

Abstract
ModelCIF (github.com/ihmwg/ModelCIF) is a data information framework developed for and by computational structural biologists to enable delivery of Findable, Accessible, Interoperable, and Reusable (FAIR) data to users worldwide. ModelCIF describes the specific set of attributes and metadata associated with macromolecular structures modeled by solely computational methods and provides an extensible data representation for deposition, archiving, and public dissemination of predicted three-dimensional (3D) models of macromolecules. It is an extension of the Protein Data Bank Exchange / macromolecular Crystallographic Information Framework (PDBx/mmCIF), which is the global data standard for representing experimentally-determined 3D structures of macromolecules and associated metadata. The PDBx/mmCIF framework and its extensions (e.g., ModelCIF) are managed by the Worldwide Protein Data Bank partnership (wwPDB, wwpdb.org) in collaboration with relevant community stakeholders such as the wwPDB ModelCIF Working Group (wwpdb.org/task/modelcif). This semantically rich and extensible data framework for representing computed structure models (CSMs) accelerates the pace of scientific discovery. Herein, we describe the architecture, contents, and governance of ModelCIF, and tools and processes for maintaining and extending the data standard. Community tools and software libraries that support ModelCIF are also described.
Keywords: ModelCIF, PDBx/mmCIF, Data Standard, Open Access, Worldwide Protein Data Bank, wwPDB, AlphaFoldDB, ModelArchive, Machine Learning, Protein Structure Prediction, Computed Structure Models
Graphical Abstract
INTRODUCTION
Brief History of Computed Structure Models (CSMs)
Protein Data Bank (PDB) is the single global repository for three-dimensional (3D) structures of biological macromolecules determined experimentally using macromolecular crystallography (MX), nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (3DEM). It was established in 1971 as the first open-access digital data resource in biology with seven protein structures [1, 2]. At the time of writing, the archive contained >200,000 structures of proteins, nucleic acids, and their complexes with one another and with small-molecule ligands (e.g., approved drugs, investigational agents, enzyme cofactors). This metric is a testament to the collective efforts and technological advances made by structural biologists working on all inhabited continents. It also highlights a daunting reality—that 99% of protein structure space remains unexplored by experimental methods. Inspired by the work of Anfinsen in 1973 [3], computational structural biologists began trying to predict the 3D structure of a protein from its amino acid sequence.
Two distinct approaches for protein structure prediction [4] have been pursued (Figure 1). The first approach is template-based structure prediction (also known as homology modeling or comparative modeling), in which the structure of an unknown protein (target) is modeled computationally based on the similarity of its amino acid sequence to that of a protein with a known structure (template). Homology modeling is generally successful when template structures from the PDB can be identified and accurately aligned to the target sequence. The second approach is template-free structure prediction, also known as ab initio or de novo modeling, which can be applied even when reliable structural templates are not available for the protein of interest. In recent years, intramolecular residue-residue contact predictions based on coevolution data [5] have been successfully applied for template-free structure prediction [6].
Figure 1.
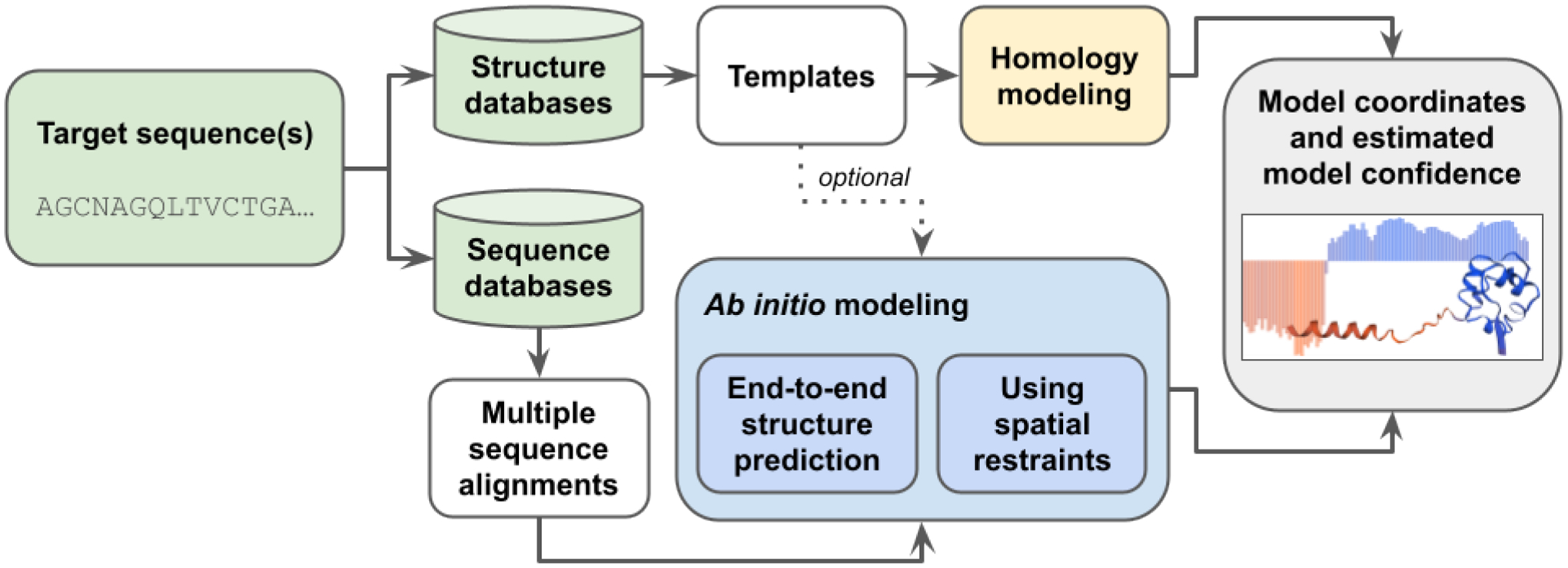
Schematic representation of modeling methods using target sequence(s), structure databases (e.g., PDB), and sequence databases (e.g., Uniclust30 [51]) as input to produce CSMs and estimates of prediction confidence. Homology modeling uses specific templates as its main input, while ab initio methods work without templates. Commonly used ab initio methods rely on multiple sequence alignments, which are either used directly as input for end-to-end structure prediction or processed to extract spatial restraints used to generate CSMs.
