

Abstract
The leading cause of death in patients with nonalcoholic fatty liver disease (NAFLD) is cardiovascular disease (CVD). However, the mechanisms are unknown. Mice deficient in hepatocyte proliferator-activated receptor-α (PPARα) (PparaHepKO) exhibit hepatic steatosis on a regular chow diet, making them prone to manifesting NAFLD. We hypothesized that the PparaHepKO mice might be predisposed to poorer cardiovascular phenotypes due to increased liver fat content. Therefore, we used PparaHepKO and littermate control mice fed a regular chow diet to avoid complications with a high-fat diet, such as insulin resistance and increased adiposity. After 30 wk on a standard diet, male PparaHepKO mice exhibited elevated hepatic fat content compared with littermates as measured by Echo MRI (11.95 ± 1.4 vs. 3.74 ± 1.4%, P < 0.05), hepatic triglycerides (1.4 ± 0.10 vs. 0.3 ± 0.01 mM, P < 0.05), and Oil Red O staining, despite body weight, fasting blood glucose, and insulin levels being the same as controls. The PparaHepKO mice also displayed elevated mean arterial blood pressure (121 ± 4 vs. 108 ± 2 mmHg, P < 0.05), impaired diastolic function, cardiac remodeling, and enhanced vascular stiffness. To determine mechanisms controlling the increase in stiffness in the aorta, we used state-of-the-art PamGene technology to measure kinase activity in this tissue. Our data suggest that the loss of hepatic PPARα induces alterations in the aortas that reduce the kinase activity of tropomyosin receptor kinases and p70S6K kinase, which might contribute to the pathogenesis of NAFLD-induced CVD. These data indicate that hepatic PPARα protects the cardiovascular system through some as-of-yet undefined mechanism.
Keywords: cardiac dysfunction, hepatic steatosis, hypertension, lean NAFLD, nonalcoholic fatty liver disease
INTRODUCTION
The accumulation of fat in the liver is typically benign; however, long-term lipid buildup may be exacerbated by inflammation leading to nonalcoholic fatty liver disease (NAFLD) that can engender type II diabetes and cardiovascular disease (CVD). The link between fat accumulation in the liver and CVD has been established (1–6), but the underlying mechanisms controlling NAFLD-CVD events remain elusive. Fat accumulation in the liver is arguably the initiating event that eventually causes hepatic insulin resistance that induces high circulating insulin levels leading to perturbations in peripheral tissue insulin signaling and glucose uptake (7, 8). The current problem in the field is that dissecting the effects of weight gain, adiposity, insulin resistance, and hepatic fat on a causal relationship with CVD is complicated.
NAFLD is an emerging epidemic and the leading cause of chronic liver disease globally (9–11). NAFLD can advance from hepatic steatosis to nonalcoholic steatohepatitis (NASH), which is fatty liver accompanied by inflammation, fibrosis, and hepatocyte damage that can further progress to cirrhosis or hepatocellular carcinoma (12). The pathophysiology leading to NAFLD is complex and can be influenced by several components, such as diet, genetics, and epigenetic factors (13, 14). NAFLD is often associated with other metabolic diseases, including obesity, insulin-resistant diabetes, and hyperlipidemia. One component of NAFLD that is unanimous in most scientific findings is that the fat-burning nuclear receptor peroxisome proliferator-activated receptor-α (PPARα) is usually significantly lower expressed in the liver (15–18). Not, surprisingly, the loss of PPARα in the liver and adipose tissues increases adiposity (19, 20). This circumstance likely allows for lipogenesis and hepatic fat accumulation due to metabolic disturbances associated with adiposity, weight gain, and obesity (19, 21).
Given this close association with metabolic diseases, a change in nomenclature to the metabolic (dysfunction)-associated fatty liver disease (MAFLD) has been proposed (22, 23). Although NAFLD and MAFLD are strongly associated with obesity (11), nonobese individuals with normal body mass index (BMI) also develop hepatic steatosis (24–27), which is referred to as lean NAFLD. For instance, ∼10–20% of nonobese persons of Asian and Caucasian descent develop NAFLD (28). Nonobese patients with NAFLD also exhibited an increased risk of CVD, including arterial hypertension, atherosclerosis, and cardiac dysfunction, as well as metabolic disorders like insulin resistance and elevated circulating glucose and triglyceride levels (24, 28, 29). Since both obesity and diabetes can cause CVD, the specific role of hepatic steatosis in developing CVD in these models is perplexing.
Our group has recently described liver-specific PPARα knockout mice (PparaHepKO) spontaneously develop hepatic steatosis under regular chow feeding without obesity and insulin resistance (19). Therefore, the present study was designed to characterize their cardiovascular phenotypes. Our findings show that the hepatic loss of PPARα induces CVD to the level described in humans with NAFLD (19). This indicates promise for these mice as a model of understanding better the mechanisms that NAFLD induces CVD without obesity or insulin resistance. Furthermore, our data indicate that hepatic PPARα protects from CVD by a liver-dependent mechanism, which is the first to report this finding.
RESEARCH DESIGN AND METHODS
Animals
The experimental procedures and protocols of this study conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center. All mice had free access to food and water ad libitum. Animals were housed in a temperature-controlled environment with a 12-h dark-light cycle. PparaHepKO and Pparafl/fl mice were bred and genotyped from our colony at the University of Mississippi Medical Center previously described (19). The mice were housed under standard temperatures between 24 and 25°C. The standard mouse chow consisted of 17% fat (Teklad 22/5 rodent diet, No. 8604, Harland Laboratories, Inc., Indianapolis, IN). Studies were performed on 30-wk-old male mice housed under standard conditions with full access to standard mouse chow and water aforementioned. Investigators were blinded to the genotypes of all mice at the time of experimentation. Mice were fasted 8 h from early morning to afternoon before euthanasia via isoflurane anesthesia, and blood and tissues were collected and stored at −80°C for further analysis.
Echocardiography
Mouse transthoracic echocardiographic measurements were performed on 30-wk-old male mice using the Vevo 3100 high-resolution imaging system (FUJIFILM VisualSonics, Toronto, ON, Canada) as previously described (30–32). Mice were initially anesthetized with 4% isoflurane in 1 L/min of O2, then placed on a heated platform (39–40°C) to maintain the animal core temperature at 37°C and maintained under anesthesia with 1–2% isoflurane. Using the ultra-high-frequency MX 400 probe, we imaged the heart in the parasternal long-axis view using the B-mode, and the following parameters were obtained from the software (Vevo LAB Software; V. 3.2.6, VisualSonics): cardiac output (CO), stroke volume (SV), ejection fraction (EF), left ventricular end-diastolic volume (LVEDV), and left ventricular mass. Using the M-mode, the following parameters were obtained from the parasternal short-axis view: internal dimensions of the left ventricular cavity left ventricular internal diameter during systole (LVIDs), left ventricular internal diameter during diastole (LVIDd), left ventricular anterior wall thickness in systole (LVAWs), left ventricular anterior wall thickness in diastole (LVAWd), left ventricular posterior wall thickness in systole (LVPWs), left ventricular posterior wall thickness in diastole (LVPWd). Recordings from B and M-modes were used to evaluate the systolic function of the left ventricle. An apical four-chamber view was obtained to determine the diastolic function, and the pulse-wave Doppler (PWD) was used to measure the following inflow velocity profile: peak velocity flow in early diastole (E), peak velocity flow in late diastole (A), and isovolumetric relaxation time (IVRT). Although they were still in the apical four-chamber view, tissue Doppler (TD) imaging was used to measure the early diastolic mitral annular velocity (e′). The ratio of E/A and E/e′ was determined. Altered E/A and E/e′ ratios were taken as markers of left ventricular diastolic dysfunction.
Vascular Stiffness
Ultrasound imaging was used for the evaluation of abdominal aorta and carotid artery stiffness. The probe was used to obtain a longitudinal view of the carotid artery and aorta, PWD was used to measure flow velocity, and the following parameters were traced and then analyzed offline from the software (Vevo LAB Software; V. 3.2.6, VisualSonics): pulsatility index (PI) and resistive index (RI). The probe was also used to obtain B-mode images of the arteries in EKV imaging mode, allowing a frame rate of 110–138 (FUJIFILM VisualSonics, Toronto, ON, Canada). The images taken were traced and analyzed with the Vevo LAB software package (Vevo vascular analysis; V. 3.2.6, VisualSonics) to determine the following parameters: pulse propagative velocity (PPV), distensibility/elasticity of the vessels, near and far intima-media thickness (IMT), and near and far wall diameters (30, 33, 34).
Measurement of Blood Pressure and Heart Rate by Radiotelemetry
Telemetry catheters were placed into the left common carotid, and the transmitter was placed subcutaneously along the left flank of the mouse as previously described (35, 36). Mice were given 7 days to recover from surgery, after which systolic, diastolic, mean arterial pressure, and heart rate were continuously recorded (sampling every 15 min for 10-s intervals) for 1 wk. Data were collected and stored using the Dataquest ART data acquisition system (Data Sciences International, St. Paul, MN) and imported into Microsoft Excel for analysis.
Kinome Array and Upstream Kinase Identification
Kinase activity was quantified using protein tyrosine kinase (PTK; Cat. No. 32508) or serine-threonine kinases (STK; Cat. No. 32501) PamChip4 porous three-dimensional (3-D) microarrays and measured using the PamStation12 (PamGene International, ’s-Hertogenbosch, The Netherlands). The substrates that are contained within each array are included in Supplemental Tables S1 and S2. The mouse aortas were pooled and measured in triplicate across three chips simultaneously for PTK and STK, as previously described (12). This approach effectively deals with large batch effects across samples, enabling the characterization of kinase activity but only in the context of analytical variance. The pooled samples were lysed using M-PER mammalian extraction buffer (Thermo Fisher Scientific, Cat. No. 78503), halt phosphatase inhibitor (Thermo Fisher Scientific, Cat. No. 78428), and protease inhibitor cocktail (Sigma, Cat. No. P2714). The samples were homogenized using TissueLyser LT (Qiagen). The protein concentration was measured in triplicate using Pierce BCA Protein assay (Thermo Fisher Scientific, Cat. No. 23225). Samples were diluted to a final protein concentration of 2.5 μg/μL. Each array contained 1 μg of protein per sample for STK chips and 5 μg for PTK chips. In the presence of ATP, kinase phosphorylation activity is quantified using fluorescently labeled antibodies to detect differential phosphorylation of 196 (PTK) or 144 (STK) reporter peptides between experimental and control conditions, as previously described (37). Evolve (PamGene) software uses a charge-coupled device (CCD) camera and light-emitting diode (LED) imaging system to record relative phosphorylation levels of each unique consensus phosphopeptide sequence every 5 min for 60 min as measured by peptide signal intensities recorded across 10-, 20-, 50-, and 100-ms exposure times. The images taken during the run were analyzed using BioNavigator (PamGene). Signal ratios are used to calculate fold change (FC) for each phosphopeptide sequence averaged across three replicates. Minimum threshold values derived from previous literature (37–40) require differential phosphopeptide signals greater than or equal to 30% (FC ≥ 1.30 or FC ≤ 0.70) for differential phosphorylation to be considered. The linear regression slopes provide phosphorylation intensity signals used in differential analyses (e.g., experimental vs. control). Undetectable or nonlinear (R2 < 0.90) phosphopeptides are excluded from subsequent analyses. We performed upstream kinase identification using kinome random sampling analyzer (K.R.S.A.) (41) and upstream kinase analysis (U.K.A.) (42), as previously described (37, 43). Measurements extensively of winners (MEOW) plots were used to interrogate individual kinase activities on substrates considering the confidence of the experimental versus the control groups using the equation: [Log2 fold change (FC) of kinase substrates × Δconfidence (experimental hits/mean hits of 2,000 random sampling iterations)]. The kinome phyla tree was made using CORAL (44).
