) | 1,000 mg/dL | 487 mg/dL | 1000 mg/dL |
| Triglycerides | 1,000 mg/dL | 1000 mg/dL | 1000 mg/dL |
| Rheumatoid factor (RF) | 1,000 U/L | 1,000 U/L | 1,000 U/L |
| Human anti-mouse antibody (HAMA) | 1:640 titer | 1:347 titer | 1:320 titer |
| Acetaminophen | 5 mg/dL | 5 mg/dL | 5 mg/dL |
| Aspirin (Acetylsalicylic acid) | 1 mg/dL | 1 mg/dL | 1 mg/dL |
| Atorvastatin | 250 ng/mL | 250 ng/mL | 250 ng/mL |
| Citalopram | 0.18 mg/dL | 0.18 mg/dL | 0.18 mg/dL |
| Clonazepam | 100 ng/mL | 100 ng/mL | 100 ng/mL |
| Donepezil | 100 ng/mL | 100 ng/mL | 100 ng/mL |
| Ibuprofen | 7 mg/dL | 7 mg/dL | 7 mg/dL |
| Memantine | 40 ng/mL | 40 ng/mL | 40 ng/mL |
| Risperidone | 3.8 μg/dL | 3.8 μg/dL | 3.8 μg/dL |
| Valproic Acid | 10 mg/dL | 10 mg/dL | 10 mg/dL |
The difference between the mean values of the test and control samples was calculated, divided by the mean value of the control sample, and multiplied by 100 to calculate percent deviation. Interference was considered clinically insignificant if deviation between the spiked and non-spiked sample did not exceed 1/2 TAE.
2.5. APOE Proteotype Assay
On the day of use, QC samples, plasma samples, and C-terminal, uniformly labeled 13C15N-Arg tryptic peptide internal standards (IS) (Vivitide, Gardner, MA) were thawed. The 5 μL sample was diluted in a 96-well plate (0.45mL polypropylene, Thermo Fisher Scientific) containing 95 μL 100 mM Tris pH 8.1, 9.6 mM sodium deoxycholate (SDC), 2.3 mM tris(2-carboxyethyl) phosphine (TCEP) for protein denaturation and reduction. A second dilution was performed by transferring 8 μL of each diluted specimen to respective wells of a 96-well PCR plate (PCR half skirt, Axygen, Corning Inc., Germany) containing 40 μL of 100 mM Tris pH 8.1, 9.6 mM SDC, 2.3 mM TCEP, 10 fmol IS peptides/μL. The samples underwent a 30-minute heat denaturation/reduction step at 50°C, followed by a 30-minute alkylation step in 40 μL of 4.8 mM iodoacetamide (Thermo Fisher Scientific). After these successive incubations, 24 μL of 0.06 μg/μL mM trypsin (Gold, MS Grade; Promega Corporation, Madison, WI) were added to digest the reduced and alkylated proteins and incubated in a thermomixer at 50 °C with shaking at 700 rpm. The trypsin digestion of APOE produces the isoform-specific peptides outlined in Table 2. After 90 min digestion, the reactions were terminated by addition of 7 μL of 35.7% formic acid/71.4 mM heptafluorobutyric acid (HFBA) (Fisher Scientific) to stop trypsin activity and precipitate the SDC. The samples were then centrifuged to pellet the precipitated SDC. The digested peptides were further purified using reverse phase solid phase extraction (Waters). The samples were washed to remove potentially contaminating substances, then eluted into a 96-well collection plate (Waters). Samples were dried under vacuum and reconstituted in 2% ACN/10% FA for analysis. The digested peptide samples were separated and analyzed using LC-MS/MS on a Waters Acquity UPLC M-Class LC interfaced to a Thermo Fisher Scientific Fusion Lumos mass spectrometer. The collection plate containing the resolubilized samples was placed in a temperature-controlled autosampler and 3 μL of sample were injected onto a CSH C18 column (Waters). Following chromatographic separation, the APOE peptides were introduced into the MS inlet via electrospray ionization, where precursor ions were filtered using a quadrupole isolation window of 1.6 m/z. Product ions were detected within the orbitrap at a mass resolution of 30,000 and the AGC target was set to 5.0e5. The product ions were detected, and the peak areas of each fragment ion were determined post-acquisition using Skyline software (MacCoss Lab, University of Washington, Seattle, WA).