Several automated software tools and web servers support template-based or template-free structure prediction, including, but not limited to, SWISS-MODEL [7], Modeller [8], ROSETTA [9], I-TASSER [10], QUARK [11], AlphaFold2 [12], and RoseTTAFold [13, 14]. In the Critical Assessment of Techniques for Protein Structure Prediction (CASP14) challenge conducted in 2020 [15], AlphaFold2 demonstrated unprecedented levels of success, an achievement largely enabled by breakthroughs applying machine learning (ML) approaches to protein structure prediction. Following CASP14, another ML-based method, RoseTTAFold, was developed and subsequently applied in combination with AlphaFold2 to predict the structures of hetero-dimeric complexes of eukaryotic proteins [14]. These ML-based structure prediction methods have proven highly successful and are now capable of generating computed structure models (CSMs) with accuracies comparable to that of lower-resolution experimentally-determined structures [16].
Paralleling advances in protein structure prediction methodologies, data resources were established to provide open access to modeled structures. SWISS-MODEL Repository [17] and ModBase [18] house millions of CSMs of proteins generated using SWISS-MODEL or Modeller, respectively. In addition, the ModelArchive, developed at the Swiss Institute of Bioinformatics (SIB, https://www.modelarchive.org/), was created to archive and provide stable digital object identifiers (DOIs) for CSMs referenced in publications. ModelArchive includes CSMs which were stored in the PDB before 2006 and has been accepting new depositions since 2013. At the time of writing, the AlphaFold Protein Structure Database (AlphaFold DB) [19] held more than 200 million protein CSMs generated by AlphaFold2. They are freely available and represent virtually all of the protein sequences cataloged in UniProtKB [20].
Significance of data standards in archiving scientific data
Data standards are technical specifications describing the semantics, logical organization, and physical encoding of data and associated metadata. They serve as the foundation for collecting, processing, archiving, and distributing data in a standard format and promoting the FAIR (Findable, Accessible, Interoperable and Reusable) principles emblematic of responsible data management in the modern era [21]. In addition to representing the results of a scientific investigation, additional metadata (such as software, authors, citations, references to external data) may be required to support data exchange among different stakeholders, including data generators, archives, and data consumers. If a consistent mechanism is utilized to store such information, it can be shared using common software, agnostic of the data provider, enabling better interoperation among resources and facilitating data search, retrieval, and reuse. Involving community experts in developing and subsequently extending data standards ensures that they are readily adopted by the community and facilitates continuous update of the standards as the field evolves.
History of PDBx/mmCIF data standard for representing macromolecular structures
One of the earliest archival formats in structural biology is the legacy PDB format [22]. Developed in the 1970s, it is human and machine readable, easy to parse, and remained the PDB standard exchange format for over forty years. However, it has several drawbacks, including fixed field widths, column positions, and metadata format, which posed severe limitations for archiving large macromolecular structures, data validation, and future expansion to support newer experimental methods.
In 1990, the Crystallographic Information Framework (CIF) [23] was adopted by the International Union of Crystallography (IUCr) as a community data standard to describe small-molecule X-ray diffraction studies. Later, in 1997, the IUCr approved the mmCIF data standard [24] to support MX experiments. The original mmCIF data standard was subsequently extended by the PDB to support other experimental methods (e.g., NMR, 3DEM), and to create the PDBx/mmCIF data dictionary [25, 26]. In 2014, this standard was adopted by the worldwide PDB (wwPDB, wwpdb.org) [2, 27] as the master format for the PDB archive. The framework describing PDBx/mmCIF is regulated by Dictionary Definition Language 2 (DDL2), a generic language that supports construction of dictionaries composed of data items grouped into categories [28]. DDL2 supports primary data types (e.g., integers, real numbers, text), boundary conditions, controlled vocabularies, and linking of data items together to express relationships (e.g., parent–child relationships). Additionally, software tools have been developed to manage the PDBx/mmCIF dictionary (mmcif.wwpdb.org/docs/software-resources.html). PDBx/mmCIF overcame the limitations of the legacy PDB format and has been extended to represent small-angle solution scattering data [29] and integrative structure models [30].