Hepatic Lipid Measurements
Tissue triglyceride levels were measured using a colorimetric assay kit according to the manufacturer’s guidelines (Triglyceride Assay Kit, Abcam, Cambridge, UK) as we previously described (19, 45). Samples from individual mice were run in duplicate and averaged, and the averages of individual mice were then used to obtain group averages. The Oil Red O staining level was determined at ×40 magnification of individual stained sections using a color Axiocam 105 camera with Zen 2 software attached to a Zeiss Axioplan microscope. The Oil Red O stain percentage was then determined from each field using NIH ImageJ software. Measurements from three different sections per individual animal were averaged, and the averages of individual mice were then used to obtain the group averages as previously described (19, 45–47).
Western Blot Analysis
Western blot analyses were performed on liver samples (30 μg) as we previously described (19, 48). Membranes were incubated overnight at 4°C with the following antibodies: PPARα (anti-goat PPARα, made from the murine PPARα peptide by Pacific Immunology, PAC No.10259) or heat shock protein 90 (HSP90) (Santa Cruz Biotechnology, No. 13119). After three washes in TBS + 0.1% Tween 20, the membrane was incubated with an infrared donkey anti-goat (IRDye 800 LI-COR Biosciences, 926–32214) or goat anti-mouse (IRDye 680, LI-COR Biosciences, 926–68020) secondary antibody (LI-COR Biosciences) (1:10,000 dilution in TBS) for 2 h at 4°C. Immunoreactivity was visualized and quantified by infrared scanning in the Odyssey system (LI-COR Biosciences).
Fasting Glucose and Insulin
The fasting plasma glucose and insulin levels were measured as we previously described (49). After an 8-h fast, a blood sample was obtained via the orbital sinus under isoflurane anesthesia. Blood glucose was measured using an Accu-Chek Advantage glucometer (Roche, Mannheim, Germany). Fasting plasma insulin concentrations were determined by ELISA (Linco Insulin ELISA kit).
Measurement of Plasma Brain-Derived Neurotrophic Factor
Plasma brain-derived neurotrophic factor (BDNF) levels were measured from 30-wk-old male mice using a mouse-specific BDNF assay kit (KA0331, Novus Biologicals, Centennial, CO). Samples were diluted 1:200 and ran in duplicate along with standards provided by the manufacturer. Samples from individual mice were averaged and then the averages of the individual mice were used to obtain group averages.
Statistical Analysis
Data were analyzed with Prism 9 (GraphPad Software, San Diego, CA) using analysis of variance combined with Tukey’s posttest to compare pairs of group means or unpaired t tests. Results are expressed as means ± SE. A one-way ANOVA with a least significant difference post hoc test was used to compare mean values between multiple groups. A two-tailed and a two-way ANOVA was used in multiple comparisons, followed by the Bonferroni post hoc analysis to identify interactions. P values of 0.05 or smaller were considered significant.
RESULTS
Evaluation of Metabolic and Cardiovascular Parameters in PparaHepKO and Pparafl/fl Mice
The PparaHepKO mice were generated by crossing homozygous PparaHepKO and Pparafl/fl mice to generate PparaHepKO and Pparafl/fl littermates (Fig. 1). Measurement of hepatic Ppara by Western blot demonstrated significantly decreased levels in the livers of PparaHepKO as compared with Pparafl/fl mice (Fig. 1). The PparaHepKO mice on a regular chow diet showed no significant changes in body weight, liver weight, or heart weight compared with Pparafl/fl littermate control mice (Table 1). These data are consistent with previous studies that showed no changes in body weight in PparaHepKO fed a standard chow diet (19). Elevated fat accumulation was revealed by Oil Red O staining in the liver of lean PparaHepKO compared with lean Pparafl/fl control mice (Fig. 2A). Hepatic triglyceride levels were also elevated in PparaHepKO mice compared with Pparafl/fl mice. However, fasting blood glucose and insulin levels were similar between PparaHepKO and Pparafl/fl mice (Fig. 2B). Our results show that PparaHepKO mice on a normal diet develop hepatic steatosis independent of obesity and insulin resistance. To determine the effects of the loss of hepatic PPARα on the cardiovascular system, blood pressure was monitored 24 h a day using implantable radiotelemetry probes in the PparaHepKO and Pparafl/fl mice. The data show significantly increased mean arterial blood pressure in PparaHepKO compared with Pparafl/fl control mice in the daytime and nighttime over the 7-day recording period (Fig. 2C). Heart rate was significantly increased in the PparaHepKO mice compared with littermate controls during the day and night over the 7-day recording period (Fig. 2D). These changes in the cardiovascular occurred without changes in PPARα mRNA expression in the heart (Fig. 2E).
Figure 1.

Generation and characterization of PparaHepKO and Pparafl/fl mice. PparaHepKO mice were generated by crossing PparaHepKO mice heterozygous for Albumin-Cre with Pparafl/fl mice generating litters with Cre-positive and Cre-negative mice. Hepatic Ppara levels were determined by Western blot and normalized to the levels of HSP90. **P < 0.01 vs. Pparafl/fl; n = 3 mice per group. P < 0.01 by unpaired t test. KO, knockout, WT, wild-type.
Table 1.
Body, heart, and liver weights in PparaHepKO and Pparafl/fl mice
| Parameter | Ppara fl/fl | Ppara HepKO | P Value |
|---|---|---|---|
| Body weight, g | 33.8 ± 0.9 | 31.4 ± 1 | 0.103 |
| Body length, cm | 9.3 ± 0.12 | 9.0 ± 0.17 | 0.106 |
| Liver weight, g | 1.34 ± 0.04 | 1.30 ± 0.05 | 0.575 |
| LW:BW, g/g | 0.04 ± 0.001 | 0.04 ± 0.002 | 0.338 |
| LW:BL, g/cm | 0.14 ± 0.001 | 0.14 ± 0.007 | 0.811 |
| Heart weight, mg | 146.1 ± 3.2 | 142.9 ± 6.2 | 0.658 |
| HW:BW, mg/g | 4.4 ± 0.1 | 4.5 ± 0.2 | 0.304 |
| HW:BL, g/cm | 15.6 ± 0.3 | 15.9 ± 0.1 | 0.724 |
Values are means ± SE. n = 11 and 16 mice per group, Pparafl/fl, PparaHepKO. BL, body length; BW, body weight; HW, heart weight; LW, liver weight. No significant differences were observed between the groups using an unpairied t test.
Figure 2.
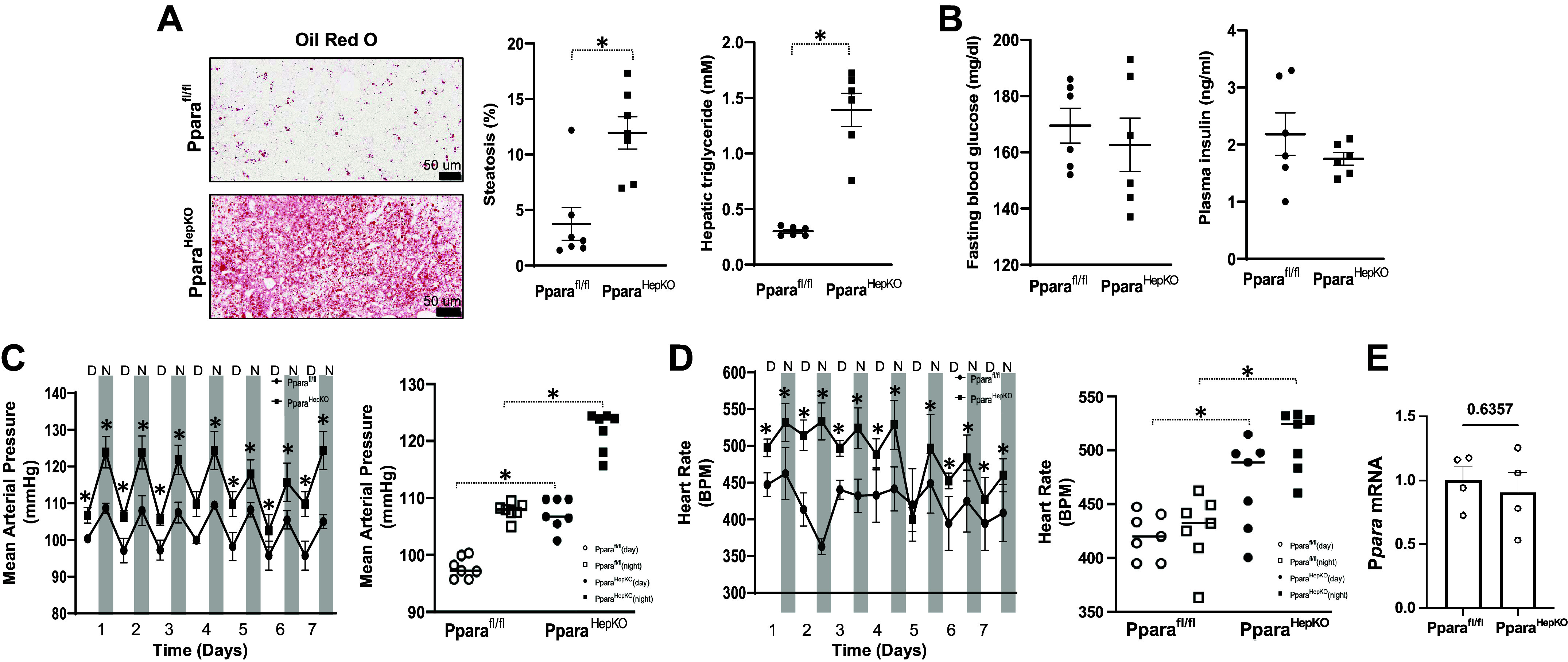
Analysis of metabolic and cardiovascular parameters in PparaHepKO and Pparafl/fl mice. Representative Oil red O staining of livers from PparaHepKO and floxed littermate control (Pparafl/fl), echo MRI steatosis measurements, and hepatic triglycerides (A), and fasting blood glucose levels and fasting plasma insulin levels (B). Values are means ± SE; *P < 0.05 vs. Pparafl/ by uparied t test; A: n = 7 mice per group; B–D: n = 6 mice per group. Scale bar = 50 μm. C: day and night mean arterial pressure (MAP) over the 7-day recording period and 7-day average MAP. D: heart rates for day and night of the 7-day recording period and 7-day average heart rate. E: real-time PCR expression of PPARα mRNA in the ventricle. Values are means ± SE; *P < 0.05 vs. Pparafl/fl (during the day) by uparied t test; n = 4 mice per group. D = day, N = night. PPARα, proliferator-activated receptor-α.
Analysis of Cardiac Function and Remodeling in PparaHepKO and Pparafl/fl Mice
Since the PparaHepKO showed cardiac dysfunction, we next wanted to further measure cardiac output in these mice and compare them to littermates. We observed that the PparaHepKO mice had left ventricular dilation with an enlarged LVID during systole and diastole compared with Pparafl/fl control mice (Fig. 3A). This was accompanied by the reduced thickness of the cardiac walls (both anterior and posterior walls) (Fig. 3B). We measured the systole and diastole in both groups of mice. The diastolic parameters, including MV E, MV A, and E/A ratio, were similar between groups (Fig. 3C). However, there was a significant decrease in the e′ and E/e′ ratio and prolonged IVRT in PparaHepKO mice compared with Pparafl/fl mice, which are indicators of diastolic dysfunction in the PparaHepKO mice (Fig. 3D). The PparaHepKO mice also exhibited reduced cardiac output and left ventricular mass, but increased left ventricular end-diastolic volume compared with Pparafl/fl control mice (Fig. 3E). Stroke volume and ejection fraction were similar in both groups (Fig. 3F), although PparaHepKO mice show a tendency to have lower values.
Figure 3.
