

Keywords: acute kidney injury, autophagy, chronic kidney disease, kidney inflammation, mitophagy
Abstract
Autophagy is a ubiquitous intracellular cytoprotective quality control program that maintains cellular homeostasis by recycling superfluous cytoplasmic components (lipid droplets, protein, or glycogen aggregates) and invading pathogens. Mitophagy is a selective form of autophagy that by recycling damaged mitochondrial material, which can extracellularly act as damage-associated molecular patterns, prevents their release. Autophagy and mitophagy are indispensable for the maintenance of kidney homeostasis and exert crucial functions during both physiological and disease conditions. Impaired autophagy and mitophagy can negatively impact the pathophysiological state and promote its progression. Autophagy helps in maintaining structural integrity of the kidney. Mitophagy-mediated mitochondrial quality control is explicitly critical for regulating cellular homeostasis in the kidney. Both autophagy and mitophagy attenuate inflammatory responses in the kidney. An accumulating body of evidence highlights that persistent kidney injury-induced oxidative stress can contribute to dysregulated autophagic and mitophagic responses and cell death. Autophagy and mitophagy also communicate with programmed cell death pathways (apoptosis and necroptosis) and play important roles in cell survival by preventing nutrient deprivation and regulating oxidative stress. Autophagy and mitophagy are activated in the kidney after acute injury. However, their aberrant hyperactivation can be deleterious and cause tissue damage. The findings on the functions of autophagy and mitophagy in various models of chronic kidney disease are heterogeneous and cell type- and context-specific dependent. In this review, we discuss the roles of autophagy and mitophagy in the kidney in regulating inflammatory responses and during various pathological manifestations.
INTRODUCTION
Autophagy is a highly dynamic and evolutionarily conserved lysosome-dependent homeostatic cellular program. Autophagy maintains intracellular quality control via recycling of damaged cellular components, including impaired organelles, protein aggregates, accumulated lipid droplets, and glycogen (1, 2). Autophagy is categorized into three main types: macroautophagy (also referred to as autophagy), microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy involves sequestration of cytosolic components into a double-membrane vesicle termed the autophagosome, which fuses with lysosomes for the degradation of superfluous cellular material. Microautophagy does not involve the formation of autophagosomes; instead, the cytosolic materials are directly sequestered into lysosomes by membrane invaginations (2). In CMA, heat shock cognate protein of 70 or 90 kDa (hsc70 or hsc90, respectively) recognizes proteins with KFERQ motifs, which are then identified by lysosome-associated membrane protein (LAMP)-2A on lysosomes and eliminated (3).
Belgian biochemist Christian de Duve first coined the term “autophagy,” which means “self-eating” in Greek. The molecular regulation of autophagy is executed by highly conserved autophagy-related (Atg) genes, which were first characterized in the yeast model by Dr. Yoshinori Ohsumi (4). Mammalian orthologs of ATG proteins have also been identified (5). Dr. Ohsumi was awarded the Nobel Prize in Physiology or Medicine in 2016 for exploring the mechanism of autophagy. The autophagy pathway has now gained significant attention due to its emerging roles during various pathological conditions (6, 7).
Basal autophagy maintains cellular homeostasis by eliminating aggregated cellular components. Basal autophagy occurs constitutively even under nutrient-rich conditions, and the mechanism of its activation has recently been shown to be different from stress-induced autophagy (8). Phosphatidylinositol 3-kinase (PI3K)-α-derived phosphatidylinositol 3-phosphate (PI3P) generated on late endosomes by inositol polyphosphate 4-phosphatase type II is essential for basal but not for starvation-induced autophagy (8). Autophagy is triggered by various stimuli such as energy starvation, hypoxia, oxidative stress, and proinflammatory cytokines including interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and damage-associated molecular patterns (DAMPs) (1, 9). Basal autophagic activities in the kidney ensure cellular homeostasis (10, 11). Loss of the basal autophagic response due to targeted deletion of Atg genes in nephrons (12), podocytes (13–15), tubules (16), or endothelial cells (17, 18) contributes to structural abnormalities in the kidney and a defective functional response. Mutation of Atg5 in the kidney epithelium resulted in severe tubulointerstitial disease with evidence of tubular injury, macrophage infiltration, myofibroblast accumulation, and tubulointerstitial fibrosis (12). Increased blood urea nitrogen (BUN) and plasma creatinine levels, hyperlipidemia, and hypoalbuminemia were observed by 4 mo of age, and these Atg5 mutant mice died of kidney failure from 5 mo of age onward (12). Podocyte-specific loss of Atg5 caused the development of albuminuria and segmental or complete glomerulosclerosis and foot process effacement (13). Furthermore, mice with podocyte-specific deletion of vacuolar protein sorting 34 (Vps34), which plays an essential role in the cellular process of autophagy, also developed albuminuria, increased BUN, and severe kidney lesions including podocyte vacuolization, proteinaceous cast formation, glomerulosclerosis, and interstitial fibrosis (14, 15). Kidney tubule cell-specific loss of Atg5 resulted in increased p62 accumulation, oxidative stress in tubular cells, and impaired kidney function (16). On the other hand, distal tubule-specific loss of Atg5 did not affect kidney function, suggesting the proximal tubule-specific importance of the basal autophagic response (16). Glomerular endothelial (17) and endothelial and hematopoietic (18) cell-specific Atg5-deficient mice displayed podocyte foot process effacement and loss of glomerular endothelial fenestrations. However, despite the structural abnormalities, there were no changes in BUN levels or the urine albumin-to-creatinine ratio (17, 18). The effects of loss of Atg5 in endothelial cells on kidney structural and functional changes were determined at 10–14 wk (17) and 4 wk (18) of age and may need confirmation at a later time point. In addition, a study by Lenoir et al. (17) highlighted that endothelium-specific Cre recombination was efficient but incomplete. Therefore, understanding endothelium-specific functions of autophagy is an area that warrants further investigation. Overall, the studies to date provide evidence of structural abnormalities in the kidney caused by loss of the Atg gene in different kidney cells and implicate the importance of basal autophagy in maintaining homeostasis in the kidney.
Activation of autophagy during stress conditions promotes cell survival by regulating oxidative stress via recycling damaged cellular components, as loss of function of the Atg gene favors apoptotic cell death (19). The removal of damaged parts of organelles such as the endoplasmic reticulum (ER) and peroxisomes are executed via ERphagy/reticulophagy and pexophagy, respectively, which also play important roles (20, 21) but are beyond the focus of this review. Here, we discuss the roles of autophagy and mitophagy in regulating kidney inflammation and in the pathogenesis of acute kidney injury (AKI) and chronic kidney disease (CKD). We will focus on the functions of autophagy and mitophagy in different models of AKI [sepsis, cisplatin, and ischemia-reperfusion injury (IRI)] and CKD [kidney fibrosis, diabetic nephropathy (DN), and podocytopathies].
AUTOPHAGY
Autophagy is a multiphasic sequential process, in which selective ATG proteins coordinate sequestration of cellular components to form autophagosomes and finally autophagolysosomes. This process begins with the formation of a phagophore, which is a double-membrane sequestering compartment, that involves initiation, nucleation (formation of the isolation membrane), elongation, closure, and maturation (1, 10, 22).
The initiation of autophagy begins with suppressive control of mechanistic target of rapamycin (mTOR) kinase complex 1 (mTORC1), which negatively regulates mammalian uncoordinated-51-like protein kinase 1 (ULK1) (22–24). ULK1, along with ATG13, ATG101, and focal adhesion kinase (FAK) family kinase-interacting protein of 200 kDa (FIP200), exists in a supramolecular complex. ULK1 drives the autophagy cascade by inducing PI3K activity of a macromolecular complex composed of beclin 1 (Vps30 in yeast), ATG14L (Atg14 in yeast), VPS15, VPS34, and PI3K regulatory subunit 4 (22–24). Ultraviolet radiation resistance-associated gene protein (UVRAG; Vps38 in yeast) can replace ATG14L and form an alternate complex with beclin 1. Autophagy beclin 1 regulator (AMBRA)-1 promotes, whereas B cell lymphoma 2 (BCL2, antiapoptotic) protein suppresses, activity of the beclin 1-ATG14L complex (22–24). Rubicon autophagy regulator (RUBCN) negatively regulates activity of the beclin 1-UVRAG complex (1). The formation of the isolation membrane involves binding of PI3P, which is generated by catalytic activity of VPS34, with WD repeat protein interacting with phosphoinositide (WIPI) and double FYVE domain-containing protein 1 (DFCP1) (22). ATG9 enables the recruitment of lipids to the nascent autophagosomal membrane (25). ATG5–ATG12 are conjugated via ATG7 (E1-like enzyme) and ATG10 (E2-like enzyme) and help in the elongation of the phagophore membrane. The membrane of the phagophore extends around the cellular components to be recycled and generates a double-membrane autophagosome (24). The endopeptidase ATG4B, by its microtubule-associated protein light chain 3 (LC3)-interacting region (LIR) motif, binds to LC3 (Atg8 in yeast), cleaves, and generates LC3-I (1, 10, 24). ATG7 and ATG10 assist with the conjugation of phospholipid phosphatidylethanolamine (PE) with unconjugated LC3-I to generate LC3-PE. LC3-PE binds with LIR-expressing autophagy substrate via adaptor protein p62/sequestosome 1 (SQSTM1) (1, 10, 22). The closed phagophore is termed the autophagosome, which ranges from 0.5 to 1.5 μm in mammals and from 0.4 to 0.9 μm in yeast (23). The autophagosome delivers its cargo for degradation by fusing with a lysosome (mammals) or vacuole (yeast) to form the autophagolysosome (mammals) or autophagic bodies (yeast), respectively (23). The cargo components are degraded by an acidic environment of the lysosome and hydrolases in the vacuole and are released back into the cytosol via lysosomal efflux permeases (22). The decrease in p62/SQSTM1 can serve as a biomarker for increased autophagy, as p62/SQSTM1 also gets degraded during the aforementioned process (22, 24). Autophagic flux is the relative rate of autophagy-mediated turnover of cargo material (1). Increased accumulation of p62 after chloroquine-mediated suppression of lysosomal activity can also serve as an indicator of increased autophagic flux (24).
MITOPHAGY
Mitochondria-specific autophagy or mitophagy is an autophagic response that selectively targets impaired, permeabilized, and cytotoxic mitochondria (26). The mitophagy pathways are executed in 1) phosphatase and tensin homolog (PTEN)-induced kinase1 (PINK1) and Parkin (E3 ubiquitin ligase, encoded by Prkn)-dependent or 2) PINK1/Parkin-independent manners. The regulators involved in PINK1/Parkin-independent mitophagy include: FUN14 domain-containing protein 1 (FUNDC1), BCL2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), 19 kDa interacting protein-3 (NIP3)-like protein X (NIX), cyclin G-associated kinase (GAK), and protein kinase C-δ (PRKCD) (26–28).
In a healthy cell, PINK1 (a serine threonine kinase) is recruited into the inner mitochondrial membrane (IMM) via translocase of the outer membrane (TOM) and translocase of the inner membrane 23 (TIM23) (26). Upon recruitment into the IMM, the mitochondrial targeting signal (MTS) of PINK1 is cleaved by matrix metalloprotease (MMP) (26). However, a depolarized mitochondrion with impaired membrane potential is unable to import PINK1, which, therefore, accumulates on the outer mitochondrial membrane (OMM). PINK1 then favors the phosphorylation of the mitochondrial fusion protein mitofusin (MFN)2, which executes an essential role in Parkin recruitment on the OMM (29, 30). Parkin ubiquitinates OMM proteins, including MFN1, MFN2, voltage-dependent anion channel (VDAC), and Miro (26, 29, 30). Ubiquitinated OMM proteins can interact with LC3 either with the help of (31) or without (32) adaptor protein p62/SQSTM1. In the case of PINK1/Parkin-independent mitophagy, LC3 directly binds with PINK1/Parkin-independent regulators of mitophagy on the OMM. The mitophagosome favors the recycling of damaged mitochondria.
Mitophagy-mediated elimination of impaired mitochondria, which exhibit low membrane potential, is closely linked to mitochondrial fusion and fission processes (26, 27). In a healthy cell, a balance between mitochondrial fusion and fission helps in maintaining the number of healthy mitochondria (26, 27, 29, 33). Mitochondrial fission promotes the segregation of damaged mitochondrial components and eliminates them via mitophagy (27, 29, 33). Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1) and fission, mitochondrial 1 (FIS1), whereas mitochondrial fusion is mediated by outer mitochondrial fusion proteins (MFN1 and MFN2) and an inner mitochondrial fusion protein [optic atrophy 1 (OPA1)] (33, 34). Mitophagy has been reported to be impaired in Mfn2- but not Mfn1-deficient states (29, 33, 34). We have confirmed a dysregulated mitophagic response in the absence of Mfn2 but not Mfn1 in macrophages by several methods: 1) electron microscopy (reduced mitophagosome formation), 2) flow cytometry (reduced colocalization of Mitotracker with Lysotracker and MtPhagy dye with Lyso dye), and 3) confocal microscopy (reduced colocalization of LC3 puncta with TIM23 and Mitotracker with Lysotracker) (34).
AUTOPHAGY AND MITOPHAGY IN THE KIDNEY
A body of emerging evidence implicates that autophagy and mitophagy in the kidney play critical physiological roles in the maintenance of homeostasis and normal kidney function (Table 1). Dysregulated autophagy leads to the development of AKI and CKD. Kidneys have the second highest number of mitochondria after the heart; therefore, a mitophagy-dependent mitochondrial quality control program is particularly essential for cellular homeostasis and regulated functioning. The recycling of damaged mitochondrial components by mitophagy prevents oxidative stress-mediated kidney damage, and studies have suggested the cytoprotective role of mitophagy mediators during AKI and CKD (27, 34, 45, 55). Here, we focus our review on recent advances in our understanding of autophagy and mitophagy in regulating kidney inflammation and in the pathogenesis of AKI and CKD.
Table 1.