History of ModelCIF and the wwPDB ModelCIF Working Group
ModelCIF provides definitions for the specific set of attributes and metadata associated with CSMs. Initial efforts to extend PDBx/mmCIF to support CSMs began in 2001 with creation of the MDB dictionary [31]. In 2006, the outcomes of a Workshop organized by the Research Collaboratory for Structural Bioinformatics (RCSB) PDB at Rutgers included recommendations to build a common portal for accessing structural models and develop data standards to support CSMs [32]. The Protein Model Portal (PMP) [33] was created at SIB in collaboration with the Protein Structure Initiative (PSI) Structural Biology Knowledgebase [34]. A collaborative project between RCSB PDB and SIB was initiated in 2016 to create data standards that represent CSMs in the PMP and the ModelArchive. These data standards were designed as an extension of PDBx/mmCIF to facilitate interoperation with PDB data. The first set of ModelCIF definitions was released on GitHub in 2018 (github.com/ihmwg/ModelCIF).
The ModelCIF Working Group (WG) was established in 2021 as a collaboration between the wwPDB partners (RCSB PDB, Protein Data Bank in Europe (PDBe), Protein Data Bank Japan (PDBj), Electron Microscopy Data Bank (EMDB), and Biological Magnetic Resonance Bank (BMRB)) and domain experts in computational structural biology (wwpdb.org/task/modelcif). In addition to wwPDB members, the WG includes representatives from ModelArchive, SWISS-MODEL, Genome3D [35], ModBase, I-TASSER, AlphaFold database, AlphaFold2/DeepMind, and RoseTTAFold. The WG is involved in development and maintenance of the ModelCIF data standard for representing and archiving CSMs and promotes its adoption across the computational biology community. The WG also promotes development of software tools supporting ModelCIF, such as the python-modelcif software library (github.com/ihmwg/python-modelcif). Feedback to the WG via email is welcome (modelcifwg@wwpdb.org).
RESULTS AND DISCUSSION
Data definitions reused from PDBx/mmCIF
In developing ModelCIF, various core PDBx/mmCIF dictionary definitions have been reused. These include representation of small-molecule ligands, polymeric macromolecules, biomolecular complexes, and their atomic coordinates, as well as related metadata definitions about modeling software used, bibliographic citations, and author names (Figure 2).
Figure 2.
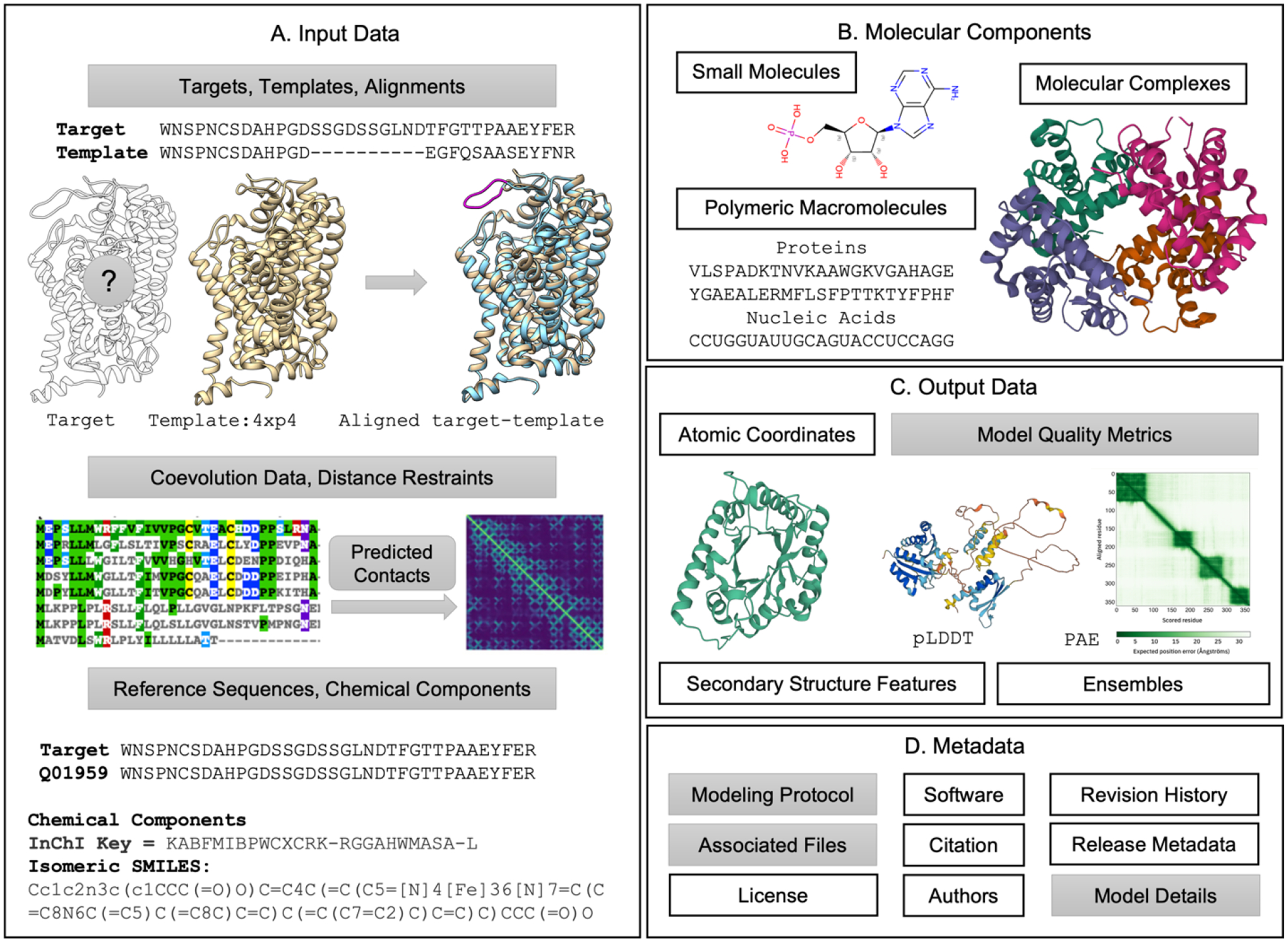
Schematic representation of the data specifications in ModelCIF. Definitions reused from PDBx/mmCIF are shown in white boxes (e.g., Atomic Coordinates) and the newly added definitions are shown in gray boxes (e.g., Model Quality Metrics). (A) Descriptions are provided for input data used in template-based and template-free modeling. (B) Representations of molecular components are retained from PDBx/mmCIF. (C) Definitions for atomic coordinates, secondary structure features, and ensembles are taken from PDBx/mmCIF; descriptions of local and global CSM quality metrics are defined in ModelCIF. (D) Several metadata definitions from PDBx/mmCIF are reused. New metadata definitions regarding modeling protocol, CSM classification (ab initio, homology, etc.) and descriptions of associated files are included in ModelCIF. Examples of CSM-specific data and metadata represented in ModelCIF are provided in the Supplementary Material.
ModelCIF data definitions
