

Abstract
Objective
Asymptomatic renal immunoglobulin A (IgA) deposition occurs in healthy subjects, but its etiologic role in disease is unclear. Galactose-deficient IgA1 (Gd-IgA1) is involved in the pathogenesis of IgA nephropathy. We investigated Gd-IgA1 deposition in transplanted kidneys that were considered healthy showing subclinical latent IgA deposition one hour after transplantation.
Methods
A total of 723 transplanted kidney specimens biopsied 1 h after kidney transplantation from 2009 to 2016 at Nagoya Red Cross Hospital were examined. A total of 81 cases of IgA deposition were extracted, and 41 were ultimately studied. Double immunofluorescence staining for Gd-IgA1 and IgA was conducted to investigate the role of Gd-IgA1 in subclinical IgA deposition.
Results
Light microscopy findings for the 41 cases indicated only minor glomerular abnormalities. Immunofluorescence analyses revealed that all cases were positive for IgA. C3, IgG, and IgM positivity rates were 78.0%, 7.3%, and 60.9%, respectively. All 41 cases were positive for Gd-IgA1, which merged with IgA deposition in immunofluorescence double staining. IgA disappeared in 26 of 40 cases (65.0%) 1 year after kidney transplantation. In contrast, IgA redeposition was observed in three cases.
Conclusion
Gd-IgA1 was demonstrated in all transplanted kidneys, with latent IgA deposition noted in otherwise healthy kidneys. Deposition of Gd-IgA1 might indicate the initial stage of IgA nephropathy; however, additional factors, such as IgG deposition, are required for the ultimate development of IgA nephropathy.
Keywords: IgA nephropathy, latent IgA deposition, galactose-deficient IgA1, donor kidney
Introduction
Immunoglobulin A (IgA) nephropathy (IgAN) is the most common form of chronic glomerulonephritis in Japan. The condition was named as such based on the pathologic characteristics of IgA deposition in the glomeruli. Patients with IgAN often present with hematuria, proteinuria, hypertension, and renal dysfunction, and 15-20% progress to end-stage renal disease (ESRD) within 10 years (1). A recent study demonstrated the efficacy of tonsillectomy in preventing progression to renal failure in IgAN patients (2); however, therapeutic strategies for treating IgAN have not been fully established (3).
A definitive diagnosis of IgAN requires a renal biopsy to demonstrate IgA immune complex deposition in the mesangial region. However, renal IgA deposition can occur in other diseases, such as chronic liver disease (4), human immunodeficiency virus infection (5), and various immune and inflammatory diseases (6). In one study, subclinical IgA deposition was also observed in 82 of 510 (16.1%) living donor kidneys in which the renal function met the donor adaptation criteria, causing them to be considered healthy kidneys (7).
In recent years, galactose-deficient IgA1 (Gd-IgA1) has attracted attention as a significant factor in the pathogenesis and progression of IgAN. KM55Ⓡ, a monoclonal antibody specific to Gd-IgA1, enables immunohistologic detection of Gd-IgA1 in kidney tissue (8). In a previous study using KM55Ⓡ, Gd-IgA1 was identified in glomeruli in patients with IgAN and purpura nephritis but not in glomeruli of kidneys of patients with other diseases involving IgA deposition, such as systemic lupus erythematosus (9). However, in another study, Gd-IgA1 was reportedly detected in secondary IgAN (10).
It is difficult to evaluate the role of subclinical latent IgA deposition in healthy kidney specimens and follow its course. Evaluating renal grafts biopsied one hour after kidney transplantation (protocol biopsies one hour after kidney transplantation) is considered a useful approach for addressing this issue and elucidating the role of latent IgA deposition in healthy kidneys.
We therefore conducted an immunohistochemical investigation of Gd-IgA1 deposition using the KM55Ⓡ antibody in donor kidneys with mesangial latent IgA deposition. Kidneys were evaluated one hour after renal transplantation to elucidate the role of Gd-IgA1 in latent mesangial IgA deposition.
Materials and Methods
Patients
In this retrospective cohort study covering the period from January 1, 2009, to December 31, 2016, a total of 723 kidney transplant biopsies (conducted 1 h after transplantation) performed at Japanese Red Cross Nagoya Daini Hospital (Nagoya, Japan) were examined. IgA nephropathy, IgA vasculitis, and lupus nephritis were also examined.
