

Abstract
The 2022 Nobel Prize in Chemistry was awarded to Professors K. Barry Sharpless, Morten Meldal, and Carolyn Bertozzi for their pioneering roles in the advent of click chemistry. Sharpless and Meldal worked to develop the canonical click reaction — the copper-catalyzed azide-alkyne cycloaddition — while Bertozzi opened new frontiers with the creation of the bioorthogonal strain-promoted azide-alkyne cycloaddition. These two reactions have revolutionized chemical and biological science by facilitating ligations that are selective, high-yielding, rapid, and clean and by providing unprecedented ways to manipulate living systems. Click chemistry has affected every aspect of chemistry and chemical biology, but few disciplines have been impacted as much as radiopharmaceutical chemistry. The importance of speed and selectivity in radiochemistry make it an almost tailor-made application of click chemistry. In this Perspective, we discuss the ways in which the copper-catalyzed azide-alkyne cycloaddition, the strain-promoted azide-alkyne cycloaddition, and a handful of ‘next generation’ click reactions have transformed radiopharmaceutical chemistry, both as tools for more efficient radiosyntheses and as linchpins of technologies that have the potential to improve nuclear medicine.
Keywords: Click chemistry, azide-alkyne cycloaddition, strain-promoted azide-alkyne cycloaddition, inverse electron-demand Diels-Alder reaction, radiopharmaceutical chemistry, radiochemistry, PET, SPECT, targeted radionuclide therapy
INTRODUCTION
The 2022 Nobel Prize in Chemistry was awarded to Professors K. Barry Sharpless, Morten Meldal, and Carolyn Bertozzi for their seminal work on the development of a now ubiquitous suite of chemical transformations known as ‘click chemistry’1. Designed to facilitate the facile and selective construction of molecules, click chemistry reactions are by design efficient, rapid, modular, clean, and water-compatible. Sharpless and Meldal independently developed what is considered the classical click ligation: the copper-catalyzed 3+2 cycloaddition between an azide and a terminal alkyne (Figure 1A)2. This reaction — known as the copper-catalyzed azide-alkyne cycloaddition (CuAAC) — represents a significant improvement to the non-catalyzed but temperature-driven 1,3-diploar Huisgen cycloaddition between the same two moieties.3 Although hailed as a breakthrough technology, the ternary nature of the CuAAC reaction and the intrinsic toxicity of copper limited its applications in biological systems. Bertozzi circumvented these issues via the creation of a two-component click ligation in which the ring strain of a cyclic octyne — rather than a catalyst — provides the driving force for a 3+2 cycloaddition with an azide (Figure 1B)4. This development initiated the era of ‘bioorthogonal’ click chemistry (i.e. the use of click ligations that are compatible with living biological systems). Excitingly, innovations in the field continue to arm chemists and biologists with an arsenal of novel click reactions — e.g. the inverse electron-demand Diels-Alder (IEDDA) reaction and the Staudinger ligation — with properties suitable for a wide variety of applications5,6.
Figure 1.
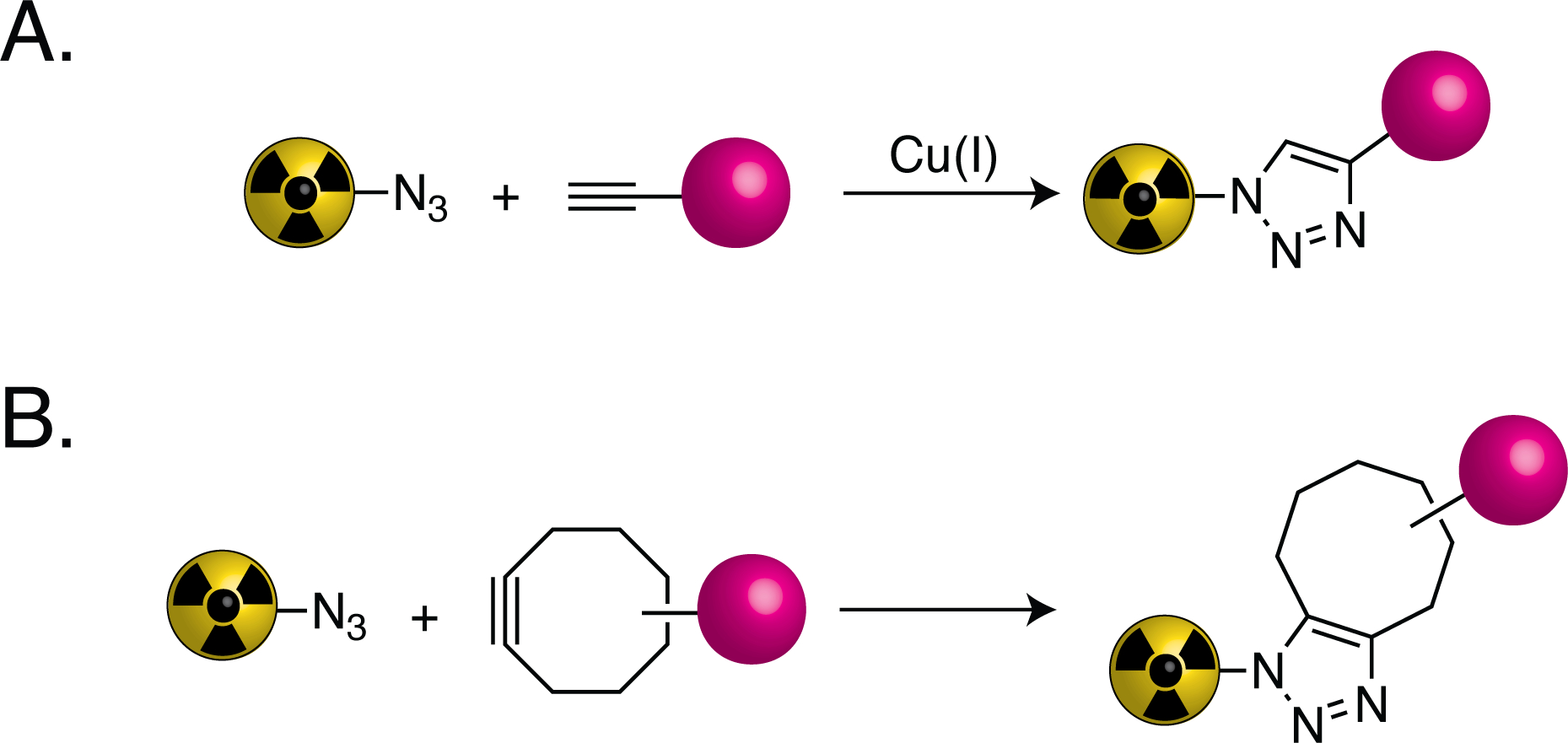
Schematic of (A) the copper-catalyzed azide-alkyne cycloaddition (CuAAC) and (B) the strain-promoted azide-alkyne cycloaddition (SPAAC). Magenta spheres represent cargoes.
Click chemistry has had a transformative effect on nearly every aspect of chemical science, but radiopharmaceutical chemistry has been impacted particularly profoundly7. Indeed, the exigencies of radiochemistry are uniquely well served by speed (a priority due to the decay of radionuclides), selectivity (critical to retain the biological activity of probes), and cleanliness (imperative due to the in vivo use of radiopharmaceuticals). As a result, radiopharmaceutical chemists have enthusiastically turned to click chemistry to solve problems in the field8–11. In this Perspective, we will discuss the practical advantages that the copper-catalyzed azide-alkyne cycloaddition (CuAAC), the strain-promoted azide-alkyne cycloaddition (SPAAC), and a host of emergent click ligations provide in radiopharmaceutical chemistry.
The Copper-Catalyzed Azide-Alkyne Cycloaddition
Surprisingly, the reaction that would later become the canonical click ligation — the copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction — is not mentioned in the original 2001 article, which coins the term ‘click chemistry’12. The CuAAC ligation relies on a Cu(I) catalyst to facilitate the reaction of an azide and a terminal alkyne to form a 1,4-disubstituted 1,2,3-triazole. One of the first applications of the CuAAC reaction to radiopharmaceutical chemistry harnessed the ligation for the synthesis of peptides labeled with the positron-emitting radiohalogen fluorine-18 (18F, t1/2 ~ 109 min)13. This use has proven enduring, as 18F-labeled synthons like 2-[18F]fluoroethylazide continue to be employed for this purpose over a decade later14. The CuAAC reaction has since been used to create radiopharmaceuticals using vectors ranging from small molecules to nanoparticles and radionuclides spanning from 11C to 225Ac. Here we highlight a selection of studies that best convey the utility of the reaction in the hand of radiochemists.