Given the variety of existing modeling methods, ModelCIF aims to be flexible regarding data representation. To fulfill this goal, new data categories were introduced to: (i) store input and intermediate results that are of relevance for existing methods; (ii) provide estimates of local and global CSM confidence; (iii) describe steps used to generate CSMs; and (iv) refer to data stored in associated files. New ModelCIF definitions are summarized in Figure 2.
In addition to CSM atomic coordinates, two sets of data items are mandatory: (i) details regarding modeled targets and (ii) list of CSMs included in the file. New definitions are provided for capturing information pertaining to the origin of modeled molecular entities. This feature is particularly useful for cross-referencing to external databases for macromolecular sequences (e.g., UniProtKB) and small molecules (e.g., PubChem [36], ChEBI[37]). Definitions supporting inclusion of small molecules that are not already specified in the wwPDB chemical component dictionary (CCD) [38] are also provided.
In ModelCIF, CSMs can be combined into groups that may belong to an ensemble (or cluster). Structural assemblies must be homogeneous (i.e., every CSM in an entry must have identical composition of molecules). Each CSM can be classified as “homology model”, “ab initio model” or “other” if neither descriptor is appropriate. The “homology model” category is used for any modeling method (including comparative modeling and protein threading) where the main inputs for generating the CSM are sequence alignments to templates. CSMs generated without templates (or where templates are not considered dominant inputs) are classified as “ab initio model” (including fragment sampling and ML-based methods).
Homology modeling methods, as used by SWISS-MODEL and Modeller for example, typically consist of three steps: (i) template identification; (ii) target-template alignment; and (iii) atomic coordinate generation. ModelCIF includes data categories to store the most relevant intermediate results in a standardized way, including a summarized version of the template search results with cross-references to relevant structure databases (e.g., PDB) and detailed information regarding template structures and target-template alignments used for modeling.
Ab initio methods start from sequence information without relying on structural templates. Methods such as I-TASSER generate CSMs using folding simulations guided by deep learning predicted spatial restraints extracted from multiple sequence alignments (MSAs) and corresponding co-evolutionary features. The spatial restraints from deep learning predictors could be residue-residue contacts, distances, dihedral angles, torsion angles, or hydrogen-bonding networks. ModelCIF enables storage of MSAs, homologous templates (optionally used as input structures for ab initio methods), and derived spatial restraints, used by ab initio folding simulations to model CSMs. ML-based ab initio methods such as AlphaFold2 and RoseTTAFold do not rely on features extracted from templates or MSAs but can instead use them as raw input to an “end-to-end” neural network that directly generates the atomic coordinates. Consequently, ModelCIF allows for inclusion of simplified descriptions of relevant input data and intermediate results. ModelCIF can also store information about sequence databases used to construct MSAs (including versions and download URLs) and minimal details regarding any input structures utilized.
