

Abstract
An Ethics Committee (EC) is an independent body composed of members with expertise in both scientific and nonscientific arenas which functions to ensure the protection of human rights and the well-being of research subjects based on six basic principles of autonomy, justice, beneficence, nonmaleficence, confidentiality, and honesty. MEDLINE, Scopus, and Directory of Open Access Journals were searched for studies relevant to this topic. This review is focused on the types of research articles that need EC approval, the submission process, and exemptions. It further highlights the constitution of ECs, their duties, the review process, and the assessment of the risk-benefit of the proposed research including privacy issues. It’s pertinent for academicians and researchers to abide by the rules and regulations put forth by ECs for upholding of human rights and protecting research subjects primarily, as well as avoiding other issues like retraction of publications. Despite various issues of cost, backlogs, lack of expertise, lesser representation of laypersons, need for multiple approvals for multisite projects, conflicts of interest, and monitoring of ongoing research for the continued safety of participants, the ECs form the central force in regulating research and participant safety. Data safety and monitoring boards complement the ECs for carrying out continuous monitoring for better protection of research subjects. The establishment of ECs has ensured safe study designs, the safety of human subjects along with the protection of researchers from before the initiation until the completion of a study.
Keywords: Privacy; Beneficence; Research Subjects; Confidentiality; Human Rights; Ethics Committees, Research
Graphical Abstract
INTRODUCTION
The journey of the role of ethics in biomedical research began with “The Doctor’s Trial” post-World War II in which 23 doctors and administrators were tried for war crimes, crimes against humanity, and conducting research without informed consent. This judgment, known as the “Nuremberg Code” was one of the first international ethical standards which gave a ten-point rule with respect to the protection of human research participants. The core principle was the requirement of voluntary consent of human subjects and respecting human autonomy.1,2
However, some researchers continued to ignore the code and violations like the Willow Brook Hepatitis Study (1956), Jewish Chronic Disease Study (1963), and 22 others were highlighted by Beecher in 1966.3,4 This led to the composition of the Declaration of Helsinki by the World Medical Association in Finland in 1964 with revisions at regular intervals.5 This affirmed the principles highlighted in the Nuremberg Code stating that research should be conducted upholding the interests and rights of the human subjects. It proposed for the first time, the submission of a research protocol to an ethics committee (EC) before the initiation of the study (Fig. 1).6
Fig. 1. Timelines of events that led to the establishment of Ethics Committees.

EC refers to “Committees established by professional societies, health facilities, or other institutions to consider decisions that have bioethical implications. The role of these committees may include consultation, education, mediation, and/or review of policies and practices.” Committees that consider the ethical dimensions of patient care are Clinical ECs whereas committees established to protect the welfare of research subjects are Research ECs.7 In this review, we will be using the terms ECs and Research ECs interchangeably.
It was in the 1960s that most nations developed guidelines regarding the formation of ECs with the main task of protection of human subjects.8 EC is an independent body composed of members with expertise in both scientific and nonscientific arenas which functions to ensure the protection of human rights and the well-being of research subjects. ECs can be of two types—Institutional Review Boards (IRBs) or Institutional ECs (IECs) (referred to IRB or IEC by different countries) that are formally constituted by an institution to review research projects for that institute. An independent EC is an autonomous EC that is not part of any institute and performs the same functions independently. It is helpful for institutes that don’t have an IRB.
Despite these regulations, the unethical standards of the Tuskegee Syphilis study emerged in 1972 in which treatment was denied to the participants in order to study the natural course of the disease. This was followed by the conversion of the National Research Act in the USA into law (1974) and the setting up of the national commission of ‘International Ethical Guidelines for Biomedical Research Involving Human Subjects’ that submitted the Belmont report in 1979. The Belmont report described the role of assessment of risk-benefit of research involving human subjects, appropriate guidelines for selection of human subjects, and definition of informed consent. It was based on the three pillars of ethics- respect, beneficence, and justice.9,10 It stressed the need for the approval of studies by an EC in accordance with the 1975 revision of the World Medical Association in Tokyo. Subsequently, countries like China, India, and South Korea adopted and legalized the need for submission of protocols to ECs from the 1980s onwards.11,12,13,14
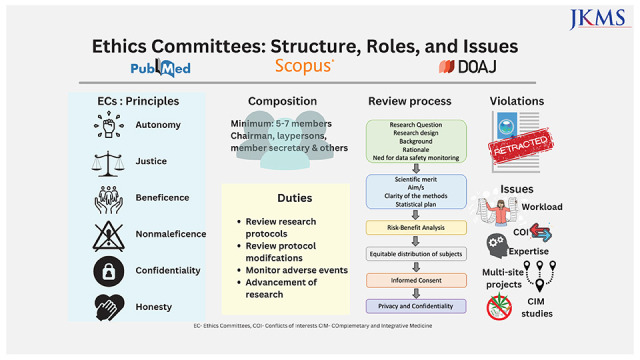
ECs function on six basic principles15:
1. Autonomy: respect the patient’s right to act on his/her own value and choice.
2. Justice: fair treatment of the research subjects.
3. Beneficence: work for the benefit of the patient.
4. Nonmaleficence: primum non-nocere or first do no harm to the patient.
5. Confidentiality: privacy protection.
6. Honesty: truthfulness in terms of the study.
Ethics approval is required for most research studies to uphold the above-mentioned principles, and protect the participants as well as the researcher.16
In this narrative review, we aim to study the structure and function of ECs or IRBs with a focus on the composition, role, violations, and development perspectives of ECs.
METHODS
Searches through MEDLINE (PubMed) and Scopus were performed in line with previously published recommendations.17
Articles published till March 15, 2023 were reviewed using the following keywords: ("Ethics Committees, Clinical/classification"[Mesh] OR "Ethics Committees, Clinical/economics"[Mesh] OR "Ethics Committees, Clinical/ethics"[Mesh] OR "Ethics Committees, Clinical/history"[Mesh] OR "Ethics Committees, Clinical/legislation and jurisprudence"[Mesh] OR "Ethics Committees, Clinical/organization AND administration"[Mesh] OR "Ethics Committees, Clinical/standards"[Mesh] OR "Ethics Committees, Clinical/statistics and numerical data"[Mesh] OR "Ethics Committees, Clinical/trends"[Mesh]). Additional searches about subtopics were also carried out (“Data Safety Monitoring Boards” OR “Independent Data Review Committees”, “Institutional Review Boards” OR “Ethics Committees” and “Problems” OR “Issues”).