The most common use of the CuAAC ligation in radiochemistry is the assembly of radiopharmaceuticals. In one particularly elegant example, three different CuAAC reactions were used to create a bifunctional probe for positron emission tomography (PET) and near-infrared fluorescence imaging (NIRF) (Figure 2A)15. More specifically, a pair of CuAAC reactions was first leveraged to append two cyclic integrin αvβ3-targeting peptides to a central near-infrared fluorophore. Next, following the modification of the fluorophore with yet another azide, a third CuAAC ligation was used to attach a 18F-bearing synthon. Remarkably, this seemingly complicated synthesis was accomplished in up to 90% yield in one-pot in under an hour. Furthermore, the completed imaging agent — dubbed ‘[18F]F-NIR-cRGD2’ — effectively delineated U-87 MG human glioblastoma xenografts in nude mice, suggesting that it may have potential as a tool for both PET and intraoperative NIRF imaging in patients with integrin αvβ3-expressing malignancies.
Figure 2.
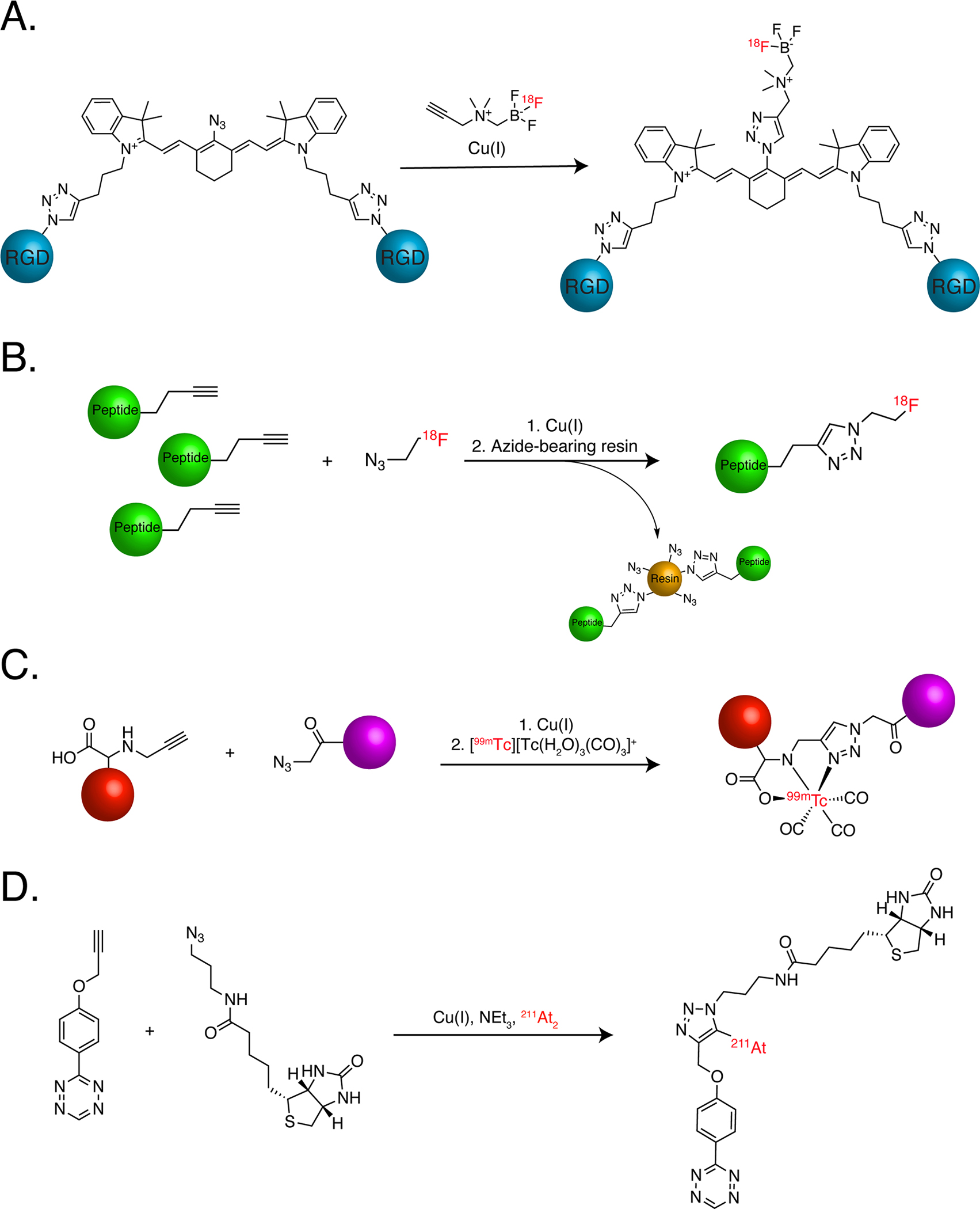
The use of the CuAAC ligation to (A) synthesize an integrin-targeted multimodal PET/NIRF imaging agent, (B) increase the specific activity of 18F-labeled peptides, (C) radiolabel a vector with a [99mTc]Tc(CO)3 core via the ‘click-to-chelate’ strategy, and (D) create an 211At-labeled probe in which the radiohalogen is appended to the triazole moiety. Red and purple spheres represent cargoes.
Another way in which the CuAAC ligation can be harnessed for radiopharmaceutical chemistry relies on a critical yet often underappreciated property of radiotracers: their molar activity, i.e. the amount of radioactivity per mole of radiopharmaceutical. Molar activity is important because it denotes the fraction of the total molecules in a formulation of radiopharmaceutical that is radiolabeled. If the molar activity of a given radiotracer is too low (i.e. only a very small fraction of the molecules is radiolabeled), unlabeled molecules will outcompete radiolabeled ones for binding sites, lowering target uptake and target-to-background signal ratios. Molar activity is especially important in the context of low abundance targets, as the risk of being outcompeted by unlabeled molecules is particularly high. Against this backdrop, the CuAAC reaction was used to quickly and easily label a molar excess of alkyne-modified HER2-targeting peptides with [18F]fluoroethylazide (Figure 2B)16. Subsequently, more than 98% of the unreacted, excess alkyne-bearing peptides was removed via the CuAAC ligation using an inexpensive azide-modified resin. In this manner, the 18F-labeled peptides with molar activities greater than 200 GBq/μmol were isolated, a 200-fold increase compared to the peptides that had not been ‘purified via click’. Importantly, this boost in molar activity provided practical benefits, as the radiopeptides with high molar activity produced better PET images than their counterparts with lower molar activity in murine models of ovarian adenocarcinoma and triple-negative breast cancer.
In the first two examples, the radionuclides in question simply acted as cargoes, small parts of synthons to be clicked to other components within the tracer. Yet this need not be the case. Indeed, in the next two applications of the CuAAC reaction, the radionuclides themselves interact far more intimately with the ligation’s 1,2,3-triazole product. The “click-to-chelate” strategy represents a way to radiolabel molecules with technetium- and rhenium-tricarbonyl cores17. In this approach, a one-pot-two-step reaction creates a triazole that not only forms a linkage to the targeting vector but also becomes an integral part of the coordination environment of the 99mTc/Re(CO)3 core via its electron-rich N3 nitrogen (Figure 2C). Given 99mTc’s status as a workhorse of single photon emission computed tomography (SPECT), this synthetic strategy can be used to streamline the development of novel 99mTc-labeled radiopharmaceuticals as well as radiotherapeutics labeled with its radiocongeners 186Re and 188Re. More recently, the CuAAC ligation has been harnessed to create a straightforward strategy for radiolabeling with 211At, a short-lived α-emitting radiohalogen that holds great potential for the targeted radionuclide therapy (TRT) of metastatic lesions but remains underutilized due to its mercurial labeling chemistry and supply chain challenges (Figure 2D). In this approach — which is based on prior work with 125I18 — the CuAAC ligation is run in the presence of the anionic radiohalogen as well as excess Cu(II).19 In addition to catalyzing the click reaction, Cu(II) oxidizes the radiohalogen to 211At+, which subsequently undergoes an electrophilic substitution with an intermediate of the cycloaddition, ultimately producing a 5-[211At]astatotriazole. Preliminary results suggest that this strategy may yield more stable 211At-labeled radiopharmaceuticals than other synthetic approaches, an important development given the radionuclide’s propensity for in vivo dehalogenation.