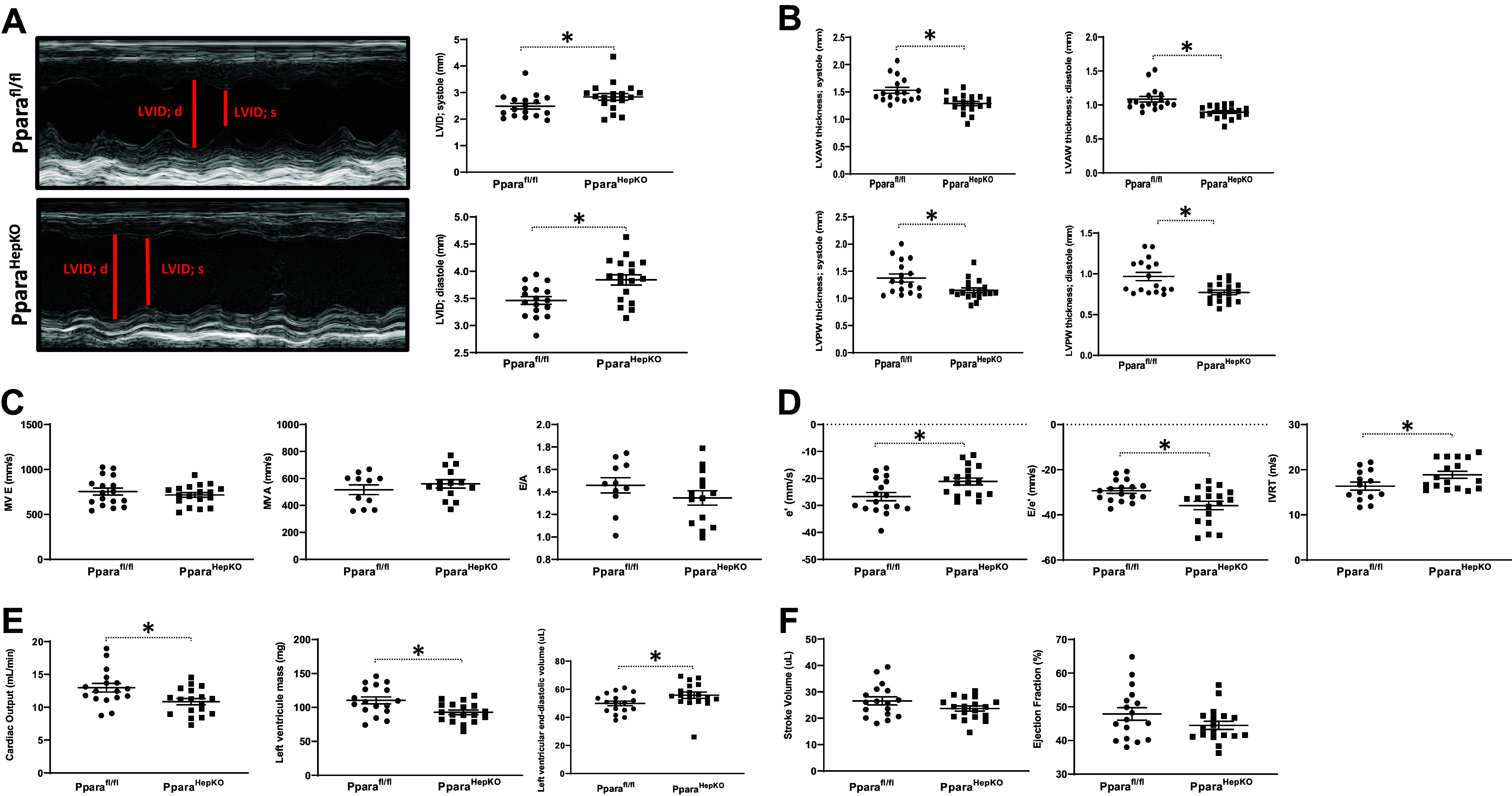
Assessment of cardiac function and remodeling in PparaHepKO and Pparafl/fl mice. A: a representative image of the dilated left ventricular chamber, LVID during systole, LVID during diastole, and LVAW thickness during systole in PparaHepKO and Pparafl/fl mice. B: quantification of the LVAW thickness during diastole, LVPW thickness during systole, and LVPW thickness during diastole in PparaHepKO and Pparafl/fl mice. Values are means ± SE; *P < 0.05 vs. Pparafl/fl; n = 17 and n = 18 mice per group, Pparafl/fl, PparaHepKO. Measurements of MV E, MV A, and E/A ratio (C), and evaluation of e′, E/e′, and IVRT in PparaHepKO and Pparafl/fl mice (D). Values are means ± SE; *P < 0.05 vs. Pparafl/fl by unpaired t test; n = 17 and n = 18 mice per group, Pparafl/fl, PparaHepKO. Cardiac output, left ventricular mass, and left-ventricular end-diastolic volume (E), and stroke volume and ejection fraction in PparaHepKO and Pparafl/fl mice (F). Values are means ± SE; *P < 0.05 vs. PPARα floxed control (Pparafl/fl) by unpaired t test; n = 17 and n = 18 mice per group, Pparafl/fl, PparaHepKO. A, peak flow velocity in late diastole; e′, early diastolic mitral annular velocity; E, peak flow velocity in early diastole; IVRT, isovolumetric relaxation time; LVAW, left ventricular anterior wall; LVID, left ventricular internal diameter; LVPW, left ventricular posterior wall; MV, mitral valve; PPARα, proliferator-activated receptor-α.
Assessment of Vascular Stiffness in PparaHepKO and Pparafl/fl Mice
The PparaHepKO mice exhibited increased aortic stiffness and decreased distensibility/elasticity compared with Pparafl/fl control mice. This was manifested as increased abdominal aortic pulsatility index, resistive index, and near abdominal aortic intima-media wall thickness in PparaHepKO mice versus Pparafl/fl control mice (Fig. 4, A–C). PparaHepKO exhibited reduced aortic distensibility/elasticity compared with Pparafl/fl control mice (Fig. 4D). No differences in abdominal aortic pulse propagation velocity, abdominal intima-media wall thicknesses far, and abdominal wall thicknesses both near and far were found between the groups (Fig. 4, E–H). Carotid pulsatility index, resistive index, the intima-media thickness of the near wall, and carotid arterial fall wall thickness were increased in PparaHepKO as compared with Pparafl/fl control mice (Fig. 5, A–D). There were no differences in carotid artery pulse propagation velocity, intima-media thickness-far, the carotid arterial wall near thickness, or distensibility/elasticity between the groups (Fig. 5, E–H).
Figure 4.
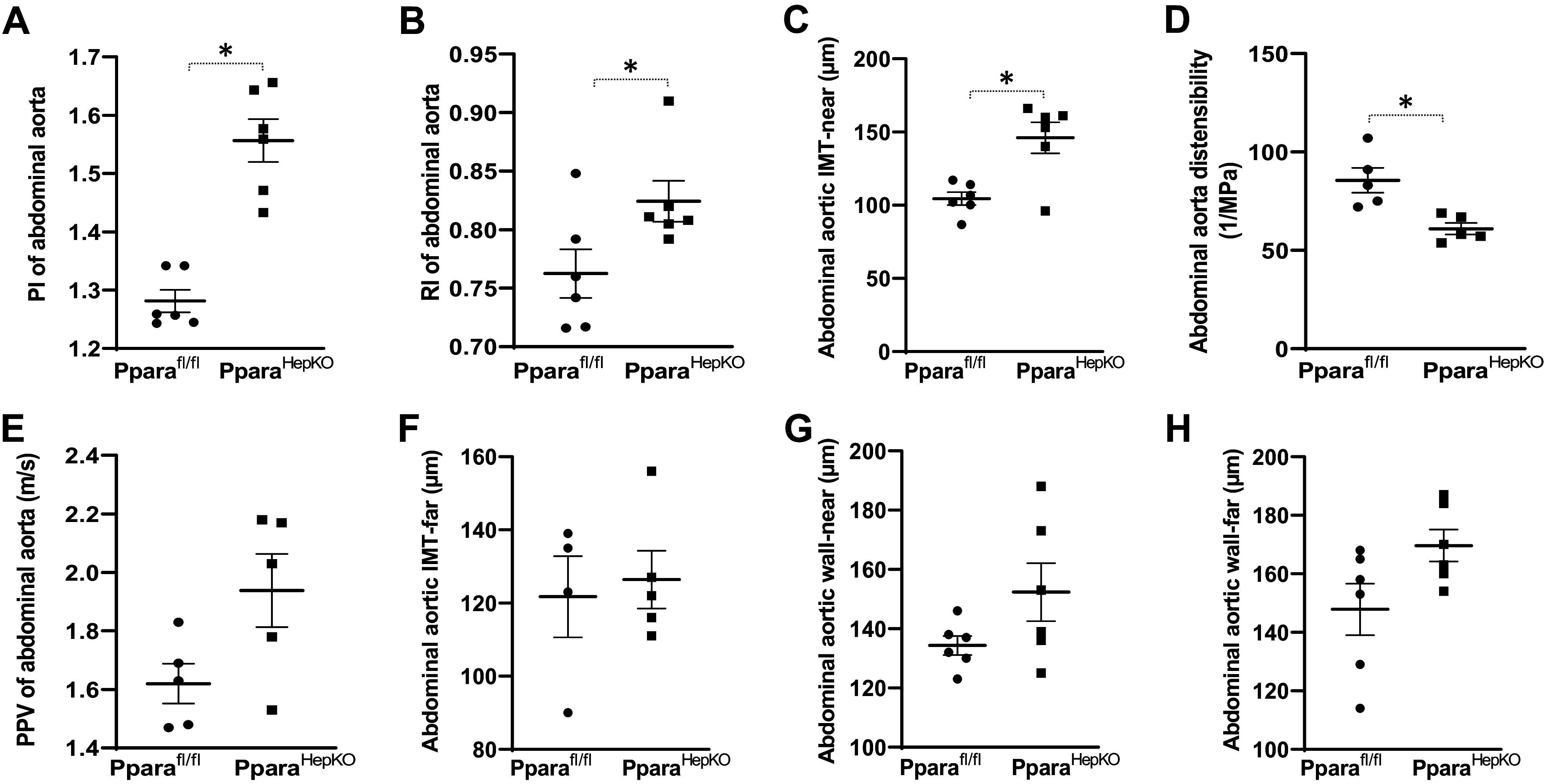
Phenotypes of the abdominal aortic in PparaHepKO and Pparafl/fl mice. Measurements of PI (A), RI (B), IMT-near (C), distensibility (D), PPV (E), IMT-far (F), wall-near thickness (G), and wall-far thickness (H) in PparaHepKO and Pparafl/fl mice. Values are means ± SE; *P < 0.05 vs. Pparafl/fl by unpaired t test; A–H; n = 6 mice per group. IMT, intima-media thickness; PI, pulsatility index; PPV, pulse propagation velocity; RI, resistive index.
Figure 5.