Table 2:
APOE peptide signatures used for proteotyping
| CLAVYQAGAR | LAVYQAGAR | LGADMEDVCGR | LGADMEDVR | APOE Proteotype = Genotype |
|---|---|---|---|---|
| Present | Absent | Present | Absent | APOE2/2 |
| Present | Present | Present | Absent | APOE2/3 |
| Present | Present | Present | Present | APOE2/4 |
| Absent | Present | Present | Absent | APOE3/3 |
| Absent | Present | Present | Present | APOE3/4 |
| Absent | Present | Absent | Present | APOE4/4 |
APOE proteotyping for each of the six APOE genotypes (APOE2/2, APOE2/3, APOE2/4, APOE3/3, APOE3/4, APOE4/4) used a combination of the presence or absence of the four targeted APOE isoform-specific tryptic peptides. The presence or absence of an APOE isoform-specific peptide was determined by monitoring the peak areas for each peptide. Peak areas above the previously determined Limit of Detection (LoD) were considered present, and peak areas below the LoD were considered absent. An R script was utilized to generate the APOE proteotypes based upon the input peak areas (or their absence) for each isoform-specific peptide (The R Foundation for Statistical Computing, https://www.r-project.org/). The combinations of present and absent peptides generate a signature for each of the six APOE proteotypes (Table 2).
2.6. APOE Quality Control Materials
Each validation run included six QC samples encompassing all six common proteotypes: APOE2/2, APOE2/3, APOE2/4, APOE3/3, APOE3/4, APOE4/4. QC were prepared from native, human plasma with APOE isoforms determined by genotyping. Acceptance of the QC sample data was determined by the presence or absence of the known peptides that correspond to each APOE proteotype.
2.7. APOE Performance Validation
2.7.1. Sensitivity
Although the APOE assay is qualitative, we chose to assess the sensitivity of the assay for each APOE isoform-specific peptide using the peak area ratio (PAR), the ratio of endogenous peptide to the IS, for positive or negative peptide detection. To determine the Limit of the Blank (LoB), the study used 30 blank samples analyzed over three days on two instruments, with two replicates per sample, and with two different reagent lots (one lot paired with one instrument) for a total of 60 measurements per day. The blank samples were native human plasma with known APOE proteotypes different from the target proteotype or bovine plasma (see Table 3). For each reagent lot, the false-positive rate (FPR) was calculated:
Table 3:
Plasma matrices for APOE blank samples.
| Tryptic Peptide | Isoform | Blank Matrices |
|---|---|---|
| LGADMEDVR | APOE4 | Bovine Plasma |
| LGADMEDVCGR | APOE2/3 | APOE4/4 |
| LAVYQAGAR | APOE3/4 | Bovine Plasma |
| CLAVYQAGAR | APOE2 | APOE4/4 |
FPR = (# Positive results/Total negative sample replicates) * 100
If the FPR did not exceed 100α% (α = 0.0125), then the 100(1-α) % was zero and confirmed that the LoB = zero.
The LoD study followed the Probit approach, where detection of the APOE isoform-specific peptides was assessed in terms of proportion (i.e., the number of positive results with respect to the total number of replicate tests). Serial dilutions were prepared from a starting sample (e.g., APOE4) with a known peak area ratio. A test sample was diluted with bovine plasma (that does not contain human APOE proteins) with four dilutions and 20 replicates per dilution level. The samples were analyzed over three days on two instruments with two different reagent lots (one lot for each instrument). These results were also used to assess repeatability. Proteotypes were expected to match previously assigned genotypes for all dilutions and replicates.
2.7.2. Repeatability
Six plasma samples representing the APOE proteotypes were analyzed over 10 days, with two runs per day, and two replicates of each test sample per run. Enough sample was prepared for 40 replicates. Test samples and QCs were assayed twice a day, in duplicate, across 10 days (2 × 2 × 10) with a single reagent lot on one instrument. Hit rates for each sample that match 100% of the expected proteotypes were considered acceptable.