All procedures performed in this study were in accordance with the ethical standards of the Ethics Committee for Human Research of Japanese Red Cross Nagoya Daini Hospital (Nagoya, Japan) and Aichi Medical University Hospital (Nagakute, Japan), where the studies were conducted (approval numbers: 2019-190 and 1295S, respectively), and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
A flow diagram illustrating the patient selection process is shown in Fig. 1. A total of 54 specimens were excluded because the frozen tissues did not contain glomeruli, which precluded routine immunofluorescence studies, including the analysis of IgA. Based on the criteria for selecting donor kidneys with “mesangial subclinical IgA deposition” evaluated by immunofluorescence microscopy on frozen sections in the absence of urinary abnormalities (urine protein [-] and <5 red blood cells per high-power field) and the absence of glomerular abnormalities, 81 cases were extracted. We excluded specimens from three patients due to hematuria and proteinuria. In addition, specimens from 32 patients were excluded due to loss of paraffin sections or an insufficient amount of preserved sections, and specimens from 5 patients were also excluded due to a lack of follow-up information. Ultimately, specimens from 41 patients were included in this study (Fig. 1).
Figure 1.
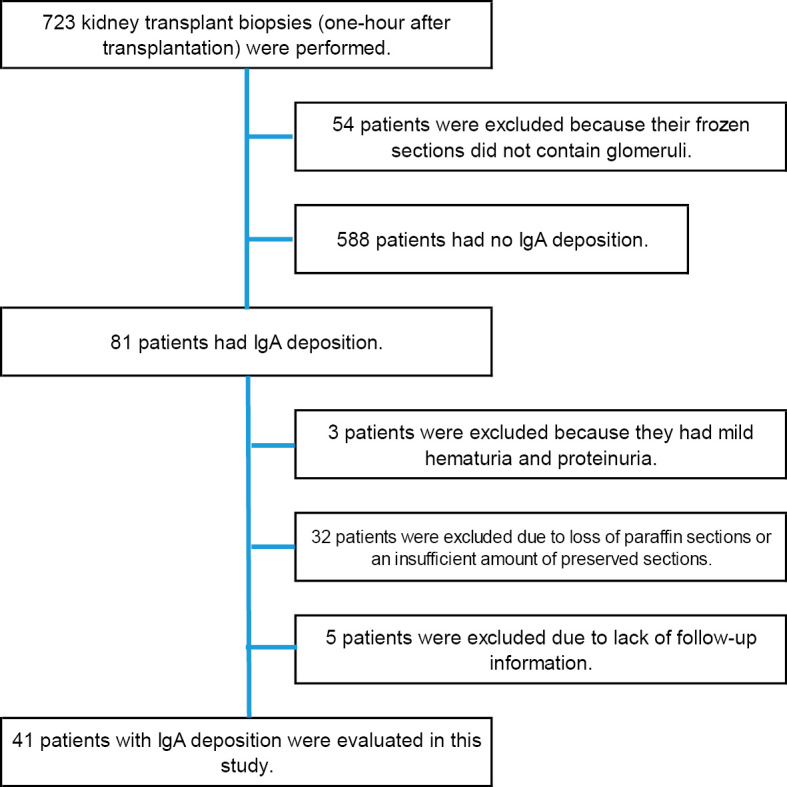
Flowchart of the study design. Eighty-one cases involving IgA deposition were extracted from a total of 723 transplanted kidney biopsies conducted 1 h after kidney transplantation. After exclusion of cases of abnormal urinalysis, loss of paraffin sections, insufficient amount of paraffin sections, and lack of follow-up data, 41 cases were included in the study. IgA: immunoglobulin A
Donor data were collected from the medical records and included the age, gender, serum creatinine level, estimated glomerular filtration rate (eGFR), medications, and history of chronic inflammatory diseases or appendicitis. Recipient data were also collected and included the age, gender, and the causes of end-stage kidney diseases. The rates of donor-recipient consanguinity and blood type incompatibility were assessed to examine the relationship between donors and recipients. Hypertension was defined by a systolic blood pressure ≥140 mmHg and/or diastolic blood pressure ≥90 mmHg based on the criteria of the Japanese Society of Hypertension Guidelines (11). The presence of diabetes mellitus was defined according to the Report of the Committee of the Japan Diabetes Society on the Classification and Diagnostic Criteria of Diabetes Mellitus (12).
Pathological analyses
Routine pathology evaluations of the kidney specimens biopsied one hour after kidney transplantation were conducted by light microscopy and immunofluorescence microscopy analyses of frozen sections. Total sclerotic glomeruli were counted under light microscopy. Tubular atrophy/interstitial fibrosis was classified semi-quantitatively according to the percentage of the renal cortical area involved as follows: non-involved, mild (≤25%), moderate (26-50%), or severe (>50%). Arteriosclerosis was classified semi-quantitatively as no intimal thickening, intimal thickening less than the thickness of the media, and intimal thickening greater than the thickness of the media, which was judged by the most prominent artery.