While CSMs generated with the newest techniques have become increasingly accurate, it is critical that they are accompanied by estimates of model quality (or prediction confidence). Quality estimates are used to evaluate models and assess their suitability for specific downstream applications. ModelCIF includes flexible support to define any number of quality assessment values. These are classified according to how they are to be interpreted (e.g., probabilities, distances, energies) or as a prediction of the similarity to the correct structure according to well defined metrics such as the TM-score [39] or lDDT [40]. Quality estimate values can be provided globally per CSM and locally per residue, to identify high- and low-quality regions, and per residue-pair, to enable assessment of contacts and domain orientations.
To facilitate reproducibility of structure prediction and to acknowledge use of publicly available software and web services, ModelCIF allows inclusion of generic definitions describing modeling protocols. Minimally, such definitions may include a free-text description of the modeling protocol as a single step. Ideally, however, multiple steps involved in structure modeling can be described. These steps can be linked to input data (e.g., target sequences, template structures, alignments, predicted contacts), software used (including parameters, version information), and output generated (e.g., CSMs), allowing to capture intermediate results obtained at each step. To keep data file sizes manageable, ModelCIF provides metadata definitions supporting description of one or more associated files. The data content of associated files can be large intermediate results, such as MSAs or quality estimates for residue-pairs. A variety of generic file formats are allowed for associated files.
Supporting software tools and resources
Table 1 provides a list of software tools and CSM resources that support ModelCIF. Additional details concerning these tools and resources are included in the supplementary material.
Table 1.
Software tools and CSM resources supporting ModelCIF.
| Software / Resource Name | URL | Description |
|---|---|---|
| Software Tools for Reading, Writing, Conversion, and Validation of ModelCIF Files | ||
| python-modelcif | https://github.com/ihmwg/python-modelcif | Software library that supports reading, writing, and validating ModelCIF files and conversion between mmCIF and BinaryCIF |
| ModelCIF-converters | https://git.scicore.unibas.ch/schwede/modelcif-converters | Collection of ModelCIF conversion tools based on python-modelcif |
| wwPDB mmCIF software resources webpage | https://mmcif.wwpdb.org/docs/software-resources.html | Website that lists community-developed software libraries and tools that support PDBx/mmCIF, many of which also support ModelCIF (e.g., ciftools-java, py-mmcif) |
| Modeling Applications and CSM Repositories | ||
| ModelArchive | https://www.modelarchive.org | Repository for CSMs contributed by modelers |
| SWISS-MODEL [7] | https://swissmodel.expasy.org | Fully automated protein structure homology modeling server and repository |
| Modeller [8] | https://salilab.org/modeller/ | Software used for comparative modeling of protein 3D structures |
| Zhang-Group servers (I-TASSER [10], QUARK [11] |
https://zhanggroup.org/D-I-TASSER/
https://zhanggroup.org/C-QUARK/ |
Ab initio and homology modeling servers for protein structure prediction, protein peptide folding, and structure-based function annotation |
| AlphaFold DB [19] | https://alphafold.ebi.ac.uk | Repository for 3D structures of proteins predicted using AlphaFold2 |
| RoseTTAFold† [13, 14] | https://robetta.bakerlab.org | Software tool that uses a three-track neural network to predict protein structures |
| Visualization Software | ||
| Mol* [52] | https://molstar.org | Web-based structure visualization and analysis tool |
| ChimeraX [53] | https://www.cgl.ucsf.edu/chimerax/ | Desktop-based structure visualization and analysis tool |
At the time of publication, RoseTTAFold module of the Robetta structure prediction server will support ModelCIF.
Advantages of ModelCIF
The value and benefits of ModelCIF are most readily recognized through its support for the FAIR principles. ModelCIF provides foundational data standards for archiving CSMs, making them freely available, and enabling seamless data exchange. Moreover, extending PDBx/mmCIF to establish ModelCIF as a data standard in its own right provides the following advantages: (a) existing definitions in PDBx/mmCIF for representing the atomic structures of biological macromolecules, small-molecules, and molecular complexes can be reused; (b) software tools developed to support PDBx/mmCIF can be reused and extended to support the extension; (c) ModelCIF can be extended to address evolving needs of the structure prediction community (e.g., protein sequence embedding, neural network model metadata); and (d) the extension facilitates interoperation with other structural biology data resources (e.g., PDB). For example, recent updates to the RCSB.org web portal to include >1,000,000 CSMs available freely from AlphaFoldDB and the ModelArchive was facilitated by ModelCIF [41]). To achieve improved parsing performance and compression, ModelCIF files can be readily converted to BinaryCIF format [42].