2.7.3. Accuracy
Accuracy of APOE proteotyping was assessed by agreement with genotyping. A comparison study included 20 patient plasma samples each with APOE2/3, APOE3/3, APOE3/4, 15 with APOE4/4, five (5) with APOE2/4, and three (3) with APOE2/2 genotypes. Genotyping was performed at the University of California, San Diego, or the University of Wisconsin AD Research Center (Madison, WI) using real time PCR Restriction Fragment Length Polymorphism analysis. APOE proteotyping accuracy was assessed over five runs, one per day with genotypes unknown to the operators. Data analysis consisted of determining the true positive and false negative rates for the APOE proteotyping method. To evaluate the performance of the candidate method, numerical estimates of positive percent agreement (PPA) were calculated. Acceptable agreement was defined as a PPA greater than 99%.
2.7.4. Carryover
Evaluation of carryover was performed to determine the extent of cross-contamination observed from sample to sample. For each APOE isoform-specific peptide, two sets of samples were included: one approximately five-times higher in peak area ratio than the highest PAR empirically measured in our lab and one slightly above the LoD of the assay.
The high concentration sample was created by spiking either recombinant APOE2 or APOE4 into native plasma, and the low concentration sample was created by diluting native plasma with bovine plasma to obtain final concentrations of APOE near the LoD of the assay. Testing was performed in a single run in the following sequence: five replicates of the low concentration samples (protected low) followed by one replicate of the high concentration sample followed by one replicate of the low concentration sample (unprotected low). This “high, low” sequence was repeated five times.
An unpaired, one-tailed t-test was performed to compare the peak area ratios of the protected low samples to unprotected low samples. Percentage carryover was calculated on an individual sample basis for each of the 5 unprotected low samples as follows:
% Carryover = (PAR of unprotected low sample - average PAR of protected low samples)/(average PAR of high concentration samples) × 100%.
Carryover was considered clinically significant if p ≤ 0.05 and/or percentage carryover ≥ 2%.
2.7.5. Interference
Potential interferents, at test concentrations recommended in CLSI EP37 [20,21] were spiked into samples representing all known APOE proteotypes with high PARs. Each sample was tested in triplicate and compared to a control sample spiked with appropriate solvent in place of interferent. Details of interference testing are similar to previous description for Aβ testing. Refer to Table 1 for the list of potential interferents tested. Interference was considered clinically insignificant if the proteotypes determined for the spiked and non-spiked samples agreed 100%.
3. Results
3.1. Aβ40 and Aβ42 Measurement
3.1.1. Precision
Precision results for five samples, analyzed over 10 days, with two runs per day, and two replicates of each test sample per run, are presented in Table 4. Total imprecision (within-lab) for Aβ40 varied from 2.7% to 7.7%. Total imprecision (within-lab) for Aβ42 varied from 3.1% to 9.5%. The greatest variability for both Aβ40 and Aβ42 was observed at very low concentrations, lower than the concentrations expected for the majority of patient specimens. Within-Day imprecision (Repeatability) varied from 1.5% to 3.0% for Aβ40 and 2.5% to 8.4% for Aβ42. Again, the greatest variability was observed at very low concentrations.
Table 4:
Plasma Aβ Precision Study
| Aβ 40 | Mean | Within-Lab (Total) | With-In Day (Repeatability) | |||
|---|---|---|---|---|---|---|
| pg/mL | SD, pg/mL | CV | SD, pg/mL | CV | ||
| TS1 | 193.5 | 6.3 | 3.2% | 2.9 | 1.5% | |
| TS2 | 618.7 | 17.0 | 2.7% | 9.6 | 1.5% | |
| TS3 | 1128.0 | 35.6 | 3.2% | 20.3 | 1.8% | |
| TS4 | 156.2 | 6.8 | 4.4% | 4.0 | 2.6% | |
| TS5 | 45.1 | 3.5 | 7.7% | 1.4 | 3.0% | |
| Aβ 42 | Mean | Within-Lab (Total) | With-In Day (Repeatability) | |||
|---|---|---|---|---|---|---|
| pg/mL | SD, pg/mL | CV | SD, pg/mL | CV | ||
| TS1 | 17.6 | 0.78 | 4.4% | 0.72 | 4.1% | |
| TS2 | 70.5 | 2.7 | 3.9% | 2.1 | 3.0% | |
| TS3 | 160.3 | 5.0 | 3.1% | 4.0 | 2.5% | |
| TS4 | 42.9 | 1.6 | 3.7% | 1.2 | 2.9% | |
| TS5 | 6.3 | 0.60 | 9.5% | 0.53 | 8.4% | |
3.1.2. Accuracy
Trueness for plasma Aβ40 and Aβ42 was assessed by recovery experiments using a surrogate matrix spiked with commercially available full length recombinant Aβ40 and Aβ42 protein standards that were value-assigned by a USP-traceable amino acid analysis. The measured concentrations (Obs.) was compared to the expected concentration to calculate percent recovery. Recovery was within acceptance limits (90% to 110%); see Table 5.