Immunofluorescence studies were conducted using polyclonal fluorescein isothiocyanate (FITC)-conjugated antibodies to IgA, IgG, IgM, C1q, C3, C4, and fibrinogen (Dako, Tokyo, Japan each diluted 1:40). These factors were evaluated in order to quantify the level of deposition in the mesangial area, and mesangial deposits were classified as 0-3+, as follows: 0, negative; 1+, weak; 2+, moderate; and 3+, strong staining. In this study, no glomerular changes with presence of moderate (2+) to strong (3+) IgA deposition or weak (1+) positive IgA with positive C3 deposition was defined as “IgA deposition”. Protocol kidney biopsies for transplanted kidneys were conducted one hour, three weeks, and one year after transplantation. Double immunofluorescence staining for IgA and Gd-IgA1 was performed on paraffin sections from 41 patients.
Double immunofluorescence staining
Immunofluorescence staining of Gd-IgA1 in renal biopsy specimens was conducted using formalin-fixed, paraffin-embedded sections of 3 μm in thickness. After deparaffinization using xylene and ethanol concentration series and subsequent rehydration, antigen retrieval using 0.05% bacterial protease subtilisin A (Sigma-Aldrich, Tokyo, Japan) dissolved in phosphate-buffered saline (PBS) was performed at room temperature for 2 h. Samples were then rinsed with distilled water 3 times and blocked with 10% goat serum (Nichirei Bioscience, Tokyo, Japan) at room temperature for 30 minutes, followed by incubation with KM55Ⓡ antibody (Immuno-Biological Laboratories, Fujioka, Japan) at 37°C for 60 minutes. After 3 washes with PBS, samples were incubated with Alexa Fluor 555-conjugated goat anti-rat IgG (1:1,000; Thermo Fisher, Tokyo, Japan) at 37°C for 30 minutes. Samples were washed with PBS and incubated with FITC-conjugated polyclonal goat anti-human IgA (1:40; MBL, Nagoya, Japan) at 37°C for 30 minutes. After being washed again with PBS, the slides were sealed. Tissues were observed under an LSM710 immunofluorescent microscope (Carl Zeiss, Oberkochen, Germany). The specificity of KM55Ⓡ antibody against Gd-IgA1 was previously demonstrated by immunohistochemistry and a neutralization assay (9).
Analyses of Gd-IgA1 deposition
We evaluated the positive patterns of IgA and Gd-IgA1 (diffuse: >80% of the total glomeruli; focal: <80%; global: ≥2/3 of the individual glomeruli; segmental: <2/3 of the individual glomeruli) by double immunofluorescence staining. Kidney biopsy specimens derived from 4 patients with donor kidney biopsy diagnosed as minor glomerular abnormalities were used as controls. Four cases of IgAN, three of IgA vasculitis, and seven of lupus nephritis [three cases of class III, two cases of class IV, and two cases of class V according to the International Society of Nephrology/Renal Pathology Society classification (13)] were also stained and evaluated.
Results
Clinical characteristics of donors and recipients
Table 1 summarizes the clinical characteristics of the donors with IgA deposition and recipients who were included in the study. Among a total of 41 living donors, the median age was 58.0 years old, and 23 (56.1%) were men. The median serum creatinine level was 0.82 mg/dL, and the eGFR was 68.1 mL/min/1.73 m2. There was no history of tonsillectomy, but six donors had a history of appendicitis. Four patients with borderline diabetes mellitus showed abnormalities in the 75-g oral glucose tolerance test, even though their HbA1c was <6.5%. Three of the patients had stable blood glucose control with only lifestyle modification, and the other patient received diabetes medication. Twelve patients were diagnosed with hypertension, and 11 of them were receiving antihypertensive medication.
Table 1.
Clinical Characteristics of Kidney Donors according to the Presence of IgA Deposits.