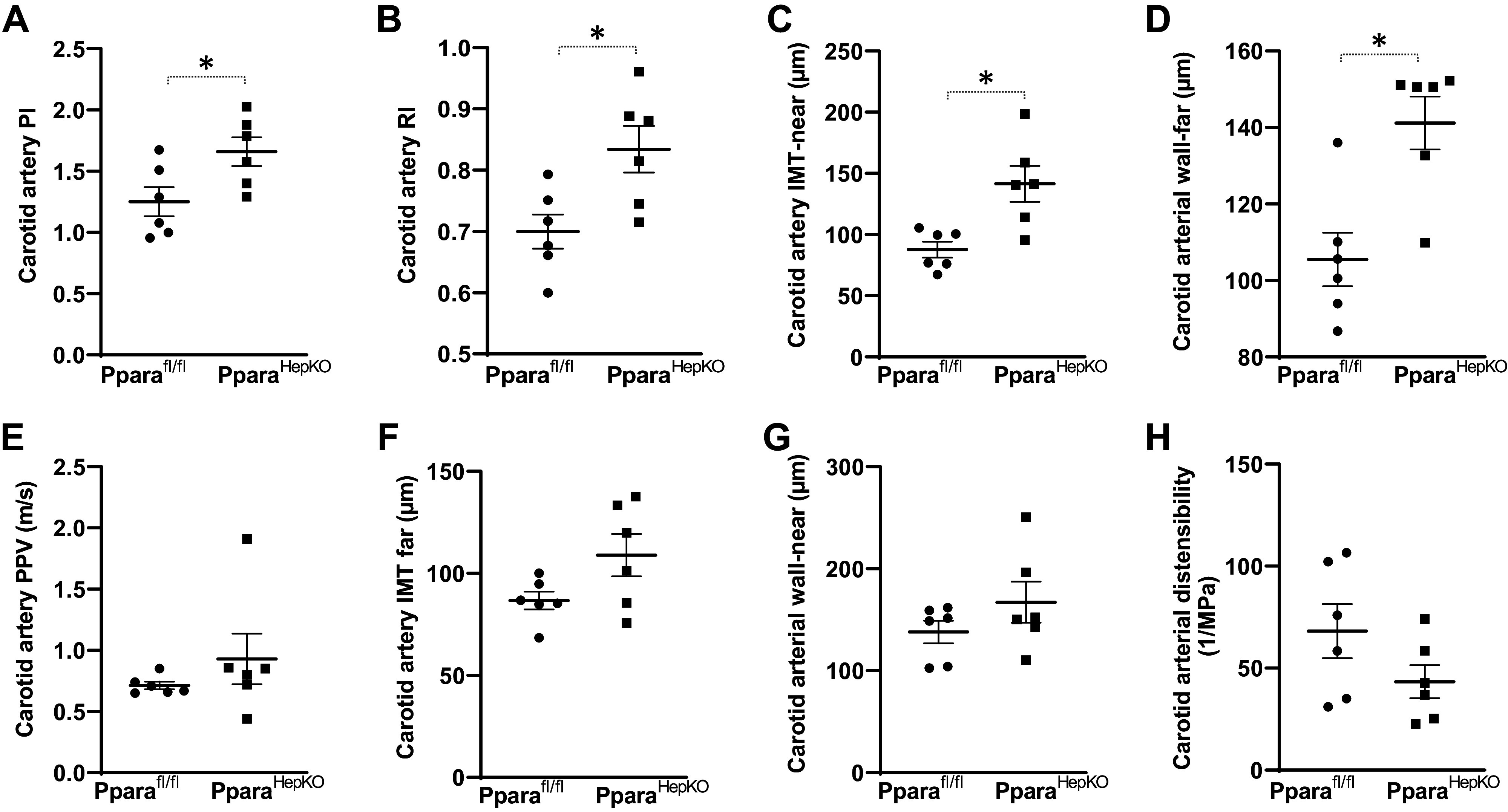
Common carotid artery phenotype in PparaHepKO and Pparafl/fl mice. Measurements of PI (A), RI (B), IMT-near (C), wall-far thickness (D), PPV (E), IMT-far (F), wall-near thickness (G), and distensibility (H) in PparaHepKO and Pparafl/fl mice. Values are means ± SE; *P < 0.05 vs. Pparafl/fl by unpaired t test; A–H; n = 6 mice per group. IMT, intima-media thickness; PI, pulsatility index; PPV, pulse propagative velocity; RI, resistive index.
Pathways in the Aortas of the PparaHepKO and Pparafl/fl Mice
Since the PparaHepKO mice exhibited phenotypes of cardiovascular dysfunction, we wanted to determine the signaling pathways in the aortas that might be changed due to the hepatic loss of PPARα. To quantitate the activity of the tyrosine kinase (TK) families that might be changed in the aortas, we compiled data from differentially phosphorylated substrates from the PamGene PamStation TK microarray chip [196 TK substrates] analysis into a heatmap (Fig. 6A) and waterfall plot of the phosphorylation level of all substrates in the PparaHepKO compared with Pparafl/fl controls (Fig. 6B). The data show that the aortas in the PparaHepKO mice have significantly less TK signaling than in Pparafl/fl mice.
Figure 6.
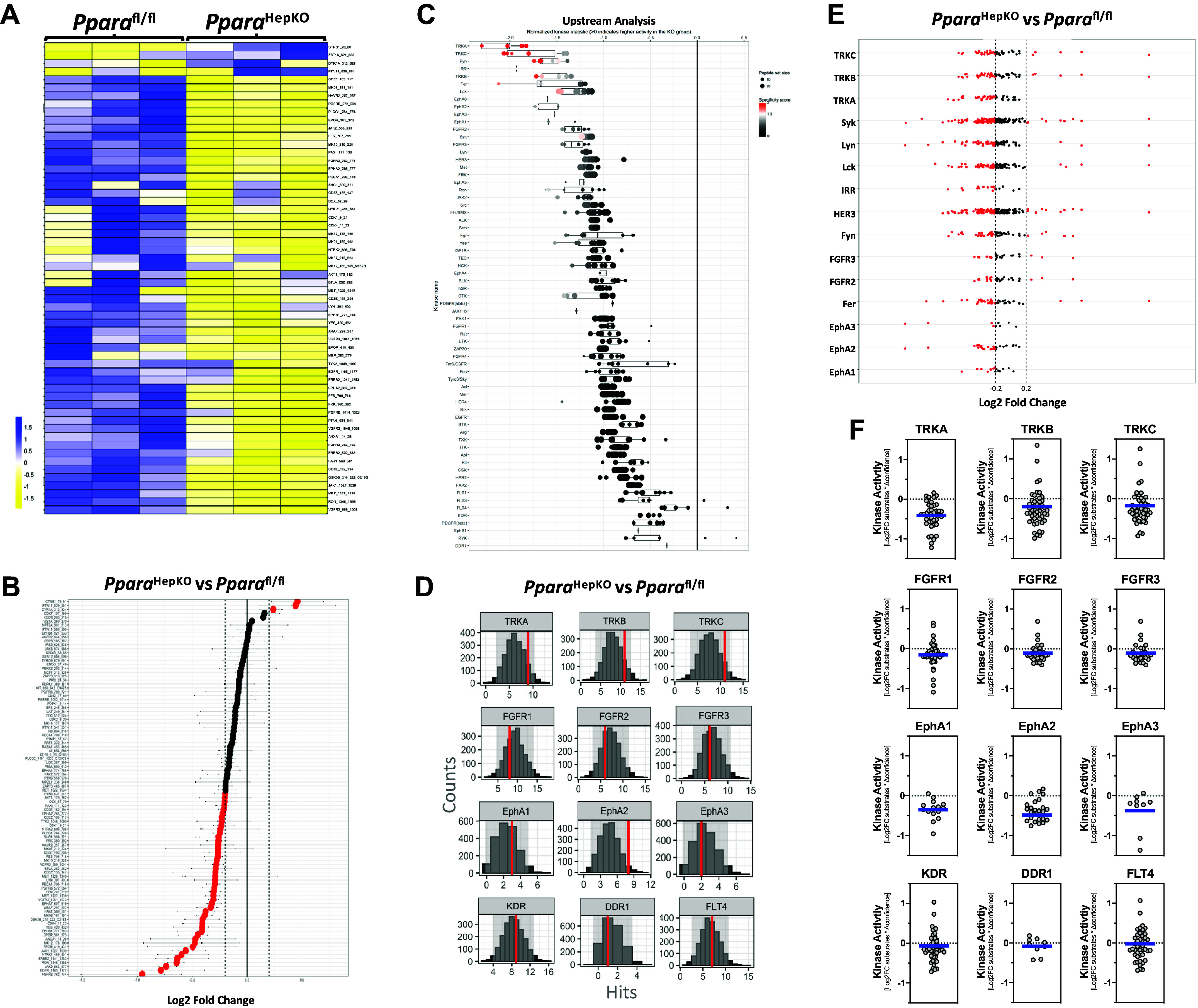
Tyrosine kinase signaling pathways in the aortas of PparaHepKO and Pparafl/fl mice. A and B: heatmap of substrate phosphorylation levels for tyrosine kinases and waterfall plot for comparison of kinase activity in PparaHepKO and Pparafl/fl mice. C: upstream tyrosine kinase analysis is represented by a waterfall plot. D: quantification of tyrosine kinases by histogram peacock plots comparing PparaHepKO vs. Pparafl/fl mice. E: tyrosine kinase substrate activity for each subfamily of tyrosine kinases. The red dots of the Log2 fold change images indicate increased or decreased activity for each individual kinase in the aortas of the PparaHepKO and Pparafl/fl mice. Red coloring indicates higher specificity. F: differentially active individual tyrosine kinases in the aortas of the PparaHepKO and Pparafl/fl mice. The blue line represents the average of the activity for each subfamily of kinases.
As previously described in literature (12, 43), we used bioinformatic analyses to identify upstream kinases responsible for the observed differential phosphorylation patterns of peptide substrates (Supplemental Tables S1 and S2; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.20982826.v1). An aggregation of the data according to the TK family upstream analysis waterfall plots in Fig. 6C showed that the tropomyosin receptor kinase A (TRKA) and the C isoform (TRKC) were the most differentially active tyrosine kinases. We next wanted to ascertain whether the differentially phosphorylated peptide substrates targeted by each tyrosine kinase against 2,000 random sampling iterations using the peacock plot analysis as we have previously described (12). The peacock plot data are represented as an annotated histogram that represents the differentially phosphorylated peptide substrates found in the experimental group (redline) targeted by a given kinase or kinase family in relation to the number of randomly selected peptide substrates targeted by that same kinase or kinase family in each of the thousands of random sampling events (gray histogram). A shift in the redline toward the right indicates higher confidence in the experimental group compared with the control and a change in kinase activity due to more differentially phosphorylated substrates compared with what is expected by random sampling (center of the histogram) for the given kinase. The peacock plots in Fig. 6D show that the TRK family (TRKA, TRKB, and TRKC) indicate statistical confidence that the signaling of this family is altered in the PparaHepKO mice compared with Pparafl/fl mice. However, the FGFR and Eph TK families indicate that they are likely not changed. One exception is EphA2, which the data indicate confidence that it is altered in the PparaHepKO mice. The others at the bottom of the waterfall plot in Fig. 5C show that KDR, DDR1, and FLT4 tyrosine kinases were not changed. To further analyze the activity of the TK group, we interrogated the Log2-fold change (Log2FC) of individual substrates for a given tyrosine kinase and the change in the peacock plot experimental group hits (redline) compared with the mean hits of 2,000 random sampling iterations (described further in the Supplemental Methods). These data are represented here as scatter plots with a blue line to represent the mean values herein referred to as MEOW plots. The analysis shows that TRKA and TRKB may be the most changed TKs in the aortas of the PparaHepKO compared with Pparafl/fl mice (Fig. 6, E and F).
The serine/threonine kinases (STK) usually signal after a TK signaling event has been provoked or by intracellular mechanisms that activate signaling mechanisms. Similar to the analysis described for tyrosine kinases above, we determined the STK signaling events that might be altered in the PparaHepKO compared with Pparafl/fl mice. We also found that several of the serine/threonine kinases had reduced activity at the substrates on the PamGene PamStation STK microarray chip (144 STK substrates) (Fig. 7, A and B). The upstream bioinformatic analysis showed that p70S6Kβ, p38δ, and IKKα were the most significantly changed STK signaling events (Fig. 7C). The peacock plot data for the most differentially active kinases show that the p70S6Kβ kinase has the most statistical confidence in being altered (Fig. 7D). To further analyze the STK group, we used the MEOW plot analysis to evaluate individual serine/threonine kinases. The results suggest that p70S6Kβ maybe be the most statistically changed STK in the aortas of the PparaHepKO compared with Pparafl/fl mice (Fig. 7, E and F).
Figure 7.