Articles in languages other than English, and reviews, conference proceedings, and editorials were excluded. Relevant articles searchable at the Directory of Open Access Journals and references of included articles were also processed for eligibility and inclusion for this narrative review.18,19,20,21
RESEARCH, SUBMISSION PROCESS, AND EXEMPTIONS
IRB approval is required for most research to protect human rights and assess the scientific soundness of the research. For this, we first need to understand what research is. Research is defined as “a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge” (Table 1).8
Table 1. Type of research studies and approvals/exemptions.
| Type of studies | Details about the need for approvals | |
|---|---|---|
| Studies involving interaction or intervention | ||
| 1. Clinical Trials- pilot, all phases and designs | A pilot study with an intention to contribute to general knowledge (fits the criteria for research) | |
| All phases of clinical trials require separate approvals. | ||
| 2. All interventional studies | Even those using standard of care or non-pharmacologic interventions need approval. | |
| 3. Diagnostic tests and devices | Analysis of data and biologic specimens need approvals. | |
| 4. Medical records review | Those identifying private identifying information need approvals. | |
| 5. Case reports | 1–3 cases don’t require IRB approvals, preferably patient consent should be taken for it nevertheless. | |
| Case series need approvals as they are hypothesis testing. | ||
| 6. Quality improvement and cost benefit analysis | Whenever it fits the definition of research, i.e., intent to generalize knowledge present. | |
| 7. Product evaluations | Whenever it fits the definition of research, i.e., intent to generalize knowledge present. | |
| 8. Public health surveillance | Those mandated by law, e.g., reporting of communicable diseases are exempt from IRB review. | |
| Studies not involving direct interaction or intervention | ||
| 1. Use of preexisting medical records or stored specimens | IRB review required if identifying information is recorded. | |
| 2. Databases, registries, biobanks of biomedical specimens | IRB review required for confidentiality purposes. | |
| Surveys/Interviews | Collection of identifying or sensitive information requires IRB approval. | |
IRB = Institutional Review Board.
An EC approval is required for studies with more than minimal risk to the subjects where the intention is to publish findings or contribute to the scientific knowledge, studies involving the compilation or analysis of data containing patient identifying information, studies with any risk of physical or mental discomfort to participants or their families, and studies on vulnerable groups.22 Minimal risk refers to the probability of discomfort posed by the research is not greater than that ordinarily encountered in routine daily life activities of an average healthy individual.5,8,22
Thus, even surveys and archived data that contain patient identifying information (name, age, address) and sensitive information (illicit drug use, comorbidities, communicable diseases, e.g., HIV AIDS) need ethical approval to uphold the privacy and anonymity of the participants as well as protection the possibility of psychological discomfort to them.10,23,24,25
Some studies may be exempted from ethical approval including most educational research, case reports on one to three patients (without any hypothesis testing), those that pose no risk to the participants, involve information freely available in the open domain for the community, analysis of open-source datasets or anonymized datasets obtained from other researchers with due informed consent taken at the time of primary data collection, research evaluating the public health programs or government public schemes.26,27 However, a formal exemption is to be decided by the IRB and not the investigator.8,28
For projects requiring an EC approval, the type of reviews includes expedited and a full board review. Expedited review is for research involving no more than minimal risk to the subjects, minor revisions of an already approved study, and is usually conducted by an experienced person or the chair of the IRB. A full board review on the other hand is for research with greater than minimal risk to the subjects or those involving vulnerable populations. This is reviewed extensively by a full IRB meeting.
The documents usually required for a full ethics review include the name of the applicant with designation, approval of the head of the department, research/trial protocol, ethical issues if any, and plans to address them, written informed consent form (and assent forms) in the language the participant understands, data collection tools, patient information sheet, regulatory clearances (e.g., Drug Controller General of India in India for drug trials), finance and funding details, Insurance, statement of conflicts of interest, information about payment or compensation to the subjects, scientific or departmental review board permission, Curriculum Vitae of the investigators, declaration of interests and any other relevant information.29,30 Waivers of consent may be provided for no more than minimal risk to the subjects when the waiver will not endanger the rights and welfare of the subjects like retrospective studies, secondary analysis of data wherein consent had been taken previously, use of open access databases with anonymized data, and emergency research as seen fit by the EC.31,32 In emergencies like the coronavirus disease 2019 pandemic, waivers may be provided if the patient is incapacitated or in life-threatening situations where there is no time for informed consent. Pandemics like these may even call for common documents for risk disclosure and audio/video/electronic consent.33,34
EC PERSONNEL, THEIR EXPERIENCE, AND DUTIES
ECs have the primary responsibility of reviewing research and its alignment with the Good Clinical Practice (GCP) guidelines.35 The research design must be scientifically sound and conducted in an ethical way to include human subjects with voluntary informed consent.
Personnel
The composition of ECs varies depending on the country, center, volume, and nature of the research reviewed. However, there are some basic recommendations laid down by national authorities and GCP.30
a) Most countries in Europe, the USA, and South Korea have a requirement of at least five members whereas recommendations in China and India need a minimum of seven members and a maximum of 12–15 members.14,36,37,38,39
b) At least one member who is autonomous, independent of the institution or trial site. It is mandatory that the chairperson of the EC is not part of the institution where the research is to be conducted.
c) At least one member who is from the non-scientific community (Fig. 2).
Fig. 2. Recommended composition of the Ethics Committee.

Others include a member secretary from within the institution and members from the scientific field. The composition should have an adequate gender and age representation with a blend of basic scientists, clinician scientists, one legal expert, one social scientist, one philosopher, and one layperson. Review of research involving vulnerable populations like children, pregnant women, handicapped, prisoners, etc., must involve one member with expertise in dealing with that population.40,41 It is also desirable to have a member or expert advisor for special areas of research who has proficiency in that field.
Responsibilities