The radiopharmaceuticals synthesized using the CuAAC reaction have been successfully translated to the clinic. In 2011, the first-in-human evaluation of an integrin-targeted radiotracer synthesized via the CuAAC ligation — [18F]F-RGD-K5 — was reported in healthy volunteers20. The first-in-human trial of a click-based somatostatin receptor-targeting probe — [18F]fluoroethyltriazole-Tyr3-octreotate ([18F]FET-βAG-TOCA) — in patients with neuroendocrine tumors was published in 201621. Recently, the CuAAC ligation was used to synthesize [68Ga]Ga-Trivehexin, a novel imaging agent composed of the Ga-chelator TRAP linked via triazoles to a trio of cyclic αvβ6-targeting peptides (Figure 3A)22. This multimeric construct boasts increased target avidity compared to its monomeric cousins and has been used for the PET imaging of both pancreatic adenocarcinoma and head and neck cancer. Indeed, first-in-human data suggests that [68Ga]Ga-Trivehexin is capable of effectively mapping the expression of αvβ6-integrin in a clinical setting (Figure 3B)22.
Figure 3.
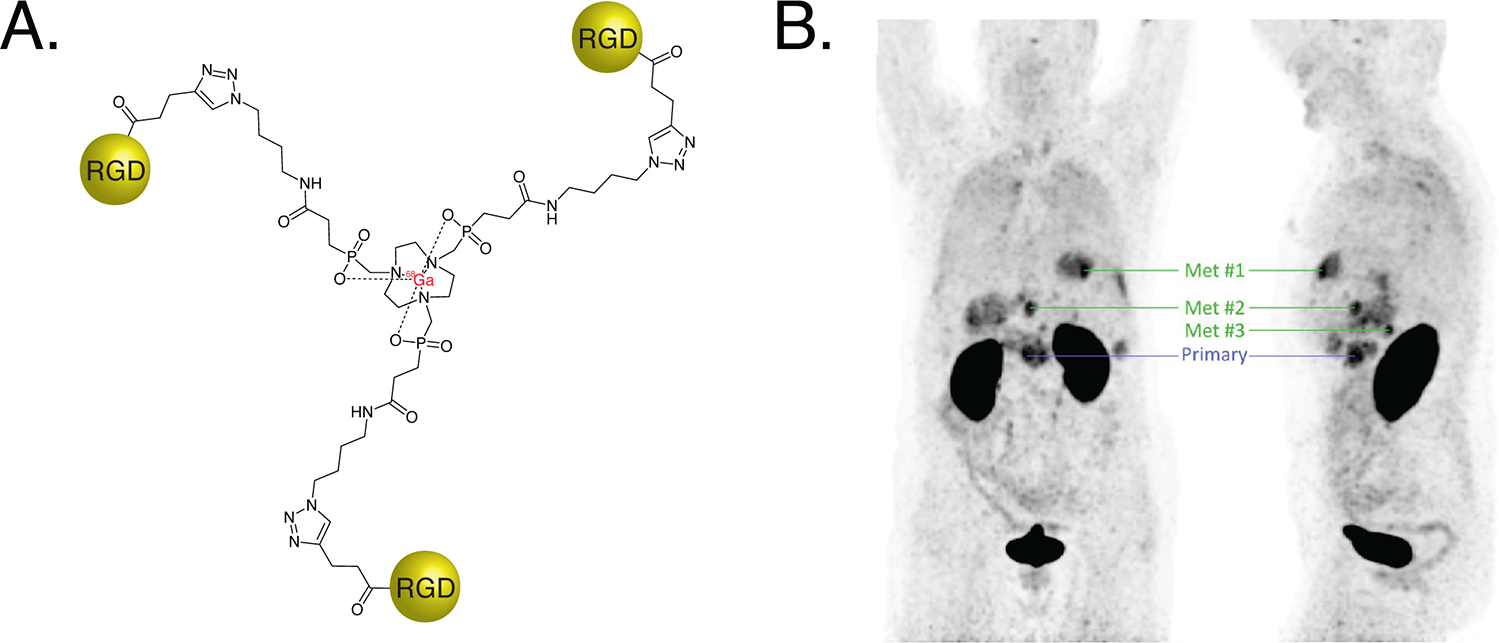
(A) The structure of [68Ga]Ga-Trivehexin; (B) PET images acquired 120 minutes after the administration of 105 MBq of [68Ga]Ga-Trivhexin to a patient with αvβ6-expressing pancreatic ductal adenocarcinoma showing uptake in a primary tumor as well as several metastatic lesions. Figure 3B was reprinted under the Creative Commons Attribution 4.0 International License (CC BY 4.0) terms from reference 22.
The Strain-Promoted Azide-Alkyne Cycloaddition
While the CuAAC ligation has had a tremendous impact on radiopharmaceutical chemistry, it nonetheless has limitations. Most notably, the Cu(I) catalyst can interfere with the use of radiometals, outcompeting the radionuclides — which are often used at only nanomolar levels — for chelators. Furthermore, the presence of high concentrations of Cu(I) can threaten the structural integrity of more sensitive biomolecules such as proteins and immunoglobulins. Consequently, many radiopharmaceutical chemists have turned to Bertozzi’s copper-free SPAAC ligation23.
In 2011, Campbell-Verduyn, et al. reported one of the first uses of the SPAAC reaction in radiopharmaceutical chemistry24. Specifically, the investigators successfully labeled a cyclooctyne-bearing variant of the gastrin-releasing peptide receptor (GRPR)-targeting peptide bombesin with various 18F-labeled azide moieties (Figure 4A). This reaction yielded several 18F-labeled peptides with high affinity for GRPR, demonstrating the promise of SPAAC for the reliable production of radiotracers. The SPAAC ligation has also been used to radiolabel azide-containing nanoparticles (SCK-NP) with a [64Cu]Cu-DOTA-labeled variant of dibenzocyclooctyne (DBCO)25, producing [64Cu]Cu-DOTA-SCK-NP with exceptionally high molar activities of ~36 TBq/μmol (Figure 4B). Yet a handful of PET imaging and biodistribution experiments have illustrated one potential drawback of the SPAAC ligation as a radiosynthetic tool: its ‘footprint’. To wit, 64Cu-labeled somatostatin receptor-targeting peptides synthesized via the SPAAC reaction exhibited significantly slower clearance from healthy tissues than those synthesized using traditional methods, a phenomenon attributed to the hydrophobicity of the bulky, tetracyclic product of the SPAAC ligation (Figure 4C)26. The same study also addressed another easily overlooked complication of the SPAAC reaction: unlike the CuAAC ligation — which forms predominantly 1,4-disubstituted triazoles — the SPAAC reaction is not regioselective, forming both 1,5- and 1,4-disubstituted triazole products that must be separated. It is important to note, however, that both obstacles (i.e. hydrophobicity and regioselectivity) will exert a much greater impact with small vectors than with larger biomolecules.
Figure 4.
The use of the SPAAC ligation to create (A) 18F-labeled GRPR-targeting peptides, (B) 64Cu-labeled nanoparticles, and (C) 64Cu-labeled somatostatin receptor-targeting peptides.