Kidney disease phenotype after genetic ablation of autophagy/mitophagy-related genes in models of AKI and CKD
| Setting | Targeted Pathway | Model | Targeted Gene | Targeted Tissue/Cell | Kidney Phenotypical Changes | Reference |
|---|---|---|---|---|---|---|
| AKI | Autophagy | Sepsis: LPS | Atg7 | Proximal tubules | Exacerbated proximal tubular cell injury and worsening kidney function | 35 |
| Atg7 | Proximal tubules | Tubular cell apoptosis and impaired kidney function | 36 | |||
| Atg5 | Podocytes | Increased transient albuminuria | 13 | |||
| Mitophagy | Sepsis: CLP, LPS | Pink1 or Prkn | Global | Increased mitochondrial fragmentation, tubular damage, apoptosis, serum creatinine, and BUN | 37 | |
| Autophagy | Cisplatin | Atg5 | Proximal tubules | Increased DNA damage, apoptosis, oxidative stress, number of protein aggregates, tissue damage, and impaired kidney function | 38 | |
| Atg7 | Proximal tubules | Exacerbated cell death, tissue damage, and impaired kidney function | 39 | |||
| Mitophagy | Cisplatin | Pink1 or Prkn | Global | Increased tubular injury, apoptosis, serum creatinine, and BUN | 40 | |
| Pink1 | Global | Reduced DRP1, tubular apoptosis, and kidney injury | 41 | |||
| Autophagy | IRI | Atg5 | Tubules | Increased tubular injury, apoptosis, and serum creatinine | 16 | |
| Atg5 | Proximal tubules | Increased number of abnormal mitochondria, tubular cell death, kidney damage, serum creatinine, and BUN | 42 | |||
| Atg7 | Proximal tubules | Increased serum creatinine and BUN | 39 | |||
| Atg7 | Tubules | Reduced FGF2 expression and fibrotic responses | 43 | |||
| Atg5 | Tubules | Exacerbated kidney injury and tubulointerstitial fibrosis | 44 | |||
| Atg5 | Distal tubules | Increased fibrotic responses | 44 | |||
| Becn1 | Tubules | Exacerbated tissue damage, inflammation, tubulointerstitial fibrosis, and impaired kidney function | 44 | |||
| Becn1 | Distal tubules | Worsening kidney function | 44 | |||
| Mitophagy | IRI | Pink1 or Prkn | Global | Exacerbated oxidative stress, inflammation, apoptosis, and mitochondrial damage in proximal tubules | 45 | |
| Mfn2 | Proximal tubules | Increased cell survival and improved kidney function | 46 | |||
| CKD | Autophagy | Kidney fibrosis/UUO | Map1lc3a | Global | Increased TGF-β1 expression, collagen accumulation, and kidney fibrosis | 47 |
| Atg5 | Proximal tubules | Cell cycle arrested at the G2/M phase and increased tubulointerstitial fibrosis | 48 | |||
| Atg5 | Proximal tubules | Increased inflammation, NF-κB activation, infiltration of macrophages and T cells in the kidney | 49 | |||
| Atg7 | Distal tubules | Increased accumulation of abnormal mitochondria, apoptotic markers, NLRP3 inflammasome formation, and kidney fibrosis | 50 | |||
| Atg7 | Proximal tubules | Reduced tubular atrophy, nephron loss, macrophage infiltration, and kidney fibrosis | 51 | |||
| Becn1 | Global | Increased TGF-β1 expression and collagen accumulation | 47 | |||
| Becn1 | Tubules | Exacerbated collagen deposition and kidney fibrosis. | 44 | |||
| Mitophagy | Kidney fibrosis/UUO or adenine diet | Pink1 or Prkn | Global | Increased kidney macrophage-derived mROS, tubulointerstitial fibrosis, and impaired kidney function | 30 | |
| Mfn2 | Myeloid lineage | Impaired macrophage mitochondrial biogenesis, increased macrophage-derived fibrotic responses, kidney fibrosis, serum creatinine, and BUN | 34 | |||
| Autophagy | DN/Akita mice | Atg7 | Proximal tubules | Increased tubular hypertrophy, macrophage infiltration, and kidney fibrosis | 52 | |
| DN/high-fat diet | Atg5 | Podocytes | Loss of podocytes and massive albuminuria | 53 | ||
| DN/high glucose | Atg5 | Podocytes | Podocyte apoptosis, leaky GFB, and glomerulosclerosis | 17 | ||
| DN/STZ | Atg5 | Endothelial cells | Capillary rarefactions, loss of fenestrations, and glomerular endothelial lesions | 17 | ||
| Autophagy | Podocytopathies/PAN, adriamycin | Atg5 | Podocytes | Increased accumulation of oxidized proteins linked to proteinuria, loss of podocytes, and glomerulosclerosis | 13 | |
| Podocytopathies/ unilateral nephrectomy | Atg7 | Podocytes | ER stress, cytoskeletal destabilization, foot process effacement, and albuminuria | 54 |
AKI, acute kidney injury; atg, autophagy-related; Becn1, beclin 1; BUN, blood urea nitrogen; CKD, chronic kidney disease; CLP, cecal ligation and puncture; DN, diabetic nephropathy; DRP1, dynamin-related protein 1; ER stress, endoplasmic reticulum-derived stress; FGF2, fibroblast growth factor 2; GFB, glomerular filtration barrier; IRI, ischemia-reperfusion injury; LPS, lipopolysaccharides; Map1lc3a, microtubule-associated protein 1 light chain 3α (LC3A); mROS, mitochondria-derived reactive oxygen species; PAN, puromycin aminonucleoside; Pink1, phosphatase and tensin homolog-induced kinase 1; Prkn, Parkin; STZ, streptozotocin; TGF-β1, transforming growth factor-β1; UUO, unilateral ureteral obstruction.
AUTOPHAGY, MITOPHAGY, AND KIDNEY INFLAMMATION
Autophagy and mitophagy execute a central role in preventing inflammatory events and in controlling both innate and adaptive immune responses (56–59). Inflammation plays a fundamental role in causing structural and functional damage during AKI (60). Unresolved inflammation acts as a driver in the pathophysiological transition from AKI to CKD. The failure of repair due to excessive inflammation-mediated tissue damage leads to kidney fibrosis (59, 60). The autoamplification loop between kidney injury and inflammation is known as necroinflammation. It promotes kidney injury directly via immune cell recruitment or indirectly through proinflammatory cytokines (56, 61). In the following subsections, we discuss studies on the roles of autophagy and mitophagy in regulating inflammatory events in the kidney.
Innate and Adaptive Immune Responses and Autophagy
Cells of both innate (59, 60) and adaptive (62) immune systems exhibit crucial roles in influencing the inflammatory microenvironment in the kidney. Pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), after binding with DAMPs, activate inflammatory signaling in the kidney (63, 64). Hyperactivated PRR signaling-mediated chronic inflammation promotes the progression of kidney diseases via chemokine-dependent infiltration of immune cells, including neutrophils, macrophages, and dendritic cells (63–65). A direct link between PRR signaling and autophagy has been reported (66). Lipopolysaccharide (LPS)-mediated activation of TLR4 induces autophagy in macrophages via myeloid differentiation factor 88 (Myd88) and the Toll/interleukin-1 receptor (TIR) domain-containing adaptor-inducing IFN-β (TRIF)-p38 axis (67). TLR2−TLR4 have been shown to enhance the autophagy-dependent phagocytic response by macrophages (68). LC3-associated phagocytosis (LAP) is the noncanonical form of autophagy, which involves conjugation of LC3 with the phagosome membrane. LAP has also been reported to prevent systemic inflammation (69). Activation of TLR1, TLR3, TLR5, TLR6, and TLR7 has been shown to stimulate autophagy. The interaction between adaptor protein of the TLR signaling pathway with beclin 1 activates autophagy by disrupting the beclin 1 interaction with BCL2 (Fig. 1) (66, 67). The reduced mRNA expression of Atg genes such as Map1lc3a (which encodes LC3) and Atg5 in the kidney from Tlr2 genetically deleted mice supports the role of TLR2 signaling in inducing the autophagic response (70). TLR2-mediated activation of autophagic response has been shown to attenuate both worsening kidney function and tissue injury in experimental AKI (70).
Figure 1.

Autophagy and mitophagy in immunity and inflammation. Autophagy is activated via the induction of Toll-like receptor (TLR) signaling. TLR binds to the damage-associated molecular pattern (DAMP) and adaptor proteins: myeloid differentiation primary response 88 (MyD88) and Toll-interleukin-1 receptor domain-containing adapter inducing interferon (IFN)-γ (TRIF) activate downstream signaling pathways. MyD88 and TRIF stimulate autophagy by suppressing the interaction between beclin 1 and B cell lymphoma 2 (BCL2) proteins. Autophagy involves engagement of phosphatidylinositol 3-phosphate (PI3P)-binding protein of WD-repeat protein interacting with phosphoinositide (WIPI) and double FYVE domain-containing protein 1 (DFCP1) with PI3P and phagophore formation. Autophagy-related genes (ATG5–ATG12) are conjugated by ATG7 and ATG10. ATG4B via its microtubule-associated protein light chain 3 (LC3)-interacting region (LIR) motif binds to LC3, cleaves, and generates LC3-I. ATG7 and ATG10 help in the conjugation of phospholipid phosphatidylethanolamine (PE) with unconjugated LC3-I and generate LC3-PE. LC3-PE binds with LIR-expressing autophagy substrate via p62/sequestosome1 (SQSTM1). The closed phagophore/autophagosome fuses with a lysosome to generate an autophagolysosome, which degrades cargo components. TLR signaling also activates receptor-interacting protein kinase (RIPK)1, resulting in RIPK3-mediated phosphorylation of mixed-lineage kinase domain-like pseudokinase (MLKL) and necroptosis. Autophagy inhibits necroptosis. Autophagy processes antigens and also participates in modulating T cell effector functions via major histocompatibility complex class II (MHC II)-dependent antigen presentation to the T cell receptor (TCR). Mitophagy involves the formation of a mitophagosome. Phosphatase and tensin homolog (PTEN)-induced kinase1 (PINK1) phosphorylates mitofusin (MFN)2, which recruits Parkin to the damaged outer mitochondrial membrane (OMM). Parkin polyubiquitinates (Ub) OMM proteins, including MFN1, MFN2, MIRO, and voltage-dependent anion channel (VDAC), which then bind with LC3-PE via p62. In PINK1-independent mitophagy, LC3-PE directly binds with OMM proteins: NIP3-like protein X (NIX), BCL2 adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), and FUN14 domain-containing 1 (FUNDC1), through LIR. Mitophagy by inhibiting the release of mitochondrial DAMPs (mtDAMPs), including mitochondria-derived reactive oxygen species (mROS), mitochondrial DNA (mtDNA), and cytochrome c (Cyt C), suppresses the production of type I IFNs and inflammatory cytokines. TLR signaling also activates the NF-κB signaling pathway, through tumor necrosis factor receptor-associated factor 6 (TRAF6), production of inflammatory cytokines, and formation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome. Autophagy inhibits NLRP3 inflammasome formation. The mitophagy mediator PINK1 also facilitates the loading of mtDAMPs to the late endosome. Endosome-dependent release of mtDAMPs prevents neutrophil chemotaxis.
Autophagy, in addition to recycling the cellular components, is also believed to facilitate the loading of the processed antigen to major histocompatibility complex (MHC) class II (71). MHC class I is expressed by all nucleated cells. However, MHC class II is expressed chiefly by professional antigen-presenting cells (APCs), such as macrophages, dendritic cells, and B cells. NOD2-dependent stimulation of the autophagic response in human dendritic cells also promotes antigen presentation (72). Professional APCs are known to activate effector T cell functions by MHC class II-mediated antigen presentation (73). Nonimmune cells of the kidney, including tubular epithelial cells, podocytes, endothelial cells, and mesangial cells, have also been reported to express MHC class II (74–76). These resident kidney nonimmune cells can behave as nonconventional MHC class II-expressing amateur APCs (77). In the experimental ovalbumin-induced model of nephritis, mouse podocytes act as APCs (77). These podocytes have been reported to promote T cell-derived production of IFN-γ and interleukin (IL)-17 by presenting processed ovalbumin antigen to T cells (77). Autophagy-dependent antigen processing and presentation by MHC class II via nonimmune kidney resident cells can regulate adaptive immune responses in the kidney via controlling T cell effector functions (56). The impaired autophagic response contributes to defective antigen processing, which results in a dysregulated adaptive immune response.
Cell Death and Autophagy
Autophagy plays a cardinal role in deciding the fate of a cell during nutrient-deprived or stress conditions and also prevents a cell from undergoing apoptotic (programmed)-, necrotic-, and necroptotic (programmed necrosis)-mediated cell deaths (78). Macrophage-derived production of IL-1β and IL-18 increased after the loss of Atg16-like 1 (Atg16l1) in hematopoietic cells (79). IL-1β and IL-18 are potent proinflammatory cytokines that favor NLR family pyrin domain containing 3 (NLRP3) inflammasome formation. NLRP3 inflammasome activation is known to promote different types of cell death pathways, including apoptosis, necroptosis, and pyroptosis (80). Deficiency of autophagy regulators (LC3 and beclin 1) is also directly linked to an increase in caspase 1-mediated production of inflammatory cytokines and accumulation of damaged mitochondria in the cytoplasm of macrophages (81). Caspase 1 favors pyroptosis, the inflammatory form of programmed cell death. Autophagy by targeting proapoptotic members of the BCL family prevents TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis (82). Excessive autophagy can also cause apoptotic (82)- or necroptotic (83)-dependent cell death. The chances of cell survival with autophagy are highest, followed by apoptosis and necroptosis, whereas a cell undergoing necrosis has the lowest chance of survival (78). BCL2 family of proteins that regulate apoptosis by promoting mitochondrial outer membrane permeabilization (MOMP) are known to suppress both autophagy (84) and PINK1/Parkin-dependent mitophagy (85).
A defective autophagic response favors both receptor-interacting protein kinase (RIPK)1-dependent and -independent forms of death in macrophages (86). Necroptosis, which is induced by RIPK1, requires RIPK3-dependent phosphorylation of mixed-lineage kinase domain-like pseudokinase (MLKL) (87). Autophagy prevents necroptosis, as impaired autophagic activity contributes to an increase in necroptosis via hyperactivated TNF and TLR signaling (86). In a sepsis model of AKI, our group has observed that RIPK3 induces mitochondria-derived reactive oxygen species (mROS) production and reduces mitochondrial complexes I and III in kidney tubular cells (88). RIPK3/MLKL signaling also promotes NLRP3 inflammasome formation (87, 89). Autophagy is known to prevent NLRP3-induced inflammation by preserving mitochondrial integrity (81).
Cell Death, Mitochondrial DAMPs, and Mitophagy
The failure of recycling of damaged mitochondrial components or mitochondrial DAMPs (mtDAMPs), including mitochondrial DNA (mtDNA), transcription factor A, mitochondrial (TFAM), and mROS can cause cell death (90–93). Mitophagy, by recycling mtDAMPs, helps in negating inflammatory signals and ensuring cell survival (33, 91–94). Mitophagy is specifically crucial for kidney tubular cells, which are rich in mitochondria. Mitochondria exhibit crucial roles during both apoptotic and necrotic forms of regulated cell death, which are driven by MOMP and mitochondrial permeability transition (MPT) (91). MOMP involves the release of mtDAMPs, including cytochrome c (Cyt C) into the cytosol, which promotes apoptosis (91). Both MOMP and MPT cause mitochondrial depolarization. mtDNA, which is released from damaged mitochondria, promotes inflammation. Naked mtDNA has been shown to activate endocrine, autocrine, or paracrine signaling to exhibit immunostimulatory effects chiefly by functioning as a TLR9 agonist (90–92). Necroptosis-mediated rupture of damaged cells results in the extracellular release of cellular components, including mtDNA (87, 91–93). mtDNA in the extracellular environment acts as DAMPs to promote tubulointerstitial inflammation and kidney injury through various pathways (90, 95–97). These pathways include NLRP3 inflammasome formation (98), the cyclic guanosine monophosphate-adenosine monophosphate (cGMP-AMP) synthase (cGAS) stimulator of IFN genes (STING) pathway (99), and TLR9-mediated activation of Myd88 and NF-κB signaling (100, 101). The increase in urinary mtDNA is directly related to the progression of disease severity in patients with AKI and DN (102–104). However, a major limitation of this study by Whitaker et al. (102) is that the progression of AKI was determined based on serum creatinine levels, which may have underestimated kidney dysfunction at baseline and hence affect the predictive value of urinary mtDNA for AKI progression. In patients with DN, urinary mtDNA correlated inversely with the estimated glomerular filtration rate (eGFR) and positively with interstitial fibrosis, suggesting that mitophagy is impaired in the kidney (104). The leakage of mtDNA from dying kidney parenchymal cells and cell-free circulating mtDNA (which can be filtered via kidneys) potentially contribute toward an increase in urinary mtDNA levels. In line with this, a previously published study reported reduced copy numbers of circulating mtDNA in patients with CKD and a low circulating copy number as a positive predictor of mortality (105). Interestingly, a recent study by He et al. (106) observed that reduced circulating mtDNA was associated with an increased risk of CKD progression, and there was no association of mtDNA copy number with mortality. The seemingly contradictory findings may be due to various factors that can influence the reporting of mtDNA copy numbers, including methodology (cell free vs. cellular), target mitochondrial gene, and patient cohorts (94). Cell-free mtDNA acts as DAMPs, but intracellular naked mtDNA, through activating endosomal TLR9-mediated signaling, can also promote proinflammatory cytokine production via stimulating the NF-κB transcription factor (91–93). In an experimental model of sepsis, release of mtDNA-containing vesicles, but not naked mtDNA, by LPS-treated human monocytes has been reported to attenuate inflammation by preventing neutrophil chemotaxis (107). The mitophagy regulator PINK1, upon sensing mitochondrial damage, interacts with late endosomes. PINK1 helps in loading mtDNA into endosomal vesicles and communicating stress signals in a paracrine manner via TLR9-mediated signaling (94, 108). Mitophagy has also been reported to potentially suppress packaging of the damaged mitochondrial components into the mitochondria-derived vesicle (MDV) and regulate inflammatory events (91).