Table 5:
Plasma Aβ Recovery Experiment
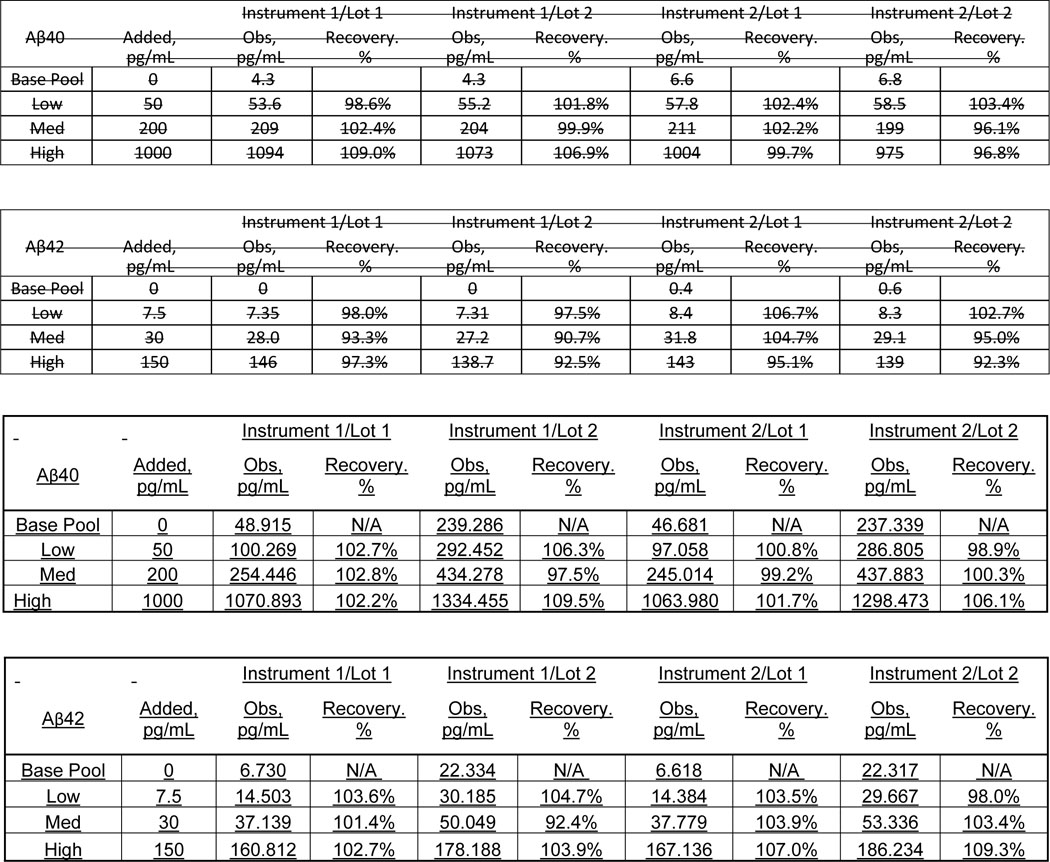
|
3.1.3. Sensitivity
The LoB was 3 pg/mL and 0 pg/mL for Aβ40 and Aβ42, respectively. The LoD was 11 pg/mL and 2 pg/mL for Aβ40 and Aβ42, respectively. For both Aβ40 and Aβ42, the LoQ was equal to the LoD. These detection limits are much lower than the concentrations observed in patient samples (lowest concentrations measured for Aβ40 and Aβ42 were 163 pg/mL and 12 pg/mL, respectively in thousands of clinical samples) and allow the discrimination of patients with and without amyloid pathology.