Easily one of the most common applications of the SPAAC ligation in radiochemistry is the synthesis of radioimmunoconjugates. Historically, the overwhelming majority of radiolabeled antibodies have been created via the stochastic attachment of chelators and prosthetic groups to the lysines of immunoglobulins. This approach is facile, but it frequently yields poorly defined and heterogenous products with suboptimal in vitro and in vivo performance27. In response, the community has dedicated considerable resources to the development of novel strategies for the attachment of cargoes to discrete sites within antibodies, a process called ‘site-specific bioconjugation’. The selectivity and bioorthogonal nature of the SPAAC reaction makes it perfectly suited for the task. For example, a genetic code expansion technique was used to integrate an azide-bearing unnatural amino acid — Nε−2-azideoethyloxycarbonyl-L-lysine (NEAK) — into the anti-CD20 mAb rituximab to yield an immunoconjugate dubbed ‘A122NEAK-rituximab’ (Figure 5A)28. This immunoconjugate was then conjugated with a DBCO-bearing variant of DOTA and labeled with 64Cu, producing a 64Cu-labeled radioimmunoconjugate with excellent in vivo behavior in a murine model of B cell lymphoma. In another case, a cell-free expression system was used to create a variant of trastuzumab containing a quartet of para-azidomethyl phenylalanine (pAMF) residues29. These unnatural amino acids were modified with DBCO-bearing versions of desferrioxamine (DFO) and 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid (DO3A), and the immunoconjugates were labeled with 89Zr and 111In, respectively (Figure 5B). The in vivo performance of the 89Zr- and 111In-labeled radioimmunoconjugates was interrogated via PET and SPECT imaging, respectively, in a murine model of ovarian cancer, with both radiotracers yielding excellent tumor uptake and tumor-to-background activity concentration ratios.
Figure 5.
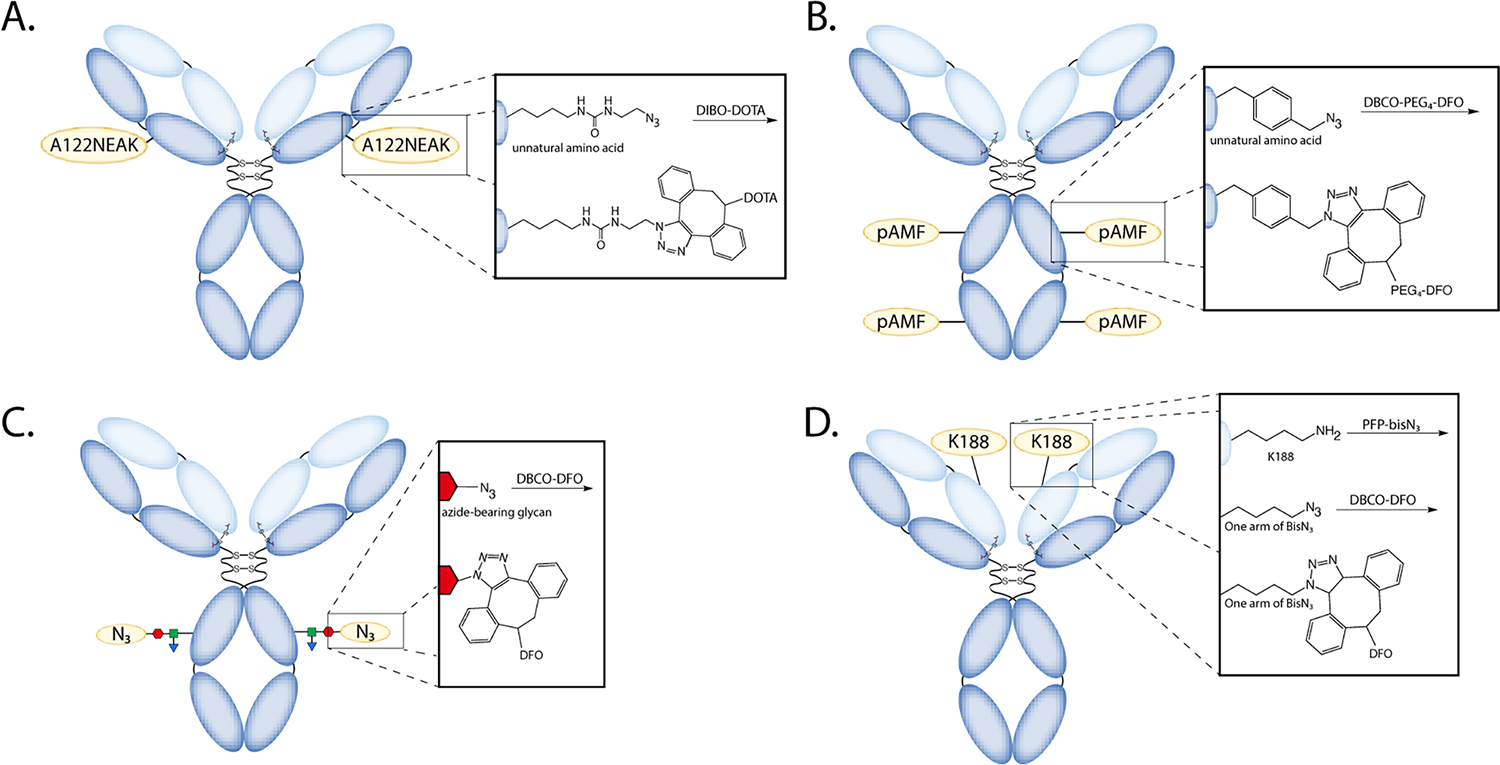
Four approaches that harness the SPAAC ligation to facilitate the site-specific bioconjugation of antibodies. In each case, the site-specificity is predicated on (A) the incorporation of the unnatural amino acid Nε−2-azideoethyloxycarbonyl-L-lysine, (B) the incorporation of the unnatural amino acid p-azidomethyl phenylalanine, (C) the chemoenzymatic manipulation of the heavy chain glycans to append azide-modified sugars, and (D) the selective ligation of a branched, azide-containing perfluorophenyl ester with the K188 residues of the light chain.
The SPAAC ligation can also be used for site-specific bioconjugation without resorting to expensive and complex genetic engineering. For example, a chemoenzymatic strategy featuring a promiscuous galactosyltransferase was employed to incorporate a pair of azide-modified sugars into the heavy chain glycans of the HER2-targeted mAb pertuzumab (Figure 5C)30. The azide-modified pertuzumab was then clicked to a DBCO-bearing variant of DFO and labeled with 89Zr, yielding an immunoPET probe that boasted dramatically improved performance compared to a stochastically labeled analog in both NSG and humanized NSG mice bearing HER2-expressing human breast cancer xenografts. This radioimmunoconjugate — [89Zr]Zr-ssDFO-pertuzumab — is currently the subject of a first-in-human clinical trial at Memorial Sloan Kettering Cancer Center (NCT04692831) designed to evaluate its potential for the PET imaging of patients with metastatic, HER2-positive breast, bladder, and renal cancer. A recent purely chemical approach to SPAAC-mediated site-specific bioconjugation eschews both genetic engineering and enzymatic transformations by relying upon the unique selectivity of a branched, azide-bearing perfluorophenyl ester reagent — PFP-bisN3 — for the K188 sites within the light chains of κIgG1 antibodies (Figure 5D)31. Using this strategy, the HER2-targeting mAb trastuzumab was modified with a quartet of azides that were then conjugated via SPAAC with a DBCO-modified variant of DFO. The subsequent labeling of the immunoconjugate with 89Zr produced a radioimmunoconjugate with high stability, high immunoreactivity, and excellent in vivo performance in a murine model of breast cancer. Each of these four SPAAC-mediated methods produces well-defined and homogeneous radioimmunoconjugates. While the first-in-human study of [89Zr]Zr-ssDFO-pertuzumab will certainly prove valuable for the field in demonstrating the clinical viability of site-specifically labeled radioimmunoconjugates, the recently developed PFP-bisN3 approach may ultimately prove to be the simplest and most readily translatable strategy.