We have also reported that the ablation of mitophagy regulators (Pink1 or Prkn) negatively impacts macrophage mitochondrial respiration and membrane potential (30). We observed increased oxidative stress and inflammatory responses in the kidney after the loss of mitophagy regulators in experimental models of CKD (30). Mitochondrial depolarization can cause cell death by disrupting cellular redox potential (33). In the adenine model of CKD, we have also reported that mitophagy in Mfn2 (but not Mfn1)-deficient myeloid cells was downregulated (34). The decrease in mitophagy in Mfn2-deficient kidney macrophages was associated with increases in mROS production, mitochondrial swelling, inflammatory, and fibrotic responses (34). Increased macrophage infiltration in the kidney after myeloid-specific Mfn2 ablation was also associated with augmented urinary monocyte chemoattractant protein (MCP)-1 levels.
Mitophagy is also crucial for the regulated functioning of kidney parenchymal cells, specifically tubular cells, which also exhibit a central role in attenuating local and systemic immune responses after kidney injury (27, 33, 45, 55, 109, 110). Proximal tubules, which are metabolically highly active, exhibit higher basal levels of mitophagy than other cells in the kidney (111–113). Mitophagy induction in proximal tubular cells can help in reducing local and systemic inflammatory responses by preventing the extracellular release of mtDNA and its availability for binding to PRRs (27, 33, 63). Activation of PRRs signaling also favors NLRP3 inflammasome formation, which contributes to worsening of the progression of AKI and CKD (63).
The aforementioned studies highlight the role of autophagy and mitophagy in regulating cellular homeostasis in the kidney, potentially by attenuating immune and nonimmune cell-derived inflammatory responses. Further research is still needed to fully understand the effects of immune or kidney resident nonimmune cell-specific loss of autophagy/mitophagy on innate and adaptive immune signals in the kidney during AKI and CKD.
AUTOPHAGY AND MITOPHAGY IN AKI
Regulated autophagic (60, 114) and mitophagic (45, 55) responses exhibit crucial roles in attenuating oxidative stress and inflammation-mediated tissue damage, which are commonly reported manifestations of AKI (115). Studies targeting autophagy (38, 39, 42, 44, 116–120) and mitophagy (37, 121, 122), using genetic (knockout/knockin) and pharmacological (inhibition/activation) approaches, have demonstrated their protective functions against AKI. However, suppression of autophagic flux (123) or mitophagy regulators (41, 46) have also been shown to prevent apoptosis and improve kidney function during AKI. This suggests that hyperactivated autophagy/mitophagy can be deleterious. In the following subsections, we discuss studies that focused on the roles of autophagy and mitophagy in various models of AKI (Fig. 2).
Figure 2.

Autophagy and mitophagy functions in acute kidney injury (AKI). Shown is a schematic presentation illustrating the roles of autophagy and mitophagy in various models of AKI: sepsis, cisplatin, and ischemia-reperfusion-injury (IRI)-mediated AKI. Autophagy and mitophagy protect against inflammation, tubular cell apoptosis, and kidney damage during AKI. However, an excessive autophagic response has been reported to promote inflammation, tissue injury, and AKI to CKD transition via fibroblast activation. cGAS, cyclic guanosine monophosphate-adenosine monophosphate synthase; FGF2, fibroblast growth factor 2; IFN-γ, interferon-γ; mROS, mitochondria-derived reactive oxygen species; mtDNA, mitochondrial DNA; NLRP3, NLR family pyrin domain containing 3; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; SIRT, sirtuin; STING, stimulator of interferon genes; TFEB, transcription factor EB; 3MA, 3-methyladenine; TSP1, thrombospondin-1.
Autophagy in Sepsis-Induced AKI
Sepsis is an infection-mediated systemic inflammatory response with an outcome of multiorgan failure and is the leading cause of mortality in patients with AKI admitted to the intensive care unit (124). Cecal ligation puncture (CLP) and LPS injection are two widely used experimental models of sepsis that mimic multiorgan damage and inflammatory responses (125). Expression of the autophagy regulatory protein LC3 was transiently induced in proximal tubular cells 3 h after CLP but declined at 9–18 h of CLP-mediated sepsis (116, 126). Similarly, LC3-II expression in the kidney initially increased at 12 or 24 h but decreased to the basal level at 48 h after LPS injection (36, 127). The decrease in the autophagic response with increased duration of CLP-induced sepsis was associated with increases in the disruption of brush borders of tubular epithelial cells and apoptosis (128). Activation of autophagy using rapamycin improved, whereas its inhibition using 3-methyladenine (3MA) or chloroquine worsened, CLP-induced kidney injury (116). The tumor suppressor gene transformation-related protein 53 (Trp53, also known as p53) also emerged as a critical regulator of autophagy during AKI (116, 129–131). Stress-responsive histone deacetylase sirtuin (SIRT)1-dependent deacetylation of p53 attenuated sepsis-induced kidney damage via activating the autophagic response (116). SIRT6, by stimulating autophagy, also protected against LPS-induced increases in proinflammatory cytokines (TNF-α and IL-6) and apoptosis in human kidney (HK)-2 tubular cells (127). AMP kinase, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), or temsirolimus (mTOR inhibitor)-mediated induction of autophagy improved kidney function after CLP or LPS injection (130). These studies suggested that chemical-mediated induction of the autophagic response attenuated CLP or LPS-induced increases in serum cystatin C and BUN levels. Autophagy decreased after compound C-mediated suppression of AMP-activated protein kinase (AMPK) (131). The decrease in autophagy contributed to increases in circulating proinflammatory cytokine (IL-6) and apoptotic cell death in the kidney after CLP (131). Autophagy suppression using proximal tubule-specific genetic ablation of Atg7 or pharmacologically using chloroquine (at the autolysosomal level) accelerated LPS-induced increases in BUN and apoptosis (36). Induction of autophagy using ursolic acid, a natural plant-derived antioxidant, in macrophages also prevented LPS-induced activation of TLR4 signaling and the inflammatory response (132). TLRs are among the widely studied PRRs in the kidney, as TLR1–TLR5, TLR7–TLR9, and TLR11 have been reported to be expressed in the kidney (63). TLR4 acts as a sensor for LPS, and it is expressed in parenchymal cells of both mouse and human kidneys (133, 134). TLR4, upon binding with LPS, activates autophagy in macrophages, through its signal adaptor proteins MyD88 and TRIF, by disrupting the interaction between beclin 1 and BCL2 (67). Moreover, Atg7-deficient kidney tubular epithelial cells displayed increased LPS-induced production of proinflammatory (IL-6) cytokine (35). These findings highlight the cytoprotective role of autophagy against sepsis-induced cellular stress in the kidney, by attenuating inflammatory responses and regulating kidney function.
Mitophagy in Sepsis-Induced AKI
Mitophagy in different models of sepsis was assessed by colocalization of LC3 puncta with Cyt C oxidase subunit IV (COX IV; a mitochondrial marker) or via expression of its regulators (PINK1 and Parkin) in the kidney. These mitophagy markers are transiently increased at 4 and 8 h but start to decline at 12 h and are further reduced at 24 h after CLP (135, 136). An impaired mitophagic response at 24 h of CLP is associated with increases in NLRP3 expression and caspase-mediated cell death (135). Genetic ablation of mitophagy regulators (Pink1 or Prkn) resulted in increased CLP- or LPS-induced tubular cell apoptosis, tubulointerstitial damage, serum creatinine, and BUN (37). Polydatin, a plant-derived component, has also been shown to alleviate CLP-induced NLRP3 inflammasome activation in the kidney and tissue injury, by inducing Parkin expression, through activating SIRT1 (136). Increased expression of mitophagy regulators after ascorbic acid treatment also exerts protection against LPS-mediated tubular apoptosis (137). These studies indicate that mitophagy helps in attenuating sepsis-induced tubulointerstitial inflammation and apoptosis in the kidney.
Autophagy in Cisplatin-Mediated AKI
Cisplatin causes injury to the S3 segment of proximal tubules (10) and nephrotoxicity by promoting apoptotic and necrotic cell death (138, 139). Cisplatin also activates autophagy, which, via attenuating caspase-dependent apoptotic cell death, protects against cisplatin-mediated kidney damage (118, 140). Cisplatin treatment promoted cleavage and degradation of autophagy regulatory proteins (beclin 1, ATG5, and ATG12) in kidney tubular epithelial cells (119). Decreased mRNA expression of autophagy regulators in mouse kidneys and cultured tubular cells after cisplatin treatments has also been reported (70). T cell and neutrophil-derived IFN-γ activates lysosomal cathepsin D, which promotes autophagic flux (141). The IFN-γ-mediated activation of autophagic flux prevents cisplatin-induced increases in BUN and serum creatinine in experimental mice and apoptotic cell death in proximal tubular cells (141). Proximal tubule-specific deletions of Atg5 (38) or Atg7 (39) are associated with increases in apoptosis and accumulation of damaged mitochondria, which exhibit increased mROS production in the proximal tubules. Autophagy suppression via knockdown of Atg5 or Becn1 (which encodes beclin 1) or chemically (3MA or bafilomycin A1) promoted caspase-dependent cell death in cisplatin-treated pig epithelial cells (117) and rat proximal tubular cells (118). Blockade of autophagic flux using chloroquine (lysosomotropic agent) further increases BUN and serum creatinine levels after cisplatin treatment (119). Overexpression of autophagy regulators, Atg5 and Becn1, protected against cisplatin-induced caspase-mediated tubular cell death (119). Inhibition of the protein kinase C-δ-activated autophagic response prevented cisplatin-mediated nephrotoxicity via inhibiting the mTOR signaling pathway (142). TLR2, but not TLR4, has been reported to activate autophagy after cisplatin treatment via the PI3K/AKT signaling pathway (70, 143). Lower mRNA expression of Atg genes (Map1lc3a and Atg5) in the kidney from cisplatin-treated Tlr2 deleted than wild-type or Tlr4-deficient mice was associated with higher tissue damage, BUN, and serum creatinine levels (70). Cisplatin-mediated activation of autophagy acts as a counteractive mechanism to attenuate TLR-mediated NF-kB-dependent induction of the inflammatory response. Becn1 haploinsufficiency accelerated, whereas enhanced Becn1 activity (gain of function mutant F121A Becn1 by knockin) reduced cisplatin-mediated increases in BUN, plasma creatinine, and kidney fibrosis (44). These findings indicate the protective functions of autophagy regulatory proteins against cisplatin-induced kidney damage. The therapeutic potential of autophagy-dependent and -independent functions of beclin 1 against the progression of AKI to CKD needs further confirmation (144). Taken together, the aforementioned findings suggest that autophagy exhibits protective functions and helps in maintaining cellular homeostasis in the kidney after cisplatin-mediated AKI to mitigate the AKI to CKD transition.
Mitophagy in Cisplatin-Mediated AKI
Cisplatin causes mitochondrial damage, which results in the loss of mitochondrial membrane potential and an increase in mROS production, and it also represses mitophagy in the kidney (40, 145, 146). Mitophagy has been shown to play a protective role against cisplatin-mediated kidney tubular injury (121). The reactivation of mitophagy and lysosomal biogenesis after peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) overexpression attenuated cisplatin-induced kidney injury (145). PGC-1α promotes lysosome biogenesis in kidney tubular cells via activating transcription factor EB (TFEB) (145). The increased expression of mitophagy regulators (PINK1 and Parkin) in the kidney after cisplatin treatment has also been reported. Studies have supported the cytoprotective functions of mitophagy against cisplatin-mediated nephrotoxicity (40, 121). Genetic ablation of mitophagy regulators (Pink1 or Prkn) resulted in worsening kidney function, increases in apoptosis, and kidney damage after cisplatin treatment (40). However, Zhou et al. (41) showed that plasma creatinine levels and tubular injury in cisplatin-treated rats were reduced after Pink1 ablation. This study also reported reduced expression of DRP1 in the kidneys of cisplatin-treated Pink1-deficient mice, suggesting that the protection against cisplatin-mediated AKI is partly mediated by suppressed mitochondrial fission. Knockdown of DRP1 in HK-2 cells resulted in reduced apoptosis and mitochondrial damage after cisplatin treatment (121). Release of mtDNA after cisplatin-induced apoptosis has been shown to accelerate necroinflammation in the kidney, through activating the innate immune response, via the cGAS STING pathway (64). Considering the contradictory reported findings, further studies are warranted to confirm if induction/suppression of mitophagy during cisplatin-induced AKI exhibits protection/damage.
Autophagy in IRI-Induced AKI
Kidney injury due to ischemia or interruption of blood flow and reperfusion or reoxygenation is unavoidable in patients undergoing kidney transplantation and can delay graft function (147). IRI involves immune cell infiltration, accumulation of damaged mitochondria, tubular cell apoptosis, and tissue damage in the kidney (16, 148). Studies have demonstrated that the expression of autophagy regulators (LC3-II and beclin 1) and autophagosome formation increased in the kidney after IRI and in cultured proximal tubular epithelial cells after hypoxia (44, 149, 150). Suzuki et al. (149) also observed increases in autophagosomes in human kidney transplant biopsy specimens. Hypoxia-mediated induction of autophagy promotes cell survival by recycling cellular damaged components; however, an excessive autophagic response can exaggerate kidney injury by type II programmed cell death (149). IRI-mediated tubular cell apoptosis and worsening of kidney function increased after suppression of autophagy pharmacologically (3MA or chloroquine) (44). Hypoxia-induced caspase activation in proximal tubular cells also increased after knockdown of Becn1 or Atg5 (44). However, inhibition of autophagic flux using bafilomycin A1 attenuated cold-ischemia-induced caspase 3 activation and apoptosis in the kidney (123). Proximal tubule-specific ablation of Atg5 resulted in increased IRI-induced accumulation of abnormal mitochondria, levels of BUN, serum creatinine, and kidney damage (42). Caspase 3-dependent apoptosis, kidney injury, increases in BUN, and serum creatinine after IRI were reduced after rapamycin-mediated autophagy stimulation (151). However, 3MA-dependent inhibition of autophagy aggravated the aforementioned changes (151). Suppression of autophagic activity in the kidney after IRI has also been reported. Preconditioning with calorie restriction-dependent induction of autophagy attenuated IRI in the kidney, through abrogating IRI-induced decreases in renal endothelial nitric oxide synthase (eNOS) and PGC-1α expression (152). Blockade of CD47, a ubiquitously expressed cell surface receptor for thrombospondin-1 (TSP1), has been linked to the induction of autophagy and attenuation of cell death (153). Increased expression of ATG5, ATG7, beclin 1, and LC3 in the kidney from CD47-deficient mice was also associated with an improvement in kidney function and reduction of kidney damage after IRI (154). Heterozygous pan-kidney tubular Becn1-deficient mice displayed worse kidney injury than homozygous distal tubular Becn1-null mice 1 day after IRI (44). This underscores the proximal tubule-specific protective functions of beclin 1 against IRI. Heterozygous and homozygous deficiencies of Becn1, respectively, in pan-kidney tubular and distal tubular cells also contributed to an increase in IRI-mediated tissue damage by 14 days (44). In addition, injection of beclin 1 peptide both before and after IRI attenuated kidney damage and fibrotic responses (44). These studies have suggested a therapeutic potential of beclin 1 against AKI to CKD progression (144). However, recently, Livingston et al. (43) reported that tubular cell-specific targeted deletion of Atg7 diminished tubular cell-derived fibroblast growth factor 2 (FGF2)-mediated stimulation of fibroblasts and fibrosis after IRI. Their findings suggest that the persistent activation of autophagy exhibited deleterious effects and promoted fibrosis and IRI-mediated AKI to CKD transition. The preponderance of evidence support that autophagy exerts cytoprotective functions during IRI. However, findings also confirmed that persistent stimulation of autophagy can be deleterious and promote tubular cell apoptosis and progression of IRI-mediated AKI.