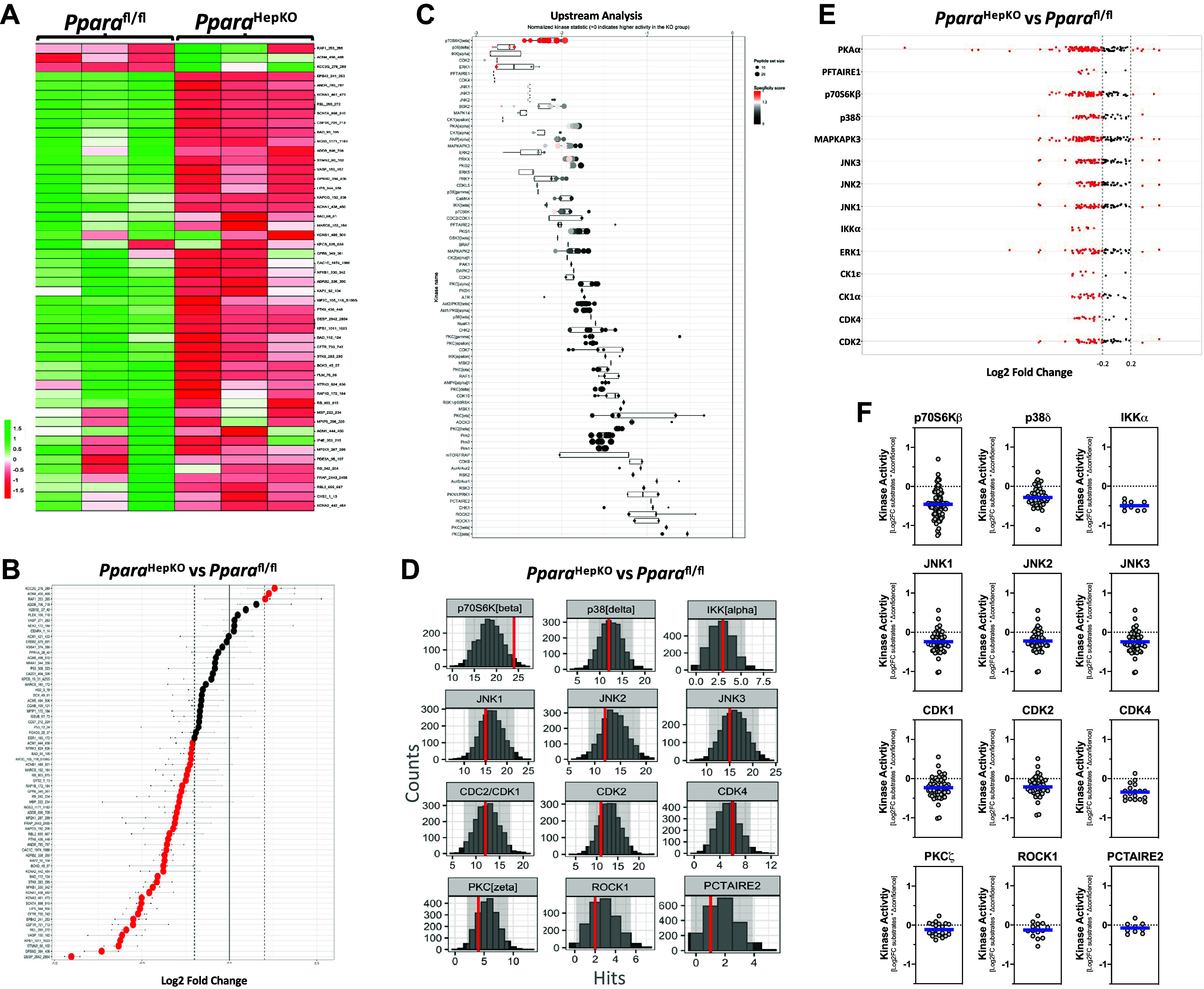
Serine/threonine kinase signaling pathways in the aortas of PparaHepKO and Pparafl/fl mice. A and B: heatmap of substrate phosphorylation levels for serine/threonine kinases and waterfall plot for comparison of kinase activity in PparaHepKO and Pparafl/fl mice. C: upstream serine/threonine kinase analysis represented by waterfall plot. D: quantification of serine/threonine kinases by histogram peacock plots comparing PparaHepKO vs. Pparafl/fl mice. E: serine/threonine kinase substrate activity for each subfamily of kinases. Red dots of the Log2 fold change images indicate increased or decreased activity for each individual kinase in the aortas of the PparaHepKO and Pparafl/fl mice. Red coloring indicates higher specificity. F: differentially active individual serine/threonine kinases in the aortas of the PparaHepKO and Pparafl/fl mice. Blue line represents the average of the activity for each subfamily of kinases.
To validate our finding of reduced TRK kinase activity, as reflected in the levels in the PparaHepKO compared with Pparafl/fl mice, we measured the plasma levels of the TRKB ligand, brain-derived neurotrophic factor (BDNF). The PparaHepKO mice exhibited significantly reduced plasma levels of BDNF as compared with Pparafl/fl mice (Fig. 8). The liver and brain supply BDNF to the circulation (50). These results demonstrate that lower TRK signaling resulted from decreased ligands in the plasma contributing to the increased vascular stiffness observed in the PparaHepKO mice.
Figure 8.

Plasma brain-derived neurotrophic factor (BDNF) in PparaHEPKO and Pparafl/fl mice. Plasma BDNF levels were measured in 30-wk-old male PparaHEPKO and Pparafl/fl mice. *P < 0.05 vs. Pparafl/fl by unpaired t test. n = 4 mice per group.
We compiled the results from the TK and STK PamGene PamStation microarray chip analysis (340 kinase peptide substrates total) and plotted them into a phyla tree of kinases to visually represent changes in kinase activity (Fig. 9). The kinase activity results were charted as bubble plots with node size based on the mean final kinase score in red (highest changed, mean node = 3), pink (mean node = 1.5), or gray (mean node = 1) circles to visually indicate potential clusters of kinase families that are changed in the aortas of the PparaHepKO compared with Pparafl/fl mice. The kinase tree data show clusters for the TK and the serine/threonine CMGC kinase families and, to a lesser extent, the CAMK and AGC kinase families. These results indicate that the hepatic loss of PPARα induces CVD in mice by controlling TK and STK signaling events.
Figure 9.

Kinase phyla tree analysis in the aortas of PparaHepKO and Pparafl/fl mice. The paralogous phylogenic relationships between differentially altered kinases in aortas of PparaHepKO and Pparafl/fl mice. Node color and size of the bubble plot on the paralogous phylogenic tree correspond to the final kinase score from the PTK and STK upstream kinase analysis. PTK, protein tyrosine kinase; STK, serine-threonine kinase.
DISCUSSION
In the present study, we investigated the impact of the loss of hepatocyte PPARα-induced NAFLD on cardiovascular dysfunction in nonobese mice. Consistent with our previous study, we found that PparaHepKO mice exhibited excessive fat accumulation in the liver on a standard chow diet (Fig. 1A) (19). The PparaHepKO mice also displayed elevated blood pressure, raised heart rate, vascular stiffness, and indices of cardiac dysfunction (Fig. 1, C and D, Figs. 3, 4, and 5). The aortas of the PparaHepKO mice had significantly less kinase signaling that was most compromised by the TRKA and TRKB tyrosine kinases and p70S6Kβ for serine/threonine kinases (Figs. 6 and 7). The PparaHepKO mice are lean and did not show metabolic abnormalities at 30 wk of age when the mice were studied. It is important to emphasize that these observed phenotypes occurred independently of obesity, hyperglycemia, and hyperinsulinemia in the PparaHepKO mice.
Elevated blood pressure is a significant risk factor for heart failure (51). Heart failure is associated with abnormalities in the left ventricular structure and function (52). In humans, heart failure has been characterized by chamber dilation, reduced cardiac muscle contractility, and lessened cardiac output (53, 54). In the present study, we observed enhanced left ventricle dilation that was associated with thinning of the cardiac walls and increased left ventricular end-diastolic volume in the PparaHepKO mice compared with littermates (Fig. 3). Dilated cardiomyopathy is characterized by left ventricular dilation, increased end-diastolic volume, thinning and weakening of the heart muscles, and an impaired ability to contract (55, 56). The structural alterations observed in the phenotypic presentation of the left ventricle of the PparaHepKO mice resemble the condition of dilated cardiomyopathy. It is one of the most common causes of heart failure. Its associated structural and functional abnormalities can lead to mortality, making it responsible for an annual mortality rate of 5–10% (57).
The PparaHepKO mice showed signs of vascular stiffness as indicated by increased pulsatility index, resistive index, and near intima-media thickness in both the abdominal aorta and common carotid artery (Figs. 4 and 5). Our results show that the reduction in distensibility/elasticity of the abdominal aorta is more pronounced and significant than that observed in the carotid artery. Both systolic and diastolic dysfunction elicit compensatory changes in the vascular system to reflect an increase in vascular resistance, decrease in arterial compliance, and elevated preload and afterload, causing high wall stress (58). Pulsatility and resistive indexes are noninvasive methods to measure the resistance of the vascular wall using Doppler ultrasonography, and their elevation is suggestive of arterial stiffness and reduced compliance (59, 60). Earlier studies have shown that elevated blood pressure increases pulsatile arterial wall stress contributing to the development and progression of arterial stiffness (61, 62). On the other hand, studies have also shown that stiffness of the aortic or carotid is linked with the progression of high blood pressure (63, 64). Hence, this study’s elevated blood pressure and vascular stiffness suggest a bidirectional relationship between hypertension and arterial stiffness, which is regulated by hepatic fat content and PPARα in the liver. In addition, in our present study, increased intima-media thickness, an indicator of cardiovascular risk, was observed in the PparaHepKO mice compared with the littermates. A novel aspect of the present finding is the relationship between vascular TK and STK signaling. Alterations in hepatic PPARα have not been previously reported to affect vascular TK or STK signaling. The results of our TK signaling analysis demonstrated alterations in kinase activity and phosphorylation of the members of the TRK family (Fig. 6). In support of this finding, we also found decreased levels of the TRK ligand BDNF in the plasma of PparaHepKO mice. It is believed that plasma levels of BDNF are derived from the brain as it can easily cross the blood-brain barrier (65). BDNF is produced from many origins, one being the liver, (50), and it has been shown to be transcriptionally controlled by PPARα (66). Our findings suggest that PPARα signaling in the liver to the aorta regulates vascular stiffness. However, the mechanism(s) by which this is regulated needs further investigation and validation.
Alterations in ventricular wall thickness increase the left ventricular end-diastolic volume due to overly stretched and enlarged ventricles, causing an increase in preload, further weakening the heart muscle and depressing contractility. The PparaHepKO mice presented impaired diastolic function as indicated by high peak velocity flow in early diastole/early diastolic mitral annular velocity (E/e′) ratio, prolonged IVRT, and e′ diminished. Heart failure with preserved ejection fraction, also known as diastolic heart failure, is characterized by ventricular stiffening that diminishes relaxation during diastole (67). The IVRT, which is the time interval between the closure of the aortic valve and opening of the mitral valve, becomes prolonged when there is impaired left ventricular relaxation, implying increased stiffness of the left ventricle, which is an indicator of diastolic dysfunction (68). The E/e′ ratio provides a useful tool for the noninvasive diagnosis of diastolic dysfunction and is the best echocardiographic parameter used to detect diastolic dysfunction in heart failure with preserved ejection fraction (69). Patients with NAFLD-associated CVD have been reported to have heart failure with preserved ejection fraction (70, 71). Hypertension is observed in ∼77% of patients with heart failure with preserved ejection fraction (72–74). Our results demonstrate that PparaHepKO mice exhibit significant adverse cardiac remodeling despite preserved left ventricular ejection fraction. Whether the observed dilated cardiomyopathy in PparaHepKO mice is due to myocardial ischemia or mediated by enhanced cardiomyocyte apoptosis needs to be investigated in future studies. The activation of TRKA in cardiac fibroblast of C57BL/6 mice prevents the death of cardiomyocytes (75). Feng et al. (76) demonstrated that TRKB signaling is essential for normal cardiac contraction and relaxation. In a murine model of myocardial ischemia, the p70S6K kinase was significantly reduced in the phosphorylation (77). Our data in this study suggest that the loss of hepatic PPARα induces alterations in the activation of the TRKA/TRKB/p70S6K kinases that might contribute to the pathogenesis of CVD in conditions of lean NAFLD.
Previous studies show that alterations in the left ventricular structure and function are observed in patients with NAFLD. However, without common risk factors for cardiac dysfunction, such as obesity, investigating the mechanisms linking NAFLD with CVD independent of other concatenate risk factors has been challenging. One of the main difficulties has been the lack of a suitable animal model where NAFLD develops without other confounding CVD risk factors like obesity and accompanying metabolic derangements such as diabetes and hyperlipidemia. Indeed, most animal models of NAFLD are based upon dietary models of high-fat feeding that provokes obesity and diabetes in addition to hepatic steatosis (78, 79).