Next Generation Click Reactions
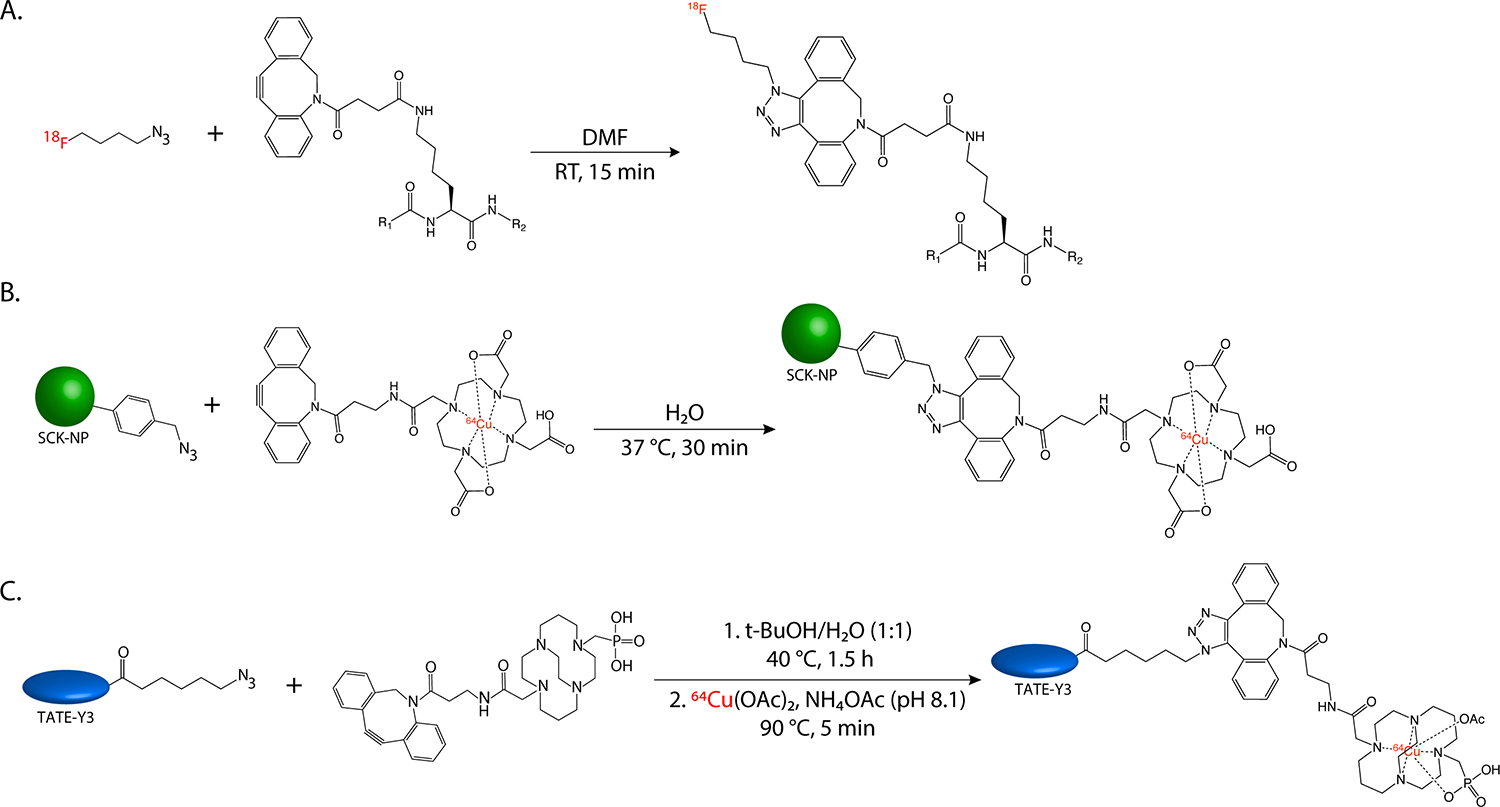
In the years since Sharpless, Meldal, and Bertozzi’s seminal discoveries, the field has continued to innovate and has developed a host of novel click ligations (and especially bioorthogonal click ligations) that radiopharmaceutical chemists have enthusiastically adopted. For example, Bertozzi’s own laboratory successfully used the Staudinger ligation to facilitate the labeling of azide-bearing biomolecules with triarylphosphine-modified fluorophores in live cells (Figure 6A). This reaction has been exploited for radiochemistry — most notably for the development of an arsenal of 18F-labeled phosphines for the mild radiolabeling of azide-bearing constructs — but its widespread adoption has been limited both by its sluggish kinetics (k2 ~ 10−3 M−1s−1) as well as the intrinsic instability of triarlyphosphines32. In another example, the click reaction between dibenzoazacyclooctyne (DIBAC) groups and sydnones (1,2,3-oxadiazoles) was used for synthesizing 18F-labeled peptides via the ligation between 4-[18F]fluorophenyl sydnone and DIBAC-bearing biomolecules (Figure 6B)33.
Figure 6.
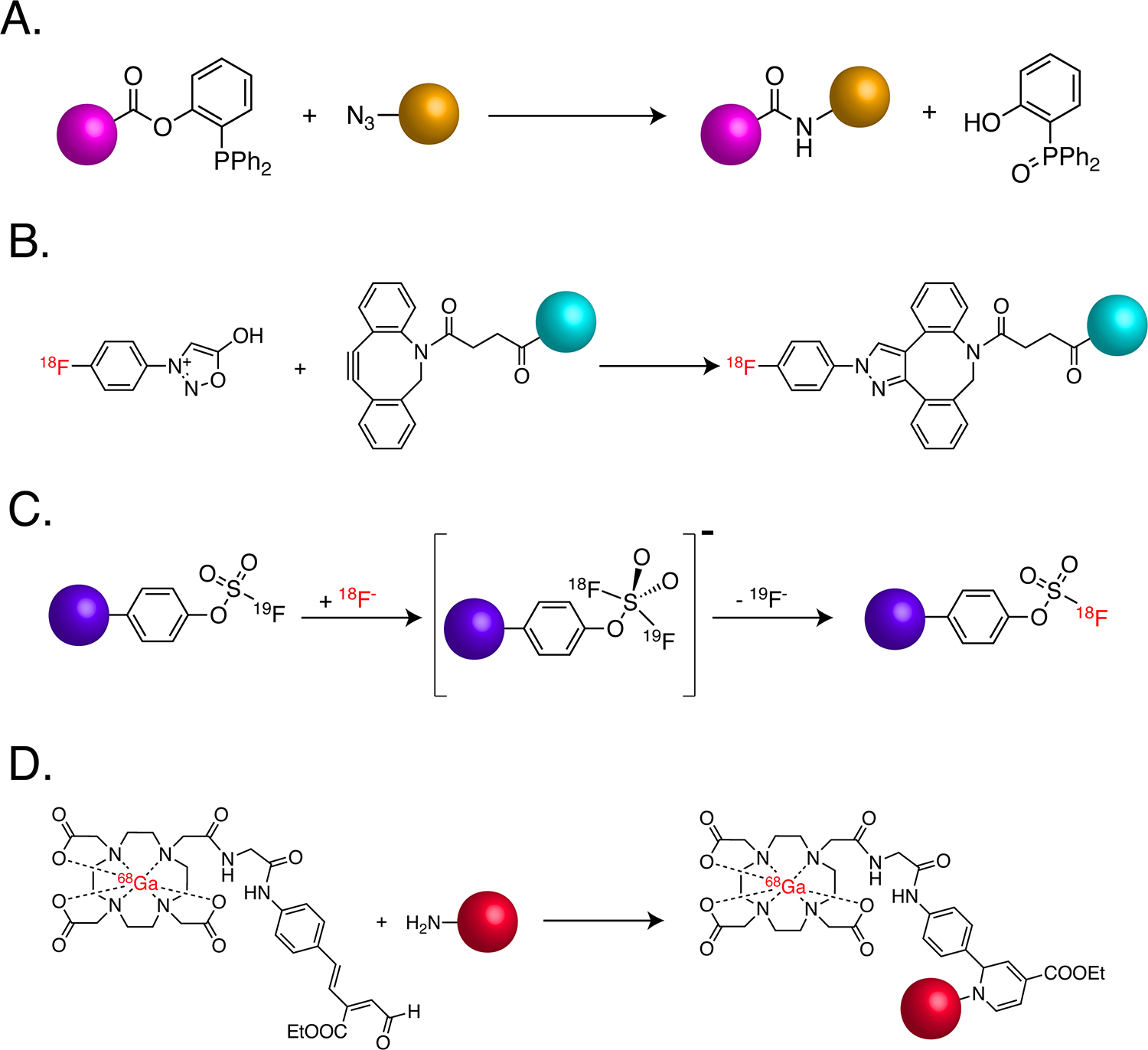
(A) The traceless Staudinger ligation; (B) 18F-labeling via the sydnone-alkyne cycloaddition; (C) 18F-labeling via sulfur-fluoride exchange (SuFEx) chemistry; (D) the bioconjugation of a [68Ga]Ga-DOTA-moiety via the RIKEN reaction. Colored spheres represent cargoes.