Mitophagy in IRI-Induced AKI
Mitophagy in proximal tubules has been reported to be induced after IRI (45). Hypoxia has been shown to also promote the mitophagic response (155). Tang et al. (45) observed that IRI-induced oxidative stress, inflammatory responses, apoptosis, and accumulation of damaged mitochondria in proximal tubules increased after ablation of the mitophagy regulators Pink1 or Prkn. Expression of BNIP3, a regulator of PINK1/Parkin-independent mitophagy, increased in kidney tubules following IRI and in cultured proximal tubular cells after oxygen deprivation (122). IRI-mediated inflammation, apoptosis, and accumulation of damaged mitochondria in the kidney also increased after genetic deletion of Bnip3 (122). MFN2 is known to play an important role in the regulation of PINK1/Parkin-dependent mitophagy (29, 30, 34). Gall et al. (156) observed that kidney-specific Mfn2 deficiency resulted in mitochondrial fragmentation in proximal tubules. However, they also reported that conditional deletion of Mfn2 in proximal tubular epithelial cells resulted in increased rat sarcoma virus (Ras)-extracellular signal-regulated kinase (ERK) (Ras-ERK) pathway-dependent cell survival and decreased BUN and serum creatinine after IRI (46). Loss of DRP1 is associated with the induction of mitophagy (157). The protection against IRI-mediated tubular apoptosis and inflammation after proximal tubular cell-targeted deletion of Drp1 might potentially be mediated by an activated mitophagic response (158). These findings suggest that mitophagy attenuates IRI-induced inflammation, oxidative stress, and cell death in the kidney.
AUTOPHAGY AND MITOPHAGY IN CKD
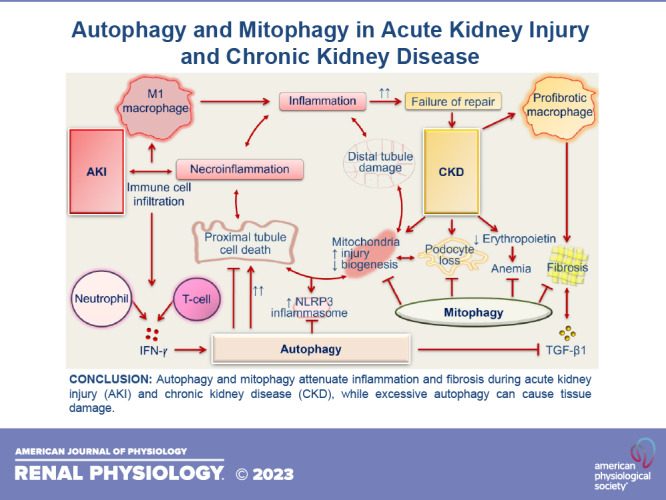
CKD is characterized by an impairment of kidney function due to loss of functional nephrons and the development of progressive fibrosis. Kidney fibrosis is the final common pathological manifestation of CKD that leads to end-stage kidney failure (159). The functions of autophagy (1, 9, 10) and mitophagy (27, 30, 34, 160, 161) in CKD have been rigorously investigated. As in the case of AKI, both protective (44, 47–50, 162–164) and deleterious (51, 165, 166) functions of autophagy in experimental models of CKD have been reported. Insights into the role of mitophagy during CKD are also emerging. Previously published studies, including by our group, have demonstrated its cytoprotective effects against CKD progression (30, 34, 146, 167–170). Here, we summarize the growing body of evidence for the role of autophagy and mitophagy during various pathological conditions of CKD (Fig. 3).
Figure 3.

Autophagy and mitophagy during chronic kidney disease (CKD). Shown is a graphical presentation showing the functions of autophagy and mitophagy in different models of CKD: kidney fibrosis, diabetic nephropathy, and podocytopathies. Autophagy prevents tubulointerstitial inflammation, tubular hypertrophy, loss of podocytes, and kidney fibrosis during CKD. Excessive autophagy can also exaggerate tubular injury and tubulointerstitial fibrosis. Mitophagy attenuates kidney macrophage-derived fibrotic responses, podocyte-, and tubular cell-derived mitochondria-specific oxidative stress, associated cell death, collagen deposition, and CKD-related anemia. AGEs, advanced glycation end products; AS-IV, astragaloside IV; CoQ10, coenzyme Q10; EPO, erythropoietin; ER, endoplasmic reticulum; FP, foot process; HDAC4, histone deacetylase-4; MFN2, mitofusin 2; MitoQ, mitoquinone; mROS, mitochondria-derived reactive oxygen species; mTORC1, mechanistic target of rapamycin (mTOR) kinase complex 1; NLRP3, NLR family pyrin domain containing 3; Nrf2, nuclear erythroid 2-related factor 2; PINK1, phosphatase and tensin homolog (PTEN)-induced kinase1; SGLT2, Na+-glucose cotransporter-2; SIRT, sirtuin; STAT1, signal transducer and activator of transcription 1; TFEB, transcription factor EB; TGF-β1, transforming growth factor-β1; 3MA, 3-methyladenine; ULK1, uncoordinated-51-like protein kinase 1.
Autophagy in Kidney Fibrosis
Kidney fibrosis, characterized by relentless extracellular matrix (ECM) deposition, is the hallmark of progressive CKD regardless of the underlying etiology. The functions of autophagy in the context of kidney fibrosis have been studied using experimental rodent models such as the unilateral ureteral obstruction (UUO) or adenine diet. In cell culture settings, transforming growth factor (TGF)-β1 treatment has been used to study the effects of increased fibrotic responses on autophagy. Autophagy has been reported to be capable of both preventing (44, 47, 48, 162, 163) and promoting (51) kidney fibrosis. Expression of LC3-II and its puncta formation initially increased in the obstructed kidneys on days 3 and 7 after UUO (47) and declined to the near-basal level on day 14 (48, 49, 162). Inhibition of autophagy genetically by knocking out Map1lc3b or Becn1 (44, 47), by knockdown of Becn1 (44, 163), or pharmacologically using 3MA (162) exaggerated UUO or TGF-β1-induced fibrotic responses. Induction of autophagy using knockin of Becn1 (44) or rapamycin (48) attenuated UUO-induced collagen type I expression and ECM deposition. Trifluoperazine-dependent stimulation of autophagy attenuated TGF-β1-induced collagen type I production by mouse mesangial cells (163). However, 3MA-mediated suppression of the autophagic response promoted tubular cell apoptosis (162). Our group also reported that autophagy, by degrading intracellular collagen type I and its aggregated components, exhibits antifibrotic functions (163). TGF-β1-induced stimulation of autophagy in HK-2 cells negatively regulated fibrotic responses by promoting degradation of mature TGF-β1 (47). Proximal tubule-specific deletion of Atg5 arrested the tubular cell cycle at the G2/M phase and was associated with increased UUO-induced tubulointerstitial fibrosis (48). Angiotensin II-treated Atg5−/− proximal tubular cells also displayed G2/M cell cycle arrest and increased fibrotic responses, suggesting a cytoprotective role of ATG5-dependent autophagy (48). Ablation of Atg5 also promoted UUO-induced inflammation via NF-κB-dependent production of proinflammatory cytokines and infiltration of macrophages and T cells in the obstructed kidney (49). Distal tubule-specific conditional deletion of Atg7 resulted in increased IL-1β, NLRP3 inflammasomes, caspase 1-mediated apoptosis, and mitochondrial damage in the obstructed kidney (50). This further confirms that ATG7-dependent autophagy exhibits protection against kidney fibrosis. miR-376b has been shown to negatively regulate Atg5 (164). Suppression of miR-376b attenuated adenine diet-induced kidney fibrosis by inducing Atg5-mediated autophagic responses in macrophages (164). The aforementioned studies highlight that autophagy prevents the progression of kidney fibrosis. However, contradictory findings also exist that show that obstructed kidneys from proximal tubule-specific Atg7-deleted mice displayed reduced tubular atrophy, nephron loss, macrophage recruitment, and tubulointerstitial fibrosis after UUO (51). This study emphasized that persistent activation of autophagy accelerated UUO-induced kidney fibrosis (51). Receptor for advanced glycation end product (RAGE)-mediated induction of ATG7 and autophagy in TGF-β1-treated mouse proximal tubular cells has been reported to exaggerate fibrotic responses (165). TGF-β1-mediated induction of autophagy has also been shown to exert proapoptotic effects (166). Considering the contrasting reported observations, it is still unclear whether autophagy exerts protective or deleterious functions during kidney fibrosis. It is plausible that the roles of autophagy during kidney fibrosis may be temporal- and context dependent, which need to be further evaluated in the cell type(s) involved.
Mitophagy in Kidney Fibrosis
We have previously reported that expression of mitophagy regulators (PINK1, MFN2, and Parkin) decreased in the kidneys from experimental mouse models of kidney fibrosis and human kidney biopsy samples with proven interstitial fibrosis and tubular atrophy (30, 34). We also observed reduced expression of mitophagy regulators in peripheral blood mononuclear cells (PBMCs) from patients with CKD and human primary kidney macrophages after TGF-β1-treatment (30). Studies by others have also demonstrated decreased PINK1, MFN2, and Parkin mRNA expression in PBMCs from patients with CKD (167). We delineated the protective function of PINK1/MFN2/Parkin-mediated mitophagy against kidney fibrosis (30). We suggested that defective mitophagy due to loss of Pink1 or Prkn or myeloid-specific Mfn2 resulted in increased kidney macrophage-derived mROS production and profibrotic responses (30). The profibrotic macrophages then drive the progression of kidney fibrosis after UUO or adenine diet and promote worsening of kidney function, as evaluated by increases in BUN and serum creatinine levels (30, 34). However, increased PINK1 and Parkin expression and colocalization of LC3-II with mitochondria, indicating activated mitophagy, but reduced MFN2 expression in the kidneys after UUO have also been reported (168, 169). Li et al. (168) confirmed that mitophagy conferred protective functions against UUO-induced increases in oxidative stress, mitochondrial damage, and TGF-β1 expression. Mitophagy also mitigated CKD-related anemia during kidney fibrosis by attenuating inflammation and promoting erythropoietin (EPO) production in the kidney (146). We uncovered that myeloid-specific ablation of Mfn2, but not Mfn1, impaired the kidney macrophage mitophagic response and mitochondrial biogenesis (34). It resulted in the accumulation of abnormal mitochondria, increased oxidative stress, collagen deposition in the kidney, and deterioration of kidney function (34). We have also reported that the mitochondrial fusion proteins (MFN1, MFN2, and OPA1) were downregulated, whereas fission proteins (DRP1 and phosphorylated DRP1) were upregulated, in the kidney during adenine diet-induced kidney fibrosis (34). UMI-77-mediated activation of mitophagy exhibited protective effects against UUO-induced kidney fibrosis by attenuating inflammation, apoptosis, and activation of NF-κB and TGF-β/Smad signaling pathways (169). Stimulation of mitophagy after UMI-77 treatment also attenuated TGF-β1-induced fibrotic responses in HK-2 cells (169). Quercetin, a natural antioxidant, has also been shown to exhibit antifibrotic effects via activating mitophagy (170). Quercetin prevents angiotensin II- or UUO-induced senescence in kidney tubular epithelial cells through promoting PINK1-dependent mitophagy via activating SIRT1 (170). The aforementioned findings suggest that mitophagy during kidney fibrosis provides cytoprotection by recycling damaged mitochondria, to negatively regulate oxidative stress, inflammation, and fibrosis.
Autophagy in DN
Autophagy in proximal tubular cells (52) and podocytes (171) has been reported to be suppressed in experimental models of diabetic kidney disease (DKD). It has been suggested that an impaired autophagic response plays a critical role in the progression of DN (1, 11, 24). Autophagy is downregulated in the kidneys of patients with DKD and experimental models of type I diabetes: streptozotocin (STZ) treatment and Akita mice (52). However, high glucose-mediated induction of autophagosome formation, LC3-II, beclin 1 expression, and autophagic flux in podocytes have also been reported (17, 172). Whether autophagy is activated or suppressed during DN is unclear; previously published studies have chiefly highlighted the protective functions of autophagy against the progression of DN (52, 171–175).
Proximal tubule-specific Atg7-deficient Akita mice displayed increased tubular hypertrophy, tissue damage, macrophage infiltration, and fibrosis in the kidney (52). The impaired autophagic activity was also associated with an increase in miR-214-mediated reduction of ULK1 expression in tubular cells (52). Suppression of miR-214, directly via genetic deletion in proximal tubular cells or indirectly via blockade of p53, attenuated STZ-induced tubular hypertrophy and albuminuria via restoring ULK1 expression and autophagy (52). Moreover, TFEB, which is known to induce transcriptional activation of autophagy, was also downregulated in the kidneys of patients and experimental model of DKD (173). Treatment with tubastatin A, a small-molecule inhibitor of histone deacetylase (HDAC)6, via acetylation of TEFB, promoted transcriptional activation of autophagy (173). Tubastatin A-mediated activation of autophagy was associated with reductions in tubular apoptosis and tubulointerstitial fibrosis in subtotally nephrectomized rats (173). Deletion of Na+-glucose cotransporter-2 (SGLT2), which mediates glucose uptake, resulted in reduced p62 accumulation in the kidney, indicating activated autophagic response (174). Inhibition of SGLT2 also improved STZ-induced hyperglycemia (174). In line with this, empagliflozin-mediated selective inhibition of SGLT2 reduced oxidative stress and apoptotic and fibrotic responses in hyperglycemic kidney proximal tubular cells by inducing autophagic responses (175). Advanced glycation end products (AGEs) are heterogeneous irreversibly glycated cross-linked sugar-derived proteins; in patients with diabetes, urinary levels of AGEs positively correlated with albuminuria (176). AGEs also promoted expression of LC3 II in rat mesangial cells in a dose-dependent manner (177). Autophagy has been shown to prevent hyperglycemia-induced accumulation of AGEs, which are also known to exaggerate inflammation and injury in proximal tubular epithelial cells (178).
Podocytes exhibit a higher basal level of autophagy than proximal tubular cells (11). The suppression of autophagy in the kidneys in patients with DN was also associated with increases in metabolic regulators (mTOR and AMPK), glomerular hypertrophy, and disruption of glomerular filtration barrier (179). Similarly, induction of mTORC1 activity indirectly via Tsc1 deletion in podocytes, exaggerated diabetic-induced podocyte damage, effacement of foot processes, and albuminuria (180). Rapamycin-mediated inhibition of mTORC1 activated autophagy and reversed diabetic-induced pathological damage in the podocyte-specific Tsc1-deleted mice (180). Rapamycin has also been shown to improve STZ-mediated proteinuria and podocyte apoptosis by inducing autophagic response (181). However, podocyte-specific regulatory-associated protein of mTOR (Raptor)-deleted mice developed progressive glomerulosclerosis, proteinuria at 8 wk of age, and foot process effacement at 4 and 12 mo of age (179). Interestingly, db/db mice with podocyte-specific hemizygous ablation of Raptor displayed reduced glomerular injury, thickening of basement membrane, and proteinuria at 40 wk of age (180). These studies indicate that optimal mTORC1 activity is required to prevent excessive autophagy-mediated tissue damage. However, upregulated mTORC1 can impair podocyte homeostasis by suppressing autophagic activities. Podocyte-specific Atg5 deletion accelerated DN due to increases in podocytopathy, disruption of the filtration barrier, and glomerulosclerosis (17). Podocyte-specific Atg5 deficient high-fat diet-fed diabetic mice also exhibited massive albuminuria and loss of podocytes (53). Treatment of cultured podocytes with sera from patients with DN and diabetic rats resulted in an increased expression of p62 protein, suggesting reduced autophagy, which was associated with increased apoptosis (53). Astragaloside IV (AS-IV), a plant-derived antioxidant, attenuated STZ or high-glucose-induced ER stress and apoptosis in the podocytes by accelerating autophagy (171). Long-term exposure to high glucose suppressed autophagic activity and resulted in insulin resistance and podocyte apoptosis (182). Inhibition of autophagy genetically (using Becn1 or Atg5 siRNAs) or pharmacologically (using 3MA), increased mouse podocyte sensitivity toward apoptosis (172). Inactivation of heme oxygenase (HO)-1 has been shown to inhibit autophagy in an AMPK-dependent manner and aggravate podocyte damage (172).