| Donor characteristics | ||
| IgA deposit, n | 41 | |
| Donor age, years, median (IQR) | 58 (51.0, 65.0) | |
| Donor gender male, n (%) | 23 (56.1) | |
| Living donor, n (%) | 41(100) | |
| Donor serum creatinine, mg/dL, median (IQR) | 0.82 (0.66, 0.86) | |
| Donor eGFR (creatinine), mL/min/1.73 m2, median (IQR) | 68.1 (60.1, 72.0) | |
| Donor hypertension, n (%) | 12 (26.8) | |
| of antihypertensive medication, n | 11 | |
| Donor borderline diabetes, n (%) | 4 (9.7) | |
| of medication, n | 1 | |
| Donor appendectomy, n (%) | 6 (14.6) | |
| Recipient characteristics | ||
| n | 41 | |
| Age (y), median (IQR) | 44 (31.0, 59.0) | |
| Male gender, n (%) | 22 (53.7) | |
| Cause of end-stage kidney disease, n (%) | ||
| Chronic glomerulonephritis unspecified | 8 (19.5) | |
| IgA nephropathy | 8 (19.5) | |
| Diabetes | 5 (12.2) | |
| Polycystic kidneys | 2 (4.9) | |
| Congenital anomalies of kidney and urinary tract | 2 (4.9) | |
| Lupus nephritis | 2 (4.9) | |
| Hypertensive nephrosclerosis | 1 (2.4) | |
| Focal glomerular sclerosis | 1 (2.4) | |
| Fibrillary glomerular nephritis | 1 (2.4) | |
| Fabry disease | 1 (2.4) | |
| Denys–Drash syndrome | 1 (2.4) | |
| Cryoglobulinemia | 1 (2.4) | |
| Unknown | 8 (19.5) | |
| Recipient and Donor | ||
| Related by blood, n (%) | 24 (58.5) | |
| Parent to children | 19 | |
| Sibling | 5 | |
| Not related by blood, n (%) | 17 (41.5) | |
| Intercouple | 17 | |
| Blood type compatibility, n (%) | 28 (68.3) |
Among a total of 41 recipients, the median age was 44 years old, and 22 (53.7%) were men. Primary kidney diseases in ESRD included chronic glomerulonephritis in 8 cases (19.5%), IgAN in 8 cases (19.5%); diabetes mellitus in 5 cases (12.2%); lupus nephritis, polycystic kidney disease, and congenital anomalies of the kidney and urinary tract in 2 cases each (4.9%); hypertensive nephrosclerosis, focal glomerular sclerosis, Fabry disease, Denys–Drash syndrome, fibrillary glomerulonephritis, and cryoglobulinemia in 1 case each (2.4%); and unknown disease in 8 cases (19.5%). Donor-recipient consanguinity was observed in 24 cases (58.5%), and blood group compatibility was observed in 28 cases (68.3%) (Table 1).
Pathological findings of donor kidney grafts one hour after transplantation
The results of a light microscopy evaluation of the pathology of renal grafts one hour after transplantation are shown in Table 2. No mesangial proliferative lesions or nephritis were observed. Findings were normal for 20 donors. Two donors showed mild glomerular changes, including one case of focal segmental glomerular sclerosis and one case of hypertrophic glomerulopathy. Seven donors had mild tubular atrophy, and 17 had mild arteriolosclerosis. There were no characteristic findings of diabetic nephropathy in the four donors with borderline diabetes mellitus. Immunofluorescence studies revealed that C3 was positive in 32 of 41 cases (78.0%), and IgM was positive in 25 cases (60.9%), whereas IgA was detected in all cases. Notably, IgG was only detected in 3 patients (7.3%), and the intensity of IgG staining was weak even in these positive cases.
Table 2.
Light Microscopy Findings One Hour after Kidney Transplantation.
| Number of observed glomeruli median (IQR) | 19 (15, 23) | ||||
| glomerular sclerosis median (IQR) | 1 (0, 3) | ||||
| Pathological findings | n=41 | ||||
| No abnormal findings | 20/41 | ||||
| Glomerular changes | Focal segmental glomerular sclerosis | 1/41 | |||
| Hypertrophic glomerulopathy | 1/41 | 2/41 | |||
| Tubular injury, mild | 7/41 | ||||
| Arterioscrelosis, intimal thickening less than thickness of media | 17/41 | ||||
| Fluorescence findings | |||||
| Negative | + | 2+ | 3+ | No glomerulus | |
| IgA | 12 | 26 | 3 | ||
| IgG | 38 | 3 | |||
| IgM | 16 | 22 | 3 | ||
| C1q | 38 | 1 | 1 | 1 | |
| C3 | 9 | 12 | 17 | 3 | |
| C4 | 39 | 1 | 1 | ||
| Fibrinogen | 35 | 5 | 1 | ||
IQR: interquartile range, Ig: immunoglobulin
Detection of IgA and Gd-IgA1 in donor kidneys with IgA deposition, IgAN, IgA vasculitis, and lupus nephritis
All 41 cases were positive for Gd-IgA1, which merged with IgA deposition under double immunofluorescence staining on paraffin sections. Co-deposition of IgA and Gd-IgA1 was observed in diffuse and global glomerular mesangial areas in 23 cases (56.1%), in diffuse and segmental areas in 5 cases (12.2%), in focal and global areas in 4 cases (9.8%), and in focal and segmental areas in 9 cases (22.0%). In all cases, IgA-positive areas were also positive for Gd-IgA1 (Table 3) (Supplementary material 1). Fig. 2 shows representative images of IgA and Gd-IgA1 deposition by double immunofluorescence staining in cases of renal grafts with IgA deposition and other comparative cases involving kidney diseases, including IgAN (Fig. 2G-I), IgA vasculitis (Fig. 2J-L), and lupus nephritis (Fig. 2M-O). Cases of renal grafts without IgA deposition were used as negative staining controls (Fig. 2A-C). In cases of IgA deposition, IgAN, and IgA vasculitis, both IgA and Gd-IgA1 were co-detected in the mesangial region. In contrast, Gd-IgA1 was not detected in most cases of lupus nephritis with mild IgA deposition (Fig. 2M-O). Faint deposition of Gd-IgA1 was observed in only one case of lupus nephritis.