The chairperson has the primary responsibility of independent and smooth functioning of the EC, ensuring the participation of all members, seeking Conflict of Interests from all members, and handling complaints against the researchers and EC members.39 It’s the responsibility of the member secretary to schedule EC meetings, handle documentation, organize an effective review of proposals, define and maintain adherence to standard operating procedures (SOPs), train EC members, and assess the need for expedited reviews/exemption from review.39 The members of the scientific community have the primary responsibility of reviewing the research protocols and their scientific soundness. The non-scientist member is crucial to safeguard the human subjects and practical issues of the research.40,41 However, studies have shown lesser participation by laypersons as compared to scientific members. A study conducted across 10 academic centers across the USA with 20 IRB meetings recorded noted that 29 community members were present in 17 of those meetings. They were primary reviewers in only two of the 93 submitted protocols due to refusal on grounds of lack of knowledge regarding medical research. Even as secondary and tertiary reviewers, they were less active and were more likely to focus on issues related to confidentiality. However, they played a greater role when they were not designated reviewers.42
Duties
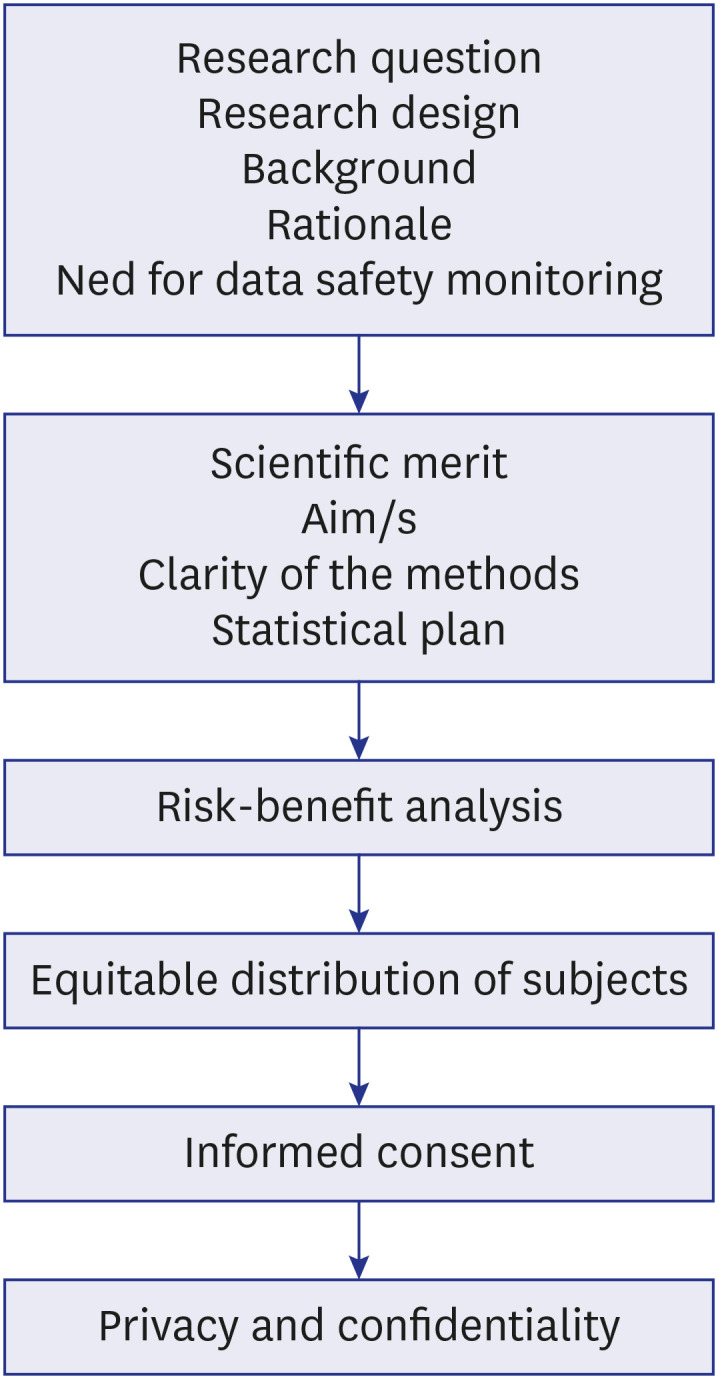
The EC or IRBs function to review and approve research protocols, monitor ongoing research involving human subjects with the aims of continual protection of human volunteers, advancement of research, and protecting the institute from litigation. Its main role is the protection of the human rights, autonomy, confidentiality, and welfare of the research subjects especially vulnerable populations. The GCP recommends the following for duties of the IRB (Table 2, Fig. 3)35:
Table 2. Review process of a study by the Ethics Committee8 .
| Review process | Details | |
|---|---|---|
| Ensuring that risks to the subject are minimized | ||
| 1. Eligibility criteria | Drug X if metabolized through the kidney, will patients with reduced renal function be excluded? | |
| If not, what is the rationale for inclusion? | ||
| 2. Subject monitoring | If the drug has possible hematologic side effects, how and at what interval is the monitoring planned? | |
| 3. Are the proposed procedures well justified? | If a renal biopsy is planned at the end of the study to look for histopathologic remission, is it justified? Is it part of routine clinical care? | |
| 4. Comparison of risks with SOC | Additional risks conferred by the drug X as compared to SOC like MMF or CYC. | |
| 5. Are the research personnel qualified? | Is training in medical ethics and GCP done? | |
| Are trained personnel (rheumatologist/nephrologists in this case) managing the research participants? | ||
| 6. Are the SOPs in place for the exclusion of subjects? | What is the level of cytopenia or severity of infection at which drug X will be stopped? | |
| 7. Appropriate location | Rheumatology and Nephrology set-up, availability of ICU backup | |
| Risk benefit assessment | ||
| 1. Risks different from SOC | Is the risk of cytopenia or infections the same as SOC? If yes, easier for IRB to approve. | |
| 2. Frequency, severity and reversibility of side effects. Potential delayed effects | Short-lasting adverse events like reversible cytopenia is acceptable as long as the benefits outweigh the risk. Any chance of delayed renal dysfunction would be difficult for the IRB to assess. | |
| 3. Vulnerable populations | If the researcher plans to include vulnerable subjects like children, elderly, pregnant women, it is difficult for the IRB to approve. | |
| 4. Perception of risks amongst subjects | Risk of death with the drug is not acceptable. | |
| 5. Preclinical and clinical studies | Results of animal and phase 1/phase 2 data about drug X to assess safety and efficacy in favor of the drug X will help the approval process. | |
| 6. Continuation of SOC/Drug wash out | Will SOC be continued along with drug X? | |
| Will a wash out period be given for SOC before initiation of the study? | ||
| 7. Type of study design | RCT preferred over open label studies to assess safety and efficacy in an unbiased manner. | |
| 8. Potential for direct benefit or indirect benefit to science or society | In this case, there is a potential benefit to the subject and society. | |
| Equitable selection of subjects | ||
| 1. Target population | Ethnic representation as different severity of lupus nephritis across Caucasians, African Americans, Asians. | |
| 2. Inclusion and exclusion | Inclusion or exclusion of children, pregnant women, chronic kidney disease, etc., need to be confirmed. | |
| Informed consent | ||
| 1. Description of the informed consent process | Information, nature, time given to the subject for providing the information, etc., need to be assessed. | |
| 2. Appropriateness of the process | Assent in case of children, appropriateness of the language. | |
| 3. Components | Patient information about lupus nephritis and drugs, potential risks, potential benefits, confidentiality, compensation, voluntary process need to be assessed. | |
| Monitoring data | ||
| 1. Data safety monitoring plan as to how adverse events will be monitored and action taken | Frequency and method need to be assessed. | |
| Protection of subject privacy and confidentiality | ||
| 1. Location of data collection | Adequate space for interviewing, minimal personnel with access to patient data. | |
| 2. Sensitive data | Any data involving stigmatizing medical conditions, behaviors, and genetic information. | |
| 3. Data storage | Method and plans for data protection. | |
| 4. Data recording | Anonymous/coded. | |
| 5. Accessing patient records | Personnel with valid access. | |
| 6. Information in the consent forms about the measures planned and risks of breach in confidentiality | ||
| Vulnerable populations | Children, prisoners, mentally disabled people, or economically or educationally disadvantaged people, should explicitly describe what measures are in place to ensure that the subjects’ rights and welfare are adequately protected. | |
e.g., An immunosuppressive drug “X” being evaluated for patients with Lupus Nephritis.
IRB = Institutional Review Board, SOC = standard of care, GCP = Good Clinical Practice, MMF = mycophenolate mofetil, CYC = cyclophosphamide, ICU = intensive care unit, SOP = standard operating procedure, RCT = randomized controlled trial.
Fig. 3. The factors assessed in the process of review of a study by an Ethics Committee.
• The IRB should obtain and review all the necessary documents for the research/trial within a reasonable time and document its views following standardized operating procedures with clear identification of the dates for approval, modifications, disapproval, or termination of an ongoing trial that was initially approved in writing.
• Qualification of the investigators should be considered for the proposed research.
• Reviewing of ongoing research as appropriate to the risks involved (at least once a year).
• Protocols indicating exemption of prior consent of the subject or their legally acceptable representative (e.g., emergency situations) should be assessed in detail for all the regulatory needs.
• Review the sum and method of compensatory payment to subjects if required.
• Functions should be performed as per written SOPs which should comply with the GCP guideline.
Most IRBs conduct meetings regularly (one–two per month depending on the number of protocols) and SOPs are followed as per the national governing authority.