The HDAC4, which is known to suppress autophagy was upregulated in the podocytes under high-glucose conditions, and in patients with DN, where HDAC4 negatively correlated with GFR (183, 184). HDAC4 has been shown to promote apoptosis by repressing autophagy via activating signal transducer and activator of transcription 1 (STAT1) pathway (184).
Resveratrol, an inducer of SIRT1 reduced podocyte apoptosis in db/db mice and high glucose-treated cells by activating autophagy via downregulating miR-393-5p (185). Induction of autophagy after resveratrol treatment also protected against STZ-induced albuminuria and kidney fibrosis (186). The inhibition of autophagy using Atg5 short hairpin RNA or 3MA reversed the resveratrol-mediated protection and accelerated podocyte cell death (185). The protective effects of resveratrol against high-glucose-mediated podocyte damage and production of vascular endothelial growth factor (VEGF) were suppressed after the knockdown of Sirt1 (186). These findings suggest that resveratrol-dependent autophagy induction is mediated by SIRT1, and it exhibits cytoprotective functions.
Vascular endothelial cell-specific ablation of Atg5 promoted the progression of STZ-induced DN by increasing capillary rarefactions, loss of fenestrations, and glomerular endothelial lesions (17).
AGEs also promoted the expression of LC3 II in rat mesangial cells in a dose-dependent manner (187). AGEs-mediated induction of ER stress promoted autophagy in mesangial cells, and activation of autophagy protects against apoptosis (187). Suppression of autophagy using Atg5 siRNA in mesangial cells resulted in increased AGE-induced cell death (187). Long noncoding RNA (lncRNA) SOX2 overlapping transcript (SOX2OT) via inducing autophagy through AKT/mTOR pathway prevented high-glucose-induced mesangial cell proliferation and fibrosis (188). The aforementioned studies in the experimental models of DN highlight the protective roles of autophagy in attenuating oxidative stress, cell death, and fibrotic responses.
Mitophagy in DN
High-glucose-treated kidney tubular cells (189–191), podocytes (192, 193), mesangial cells (194, 195), and endothelial cells (196) exhibited reduced expression of mitophagy regulators (PINK1, Parkin, LC3 II). High-fat diet-fed and STZ-treated mice models of DKD displayed decreased kidney mitophagic response (197). STZ-induced DKD also resulted in a decrease in Parkin expression in the kidney tubular epithelial cells (198). The reduced expression of PINK1 and Parkin in the tubular cells of db/db mice was also associated with increased mitochondrial fragmentation, mROS production, and apoptosis (190). However, contrasting findings also demonstrated an increase in the PINK1/Parkin-mediated mitophagy in the kidneys of db/db mice (194), high-fat diet-fed rats, and in palmitic acid-treated podocytes (199). The induction of BNIP3-dependent mitophagy in kidney tubular cells after STZ-induced DKD has also been reported (189). An increase in LC3 II expression in kidney tubular cells of patients with DN, and STZ-induced diabetic rat model might be due to a reduction in clearance of mitophagosomes, because of defective mitophagic flux (189). PBMCs from patients with DN displayed impaired mitochondrial metabolism with reduced maximal respiration rate, reserve capacity, and bioenergetic health index than patients with diabetes without kidney disease (195). It suggests that mitochondrial quality control is impaired during DKD.
Mitoquinone (MitoQ), a mitochondrial-specific antioxidant, has been shown to partly restore PINK1 and Parkin expression in high-glucose-treated tubular cells (190). An increase in the mitophagy regulators after MitoQ treatment was also associated with improvement in glomerular and tubular functions and attenuation of interstitial fibrosis in the heterozygous Ins2AkitaJ mouse model of type I diabetes (200). MitoQ also attenuated NLRP3 inflammasome-mediated tubular injury in the kidney of db/db mice and high-glucose-treated tubular epithelial cells (201). Overexpression of optineurin (OPTN) in kidney tubular epithelial cells also attenuated NLRP3 inflammasome formation, oxidative stress, and kidney injury by promoting mitophagy (202). Decreased Parkin in the kidneys of STZ-induced DN mice was inversely correlated with increased IL-6, TGF-β1, and GATA binding factor 4 (GATA-4) expression (202). Overexpression of Parkin prevented the progression of DN by promoting ubiquitination of GATA4 (202). Suppressing thioredoxin interaction protein (TXNIP) reversed the decrease in mitophagy, increases in fibrotic responses, and mROS production in high-glucose-treated HK-2 cells and kidneys of diabetic rats (189). Transplantation of SIRT3 overexpressing amniotic fluid stem cells in the kidney of db/db mice also protected against increases in inflammation, apoptosis, worsening of kidney function, and fibrosis by promoting mitophagy (203). Metformin, an AMPK agonist, by activating mitophagy attenuated oxidative stress and fibrotic responses in the kidney from high-fat diet and STZ-treated diabetic mice and in high-glucose/high-fatty acid-treated tubular epithelial cells (197).
Induction of forkhead box class O1 (FoxO1) and PINK1/Parkin-mediated mitophagy exhibited antioxidative effects and attenuated STZ-mediated podocyte damage (192). The silencing of lncRNAs also prevented podocyte apoptosis by restoring the expression of Parkin and mitophagy and downregulating mammalian sterile 20-like kinase 1 (Mst1) (193).
Defective mitophagy in high-glucose-treated human mesangial cells also contributed to an increase in the number of fragmented mitochondria (195). Ursolic acid, a mitophagy stimulator, reversed high glucose-mediated increases in collagen I expression and hypertrophy in mesangial cells by inducing PINK1 and LC3 II expression (194). The genetic ablation of miR-379 promotes adaptive mitophagy, via suppressing FIS1 (204). The miR-379-induced mitophagy attenuated STZ-mediated decrease in mitochondrial respiration in mesangial cells, mesangial matrix expansion, podocyte effacement, and thickening of glomerular basement membrane (204).
Coenzyme Q10 (CoQ10) partly protected against high-glucose-induced glomerular endothelial cell death, mitochondrial damage, and mROS production by stimulating mitophagy (196). CoQ10 activates mitophagy by restoring antioxidant response element, nuclear erythroid 2-related factor 2 (Nrf2) signaling (196). It is still unclear whether mitophagy is activated or suppressed during DN, due to contradictory published findings. Collectively, the studies strongly support protective functions of mitophagy-mediated mitochondrial quality control against progression of DN.
Autophagy in Podocytopathies
Podocytopathies arise from direct or indirect insults, due to genetic defect(s), toxin, infection-related, or immune-mediated damage to the podocytes, resulting in proteinuria or nephrotic syndrome (NS), which can lead to kidney failure (1). Proteinuria can be due to glomerular (disruption of glomerular filtration barrier) or nonglomerular (impaired reabsorption by proximal tubular cells) injuries (205). Autophagy is highly activated in glomeruli of the mouse model of bovine serum albumin (BSA)-overload-induced proteinuria and in patients with acquired proteinuric kidney disease (13). The ultrastructural analysis of human kidney biopsy samples also suggested induction of autophagic activity in podocytes (206). Podocyte-specific expression of beclin 1 has been reported to be higher in patients with minimal change disease (MCD) than focal segmental glomerulosclerosis (FSGS), two common causes of idiopathic NS (207). Studies have reported that patients with FSGS have worse outcomes and poor survival than patients with MCD and develop end-stage kidney disease sooner (208). Interestingly, follow-up kidney biopsies of patients with MCD, who progressed to FSGS, had lower podocyte autophagic activity compared with those who maintained high autophagic activity and retained MCD status (207). These findings suggest a protective role of podocyte autophagy against NS and progression of podocytopathies (207). Moreover, in patients with MCD, the number of autophagosomes also correlated positively with eGFR (209). The LC3 puncta formation in podocytes also increased in adriamycin-induced nephropathy, a model that mimics human FSGS (210). However, a transient decrease in autophagy in podocytes after unilateral nephrectomy has also been reported (54).
HDAC4, which suppresses autophagy and promotes apoptosis via activating STAT1 pathway, was induced in patients with FSGS (183, 184). The HDAC4 expression negatively correlated with kidney function (183, 184). Puromycin aminonucleoside (PAN)-induced podocyte apoptosis and foot process effacement increased after suppressing autophagy using Becn1 siRNA or pharmacological agents (3MA or chloroquine) (207). Rapamycin-mediated induction of autophagy reversed the aforementioned changes (207). The deposition of oxidized and ubiquitinated proteins after podocyte-specific Atg5 deletion (Atg5Δpodocyte) correlated with increases in PAN and adriamycin-induced proteinuria (13). The deposition also positively correlated with glomerulopathy, loss of podocytes, and late-onset glomerulosclerosis in aged mice (13). Conditional deletion of Atg7 in podocytes dramatically increased ER stress, ubiquitinated protein accumulation, foot process effacement, cytoskeletal destabilization, and proteinuria after unilateral nephrectomy (54). Nephron-specific deletion of Atg5 or Atg7 also resulted in the mild loss of podocyte function by 2 mo, pathological changes (similar to human FSGS) in glomeruli by 4 mo of age (12). These changes resulted in kidney failure by 6 mo of age (12). Proximal tubular cells from nephron-specific Atg5 or Atg7 deleted mice also exhibited an increased number of abnormal mitochondria and oxidative stress.
Podocyte-specific inhibition of mTOR either by selective deletion of the Mtor in podocytes (Mtor podo-KO) or via rapamycin resulted in impaired autophagic flux in podocytes (211). The decrease in autophagic flux contributed to an accumulation of autophagosomes and autophagolysosomes (211). Nutrient starvation stimulates autophagy via inhibition of mTOR, while persistent starvation can also activate mTOR (211). The accumulation of autophagic vesicles in the Mtor deficient podocytes can be due to absence of the negative feedback loop. The defective autophagic flux in podocytes of Mtor podo-KO mice was associated with proteinuria at 3 wk and end-stage kidney failure at 5 wk of age (211). These findings highlight the protective role of autophagy in regulating podocyte functions. The failure of fusion of autophagosome and lysosome contributed to their accumulation, with an increase in protein p62 expression after podocyte-specific ablation of prorenin receptor (212). Podocyte-specific prorenin receptor-deleted mice died between 2 and 3 wk of age, due to NS-associated complications. These complications include podocyte foot process effacement, deranged cytoskeleton, and albuminuria. These findings suggest a protective role of autophagy in maintaining podocyte structure and functions (212). PAN-mediated podocyte cytoskeleton destabilization and cell death in human cultured podocytes diminished after trehalose (an mTOR-independent stimulator of autophagy) treatment (213). Podocyte-specific conditional Ctsd (encodes cathepsin D)-deleted mice displayed albuminuria at 5 mo of age and end-stage kidney failure by 20–22 mo (214). These complications were also associated with reduced autophagic response, due to defective lysosomal activities (214). This resulted in the accumulation of autophagosomes and toxic subunit c-positive lipofuscins, which are the byproducts of failed intracellular catabolism in the lysosomes (214). Genetic variants of apolipoprotein L1 (APOL1) have been reported in patients with FSGS and linked to albuminuria, loss of podocytes, and end-stage kidney disease (215). Mice overexpressing risk variants of APOL1 also developed kidney disease (216). APOL1 risk variant-associated cytotoxicity may be resolved by correcting the functional blockade of autophagic flux (217). The aforementioned studies indicate that activation of autophagic response helps in attenuating proteinuria and glomerulosclerosis during podocytopathies.
Mitophagy in Podocytopathies
The presence of abnormal swollen mitochondria in kidney biopsy samples from patients with FSGS, and podocytes and proximal tubular cells from nephron-specific Atg5 or Atg7 deleted mice, highlights the importance of mitochondrial quality control (12). In the albumin overload-induced mouse model of proteinuria, the reduced NIX or BNIP3L-dependent mitophagy in kidney tubular cells led to increases in mitochondrial fragmentation, tubular epithelial cell apoptosis, and loss of kidney function (218). The overexpression of NIX attenuated, whereas NIX suppression using siRNA aggravated apoptosis in albumin-treated kidney tubular epithelial cells (218). Albumin-overload-mediated mitochondrial swelling, loss of membrane potential, increased mROS production and associated apoptosis have also been reported in proximal tubular epithelial cells (219, 220). Albumin-overload-induced decreases in mitochondrial membrane potential, ATP generation, and an increase in mROS production in proximal tubular epithelial cells escalated after knockdown of PRKN (221). However, Parkin overexpression reversed the aforementioned changes (221). These findings highlight the importance of mitophagy-mediated quality control mechanism against the progression of podocytopathies.
CONCLUSIONS AND PERSPECTIVES
Molecular machinery of autophagy and mitophagy can be manipulated to control the progression of kidney diseases. The state of activation/suppression of autophagic and mitophagic responses during a particular pathologic condition must be considered when thinking about manipulating the signaling pathways for therapeutic purposes. The activated autophagy and suppressed mitophagy or vice versa can also coexist during a disease condition. For example, after 24 h of CLP or LPS-mediated sepsis, autophagy has been reported to be activated (36, 127), whereas mitophagy has been found to be suppressed in the kidney (135). Therefore, stimulating mitophagic than autophagic response, at a specific time-point, might be more effective in such a scenario and less deleterious, and help in preventing kidney damage. When considering induction/suppression of autophagy/mitophagy signaling via a therapeutic agent, not only the pathologic context, but the cell-specific targeting is equally critical. For example, podocytes exhibit higher autophagy, but lower mitophagy at the basal level than proximal tubular cells (111, 222). The spatiotemporal analysis of activation/suppression of autophagy and mitophagy signaling pathways in different models of AKI and CKD still warrants further confirmation.
GRANTS
D.B. is supported by National Heart, Lung, and Blood Institute Grant T32HL134629. M.E.C is supported by National Heart, Lung, and Blood Institute Grants R01HL133801 and R01HL055330.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.B. prepared figures; D.B. drafted manuscript; D.B. and M.E.C. edited and revised manuscript; D.B. and M.E.C. approved final version of manuscript.