3.1.4. Interference
No interference was observed from the compounds tested up to the concentrations provided in Table 1, including common endogenous interferents, rheumatoid factor, human anti-mouse antibodies, and drugs. Lipemia (triglycerides up to 1000 mg/dL), hemolysis, (hemoglobin up to 1000 mg/dL), icterus (bilirubin up to 2 mg/dL), and proteins up 12.5 g/dL did not affect the assay. However, clinically significant bilirubin concentrations greater than 2 mg/dL interfered with Aβ quantitation.
3.1.5. Analytical Measurement Range and Clinically Reportable Range (CRR)
Aβ40 concentrations were linear from 10 to 1780 pg/mL, and Aβ42 concentrations were linear from 2 to 254 pg/mL (Figure 1). To determine CRR, four dilutions were tested: 1/4, 1/8, and 1/16, each measured in duplicate. All dilutions were acceptable with recovery within the ½ TAE acceptance criteria (Table 6). Therefore, based on the LoD, linearity, 16-fold dilution, and current concentrations of the highest calibrator (245 and 1650 pg/mL for Aβ42 and Aβ40, respectively), we can define the CRR for Aβ40 as 11 – 26,400 pg/mL, and for Aβ42 as 2– 3,920 pg/mL.
Figure 1:

Aβ40 and Aβ42 Linearity
Table 6:
Aβ42 and Aβ40 Dilution and Recovery Study
| Dilution | Expected concentration (pg/mL) | Measured concentration (pg/mL) | Recovery |
|---|---|---|---|
| Undiluted | 4950.00 | ||
| 1:4 | 1237.50 | 1265.24 | 102.2% |
| 1:8 | 618.75 | 652.81 | 105.5% |
| 1:16 | 309.38 | 317.88 | 102.7% |
| Dilution | Expected concentration (pg/mL) | Measured concentration (pg/mL) | Recovery |
| Undiluted | 735.00 | ||
| 1:4 | 183.75 | 184.95 | 100.7% |
| 1:8 | 91.88 | 94.84 | 103.2% |
| 1:16 | 45.94 | 46.07 | 100.3% |
3.1.6. Carryover
Carryover was considered not significant at 0.05% and 0.17% for Aβ40 and Aβ42, respectively.
3.2. APOE Proteotype Determination
3.2.1. Sensitivity and Repeatability
The LoB was 0.0 for all four APOE isoform-specific peptides used for proteotyping. The LoD for each peptide (Table 7) are in the femtomolar range; much lower than the millimolar concentrations observed in human plasma. Repeatability was 100%; all samples at all dilutions tested recovered the correct proteotype.
Table 7:
APOE Limits of Detection
| Peptide | LoD (PAR) | LoD (fmol) |
|---|---|---|
| LAVYQAGAR | 0.0677 | 27.1 |
| LGADMEDVR | 0.0344 | 13.8 |
| CLAVYQAGAR | 0.0275 | 11.0 |
| LGADMEDVCGR | 0.0301 | 12.0 |
3.1.8. Accuracy
Every specimen matched the expected genotype for a positive percent agreement (PPA) of 100% (Table 8). Interestingly, three of the samples originally genotyped as APOE3/3 were, in fact, APOE3/4 individuals by proteotyping. These samples were re-genotyped and verified to be APOE3/4.
Table 8:
Comparison of APOE Proteotype with APOE Genotype: Positive Percent Agreement (PPA).
| Genotype | Expected # of Samples | Actual # of Samples | PPA (%) |
|---|---|---|---|
| APOE2/3 | 100 | 100 | 100 |
| APOE2/4 | 25 | 25 | 100 |
| APOE3/3 | 100 | 100 | 100 |
| APOE3/4 | 115 | 115 | 100 |
| APOE4/4 | 75 | 75 | 100 |
3.1.9. Carryover
Carryover was ≤0.4% for all peptides and was not considered clinically significant.