High-fat diet feeding in preclinical models of NAFLD induces a state of positive energy balance leading to obesity and its consequent risk of other metabolic diseases such as insulin-resistant diabetes, hyperlipidemia, and hypertriglyceridemia (80–83). Previous studies have reported that high-fat diet-induced NAFLD in mice was associated with increased blood pressure (84, 85). These metabolic changes are correlated with CVD making the specific contribution of hepatic steatosis challenging to dissect from these models. Since PparaHepKO mice in the present study were fed a regular chow diet, we could determine the effect of hepatic steatosis on cardiovascular phenotypes in this model independent of any changes in body weight and fasting glucose and insulin levels. In addition, PparaHepKO mice have normal plasma triglyceride and cholesterol levels compared with Pparafl/fl on a regular diet eliminating any interfering effects of alterations in plasma lipids (19). The PparaHepKO mice fed a standard chow diet are a model reflective of a subgroup of patients that develop NAFLD without obesity and diabetes (86, 87). These patients are also at greater risk of CVD; however, the mechanisms by which this occurs are not fully understood (88).
Activation of PPARα in the obese reduces liver fat content and reorganizes the hepatic lipid species (89, 90). Recent findings have shown that bilirubin activates PPARα, which reduces NAFLD and improves liver function (46, 48, 91–95). Interestingly, plasma bilirubin levels are decreased in humans with obesity (96) and increasing levels have been shown to be heart healthy (3, 5, 97). However, whether bilirubin’s cardioprotective function is directly on the cardiovascular system or via a PPARα liver-mediated mechanism remains to be answered. It is important to note that female PparaHepKO mice do not exhibit any significant increases in hepatic steatosis on a normal fat diet at 30 wk of age (data not shown). It is possible that hepatic steatosis in female PparaHepKO takes longer to develop than in males. Future studies with aged female PparaHepKO mice are necessary to determine whether this process is delayed or if female PparaHepKO are protected against the development of hepatic steatosis as compared with males.
In conclusion, a reduction in hepatic PPARα manifests events that lead to poorer cardiovascular phenotypes in mice that present to the level of NAFLD-induced CVD in humans. The association between NAFLD and CVD has been an area of intense investigation in recent years (24–27). Clinical and epidemiological studies have established that NAFLD is linked to adverse cardiovascular events such as coronary artery disease, cardiac remodeling, left ventricular diastolic dysfunction, cardiac rhythm disturbances, and ischemic stroke (28, 29, 98, 99). In fact, cardiovascular events and complications of CVD are the leading cause of death in patients with NAFLD, even more than the liver disease itself (27, 100). Structural remodeling of the heart and an increased risk of cardiomyopathy is observed in patients who manifest NAFLD (101–104). Interestingly, alterations in the left ventricular structure and function are observed in patients with NAFLD without typical risk factors such as obesity, suggesting a potential role for hepatic steatosis in developing CVD (70, 88). Future studies to reveal the hepatic mechanisms that cause these events, like hepatic hormones such as fibroblast growth factor 21 (FGF21) and many others that are regulated by PPARα, warrant further investigation into the link between NAFLD and CVD.
Perspectives and Significance
We know that PparaHepKO mice exhibit increased hepatic fat accumulation and signs of CVD even when fed a standard normal-fat chow diet. Our study further demonstrates that increased hepatic fat accumulation in PparaHepKO mice is associated with elevated blood pressure, cardiac remodeling, systolic and diastolic dysfunction, and vascular stiffness, which might be related to the liver-induced kinase signaling to the aorta among various other pathways. To the best of our knowledge, this is the first study describing the cardiac and vascular phenotype in hepatocyte-specific PPARα knockout mice. The PparaHepKO mice are a favorable model supporting investigations on the specific role of NAFLD-induced CVD and could represent a useful tool in research for therapeutic targets for CVD, especially in patients with NAFLD in the absence of obesity and diabetes. More work is needed to reveal the hepatic mechanisms that PPARα controls to signal to peripheral tissues, such as the heart and vasculature, to activate pathways that cause deleterious effects that lead to CVD.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Tables S1 and S2 and Supplemental Material: https://doi.org/10.6084/m9.figshare.20982826.v1.
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Grants 1R01DK121748-01A1 (to D. E. Stec), 1R01DK121797-01A1 (to T. D. Hinds); the National Heart, Lung and Blood Institute Grants P01 HL05197-11 (to D. E. Stec) and K01HL125445 (to T. D. Hinds); and the National Institute of General Medical Sciences Grant P20GM104357-02 (to D. E. Stec). This study was also supported by an American Heart Association Postdoctoral Award (23POST1020493) (to O. O. Badmus).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
O.O.B., T.D.H., and D.E.S. conceived and designed research; O.O.B., Z.A.K., E.A.B., A.A.d.S., L.C.T., G.J.M., W-H.L., J.F.C., and D.E.S. performed experiments; O.O.B., Z.A.K., E.A.B., A.A.d.S., L.C.T., G.J.M., W-H.L., J.F.C., T.D.H., and D.E.S. analyzed data; O.O.B., Z.A.K., E.A.B., A.A.d.S., L.C.T., G.J.M., W-H.L., J.F.C., T.D.H., and D.E.S. interpreted results of experiments; O.O.B., Z.A.K., E.A.B., A.A.d.S., G.J.M., T.D.H., and D.E.S. prepared figures; D.E.S. and T.D.H. drafted manuscript; O.O.B., Z.A.K., E.A.B., A.A.d.S., L.C.T., G.J.M., W-H.L., J.F.C., T.D.H., and D.E.S. edited and revised manuscript; O.O.B., Z.A.K., E.A.B., A.A.d.S., L.C.T., G.J.M., W-H.L., J.F.C., T.D.H., and D.E.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Matthew Hazzard at the University of Kentucky for creating the illustration of the mice used in Fig. 1. The authors acknowledge the analytical and assay core in the Department of Physiology & Biophysics at the University of Mississippi Medical Center.
REFERENCES
- 1. Muzurović E, Peng CC, Belanger MJ, Sanoudou D, Mikhailidis DP, Mantzoros CS. Nonalcoholic fatty liver disease and cardiovascular disease: a review of shared cardiometabolic risk factors. Hypertension 79: 1319–1326, 2022. doi: 10.1161/HYPERTENSIONAHA.122.17982. [DOI] [PubMed] [Google Scholar]
- 2. Ismaiel A, Dumitraşcu DL. Cardiovascular risk in fatty liver disease: the liver-heart axis-literature review. Front Med (Lausanne) 6: 202, 2019. doi: 10.3389/fmed.2019.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hinds TD Jr, Stec DE. Bilirubin safeguards cardiorenal and metabolic diseases: a protective role in health. Curr Hypertens Rep 21: 87, 2019. doi: 10.1007/s11906-019-0994-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weaver L, Hamoud AR, Stec DE, Hinds TD Jr. Biliverdin reductase and bilirubin in hepatic disease. Am J Physiol Gastrointest Liver Physiol 314: G668–G676, 2018. doi: 10.1152/ajpgi.00026.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hinds TD Jr, Stec DE. Bilirubin, a cardiometabolic signaling molecule. Hypertension 72: 788–795, 2018. [Erratum in Hypertension 72: e95, 2018]. doi: 10.1161/HYPERTENSIONAHA.118.11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamoud AR, Weaver L, Stec DE, Hinds TD Jr. Bilirubin in the liver-gut signaling axis. Trends Endocrinol Metab 29: 140–150, 2018. doi: 10.1016/j.tem.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heinrich G, Ghadieh HE, Ghanem SS, Muturi HT, Rezaei K, Al-Share QY, Bowman TA, Zhang D, Garofalo RS, Yin L, Najjar SM. Loss of hepatic CEACAM1: a unifying mechanism linking insulin resistance to obesity and non-alcoholic fatty liver disease. Front Endocrinol (Lausanne) 8: 8, 2017. doi: 10.3389/fendo.2017.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Russo L, Lester SG, Khuder SS, Hinds TD, Friedman SL, Najjar SM. Loss of liver carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) causes insulin resistance and NASH pathogenesis in mice (Abstract). Hepatology 58: 534A, 2013. [Google Scholar]
- 9. Liu Y, Zhong GC, Tan HY, Hao FB, Hu JJ. Nonalcoholic fatty liver disease and mortality from all causes, cardiovascular disease, and cancer: a meta-analysis. Sci Rep 9: 11124, 2019. doi: 10.1038/s41598-019-47687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Younossi ZM, Stepanova M, Negro F, Hallaji S, Younossi Y, Lam B, Srishord M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine (Baltimore) 91: 319–327, 2012. doi: 10.1097/MD.0b013e3182779d49. [DOI] [PubMed] [Google Scholar]
- 11. Badmus OO, Hillhouse SA, Anderson CD, Hinds TD, Stec DE. Molecular mechanisms of metabolic associated fatty liver disease (MAFLD): functional analysis of lipid metabolism pathways. Clin Sci (Lond) 136: 1347–1366, 2022. doi: 10.1042/CS20220572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Creeden JF, Kipp ZA, Xu M, Flight RM, Moseley HNB, Martinez GJ, Lee WH, Alganem K, Imami AS, McMullen MR, Roychowdhury S, Nawabi AM, Hipp JA, Softic S, Weinman SA, McCullumsmith R, Nagy LE, Hinds TD Jr. Hepatic kinome atlas: an in-depth identification of kinase pathways in liver fibrosis of humans and rodents. Hepatology 76: 1376–1388, 2022. doi: 10.1002/hep.32467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carr RM, Oranu A, Khungar V. Nonalcoholic fatty liver disease: pathophysiology and management. Gastroenterol Clin North Am 45: 639–652, 2016. doi: 10.1016/j.gtc.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pal P, Palui R, Ray S. Heterogeneity of non-alcoholic fatty liver disease: implications for clinical practice and research activity. World J Hepatol 13: 1584–1610, 2021. doi: 10.4254/wjh.v13.i11.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F, Barquissau V, Régnier M, Lukowicz C, Benhamed F, Iroz A, Bertrand-Michel J, Al Saati T, Cano P, Mselli-Lakhal L, Mithieux G, Rajas F, Lagarrigue S, Pineau T, Loiseau N, Postic C, Langin D, Wahli W, Guillou H. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65: 1202–1214, 2016. doi: 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Komatsu M, Kimura T, Yazaki M, Tanaka N, Yang Y, Nakajima T, Horiuchi A, Fang ZZ, Joshita S, Matsumoto A, Umemura T, Tanaka E, Gonzalez FJ, Ikeda S, Aoyama T. Steatogenesis in adult-onset type II citrullinemia is associated with down-regulation of PPARα. Biochim Biophys Acta 1852: 473–481, 2015. doi: 10.1016/j.bbadis.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, Van Hul W, Mertens I, Hubens G, Van Marck E, Michielsen P, Van Gaal L, Staels B. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol 63: 164–173, 2015. doi: 10.1016/j.jhep.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 18. Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD Jr. Glucocorticoid receptor β induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem 291: 25776–25788, 2016. doi: 10.1074/jbc.M116.752311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stec DE, Gordon DM, Hipp JA, Hong S, Mitchell ZL, Franco NR, Robison JW, Anderson CD, Stec DF, Hinds TD Jr. Loss of hepatic PPARα promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am J Physiol Regul Integr Comp Physiol 317: R733–R745, 2019. doi: 10.1152/ajpregu.00153.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hinds TD Jr, Kipp ZA, Xu M, Yiannikouris FB, Morris AJ, Stec DF, Wahli W, Stec DE. Adipose-specific PPARα knockout mice have increased lipogenesis by PASK-SREBP1 signaling and a polarity shift to inflammatory macrophages in white adipose tissue. Cells 11: 4, 2021. doi: 10.3390/cells11010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Régnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, Fouché E, Lasserre F, Naylies C, Bétoulières C, Barquissau V, Mouisel E, Bertrand-Michel J, Batut A, Saati TA, Canlet C, Tremblay-Franco M, Ellero-Simatos S, Langin D, Postic C, Wahli W, Loiseau N, Guillou H, Montagner A. Hepatocyte-specific deletion of PPARα promotes NAFLD in the context of obesity. Sci Rep 10: 6489, 2020. doi: 10.1038/s41598-020-63579-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, Zelber-Sagi S, Wai-Sun Wong V, Dufour JF, Schattenberg JM, Kawaguchi T, Arrese M, Valenti L, Shiha G, Tiribelli C, Yki-Järvinen H, Fan JG, Grønbæk H, Yilmaz Y, Cortez-Pinto H, Oliveira CP, Bedossa P, Adams LA, Zheng MH, Fouad Y, Chan WK, Mendez-Sanchez N, Ahn SH, Castera L, Bugianesi E, Ratziu V, George J.. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 73: 202–209, 2020. doi: 10.1016/j.jhep.2020.03.039. [DOI] [PubMed] [Google Scholar]