REFERENCES
- 1. Choi ME. Autophagy in kidney disease. Annu Rev Physiol 10: 297–322, 2020. doi: 10.1146/annurev-physiol-021119-034658. [DOI] [PubMed] [Google Scholar]
- 2. Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol 24: 186–203, 2023. doi: 10.1038/s41580-022-00529-z. [DOI] [PubMed] [Google Scholar]
- 3. Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem 293: 5414–5424, 2018. doi: 10.1074/jbc.R117.818237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 119: 301–311, 1992. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42, 2019. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van Noorden R, Ledford H. Medicine Nobel for research on how cells ‘eat themselves’. Nature 538: 18–19, 2016. doi: 10.1038/nature.2016.20721. [DOI] [PubMed] [Google Scholar]
- 7. Aman Y, Schmauck-Medina T, Hansen M, Morimoto RI, Simon AK, Bjedov I, Palikaras K, Simonsen A, Johansen T, Tavernarakis N, Rubinsztein DC, Partridge L, Kroemer G, Labbadia J, Fang EF. Autophagy in healthy aging and disease. Nat Aging 1: 634–650, 2021. doi: 10.1038/s43587-021-00098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodgers SJ, Jones EI, Arumugam S, Hamila SA, Danne J, Gurung R, Eramo MJ, Nanayakkara R, Ramm G, McGrath MJ, Mitchell CA. Endosome maturation links PI3Kα signaling to lysosome repopulation during basal autophagy. EMBO J 41: e110398, 2022. doi: 10.15252/embj.2021110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leventhal JS, Wyatt CM, Ross MJ. Recycling to discover something new: the role of autophagy in kidney disease. Kidney Int 91: 4–6, 2017. doi: 10.1016/j.kint.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 10. Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol 16: 489–508, 2020. doi: 10.1038/s41581-020-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhatia D, Choi ME. Autophagy in kidney disease: advances and therapeutic potential. Prog Mol Biol Transl Sci 172: 107–133, 2020. doi: 10.1016/bs.pmbts.2020.01.008. [DOI] [PubMed] [Google Scholar]
- 12. Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, Shankland SJ, D'Agati VD, Duffield JS. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26: 1040–1052, 2015. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstädt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 120: 1084–1096, 2010. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bechtel W, Helmstädter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, Kerjaschki D, Walz G, Huber TB. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol 24: 727–743, 2013. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Chen MX, Fogo AB, Harris RC, Chen JK. mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. J Am Soc Nephrol 24: 198–207, 2013. doi: 10.1681/ASN.2012010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, Huber TB. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 8: 826–837, 2012. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 17. Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A, Massé JM, Souyri M, Huber TB, Tharaux PL. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 11: 1130–1145, 2015. doi: 10.1080/15548627.2015.1049799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsuda J, Namba T, Takabatake Y, Kimura T, Takahashi A, Yamamoto T, Minami S, Sakai S, Fujimura R, Kaimori JY, Matsui I, Hamano T, Fukushima Y, Matsui K, Soga T, Isaka Y. Antioxidant role of autophagy in maintaining the integrity of glomerular capillaries. Autophagy 14: 53–65, 2018. doi: 10.1080/15548627.2017.1391428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25: 1025–1040, 2005. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol 13: 681–696, 2017. doi: 10.1038/nrneph.2017.129. [DOI] [PubMed] [Google Scholar]
- 21. Vasko R, Ratliff BB, Bohr S, Nadel E, Chen J, Xavier S, Chander P, Goligorsky MS. Endothelial peroxisomal dysfunction and impaired pexophagy promotes oxidative damage in lipopolysaccharide-induced acute kidney injury. Antioxid Redox Signal 19: 211–230, 2013. doi: 10.1089/ars.2012.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ryter SW, Bhatia D, Choi ME. Autophagy: a lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid Redox Signal 30: 138–159, 2019. doi: 10.1089/ars.2018.7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460–473, 2014. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P , et al. Autophagy in major human diseases. EMBO J 40: e108863, 2021. doi: 10.15252/embj.2021108863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mailler E, Guardia CM, Bai X, Jarnik M, Williamson CD, Li Y, Maio N, Golden A, Bonifacino JS. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun 12: 6750, 2021. doi: 10.1038/s41467-021-26999-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J 40: e104705, 2021. doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhatia D, Choi ME. The emerging role of mitophagy in kidney diseases. J Life Sci (Westlake Village) 1: 13–22, 2019. doi: 10.36069/jols/20191203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lei Y, Klionsky DJ. New regulators of PRKN-independent mitophagy. Autophagy 18: 1–3, 2022. doi: 10.1080/15548627.2021.2012867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Y, Dorn GW 2nd.. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475, 2013. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhatia D, Chung KP, Nakahira K, Patino E, Rice MC, Torres LK, Muthukumar T, Choi AM, Akchurin OM, Choi ME. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 4: e132826, 2019. doi: 10.1172/jci.insight.132826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131, 2010. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 32. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106, 2010. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhatia D, Capili A, Choi ME. Mitochondrial dysfunction in kidney injury, inflammation, and disease: potential therapeutic approaches. Kidney Res Clin Pract 39: 244–258, 2020. doi: 10.23876/j.krcp.20.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhatia D, Capili A, Nakahira K, Muthukumar T, Torres LK, Choi AMK, Choi ME. Conditional deletion of myeloid-specific mitofusin 2 but not mitofusin 1 promotes kidney fibrosis. Kidney Int 101: 963–986, 2022. doi: 10.1016/j.kint.2022.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leventhal JS, Ni J, Osmond M, Lee K, Gusella GL, Salem F, Ross MJ. Autophagy limits endotoxemic acute kidney injury and alters renal tubular epithelial cell cytokine expression. PLoS One 11: e0150001, 2016. doi: 10.1371/journal.pone.0150001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mei S, Livingston M, Hao J, Li L, Mei C, Dong Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep 6: 22171, 2016. doi: 10.1038/srep22171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y, Zhu J, Liu Z, Shu S, Fu Y, Liu Y, Cai J, Tang C, Liu Y, Yin X, Dong Z. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol 38: 101767, 2021. doi: 10.1016/j.redox.2020.101767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, Yoshimori T, Isaka Y, Rakugi H. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 180: 517–525, 2012. doi: 10.1016/j.ajpath.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 39. Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82: 1271–1283, 2012. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y, Tang C, Cai J, Chen G, Zhang D, Zhang Z, Dong Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis 9: 1113, 2018. doi: 10.1038/s41419-018-1152-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou L, Zhang L, Zhang Y, Yu X, Sun X, Zhu T, Li X, Liang W, Han Y, Qin C. PINK1 deficiency ameliorates cisplatin-induced acute kidney injury in rats. Front Physiol 10: 1225, 2019. doi: 10.3389/fphys.2019.01225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22: 902–913, 2011. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Livingston MJ, Shu S, Fan Y, Li Z, Jiao Q, Yin XM, Venkatachalam MA, Dong Z. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 19: 256–277, 2023. doi: 10.1080/15548627.2022.2072054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi M, Maique J, Shepard S, Li P, Seli O, Moe OW, Hu MC. In vivo evidence for therapeutic applications of beclin 1 to promote recovery and inhibit fibrosis after acute kidney injury. Kidney Int 101: 63–78, 2022. doi: 10.1016/j.kint.2021.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, He L, Tan J, Liu Y, Liu H, Sun L, Duan S, Peng Y, Liu F, Yin XM, Zhang Z, Dong Z. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 14: 880–897, 2018. doi: 10.1080/15548627.2017.1405880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gall JM, Wang Z, Bonegio RG, Havasi A, Liesa M, Vemula P, Borkan SC. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J Am Soc Nephrol 26: 1092–1102, 2015. doi: 10.1681/ASN.2014010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding Y, Kim S, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 25: 2835–2846, 2014. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H, Peng X, Wang Y, Cao S, Xiong L, Fan J, Wang Y, Zhuang S, Yu X, Mao H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 12: 1472–1486, 2016. doi: 10.1080/15548627.2016.1190071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peng X, Wang Y, Li H, Fan J, Shen J, Yu X, Zhou Y, Mao H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis 10: 253, 2019. doi: 10.1038/s41419-019-1483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nam SA, Kim WY, Kim JW, Park SH, Kim HL, Lee MS, Komatsu M, Ha H, Lim JH, Park CW, Yang CW, Kim J, Kim YK. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis 10: 78, 2019. doi: 10.1038/s41419-019-1356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12: 976–998, 2016. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma Z, Li L, Livingston MJ, Zhang D, Mi Q, Zhang M, Ding HF, Huo Y, Mei C, Dong Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest 130: 5011–5026, 2020. doi: 10.1172/JCI135536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K, Kim EH, Haneda M, Kajiwara N, Hayashi K, Ohashi H, Ugi S, Maegawa H, Uzu T. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 65: 755–767, 2016. doi: 10.2337/db15-0473. [DOI] [PubMed] [Google Scholar]
- 54. Oliva Trejo JA, Asanuma K, Kim EH, Takagi-Akiba M, Nonaka K, Hidaka T, Komatsu M, Tada N, Ueno T, Tomino Y. Transient increase in proteinuria, poly-ubiquitylated proteins and ER stress markers in podocyte-specific autophagy-deficient mice following unilateral nephrectomy. Biochem Biophys Res Commun 446: 1190–1196, 2014. doi: 10.1016/j.bbrc.2014.03.088. [DOI] [PubMed] [Google Scholar]
- 55. Su L, Zhang J, Gomez H, Kellum JA, Peng Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 19: 401–414, 2023. doi: 10.1080/15548627.2022.2084862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leventhal JS, He JC, Ross MJ. Autophagy and immune response in kidneys. Semin Nephrol 34: 53–61, 2014. doi: 10.1016/j.semnephrol.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 57. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy 14: 243–251, 2018. doi: 10.1080/15548627.2017.1402992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Crotzer VL, Blum JS. Autophagy and adaptive immunity. Immunology 131: 9–17, 2010. doi: 10.1111/j.1365-2567.2010.03321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation. Autophagy 13: 997–1003, 2017. doi: 10.1080/15548627.2017.1309485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gong L, Pan Q, Yang N. Autophagy and inflammation regulation in acute kidney injury. Front Physiol 11: 576463, 2020. [Erratum in Front Physiol 11: 639938, 2021]. doi: 10.3389/fphys.2020.576463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol 27: 27–39, 2016. doi: 10.1681/ASN.2015040405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rabb H. The T cell as a bridge between innate and adaptive immune systems: implications for the kidney. Kidney Int 61: 1935–1946, 2002. doi: 10.1046/j.1523-1755.2002.00378.x. [DOI] [PubMed] [Google Scholar]
- 63. Leemans JC, Kors L, Anders HJ, Florquin S. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol 10: 398–414, 2014. doi: 10.1038/nrneph.2014.91. [DOI] [PubMed] [Google Scholar]
- 64. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, Fujii R, Ishidate F, Tanaka T, Tanaka Y, Hirokawa N, Nangaku M, Inagi R. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 29: 1261–1273.e6, 2019. doi: 10.1016/j.celrep.2019.09.050. [DOI] [PubMed] [Google Scholar]
- 65. Liu Y, Liu X, Zhou S, Xu R, Hu J, Liao G, Liao J, Guo Z, Li Y, Yang S, Li S, Chen H, Guo Y, Li M, Fan L, Li L, Zhao M, Liu D. Single-cell profiling of kidney transplant recipients with immunosuppressive treatment reveals the dynamic immune characteristics. Front Immunol 12: 639942, 2021. doi: 10.3389/fimmu.2021.639942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Oh JE, Lee HK. Pattern recognition receptors and autophagy. Front Immunol 5: 300, 2014. doi: 10.3389/fimmu.2014.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27: 135–144, 2007. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang M, Qi Y, Cao Y, Zhang X, Wang Y, Liu Q, Zhang J, Zhou G, Ai Y, Wei S, Wang L, Liu G, Lian Z, Han H. Domain fusion TLR2-4 enhances the autophagy-dependent clearance of Staphylococcus aureus in the genetic engineering goat. eLife 11: e78044, 2022. doi: 10.7554/eLife.78044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wan J, Weiss E, Ben Mkaddem S, Mabire M, Choinier PM, Picq O, Thibault-Sogorb T, Hegde P, Pishvaie D, Bens M, Broer L, Gilgenkrantz H, Moreau R, Saveanu L, Codogno P, Monteiro RC, Lotersztajn S. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci Transl Med 12: eaaw8523, 2020. doi: 10.1126/scitranslmed.aaw8523. [DOI] [PubMed] [Google Scholar]
- 70. Andrade-Silva M, Cenedeze MA, Perandini LA, Felizardo RJF, Watanabe IKM, Agudelo JSH, Castoldi A, Gonçalves GM, Origassa CST, Semedo P, Hiyane MI, Foresto-Neto O, Malheiros DMAC, Reis MA, Fujihara CK, Zatz R, Pacheco-Silva A, Câmara NOS, de Almeida DC. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin Sci (Lond) 132: 1725–1739, 2018. doi: 10.1042/CS20170262. [DOI] [PubMed] [Google Scholar]
- 71. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307: 593–596, 2005. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 72. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90–97, 2010. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 73. Schuijs MJ, Hammad H, Lambrecht BN. Professional and 'amateur' antigen-presenting cells in type 2 immunity. Trends Immunol 40: 22–34, 2019. doi: 10.1016/j.it.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Müller CA, Markovic-Lipkovski J, Risler T, Bohle A, Müller GA. Expression of HLA-DQ, -DR, and -DP antigens in normal kidney and glomerulonephritis. Kidney Int 35: 116–124, 1989. doi: 10.1038/ki.1989.16. [DOI] [PubMed] [Google Scholar]
- 75. Baudeau C, Delarue F, Hé CJ, Nguyen G, Adida C, Peraldi MN, Sraer JD, Rondeau E. Induction of MHC class II molecules HLA-DR, -DP and -DQ and ICAM 1 in human podocytes by γ-interferon. Exp Nephrol 2: 306–312, 1994. [PubMed] [Google Scholar]
- 76. Gluhovschi C, Gluhovschi G, Potencz E, Lazar E, Petrica L, Bozdog G, Gadalean F, Bob F, Cioca D, Velciov S. What is the significance of HLA-DR antigen expression in the extraglomerular mesangium in glomerulonephritis? Hum Immunol 73: 1098–1101, 2012. doi: 10.1016/j.humimm.2012.07.326. [DOI] [PubMed] [Google Scholar]
- 77. Li S, Liu Y, He Y, Rong W, Zhang M, Li L, Liu Z, Zen K. Podocytes present antigen to activate specific T cell immune responses in inflammatory renal disease. J Pathol 252: 165–177, 2020. doi: 10.1002/path.5508. [DOI] [PubMed] [Google Scholar]