CONCLUSION AND PERSPECTIVES
Computational structural biology is rapidly advancing before our eyes as a discipline. During manuscript preparation, Meta AI announced the development of their own ML-based method for protein structure prediction and used it to generate more than 600 million CSMs that are now publicly available [43]. It is also likely that additional open-access resources distributing CSMs of proteins will emerge before this paper appears in print. Ideally, every one of these newly established databases of predicted structures will embrace the ModelCIF data standard for deposition, archiving, and dissemination of CSMs. The wwPDB ModelCIF Working Group is committed to maintaining and updating the data standard as new approaches to computational structure modeling of biological macromolecules emerge and are validated. The wwPDB is also supporting community efforts, such as the 3D-Beacons network [44], to encourage adoption of common data standards and facilitate access to 3D-structure information.
Looking ahead, CSMs of large, intricately folded ribonucleic acid (RNA) chains may be of particular importance to basic and applied researchers working across fundamental biology, biomedicine, biotechnology/bioengineering, and the energy sciences. Progress in this field is driven by the development of several RNA structure prediction and model quality assessment tools (e.g., SimRNA [45], RNAComposor [46], FARFAR2 [47], Vfold [48], NAST [49], ARES [50]). Community-organized blind challenges such as CASP will continue to be important in accelerating technical developments in de novo structure prediction for both proteins and nucleic acids.
Supplementary Material
Research Highlights.
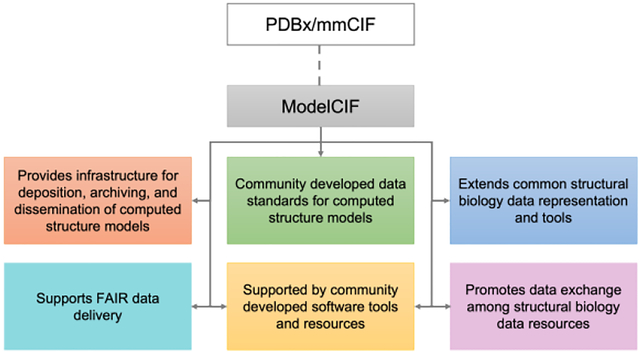
Community-driven data representation for computed structure models (CSMs) of biological macromolecules.
Provides common data representation and tools that enable interoperation of structural biology data resources.
Provides data and software infrastructure for deposition, archiving, and public dissemination of CSMs.
Provides data infrastructure that supports FAIR (Findable, Accessible, Interoperable, Reusable) data delivery and accelerates scientific discovery.
ACKNOWLEDGEMENTS
The authors thank the tens of thousands of researchers worldwide who enable computational structure modeling of proteins by depositing ~200,000 experimentally-determined structures to the PDB since 1971. We also gratefully acknowledge contributions to the PDBx/mmCIF data standard made by past members of Worldwide Protein Data Bank partner organizations (RCSB PDB, PDBe, PDBj, EMDB, and BMRB) and members of the structural biology community. G.T., S.B., and T.S. acknowledge the contributions of Andrew Mark Waterhouse and Dario Behringer to ModelArchive and support of ModelCIF.
FUNDING
RCSB PDB core operations are jointly funded by the National Science Foundation (DBI-1832184, PI: S.K. Burley), the US Department of Energy (DE-SC0019749, PI: S.K. Burley), and the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and the National Institute of General Medical Sciences of the National Institutes of Health (R01GM133198, PI: S.K. Burley). Other funding awards to RCSB PDB by the NSF and to PDBe by the UK Biotechnology and Biological Research Council are jointly supporting development of a Next Generation PDB archive (DBI-2019297, PI: S.K. Burley; BB/V004247/1, PI: Sameer Velankar) and new Mol* features (DBI-2129634, PI: S.K. Burley; BB/W017970/1, PI: Sameer Velankar). B. Vallat acknowledges funding from NSF (NSF DBI-2112966, PI: B. Vallat; NSF DBI-1756248, PI: B. Vallat). A. Sali acknowledges funding from NIH and NSF (NIH R01GM083960, PI: A. Sali; NSF DBI-2112967, PI: A. Sali; NSF DBI-1756250, PI: A. Sali; NIH P41GM109824, PI: M.P. Rout). PDBj is supported by grants from the Database Integration Coordination Program from the department of NBDC program, Japan Science and Technology Agency (JPMJND2205, PI: G. Kurisu), and partially supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number 22ama121001. The Protein Data Bank in Europe is supported by European Molecular Biology Laboratory-European Bioinformatics Institute and the AlphaFold Database work is additionally funded by DeepMind. The 3D-Beacons work was supported by funding from the UK Biotechnology and Biological Research Council to PDBe and Christine Orengo group (BB/S020144/1, BB/S020071/1). J. Hoch acknowledges funding from NIH (R01GM109046) for BMRB. G.T., S.B., and T.S. acknowledge funding from NIH and National Institute of General Medical Sciences (U01 GM093324-01), ELIXIR (3D-BioInfo), and the SIB Swiss Institute of Bioinformatics. Y.Z. acknowledges support from the Extreme Science and Engineering Discovery Environment (XSEDE), which is funded by the National Science Foundation (ACI-1548562). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT Authorship Contribution Statement
Brinda Vallat: Conceptualization, Methodology, Software, Writing – original draft, Visualization, Project administration, Funding acquisition
Gerardo Tauriello: Conceptualization, Methodology, Software, Writing – original draft, Visualization
Stefan Bienert: Methodology, Software
Juergen Haas: Methodology
Benjamin M. Webb: Conceptualization, Methodology, Software, Writing – original draft