- 23. Eslam M, Sanyal AJ, George J; International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158: 1999–2014.e1, 2020. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 24. Mahfood Haddad T, Hamdeh S, Kanmanthareddy A, Alla VM. Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: a systematic review and meta-analysis. Diabetes Metab Syndr 11 Suppl 1: S209–S216, 2017. doi: 10.1016/j.dsx.2016.12.033. [DOI] [PubMed] [Google Scholar]
- 25. Tana C, Ballestri S, Ricci FD, Vincenzo A, Ticinesi A, Gallina S, Giamberardino MA, Cipollone F, Sutton R, Vettor R, Fedorowski A, Meschi T. Cardiovascular risk in non-alcoholic fatty liver disease: mechanisms and therapeutic implications. Int J Environ Res Public Health 16, 3104, 2019. doi: 10.3390/ijerph16173104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pellicori P, Vaduganathan M, Ferreira JP, Zannad F, Sanyal AJ. Cross-talk between non-alcoholic fatty liver disease and cardiovascular disease: implications for future trial design. Diabetes Metab 48: 101281, 2022. doi: 10.1016/j.diabet.2021.101281. [DOI] [PubMed] [Google Scholar]
- 27. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 363: 1341–1350, 2010. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 28. Mellinger JL, Pencina KM, Massaro JM, Hoffmann U, Seshadri S, Fox CS, O'Donnell CJ, Speliotes EK. Hepatic steatosis and cardiovascular disease outcomes: an analysis of the Framingham Heart Study. J Hepatol 63: 470–476, 2015. doi: 10.1016/j.jhep.2015.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. VanWagner LB, Wilcox JE, Colangelo LA, Lloyd-Jones DM, Carr JJ, Lima JA, Lewis CE, Rinella ME, Shah SJ. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: a population-based study. Hepatology 62: 773–783, 2015. doi: 10.1002/hep.27869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang B, Liu B, McNeill KL, Chowienczyk PJ. Measurement of pulse wave velocity using pulse wave Doppler ultrasound: comparison with arterial tonometry. Ultrasound Med Biol 34: 509–512, 2008. doi: 10.1016/j.ultrasmedbio.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 31. do Carmo JM, Omoto ACM, Dai X, Moak SP, Mega GS, Li X, Wang Z, Mouton AJ, Hall JE, da Silva AA. Sex differences in the impact of parental obesity on offspring cardiac SIRT3 expression, mitochondrial efficiency, and diastolic function early in life. Am J Physiol Heart Circ Physiol 321: H485–H495, 2021. doi: 10.1152/ajpheart.00176.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. da Silva AA, Moak SP, Dai X, Borges GC, Omoto ACM, Wang Z, Li X, Mouton AJ, Hall JE, do Carmo JM. Parental obesity alters offspring blood pressure regulation and cardiovascular responses to stress: role of P2X7R and sex differences. Am J Physiol Regul Integr Comp Physiol 322: R421–R433, 2022. doi: 10.1152/ajpregu.00300.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stone K, Fryer S, Faulkner J, Meyer ML, Zieff G, Paterson C, Burnet K, Kelsch E, Credeur D, Lambrick D, Stoner L. Acute changes in carotid-femoral pulse-wave velocity are tracked by heart-femoral pulse-wave velocity. Front Cardiovasc Med 7: 592834, 2020. doi: 10.3389/fcvm.2020.592834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Graham MR, Evans P, Davies B, Baker JS. Arterial pulse wave velocity, inflammatory markers, pathological GH and IGF states, cardiovascular and cerebrovascular disease. Vasc Health Risk Manag 4: 1361–1371, 2008. doi: 10.2147/vhrm.s3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Butz GM, Davisson RL. Long-term telemetric measurement of cardiovascular parameters in awake mice: a physiological genomics tool. Physiol Genomics 5: 89–97, 2001. doi: 10.1152/physiolgenomics.2001.5.2.89. [DOI] [PubMed] [Google Scholar]
- 36. Carlson SH, Wyss JM. Long-term telemetric recording of arterial pressure and heart rate in mice fed basal and high NaCl diets. Hypertension 35: E1–5, 2000. doi: 10.1161/01.hyp.35.2.e1. [DOI] [PubMed] [Google Scholar]
- 37. Creeden JF, Alganem K, Imami AS, Brunicardi FC, Liu SH, Shukla R, Tomar T, Naji F, McCullumsmith RE. Kinome array profiling of patient-derived pancreatic ductal adenocarcinoma identifies differentially active protein tyrosine kinases. Int J Mol Sci 21: 8679, 2020. doi: 10.3390/ijms21228679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGuire JL, Depasquale EA, Funk AJ, O'Donnovan SM, Hasselfeld K, Marwaha S, Hammond JH, Hartounian V, Meador-Woodruff JH, Meller J, McCullumsmith RE. Abnormalities of signal transduction networks in chronic schizophrenia. NPJ Schizophr 3: 30, 2017. doi: 10.1038/s41537-017-0032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Appuhamy JA, Nayananjalie WA, England EM, Gerrard DE, Akers RM, Hanigan MD. Effects of AMP-activated protein kinase (AMPK) signaling and essential amino acids on mammalian target of rapamycin (mTOR) signaling and protein synthesis rates in mammary cells. J Dairy Sci 97: 419–429, 2014. doi: 10.3168/jds.2013-7189. [DOI] [PubMed] [Google Scholar]
- 40. Dorsett CR, McGuire JL, Niedzielko TL, DePasquale EA, Meller J, Floyd CL, McCullumsmith RE. Traumatic brain injury induces alterations in cortical glutamate uptake without a reduction in glutamate transporter-1 protein expression. J Neurotrauma 34: 220–234, 2017. doi: 10.1089/neu.2015.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DePasquale EAK, Alganem K, Bentea E, Nawreen N, McGuire JL, Naji F, Hilhorst R, Meller J, McCullumsmith RE. KRSA: network-based prediction of differential kinase activity from kinome array data. PLoS One 16: e0260440, 2020. doi: 10.1371/journal.pone.0260440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chirumamilla CS, Fazil M, Perez-Novo C, Rangarajan S, de Wijn R, Ramireddy P, Verma NK, Vanden Berghe W. Profiling activity of cellular kinases in migrating T-cells. Methods Mol Biol 1930: 99–113, 2019. doi: 10.1007/978-1-4939-9036-8_13. [DOI] [PubMed] [Google Scholar]
- 43. Bates EA, Kipp ZA, Martinez GJ, Badmus OO, Soundarapandian MM, Foster D, Xu M, Creeden JF, Greer JR, Morris AJ, Stec DE, Hinds TD Jr. Suppressing hepatic UGT1A1 increases plasma bilirubin, lowers plasma urobilin, reorganizes kinase signaling pathways and lipid species and improves fatty liver disease. Biomolecules 13: 252, 2023. doi: 10.3390/biom13020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Metz KS, Deoudes EM, Berginski ME, Jimenez-Ruiz I, Aksoy BA, Hammerbacher J, Gomez SM, Phanstiel DH. Coral: clear and customizable visualization of human kinome data. Cell Syst 7: 347–350.e1, 2018. doi: 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hinds TD Jr, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem 291: 25179–25191, 2016. doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hinds TD Jr, Creeden JF, Gordon DM, Stec DF, Donald MC, Stec DE. Bilirubin nanoparticles reduce diet-induced hepatic steatosis, improve fat utilization, and increase plasma β-hydroxybutyrate. Front Pharmacol 11: 594574, 2020. doi: 10.3389/fphar.2020.594574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hinds TD Jr, Hosick PA, Chen S, Tukey RH, Hankins MW, Nestor-Kalinoski A, Stec DE. Mice with hyperbilirubinemia due to Gilbert's syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am J Physiol Endocrinol Metab 312: E244–E252, 2017. doi: 10.1152/ajpendo.00396.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gordon DM, Neifer KL, Hamoud AA, Hawk CF, Nestor-Kalinoski AL, Miruzzi SA, Morran MP, Adeosun SO, Sarver JG, Erhardt PW, McCullumsmith RE, Stec DE, Hinds TD Jr. Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator-activated receptor α. J Biol Chem 295: 9804–9822, 2020. doi: 10.1074/jbc.RA120.013700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stec DE, Gordon DM, Nestor-Kalinoski AL, Donald MC, Mitchell ZL, Creeden JF, Hinds TD Jr. Biliverdin reductase A (BVRA) knockout in adipocytes induces hypertrophy and reduces mitochondria in white fat of obese mice. Biomolecules 10: 387, 2020. doi: 10.3390/biom10030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang B, Ren Q, Zhang JC, Chen QX, Hashimoto K. Altered expression of BDNF, BDNF pro-peptide and their precursor proBDNF in brain and liver tissues from psychiatric disorders: rethinking the brain-liver axis. Transl Psychiatry 7: e1128, 2017. doi: 10.1038/tp.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fuchs FD, Whelton PK. High blood pressure and cardiovascular disease. Hypertension 75: 285–292, 2020. doi: 10.1161/HYPERTENSIONAHA.119.14240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reis Filho JR, Cardoso JN, Cardoso CM, Pereira-Barretto AC. Reverse cardiac remodeling: a marker of better prognosis in heart failure. Arq Bras Cardiol 104: 502–506, 2015. doi: 10.5935/abc.20150025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Feldman AM, Kubota T, Li YY, Wagner D, Combes A, McTiernan C. Evidence of phenotypic alteration as a cause of systolic dysfunction in the failing heart. Cardiol Clin 16: 677–689, ix, 1998. doi: 10.1016/s0733-8651(05)70044-8. [DOI] [PubMed] [Google Scholar]
- 54. Dorn GW 2nd, Molkentin JD. Manipulating cardiac contractility in heart failure: data from mice and men. Circulation 109: 150–158, 2004. doi: 10.1161/01.CIR.0000111581.15521.F5. [DOI] [PubMed] [Google Scholar]
- 55. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 121: 731–748, 2017. doi: 10.1161/CIRCRESAHA.116.309396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ramaccini D, Montoya-Uribe V, Aan FJ, Modesti L, Potes Y, Wieckowski MR, Krga I, Glibetic M, Pinton P, Giorgi C, Matter ML. Mitochondrial function and dysfunction in dilated cardiomyopathy. Front Cell Dev Biol 8: 624216, 2020. doi: 10.3389/fcell.2020.624216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sen-Chowdhry S, McKenna WJ. Sudden death from genetic and acquired cardiomyopathies. Circulation 125: 1563–1576, 2012. doi: 10.1161/CIRCULATIONAHA.111.025528. [DOI] [PubMed] [Google Scholar]
- 58. Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, Liu PP, Matsumori A, Mazzanti A, McMurray J, Priori SG. Dilated cardiomyopathy. Nat Rev Dis Primers 5: 32, 2019. doi: 10.1038/s41572-019-0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jankowski P, Kawecka-Jaszcz K, Czarnecka D. Ascending aortic blood pressure waveform is related to coronary atherosclerosis in hypertensive as well as in normotensive subjects. Blood Press 16: 246–253, 2007. doi: 10.1080/08037050701428125. [DOI] [PubMed] [Google Scholar]