3.1.10. Interference
Because the stock material for triglycerides, rheumatoid factor (RF), and human anti-mouse antibody (HAMA) are derived from human sources with trace amounts of APOE protein, interference test materials required a correction by blanking the interference materials. Three replicates of a bovine plasma matrix blank sample were prepared with these sample sets. Bovine plasma was previously tested for the presence of human APOE proteins and none were observed. None of the potential interferents tested interfered with accurate APOE proteotyping up to the concentrations tested (Table 1).
4. Discussion
The National Institute on Aging and the Alzheimer’s Association (NIA-AA) recognize the value of amyloid PET and other imaging procedures along with CSF Aβ as AD biomarkers [22], and currently amyloid PET is the only FDA approved diagnostic for detection of brain amyloid. However, these methods remain underutilized, perhaps because of cost, unavailability in rural areas, complexity, and lack of perceived usefulness by clinicians. Many believe PET is unlikely to be widely implemented in clinical practice due to logistics and access challenges while lumbar puncture is considered complicated, time-consuming, and invasive [23]. Plasma-based biomarkers offer practical advantages over imaging and CSF measurements, making plasma tests more accessible to clinicians and patients.
To meet the challenge for more practical and accessible AD biomarkers many investigators have focused greater attention on the development of blood-based biomarkers. Pathologically, AD is characterized by an imbalance in the production and clearance of amyloid-beta in the brain leading to plaque formation. The correlation of CSF and plasma Aβ with amyloid plaque burden revealed plasma Aβ concentration as a strong biomarker candidate. Initially, efforts to translate the results found in CSF into a blood test were largely unsuccessful due to methodological difficulties, lack of standardization, and the concern that, although most Aβ peptides are derived from neuronal tissue, there is contribution from peripheral organs and platelets [24,25]. The Aβ42 peptide is a particularly difficult peptide to work with due to its tendency to aggregate, a characteristic that relates to in vivo plaque deposition in the brain [23]. This is a particular concern for in vitro assays, where clumping can affect chromatographic separation and inhibit antibody capture. Additionally, plasma provides a more complex matrix than CSF with greater protein diversity, and much lower Aβ peptide concentrations than in CSF, requiring more sensitive and robust assays [24]. Plasma also contains proteases that may affect plasma protein stability, including Aβ peptides [24,26].
Despite early studies showing irreproducible data and a concerning lack of correlation of plasma and CSF amyloid peptides, likely due to method-specific flaws [26], numerous recent studies have demonstrated that plasma Aβ42/40 ratio is a reliable tool for the assessment of amyloid pathology in the brain [13,14,27–29]. Immunoassays, and to a greater extent, immunoprecipitation/mass spectrometry assays have clearly demonstrated that a decrease in Aβ42 or Aβ42/40 ratio is associated with clinical AD with diagnostic accuracy similar to CSF. The most common methods for Aβ measurement include liquid chromatography/mass spectroscopy (LC-MS) and immunoassays.
While immunoassays are less complex and relatively easier to perform than LC-MS-based assays, a recent study demonstrated considerably better diagnostic performance using LC-MS [30]. For Aβ42 and Aβ40 the correlation coefficients between the results for single bead-based immunoassays versus LC-MS were 0.207 and 0.404, respectively [30]. For the Aβ42/40 ratio the correlation was only 0.189 [30]. In terms of plasma Aβ42/40 ratio concordance with amyloid PET status the area under the receiver operating characteristic curve (AUC) was 0.620 for the single bead-based immunoassay, and 0.817 for the LC-MS-based assay [30]. When added to an algorithm with age, sex, and APOE genotype, only the addition of the LC-MS Aβ42/40 ratio significantly improved the AUC for discrimination of positive amyloid PET status [30].
Compared to the most common APOE allele, APOE3, individuals with an APOE4 allele have higher serum cholesterol concentrations and an increased risk for both coronary heart disease (CHD) and AD [31–34]. Individuals with an APOE2 allele tend to have higher serum triglycerides and a lower AD risk profile [31]. APOE2 homozygosity is associated with familial type III hyperlipoproteinemia and an increased CHD risk [31]. Given the clinical significance of APOE, accurate and reliable proteotyping or genotyping methods are required.