An EC review is a continuous process and is needed before the initiation of research, before the extension of the approval period, prior to modifications to an already approved study, for monitoring of any adverse events, and until all the data collection and analysis is complete.8 An oversight to the monitoring of trials (usually single center, early phase, less risky) is provided by the IRBs through annual reviews, adverse event monitoring, and reporting of undue events by the principal investigator (PI). However, complex clinical trials and/or multicenter, randomized controlled trials, interventional studies with pre-existing concerns about safety, or study participants who might need additional protection through an additional committee referred to as the Data Safety Monitoring Board (DSMB).43,44
RISKS, BENEFITS, CONFIDENTIALITY, AND PRIVACY ISSUES IN RESEARCH PROTOCOLS
The role of the EC is not only to provide direct protection to human subjects from physical or mental harm but also to weigh the risks and benefits involved in the research. It must be assessed if the study is designed to add to the current scientific knowledge base and help society.8
The research protocol is the document that includes the research question, aims and objectives, a critical literature review, methodology, and statistical plan. It is pertinent that the IRB reviews the protocol with respect to the clarity and focus of the research question; and whether the study design is suitable to answer the same. This is decided by the chair or a special departmental committee (Table 2, Fig. 3).
Privacy and confidentiality are a part and parcel of every physician-patient relationship. Needless to say, this must be maintained in a researcher-human subject relationship as well. It helps build trust, curbs participant anxiety, maintains their dignity, and above all their autonomy.10 The International Committee of Medical Journal Editors recommends that authors must ensure that nonessential information like names, initials hospital record numbers, etc., are omitted during data collection, storage, and publication whenever possible.45 However, there’s an extent to which this confidentiality can be maintained. Information required for scientific purposes (e.g., clinical photographs) or those with mandated legal reporting may breach participant privacy. This needs to be explained to the participant and recorded in written informed consent (Table 2).
The role of the IRB with respect to privacy and confidentiality is to:
• Review the consent document and assess the sensitivity of the information, the duration for which it will be held, the usefulness of the information, and the ability to protect it.
• For multicenter projects, review the measures taken by the research team to maintain the privacy of the research subjects including the number of personnel with access to the information, data storage, and transfer.
• An ongoing review of the research must include monitoring of confidentiality issues to check for maintenance of the same and the need for a revised privacy protection plan.
• Educate researchers and IRB members regarding the data privacy and protection process.46
Review of informed consent by IRBs is especially important in low-middle-income countries. There are various issues related to the lack of understanding of the information provided, maintaining privacy due to interference by family members, and the inability to assess risk and benefit by the research participant. IRBs have an additional responsibility to ensure that studies have minimal/no risk to the participant, the consent forms are clear and simple to understand and ensure the proper process of obtaining informed consent is being followed without undue pressure or coercion to participate in the study.47
VIOLATIONS OF ETHICS APPROVAL RULES AND REGULATIONS
Violations of IRB approval rules like lack of approval, lack of approval of modifications to the protocol, and lack of informed consent can result in dire aftermaths for the authors. It can result in the withdrawal of the article if it’s still in press, retraction if it’s already published, and even removal if it has legal consequences. The number of papers retracted as searched on the retraction database48 is steadily increasing by the decade from 474 in the 1990s to 6120 in the 2010s. The most common reason for retraction is plagiarism whereas violation of IRB rules accounts for 4–5% of all retractions.49,50 When consultations for ethical inquiries to the Korean Association of Medical Journal Editor were analyzed, the most common reason was duplicate publications (12 of 80) with issues with IRB approval (5 of 80) and informed consent (6 of 80).51 Some of the examples of types of studies and their reasons for retractions have been summarized in Table 3.
Table 3. Subject wise analysis of examples of retractions secondary to lack of IRB approvals/ethical issues.
| No. | Health science (750) | Title | Type | Reasons | Date of publication | Date of retraction |
|---|---|---|---|---|---|---|
| 1 | Alternative medicine (15) | Retraction note: Improved treatment of asthma by using natural sources of antioxidants83 | Clinical Trial | • Lack of IRB approval | 06/26/2013 | 09/25/2014 |
| • Informed consent: none82 | ||||||
| 2 | Anesthesia (183) | Retraction note: Efficacy of dexmedetomidine as an adjunct to ropivacaine in bilateral dual-transversus abdominis plane blocks in patients with ovarian cancer who underwent cytoreductive surgery84 | Clinical Trial | • Lack of IRB approval for modifications | 08/18/2021 | 08/11/2022 |
| 3 | Cardiology/Cardiothoracic surgery (77) | Virtual reality-guided aortic valve leaflet reconstruction for type 0 bicuspid aortic stenosis85 | Case Report | • Lack of IRB approval for using a device not approved by the national authority | 06/01/2022 | 11/08/2022 |
| - Lack of patient consent | ||||||
| 4 | Dermatology (13) | Influence of maternal diet during lactation and use of formula feeds on development of atopic eczema in high risk infants86 | Clinical Trial | • Concerns/issues about authorship | 07/22/1989 | 10/28/2015 |
| • Ethical violations by author | ||||||
| • Falsification/fabrication of data | ||||||
| • Investigation by company/institution | ||||||
| • Misconduct: official investigation/finding | ||||||
| • Misconduct by author | ||||||
| • Upgrade/update of prior notice | ||||||
| 5 | Endocrinology (21) | Relationship between chronic kidney disease staging and vitamin D deficiency: a retrospective study87 | Medical Records Review | + Concerns/issues about authorship | 01/13/2022 | 03/17/2022 |
| + Concerns/issues about data | ||||||
| + False/forged authorship | ||||||
| + Investigation by journal/publisher | ||||||
| + Lack of IRB/IACUC approval | ||||||
| 6 | Gastroenterology (77) | The profile of the key pro-inflammatory cytokines in the serum of patients with CD and their association with the disease severity and activity88 | Prospective Observational Study | + Concerns/issues about authorship | 11/21/22 | 03/02/2023 |
| + Lack of IRB/IACUC approval | ||||||
| + Paper mill | ||||||
| + Unreliable results | ||||||
| 7 | Immunology (55) | Immunotherapy of HIV-infected patients with Gc protein-derived macrophage activating factor (GcMAF)89 | Clinical Study | • Lack of IRB approval | 11/28/2008 | 08/11/2014 |
| 8 | Neurology (76) | The association of interleukin-16 gene polymorphisms with IL-16 serum levels and risk of multiple sclerosis90 | Observational Study | + Concerns/issues about data | 02/02/17 | 01/06/21 |
| + Lack of IRB/IACUC approval | ||||||
| 9 | Obstetrics and gynecology (64) | Retraction note: Is early intervention using Mansoura-VV uterine compression sutures an effective procedure in the management of primary atonic postpartum hemorrhage?: a prospective study91 | Prospective Observational Study | • Lack of IRB approval | 05/31/17 | 02/04/2023 |
| 10 | Pediatrics (57) | Childhood iron deficiency anemia leads to recurrent respiratory tract infections and gastroenteritis92 | Prospective Observational Study | • Lack of IRB approval | 09/02/2019 | 05/11/2018 |
IRB = Institutional Review Board.