- 78. Chen Q, Kang J, Fu C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct Target Ther 3: 18, 2018. doi: 10.1038/s41392-018-0018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268, 2008. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 80. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol 18: 2114–2127, 2021. doi: 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, Thorburn A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep 7: 45–52, 2014. doi: 10.1016/j.celrep.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18: 571–580, 2011. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell 55: 451–466, 2014. doi: 10.1016/j.molcel.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 86. Lim J, Park H, Heisler J, Maculins T, Roose-Girma M, Xu M, Mckenzie B, van Lookeren Campagne M, Newton K, Murthy A. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife 8: e44452, 2019. doi: 10.7554/eLife.44452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 4: e128834, 2019. doi: 10.1172/jci.insight.128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sureshbabu A, Patino E, Ma KC, Laursen K, Finkelsztein EJ, Akchurin O, Muthukumar T, Ryter SW, Gudas L, Choi AMK, Choi ME. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight 3: e98411, 2018. doi: 10.1172/jci.insight.98411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chen J, Wang S, Fu R, Zhou M, Zhang T, Pan W, Yang N, Huang Y. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J Transl Med 16: 233, 2018. doi: 10.1186/s12967-018-1606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hepokoski M, Singh P. Mitochondria as mediators of systemic inflammation and organ cross talk in acute kidney injury. Am J Physiol Renal Physiol 322: F589–F596, 2022. doi: 10.1152/ajprenal.00372.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol 23: 159–173, 2023. doi: 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol 23: 487–500, 2022. doi: 10.1038/s41590-022-01132-2. [DOI] [PubMed] [Google Scholar]
- 94. Rabas N, Palmer S, Mitchell L, Ismail S, Gohlke A, Riley JS, Tait SWG, Gammage P, Soares LL, Macpherson IR, Norman JC. PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J Cell Biol 220: e202006049, 2021. doi: 10.1083/jcb.202006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jin L, Yu B, Armando I, Han F. Mitochondrial DNA-mediated inflammation in acute kidney injury and chronic kidney disease. Oxid Med Cell Longev 2021: 9985603, 2021. doi: 10.1155/2021/9985603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kronenberg F, Eckardt KU. Mitochondrial DNA and kidney function. Clin J Am Soc Nephrol 17: 942–944, 2022. doi: 10.2215/CJN.05820522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Eirin A, Saad A, Woollard JR, Juncos LA, Calhoun DA, Tang H, Lerman A, Textor SC, Lerman LO. Glomerular hyperfiltration in obese African American hypertensive patients is associated with elevated urinary mitochondrial-DNA copy number. Am J Hypertens 30: 1112–1119, 2017. [Erratum in Am J Hypertens 30: 1145, 2017]. doi: 10.1093/ajh/hpx103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414, 2012. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, Palmer M, Susztak K. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab 30: 784–799.e5, 2019. doi: 10.1016/j.cmet.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of mitochondrial DNA in septic AKI via Toll-like receptor 9. J Am Soc Nephrol 27: 2009–2020, 2016. doi: 10.1681/ASN.2015040376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Han SJ, Williams RM, D'Agati V, Jaimes EA, Heller DA, Lee HT. Selective nanoparticle-mediated targeting of renal tubular Toll-like receptor 9 attenuates ischemic acute kidney injury. Kidney Int 98: 76–87, 2020. doi: 10.1016/j.kint.2020.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Whitaker RM, Stallons LJ, Kneff JE, Alge JL, Harmon JL, Rahn JJ, Arthur JM, Beeson CC, Chan SL, Schnellmann RG. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney Int 88: 1336–1344, 2015. doi: 10.1038/ki.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Eirin A, Saad A, Tang H, Herrmann SM, Woollard JR, Lerman A, Textor SC, Lerman LO. Urinary mitochondrial DNA copy number identifies chronic renal injury in hypertensive patients. Hypertension 68: 401–410, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wei PZ, Kwan BC, Chow KM, Cheng PM, Luk CC, Li PK, Szeto CC. Urinary mitochondrial DNA level is an indicator of intra-renal mitochondrial depletion and renal scarring in diabetic nephropathy. Nephrol Dial Transplant 33: 784–788, 2018. doi: 10.1093/ndt/gfx339. [DOI] [PubMed] [Google Scholar]
- 105. Fazzini F, Lamina C, Fendt L, Schultheiss UT, Kotsis F, Hicks AA, Meiselbach H, Weissensteiner H, Forer L, Krane V, Eckardt KU, Köttgen A, Kronenberg F; GCKD Investigators. Mitochondrial DNA copy number is associated with mortality and infections in a large cohort of patients with chronic kidney disease. Kidney Int 96: 480–488, 2019. doi: 10.1016/j.kint.2019.04.021. [DOI] [PubMed] [Google Scholar]
- 106. He WJ, Li C, Huang Z, Geng S, Rao VS, Kelly TN, Hamm LL, Grams ME, Arking DE, Appel LJ, Rebholz CM; CRIC Study Investigators. Association of mitochondrial DNA copy number with risk of progression of kidney disease. Clin J Am Soc Nephrol 17: 966–975, 2022. doi: 10.2215/CJN.15551121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Konecna B, Park J, Kwon WY, Vlkova B, Zhang Q, Huang W, Kim HI, Yaffe MB, Otterbein LE, Itagaki K, Hauser CJ. Monocyte exocytosis of mitochondrial danger-associated molecular patterns in sepsis suppresses neutrophil chemotaxis. J Trauma Acute Care Surg 90: 46–53, 2021. doi: 10.1097/TA.0000000000002973. [DOI] [PubMed] [Google Scholar]
- 108. Lee BL, Barton GM. Trafficking of endosomal Toll-like receptors. Trends Cell Biol 24: 360–369, 2014. doi: 10.1016/j.tcb.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, Ichimura T, Kuchroo V, Bonventre JV. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest 125: 1620–1636, 2015. doi: 10.1172/JCI75417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gerhardt LMS, Liu J, Koppitch K, Cippà PE, McMahon AP. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci USA 118: e2026684118, 2021. doi: 10.1073/pnas.2026684118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214: 333–345, 2016. doi: 10.1083/jcb.201603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hall AM, Unwin RJ, Parker N, Duchen MR. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol 20: 1293–1302, 2009. doi: 10.1681/ASN.2008070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol 40: 123–137, 2013. doi: 10.1111/1440-1681.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sureshbabu A, Ryter SW, Choi ME. Oxidative stress and autophagy: crucial modulators of kidney injury. Redox Biol 4: 208–214, 2015. doi: 10.1016/j.redox.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers 7: 52, 2021. doi: 10.1038/s41572-021-00284-z. [DOI] [PubMed] [Google Scholar]
- 116. Sun M, Li J, Mao L, Wu J, Deng Z, He M, An S, Zeng Z, Huang Q, Chen Z. p53 deacetylation alleviates sepsis-induced acute kidney injury by promoting autophagy. Front Immunol 12: 685523, 2021. doi: 10.3389/fimmu.2021.685523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 294: F777–F787, 2008. doi: 10.1152/ajprenal.00590.2007. [DOI] [PubMed] [Google Scholar]
- 118. Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74: 631–640, 2008. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 119. Herzog C, Yang C, Holmes A, Kaushal GP. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am J Physiol Renal Physiol 303: F1239–F1250, 2012. doi: 10.1152/ajprenal.00659.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 176: 1181–1192, 2010. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhao C, Chen Z, Qi J, Duan S, Huang Z, Zhang C, Wu L, Zeng M, Zhang B, Wang N, Mao H, Zhang A, Xing C, Yuan Y. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 8: 20988–21000, 2017. doi: 10.18632/oncotarget.15470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tang C, Han H, Liu Z, Liu Y, Yin L, Cai J, He L, Liu Y, Chen G, Zhang Z, Yin XM, Dong Z. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis 10: 677, 2019. doi: 10.1038/s41419-019-1899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Turkmen K, Martin J, Akcay A, Nguyen Q, Ravichandran K, Faubel S, Pacic A, Ljubanović D, Edelstein CL, Jani A. Apoptosis and autophagy in cold preservation ischemia. Transplantation 91: 1192–1197, 2011. doi: 10.1097/TP.0b013e31821ab9c8. [DOI] [PubMed] [Google Scholar]
- 124. Kuwabara S, Goggins E, Okusa MD. The pathophysiology of sepsis-associated AKI. Clin J Am Soc Nephrol 17: 1050–1069, 2022. doi: 10.2215/CJN.00850122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Korneev KV. Mouse models of sepsis and septic shock. Mol Biol (Mosk) 53: 799–814, 2019. doi: 10.1134/S0026898419050100. [DOI] [PubMed] [Google Scholar]
- 126. Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, Hsu C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 37: 289–296, 2012. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- 127. Zhang Y, Wang L, Meng L, Cao G, Wu Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle 18: 425–436, 2019. doi: 10.1080/15384101.2019.1568746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int 89: 779–791, 2016. doi: 10.1016/j.kint.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Molitoris BA, Dagher PC, Sandoval RM, Campos SB, Ashush H, Fridman E, Brafman A, Faerman A, Atkinson SJ, Thompson JD, Kalinski H, Skaliter R, Erlich S, Feinstein E. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol 20: 1754–1764, 2009. doi: 10.1681/ASN.2008111204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Howell GM, Gomez H, Collage RD, Loughran P, Zhang X, Escobar DA, Billiar TR, Zuckerbraun BS, Rosengart MR. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One 8: e69520, 2013. doi: 10.1371/journal.pone.0069520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Escobar DA, Botero-Quintero AM, Kautza BC, Luciano J, Loughran P, Darwiche S, Rosengart MR, Zuckerbraun BS, Gomez H. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J Surg Res 194: 262–272, 2015. doi: 10.1016/j.jss.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zhao J, Zheng H, Sui Z, Jing F, Quan X, Zhao W, Liu G. Ursolic acid exhibits anti-inflammatory effects through blocking TLR4-MyD88 pathway mediated by autophagy. Cytokine 123: 154726, 2019. doi: 10.1016/j.cyto.2019.05.013. [DOI] [PubMed] [Google Scholar]
- 133. Krüger B, Krick S, Dhillon N, Lerner SM, Ames S, Bromberg JS, Lin M, Walsh L, Vella J, Fischereder M, Krämer BK, Colvin RB, Heeger PS, Murphy BT, Schröppel B. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci USA 106: 3390–3395, 2009. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC, Au WS, Chan KW, Lai KN, Tang SC. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol 23: 86–102, 2012. doi: 10.1681/ASN.2010111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Liu JX, Yang C, Zhang WH, Su HY, Liu ZJ, Pan Q, Liu HF. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci 235: 116828, 2019. doi: 10.1016/j.lfs.2019.116828. [DOI] [PubMed] [Google Scholar]
- 136. Gao Y, Dai X, Li Y, Li G, Lin X, Ai C, Cao Y, Li T, Lin B. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J Transl Med 18: 114, 2020. doi: 10.1186/s12967-020-02283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Chen ZD, Hu BC, Shao XP, Hong J, Zheng Y, Zhang R, Shao ZQ, Liu JQ, Yang XH, Sun RH, Mo SJ. Ascorbate uptake enables tubular mitophagy to prevent septic AKI by PINK1-PARK2 axis. Biochem Biophys Res Commun 554: 158–165, 2021. doi: 10.1016/j.bbrc.2021.03.103. [DOI] [PubMed] [Google Scholar]
- 138. Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 285: F610–F618, 2003. doi: 10.1152/ajprenal.00101.2003. [DOI] [PubMed] [Google Scholar]
- 139. Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, Chen X, Han J. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol 26: 2647–2658, 2015. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wang Y, Liu Z, Shu S, Cai J, Tang C, Dong Z. AMPK/mTOR signaling in autophagy regulation during cisplatin-induced acute kidney injury. Front Physiol 11: 619730, 2020. doi: 10.3389/fphys.2020.619730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kimura A, Ishida Y, Inagaki M, Nakamura Y, Sanke T, Mukaida N, Kondo T. Interferon-γ is protective in cisplatin-induced renal injury by enhancing autophagic flux. Kidney Int 82: 1093–1104, 2012. doi: 10.1038/ki.2012.240. [DOI] [PubMed] [Google Scholar]
- 142. Zhang D, Pan J, Xiang X, Liu Y, Dong G, Livingston MJ, Chen JK, Yin XM, Dong Z. Protein kinase Cδ suppresses autophagy to induce kidney cell apoptosis in cisplatin nephrotoxicity. J Am Soc Nephrol 28: 1131–1144, 2017. doi: 10.1681/ASN.2016030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Shen Q, Zhang X, Li Q, Zhang J, Lai H, Gan H, Du X, Li M. TLR2 protects cisplatin-induced acute kidney injury associated with autophagy via PI3K/Akt signaling pathway. J Cell Biochem 120: 4366–4374, 2019. doi: 10.1002/jcb.27722. [DOI] [PubMed] [Google Scholar]
- 144. Minami S, Nakamura S. Therapeutic potential of beclin1 for transition from AKI to CKD: autophagy-dependent and autophagy-independent functions. Kidney Int 101: 13–15, 2022. doi: 10.1016/j.kint.2021.10.021. [DOI] [PubMed] [Google Scholar]
- 145. Lynch MR, Tran MT, Ralto KM, Zsengeller ZK, Raman V, Bhasin SS, Sun N, Chen X, Brown D, Rovira II, Taguchi K, Brooks CR, Stillman IE, Bhasin MK, Finkel T, Parikh SM. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 5: e126749, 2019. [Erratum in JCI Insight 5: e142898, 2020]. doi: 10.1172/jci.insight.126749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Geng G, Liu J, Xu C, Pei Y, Chen L, Mu C, Wang D, Gao J, Li Y, Liang J, Zhao T, Zhang C, Zhou J, Chen Q, Zhu Y, Shi L. Receptor-mediated mitophagy regulates EPO production and protects against renal anemia. Elife 10: e64480, 2021. doi: 10.7554/eLife.64480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Tammaro A, Kers J, Scantlebery AML, Florquin S. Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: the fine balance between adaptive repair and tissue degeneration. Front Immunol 11: 1346, 2020. doi: 10.3389/fimmu.2020.01346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lau A, Rahn JJ, Chappellaz M, Chung H, Benediktsson H, Bihan D, von Mässenhausen A, Linkermann A, Jenne CN, Robbins SM, Senger DL, Lewis IA, Chun J, Muruve DA. Dipeptidase-1 governs renal inflammation during ischemia reperfusion injury. Sci Adv 8: eabm0142, 2022. doi: 10.1126/sciadv.abm0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, Imai E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 368: 100–106, 2008. doi: 10.1016/j.bbrc.2008.01.059. [DOI] [PubMed] [Google Scholar]
- 150. Isaka Y, Suzuki C, Abe T, Okumi M, Ichimaru N, Imamura R, Kakuta Y, Matsui I, Takabatake Y, Rakugi H, Shimizu S, Takahara S. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant Proc 41: 52–54, 2009. doi: 10.1016/j.transproceed.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 151. Zhang YL, Zhang J, Cui LY, Yang S. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp Biol Med (Maywood) 240: 1590–1598, 2015. doi: 10.1177/1535370215581306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lempiäinen J, Finckenberg P, Mervaala EE, Sankari S, Levijoki J, Mervaala EM. Caloric restriction ameliorates kidney ischaemia/reperfusion injury through PGC-1α-eNOS pathway and enhanced autophagy. Acta Physiol (Oxf) 208: 410–421, 2013. doi: 10.1111/apha.12120. [DOI] [PubMed] [Google Scholar]
- 153. Soto-Pantoja DR, Ridnour LA, Wink DA, Roberts DD. Blockade of CD47 increases survival of mice exposed to lethal total body irradiation. Sci Rep 3: 1038, 2013. doi: 10.1038/srep01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. El-Rashid M, Ghimire K, Sanganeria B, Lu B, Rogers NM. CD47 limits autophagy to promote acute kidney injury. FASEB J 33: 12735–12749, 2019. doi: 10.1096/fj.201900120RR. [DOI] [PubMed] [Google Scholar]
- 155. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185, 2012. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 156. Gall JM, Wang Z, Liesa M, Molina A, Havasi A, Schwartz JH, Shirihai O, Borkan SC, Bonegio RG. Role of mitofusin 2 in the renal stress response. PLoS One 7: e31074, 2012. doi: 10.1371/journal.pone.0031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, Youle RJ. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 216: 3231–3247, 2017. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Perry HM, Huang L, Wilson RJ, Bajwa A, Sesaki H, Yan Z, Rosin DL, Kashatus DF, Okusa MD. Dynamin-related protein 1 deficiency promotes recovery from AKI. J Am Soc Nephrol 29: 194–206, 2018. doi: 10.1681/ASN.2017060659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, Massy Z, Wanner C, Anders HJ. Chronic kidney disease. Nat Rev Dis Primers 3: 17088, 2017. doi: 10.1038/nrdp.2017.88. [DOI] [PubMed] [Google Scholar]