- 60. Morillas P, Quiles J, Mateo I, Bertomeu-González V, Castillo J, de Andrade H, Roldan J, Miralles B, Masiá MD, Carrillo P, Bertomeu-Martínez V. Carotid resistive index in treated hypertensive patients: relationship with target organ damage. Blood Press 21: 360–366, 2012. doi: 10.3109/08037051.2012.694181. [DOI] [PubMed] [Google Scholar]
- 61. Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension 64: 210–214, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mitchell GF. Aortic stiffness, pressure and flow pulsatility, and target organ damage. J Appl Physiol (1985) 125: 1871–1880, 2018. doi: 10.1152/japplphysiol.00108.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liao D, Arnett DK, Tyroler HA, Riley WA, Chambless LE, Szklo M, Heiss G. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension 34: 201–206, 1999. doi: 10.1161/01.hyp.34.2.201. [DOI] [PubMed] [Google Scholar]
- 64. Dernellis J, Panaretou M. Aortic stiffness is an independent predictor of progression to hypertension in nonhypertensive subjects. Hypertension 45: 426–431, 2005. doi: 10.1161/01.HYP.0000157818.58878.93. [DOI] [PubMed] [Google Scholar]
- 65. Karege F, Schwald M, Cisse M. Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci Lett 328: 261–264, 2002. doi: 10.1016/s0304-3940(02)00529-3. [DOI] [PubMed] [Google Scholar]
- 66. Patel D, Roy A, Raha S, Kundu M, Gonzalez FJ, Pahan K. Upregulation of BDNF and hippocampal functions by a hippocampal ligand of PPARα. JCI Insight 5: e136654 2020. doi: 10.1172/jci.insight.136654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Obokata M, Reddy YNV, Borlaug BA. Diastolic dysfunction and heart failure with preserved ejection fraction: understanding mechanisms by using noninvasive methods. JACC Cardiovasc Imaging 13: 245–257, 2020. doi: 10.1016/j.jcmg.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Henein MY, Lindqvist P. Diastolic function assessment by echocardiography: a practical manual for clinical use and future applications. Echocardiography 37: 1918–2020, 1908. doi: 10.1111/echo.14698. [DOI] [PubMed] [Google Scholar]
- 69. Kasner M, Westermann D, Steendijk P, Gaub R, Wilkenshoff U, Weitmann K, Hoffmann W, Poller W, Schultheiss HP, Pauschinger M, Tschöpe C. Utility of Doppler echocardiography and tissue Doppler imaging in the estimation of diastolic function in heart failure with normal ejection fraction: a comparative Doppler-conductance catheterization study. Circulation 116: 637–647, 2007. doi: 10.1161/CIRCULATIONAHA.106.661983. [DOI] [PubMed] [Google Scholar]
- 70. Fudim M, Zhong L, Patel KV, Khera R, Abdelmalek MF, Diehl AM, McGarrah RW, Molinger J, Moylan CA, Rao VN, Wegermann K, Neeland IJ, Halm EA, Das SR, Pandey A. Nonalcoholic fatty liver disease and risk of heart failure among medicare beneficiaries. J Am Heart Assoc 10: e021654, 2021. doi: 10.1161/JAHA.121.021654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Salah HM, Pandey A, Soloveva A, Abdelmalek MF, Diehl AM, Moylan CA, Wegermann K, Rao VN, Hernandez AF, Tedford RJ, Parikh KS, Mentz RJ, McGarrah RW, Fudim M. Relationship of nonalcoholic fatty liver disease and heart failure with preserved ejection fraction. JACC Basic Transl Sci 6: 918–932, 2021. doi: 10.1016/j.jacbts.2021.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yancy CW, Lopatin M, Stevenson LW, De Marco T, Fonarow GC; ADHERE Scientific Advisory Committee and Investigators. Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the Acute Decompensated Heart Failure National Registry (ADHERE) Database. J Am Coll Cardiol 47: 76–84, 2006. [Erratum in J Am Coll Cardiol 47: 1502, 2006]. doi: 10.1016/j.jacc.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 73. Bhuiyan T, Maurer MS. Heart failure with preserved ejection fraction: persistent diagnosis, therapeutic enigma. Curr Cardiovasc Risk Rep 5: 440–449, 2011. doi: 10.1007/s12170-011-0184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Oh GC, Cho HJ. Blood pressure and heart failure. Clin Hypertens 26: 1, 2020. doi: 10.1186/s40885-019-0132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aridgides D, Salvador R, PereiraPerrin M. Trypanosoma cruzi coaxes cardiac fibroblasts into preventing cardiomyocyte death by activating nerve growth factor receptor TrkA. PLoS One 8: e57450, 2013. doi: 10.1371/journal.pone.0057450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Feng N, Huke S, Zhu G, Tocchetti CG, Shi S, Aiba T, Kaludercic N, Hoover DB, Beck SE, Mankowski JL, Tomaselli GF, Bers DM, Kass DA, Paolocci N. Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proc Natl Acad Sci USA 112: 1880–1885, 2015. [Erratum in Proc Natl Acad Sci USA 112: E1691, 2015]. doi: 10.1073/pnas.1417949112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Demeulder B, Zarrinpashneh E, Ginion A, Viollet B, Hue L, Rider MH, Vanoverschelde JL, Beauloye C, Horman S, Bertrand L. Differential regulation of eEF2 and p70S6K by AMPKα2 in heart. Biochim Biophys Acta 1832: 780–790, 2013. doi: 10.1016/j.bbadis.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 78. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 8: 35–44, 2011. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 79. Im YR, Hunter H, de Gracia Hahn D, Duret A, Cheah Q, Dong J, Fairey M, Hjalmarsson C, Li A, Lim HK, McKeown L, Mitrofan CG, Rao R, Utukuri M, Rowe IA, Mann JP. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 74: 1884–1901, 2021. doi: 10.1002/hep.31897. [DOI] [PubMed] [Google Scholar]
- 80. Recena Aydos L, Aparecida do Amaral L, Serafim de Souza R, Jacobowski AC, Freitas Dos Santos E, Rodrigues Macedo ML. Nonalcoholic fatty liver disease induced by high-fat diet in C57bl/6 models. Nutrients 11: 3067, 2019. doi: 10.3390/nu11123067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Van Herck MA, Vonghia L, Francque SM. Animal models of nonalcoholic fatty liver disease – a starter’s guide. Nutrients 9: 1072, 2017., doi: 10.3390/nu9101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 285: 17271–17276, 2010. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res 116: 991–1006, 2015. doi: 10.1161/CIRCRESAHA.116.305697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tao X, He H, Peng J, Xu R, Fu J, Hu Y, Li L, Yang X, Feng X, Zhang C, Zhang L, Yu X, Shen A, Huang K, Fu Q. Overexpression of PDE4D in mouse liver is sufficient to trigger NAFLD and hypertension in a CD36-TGF-β1 pathway: therapeutic role of roflumilast. Pharmacol Res 175: 106004, 2022. doi: 10.1016/j.phrs.2021.106004. [DOI] [PubMed] [Google Scholar]
- 85. Zhong F, Zhou X, Xu J, Gao L. Rodent models of nonalcoholic fatty liver disease. Digestion 101: 522–535, 2020. doi: 10.1159/000501851. [DOI] [PubMed] [Google Scholar]
- 86. VanWagner LB, Armstrong MJ. Lean NAFLD: a not so benign condition? Hepatol Commun 2: 5–8, 2018. doi: 10.1002/hep4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Younes R, Bugianesi E. NASH in lean individuals. Semin Liver Dis 39: 86–95, 2019. doi: 10.1055/s-0038-1677517. [DOI] [PubMed] [Google Scholar]
- 88. Kim Y, Han E, Lee JS, Lee HW, Kim BK, Kim MK, Kim HS, Park JY, Kim DY, Ahn SH, Lee BW, Kang ES, Cha BS, Lee YH, Kim SU. Cardiovascular risk is elevated in lean subjects with nonalcoholic fatty liver disease. Gut Liver 16: 290–299, 2022. doi: 10.5009/gnl210084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Abdelmoneim D, El-Adl M, El-Sayed G, El-Sherbini ES. Protective effect of fenofibrate against high-fat-high-fructose diet induced non-obese NAFLD in rats. Fundam Clin Pharmacol 35: 379–388, 2021. doi: 10.1111/fcp.12597. [DOI] [PubMed] [Google Scholar]
- 90. Yavarow ZA, Kang HR, Waskowicz LR, Bay BH, Young SP, Yen PM, Koeberl DD. Fenofibrate rapidly decreases hepatic lipid and glycogen storage in neonatal mice with glycogen storage disease type Ia. Hum Mol Genet 29: 286–294, 2020. doi: 10.1093/hmg/ddz290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kipp ZA, Martinez GJ, Bates EA, Maharramov AB, Flight RM, Moseley HNB, Morris AJ, Stec DE, Hinds TD Jr. Bilirubin nanoparticle treatment in obese mice inhibits hepatic ceramide production and remodels liver fat content. Metabolites 13: 215: 2023. doi: 10.3390/metabo13020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gordon DM, Hong SH, Kipp ZA, Hinds TD Jr. Identification of binding regions of bilirubin in the ligand-binding pocket of the peroxisome proliferator-activated receptor-A (PPARα). Molecules 26: 2975, 2021. doi: 10.3390/molecules26102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gordon DM, Blomquist TM, Miruzzi SA, McCullumsmith R, Stec DE, Hinds TD Jr. RNA sequencing in human HepG2 hepatocytes reveals PPAR-α mediates transcriptome responsiveness of bilirubin. Physiol Genomics 51: 234–240, 2019. doi: 10.1152/physiolgenomics.00028.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, Hinds TD Jr. Bilirubin binding to PPARα inhibits lipid accumulation. PLoS One 11: e0153427, 2016. doi: 10.1371/journal.pone.0153427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Stec DE, Hinds TD Jr. Natural product heme oxygenase inducers as treatment for nonalcoholic fatty liver disease. Int J Mol Sci 21: 9493, 2020. doi: 10.3390/ijms21249493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kipp ZA, Xu M, Bates EA, Lee WH, Kern PA, Hinds TD Jr. Bilirubin levels are negatively correlated with adiposity in obese men and women, and its catabolized product, urobilin, is positively associated with insulin resistance. Antioxidants (Basel) 12: 170, 2023. doi: 10.3390/antiox12010170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Creeden JF, Gordon DM, Stec DE, Hinds TD Jr. Bilirubin as a metabolic hormone: the physiological relevance of low levels. Am J Physiol Endocrinol Metab 320: E191–E207, 2021. doi: 10.1152/ajpendo.00405.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hu J, Xu Y, He Z, Zhang H, Lian X, Zhu T, Liang C, Li J. Increased risk of cerebrovascular accident related to non-alcoholic fatty liver disease: a meta-analysis. Oncotarget 9: 2752–2760, 2018. doi: 10.18632/oncotarget.22755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stine JG, Niccum BA, Zimmet AN, Intagliata N, Caldwell SH, Argo CK, Northup PG. Increased risk of venous thromboembolism in hospitalized patients with cirrhosis due to non-alcoholic steatohepatitis. Clin Transl Gastroenterol 9: 140, 2018. doi: 10.1038/s41424-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J Hepatol 49: 608–612, 2008. doi: 10.1016/j.jhep.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 101. Hallsworth K, Hollingsworth KG, Thoma C, Jakovljevic D, MacGowan GA, Anstee QM, Taylor R, Day CP, Trenell MI. Cardiac structure and function are altered in adults with non-alcoholic fatty liver disease. J Hepatol 58: 757–762, 2013. doi: 10.1016/j.jhep.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 102. Anstee QM, Mantovani A, Tilg H, Targher G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 15: 425–439, 2018. doi: 10.1038/s41575-018-0010-0. [DOI] [PubMed] [Google Scholar]
- 103. Chiu LS, Pedley A, Massaro JM, Benjamin EJ, Mitchell GF, McManus DD, Aragam J, Vasan RS, Cheng S, Long MT. The association of non-alcoholic fatty liver disease and cardiac structure and function – Framingham Heart Study. Liver Int 40: 2445–2454, 2020. doi: 10.1111/liv.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. VanWagner LB, Wilcox JE, Ning H, Lewis CE, Carr JJ, Rinella ME, Shah SJ, Lima JAC, Lloyd-Jones DM. Longitudinal association of non-alcoholic fatty liver disease with changes in myocardial structure and function: the CARDIA Study. J Am Heart Assoc 9: e014279, 2020. doi: 10.1161/JAHA.119.014279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1 and S2 and Supplemental Material: https://doi.org/10.6084/m9.figshare.20982826.v1.
Data Availability Statement
Data will be made available upon reasonable request.