Violations can be assessed before the studies are published for those with IRB approval. It is the responsibility of the IRBs to monitor whether ongoing studies are abiding by the ethical regulations and whether the approved protocol is being followed. A study conducted in India by an IRB at a tertiary care hospital in Mumbai monitored 12 clinical trials from 2011–2017. The most common violations were related to informed consent, followed by a lack of understanding of protocol and protocol deviations. This was corrected by re-taking of the informed consent and retraining in GCP by the IRB.52 A similar study in Uganda done from 2007–2010 with monitoring of 40 research projects also found a similar frequency and reasons for violations.53
Journal editors routinely check if a statement mentioning whether ethics approval was sought has been mentioned in the manuscript. Depending on the journal and type of article, further details of the EC approval can be sought by the journal editorial board.54
ISSUES AND ONGOING DEVELOPMENTS
ECs were developed to provide ethical oversight to clinical research. But here are various issues associated with the functioning of IRBs.
• Composition: Most studies indicate a skewed gender representation in the structure IRBs. Further, the participation of laypersons on the board is minimal.14,42,55,56,57
• Overburdened IRBs, delays, and operational costs: The IRB reviews have been associated with delays from over 4 to 7 months on average from surveys conducted across the USA.58,59 A delay in biomedical research can translate into more than monetary loss as biomedical research saves lives and a delay in the approvals can result in greater loss of life.60 An older survey conducted across 63 institutions (with 20 being low volume, 24 intermediate volume, and 19 being high volume centers) in the USA in 2005 reported the median amount spent by academic medical centers on IRB was $750,000/year with an average of $559 per review. The main costs are divided across staff salary, board salary, space, outsourcing of the reviews, travel, supplies, and equipment.61 Over the years, there is a definite increase in the number of ongoing research projects thus increasing these costs further. Furthermore, documentation of Food and Drug Administration (FDA) warning letters to IRBs was predominantly related to paperwork stressing on documentation of reviews and meetings rather than ethical issues.62 Increasing paperwork further results in delays and added costs. These deficiencies are more marked in developing nations like India and China dealing with issues like lack of regulation, informal ethics reviews, lack of supervision, and insufficient ethics review capacity.63,64
• Multi-site projects: With multicenter projects on the rise, a single protocol is often reviewed by multiple IRBs. In a review of 17 articles reported from UK, USA and Europe, which underwent multiple IRB reviews of the same protocol there were discrepancies in the judgment. Five of 26 reported rejection at some and acceptance by some IRBs. However, there were great differences in the protocol revisions, consent, patient information sheets, risk-benefit assessment, and compensation arrangements.65 Keeping these issues in mind, the Common Rule in the USA was revised in 2017 with IRB approval required only from one center for multisite projects.66 This may be extrapolated to other nations or consideration of an expedited review at other sites when fully reviewed at one IRB can be considered.
• Independent EC and IEC: Independent ECs have inherent tissues of limitation of knowledge about the local community and use of these may promote IRB shopping. Whereas, local IRBs can have conflicts of interest as colleagues of investigators may be on the review board. Thus, a central IRB can alleviate some of these concerns by avoiding repetitive reviews, minimizing conflicts, and establishing a centralized adverse event reporting system.67,68,69 A central IRB can be formed by experts on a particular subject or by a group of institutes like the National Cancer Institute’s Central IRB and the Biomedical Research Alliance of New York respectively.70,71
• Scientific expertise of the IRB reviewers: The IRB reviewers may lack the scientific expertise to review sophisticated research projects that may affect the quality of the research.11,14,57,72 Regular training in research ethics and GCP along with adequate consultations with external experts is needed. This can be done at a national, regional, and international level. First, by identifying core issues and then solutions for them by focused training.73 Training of EC members is conducted across Central Asia and Eastern Europe under the framework of Forum for Ethics Committees in the Confederation of Independent States and Strategic Initiative for Developing Capacity in Ethical Review program that train members regarding GCP, bioethics, the establishment of an EC, review processes and SOPs, choosing independent consultants, and confidentiality agreements.74
• Review of studies involving complementary and integrative medicine (CIM) is a challenge due to the lack of quality evidence to support the basis for their use. Moreover, most international regulatory bodies and research regulations do not address CIM, thus leaving the review process and decision-making to the IRBs. However, it is to be emphasized here studies irrespective of the type (modern or CIM) must be reviewed using the same principles of respect, beneficence, and justice. Well-designed studies on CIM are essential to ascertain the health and safety of patients.75
DSMB
DSMB is defined by the FDA, USA as “a group of individuals with pertinent scientific expertise that review research data of an ongoing trial on a regular basis, advises the sponsor/or researcher regarding the continuing safety of research subjects and those yet to be recruited into the research trial, and advises as to the continuing validity and scientific merit of the trial.”76 It’s an autonomous entity independent of the researchers, sponsors, and the IRB so as to control data sharing and protect the authenticity of the clinical trial from unfavorable impact.35 It was first developed in the USA in the 1960s as the NIH began sponsoring multicenter trials, the first trial was the Coronary Drug Project which used a DSMB for monitoring.77 Over time, it became a common practice for the sponsors to have experienced scientific personnel serving on these committees. Although the FDA does not mandate DSMB for all trials, DSMBs are generally recommended for large, multi-site studies evaluating treatments that intend to reduce mortality and morbidity.
DSMBs are usually constituted for:
• The study outcome is such that a highly encouraging or detrimental result is a possibility in an interim analysis that may require an early termination of the study on ethical grounds.
• When the safety concerns are high, e.g., invasive therapy is administered.
• Previous data suggesting serious toxicity with the study treatment.
• Studies involving vulnerable populations.
• Studies including subjects at an increased risk of death or serious outcomes.
• Large, multisite, long-duration studies.
In India, it is recommended by the Indian GCP guidelines that the sponsor may establish a DSMB to assess the progress of the trial, and in 2006 Indian Council of Medical Research (ICMR) mandated a DSMB to review data emerging from research on interventions in the emergency setting.39 These were updated in 2012 by the ICMR to include all stem cell research involving human subjects. The SOPs for the constitution and responsibilities of the DSMB are laid down by the World Health Organization and are similar across USA, Europe, and South Korea.78,79
DSMBs are constituted by scientific members and are appointed by the funding agency, before the recruitment of the first subject in the trial. It can consist of as few as three members and is typically constituted of clinicians and at least one biostatistician. Others that may be included are medical ethicists, other scientists, etc. The most important requisite is that the members should be independent of the sponsors, investigators IRBs, regulatory authorities, and site or study staff. They should have no conflicts of interest with the sponsors, researchers, or study staff.
The functions of the DSMB are:
• To uphold participant safety.
• Ensure credibility and integrity of the trial for future subjects.
• Ensure the timely conclusion of the study so that the results can be disseminated.
• Identify protocol violations if any.
• Identify unexpectedly high dropouts and evaluate for the same.
• Ensure the validity of the results.
The above functions are carried out by an initial organizational meeting to understand the protocol and safety monitoring plan followed by an early safety review meeting to review early safety information. Continuing periodic reviews to assess safety, efficacy, and the progress of the trial are then carried out with reporting of serious adverse events.44 A final meeting is to be held at the termination of a study. DSMBs function independently of the IRBS but the PIs must submit DSMB reports or minutes to the IRB.