Sulfur fluoride exchange (SuFEx) chemistry, first described in the chemical literature almost 100 years ago, has more recently gained traction as a useful click ligation for radiochemistry34. SuFEx chemistry has facilitated the synthesis of 18F-labeled probes via isotope exchange (Figure 6C)35, as demonstrated by the labeling of micrograms of 19F-bearing precursors with 18F in seconds at room temperature to produce high purity, isotopologous radiotracers in molar activities up to 280 GBq/μmol that did not require purification via HPLC. Silicon fluoride acceptor (SiFA) chemistry is a more recent iteration of this isotopic exchange-based approach, and it offers similar benefits. ‘RIKEN’ click chemistry — the rapid and mild 6π-azaelectrocyclization between α, β, γ, ∂-unsaturated aldehydes and primary amines — has also been exploited for the creation of PET imaging agents36. Specifically, this ligation has been used to append a 68Ga-labeled variant of DOTA bearing an α, β, γ, ∂-unsaturated aldehyde to the lysine residue of a cyclic RGD peptide to create a probe for imaging the expression of αvβ3 integrin (Figure 6D).
While each of these ligations has a place in radiopharmaceutical chemistry, the next-generation click reaction that has had the biggest impact on the field is, without question, the inverse electron-demand Diels-Alder (IEDDA) ligation between tetrazine (Tz) and trans-cyclooctene (TCO) (Figure 7A). The IEDDA reaction’s earliest applications in radiochemistry centered on the labeling of peptides and antibodies37,38. Along these lines, one of the more creative uses of the IEDDA ligation was the repurposing of 2-[18F]deoxyfluoroglucose to create 18F-labeled tetrazines for the site-specific labeling of single-domain antibodies bearing C-terminal TCO moieties (Figure 7B)39. Yet even from these earliest days, it was clear that the exceptional speed of the IEDDA ligation (k2 values of more than 100,000 M−1s−1) made it nearly perfect for one application in particular: in vivo pretargeting.
Figure 7.
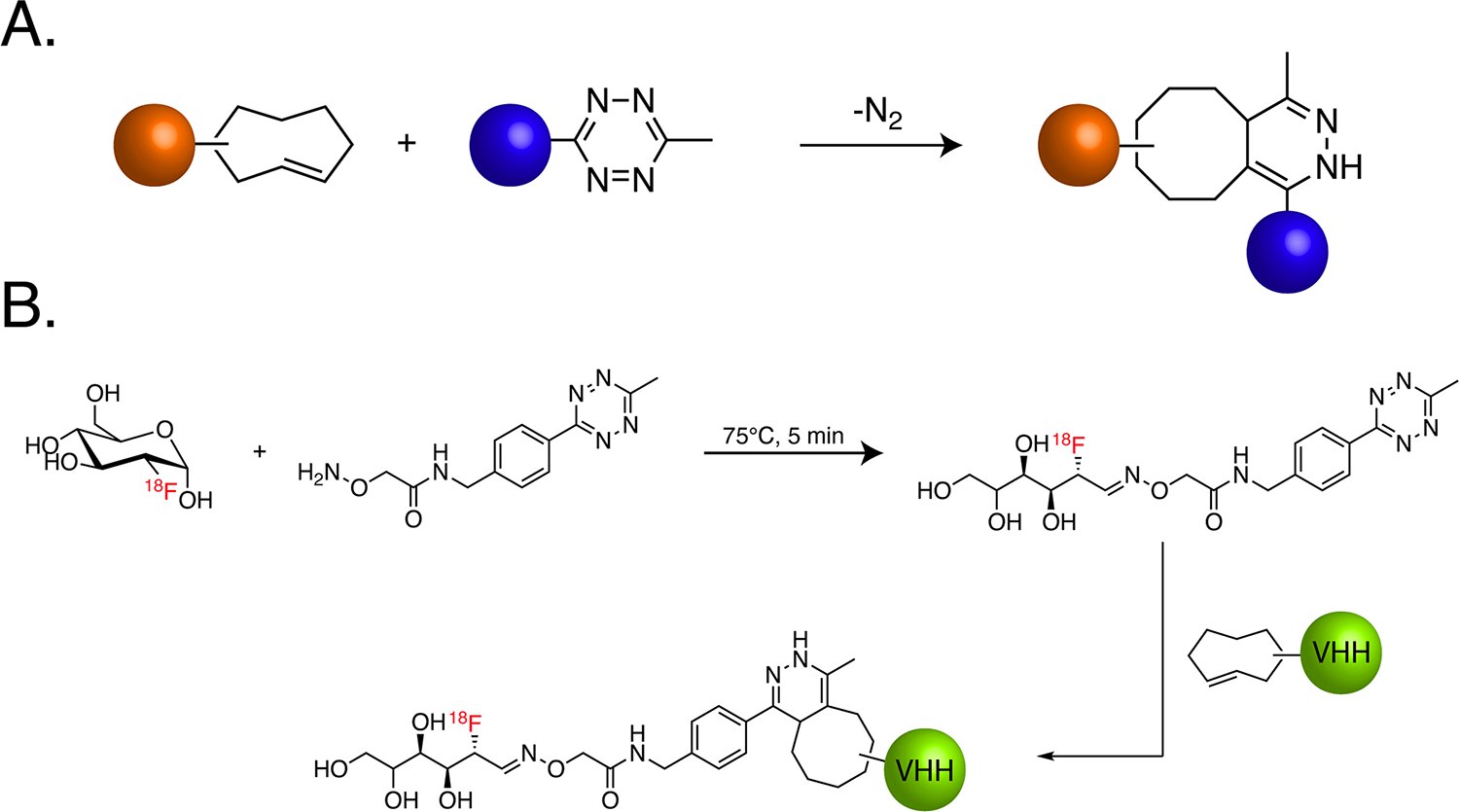
(A) Schematic of the inverse electron-demand Diels-Alder (IEDDA) ligation; (B) schematic of the method devised by Ploegh and coworkers that harnesses [18F]FDG and the IEDDA ligation for the site-specific radiolabeling of antibody fragments. Orange and blue spheres represent cargoes.
Briefly, in vivo pretargeting is an approach to nuclear imaging and radioimmunotherapy designed to circumvent the high radiation doses to healthy tissues associated with traditional, directly labeled radioimmunoconjugates40. In pretargeting, the radionuclide and antibody are decoupled and injected sequentially (antibody first; radioligand second), and a selective ligation facilitates the in vivo combination of the two components at the tumor site (Figure 8A). In the ligation’s first foray into in vivo pretargeting, a colorectal cancer xenograft-bearing mouse was injected with a TCO-modified variant of the TAG72-targeting antibody CC49 (CC49-TCO) and then, 24 hours later, a 111In-labeled dipyridyltetrazine radioligand41. SPECT imaging revealed the remarkable efficacy of this approach, as it produced selective tumor uptake and high tumor-to-background image contrast. Following this initial proof-of-concept, the approach has been expanded to different antibody/antigen systems as well as an array of radionuclides — most notably 68Ga, 18F, and 64Cu42–44. But the most promising work to date has been in the context of pretargeted radioimmunotherapy (PRIT). The earliest work here was performed with 177Lu, but the 225Ac-PRIT investigations best demonstrate the potential of the strategy45. Here, a CA19.9-targeting, TCO-bearing immunoconjugate (5B1-TCO) and an 225Ac-labeled tetrazine radioligand ([225Ac]Ac-DOTA-PEG7-Tz) were used in mice bearing pancreatic ductal adenocarcinoma xenografts (Figure 8B). The strategy delivered high concentrations of the α-emitting radiometal to tumor tissue, ultimately producing higher dose rates to the tumor and better therapeutic indices than directly labeled [225Ac]Ac-DOTA-5B1. Furthermore, it has been demonstrated that sequential injections of two tetrazine radioligands labeled positron- and β-emitting isotopologues of copper (64Cu and 67Cu, respectively) can also be used to create a theranostic approach to PRIT in which the PET images acquired with the former can be used to predict the response to therapy with the latter (Figure 8C)46. Although IEDDA-based pretargeting has not yet been translated to the clinic, a recent pretargeted PET study in companion dogs with osteodestructive lesions suggests that the approach could work in the larger blood volumes of humans47. The first-in-human demonstration of this technology is anticipated in the near future.