Augustin Žídek: Software, Writing – Review & Editing
Wei Zheng: Software, Writing – Review & Editing
Ezra Peisach: Methodology, Writing – original draft
Dennis W. Piehl: Writing – original draft
Ivan Anischanka: Software, Writing – Review & Editing
Ian Sillitoe: Writing – Review & Editing
James Tolchard: Writing – Review & Editing
Mihaly Varadi: Writing – Review & Editing
David Baker: Writing – Review & Editing
Christine Orengo: Funding acquisition, Writing – Review & Editing
Yang Zhang: Funding acquisition
Jeffrey C. Hoch: Funding acquisition, Writing – Review & Editing
Genji Kurisu: Funding acquisition, Writing – Review & Editing
Ardan Patwardhan: Writing – Review & Editing
Sameer Velankar: Funding acquisition, Writing – Review & Editing
Stephen K. Burley: Conceptualization, Supervision, Funding acquisition, Writing – original draft
Andrej Sali: Funding acquisition, Writing – Review & Editing
Torsten Schwede: Funding acquisition, Writing – Review & Editing
Helen M. Berman: Funding acquisition, Supervision, Writing – Review & Editing
John D. Westbrook: Conceptualization, Methodology, Software, Supervision
REFERENCES
- [1].Protein Data Bank. (1971). Crystallography: Protein Data Bank. Nature (London), New Biol. 233, 223–223.16063295 [Google Scholar]
- [2].wwPDB consortium. (2019). Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 47, D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Anfinsen CR. (1973). Principles that govern the folding of protein chains. Science. 181, 223–230. [DOI] [PubMed] [Google Scholar]
- [4].Baker D, Sali A. (2001). Protein structure prediction and structural genomics. Science. 294, 93–96. [DOI] [PubMed] [Google Scholar]
- [5].Gobel U, Sander C, Schneider R, Valencia A. (1994). Correlated mutations and residue contacts in proteins. Proteins. 18, 309–317. [DOI] [PubMed] [Google Scholar]
- [6].Abriata LA, Tamo GE, Monastyrskyy B, Kryshtafovych A, Dal Peraro M. (2018). Assessment of hard target modeling in CASP12 reveals an emerging role of alignment-based contact prediction methods. Proteins. 86 Suppl 1, 97–112. [DOI] [PubMed] [Google Scholar]
- [7].Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sali A, Blundell TL. (1993). Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- [9].Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, et al. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat Methods. 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mortuza SM, Zheng W, Zhang C, Li Y, Pearce R, Zhang Y. (2021). Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions. Nature communications. 12, 5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature. 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science. 373, 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Humphreys IR, Pei J, Baek M, Krishnakumar A, Anishchenko I, Ovchinnikov S, et al. (2021). Computed structures of core eukaryotic protein complexes. Science. 374, eabm4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. (2021). Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins. 89, 1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shao C, Bittrich S, Wang S, Burley SK. (2022). Assessing PDB macromolecular crystal structure confidence at the individual amino acid residue level. Structure. 30, 1385–1394 e1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bienert S, Waterhouse A, de Beer TA, Tauriello G, Studer G, Bordoli L, et al. (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 45, D313–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pieper U, Webb BM, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, et al. (2014). ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 42, D336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].UniProt Consortium. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, et al. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Westbrook JD, Fitzgerald PMD. (2009). Chapter 10 The PDB format, mmCIF formats, and other data formats. In Structural Bioinformatics, Second Edition, (Bourne PE, Gu J, eds), p. 271–291, John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
- [23].Hall SR, Allen FH, Brown ID. (1991). The crystallographic information file (CIF): a new standard archive file for crystallography. Acta Crystallographica Section A Foundations of Crystallography. 47, 655–685. [Google Scholar]
- [24].Fitzgerald PMD, Westbrook JD, Bourne PE, McMahon B, Watenpaugh KD, Berman HM. (2005). 4.5 Macromolecular dictionary (mmCIF). In International Tables for Crystallography G Definition and exchange of crystallographic data, (Hall SR, McMahon B, eds), p. 295–443, Springer, Dordrecht, The Netherlands. [Google Scholar]
- [25].Westbrook JD, Young JY, Shao C, Feng Z, Guranovic V, Lawson C, et al. (2022). PDBx/mmCIF Ecosystem: Foundational semantic tools for structural biology. J Mol Biol. 434, 167599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Westbrook J, Henrick K, Ulrich EL, Berman HM. (2005). 3.6.2 The Protein Data Bank exchange data dictionary. In International Tables for Crystallography, (Hall SR, McMahon B, eds), p. 195–198, Springer, Dordrecht, The Netherlands. [Google Scholar]
- [27].Berman HM, Henrick K, Nakamura H. (2003). Announcing the worldwide Protein Data Bank. Nature Structure Biology. 10, 980. [DOI] [PubMed] [Google Scholar]