Dramatic instances in which trials have been stopped prematurely on the recommendation of the DSMB include the withdrawal of rofecoxib and celecoxib in two trials on the prevention of colonic polyps due to increased cardiovascular events.80,81
CONCLUSION
We have come a long way from the horrific ethical compromises in clinical studies in history to establishing adequate safety for the human subjects participating in clinical research today. The establishment of the IRB or EC has ensured safe study designs and the safety of human subjects right from before the study initiation until its completion. This is further supplanted by additional boards like DSMBs. However, we still need studies assessing the outcomes of the ECs on a global basis and addressing various issues that are still pertinent to the working of the ECs.82
Footnotes
Disclosures: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Mehta P, Zimba O, Gasparyan AY, Seiil B, Yessirkepov M.
- Data curation: Mehta P.
- Writing - original draft: Mehta P.
- Writing - review & editing: Mehta P, Zimba O, Gasparyan AY, Seiil B, Yessirkepov M.
References
- 1.United States Holocaust Memorial Museum. The doctors trial: the medical case of the subsequent nuremberg proceedings. [Updated 2023]. [Accessed March 17, 2023]. https://encyclopedia.ushmm.org/content/en/article/the-doctors-trial-the-medical-case-of-the-subsequent-nuremberg-proceedings .
- 2.Nuremberg Military Tribunal. The nuremberg code. JAMA. 1996;276(20):1691. [PubMed] [Google Scholar]
- 3.Krugman S. The Willowbrook hepatitis studies revisited: ethical aspects. Rev Infect Dis. 1986;8(1):157–162. doi: 10.1093/clinids/8.1.157. [DOI] [PubMed] [Google Scholar]
- 4.Beecher HK. Ethics and clinical research. N Engl J Med. 1966;274(24):1354–1360. doi: 10.1056/NEJM196606162742405. [DOI] [PubMed] [Google Scholar]
- 5.World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA. 1997;277(11):925–926. [PubMed] [Google Scholar]
- 6.World Medical Association. WMA DOH 1964-2014. [Updated 2023]. [Accessed March 17, 2023]. https://www.wma.net/publications/wma-doh-1964-2014/
- 7.National Center for Biotechnology Information. Ethics Committees. [Updated 2023]. [Accessed April 5, 2023]. https://www.ncbi.nlm.nih.gov/mesh/68017041 .
- 8.Code of Federal Regulations. Part 46 - Protection of human subjects. [Updated 2023]. [Accessed March 26, 2023]. https://www.ecfr.gov/on/2018-07-19/title-45/subtitle-A/subchapter-A/part-46 .
- 9.Office for Human Research Protections. Federal policy for the protection of human subjects (‘Common Rule’) [Updated 2009]. [Accessed March 17, 2023]. https://www.hhs.gov/ohrp/regulations-and-policy/regulations/common-rule/index.html .
- 10.Office for Human Research Protections. Read the Belmont report. [Updated 2018]. [Accessed March 17, 2023]. https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/read-the-belmont-report/index.html .
- 11.Hennig W. Bioethics in China: although national guidelines are in place, their implementation remains difficult. EMBO Rep. 2006;7(9):850–854. doi: 10.1038/sj.embor.7400794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar NK. Bioethics activities in India. East Mediterr Health J. 2006;12(Suppl 1):S56–S65. [PubMed] [Google Scholar]
- 13.Thatte UM, Marathe PA. Ethics Committees in India: past, present and future. Perspect Clin Res. 2017;8(1):22–30. doi: 10.4103/2229-3485.198549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim OJ, Park BJ, Sohn DR, Lee SM, Shin SG. Current status of the Institutional Review Boards in Korea: constitution, operation, and policy for protection of human research participants. J Korean Med Sci. 2003;18(1):3–10. doi: 10.3346/jkms.2003.18.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beauchamp TL, Childress JF. Principles of Biomedical Ethics. 8th ed. Oxford, UK: Oxford University Press; 2019. [Google Scholar]
- 16.von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle. 2019;10(5):1143–1145. doi: 10.1002/jcsm.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasparyan AY, Ayvazyan L, Blackmore H, Kitas GD. Writing a narrative biomedical review: considerations for authors, peer reviewers, and editors. Rheumatol Int. 2011;31(11):1409–1417. doi: 10.1007/s00296-011-1999-3. [DOI] [PubMed] [Google Scholar]
- 18.Lapid MI, Clarke BL, Wright RS. Institutional Review Boards: what clinician researchers need to know. Mayo Clin Proc. 2019;94(3):515–525. doi: 10.1016/j.mayocp.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 19.Dutton JJ. Institutional Review Boards, Declaration of Helsinki, and HIPAA regulations. Ophthal Plast Reconstr Surg. 2013;29(5):335–340. doi: 10.1097/IOP.0b013e3182a1fd39. [DOI] [PubMed] [Google Scholar]
- 20.Wagner RM. Ethical review of research involving human subjects: when and why is IRB review necessary? Muscle Nerve. 2003;28(1):27–39. doi: 10.1002/mus.10398. [DOI] [PubMed] [Google Scholar]
- 21.Byerly WG. Working with the Institutional Review Board. Am J Health Syst Pharm. 2009;66(2):176–184. doi: 10.2146/ajhp070066. [DOI] [PubMed] [Google Scholar]
- 22.World Health Organization (WHO) Centre for Health Development. WHO guidance on research methods for health emergency and disaster risk management. [Updated 2021]. [Accessed March 30, 2023]. https://extranet.who.int/kobe_centre/en/project-details/GUIDANCE_ResearchMethods_HealthEDRM .
- 23.Orvis AK, Dellavalle RP. Institutional Review Board approval for surveys: why it is necessary. J Am Acad Dermatol. 2008;59(4):718–719. doi: 10.1016/j.jaad.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 24.Gaur PS, Zimba O, Agarwal V, Gupta L. Reporting survey based studies - a primer for authors. J Korean Med Sci. 2020;35(45):e398. doi: 10.3346/jkms.2020.35.e398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey E, Mühlmann C, Rice S, Nedeljkovic M, Alvarez-Jimenez M, Sander L, et al. Ethical issues and practical barriers in internet-based suicide prevention research: a review and investigator survey. BMC Med Ethics. 2020;21(1):37. doi: 10.1186/s12910-020-00479-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balon R, Guerrero AP, Coverdale JH, Brenner AM, Louie AK, Beresin EV, et al. Institutional Review Board approval as an educational tool. Acad Psychiatry. 2019;43(3):285–289. doi: 10.1007/s40596-019-01027-9. [DOI] [PubMed] [Google Scholar]
- 27.Panda M, Heath GW, Desbiens NA, Moffitt B. Research status of case reports for medical school Institutional Review Boards. JAMA. 2007;298(11):1277–1278. doi: 10.1001/jama.298.11.1277. [DOI] [PubMed] [Google Scholar]
- 28.Loe JD, Winkelman DA, Robertson CT. An assessment of the human subjects protection review process for exempt research. J Law Med Ethics. 2016;44(3):481–491. doi: 10.1177/1073110516667944. [DOI] [PubMed] [Google Scholar]
- 29.World Health Organization. Guidance for submissions of documents. [Updated 2021]. [Accessed March 17, 2023]. https://www.who.int/groups/research-ethics-review-committee/guidance-for-submissions-of-documents .
- 30.European Medicines Agency. 3. Institutional Review Board/Independent Ethics Committee (IRB/IEC): ICH E6(R2) Good Clinical Practice. [Updated 2018]. [Accessed March 26, 2023]. https://ichgcp.net/3-institutional-review-boardindependent-ethics-committee-irbiec .