Figure 8.
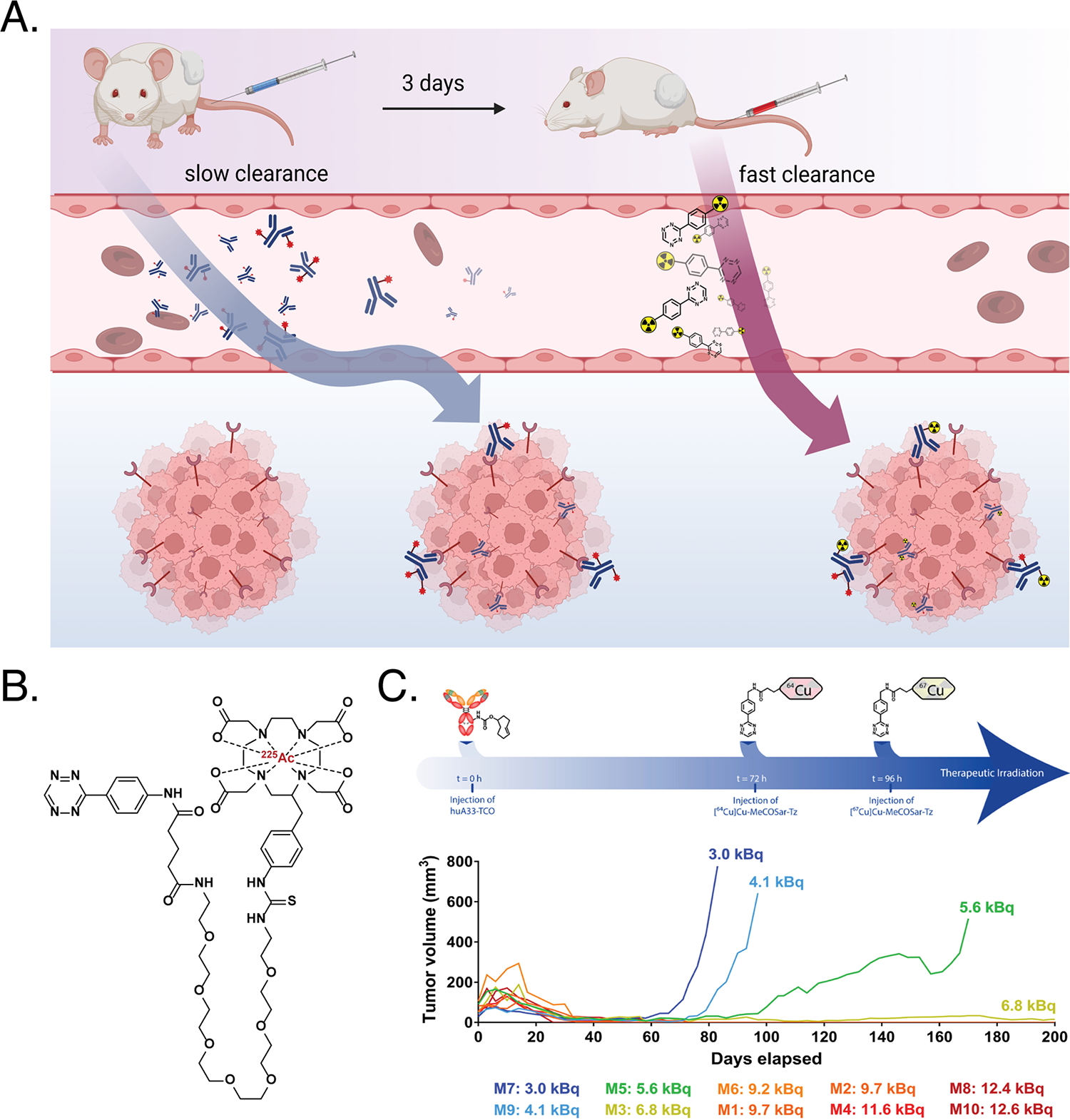
(A) Schematic of in vivo pretargeting based on the IEDDA ligation; (B) structure of [225Ac]Ac-DOTA-PEG7-Tz; (C) timeline of an approach to theranostic pretargeting that leverages a 64Cu-labeled tetrazine for PET imaging and a 67Cu-labeled tetrazine for targeted radionuclide therapy (top) as well as the relationship between the image-derived cumulative activity of the 64Cu-labeled tetrazine in the tumor and the tumor’s response to therapy with the 67Cu-labeled tetrazine (bottom). Figure 8A is an original figure created with BioRender.com. Figure 8C was reprinted under the Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND 4.0) terms of reference 46.
Outlook
Over the past twenty years, click chemistry has transformed radiopharmaceutical chemistry. These reactions have not only enabled the synthesis of radiotracers with heretofore unattainable levels of precision and efficiency but have also emerged as key components of technologies — such as site-specific bioconjugation and in vivo pretargeting — that have the potential to fundamentally alter the way the field operates. Yet we believe the next decade of work at the intersection of click chemistry and radiochemistry may be the most exciting of all. Along these lines, two nascent trends stand out. First, radiochemists and organic chemists are increasingly collaborating on the development of new click transformations (e.g. SuFEx chemistry) with radiochemical applications in mind. Second, and even more remarkably, an ever-expanding array of click-based radiopharmaceuticals are being translated in first-in-human studies, expanding click chemistry’s impact on nuclear medicine from the laboratory to the clinic. As we move forward in the wake of last year’s Nobel Prize, we are eager to see the ways in which the work of the three laureates and those they have inspired continue to impact our field and drive the development of novel radiopharmaceutical technologies for the patients that depend on them.
Acknowledgements:
The authors are grateful to the NIH (R35CA232130 to JSL; R01CA204167 and U01CA221046 to JSL and BMZ; R01CA240963 and R01CA244327 to BMZ) and the Tow Foundation (DB) for their generous support.
Footnotes
Competing Interests: BMZ and JSL hold intellectual property related to the application of click chemistry to radiopharmaceutical chemistry. DB and SMS declare no competing interests.
REFERENCES
- 1.The Nobel Prize in Chemistry 2022., <www.nobelprize.org/prizes/chemistry/2022/summary> (2022).