Our findings demonstrate that this LC-MS/MS analytical platform accurately and reliably; a) quantified plasma Aβ42, Aβ40 concentrations, b) plasma Aβ42/40 ratio, and c) identified the presence or absence of plasma APOE isoform-specific peptides used to determine an individual’s APOE genotype. These analytical validation experiments document excellent precision, a wide linear range, low limit of detection, and independence from common interferents, including commonly used medications. Although there is no certified reference material for plasma Aβ isoforms or recognized reference methods, our innovative, state-of-the-art LC-MS/MS application accurately recovered and quantified known recombinant Aβ protein amounts and concentrations (assigned by USP-traceable AAA). Likewise, the LC-MS/MS identification of APOE isoform–specific peptides resulted in 100% agreement with assigned genotypes; in fact, where discrepancies were seen, repeat genotyping showed that the LC-MS/MS peptide analysis and APOE proteotype assignment was correct. Clinical studies to assess diagnostic accuracy with the use of these biomarkers and to determine appropriate cut points for interpretation have recently been completed and a manuscript is being drafted.
Highlights:
There is a need for a more accessible, less-invasive method to aid in the diagnosis of Alzheimer’s disease and facilitate clinical trial enrollment.
The C2N PrecivityAD™ LC-MS/MS assays for plasma amyloid-beta peptides and APOE proteotypes meet this urgent need.
The C2N PrecivityAD™ LC-MS/MS assays are precise, sensitive, accurate, and linear over a wide analytical range, and suitable for use in the clinical laboratory.
Acknowledgments
Funding: This work was supported by NIH R44 AG059489 and BrightFocus CA2016636, The Gerald and Henrietta Rauenhorst (GHR) Foundation, and the Alzheimer’s Drug Discovery Foundation (ADDF# GC-201711-2013978).
Nonstandard Abbreviations:
- PAR
peak area ratio
- Aβ
amyloid beta
- APOE
apolipoprotein E
- CSF
cerebrospinal fluid
- AAA
amino acid analysis
- USP
United States Pharmacopeia
- AMR
analytical measurement range
- TAE
total allowable error
- SDC
sodium deoxycholate
- TCEP
tris(2-carboxyethyl) phosphine
- IS
internal standards
- CRR
clinically reportable range
Footnotes
Competing Interests
All authors are full time employees or advisors to C2N Diagnostics, receive equity or equity options, and contributed to the development of the PrecivityAD™ test.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aisen P, Touchon J, Andrieu S, Boada M, Doody R, Nosheny RL, et al. Registries and Cohorts to Accelerate Early Phase Alzheimer’s Trials. A Report from the E.U./U.S. Clinical Trials in Alzheimer’s Disease Task Force. J Prev Alzheimer’s Dis 2016;3:68–74. [DOI] [PubMed] [Google Scholar]
- 2.Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993; 261:921–3. [DOI] [PubMed] [Google Scholar]
- 4.Liu CC1, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9:106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verghese PB, Castellano JM, and Holtzman DM, Roles of Apolipoprotein E in Alzheimer’s Disease and Other Neurological Disorders. Lancet Neurol 2011; 10: 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis RJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996; 46:1592–1596. [DOI] [PubMed] [Google Scholar]
- 7.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991; 541:163. [DOI] [PubMed] [Google Scholar]
- 8.Kok E, et al. Apolipoprotein E–dependent accumulation of Alzheimer disease–related lesions begins in middle age. Ann Neurol. 2009; 65:650–657. [DOI] [PubMed] [Google Scholar]
- 9.Polvikoski T, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995; 333:1242–7. [DOI] [PubMed] [Google Scholar]
- 10.Schmechel DE, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993; 90:9649–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000; 1:507–37. [DOI] [PubMed] [Google Scholar]
- 12.Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol 2021; 20:68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement 2017; 13:841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018; 554:249–254. [DOI] [PubMed] [Google Scholar]