- 31.Dal-Ré R. Waivers of informed consent in research with competent participants and the Declaration of Helsinki. Eur J Clin Pharmacol. 2023;79(4):575–578. doi: 10.1007/s00228-023-03472-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebers S, Aaronson NK, van Leeuwen FE, Schmidt MK. Exceptions to the rule of informed consent for research with an intervention. BMC Med Ethics. 2016;17(1):9. doi: 10.1186/s12910-016-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones XM, Zimba O, Gupta L. Informed consent for scholarly articles during the COVID-19 pandemic. J Korean Med Sci. 2021;36(3):e31. doi: 10.3346/jkms.2021.36.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards AD. Ethical guidelines for deliberately infecting volunteers with COVID-19. J Med Ethics. 2020;46(8):502–504. doi: 10.1136/medethics-2020-106322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.European Medicines Agency. ICH harmonised guideline integrated addendum to ICH E6(R1): Guideline for Good Clinical Practice ICH E6(R2) [Updated 2018]. [Accessed April 12, 2023]. https://ichgcp.net/home.
- 36.Council for International Organizations of Medical Sciences. International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva, Switzerland: Council for International Organizations of Medical Sciences; 2002. [PubMed] [Google Scholar]
- 37.Qiao H. A brief introduction to Institutional Review Boards in the United States. Pediatr Investig. 2018;2(1):46–51. doi: 10.1002/ped4.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hemminki E. Research Ethics Committees in the regulation of clinical research: comparison of Finland to England, Canada, and the United States. Health Res Policy Syst. 2016;14(1):5. doi: 10.1186/s12961-016-0078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Indian Council of Medical Research. Guidelines. [Updated 2017]. [Accessed March 30, 2023]. https://main.icmr.nic.in/guidelines?field_select_disease_tid=97 .
- 40.Association for Accreditation of Human Research Protection Programs. The AAHRPP accreditation program. [Updated 2019]. [Accessed March 20, 2023]. http://www.aahrpp.org/resources/for-accreditation/procedure/procedure-doc-1/the-aahrpp-accreditation-program .
- 41.Office for Human Research Protections. Regulations, policy & guidance. [Updated 2020]. [Accessed March 20, 2023]. https://www.hhs.gov/ohrp/regulations-and-policy/index.html .
- 42.Lidz CW, Simon LJ, Seligowski AV, Myers S, Gardner W, Candilis PJ, et al. The participation of community members on medical Institutional Review Boards. J Empir Res Hum Res Ethics. 2012;7(1):1–6. doi: 10.1525/jer.2012.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.National Institutes of Health. NIH policy for data and safety monitoring. [Updated 1998]. [Accessed March 29, 2023]. https://grants.nih.gov/grants/guide/notice-files/not98-084.html .
- 44.Ellenberg SS, Fleming TR, DeMets DL. Data Monitoring Committees in Clinical Trials: A Practical Perspective. 2nd ed. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2019. [Google Scholar]
- 45.International Committee of Medical Journal Editors. Homepage of ICMJE. [Updated 2023]. [Accessed March 26, 2023]. https://www.icmje.org/
- 46.Easter MM, Davis AM, Henderson GE. Confidentiality: more than a linkage file and a locked drawer. IRB. 2004;26(2):13–17. [PubMed] [Google Scholar]
- 47.Regmi PR, Aryal N, Kurmi O, Pant PR, van Teijlingen E, Wasti SP. Informed consent in health research: challenges and barriers in low-and middle-income countries with specific reference to Nepal. Developing World Bioeth. 2017;17(2):84–89. doi: 10.1111/dewb.12123. [DOI] [PubMed] [Google Scholar]
- 48.Center For Scientific Integrity, Inc. Retraction Watch Database. [Updated 2023]. [Accessed March 29, 2023]. http://retractiondatabase.org/RetractionSearch.aspx?
- 49.Moylan EC, Kowalczuk MK. Why articles are retracted: a retrospective cross-sectional study of retraction notices at BioMed Central. BMJ Open. 2016;6(11):e012047. doi: 10.1136/bmjopen-2016-012047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campos-Varela I, Villaverde-Castañeda R, Ruano-Raviña A. Retraction of publications: a study of biomedical journals retracting publications based on impact factor and journal category. Gac Sanit. 2020;34(5):430–434. doi: 10.1016/j.gaceta.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 51.Kim YS, Ham DS. Analysis of consultations by the Committee for Publication Ethics of the Korean Association of Medical Journal Editors. Sci Ed. 2020;7(2):184–188. [Google Scholar]
- 52.Shetty YC, Singh KN, Marathe PA, Jalgaonkar SV, Gajbhiye S, Katkar J, et al. Reports of site monitoring visits by Institutional Ethics Committees in an Indian tertiary care hospital: a retrospective analysis. Indian J Med Ethics. 2019;4(3):178–183. doi: 10.20529/IJME.2019.042. [DOI] [PubMed] [Google Scholar]
- 53.Ochieng J, Ecuru J, Nakwagala F, Kutyabami P. Research site monitoring for compliance with ethics regulatory standards: review of experience from Uganda. BMC Med Ethics. 2013;14(1):23. doi: 10.1186/1472-6939-14-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bain LE. Ethics approval: responsibilities of journal editors, authors and Research Ethics Committees. Pan Afr Med J. 2017;28:200. doi: 10.11604/pamj.2017.28.200.14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Veras Santos EP, Guerriero IC. Professional and academic profile of the Brazilian Research Ethics Committees. BMC Med Ethics. 2022;23(1):109. doi: 10.1186/s12910-022-00847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salim M, Hamid S. Independent Review of research proposals from ethical point of view in Pakistan. J Ayub Med Coll Abbottabad. 2018;30(4):588–591. [PubMed] [Google Scholar]
- 57.Brahme R, Mehendale S. Profile and role of the members of Ethics Committees in hospitals and research organisations in Pune, India. Indian J Med Ethics. 2009;6(2):78–84. doi: 10.20529/IJME.2009.026. [DOI] [PubMed] [Google Scholar]
- 58.Varley PR, Feske U, Gao S, Stone RA, Zhang S, Monte R, et al. Time required to review research protocols at 10 veterans affairs Institutional Review Boards. J Surg Res. 2016;204(2):481–489. doi: 10.1016/j.jss.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silberman G, Kahn KL. Burdens on research imposed by Institutional Review Boards: the state of the evidence and its implications for regulatory reform. Milbank Q. 2011;89(4):599–627. doi: 10.1111/j.1468-0009.2011.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitney SN, Schneider CE. Viewpoint: a method to estimate the cost in lives of ethics board review of biomedical research. J Intern Med. 2011;269(4):396–402. doi: 10.1111/j.1365-2796.2011.02351_2.x. [DOI] [PubMed] [Google Scholar]
- 61.Sugarman J, Getz K, Speckman JL, Byrne MM, Gerson J, Emanuel EJ, et al. The cost of Institutional Review Boards in academic medical centers. N Engl J Med. 2005;352(17):1825–1827. doi: 10.1056/NEJM200504283521723. [DOI] [PubMed] [Google Scholar]
- 62.Bramstedt KA, Kassimatis K. A study of warning letters issued to Institutional Review Boards by the United States Food and Drug Administration. Clin Invest Med. 2004;27(6):316–323. [PubMed] [Google Scholar]
- 63.Kadam R, Karandikar S. Ethics Committees in India: facing the challenges! Perspect Clin Res. 2012;3(2):50–56. doi: 10.4103/2229-3485.96444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou J. Problems and development strategies for Research Ethics Committees in China’s higher education institutions. J Med Ethics. 2020:medethics-2020-106768. doi: 10.1136/medethics-2020-106768. [DOI] [PubMed] [Google Scholar]
- 65.Edwards SJ, Stone T, Swift T. Differences between Research Ethics Committees. Int J Technol Assess Health Care. 2007;23(1):17–23. doi: 10.1017/S0266462307051525. [DOI] [PubMed] [Google Scholar]
- 66.Office for Human Research Protections. Revised Common Rule. [Updated 2017]. [Accessed March 27, 2023]. https://www.hhs.gov/ohrp/regulations-and-policy/regulations/finalized-revisions-common-rule/index.html .