- [28].Westbrook JD, Berman HM, Hall SR. (2005). 2.6 Specification of a relational Dictionary Definition Language (DDL2). In International Tables for Crystallography, (Hall SR, McMahon B, eds), p. 61–72, Springer, Dordrecht, The Netherlands. [Google Scholar]
- [29].Malfois M, Svergun DI. (2000). sasCIF: an extension of core Crystallographic Information File for SAS. Journal of Applied Crystallography. 33, 812–816. [Google Scholar]
- [30].Vallat B, Webb B, Westbrook JD, Sali A, Berman HM. (2018). Development of a Prototype System for Archiving Integrative/Hybrid Structure Models of Biological Macromolecules. Structure. 26, 894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Migliavacca E, Adzhubei AA, Peitsch MC. (2001). MDB: a database system utilizing automatic construction of modules and STAR-derived universal language. Bioinformatics. 17, 1047–1052. [DOI] [PubMed] [Google Scholar]
- [32].Berman HM, Burley SK, Chiu W, Sali A, Adzhubei A, Bourne PE, et al. (2006). Outcome of a workshop on archiving structural models of biological macromolecules. Structure. 14, 1211–1217. [DOI] [PubMed] [Google Scholar]
- [33].Haas J, Roth S, Arnold K, Kiefer F, Schmidt T, Bordoli L, et al. (2013). The Protein Model Portal--a comprehensive resource for protein structure and model information. Database (Oxford). 2013, bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gabanyi MJ, Adams PD, Arnold K, Bordoli L, Carter LG, Flippen-Andersen J, et al. (2011). The Structural Biology Knowledgebase: a portal to protein structures, sequences, functions, and methods. J Struct Funct Genomics. 12, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sillitoe I, Andreeva A, Blundell TL, Buchan DWA, Finn RD, Gough J, et al. (2020). Genome3D: integrating a collaborative data pipeline to expand the depth and breadth of consensus protein structure annotation. Nucleic Acids Res. 48, D314–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. (2021). PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hastings J, Owen G, Dekker A, Ennis M, Kale N, Muthukrishnan V, et al. (2016). ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 44, D1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Westbrook JD, Shao C, Feng Z, Zhuravleva M, Velankar S, Young J. (2015). The chemical component dictionary: complete descriptions of constituent molecules in experimentally determined 3D macromolecules in the Protein Data Bank. Bioinformatics. 31, 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang Y, Skolnick J. (2004). Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702–710. [DOI] [PubMed] [Google Scholar]
- [40].Mariani V, Biasini M, Barbato A, Schwede T. (2013). lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 29, 2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Burley SK, Bhikadiya C, Bi C, Bittrich S, Chao H, Chen L, et al. (2023). RCSB Protein Data Bank (RCSB.org): Delivery of Experimentally-Determined PDB Structures Alongside One Million Computed Structure Models of Proteins from Artificial Intelligence/Machine Learning. Nucleic Acids Res. 51, D488–D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sehnal D, Bittrich S, Velankar S, Koča J, Svobodová R, Burley SK, et al. (2020). BinaryCIF and CIFTools––Lightweight, Efficient and Extensible Macromolecular Data Management. PLoS Comput Biol. 16, e1008247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. (2022). Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv, doi: 10.1101/2022.07.20.500902. [DOI] [PubMed] [Google Scholar]
- [44].Varadi M, Nair S, Sillitoe I, Tauriello G, Anyango S, Bienert S, et al. (2022). 3D-Beacons: decreasing the gap between protein sequences and structures through a federated network of protein structure data resources. GigaScience. 11, giac118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Boniecki MJ, Lach G, Dawson WK, Tomala K, Lukasz P, Soltysinski T, et al. (2016). SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 44, e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Biesiada M, Purzycka KJ, Szachniuk M, Blazewicz J, Adamiak RW. (2016). Automated RNA 3D Structure Prediction with RNAComposer. Methods Mol Biol. 1490, 199–215. [DOI] [PubMed] [Google Scholar]
- [47].Watkins AM, Rangan R, Das R. (2020). FARFAR2: Improved De Novo Rosetta Prediction of Complex Global RNA Folds. Structure. 28, 963–976 e966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li J, Zhang S, Zhang D, Chen SJ. (2022). Vfold-Pipeline: a web server for RNA 3D structure prediction from sequences. Bioinformatics. 38, 4042–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, et al. (2009). Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 15, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Townshend RJL, Eismann S, Watkins AM, Rangan R, Karelina M, Das R, et al. (2021). Geometric deep learning of RNA structure. Science. 373, 1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mirdita M, von den Driesch L, Galiez C, Martin MJ, Soding J, Steinegger M. (2017). Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sehnal D, Bittrich S, Deshpande M, Svobodova R, Berka K, Bazgier V, et al. (2021). Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 49, W431–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.