Table 3.
Biopsy Findings of Double Staining One Hour after Kidney Transplantation.
| Numbers of glomeruli, median (IQR) | 12 (9,17) | |
| Localization of IgA and Gd-IgA1 staining | ||
| Diffuse and global, n (%) | 23 (56.1) | |
| Diffuse and segmental, n (%) | 5 (12.2) | |
| Focal and global, n (%) | 4 (9.8) | |
| Focal and segmental, n (%) | 9 (22.0) | |
| The intensity of staining | ||
| IgA > Gd-IgA1, n (%) | 7 (17.1) | |
| <, n (%) | 7 (17.1) | |
| =, n (%) | 27 (65.9) |
IQR: interquartile range, IgA: immunoglobulin A, Gd-IgA1: galactose-deficient IgA1
Figure 2.

Double immunofluorescence staining for IgA and Gd-IgA1 in various kidney diseases. Representative images of double immunofluorescence staining for immunoglobulin A (IgA) and galactose-deficient IgA1 (Gd-IgA1) in various kidney disease specimens are shown. (A-C): Donor kidney without IgA deposition (IgAD) 1 hour after transplantation. (D-F): Donor kidney with IgAD 1 hour after transplantation. (G-I): Case of IgA nephropathy (IgAN). (J-L): Case of IgA vasculitis (IgAV). (M-O): Case of lupus nephritis (LN). (A, D, G, J, and M): IgA staining. (B, E, H, K, and N): Gd-IgA1 staining. (C, F, I, L, and O): Merged images. Gd-IgA1 was not observed in the donor kidney without IgA deposition one hour after transplantation (B). Gd-IgA was detected and merged with IgA in the case of IgAD (D-F), IgAN (G-I), and IgAV (J-L). In contrast, Gd-IgA was not detected in the case of LN, although IgA was detected in glomerular mesangial areas (M-O).
Changes in IgA deposition after transplantation
In patients with IgA and Gd-IgA deposition on 1-h post-transplant kidney biopsies, IgA had disappeared in 15 (37.5%) and 26 (65.0%) of 40 cases at 3 weeks and 1 year after transplantation, respectively (Fig. 3). One case was not biopsied after transplantation. We then classified the cases as C3-positive (n=32) or C3-negative (n=9) as described above (Table 2) and assessed the rate of IgA deposit disappearance. Among the 32 C3-positive cases, IgA deposits had disappeared in 18 cases (56.3%) at the 1-year protocol biopsy. Among the nine C3-negative cases, one patient did not undergo subsequent renal biopsies, and IgA deposition had disappeared in all patients by one year after transplantation (Fig. 3).
Figure 3.

Changes in IgA deposition after transplantation in all patients. A total of 41 cases of donor kidneys with IgA deposition 1 h after transplantation were divided into groups according to the presence of IgA and C3 by fluorescence staining 1 h after transplantation and 1 year after transplantation. Among the 32 C3-positive cases, IgA deposition had disappeared in 19 by 1 year after transplantation. Among the nine C3-negative cases, one case did not undergo a subsequent renal biopsy, and IgA deposition had disappeared in all patients by one year after transplantation. IgA: immunoglobulin A
In eight patients whose primary kidney disease was IgAN, IgA had disappeared in five cases with IgA deposition at three weeks after kidney transplantation (Fig. 4). However, IgA redeposition was observed at the one-year protocol biopsy in the three patients with C3 deposition at one hour after transplantation and without IgA deposition at the three-week protocol biopsy (Fig. 4). Representative pictures of the time course of IgA and Gd-IgA1 staining (one hour, three weeks, and one year) are shown in Supplementary material 2.
Figure 4.

Changes in IgA deposition after transplantation in patients whose primary kidney disease was IgA nephropathy. In eight patients whose primary kidney disease was IgAN, IgA had disappeared in five with IgA deposition at three weeks after kidney transplantation. IgA redeposition was observed at the one-year protocol biopsy in three of these patients.