- 2.Meldal M & Tornoe CW Cu-catalyzed azide-alkyne cycloaddition. Chemical Reviews 108, 2952–3015, doi: 10.1021/cr0783479 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Huisgen R Centenary Lecture - 1,3-Dipolar Cycloadditions. Proc. Chem. Soc, 357–396, doi: 10.1039/ps9610000357 (1961). [DOI] [Google Scholar]
- 4.Jewett JC & Bertozzi CR Cu-free click cycloaddition reactions in chemical biology. Chemical Society Reviews 39, 1272–1279, doi: 10.1039/b901970g (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devaraj NK The future of bioorthogonal chemistry. ACS Central Science 4, 952–959, doi: 10.1021/acscentsci.8b00251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackman ML, Royzen M & Fox JM Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. Journal of the American Chemical Society 130, 13518–13519, doi: 10.1021/ja8053805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeglis BM & Lewis JS Click Here for Better Chemistry. N Engl J Med, doi: 10.1056/NEJMcibr2213596 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer JP, Adumeau P, Lewis JS & Zeglis BM Click chemistry and radiochemistry: The first 10 years. Bioconjugate Chemistry 27, 2791–2807, doi: 10.1021/acs.bioconjchem.6b00561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng D, Zeglis BM, Lewis JS & Anderson CJ The growing impact of bioorthogonal click chemistry on the development of radiopharmaceuticals. Journal of Nuclear Medicine 54, 829–832, doi: 10.2967/jnumed.112.115550 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wangler C, Schirrmacher R, Bartenstein P & Wangler B Click-chemistry reactions in radiopharmaceutical chemistry: fast & easy introduction of radiolabels into biomolecules for in vivo imaging. Current Medicinal Chemistry 17, 1092–1116, doi: 10.2174/092986710790820615 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Mamat C, Ramenda T & Wuest F Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Reviews in Organic Chemistry 6, 21–34, doi: 10.2174/157019309787316148 (2009). [DOI] [Google Scholar]
- 12.Kolb HC, Finn MG & Sharpless KB Click chemistry: diverse chemical function from a few good reactions. Angewandte Chemie International Edition 40, 2004–2021, doi: (2001). [DOI] [PubMed] [Google Scholar]
- 13.Marik J & Sutcliffe JL Click for PET: rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Letters 47, 6681–6684, doi: 10.1016/j.tetlet.2006.06.176 (2006). [DOI] [Google Scholar]
- 14.Glaser M & Arstad E “Click labeling” with 2-[18f]fluoroethylazide for positron emission tomography. Bioconjug Chem 18, 989–993, doi: 10.1021/bc060301j (2007). [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Weng J, Lin J, Ye D & Zhang Y NIR scaffold bearing three handles for biocompatible sequential click installation of multiple functional arms. Journal of the American Chemical Society 142, 2787–2794, doi: 10.1021/jacs.9b10467 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Pisaneschi F et al. Automated, resin-based method to enhance the specific activity of fluorine-18 clicked PET radiotracers. Bioconjugate Chemistry 28, 583–589, doi: 10.1021/acs.bioconjchem.6b00678 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kluba CA & Mindt TL Click-to-Chelate: development of technetium and rhenium-tricarbonyl labeled radiopharmaceuticals. Molecules 18, 3206–3226, doi: 10.3390/molecules18033206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan R et al. A one-pot three-component radiochemical reaction for rapid assembly of 125I-labeled molecular probes. J Am Chem Soc 135, 703–709, doi: 10.1021/ja307926g (2013). [DOI] [PubMed] [Google Scholar]
- 19.Denk C et al. Multifunctional clickable reagents for rapid bioorthogonal astatination and radio-crosslinking. ChemPlusChem 84, 774, doi: 10.1002/cplu.201900250 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Doss M et al. Biodistribution and radiation dosimetry of the integrin marker 18F-RGD-K5 determined from whole-body PET/CT in monkeys and humans. Journal of Nuclear Medicine 53, 787–795, doi: 10.2967/jnumed.111.088955 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubash SR et al. Clinical translation of a click-labeled 18F-octreotate radioligand for imaging neuroendocrine tumors. Journal of Nuclear Medicine 57, 1207–1213, doi: 10.2967/jnumed.115.169532 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Quigley NG et al. PET/CT imaging of head-and-neck and pancreatic cancer in humans by targeting the “Cancer Integrin” alphavbeta6 with Ga-68-Trivehexin. European Journal of Nuclear Medicine and Molecular Imaging 49, 1136–1147, doi: 10.1007/s00259-021-05559-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agard NJ, Prescher JA & Bertozzi CR A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. Journal of the American Chemical Society 126, 15046–15047, doi: 10.1021/ja044996f (2004). [DOI] [PubMed] [Google Scholar]
- 24.Campbell-Verduyn LS et al. Strain-promoted copper-free “click” chemistry for 18F radiolabeling of bombesin. Angewandte Chemie International Edition 50, 11117–11120, doi: 10.1002/anie.201105547 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Zeng D et al. 64Cu Core-labeled nanoparticles with high specific activity via metal-free click chemistry. ACS Nano 6, 5209–5219, doi: 10.1021/nn300974s (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai Z et al. 64Cu-labeled somatostatin analogues conjugated with cross-bridged phosphonate-based chelators via strain-promoted click chemistry for PET imaging: in silico through in vivo studies. Journal of Medicinal Chemistry 57, 6019–6029, doi: 10.1021/jm500416f (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal P & Bertozzi CR Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chemistry 26, 176–192, doi: 10.1021/bc5004982 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y et al. Synthesis of site-specific radiolabeled antibodies for radioimmunotherapy via genetic code expansion. Bioconjugate Chemistry 27, 2460–2468, doi: 10.1021/acs.bioconjchem.6b00412 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Ahn SH et al. Site-specific (89)Zr- and (111)In-radiolabeling and in vivo evaluation of glycan-free antibodies by azide-alkyne cycloaddition with a non-natural amino acid. Bioconjugate Chemistry 31, 1177–1187, doi: 10.1021/acs.bioconjchem.0c00100 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Vivier D et al. The Iifluence of glycans-specific bioconjugation on the FcgammaRI binding and In vivo performance of (89)Zr-DFO-pertuzumab. Theranostics 10, 1746–1757, doi: 10.7150/thno.39089 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarrett SM et al. Lysine-directed site-selective bioconjugation for the creation of radioimmunoconjugates. Bioconjugate Chemistry 33, 1750–1760, doi: 10.1021/acs.bioconjchem.2c00354 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mamat C, Gott M & Steinbach J Recent progress using the Staudinger ligation for radiolabeling applications. Journal of Labelled Compounds and Radiopharmaceuticals 61, 165–178, doi: 10.1002/jlcr.3562 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Narayanam MK et al. Positron emission tomography tracer design of targeted synthetic peptides via (18)F-sydnone alkyne cycloaddition. Bioconjugate Chemistry 32, 2073–2082, doi: 10.1021/acs.bioconjchem.1c00379 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Steinkopf W Über aromatische sulfofluoride. J Prakt Chem 117, 1–82 (1927). [Google Scholar]
- 35.Zheng Q et al. Sulfur [(18)F]fluoride exchange click chemistry enabled ultrafast late-stage radiosynthesis. Journal of the American Chemical Society 143, 3753–3763, doi: 10.1021/jacs.0c09306 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamoto Y et al. Expanding the applicability of the metal labeling of biomolecules by the RIKEN click reaction: A case study with gallium-68 positron emission tomography. ChemBioChem 19, 2055–2060, doi: 10.1002/cbic.201800335 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Zeglis BM et al. Modular strategy for the construction of radiometalated antibodies for positron emission tomography based on inverse electron demand Diels-Alder click chemistry. Bioconjugate Chemistry 22, 2048–2059, doi: 10.1021/bc200288d (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z et al. Tetrazine-trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chemical Communications 46, 8043–8045, doi: 10.1039/c0cc03078c (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rashidian M et al. The use of (18)F-2-fluorodeoxyglucose (FDG) to label antibody fragments for immuno-PET of pancreatic cancer. ACS Central Science 1, 142–147, doi: 10.1021/acscentsci.5b00121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patra M, Zarschler K, Pietzsch HJ, Stephan H & Gasser G New insights into the pretargeting approach to image and treat tumours. Chemical Society Reviews 45, 6415–6431, doi: 10.1039/c5cs00784d (2016). [DOI] [PubMed] [Google Scholar]
- 41.Rossin R et al. In vivo chemistry for pretargeted tumor imaging in live mice. Angewandte Chemie International Edition 49, 3375–3378, doi: 10.1002/anie.200906294 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Edem PE et al. Evaluation of a (68)Ga-labeled DOTA-tetrazine as a PET alternative to (111)In-SPECT pretargeted imaging. Molecules 25, doi: 10.3390/molecules25030463 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer JP et al. (18)F-Based pretargeted PET imaging based on bioorthogonal Diels-Alder click chemistry. Bioconjugate Chemistry 27, 298–301, doi: 10.1021/acs.bioconjchem.5b00504 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cook BE et al. Pretargeted PET imaging using a site-specifically labeled immunoconjugate. Bioconjugate Chemistry 27, 1789–1795, doi: 10.1021/acs.bioconjchem.6b00235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poty S et al. Leveraging bioorthogonal click chemistry to improve (225)Ac-radioimmunotherapy of pancreatic ductal adenocarcinoma. Clinical Cancer Research 25, 868–880, doi: 10.1158/1078-0432.CCR-18-1650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keinanen O et al. Harnessing (64)Cu/(67)Cu for a theranostic approach to pretargeted radioimmunotherapy. Proceedings of the National Academy Sciences of the United States of America 117, 28316–28327, doi: 10.1073/pnas.2009960117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maitz CA et al. Pretargeted PET of osteodestructive lesions in dogs. Molecular Pharmaceutics 19, 3153–3162, doi: 10.1021/acs.molpharmaceut.2c00220 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]