- 67.Emanuel EJ, Wood A, Fleischman A, Bowen A, Getz KA, Grady C, et al. Oversight of human participants research: identifying problems to evaluate reform proposals. Ann Intern Med. 2004;141(4):282–291. doi: 10.7326/0003-4819-141-4-200408170-00008. [DOI] [PubMed] [Google Scholar]
- 68.Forster D. Independent Institutional Review Boards. Seton Hall Law Rev. 2003;32(3):513–523. [PubMed] [Google Scholar]
- 69.Flynn KE, Hahn CL, Kramer JM, Check DK, Dombeck CB, Bang S, et al. Using central IRBs for multicenter clinical trials in the United States. PLoS One. 2013;8(1):e54999. doi: 10.1371/journal.pone.0054999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.National Cancer Institute (NCI) Central Institutional Review Board. Welcome to the CIRB. [Updated 2023]. [Accessed March 30, 2023]. https://ncicirb.org/
- 71.Biomedical Research Alliance of New York. IRB services. [Updated 2023]. [Accessed March 30, 2023]. https://www.brany.com/irb-services/
- 72.Borovečki A, ten Have H, Oresković S. Education of Ethics Committee members: experiences from Croatia. J Med Ethics. 2006;32(3):138–142. doi: 10.1136/jme.2005.011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davies H, Wells F, Druml C. How can we provide effective training for Research Ethics Committee members? A European assessment. J Med Ethics. 2008;34(4):301–302. doi: 10.1136/jme.2007.021485. [DOI] [PubMed] [Google Scholar]
- 74.Chaschin M, Mishatkina T, Guryleva M, Pustovit S, Zagirtdinova F, Sarymsakova B. Developing national systems for research ethics education in Eastern Europe and Central Asia. Pharmaceut Med. 2008;22(5):289–295. [Google Scholar]
- 75.Cooper JA, Borasky D, Rosenfeld S, Sugarman J. Challenges in the ethical review of research involving complementary and integrative medicine. Ther Innov Regul Sci. 2016;50(3):337–341. doi: 10.1177/2168479015620246. [DOI] [PubMed] [Google Scholar]
- 76.U.S. Food and Drug Administration. Establishment and operation of clinical trial data monitoring committees. [Updated 2021]. [Accessed April 12, 2023]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/establishment-and-operation-clinical-trial-data-monitoring-committees .
- 77.The Coronary Drug Project. Initial findings leading to modifications of its research protocol. JAMA. 1970;214(7):1303–1313. [PubMed] [Google Scholar]
- 78.Lee BR, Lee KE. Independent data monitoring committees: review of current guidelines. Korean J Clin Pharm. 2016;26(2):181–186. [Google Scholar]
- 79.UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases. Operational Guidelines for the Establishment and Functioning of Data and Safety Monitoring Boards. Geneva, Switzerland: World Health Organization; 2005. [Google Scholar]
- 80.Sibbald B. Rofecoxib (Vioxx) voluntarily withdrawn from market. CMAJ. 2004;171(9):1027–1028. doi: 10.1503/cmaj.1041606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Federal Register. Pfizer, Inc.; Withdrawal of approval of familial adenomatous polyposis indication for CELEBREX. [Updated 2012]. [Accessed March 30, 2023]. https://www.federalregister.gov/documents/2012/06/08/2012-13900/pfizer-inc-withdrawal-of-approval-of-familial-adenomatous-polyposis-indication-for-celebrex .
- 82.Coleman CH, Bouësseau MC. How do we know that Research Ethics Committees are really working? The neglected role of outcomes assessment in research ethics review. BMC Med Ethics. 2008;9(1):6. doi: 10.1186/1472-6939-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Toan N, Thi Hanh T. Retraction note: Improved treatment of asthma by using natural sources of antioxidants. Springerplus. 2014;3(1):558. doi: 10.1186/2193-1801-3-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang JP, Zhang N, Chen X, Zhou Y, Jiang Z, Gao C, et al. Retraction note: Efficacy of dexmedetomidine as an adjunct to ropivacaine in bilateral dual-transversus abdominis plane blocks in patients with ovarian cancer who underwent cytoreductive surgery. BMC Anesthesiol. 2022;22(1):188. doi: 10.1186/s12871-022-01731-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Retraction: Virtual reality-guided aortic valve leaflet reconstruction for type 0 bicuspid aortic stenosis. Interact Cardiovasc Thorac Surg. 2022;35(6):ivac281. doi: 10.1093/icvts/ivac281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Group BM. Retraction: Influence of maternal diet during lactation and use of formula feeds on development of atopic eczema in high risk infants. BMJ. 2015;351:h5682. doi: 10.1136/bmj.h5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kantas T, Avendaño Capriles CA, Babor S, Tamdin T, Al-Rihani H, Thalla A, et al. Retraction: Relationship between chronic kidney disease staging and vitamin D deficiency: a retrospective study. Cureus. 2022;14(3):r55. doi: 10.7759/cureus.r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Al Qteishat A, Kirov K, Bokov D. The profile of the key pro-inflammatory cytokines in the serum of patients with CD and their association with the disease severity and activity. BMC Gastroenterol. 2022;22(1):477. doi: 10.1186/s12876-022-02562-w. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Retraction. Immunotherapy of HIV-infected patients with Gc protein-derived macrophage activating factor. J Med Virol. 2014;86(11):1998. doi: 10.1002/jmv.24060. [DOI] [PubMed] [Google Scholar]
- 90.Statement of Retraction: The association of interleukin-16 gene polymorphisms with IL-16 serum levels and risk of multiple sclerosis. Immunol Invest. 2023;52(1):134–134. doi: 10.1080/08820139.2020.1855698. [DOI] [PubMed] [Google Scholar]
- 91.El Refaeey AE, Abdelfattah H, Mosbah A, Gamal AM, Fayla E, Refaie W, et al. Retraction note: Is early intervention using Mansoura-VV uterine compression sutures an effective procedure in the management of primary atonic postpartum hemorrhage?: a prospective study. BMC Pregnancy Childbirth. 2023;23(1):95. doi: 10.1186/s12884-023-05410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jayaweera JA, Reyes M, Joseph A. Childhood iron deficiency anemia leads to recurrent respiratory tract infections and gastroenteritis. Sci Rep. 2019;9(1):12637. doi: 10.1038/s41598-019-49122-z. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]