There were only 3 cases with IgG deposition associated with IgA deposition at one hour after transplantation (Table 2). C3 deposition was demonstrated in all three cases at one hour after transplantation, and IgA deposition was observed in two of the three cases at one year after transplantation.
Discussion
To our knowledge, this is the first study to evaluate the relationship between deposition of IgA and Gd-IgA1 in a large number of healthy individuals who were eligible to be kidney transplantation donors. IgA deposition is reportedly present in 4-10% of autopsied kidneys (14-16) and 10-30% of transplanted kidneys (7,17-19). The frequency of 12.1% (81 of 723) observed in the present study is comparable to that of previous reports. Although it is difficult to assess renal pathology in healthy subjects with latent IgA deposition, renal transplant baseline grafts are acceptable for assessing the role of latent subclinical IgA deposition, as in healthy individuals. As a novel aspect of the present study, the subjects were healthy kidney transplantation donors without significant urinary abnormalities or kidney diseases, such as IgA vasculitis, and without a history of tonsillitis or systemic diseases, including inflammatory bowel disease, which may be associated with IgAN.
Co-deposition of Gd-IgA1 was observed in all 41 cases with incidental IgA deposition, suggesting that Gd-IgA1 can be deposited in healthy kidneys with subclinical IgA deposition. Gd-IgA1 alone reportedly does not induce the proliferation of cultured mesangial cells, but immune complexes involving Gd-IgA1 were shown to induce the proliferation of mesangial cells, suggesting that immune complexes are involved in the pathogenesis of IgAN (20). Suzuki et al. proposed a multi-hit hypothesis in which IgAN develops through four processes: (1) increased production of Gd-IgA1, (2) production of Gd-IgA1-specific IgG, (3) formation of immune complexes, and (4) deposition on mesangial cells, resulting in mesangial cell activation and glomerular damage (21). It has been suggested that Gd-IgA1 deposition can be used as a disease-specific marker for IgAN and IgA vasculitis with nephritis (9). However, Cassol et al. reported positive Gd-IgA1 deposition not only in primary IgAN but also other glomerulonephritis conditions, including staphylococcal infection-induced glomerulonephritis, lupus nephritis, and secondary IgAN conditions, such as psoriasis and inflammatory bowel disease (10). Wang et al. also reported deposition of Gd-IgA1 in glomeruli associated with circulating Gd-IgA1 in patients with secondary IgAN (22). Other studies have reported secondary IgA deposits exhibiting weak Gd-IgA1 staining compared with primary IgA deposits (10,23). In our studies, Gd-IgA1 staining was not significant in lupus nephritis (Fig. 2). In the present study, there was no association between the intensity of Gd-IgA1 staining and the history of systemic or primary disease among transplant donors and recipients.
Activation of the complement system is considered an important process in the progression from deposition to tissue damage (24), and there are reports of an association between C3 deposition and renal dysfunction in IgAN (25). In our study, it is important to note that C3 was positive at 1 h after transplantation in 78% of cases (32 of 41) involving IgA deposits without nephritis. These data were comparable to a previous report in which 2/3 of renal transplant donors with IgA deposition showed positive C3 staining (26). In addition, we were able to evaluate the clinical characteristics of 22 of 41 donors 5 years after transplantation (Supplementary material 3). Unfortunately, the data for the other 19 donors were lost, primarily due to changing hospitals. The 22 evaluable donors did not show urinary abnormalities 5 years after transplantation, even though the majority of 1-h protocol biopsies showed C3 deposition (68.1%, Supplementary material 3). These findings suggest that C3 deposition alone in addition to Gd-IgA1 deposition is not sufficient for the development of nephritis (IgAN).
In the present study, IgA deposition had disappeared in 26 (65.0%) of 40 cases by 1 year after transplantation, which is consistent with a previous report showing that latent IgA deposition had disappeared in 14/20 (70.0%) graft kidneys by 1 year after transplantation (27).
In Fig. 4, three cases among eight recipients who had IgAN showed disappearance of IgA three weeks after transplantation; however, they showed redeposition of IgA one year later. However, at that evaluation one year later, the pathological findings of these three cases indicated only minor glomerular abnormalities, and they did not show any urinary abnormalities, IgG deposition, or C3 deposition. Notably, Moriyama et al. reported that 26.5% of recipients with primary IgAN developed recurrent IgAN within 5 years, and they suggested that latent IgA deposition from the donor kidney was a risk factor of recurrent IgAN (28).