- 160. Tang C, He L, Liu J, Dong Z. Mitophagy: basic mechanism and potential role in kidney diseases. Kidney Dis (Basel) 1: 71–79, 2015. doi: 10.1159/000381510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Zuo Z, Jing K, Wu H, Wang S, Ye L, Li Z, Yang C, Pan Q, Liu WJ, Liu HF. Mechanisms and functions of mitophagy and potential roles in renal disease. Front Physiol 11: 935, 2020. doi: 10.3389/fphys.2020.00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kim WY, Nam SA, Song HC, Ko JS, Park SH, Kim HL, Choi EJ, Kim YS, Kim J, Kim YK. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology (Carlton) 17: 148–159, 2012. doi: 10.1111/j.1440-1797.2011.01541.x. [DOI] [PubMed] [Google Scholar]
- 163. Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem 287: 11677–11688, 2012. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yang S, Abdulla R, Lu C, Zhang L. Inhibition of microRNA-376b protects against renal interstitial fibrosis via inducing macrophage autophagy by upregulating Atg5 in mice with chronic kidney disease. Kidney Blood Press Res 43: 1749–1764, 2018. doi: 10.1159/000495394. [DOI] [PubMed] [Google Scholar]
- 165. Liu B, Sun T, Li H, Qiu S, Li Y, Zhang D. Proximal tubular RAGE mediated the renal fibrosis in UUO model mice via upregulation of autophagy. Cell Death Dis 13: 399, 2022. doi: 10.1038/s41419-022-04856-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Xu Y, Yang S, Huang J, Ruan S, Zheng Z, Lin J. Tgf-β1 induces autophagy and promotes apoptosis in renal tubular epithelial cells. Int J Mol Med 29: 781–790, 2012. doi: 10.3892/ijmm.2012.911. [DOI] [PubMed] [Google Scholar]
- 167. Bhansali S, Bhansali A, Walia R, Saikia UN, Dhawan V. Alterations in mitochondrial oxidative stress and mitophagy in subjects with prediabetes and type 2 diabetes mellitus. Front Endocrinol (Lausanne) 8: 347, 2017. doi: 10.3389/fendo.2017.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Li S, Lin Q, Shao X, Zhu X, Wu J, Wu B, Zhang M, Zhou W, Zhou Y, Jin H, Zhang Z, Qi C, Shen J, Mou S, Gu L, Ni Z. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic Biol Med 152: 632–649, 2020. doi: 10.1016/j.freeradbiomed.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 169. Jin L, Yu B, Liu G, Nie W, Wang J, Chen J, Xiao L, Xia H, Han F, Yang Y. Mitophagy induced by UMI-77 preserves mitochondrial fitness in renal tubular epithelial cells and alleviates renal fibrosis. FASEB J 36: e22342, 2022. doi: 10.1096/fj.202200199RR. [DOI] [PubMed] [Google Scholar]
- 170. Liu T, Yang Q, Zhang X, Qin R, Shan W, Zhang H, Chen X. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci 257: 118116, 2020. doi: 10.1016/j.lfs.2020.118116. [DOI] [PubMed] [Google Scholar]
- 171. Guo H, Wang Y, Zhang X, Zang Y, Zhang Y, Wang L, Wang H, Wang Y, Cao A, Peng W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKα-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci Rep 7: 6852, 2017. doi: 10.1038/s41598-017-07061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Dong C, Zheng H, Huang S, You N, Xu J, Ye X, Zhu Q, Feng Y, You Q, Miao H, Ding D, Lu Y. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp Cell Res 337: 146–159, 2015. doi: 10.1016/j.yexcr.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 173. Brijmohan AS, Batchu SN, Majumder S, Alghamdi TA, Thieme K, McGaugh S, Liu Y, Advani SL, Bowskill BB, Kabir MG, Geldenhuys L, Siddiqi FS, Advani A. HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front Pharmacol 9: 34, 2018. doi: 10.3389/fphar.2018.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, Koepsell H, Thomson SC, Rieg T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 306: F194–F204, 2014. doi: 10.1152/ajprenal.00520.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Lee YH, Kim SH, Kang JM, Heo JH, Kim DJ, Park SH, Sung M, Kim J, Oh J, Yang DH, Lee SH, Lee SY. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am J Physiol Renal Physiol 317: F767–F780, 2019. doi: 10.1152/ajprenal.00565.2018. [DOI] [PubMed] [Google Scholar]
- 176. Coughlan MT, Patel SK, Jerums G, Penfold SA, Nguyen TV, Sourris KC, Panagiotopoulos S, Srivastava PM, Cooper ME, Burrell LM, Macisaac RJ, Forbes JM. Advanced glycation urinary protein-bound biomarkers and severity of diabetic nephropathy in man. Am J Nephrol 34: 347–355, 2011. doi: 10.1159/000331064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Chen J, Zhao D, Zhu M, Zhang M, Hou X, Ding W, Sun S, Bu W, Feng L, Ma S, Jia X. Paeoniflorin ameliorates AGEs-induced mesangial cell injury through inhibiting RAGE/mTOR/autophagy pathway. Biomed Pharmacother 89: 1362–1369, 2017. doi: 10.1016/j.biopha.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 178. Takahashi A, Takabatake Y, Kimura T, Maejima I, Namba T, Yamamoto T, Matsuda J, Minami S, Kaimori JY, Matsui I, Matsusaka T, Niimura F, Yoshimori T, Isaka Y. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes 66: 1359–1372, 2017. doi: 10.2337/db16-0397. [DOI] [PubMed] [Google Scholar]
- 179. Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstädt H, Kerjaschki D, Cohen CD, Hall MN, Rüegg MA, Inoki K, Walz G, Huber TB. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121: 2181–2196, 2011. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Xiao T, Guan X, Nie L, Wang S, Sun L, He T, Huang Y, Zhang J, Yang K, Wang J, Zhao J. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem 394: 145–154, 2014. doi: 10.1007/s11010-014-2090-7. [DOI] [PubMed] [Google Scholar]
- 182. Audzeyenka I, Rogacka D, Piwkowska A, Angielski S, Jankowski M. Viability of primary cultured podocytes is associated with extracellular high glucose-dependent autophagy downregulation. Mol Cell Biochem 430: 11–19, 2017. doi: 10.1007/s11010-017-2949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Wei Q, Dong Z. HDAC4 blocks autophagy to trigger podocyte injury: non-epigenetic action in diabetic nephropathy. Kidney Int 86: 666–668, 2014. doi: 10.1038/ki.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wang X, Liu J, Zhen J, Zhang C, Wan Q, Liu G, Wei X, Zhang Y, Wang Z, Han H, Xu H, Bao C, Song Z, Zhang X, Li N, Yi F. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int 86: 712–725, 2014. doi: 10.1038/ki.2014.111. [DOI] [PubMed] [Google Scholar]
- 185. Huang SS, Ding DF, Chen S, Dong CL, Ye XL, Yuan YG, Feng YM, You N, Xu JR, Miao H, You Q, Lu X, Lu YB. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci Rep 7: 45692, 2017. doi: 10.1038/srep45692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wen D, Huang X, Zhang M, Zhang L, Chen J, Gu Y, Hao CM. Resveratrol attenuates diabetic nephropathy via modulating angiogenesis. PLoS One 8: e82336, 2013. doi: 10.1371/journal.pone.0082336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Chiang CK, Wang CC, Lu TF, Huang KH, Sheu ML, Liu SH, Hung KY. Involvement of endoplasmic reticulum stress, autophagy, and apoptosis in advanced glycation end products-induced glomerular mesangial cell injury. Sci Rep 6: 34167, 2016. doi: 10.1038/srep34167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Chen K, Yu B, Liao J. LncRNA SOX2OT alleviates mesangial cell proliferation and fibrosis in diabetic nephropathy via Akt/mTOR-mediated autophagy. Mol Med 27: 71, 2021. doi: 10.1186/s10020-021-00310-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Huang C, Zhang Y, Kelly DJ, Tan CY, Gill A, Cheng D, Braet F, Park JS, Sue CM, Pollock CA, Chen XM. Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci Rep 6: 29196, 2016. doi: 10.1038/srep29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, Wang J, Qin Y, Liu Y, Tang C, He L, Greka A, Zhou Z, Liu F, Dong Z, Sun L. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol 11: 297–311, 2017. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chen K, Dai H, Yuan J, Chen J, Lin L, Zhang W, Wang L, Zhang J, Li K, He Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis 9: 105, 2018. doi: 10.1038/s41419-017-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Li W, Du M, Wang Q, Ma X, Wu L, Guo F, Ji H, Huang F, Qin G. FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/Parkin pathway. Endocrinology 158: 2155–2167, 2017. doi: 10.1210/en.2016-1970. [DOI] [PubMed] [Google Scholar]
- 193. Guo F, Wang W, Song Y, Wu L, Wang J, Zhao Y, Ma X, Ji H, Liu Y, Li Z, Qin G. LncRNA SNHG17 knockdown promotes Parkin-dependent mitophagy and reduces apoptosis of podocytes through Mst1. Cell Cycle 19: 1997–2006, 2020. doi: 10.1080/15384101.2020.1783481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Lu X, Fan Q, Xu L, Li L, Yue Y, Xu Y, Su Y, Zhang D, Wang L. Ursolic acid attenuates diabetic mesangial cell injury through the up-regulation of autophagy via miRNA-21/PTEN/Akt/mTOR suppression. PLoS One 10: e0117400, 2015. doi: 10.1371/journal.pone.0117400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Czajka A, Ajaz S, Gnudi L, Parsade CK, Jones P, Reid F, Malik AN. Altered mitochondrial function, mitochondrial dna and reduced metabolic flexibility in patients with diabetic nephropathy. EBioMedicine 2: 499–512, 2015. doi: 10.1016/j.ebiom.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Sun J, Zhu H, Wang X, Gao Q, Li Z, Huang H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J Endocrinol. 240: 445–465, 2019. doi: 10.1530/JOE-18-0578. [DOI] [PubMed] [Google Scholar]
- 197. Han YC, Tang SQ, Liu YT, Li AM, Zhan M, Yang M, Song N, Zhang W, Wu XQ, Peng CH, Zhang H, Yang S. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis 12: 925, 2021. doi: 10.1038/s41419-021-04184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Chen X, Liu W, Xiao J, Zhang Y, Chen Y, Luo C, Huang Q, Peng F, Gong W, Li S, He X, Zhuang Y, Wu N, Liu Y, Wang Y, Long H. FOXO3a accumulation and activation accelerate oxidative stress-induced podocyte injury. FASEB J 34: 13300–13316, 2020. doi: 10.1096/fj.202000783R. [DOI] [PubMed] [Google Scholar]
- 199. Jiang XS, Chen XM, Hua W, He JL, Liu T, Li XJ, Wan JM, Gan H, Du XG. PINK1/Parkin mediated mitophagy ameliorates palmitic acid-induced apoptosis through reducing mitochondrial ROS production in podocytes. Biochem Biophys Res Commun 525: 954–961, 2020. doi: 10.1016/j.bbrc.2020.02.170. [DOI] [PubMed] [Google Scholar]
- 200. Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(+/)-(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J 432: 9–19, 2010. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, Yang M, Yang S, Zhu X, Yuan S, Liu F, Xiao L, Kanwar YS, Sun L. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol 16: 32–46, 2018. [Erratum in Redox Biol 24: 101216, 2019]. doi: 10.1016/j.redox.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Chen K, Chen J, Wang L, Yang J, Xiao F, Wang X, Yuan J, Wang L, He Y. Parkin ubiquitinates GATA4 and attenuates the GATA4/GAS1 signaling and detrimental effects on diabetic nephropathy. FASEB J 34: 8858–8875, 2020. doi: 10.1096/fj.202000053R. [DOI] [PubMed] [Google Scholar]
- 203. Feng J, Lu C, Dai Q, Sheng J, Xu M. SIRT3 facilitates amniotic fluid stem cells to repair diabetic nephropathy through protecting mitochondrial homeostasis by modulation of mitophagy. Cell Physiol Biochem 46: 1508–1524, 2018. doi: 10.1159/000489194. [DOI] [PubMed] [Google Scholar]
- 204. Kato M, Abdollahi M, Tunduguru R, Tsark W, Chen Z, Wu X, Wang J, Chen ZB, Lin FM, Lanting L, Wang M, Huss J, Fueger PT, Chan D, Natarajan R. miR-379 deletion ameliorates features of diabetic kidney disease by enhancing adaptive mitophagy via FIS1. Commun Biol 4: 30, 2021. [Erratum in Commun Biol 4: 175, 2021]. doi: 10.1038/s42003-020-01516-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Kopp JB, Anders HJ, Susztak K, Podestà MA, Remuzzi G, Hildebrandt F, Romagnani P. Podocytopathies. Nat Rev Dis Primers 6: 68, 2020. doi: 10.1038/s41572-020-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Sato S, Kitamura H, Adachi A, Sasaki Y, Ghazizadeh M. Two types of autophagy in the podocytes in renal biopsy specimens: ultrastructural study. J Submicrosc Cytol Pathol 38: 167–174, 2006. [PubMed] [Google Scholar]
- 207. Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong Y, Zhang C, Zen K, Liu Z. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol 234: 203–213, 2014. doi: 10.1002/path.4382. [DOI] [PubMed] [Google Scholar]
- 208. Chou YH, Lien YC, Hu FC, Lin WC, Kao CC, Lai CF, Chiang WC, Lin SL, Tsai TJ, Wu KD, Chen YM. Clinical outcomes and predictors for ESRD and mortality in primary GN. Clin J Am Soc Nephrol 7: 1401–1408, 2012. doi: 10.2215/CJN.04500511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. da Silva CA, Monteiro M, Araújo LS, Urzedo MG, Rocha LB, Dos Reis MA, Machado JR. In situ evaluation of podocytes in patients with focal segmental glomerulosclerosis and minimal change disease. PLoS One 15: e0241745, 2020. doi: 10.1371/journal.pone.0241745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Yi M, Zhang L, Liu Y, Livingston MJ, Chen JK, Nahman NS Jr, Liu F, Dong Z. Autophagy is activated to protect against podocyte injury in adriamycin-induced nephropathy. Am J Physiol Renal Physiol 313: F74–F84, 2017. doi: 10.1152/ajprenal.00114.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Cinà DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, Jurisicova A, Quaggin SE. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol 23: 412–420, 2012. doi: 10.1681/ASN.2011070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, Sohn D, Sihn G, Rousselle A, Fokuhl V, Maschke U, Purfürst B, Schneider W, Rump LC, Luft FC, Dechend R, Bader M, Huber TB, Nguyen G, Muller DN. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol 22: 2193–2202, 2011. doi: 10.1681/ASN.2011020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Kang YL, Saleem MA, Chan KW, Yung BY, Law HK. Trehalose, an mTOR independent autophagy inducer, alleviates human podocyte injury after puromycin aminonucleoside treatment. PLoS One 9: e113520, 2014. doi: 10.1371/journal.pone.0113520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Yamamoto-Nonaka K, Koike M, Asanuma K, Takagi M, Oliva Trejo JA, Seki T, Hidaka T, Ichimura K, Sakai T, Tada N, Ueno T, Uchiyama Y, Tomino Y. Cathepsin D in podocytes is important in the pathogenesis of proteinuria and CKD. J Am Soc Nephrol 27: 2685–2700, 2016. doi: 10.1681/ASN.2015040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Papeta N, Kiryluk K, Patel A, Sterken R, Kacak N, Snyder HJ, Imus PH, Mhatre AN, Lawani AK, Julian BA, Wyatt RJ, Novak J, Wyatt CM, Ross MJ, Winston JA, Klotman ME, Cohen DJ, Appel GB, D'Agati VD, Klotman PE, Gharavi AG. APOL1 variants increase risk for FSGS and HIVAN but not IgA nephropathy. J Am Soc Nephrol 22: 1991–1996, 2011. doi: 10.1681/ASN.2011040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu CA, Rohacs T, Inoue K, Ishibe S, Saleem MA, Palmer MB, Cuervo AM, Kopp JB, Susztak K. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23: 429–438, 2017. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Chun J, Zhang JY, Wilkins MS, Subramanian B, Riella C, Magraner JM, Alper SL, Friedman DJ, Pollak MR. Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc Natl Acad Sci USA 116: 3712–3721, 2019. doi: 10.1073/pnas.1820414116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Xu D, Chen P, Wang B, Wang Y, Miao N, Yin F, Cheng Q, Zhou Z, Xie H, Zhou L, Liu J, Wang X, Zent R, Lu L, Zhang W. NIX-mediated mitophagy protects against proteinuria-induced tubular cell apoptosis and renal injury. Am J Physiol Renal Physiol 316: F382–F395, 2019. doi: 10.1152/ajprenal.00360.2018. [DOI] [PubMed] [Google Scholar]
- 219. Erkan E, Devarajan P, Schwartz GJ. Mitochondria are the major targets in albumin-induced apoptosis in proximal tubule cells. J Am Soc Nephrol 18: 1199–1208, 2007. doi: 10.1681/ASN.2006040407. [DOI] [PubMed] [Google Scholar]
- 220. Zhuang Y, Ding G, Zhao M, Bai M, Yang L, Ni J, Wang R, Jia Z, Huang S, Zhang A. NLRP3 inflammasome mediates albumin-induced renal tubular injury through impaired mitochondrial function. J Biol Chem 289: 25101–25111, 2014. doi: 10.1074/jbc.M114.578260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Duan P, Tan J, Miao Y, Zhang Q. PINK1/Parkin-mediated mitophagy plays a protective role in albumin overload-induced renal tubular cell injury. Front Biosci (Landmark Ed) 27: 184, 2022. doi: 10.31083/j.fbl2706184. [DOI] [PubMed] [Google Scholar]
- 222. Bork T, Liang W, Yamahara K, Lee P, Tian Z, Liu S, Schell C, Thedieck K, Hartleben B, Patel K, Tharaux PL, Lenoir O, Huber TB. Podocytes maintain high basal levels of autophagy independent of mTOR signaling. Autophagy 16: 1932–1948, 2020. doi: 10.1080/15548627.2019.1705007. [DOI] [PMC free article] [PubMed] [Google Scholar]