- 15.West T, Kirmess K, Meyer M, Holubasch M, Knapik S, Hu Y, Verghese P, Smith E, Harpstrite S, Fogelman I, Braunstein J, Yarasheski K APTUS™: Measurement of plasma Aβ42/40 concentration ratios by mass spectrometry predicts brain amyloidosis in banked samples from multiple, diverse cohorts Clinical Trials in Alzheimer’s Disease, San Diego, 2019. [Google Scholar]
- 16.CLSI. Evaluation of Precision of Quantitative Measurement Procedures; Approved Guideline− Third Edition. CLSI document EP05-A3. Wayne, PA: Clinical and Laboratory Standards Institute; 2014. [Google Scholar]
- 17.West T, Kirmess KM, Meyer MR et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegeneration 16, 30 (2021). 10.1186/s13024-021-00451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.CLSI. Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical Approach, Approved Guideline. CLSI document EP06-A. Wayne, PA: Clinical and Laboratory Standards Institute; 2003. [Google Scholar]
- 19.CLSI. Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline-Second Edition. CLSI document EP17. Wayne, PA: Clinical and Laboratory Standards Institute; 2012. [Google Scholar]
- 20.CLSI. Interference Testing in Clinical Chemistry. Third Edition. CLSI guideline EP07. Wayne, PA: Clinical and Laboratory Standards Institute; 2018. [Google Scholar]
- 21.CLSI. EP37: Supplemental Tables for Interference Testing in Clinical Chemistry, 1st Edition. Clinical and Laboratory Standards Institute; 2018. [Google Scholar]
- 22.Jack CR Jr. Bennett DA, Blennow K, Carrillod MC, Dunne B, Haeberleinfet SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018; 14: 535–562, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashton NJ, Scholl M, Heurling K, Gkanatsiou E, Portelius E, Hoglund K, et al. Update onbiomarkers for amyloid pathology in Alzheimer’s disease. Biomark Med 2018; 12: Published Online:15 Jun 2018 10.2217/bmm-2017-0433. [DOI] [PubMed] [Google Scholar]
- 24.Bateman RJ, Blennow K, Doody R, Hendrix S, Lovestone S, Salloway S, et a. Plasma biomarkers of AD emerging as essential tools for drug development: an EU/US CTAD Task Force report. J Prev Alz Dis 2019; 6:169–173. [DOI] [PubMed] [Google Scholar]
- 25.Zetterberg H, Burnham SC. Blood-based molecular biomarkers for Alzheimer’s disease. Mol Brain 2019; 12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blennow K, Zetterberg H. The past and future of Alzheimer’s disease fluid biomarkers. JAlz Dis 2018; 62: 1125–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perez-Grijalba V, Romero J, Pesini P, Sarasa L, Monleon I, San-Jose I, et al. Plasma Abeta 42/40 ratio detects early stages of Alzheimer’s disease and correlates with CSF and neuroimaging biomarkers in the AB255 Study. J Prev Alzheimers Dis 2019; 6:34–41. [DOI] [PubMed] [Google Scholar]
- 28.Nabers A, Perna L, Lange J, Mons U, Schartner J, Guldenhaupt J, et al. Amyloid blood biomarker detects Alzheimer’s disease. EMBO Mol Med 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowe CC, Bourgeat P, Ellis KA, Brown B, Lim YY, Mulligan R, et al. Predicting Alzheimer disease with β-amyloid imaging: results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann Neurol. 2013. 74:905–13. [DOI] [PubMed] [Google Scholar]
- 30.Keshaven A, Pannee J, Karikari TK, Lantero Rodriguez J, Ashton NJ, Nicholas JN, et al. Population-based blood screening for preclinical Alzheimer’s disease in a British birth cohort at age 70. Brain 2021; 10.1093/brain/awaa403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002; 155(6):487–95. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- 32.Dallongeville J, Lussier-Cacan S, Davignon J. Modulation of plasma triglycerides levels by apoE phenotype: a metaanalysis.J Lipid Res 1992;33:447–54. [PubMed] [Google Scholar]
- 33.Schiele F, DeBacquer D, Vincent-Viry M, et al. Apolipoprotein E serum concentration and polymorphism in six European countries: the ApoEurope Project. Atherosclerosis 2000; 152:475–88. [DOI] [PubMed] [Google Scholar]
- 34.Stengard JH, Zerba KE, Pekkanen J, Ehnholm C, Nissinen A. and Sing CF Apolipoprotein E polymorphism predicts death from coronary heart disease in a longitudinal study of elderly Finnish men. Circulation 1995; 91, 265–269. [DOI] [PubMed] [Google Scholar]