There were few IgG-positive cases (7.3%, 3 of 41) at 1 h after transplantation in this cohort. In addition, IgG staining was weak even in the positive cases (Table 2). Interestingly, C3 and IgA tended to be preserved at one year after transplantation in the patients with IgG deposition. Previous reports have suggested that IgG deposition contributes to the generation of inflammation in mesangial cells, thereby affecting the degree of glomerular damage and clinical outcome (29,30). IgG deposition appears to be an important factor in the development of IgAN. The findings of our investigations are in agreement with the multi-hit hypothesis.
Recent genome-wide association studies strongly indicate that many IgAN-related loci also affect other autoimmune diseases and infections, such as inflammatory bowel disease and rheumatoid arthritis (31). These studies suggest that genetic predisposition and environmental factors can cause IgA deposition in healthy individuals, leading to secondary IgAN as well as IgAN.
Several limitations associated with the present study warrant mention. First, all patients in this study were Japanese, so genetic trends could not be evaluated. Second, a long-term follow-up study by our group (Murata et al.) reported that three donor kidneys with IgA deposition subsequently developed proteinuria and hematuria, resulting in a diagnosis of IgAN by a kidney biopsy (32). Unfortunately, these three cases could not be evaluated in the present study due to insufficient tissues samples. Third, it may be difficult to predict whether or not IgAN will develop based on our data because the observation period was limited.
In summary, this study demonstrated that Gd-IgA1 is present in IgA deposition in donor kidneys one hour after transplantation. IgA and Gd-IgA1 deposition seems to be an initial event in the development of IgAN; however, Gd-IgA1 deposition alone is not sufficient to induce IgAN, and additional factors, such as complement activation and IgG deposition, are required for the development of IgAN.
The authors state that they have no Conflict of Interest (COI).
Supplementary Material
Images of double immunofluorescence staining for immunoglobulin A (IgA) and galactose-deficient IgA1 (Gd-IgA1) in three representative cases of transplanted kidneys with IgA deposition biopsied 1 hour after transplantation are shown. (A, D, and G): IgA staining. (B, E, and H): Gd-IgA1 staining. (C, F, and I): merged images. Gd-IgA1 was merged with IgA, suggesting that deposition stained by IgA antibody and Gd-IgA1 deposition were identical. Scale bars = 50 μm.
Acknowledgement
We greatly appreciate the technical assistance of Shuko Seko (Nagoya Daini Red Cross Hospital, Nagoya, Japan) and Mai Yamauchi (Aichi Medical University, Nagakute, Japan).
References
- 1. Wakai K, Kawamura T, Endoh M, et al. A scoring system to predict renal outcome in IgA nephropathy: from a nationwide prospective study. Nephrol Dial Transplant 21: 2800-2808, 2006. [DOI] [PubMed] [Google Scholar]
- 2. Hirano K, Matsuzaki K, Yasuda T, et al. Association between tonsillectomy and outcomes in patients with immunoglobulin a nephropathy. JAMA Netw Open 2: e194772, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med 368: 2402-2414, 2013. [DOI] [PubMed] [Google Scholar]
- 4. Pouria S, Barratt J. Secondary IgA nephropathy. Semin Nephrol 28: 27-37, 2008. [DOI] [PubMed] [Google Scholar]
- 5. Rollino C, Vischini G, Coppo R. IgA nephropathy and infections. J Nephrol 29: 463-468, 2016. [DOI] [PubMed] [Google Scholar]
- 6. Saha MK, Julian BA, Novak J, Rizk DV. Secondary IgA nephropathy. Kidney Int 94: 674-681, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int 63: 2286-2294, 2003. [DOI] [PubMed] [Google Scholar]
- 8. Yasutake J, Suzuki Y, Suzuki H, et al. Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol Dial Transplant 30: 1315-1321, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki H, Yasutake J, Makita Y, et al. IgA nephropathy and IgA vasculitis with nephritis have a shared feature involving galactose-deficient IgA1-oriented pathogenesis. Kidney Int 93: 700-705, 2018. [DOI] [PubMed] [Google Scholar]
- 10. Cassol CA, Bott C, Nadasdy GM, et al. Immunostaining for galactose-deficient immunoglobulin A is not specific for primary immunoglobulin A nephropathy. Nephrol Dial Transplant 35: 2123-2129, 2020. [DOI] [PubMed] [Google Scholar]
- 11. Umemura S, Arima H, Arima S, et al. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2019). Hypertens Res 42: 1235-1481, 2019. [DOI] [PubMed] [Google Scholar]
- 12. Seino Y, Nanjo K, Tajima N, et al.; Committee of the Japan Diabetes Society on the Diagnostic Criteria of Diabetes Mellitus. Report of the committee on the classification and diagnostic criteria of diabetes mellitus. J Diabetes Investig 1: 212-228, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bajema IM, Wilhelmus S, Alpers CE, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int 93: 789-796, 2018. [DOI] [PubMed] [Google Scholar]
- 14. Sinniah R. Occurrence of mesangial IgA and IgM deposits in a control necropsy population. J Clin Pathol 36: 276-279, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Varis J, Rantala I, Pasternack A, et al. Immunoglobulin and complement deposition in glomeruli of 756 subjects who had committed suicide or met with a violent death. J Clin Pathol 46: 607-610, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Waldherr R, Rambausek M, Duncker WD, Ritz E. Frequency of mesangial IgA deposits in a non-selected autopsy series. Nephrol Dial Transplant 4: 943-946, 1989. [DOI] [PubMed] [Google Scholar]
- 17. Sanfilippo F, Croker BP, Bollinger RR. Fate of four cadaveric donor renal allografts with mesangial IgA deposits. Transplantation 33: 370-376, 1982. [DOI] [PubMed] [Google Scholar]
- 18. Sugiyama S, Yamamoto T, Tsuyuki M, Ohshima S. Study of IgA nephropathy in the transplanted kidney - sequential changes in mesangial IgA deposits found one hour post-transplantation kidney biopsies. Nihon Jinzo Gakkai Shi (J Jpn Nephrol) 28: 729-737, 1986. [PubMed] [Google Scholar]
- 19. Gaber LW, Khan FN, Graviss EA, et al. Prevalence, characteristics, and outcomes of incidental IgA glomerular deposits in donor kidneys. Kidney Int Rep 5: 1914-1924, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Novak J, Tomana M, Matousovic K, et al. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int 67: 504-513, 2005. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol 22: 1795-1803, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang M, Lv J, Zhang X, Chen P, Zhao M, Zhang H. Secondary IgA nephropathy shares the same immune features with primary IgA nephropathy. Kidney Int Rep 5: 165-172, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao L, Peng L, Yang D, et al. Immunostaining of galactose-deficient IgA1 by KM55 is not specific for immunoglobulin A nephropathy. Clin Immunol 217: 108483, 2020. [DOI] [PubMed] [Google Scholar]
- 24. Tortajada A, Gutierrez E, Pickering MC, Praga Terente M, Medjeral-Thomas N. The role of complement in IgA nephropathy. Mol Immunol 114: 123-132, 2019. [DOI] [PubMed] [Google Scholar]
- 25. Wu D, Li X, Yao X, et al. Mesangial C3 deposition and serum C3 levels predict renal outcome in IgA nephropathy. Clin Exp Nephrol 25: 641-651, 2021. [DOI] [PubMed] [Google Scholar]
- 26. Wang Z, Zhang X, Han W, et al. Immune characteristics of renal allograft donors with mesangial IgA deposition. Int Immunopharmacol 91: 107282, 2021. [DOI] [PubMed] [Google Scholar]
- 27. Sofue T, Inui M, Hara T, et al. Latent IgA deposition from donor kidneys does not affect transplant prognosis, irrespective of mesangial expansion. Clin Transplant 27 (Suppl 26): 14-21, 2013. [DOI] [PubMed] [Google Scholar]
- 28. Moriyama T, Nitta K, Suzuki K, et al. Latent IgA deposition from donor kidney is the major risk factor for recurrent IgA nephropathy in renal transplantation. Clin Transplant 19 (Suppl 14): 41-48, 2005. [DOI] [PubMed] [Google Scholar]
- 29. Shin DH, Lim BJ, Han IM, et al. Glomerular IgG deposition predicts renal outcome in patients with IgA nephropathy. Mod Pathol 29: 743-752, 2016. [DOI] [PubMed] [Google Scholar]
- 30. Rizk DV, Saha MK, Hall S, et al. Glomerular immunodeposits of patients with IgA nephropathy are enriched for IgG autoantibodies specific for galactose-deficient IgA1. J Am Soc Nephrol 30: 2017-2026, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46: 1187-1196, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murata M, Takeda A, Ootsuka Y, et al. Study of glomerulopathy in donors after kidney transplantation. Nephron 144 (Suppl 1): 86-90, 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Images of double immunofluorescence staining for immunoglobulin A (IgA) and galactose-deficient IgA1 (Gd-IgA1) in three representative cases of transplanted kidneys with IgA deposition biopsied 1 hour after transplantation are shown. (A, D, and G): IgA staining. (B, E, and H): Gd-IgA1 staining. (C, F, and I): merged images. Gd-IgA1 was merged with IgA, suggesting that deposition stained by IgA antibody and Gd-IgA1 deposition were identical. Scale bars = 50 μm.